

Abstract
Regulation of GABA release is crucial for normal brain functioning, and GABAA-mediated IPSCs are strongly influenced by repetitive stimulation and neuromodulation. However, GABA exocytosis has not been examined directly in organized tissue. Important issues remain outside the realm of electrophysiological techniques or are complicated by postsynaptic factors. For example, it is not known whether all presynaptic modulators affect release from all boutons in the same way, or whether modulator effects depend on the presence of certain types of voltage-gated calcium channels (VGCCs). To address such issues, we used confocal imaging and styryl dyes to monitor exocytosis from identified GABAergic boutons in organotypic hippocampal slice cultures. Repetitively evoked IPSCs declined more rapidly and completely than exocytosis, suggesting that depletion of filled vesicles cannot fully account for IPSC depression and underscoring the usefulness of directly imaging exocytosis. Stimulation at 10 Hz produced a transient facilitation of exocytosis that was dependent on L-type VGCCs. Using specific toxins, we found that release mediated via N-type and P-type VGCCs had similar properties. Neither baclofen nor a cannabinoid receptor agonist, CP55940, affected all boutons uniformly; they slowed release from some but completely prevented detectable release from others. Increasing stimulus frequency overcame this blockade of release. However, baclofen and CP55940 did not act identically, because only baclofen reduced facilitation and affected bouton releasing via P/Q-type VGCCs. Direct observation thus revealed novel features of GABAergic exocytosis and its regulation that would have been difficult or impossible to detect electrophysiologically. These features advance the understanding of the regulation of synapses and networks by presynaptic inhibition.
Keywords: baclofen, cannabinoids, GABA, hippocampus, inhibition, FM1-43
Introduction
GABAAergic inhibition is well controlled in the normal brain, and its dysregulation can lead to pathophysiological phenomena. GABA release is regulated by ligands that mediate presynaptic inhibition (Thompson, 1994) and by retrograde signaling such as depolarization-induced suppression of inhibition (DSI) (for review, see Alger, 2002). Nevertheless, many fundamental details remain unclear: does a given ligand affect all susceptible boutons in the same way? Do different presynaptic inhibitory ligands inhibit release in the same way? Do the presynaptic inhibitory effects depend on the types of voltage-gated calcium channels (VGCCs) on the boutons? Such questions are difficult or impossible to address with electrophysiological methods, because IPSCs are generally the product of release from many boutons. Inhibitory interneurons and their axons are often diffusely scattered, and their small and inaccessible synaptic boutons are dispersed over the target cell somata. Significant postsynaptic factors, such as GABAA receptor desensitization (Brenowitz and Trussell, 2001), driving force (McCarren and Alger, 1985; Huguenard and Alger, 1986; Thompson and Gahwiler, 1989), and depolarizing GABA responses (Alger and Nicoll, 1979; Michelson and Wong, 1991; Kaila, 1994; Staley et al., 1995) complicate the indirect analysis of release by measurement of IPSCs.
GABA release differs from the release of other neurotransmitters. For example, glutamatergic and GABAergic terminals do not have the same complement of voltage-dependent Ca2+ channels (Wheeler et al., 1994; Poncer et al., 1997), baclofen does not inhibit the two types of terminals in the same way (Scanziani et al., 1992; Thompson et al., 1992), synapsin III plays different roles in glutamate and GABA terminals (Feng et al., 2002), and whereas glutamate release is typically facilitated by brief repetitive stimulation, GABA release is generally depressed. Hence, it is not always safe to extrapolate conclusions derived from one class of terminals to the other.
Therefore, to study GABA exocytosis, we adapted the technique of styryl dye-destaining (Betz and Bewick, 1992; Betz et al., 1992) to GABAergic boutons in organotypic slice cultures. Although used to identify properties of GABAergic boutons in dissociated cell culture (Kannenberg et al., 1999; Irving et al., 2000; Akaike et al., 2002; Kirischuk et al., 2002; Timmermann et al., 2002), FM1-43 (or its equivalent, Synaptogreen-C4, which we used here) has not been used in the study of regulation of GABA release. Styryl dye loading and unloading, confocal microscopy, and pharmacological tools made possible the isolation and identification of single GABAergic boutons in the rat hippocampal CA1 stratum pyramidale region. A newly developed mouse strain (Erdélyi et al., 2002) expressing an enhanced green fluorescent protein (eGFP) chimera of a synthetic enzyme for GABA, glutamic acid decarboxylase 65 (GAD65), confirmed that the boutons in rat slices were GABAergic. We determined that the stained puncta represented functioning nerve terminals by monitoring the activity-dependent loss of Synaptogreen-C4, its sensitivity to Cd2+, and stimulation frequency. We used quantitative analyses of destaining to test several hypotheses for the regulation of exocytosis: (1) that ligands of presynaptic GABAB and CB1 receptors would inhibit release from all boutons equally, and their effects would be indistinguishable; (2) that the efficacy of presynaptic inhibition of GABA release would be frequency-dependent, declining with higher stimulation frequencies; and (3) that the particular VGCCs on the boutons would influence the capability of a given presynaptic receptor to affect release, and the frequency-dependent decline in efficacy of presynaptic inhibition. The data confirmed some predictions and showed that GABA exocytosis resembles glutamate exocytosis but is not identical. The techniques used will be applicable to GABAergic synapses in other brain regions.
Materials and Methods
Preparation of organotypic slice cultures. All experiments were performed on organotypic hippocampal slice cultures (Gahwiler et al., 1998). Hippocampi were dissected from 5- or 6-d-old CO2-anesthetized rat or GAD65-eGFP mouse pups and cut into 375 μm thick transverse slices using a McIlwain tissue chopper (Brinkmann Instruments). Slices were attached to polylysine-coated glass coverslips in 20 μl of chicken plasma coagulated with thrombin. Coverslips were placed into culture tubes with 750 μl of serum-containing media and incubated in a roller-drum at 36°C. Slices were X-irradiated at the time of explantation and treated overnight with antimitotics to reduce the proliferation of glial cells. Slice cultures were maintained in vitro for >14 d before performing experiments to allow for synaptic maturation.
Electrophysiology. Cultures were perfused at ∼1 ml/min with control saline containing the following (in mm): 137 NaCl, 2.8 KCl, 2.5 CaCl2, 2.5 MgCl2, 23.2 NaHCO3, 0.4 NaH2PO4, pH to 7.2 with HEPES, 0.05 adenosine (except as noted), and 5.6 glucose at room temperature (20-22°C). Most cultures were pretreated with a 250 nm concentration of either ω-agatoxin IVA (agatoxin) or conotoxin GVIA (conotoxin) (Sigma, St. Louis, MO) for 1-3 hr before an experiment. Extracellular stimuli (100 μsec in duration; 150-250 μA) were delivered near the border between CA1 s. oriens and s. pyramidale using a concentric bipolar electrode lowered ∼25-50 μm into the slices, which are 50-100 μm thick. Postsynaptic responses were recorded using either whole-cell or extracellular recording techniques with an Axoclamp 2B amplifier (Axon Instruments, Foster City, CA) low-pass filtered at 2 kHz and digitized at 10 kHz. IPSCs were recorded with patch pipettes (5-7 MΩ) filled with the following (in mm): 90 CsCH3SO4, 50 CsCl, 1 MgCl2, 10 HEPES, 0.2 BAPTA, 2 Mg-ATP, and 5 QX-314, pH 7.2. Whole-cell recordings, during which the access resistance exceeded 30 MΩ, were discarded. Field EPSPs were recorded in s. radiatum using a patch pipette filled with extracellular saline. In some experiments, GABAB responses were blocked with CGP55485 (Tocris Cookson, Ballwin, MO). Except as noted, all other reagents were from Sigma.
Dye loading and imaging. Slices were placed in a chamber and perfused with control saline for 5 min. The perfusion was then switched to control saline containing a 10 μm concentration of either Synaptogreen-C4 or Synaptored-C2 (Biotium) for 5-7 min. The loading stimulation (1800 stimuli at 10-Hz) began after 1-2 min of dye perfusion and ended 1 min before wash. Dye was washed from the chamber with control saline containing 150 μm ADVASEP-7 (Biotium) for 15-20 min, after which the unloading stimulation protocol was applied. ADVASEP-7 removes Synaptogreen and reduces background fluorescence, leaving the punctate staining indicative of synaptic boutons. Because boutons take up the dye only through the recapture of vesicles after neurotransmitter release, boutons that do not release in the presence of the dye are not labeled.
For experiments on GABAB and CB1 receptors, the corresponding agonist or agonist-antagonist mixture was included in the ADVASEP-7-containing wash. For experiments on L-type Ca2+ channels, nifedipine was added to the wash solution. All confocal images were acquired with a Zeiss (Thornwood, NY) LSM 510 microscope using a 40× 0.8 numerical aperture water-immersion objective. Synaptogreen and eGFP were excited with an argon laser at 488 nm and imaged through a 505 low-pass filter. Synaptored was excited by a helium-neon laser at 543 nm and imaged through a 560 LP filter. For destaining experiments, images were taken every 15 sec. After collecting four baseline images, the destaining protocol (3 min of extracellular stimulation; 12 images) was initiated. After the end of the stimulation, four additional images were collected (i.e., 20 total).
GAD65-eGFP mice. A genomic clone, containing a 5.5 kb upstream region and the first six exons of the mouse GAD65 gene, was isolated from a mouse λ phage genomic library, and the eGFP marker gene without its own translation start site was fused in frame to the third exon of the GAD65 gene (Erdélyi et al., 2002). Transgenic mice were derived by standard pronuclear injection of CBA/C57B16F2 fertilized eggs. For this study, transgenic mice on the CBA/C57B16 background were used from line GAD65 3e/gfp5.5#30. In this line, a 6.5 kb segment of the GAD65 gene, which includes 5.5 kb of the five upstream regions, the first two exons, and a portion of the third exon and the introns in between, drives the expression of eGFP almost exclusively to the GABAergic neurons in many brain regions, including the neocortex and hippocampus.
Data analysis. Images were analyzed off-line using Metamorph image analysis software (Universal Imaging Corporation, West Chester, PA). Setable software parameters permit the computer to identify labeled boutons by size (0.5-2.2 μm2), shape (“shape factor” range, 0.55-1.0; i.e., approximately round), and fluorescence intensity (≥3 SDs above background). The program generates a region of interest around each identified bouton in the first image, and the fluorescence within each region was measured automatically throughout the series of 20 images. Data from experiments in which there was a shifting in the x, y, or z planes were discarded.
The total fluorescence lost from a given bouton was calculated as [(1 - Last3/First4) × 100)], where Last3 is the mean of its last three values, and First4 is the mean of its first four values. If <25% of the fluorescence was lost, or if τdestain was >300 sec, the bouton was classified as “nondestaining”. The kinetics of fluorescence loss was estimated as τdestain by fitting single exponentials, fit to the data between First4 and Last3 using Origin software (Microcal Software, Northampton, MA). The τdestain differences between experimental groups are robust, but we have not ascribed any mechanistic significance to the exponentials themselves and have not attempted multiexponential fits. The fractional release parameter, f, was calculated from ln(F1/F2)/n (Isaacson and Hille, 1997), where ln is the natural logarithm, F1 and F2 are the fluorescence intensities at times 1 and 2, respectively, and n is the number of stimuli delivered during that period. All data are presented as mean ± SEM. Data sets were compared with either ANOVA followed by Tukey-Kramer post hoc analysis or Student's t test using SYSTAT software (Systat, Evanston, IL). Distributions were compared using the Komolgorov-Smirnov (KS) test. Significance levels were p < 0.05, for ANOVAs and t tests, and p < 0.005 for KS tests.
Results
Isolation of GABAergic boutons
The study of GABA release from single boutons in brain slices requires avoiding non-GABAergic boutons. Therefore, we used rat hippocampal organotypic slice cultures (Gahwiler et al., 1998), because they have excellent optical characteristics and lack extrinsic non-GABAergic afferents. Second, we bath-applied adenosine, because it blocks glutamate (Lambert and Teyler, 1991; Scanziani et al., 1992; Thompson et al., 1992), although not GABA release in the hippocampus (Lambert and Teyler, 1991; Thompson et al., 1992). Third, we studied boutons in s. pyramidale, where glutamatergic synapses are rare and GABAergic boutons are numerous. Fourth, we selected boutons on the basis of morphological data from identified hippocampal interneuron boutons (Miles et al., 1996). Last, we compared labeled boutons in rats with identified GABAergic boutons in GAD65-eGFP mice. Our dye-loading protocol and basic data are shown in Figure 1. Slices were stimulated in the presence of Synaptogreen and, after ∼20 min of washing with ADVASEP-7-containing control saline (Kay et al., 1999), were then stimulated again to induce dye release (Fig. 1A).
Figure 1.
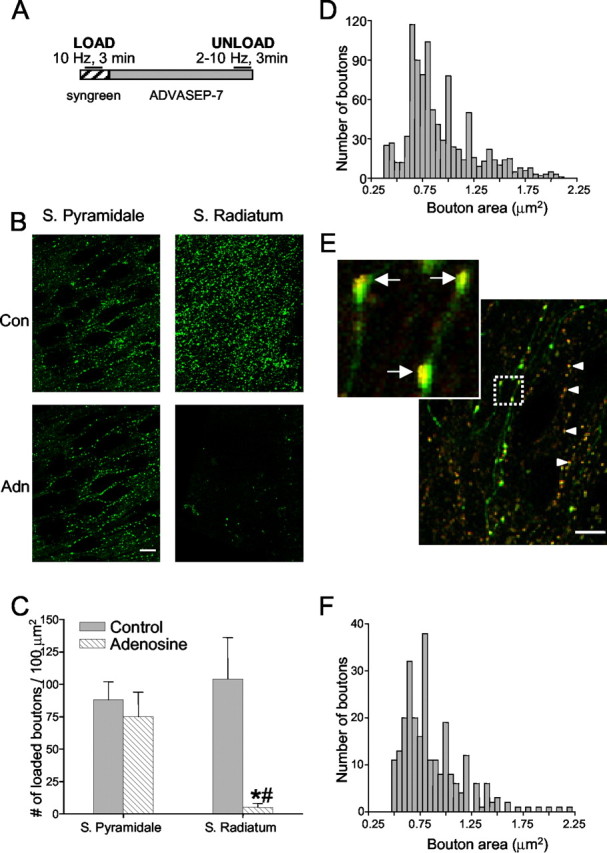
Loading of GABAergic boutons with styryl dye. A, Protocol for loading organotypic hippocampal slices with Synaptogreen-C4 dye. The dye was present in the slice for 2 min before field stimulation of 10 Hz for 3 min was given and for 2 min after stimulation to allow for uptake. The dye was removed and the chelator ADVASEP-7 was perfused for ∼20 min before an unloading stimulus train was given. B, Boutons loaded in s. pyamidale and s. radiatum in the absence and presence of adenosine. Scale bar, 20 μm. C, Group data showing numbers of labeled boutons present in s. radiatum and s. pyramidale in the absence (control) or presence of 50 μm adenosine. There was a highly significant reduction in the numbers of boutons in s. radiatum and no change in s. pyramidale. *, Significantly different from control; #, significantly different from s. pyramidale. D, Distribution of the sizes (areas) of 834 labeled boutons from nine rat slices in s. pyramidale. E, Overlay image of Synaptored-loaded boutons in GAD65-eGFP mouse culture. Arrowheads in E point to Synaptored-labeled boutons in a GAD65-eGFP mouse culture. Arrows in inset indicate yellow clusters of Synaptored-filled vesicles with eGFP-labeled boutons. Scale bar, 10 μm. F, Distribution of Synaptored-loaded bouton sizes of 255 boutons from three slices from GAD65-eGFP mice.
We confirmed that adenosine prevented labeling of glutamatergic boutons first by comparing the punctate labeling in s. radiatum caused by stimulation in the absence or presence of 50 μm adenosine (Fig. 1B,C). Adenosine reduced the number of labeled puncta per 100 μm2 patch in s. radiatum (optical thickness, ∼2 μm) by >90% (control, 104 ± 33 puncta; adenosine, 5 ± 3; n = 3 slices; p < 0.05) (Fig. 1C). Adenosine did not significantly reduce the number of puncta in s. pyramidale (control, 88 ± 14; adenosine, 75 ± 19; n = 3 slices) (Fig. 1B,C). We also confirmed (Dunwiddie and Masino, 2001) that the glutamatergic field EPSP recorded in s. radiatum was abolished by adenosine and did not recover, even if the stimulation frequency was raised to 10 Hz for 90 sec (data not shown). Besides adenosine, in most experiments, we also used a VGCC toxin, agatoxin or conotoxin, to isolate different sets of GABAergic boutons (Poncer et al., 1997; Wilson et al., 2001). Because glutamate release is strongly inhibited by both agatoxin and conotoxin (Luebke et al., 1993; Wheeler et al., 1994; Poncer et al., 1997), the combination of adenosine plus either agatoxin or conotoxin ensured that glutamatergic boutons did not take up dye.
Figure 1D shows morphometric data on 834 boutons (from nine slices) that met our selection criteria. The mean area of the loaded boutons (0.86 ± 0.05 μm2) is comparable with, but slightly smaller than, the reported mean of GABAergic synaptic boutons measured with electron microscopy (EM) (Miles et al., 1996) as expected, because the fluorescent puncta actually represent the clusters of labeled synaptic vesicles within the boutons (Sankaranarayanan et al., 2003), whereas the EM measurements are of the outer margins of the bouton membranes.
As a final test, we compared Synaptogreen-labeled boutons from rat slices with Synaptored-labeled boutons from transgenic mice expressing eGFP under the control of the GAD65 promoter (GAD65-eGFP mice) (Erdélyi et al., 2002). (Synaptored, a red-emitting styryl dye, is used instead of Synaptogreen in eGFP-expressing tissue so that the dye can be distinguished from eGFP.) Figure 1E shows an example. There were on average 28 ± 5 (mean ± SEM) eGFP-positive cells in the CA1 regions of single slices (n = 7 slices from two mice). There was no systematic difference in the location of the positive cell somata: s. oriens, s. pyramidale, and s. radiatum each had ∼30% of the total. Boutons in rat (Fig. 1D) and in GAD65-eGFP mice cultures (Fig. 1F) were the same size. The overlays of the Synaptored staining with that of eGFP labeling occasionally revealed distinct yellow clusters within green varicosities (Fig. 1E, inset), as expected if red-labeled vesicles are present within green-labeled boutons. The boutons from these mice thus provided an ideal independent measure of the size, location, and distribution of GABAergic boutons. Evidence that the puncta in rats are functioning nerve boutons is described below.
Exocytosis
Figure 2A shows images of a rat slice culture taken before, during, and after destaining. Before stimulation, there was little time-dependent decline in fluorescence. The onset of 5 Hz stimulation caused a rapid loss of fluorescence to a steady state in most boutons (Fig. 2B,C) (supplemental Fig. 1; available at www.jneurosci.org). The stimulation was continued for 3 min, and the mean time constant of destaining (τdestain) defined by the group data was 68 ± 6 sec (n = 3 slices), which is similar to the destaining rate of glutamate boutons (Zakharenko et al., 2001).
Figure 2.

Repetitive stimulation causes destaining of boutons. A, Images taken before stimulation, 15 sec after beginning stimulation, and 1 min after the end of stimulation. Arrowheads point to Synaptogreen-labeled boutons in a rat culture. B, Destaining profiles for 50 boutons from a single slice caused by 5 Hz field stimulation. Each trace is from a single bouton imaged at successive 15 sec intervals throughout the trial (nondestaining boutons are indicated in red). C, Mean data from the individual traces in B.
If destaining represents vesicular release, it should be frequency-dependent. The mean τdestain decreased as a function of stimulation frequency of 2, 5, and 10 Hz in 3 min long trains (Fig. 3A) and was well described by the same relationship between destaining rate and stimulation frequency that described glutamate release (Zakharenko et al., 2001). Increasing stimulation frequency also decreased the total amount of fluorescence remaining at the end of the trains (2 Hz, 44 ± 4%; 5 Hz, 33 ± 3%; 10 Hz, 25 ± 3%) (Fig. 3C). Photo-bleaching caused by repeated imaging of dye-loaded boutons decreases fluorescence but is unrelated to vesicular dye release. We used low-excitation light levels and obtained images every 15 sec only to reduce photo-bleaching to ∼10% of initial fluorescence (Fig. 2A). We did not correct for photo-bleaching; however, its effect is constant across all conditions and should not affect our conclusions.
Figure 3.

Frequency dependence of destaining. A, Group data showing destaining induced by different field stimulation frequencies. The numbers of boutons in each of the 14 slices (2 Hz, 5; 5 Hz, 3; 10 Hz, 6) represented ranged from 33 to 147 (mean, 78 ± 10; different slices per frequency; no significant difference in numbers of loaded boutons between slices). B, The mean time constant of destaining is frequency dependent. The symbols represent the mean time constants of decay at the indicated frequencies obtained from the best fits of exponential curves to the data in A. The solid line is the fit of the data by an empirical equation, analogous to that used by Zakharenko et al. (2001), to illustrate the kinetics of destaining of boutons in CA1 s. radiatum: τdestain = τ(min) + q/f, where τdestain is the time constant of destaining, τ(min) is the minimal time constant at 10 Hz, f is the frequency of stimulation, and q is a fit constant. C, The steady-state level of fluorescence at the end of stimulation trains declines as the frequency of stimulation during the trains goes up. The numbers of nondestaining boutons remained constant across different frequencies of stimulation. D, Destaining data from each bouton were fit by single exponentials. The bar graph plots the distribution of the time constants for the indicated numbers of boutons that made up the groups in A (total, n = 797 boutons). Note the shift in the population means (arrows) to faster time constants as the frequency increases. E, Cd2+ prevents destaining. The filled circles represent fluorescence measurements from 154 boutons in three slices that were loaded, washed, and imaged normally but received no unloading stimulation. The slight decrease in fluorescence by the end of the trial is the result of photo-bleaching. Data represented by open circles were obtained from measurements of 159 boutons in three slices loaded in normal solution, washed for 20 min in ADVASEP-7 plus 100 μm Cd2+, and then stimulated at 5 Hz (black bar) still in the presence of Cd2+. Cd2+ was then washed from the chamber for ≥20 min, and the slices were stimulated again (open diamonds). Cd2+ essentially prevented destaining (cf. open circles and diamonds); the data in Cd2+ are not significantly different from the photo-bleaching data.
The behavior of individual boutons is hidden in the group data, and yet they might not all destain in the same manner. After determining τdestain for individual boutons, we first identified a group of boutons that either lost <25% of their initial fluorescence during stimulation (∼20% of the population) or had τdestain values >300 sec (∼5% of the population). A loss of <25% was just larger than the loss caused by photo-bleaching, and 300 sec was >3 SDs slower than the mean τdestain and, hence, seemed likely to be nonphysiological. We considered boutons in these groups (∼25% of the total) to be nondestaining. As stimulus frequency was increased, there was no significant difference in the percentage of nondestaining boutons detected (Fig. 3C) (2 Hz, 26 ± 7%; 5 Hz, 22 ± 6%; 10 Hz, 25 ± 9%), and the distribution of τdestain values shifted toward faster rates of release (Fig. 3D).
If destaining represents neurotransmitter release, it should depend on Ca2+ influx into the boutons. Indeed, 50 μm Cd2+ prevented activity-dependent destaining (Fig. 3E). The percentage of nondestaining boutons greatly increased (control, 25 ± 9%; Cd2+, 85 ± 6%; data not shown) and became indistinguishable from the percentage that did not lose fluorescence in the absence of stimulation (97 ± 2%). Removal of Cd2+ significantly decreased the percentage of nondestaining boutons (to 30 ± 10%) and restored the activity-dependent destaining (Fig. 3E).
Evoked IPSCs decline more rapidly and completely than boutons destain
Figure 4A shows whole-cell IPSCs (adenosine present) evoked by 2 or 10 Hz stimulation (150 IPSCs in each train). IPSCs declined more rapidly than boutons destained. In Figure 4B (2 Hz), images were taken every 15 sec, and imaging a frame required 1 sec. Thus, to make this comparison, we averaged the IPSCs across a 1 sec interval of stimulation every 15 sec. Figure 4C shows that the same disparity is seen with 10 Hz stimulation; after 150 stimuli, the bouton fluorescence was still ∼75% of control levels, whereas ISPCs were reduced to ∼10% of control. In control experiments, loading of terminals with dye did not affect baseline IPSCs (108 ± 5% of control) and did not prevent the rapid rundown of IPSCs with 2 or 10 Hz stimulation (data not shown), indicating that the dye itself did not interfere with release. The results emphasize the difficulties with inferring details of presynaptic phenomena from postsynaptic measurements. Postsynaptic factors can, in part, account for the discrepancy between destaining and eIPSCs, although there are other possibilities (see Discussion). Nevertheless, we could directly investigate the regulation of exocytosis.
Figure 4.
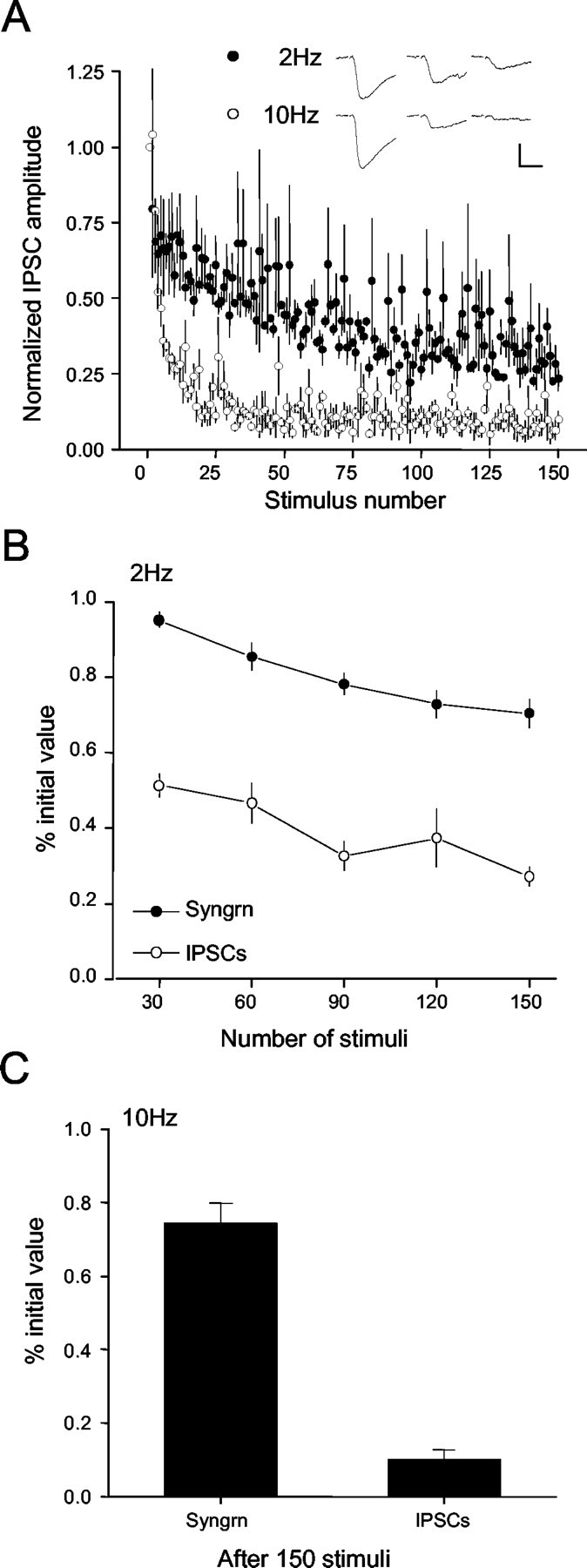
Comparison of repetitive stimulation on dye release and monosynaptic evoked IPSCs at 2 and 10 Hz. A, Decrease in IPSC amplitudes (from 3 cells, 3 slices) caused by 150 stimuli delivered at 2 Hz (•) or 10 Hz (○). Inset, Representative IPSCs, means of 3 each, at the indicated times. Calibration: 200 pA, 25 msec. B, Comparison of the destaining of Synaptogreen-loaded boutons (•) and the depression of IPSCs (○) during 2 Hz stimulation. Fluorescence measurements were made after the indicated number of stimuli. For IPSC depression, the last two IPSCs in the indicated period were averaged (see Results). C, Comparison of the destaining of Synaptogreen-loaded boutons and IPSC depression after 15 sec of 10 Hz stimulation.
GABA release via either N-type or P/Q-type VGCCs
VGCCs are keys to the regulation of transmitter release. Therefore, we asked whether the nature of the VGCCs coupled to GABA release affected destaining rates and pretreated slices with either agatoxin or conotoxin. Electrophysiological studies have implied that P/Q-type VGCCs predominantly mediate GABA release from some interneurons and N-type channels from others (Poncer et al., 1997; Wilson et al., 2001). The segregation was fairly strict, although VGCC mixtures did exist on some cells. We found that the number of loaded boutons in s. pyramidale of agatoxin-treated cultures was 65 ± 7% (n = 9 slices; mean count, 57 boutons per 100 μm2 of 2 μm-thick optical section) of the number found in toxin-untreated cultures, whereas conotoxin-treated cultures had only 33 ± 7% (n = 7 slices; mean count, 29 boutons per 100 μm2 of optical section) as many loaded boutons as did untreated cultures. In three cultures, we applied both agatoxin and conotoxin to dye-loaded slices and observed that stimulation then produced no loss in fluorescence above photo-bleaching levels (89 ± 2% nondestaining boutons; 3 slices). Thus, although the SEs in the measurements could mask a 10-15% contribution of boutons that are released through both N-type and P/Q-type terminals, it appears that the great majority of GABAergic boutons in s. pyramidale depend primarily or exclusively on Ca2+ influx through N-type channels for exocytosis, and the remainder depend on P/Q-type channels. Having isolated two main classes of GABAergic boutons, we compared their destaining rates but found no significant difference between them (supplemental Fig. 1), implying that GABA exocytosis was effectively coupled to both N-type and P/Q-type VGCCs.
Frequency-dependent facilitation of GABA release and the role of L-type Ca2+ channels
Although destaining obviously proceeded more rapidly with 10 Hz rather than 2 Hz stimulation (Fig. 3), it was not clear whether this represented a genuine facilitation of release. To address this issue, we calculated the parameter f, the fractional destaining rate per action potential (see Materials and Methods) in the presence of agatoxin, and plotted the results in Figure 5. Over the first 150 stimuli at the frequencies tested, f was essentially constant (finitial: 2 Hz, 0.26 ± 0.02%; 5 Hz, 0.20 ± 0.01%; 10 Hz, 0.17 ± 0.02%) and agreed with f reported for glutamate release (Ryan and Smith, 1995; Isaacson and Hille, 1997). During the remainder of the 3 min stimulation train, f remained constant with 2 Hz stimulation but transiently increased and then decreased with 10 Hz stimulation. Because the stimulation trains were the same length but had variable frequency, the number of stimuli in each train also varied. To compare f after equal numbers of stimuli, we replotted the data after 150 and 300 stimuli for five slices stimulated at 2 Hz and six slices at 10 Hz (292 and 403 boutons, respectively). The results from two representative slices are illustrated in Figure 5A2. Three hundred stimuli delivered at 10 Hz increased f to 0.52 ± 0.03% but did not affect f during 2 Hz stimulation (0.21 ± 0.02%). The elevation in f implies that GABA release was transiently facilitated.
Figure 5.
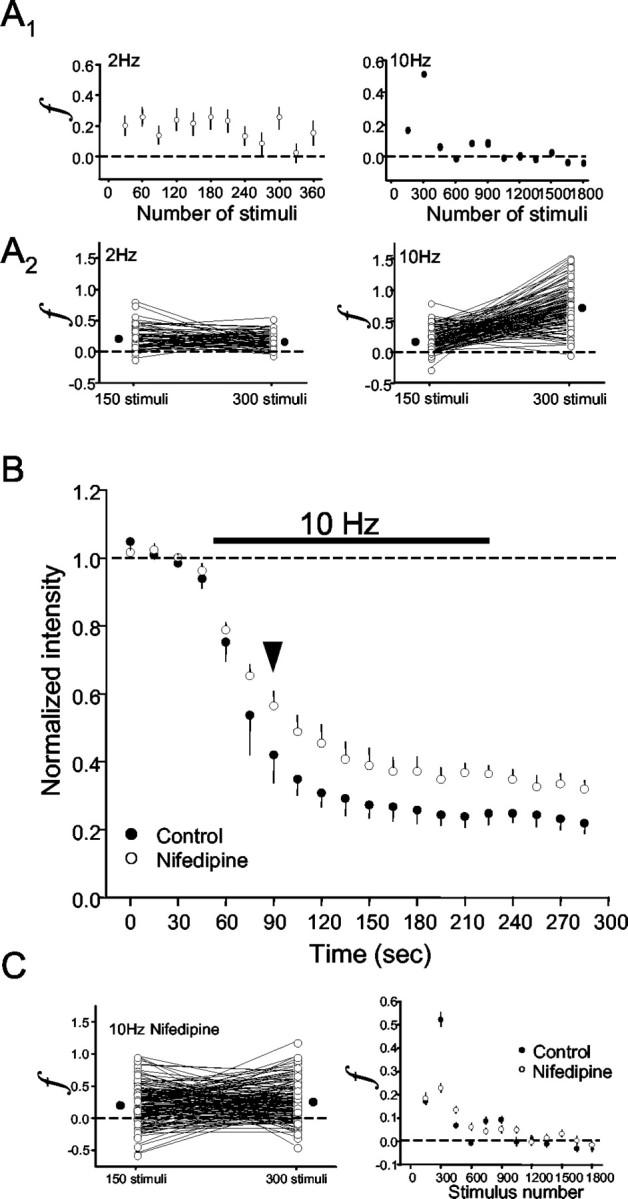
Fractional destaining of GABAergic boutons. The fractional destaining rate, f (see Materials and Methods), is an estimate of the average amount of release induced by an action potential in a given interval. A, Top graphs show group data for f as a function of stimulus number. f remained constant throughout 2 Hz stimulation (left) but transiently increased with 10 Hz stimulation (right). The stimulus trains in A1 started at t = 0; thus, the first f value that could be obtained is after 15 sec of stimulation. To compare f after equal numbers of stimuli, f was calculated after 150 and 300 stimuli for the individual boutons that made up the group data in these plots. Representative data from two slices are shown replotted in A2, where each line represents an individual bouton. Most individual values of f increase (mean f = 0.52) between 150 and 300 stimuli with 10 Hz stimulation (n = 112 boutons) but not with 2 Hz stimulation (mean f = 0.19; 63 boutons). The filled circles represent the means of the groups. B, Group data (n = 4 slices; 278 boutons) showing effects of 10 μm nifedipine on destaining induced by 10 Hz stimulation in the presence of agatoxin. Data in C are from 101 individual boutons (○) and the mean of those boutons (•) from a single slice showing that the transient enhancement of f is blocked (mean f = 0.22) by nifedipine. Graph in the bottom right shows that nifedipine (○) blocked the transient enhancement of f. Control data (•) are replotted from A (bottom) for comparison.
GABA synapses generally depress during repetitive stimulation; hence, the mechanism of facilitation of exocytosis was not clear. Ca2+ entering through L-type channels can evidently accumulate during high-frequency trains and facilitate release after the trains (Jensen and Mody, 2001). We tested the possibility that L-type channels might contribute to the facilitation of release during 10 Hz stimulation. Indeed, 10 μm nifedipine, an L-type channel antagonist, slowed destaining and prevented the increase in f without affecting finitial (Fig. 5B) or the proportion of nondestaining boutons (data not shown). The destaining curve in nifedipine essentially takes the identical shape as the control curve beginning at 30 sec of stimulation (arrow), and similarly f becomes indistinguishable between nifedipine and control at this point (Fig. 5C, bottom right graph). We confirmed that nifedipine did not affect baseline IPSCs (90 ± 7% of controls; data not shown) and had no effect on depression caused by 2 Hz stimulation, although, as reported by Jensen et al. (1999), there was a tendency for IPSC depression caused by 10 Hz stimulation to be enhanced by nifedipine. Thus, nifedipine affected primarily the brief period of facilitation, and we conclude that L-type Ca2+ channels contribute to GABA release for a limited period during relatively high-frequency stimulation.
Presynaptic inhibition by G-protein-coupled receptor agonists
In addition to control by VGCCs, transmitter release is regulated by presynaptic modulators, yet the interrelationship between these two forms of regulation is not well understood. To determine whether presynaptic inhibitors would have the same effects on both N-type- and P/Q-type-dependent release, we studied each population in isolation by pretreating slices with either agatoxin or conotoxin. We first compared 2 and 10 Hz trains on IPSCs elicited in the absence and presence of 10 μm baclofen in Figure 6, A1 (2 Hz) and A2 (10 Hz). Because baclofen reduced the control IPSCs by ∼66%, the data were normalized to the first response in the trains. This revealed that baclofen decreased the relative depression of IPSCs evoked by repetitive stimulation, and that the extent of this effect was approximately equal for the two stimulus frequencies (dashed lines). Figure 6B shows group data from 440 agatoxin-treated boutons from nine slices. Destaining in the presence of baclofen was significantly decreased compared with destaining in control slices and increased back to and beyond control by the GABAB antagonist CGP55485. The enhanced destaining caused by CGP55485 is consistent with the increase in GABAergic IPSCs on CA1 interneurons (Lei and McBain, 2003) and suggests the presence of a GABAB-mediated inhibitory tone.
Figure 6.
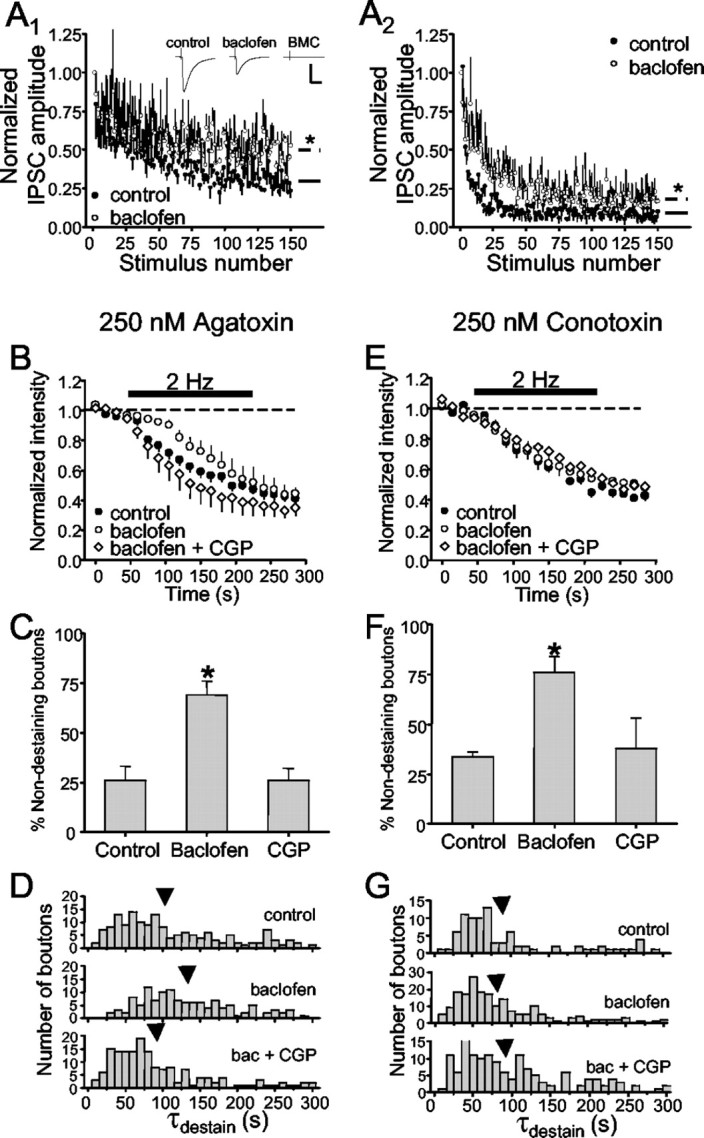
Baclofen reduces vesicular release. A, Left, Stimulation at 2 Hz induced IPSCs before (•) and after application of 10 μm baclofen (○). The solid (Fig. 4A, control) and dashed (baclofen) lines represent the steady-state level of IPSC depression computed as the average of the last 10 responses. A, Right, Same as left but with 10 Hz stimulation. Inset shows representative IPSCs (means of 3 each). B, Comparison of control group data (reproduced from Fig. 3D) from slices pretreated with 250 nm agatoxin and four slices (265 boutons) treated first with 10 μm baclofen and then with baclofen plus the GABAB antagonist, CGP55485 (CGP), at 10 μm. C, Baclofen significantly increased the number of nondestaining boutons compared with control and CGP. D, Baclofen significantly shifted the distribution of individual bouton destaining time constants (τdestain) toward longer values, an effect that was also prevented by CGP55485. Arrows indicate the means of the distributions. E, Comparison of control group data from slices pretreated with 250 nm conotoxin and six slices (290 boutons) treated first with 10 μm baclofen and then with baclofen plus CGP55485 at 10 μm. F, Baclofen significantly increased the number of nondestaining boutons compared with control and CGP55485. G, Baclofen had no significant effect on the distribution of τdestain. Stimulation is indicated by the black bar in B and E.
IPSCs represent the summated response of a stimulated population of boutons and, therefore, details of modulation of individual boutons are unknown. We tested the parsimonious hypothesis that all boutons are affected uniformly by examining the effects of baclofen on destaining. Contrary to this hypothesis, baclofen did not simply slow the rate of release from all boutons. It also significantly increased the numbers of nondestaining boutons (control, 26 ± 7%; baclofen, 69 ± 7%) (Fig. 6C), suggesting that it either blocked release entirely or reduced the probability of release (Pr) so much that release was effectively prevented in this group. There was a significant slowing in the distribution of τdestain values from the remaining boutons (Fig. 6D), whether determined by t test (control τdestain, 105 ± 6 sec; baclofen τdestain, 130 ± 6 sec; p < 0.05) or by KS test (data not shown; p < 0.005). Final steady-state values of fluorescence from these boutons were not affected by baclofen (control, 44 ± 4%; baclofen, 46 ± 5%). CGP55485 decreased the number of nondestaining boutons to 26 ± 6% (Fig. 6C) and significantly shifted the distribution of individual τdestain values to the left (mean, 88 ± 6 sec; p < 0.05) (Fig. 6C).
The data in Figure 6, B-D, were obtained in the presence of agatoxin, and we conclude that baclofen affects GABA release mediated by N-type VGCCs in two ways. The similarities in the basic properties of release mediated by N-type or P/Q-type VGCCs (supplemental Fig. 1) suggested that all fast release might be regulated in the same way by GABAB receptors, and we tested this hypothesis. However, whereas baclofen significantly increased the numbers of nondestaining boutons in conotoxin-treated slices (control, 34 ± 2%; baclofen, 76 ± 8%) (Fig. 6F), it did not alter release from the destaining boutons (Fig. 6E,G). Neither the mean τdestain (control, 91 ± 7 sec; baclofen, 81 ± 5 sec; not significant) nor final steady-state value of fluorescence (51 ± 2%) were affected by baclofen. CGP55485 decreased the percentage of nondestaining boutons to 26 ± 6% (Fig. 6F) but did not affect the τdestain distribution (mean, 95 ± 6 sec) or steady-state fluorescence. The simplest explanation for the inability of baclofen to slow release from the remaining boutons that destained in conotoxin is that these P/Q-dominant terminals do not possess GABAB receptors. Thus, the effects of baclofen on GABA release depended on the predominant VGCC present.
Presynaptic inhibition by CB1 cannabinoid receptor agonists
CB1 receptors (CB1Rs) are present almost exclusively on hippocampal interneurons that colocalize the neuropeptide cholecystokinin and release GABA only through the activation of N-type VGCCs (for review, see Alger, 2002). Moreover, depolarization-induced suppression of inhibition (DSI) is mediated by the retrograde release of an endocannabinoid that inhibits GABA release by activating the presynaptic CB1Rs (Wilson and Nicoll, 2001). We predicted that the CB1 receptor agonist, CP55940, at 1 μm would affect GABA release exactly as baclofen did. In cultures pretreated with agatoxin, CP55940 significantly lengthened individual τdestain values (CP55940, 125 ± 9 sec; p < 0.05), and this was reversed by the CB1 receptor antagonist, AM251 (4 μm; AM251, 93 ± 6 sec; not different from control) (Fig. 7A,C). CP55940 also significantly increased the percentage of nondestaining boutons during 2 Hz stimulation to 62 ± 10% of the total (Fig. 7B), and AM251 reversed this to 27 ± 9% (Fig. 7B). There was no significant difference in steady-state fluorescence intensity remaining at the end of 2 Hz stimulation (CP55940, 53 ± 4%; AM251, 37 ± 6%). The data are consistent with the observation that physiologically released endocannabinoids (Wilson et al., 2001) and DSI (Lenz and Alger, 1999) are insensitive to agatoxin, and that CB1Rs inhibit release mediated by N-type Ca2+ channels. Nevertheless, the electrophysiological evidence was necessarily based on fairly small numbers of cells, and cannabinoid effects on interneurons releasing by P/Q-type Ca2+ influx could have been missed. As predicted by the original hypothesis, we found that in cultures pretreated with conotoxin, CP55940 no longer had any effect on the number of nondestaining boutons, rate of destaining, or total amount of fluorescence remaining (Fig. 7D-F). These results strongly support the conclusion that CB1R-possessing GABAergic boutons release GABA exclusively via activation of N-type Ca2+ channels.
Figure 7.

The cannabinoid CB1 receptor agonist, CP55940, inhibits vesicular release evoked with 2 Hz stimulation. A, Comparison of control group data (Fig. 3D) from slices pretreated with 250 nm agatoxin and five slices (384 boutons) treated first with 1 μm CP55940 and then with CP55940 plus the CB1 antagonist, AM251, at 4 μm. CP55940 slowed the release elicited by 2 Hz stimulation, and this was prevented by AM251. B, CP55940 increased the percentage of nondestaining boutons over control levels. AM251 plus CP55940 treatment prevented the increase in numbers of nondestaining boutons. C, For the boutons that did destain in CP55940, the distribution of τdestain was shifted toward longer values, an effect that was also prevented by AM251. CP55940 had no effect on mean destaining (D), number of nondestaining boutons (E), or τdestain (F) in four conotoxin-treated slices (136 boutons). Control data are from Figure 6G. Stimulation is indicated by the black bar in A and D.
Frequency dependence of presynaptic inhibition of GABA release
The previous experiments revealed differences between the presynaptic inhibition by GABABR and CB1R. The efficacy of GABAB-mediated inhibition of glutamate release is inversely dependent on stimulation frequency (Isaacson and Hille, 1997), but whether this is true for G-protein-coupled receptor (GPCR) inhibition of GABA release is not known. We found that 10 Hz stimulation in agatoxin substantially diminished the inhibitory effects of baclofen (Fig. 8A-C) and, in sharp contrast to the results with 2 Hz, prevented baclofen from increasing the number of nondestaining boutons (control, 25 ± 9%; baclofen, 25 ± 15%) (Fig. 8B). Baclofen still produced a rightward shift in the distribution of τdestain (control, 35 ± 3 sec; baclofen, 55 ± 3 sec) (Fig. 8C). We could not test the effects of CGP55845 on these slices, because relatively few brightly loaded boutons remained after 10 Hz stimulation. Hence, activation of GABAB receptors was no longer able to prevent release when stimulation was given at 10 Hz, but it continued to slow the rate of release from N-type VGCC-possessing boutons.
Figure 8.

Regulation of GABA release elicited by 10 Hz stimulation. A, Agatoxin-pretreated slices. A comparison of control group data (Fig. 3A) with four slices (324 boutons) also treated with baclofen is shown. B, Baclofen does not increase the percentage of nondestaining boutons when 10 Hz stimulation is given. C, Baclofen shifts the distribution of individual destaining time constants toward longer values, although much less than for 2 Hz stimulation. D-F, Same as for A-C except on three conotoxin-pretreated slices (120 boutons). Stimulation is indicated by the black bar in A and D. G, The transient enhancement of f induced by 10 Hz stimulation is prevented by baclofen, although not by CP55940. Data are plotted for f values after 150 and 300 stimuli as in Figure 5A2. f increases in control conditions (control data calculated from data shown in Fig. 5A2, right plot; agatoxin present). H, Enhancement of f normalized to initial value for each condition.
In conotoxin-pretreated cultures stimulated at 10 Hz, baclofen no longer increased the fraction of nondestaining boutons (Fig. 8E) and still had no effect on the rate of release as assessed either by the group data (Fig. 8D) (control, 44 ± 3 sec; baclofen, 52 ± 4 sec) or the individual τdestain values (Fig. 8F). Thus, unlike its residual effects on release mediated by N-type VGCCs, baclofen completely lost its ability to inhibit release mediated by P/Q-type VGCCs when the stimulation frequency was raised to 10 Hz.
As anticipated, the efficacy of CB1R-mediated presynaptic inhibition was similarly reduced by 10 Hz stimulation in agatoxin-treated cultures. CP55940 did not increase the number of nondestaining boutons (CP55940, 35 ± 6%) but significantly slowed the loss of fluorescence from releasing boutons (to 58 ± 2 sec; n = 7 slices; 445 boutons; data not shown). Because CP55940 was ineffective in the presence of conotoxin at 2 Hz, we did not test CP55940 with 10 Hz stimulation in conotoxin. Table 1 summarizes the effects of baclofen and CP55940 on destaining of GABAergic boutons as a function of VGCC subtype and stimulation frequency.
Table 1.
Comparison of baclofen and CP55940 effects on destaining of GABAergic boutons as a function of stimulation frequency and VGCC subtype-mediating release
|
|
2 Hz stimulation |
10 Hz stimulation |
||||||
|---|---|---|---|---|---|---|---|---|
|
|
Non-DS percentage
|
τdestain
|
Non-DS percentage
|
τdestain
|
f
|
|||
| AgTx-pretreated cultures (release mediated by N-type VGCCs) | ||||||||
| GABABR | ↑ | ↑ | No change | ↑ | ↓ | |||
| CB1R | ↑ | ↑ | No change | ↑ | No change | |||
| CTx-pretreated cultures (release mediated by PQ-type VGCCs) | ||||||||
| GABABR | ↑ | No change | No change | No change | n.t. | |||
| CB1R
|
No change
|
No change
|
n.t.
|
n.t.
|
n.t.
|
|||
Non-DS percentage, percentage of boutons classified as nondestaining; τdestain, mean rate of destaining from individual boutons; f, increase in facilitation ratio measured between 150 and 300 stimuli; ↑, ↓, denote statistically significant change from control value; No change, no statistically significant change from control value; n.t., not tested.
Frequency-dependent facilitation and presynaptic inhibition
We wondered whether the facilitation of release induced by 10 Hz stimulation (Fig. 5) played a role in the frequency-dependent decrease in presynaptic inhibition and, therefore, examined the effects of baclofen and CP55940 on f. Both agonists reduced f measured after the first 150 stimuli, finitial, by ∼30 and ∼50%, respectively (Fig. 8G). Thus, to simplify the comparison between them, we replotted the data after normalizing them to finitial. The new plot, Figure 8H, shows that the increase in f, which occurred between 150 and 300 stimuli, was prevented by baclofen but not by CP55940. Because increasing stimulus frequency reduced the efficacy of both agonists, we conclude that the process represented by f is not primarily responsible for the reduction.
Discussion
This is the first report using styryl dye to study regulation of GABAergic exocytosis in organized tissue. To identify the GABAergic boutons unambiguously, we used pharmacological tools with selective effects on glutamate or GABA release, focused on a region where GABAergic terminals predominate, and compared the presumed GABAergic rat boutons with GAD65-eGFP labeled boutons from transgenic mice. The dye-loaded puncta met all of the criteria for functioning GABAergic boutons. We then investigated frequency-dependent, short-term regulation of GABA release and GPCR-mediated depression.
With 2 and 5 Hz stimulation, release from most boutons declined at uniform rates and was similar, whether induced by Ca2+-influx through N-type or P/Q-type VGCCs. Fractional release remained essentially constant, meaning that the same amount of release was induced by each action potential. Kraushaar and Jonas (2000) concluded that depression of GABAergic transmission in the dentate gyrus during prolonged 5 Hz stimulation was a presynaptic phenomenon. However, this analysis was based on the coefficient of variation of IPSCs, and a contribution of postsynaptic factors could not be entirely ruled out. We confirmed that a unitary presynaptic process dominates the decline in release at low frequencies but also showed that IPSCs depress more rapidly and completely than exocytosis. In principle, recycled vesicles refilled with transmitter but not dye might dilute the pool of dye-filled vesicles and retard the destaining rate (Ryan and Smith, 1995). However, vesicle recycling and refilling are slow processes (Ryan and Smith, 1995; Isaacson and Hille, 1997) and cannot account for the large differences between the rates of IPSC depression and bouton-destaining over a 15 sec period of stimulation (Fig. 4). Postsynaptic factors such as receptor desensitization could be responsible but so perhaps could a “kiss-and-run” model of release (Aravanis et al., 2003; Gandhi and Stevens, 2003), because partial vesicle emptying could cause both smaller postsynaptic responses and longer retention of dye in vesicles. Future work is required to explain the differences.
We found that 10 Hz stimulation caused a short-term facilitation of GABAergic exocytosis in the presence of agatoxin. High-stimulation frequencies can overcome a block of glutamatergic transmission induced by either conotoxin or agatoxin (Luebke et al., 1993; Castillo et al., 1994), but N-type and P/Q-type channels are colocalized at glutamatergic synapses (Wheeler et al., 1994; Reid et al., 1997). Presumably, the Ca2+ entering through one type can compensate for loss of influx through the other because of the highly nonlinear relationship between Ca2+ and exocytosis (Reid et al., 1998). However, the majority of somatic GABAergic boutons do not have both classes of VGCCs (Poncer et al., 1997; Wilson et al., 2001). [(The interneurons that use both VGCC types primarily mediate slow dendritic GABAA inhibition (Banks et al., 1998; Wilson et al., 2001)]. Although L-type Ca2+ influx does not participate in GABA release at low-stimulus frequencies (Takahashi and Momiyama, 1993; Ohno-Shosaku et al., 1994; Jensen et al., 1999), we observed that nifedipine blocked facilitation of GABAergic exocytosis, suggesting that, at higher frequencies, L-type VGCCs can participate. This has been a controversial issue. In hippocampal cells in dissociated culture, L-type VGCC antagonists enhance eIPSC depression during high-frequency stimulation (Jensen et al., 1999) but reportedly only affect post-tetanic potentiation, and not release during trains, in the dentate gyrus of hippocampal slices (Jensen and Mody, 2001). The reduction in facilitation by nifedipine suggests that the responsible L-channels are located near the GABA release sites. However, in an immunocytochemical study of cultured GABAergic mouse neocortical cells, Cav 1.2 and 1.3 subunits were localized to somatodendritic regions and not axons or terminals (Timmermann et al., 2002). Experimental factors could account for the discrepancy between the physiological and morphological data, or Ca2+-influx through somatodendritic L-type VGCCs might be able influence release.
Having established the basic properties of GABAergic exocytosis, we inquired into its regulation by presynaptic inhibitory CB1Rs and GABABRs. Both GPCRs are localized on presynaptic nerve terminals and were expected to affect exocytosis in the same way, although this had never been directly tested. Contrary to expectations, we found that their effects are similar but not identical (Table 1).
GABAB receptors are ubiquitous on GABAergic boutons possessing N-type VGCCs: baclofen prevented or slowed release from essentially all boutons pretreated with agatoxin and stimulated at 2 Hz. GABAB receptors are not present on all boutons that use P/Q channels for release: baclofen prevented release from most of the destainable boutons in conotoxin-pretreated slices and did not affect the rest. CB1Rs also appear to be present on all boutons that release via N-type VGCCs, but they are, as predicted (Wilson et al., 2001), absent from P/Q-possessing boutons; CP55940 did not affect release in conotoxin-pretreated slices. However, these estimates would be slightly in error if boutons possessing one type of VGCC tended to be nondestaining.
Presynaptic regulation of release by GABAB and CB1 receptors was unexpectedly complex. Although release from some terminals was reversibly slowed, release from most terminals was effectively blocked completely. In agatoxin-pretreated slices, both baclofen and CP55940 increased the number of nondestaining boutons very significantly, to ∼75% of the total population, when stimulation was given at 2 Hz (Figs. 6B, 8B). Assuming that only 75% of the population was capable of destaining (∼25% of boutons were classified as nondestaining), ∼two-thirds (i.e., 50/75%) of the total was effectively prevented from vesicular release by baclofen or CP55940. The occurrence of a significant fraction of nondestaining boutons in control conditions appears to be typical for these experiments (Isaacson and Hille, 1997; Zakharenko et al., 2001, 2002), yet the reasons for it are not known.
Variability in GABA release correlates with variability in the amplitudes of Ca2+ signals in individual boutons (Kirischuk and Grantyn, 2002). Conceivably, boutons with low intrinsic Pr, related to small amounts of Ca2+ entry, can be prevented from releasing transmitter by presynaptic inhibition, whereas release from boutons with more robust Ca2+ influx is merely reduced. The amount of Ca2+ influx can also be influenced by K+ channel activity, and K+ conductances can be increased by both GABAB (Gahwiler and Brown, 1985) and CB1 receptors (Deadwyler et al., 1995). Alternatively, baclofen can inhibit release at some synapses, although not necessarily GABAergic synapses (Scanziani et al., 1992), by acting downstream from Ca2+ influx (i.e., it blocks Ca2+- and TTX-insensitive miniature EPSCs) (Scanziani et al., 1992) and modulates vesicle priming (Sakaba and Neher, 2003). Similarly, a CB1R agonist can suppress Ca2+- and TTX-insensitive miniature IPSCs in the cerebellum (Takahashi and Linden, 2000; Diana and Marty, 2003). In the hippocampus, there is some discrepancy on this point. The effects of CB1R agonists on synaptic transmission are blocked by Cd2+ (Hoffman and Lupica, 2000) or by conotoxin (Wilson et al., 2001), and CB1R activation does suppress hippocampal N-type Ca2+ currents (Twitchell et al., 1997). Hence, blockade of Ca2+ influx might explain the CB1R-mediated suppression of GABA release. In contrast, Varma et al. (2002) reported that CB1R agonists continue to suppress ∼30% of the TTX-insensitive mIPSCs in conotoxin, which is consistent with a downstream effect of CB1R. Conceivably, the two aspects of presynaptic inhibition on Ca2+ influx and a downstream site correspond to the slowing or prevention of GABAergic exocytosis (Wu and Saggau, 1997). It is interesting that increasing stimulation frequency to 10 Hz prevented the GPCRs from increasing the numbers of nondestaining boutons (Fig. 8A). In any case, at low stimulus frequencies, the inhibitory coupling between GABAB or CB1 receptors and the release machinery is likely to involve the same mechanisms.
With 10 Hz stimulation, a difference between the two receptor systems emerged. The transient facilitation of exocytosis was blocked by baclofen but not by CP55940. Because facilitation was dependent on L-type VGCCs (Fig. 5B), we propose that baclofen prevents facilitation by blocking Ca2+ influx through L-type VGCCs in the nerve terminals, because it blocks these channels in hippocampal cell somata (Scholz and Miller, 1991). CB1R activation does not block L-type VGCCs (Twitchell et al., 1997), which is consistent with this interpretation. An important task will be to investigate the differential coupling between L-type VGCCs and these GPCRs. Although we predicted that facilitation would account for the reduction in presynaptic inhibition by 10 Hz stimulation, this was not supported. A 10 Hz stimulation overcame the effects of both GABABR and CB1R agonists, but baclofen prevented facilitation. Isaacson and Hille (1997) suggested that the ability of 20 Hz stimulation to overcome the inhibition of glutamate release of baclofen reflected either the build-up of residual Ca2+ or the increased dominance of baclofen-insensitive steps in the release process. Similar factors may be involved in the decreased ability of GPCRs to inhibit GABA release.
We conclude that regulation of GABA exocytosis is complex and depends on many factors including stimulus frequency, predominant class of VGCC present, and identity of neuromodulator, as well as undetermined factors. Differences between suppression of IPSCs and exocytosis imply that exclusive focus on IPSCs in the study of release may not be a reliable strategy. An intriguing observation was that release from many boutons was evidently entirely prevented, whereas release from others was slowed. The difference may be related to distinct mechanisms of presynaptic inhibition and will be the subject of additional investigations. Direct visualization of exocytosis should be applicable to the study of GABA regulation in other regions of the brain.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants NS30219 and DA14625. D.H.B. was supported by NIH Postdoctoral Training Grant NS07375. We are indebted to Scott Thompson for helpful discussions, and we also thank Jimok Kim for his comments on a previous draft of this manuscript.
Correspondence should be addressed to Dr. Bradley E. Alger, Department of Physiology, University of Maryland School of Medicine, 655 West Baltimore Street, Baltimore, MD 21201. E-mail: balger@umaryland.edu.
D.H. Brager's present address: Division of Neuroscience, 1 Baylor Plaza, Baylor College of Medicine, Houston, TX 77030.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310475-12$15.00/0
References
- Akaike N, Murakami M, Katsurabayashi S, Jin Y-H, Imazawa T ( 2002) Focal stimulation of single GABAergic presynaptic boutons on the rat hippocampal neuron. Neurosci Res 42: 187-195. [DOI] [PubMed] [Google Scholar]
- Alger BE ( 2002) Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol 68: 247-286. [DOI] [PubMed] [Google Scholar]
- Alger BE, Nicoll RA ( 1979) GABA-mediated biphasic inhibitory responses in hippocampus. Nature 281: 315-317. [DOI] [PubMed] [Google Scholar]
- Aravanis AM, Pyle JL, Tsien RW ( 2003) Single synaptic vesicles fusing transiently and successively without loss of identity. Nature 423: 643-647. [DOI] [PubMed] [Google Scholar]
- Banks MI, Li T-B, Pearce RA ( 1998) The synaptic basis of GABAA, slow. J Neurosci 18: 1305-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz WJ, Bewick GS ( 1992) Optical analysis of synaptic vesicle recycling at the frog neuromuscular junction. Science 255: 200-203. [DOI] [PubMed] [Google Scholar]
- Betz WJ, Mao F, Bewick GS ( 1992) Activity-dependent fluorescent staining and destaining of living vertebrate motor nerve terminals. J Neurosci 12: 363-375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz S, David J, Trussell L ( 2001) Enhancement of synoptic efficacy by presynaptic GABAB receptors. Neuron 20: 135-141. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Weisskopf MG, Nicoll RA ( 1994) The role of Ca2+ channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron 12: 261-269. [DOI] [PubMed] [Google Scholar]
- Deadwyler SA, Hampson RE, Mu J, Whyte A, Childers S ( 1995) Cannabinoids modulate voltage sensitive potassium A-current in hippocampal neurons via a cAMP-dependent process. J Pharmacol Exp Ther 273: 734-743. [PubMed] [Google Scholar]
- Diana MA, Marty A ( 2003) Characterization of depolarization-induced suppression of inhibition using paired interneuron-Purkinje cell recordings. J Neurosci 23: 5906-5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA ( 2001) The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24: 31-55. [DOI] [PubMed] [Google Scholar]
- Erdélyi F, Sekerkova G, Katarova Z, Hájos N, Pálhalmi J, Freund TF, Mugnaini E, Szabó G ( 2002) GAD65-GFP transgenic mice expressing GFP in the GABAergic nervous system. FENS Abstr 1: AO11-AO13. [Google Scholar]
- Feng J, Chi P, Blanpied TA, Xu Y, Magarinos AM, Ferreira A, Takahashi RH, Kao H-T, McEwen BS, Ryan TA, Augustine GJ, Greengard P ( 2002) Regulation of neurotransmitter release by synapsin III. J Neurosci 22: 4372-4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahwiler BH, Brown DA ( 1985) GABAB-receptor-activated K+ current in voltage-clamped CA3 pyramidal cells in hippocampal cultures. Proc Natl Acad Sci USA 82: 1558-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahwiler BH, Thompson SM, McKinney RA, Debanne D, Robertson RT ( 1998) Organotypic slice cultures of neural tissue. In: Culturing nerve cells (Banker G, Goslin K, eds), pp 461-498. Cambridge, MA: MIT.
- Gandhi SP, Stevens CF ( 2003) Three modes of synaptic vesicular recycling revealed by single-vesicle imaging. Nature 423: 607-613. [DOI] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR ( 2000) Mechanisms of cannabinoid inhibition of GABAA synaptic transmission in the hippocampus. J Neurosci 20: 2470-2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, Alger BE ( 1986) Whole-cell voltage-clamp study of the fading of GABA-activated currents in acutely dissociated hippocampal neurons. J Neurophysiol 56: 1-18. [DOI] [PubMed] [Google Scholar]
- Irving AJ, Coutts AA, Harvey J, Rae MG, Mackie K, Bewick GS, Pertwee RG ( 2000) Functional expression of cell surface cannabinoid CB1 receptors on presynaptic inhibitory terminals in cultured rat hippocampal neurons. Neuroscience 98: 253-262. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Hille B ( 1997) GABAB-mediated presynaptic inhibition of excitatory transmission and synaptic vesicle dynamics in cultured hippocampal neurons. Neuron 18: 143-152. [DOI] [PubMed] [Google Scholar]
- Jensen K, Mody I ( 2001) L-type Ca2+ channel-mediated short-term plasticity of GABAergic synapses. Nat Neurosci 4: 975-976. [DOI] [PubMed] [Google Scholar]
- Jensen K, Jensen MS, Lambert JDC ( 1999) Role of presynaptic L-type Ca2+ channels in GABAergic synaptic transmission in cultured hippocampal neurons. J Neurophysiol 81: 1225-1230. [DOI] [PubMed] [Google Scholar]
- Kaila K ( 1994) Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol 42: 489-537. [DOI] [PubMed] [Google Scholar]
- Kannenberg K, Sieghart W, Reuter H ( 1999) Clusters of GABAA receptors on cultured hippocampal cells correlate only partially with functional synapses. Eur J Neurosci 11: 1256-1264. [DOI] [PubMed] [Google Scholar]
- Kay AR, Alfonso A, Alford S, Cline HT, Holgado AM, Sakmann B, Snitsarev VA, Stricker TP, Takahashi M, Wu LG ( 1999) Imaging synaptic activity in intact brain and slices with FM1-43 in C. elegans, lamprey, and rat. Neuron 24: 809-817. [DOI] [PubMed] [Google Scholar]
- Kirischuk S, Clements JD, Grantyn R ( 2002) Presynaptic and postsynaptic mechanisms underlie paired pulse depression at single GABAergic boutons in rat collicular cultures. J Physiol (Lond) 543: 99-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraushaar U, Jonas P ( 2000) Efficacy and stability of quantal GABA release at a hippocampal interneuron-principal neuron synapse. J Neurosci 20: 5594-5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert NA, Teyler TJ ( 1991) Adenosine depresses excitatory but not fast inhibitory synaptic transmission in area CA1 of the rat hippocampus. Neurosci Lett 122: 50-52. [DOI] [PubMed] [Google Scholar]
- Lei S, McBain CJ ( 2003) GABA B receptor modulation of excitatory and inhibitory synaptic transmission onto rat CA3 hippocampal interneurons. J Physiol (Lond) 546: 439-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz RA, Alger BE ( 1999) Calcium dependence of depolarization-induced suppression of inhibition in rat hippocampal CA1 pyramidal neurons. J Physiol (Lond) 521: 147-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebke JI, Dunlap K, Turner TJ ( 1993) Multiple calcium channel types control glutamatergic synaptic transmission in the hippocampus. Neuron 11: 895-902. [DOI] [PubMed] [Google Scholar]
- McCarren M, Alger BE ( 1985) Use-dependent depression of IPSPs in rat hippocampal pyramidal cells in vitro. J Neurophysiol 53: 557-571. [DOI] [PubMed] [Google Scholar]
- Michelson HB, Wong RKS ( 1991) Excitatory synaptic responses mediated by GABAA receptors in the hippocampus. Science 253: 1420-1423. [DOI] [PubMed] [Google Scholar]
- Miles R, Toth K, Gulyas AI, Hajos N, Freund TF ( 1996) Differences between somatic and dendritic inhibition in the hippocampus. Neuron 16: 815-823. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Hirata K, Sawada S, Yamamoto C ( 1994) Contributions of multiple calcium channel types to GABAergic transmission in rat cultured hippocampal neurons. Neurosci Lett 181: 145-148. [DOI] [PubMed] [Google Scholar]
- Poncer J-C, McKinney RA, Gahwiler BH, Thompson SM ( 1997) Either N- or P-type calcium channels mediate GABA release at distinct hippocampal inhibitory synapses. Neuron 18: 463-472. [DOI] [PubMed] [Google Scholar]
- Reid CA, Clements JD, Bekkers JM ( 1997) Nonuniform distribution of Ca2+ channel subtypes on presynaptic terminals of excitatory synapses in hippocampal cultures. J Neurosci 17: 2738-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid CA, Bekkers JM, Clements JD ( 1998) N- and P/Q-type Ca2+ channels mediate transmitter release with a similar cooperativity at rat hippocampal autapses. J Neurosci 18: 2849-2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan TA, Smith SJ ( 1995) Vesicle pool mobilization during action potential firing at hippocampal synapses. Neuron 14: 983-989. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E ( 2003) Direct modulation of synaptic vesicle priming by GABAB receptor activation at a glutamatergic synapse. Nature 424: 775-778. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan S, Atluri PP, Ryan TA ( 2003) Actin has a molecular scaffolding, not propulsive, role in presynaptic function. Nat Neurosci 6: 127-135. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Capogna M, Gahwiler BH, Thompson SM ( 1992) Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron 9: 919-927. [DOI] [PubMed] [Google Scholar]
- Scholz KP, Miller RJ ( 1991) GABAB receptor-mediated inhibition of Ca2+ currents and synaptic transmission in cultured rat hippocampal neurones. J Physiol (Lond) 444: 669-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR ( 1995) Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science 269: 977-981. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Linden OJ ( 2000) Cannabinoid receptor modulation of synapses received by cerebellar Purkinje cells. J Neurophysiol 83: 1167-1180. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Momiyama A ( 1993) Different types of calcium channels mediate central synaptic transmission. Nature 366: 156-158. [DOI] [PubMed] [Google Scholar]
- Thompson SM ( 1994) Modulation of inhibitory synaptic transmission in the hippocampus. Prog Neurobiol 42: 575-609. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gahwiler BH ( 1989) Activity-dependent disinhibition. I. Repetitive stimulation reduces IPSP driving force and conductance in the hippocampus in vitro. J Neurophysiol 61: 501-511. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Haas HL, Gahwiler BH ( 1992) Comparison of the actions of adenosine at pre- and postsynaptic receptors in the rat hippocampus in vitro. J Physiol (Lond) 451: 347-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermann DB, Westenbroek RE, Schousboe A, Catterall WA ( 2002) Distribution of high-voltage-activated calcium channels in cultured γ-aminobutyric acidergic neurons from mouse cerebral cortex. J Neurosci Res 67: 48-61. [DOI] [PubMed] [Google Scholar]
- Twitchell W, Brown S, Mackie K ( 1997) Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol 78: 43-50. [DOI] [PubMed] [Google Scholar]
- Varma N, Morishita W, Lenz RA, London B, Alger BE ( 2002) Presynaptic mechanisms of DSI expression in hippocampus. Neuropharmacol 43: 550-562. [DOI] [PubMed] [Google Scholar]
- Wheeler DB, Randall A, Tsien RW ( 1994) Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science 264: 107-111. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA ( 2001) Endogenous cannabinoids mediate retrograde signaling at hippocampal synapses. Nature 410: 588-592. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA ( 2001) Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 31: 453-462. [DOI] [PubMed] [Google Scholar]
- Wu L-G, Saggau P ( 1997) Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci 20: 204-212. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA ( 2001) Visualization of changes in presynaptic function during long-term synaptic plasticity. Nat Neurosci 4: 711-717. [DOI] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA ( 2002) Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron 35: 1099-1110. [DOI] [PubMed] [Google Scholar]