

Abstract
Several human neurodegenerative diseases are associated with abnormal accumulations of aggregated tau proteins and glial degeneration in astrocytes, but the mechanism whereby tau proteins cause astrocytic degeneration is unclear. Here, we analyzed the biological consequences of overexpressing the longest human tau isoform in primary cultures of rat astrocytes using adenoviral-mediated gene transfer. Significantly, we found specific decreases in stable detyrosinated [glutamate (Glu)] microtubules (MTs) with concomitant increases in tubulin biosynthesis and the accumulation of acetylated, tyrosinated, α- and β-tubulin. The consequences of this selective reduction in stable Glu-MTs included contemporaneous decreases in kinesin levels, collapse of the intermediate filament network, progressive disruption of kinesin-dependent trafficking of organelles, fragmentation of the Golgi apparatus that culminated in atrophy, and non-apoptotic death of astrocytes. These results suggest that reduced stable Glu-MTs is a primary consequence of tau accumulation that initiates mechanisms underlying astrocyte dysfunction and death in human neurodegenerative glial tauopathies.
Keywords: Alzheimer, astrocyte, astroglial, degeneration, Golgi, microtubule, tau
Introduction
Abnormal tau accumulation in CNS neurons and glia are hallmarks of neurodegenerative tauopathies, and mounting evidence demonstrates primary roles for tau in pathogenesis of these diseases. The major neuropathological characteristics of tauopathies are neuronal and glial inclusions, dystrophic neurites, and neuropil threads formed from aggregated tau in paired helical filaments (PHFs) or straight filaments (Spillantini and Goedert, 1998; Lee et al., 2001). In addition to neuronal tau pathologies, brains of diverse tauopathies, including progressive supranuclear palsy (PSP) (Hauw et al., 1990; Yamada et al., 1992), corticobasal degeneration (CBD) (Shin et al., 1991), Pick's disease, argiophillic grain disease (Botez et al., 1999), and a subset of frontotemporal dementia (FTD) and parkinsonism linked to chromosome 17 (FTDP-17) syndromes, develop abundant fibrillary glial tau lesions (Goedert et al., 1999). Tau inclusions exist as “coiled bodies” in oligodendrocytes (Braak and Braak, 1987) or as “thorny” and “tufted” astrocytes (Hauw et al., 1990) or as astrocytic plaques (Feany and Dickson, 1995).
Tau, a microtubule-associated protein, functions to stabilize and promote microtubules (MTs) assembly, and multiple posttranslational modified forms of tubulin have been found in MTs. Both detyrosinated [glutamate (Glu)]-MTs and acetylated (Ac) MTs are found in more stable MTs, whereas tyrosinated (Tyr) MTs are more dynamic (Gundersen et al., 1984, 1987). Although tau is most abundantly expressed in CNS neurons, astrocytes and oligodendrocytes also express tau at low levels (Lee et al., 2001). However, in pathological conditions, tau accumulates in glia because of either increased synthesis or reduced clearance resulting in the fibrillization of tau and the degeneration of affected cells.
The functions of astrocytes are poorly understood, and their significance has been primarily underestimated. Recent studies demonstrate that astrocytes contribute to neuronal survival (Manthorpe et al., 1986) and control neuronal excitability and synaptic transmission (Carmignoto, 2000). Thus, degeneration of astrocytes may affect neuronal activities and survival. However, the mechanism(s) and the biological consequences of tau accumulation in astrocytes in neurodegenerative disorders remain unknown. Recently, transgenic tau mice evidenced selective deposition of tau at the distal processes of astrocytes, which recapitulates specific features of astrocytic plaques found in FTDs, such as PSP and CBD (Higuchi et al., 2002). Thus, we hypothesize that this selective accumulation of tau contributes to the collapse of the cytoskeleton and degeneration of astrocytes.
To test this hypothesis and to clarify the molecular mechanism of astrocyte degeneration in tauopathies, we developed a cell culture model system by overexpressing tau in primary rat astrocytes and investigated the pathological consequences of this overexpression. Our results demonstrate that tau accumulation in astrocytes causes a selective reduction of both Glu-MTs and kinesin, followed by disruption of kinesin-dependent organelle trafficking, collapse of the intermediate filament (IF) network, MT bundling, fragmentation of the Golgi apparatus (GA), and eventually the atrophy and death of affected cells. Together, these findings provide insights into plausible mechanisms of astrocytic pathology in a variety of neurodegenerative tauopathies.
Materials and Methods
Preparation of cortical astrocyte cultures. Enriched cultures of astrocytes were generated using methods described by McCarthy and de Vellis (1980) from rat pups at postnatal days 1-3 d. Their brains were aseptically removed and placed in sterile culture dishes containing HBSS. The midbrain, meninges, and blood vessels were dissected from the sample. The remaining cerebral cortices were mechanically dissociated and suspended in DMEM containing 10% fetal bovine serum, 7.5 mm glucose, 4 mm l-gulutamine, 1000 U/ml penicillin, and 1 ng/ml streptomycin (10% DMEM). The dissociated brain cells were then centrifuged at 2000 rpm for 5 min, resuspended in 10% DMEM, and plated in a 75 cm2 flask. The cells were maintained in 10% DMEM at 37°C in an atmosphere of 5% CO2 air. After 5 d, the cells were mechanically dissociated to generate cultures enriched for >98% astrocytes. The astrocytes were harvested by trypsinization, plated in six-well plates, and maintained in 10% DMEM for 3 d. One day before the experiments, the culture medium was switched to 0.5% DMEM.
Construction and infection with adenoviral vector. An adenovirus (AV) carrying the longest human tau isoform (T40) cDNA (T40-AV) and an AV carrying green fluorescent protein (GFP) gene (GFP-AV) driven by the human cytomegalovirus promoter were constructed using an Adeno-Quest kit (QBiogene, Quebec, Canada). Viral titer was determined by plaque assay on human embryonic kidney 293 cells as plaque formation units per milliliter. The titers of both viruses were 5.0 × 1010/ml
Infection of cultured astrocytes by virus was performed at a multiplicity of infection of ∼100 for 2 hr in DMEM without serum at 37°C, and the cells were thereafter reincubated in 0.5% DMEM.
Image analysis procedures for cell survival rate and cell area. Phase-contrast images of cultured rat astrocytes at different time points after adenoviral infection were taken, the outline of each cell was traced manually using a personal computer, and cell number and the average cell area in each image were calculated by ImageQuant software (Molecular Dynamics, Sunnyvale, CA). The survival rate was calculated based on the number of cells remaining attached to the plate that do not intercalate trypan blue as a percentage of the number of astrocytes at time 0.
Preparation of lysates from astrocytes and Western blot analyses. Cultured astrocytes were rinsed with PBS and lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris, 150 mm NaCl, 1% NP-40, 5 mm EDTA, 0.5% sodium deoxycholate, and 0.1% SDS, pH 8.0) supplemented with a mixture of protease inhibitors (l-1-tosylamide-2-phenylethylchloromethyl, N-tosyl- l-lysine chloromethyl ketone, leupeptin, pepstatin, and soybean trypsin inhibitor, each at 1 μg/ml). Cell lysates were sonicated and centrifuged at 50,000 × g for 40 min at 4°C. The supernatants were collected, and protein concentrations were determined using the bicinchoninic acid method (Pierce, Rockford, IL). The samples were resolved on 7.5% SDS-PAGE gels and transferred onto nitrocellulose membranes. Western blot analyses were performed using various primary antibodies (see below). [125I]-Labeled goat anti-mouse IgG or [125I]-labeled protein A were used as secondary reporting reagents, and the blots were exposed to PhosphorImager plates (Molecular Dynamics). The signal intensity was quantified using ImageQuant software (Molecular Dynamics).
Metabolic labeling of astrocytes and immunoprecipitation. To estimate tubulin subunit biosynthesis, astrocytes infected with T40-AV were rinsed with PBS and labeled for 2 hr with 1 ml of methionine-free DMEM supplemented with 250 μCi of [35S]methionine at 0, 4, 8, and 24 hr after infection. At the end of the radiolabeling, the medium was removed and replaced with RIPA buffer, and the lysates were centrifuged at 15,000 × g for 20 min. Equal amounts of proteins from RIPA extracts were immunoprecipitated with an anti-β-tubulin monoclonal antibody (mAb). The immunoprecipitates were resolved on a 7.5% SDS-PAGE gels, and radioactive signals were detected with PhosphorImager after the exposure for 3 d.
Preparation of cytoskeletal fractions. Astrocytes were harvested in reassembly buffer [0.1 m 2-(N- morpholino)ethanesulfonic acid, 0.5 mm MgSO4, 1 mm EGTA, and 2 mm dithiothreitol, pH 6.8] supplemented with 0.1% Triton X-100, 4 m glycerol, 2 mm GTP, and a mixture of protease inhibitors (as mentioned above) at 37°C. Cell lysates were homogenized with 15 strokes in a warm Dounce homogenizer and immediately centrifuged at 50,000 × g for 40 min at 30°C. The supernatants were removed, and the protein concentration was determined by the bicinchoninic acid method (Pierce). The resulting pellets were each resuspended in a volume of sample buffer corresponding to the total volume of supernatant after normalizing for total protein. The samples were resolved on 7.5% SDS-PAGE gels and transferred onto nitrocellulose replicas.
Indirect immunofluorescence stainings of astrocytes. Astrocytes were fixed with 0.3% glutaraldehyde in PEM buffer [80 mm piperazine-N, N′-bis (2-ethanesulfonic acid), pH 6.8, 5 mm EGTA, and 1 mm MgCl2] for 10 min and permeabilized with 0.5% Triton X-100 in PBS for 15 min. Glutaraldehyde was then quenched with 10 mg/ml sodium borohydride in PBS for 7 min, followed by 0.1 m glycine in PBS for 20 min. After rinsing with PBS, the cells were incubated with primary antibodies for 2 hr at room temperature and incubated with fluorescent-labeled secondary antibodies for 1 hr at room temperature. Secondary FITC anti-mouse and anti-rabbit, Texas Red anti-mouse, anti-rabbit, and anti-rat, and sulfo-NHS 7-amino 4-methyl coumarin-3-acetic acid anti-mouse were purchased from Molecular Probes (Eugene, OR). Samples of PSP brain were obtained and characterized in the Brain Bank of the Center for Neurodegenerative Disease Research (Philadelphia, PA) as described previously (Forman et al., 2002) and were examined here by similar methods.
Transmission electron microscopy. Transmission electron microscopy (EM) was performed on representative samples of astrocytes after fixation with 4% paraformaldehyde and 2% glutaraldehyde in PBS or with 4% paraformaldehyde and 0.25% glutaraldehyde in PBS for 60 min, followed by quenching of the samples with 0.1% sodium borohydride in Tris-buffered saline for 10 min and treatment for another 10 min with 20% ethanol. The samples were embedded in Epon-812 and polymerized at 70°C for 48 hr. Thin sections at 65 nm were cut, mounted on 200-mesh copper grids, stained with 1% uranyl acetate in 50% ethanol and bismuth subnitrite, and examined with a JEM1010 electron microscope (Jeol, Peabody, MA) at 80 kV.
Antibodies and dyes. The following antibodies were used: rabbit polyclonal anti-human recombinant tau anibody 17026 (Ishihara et al., 1999); anti-non-phosphorylated tau mAb T1 (Binder et al., 1985); anti-human tau mAb T14 (Kosik et al., 1988); anti-phosphorylation-independent tau mAb T46 (Kosik et al., 1988); anti-phosphorylated tau mAbs PHF1 and 12E8 (Seubert et al., 1995); rabbit polyclonal anti-α-tubulin antibody (Santa Cruz Biotechnology, Santa Cruz, CA); anti-β-tubulin mAb (Sigma, St. Louis, MO); anti-acetylated tubulin mAb (Sigma); anti-tyrosinated tubulin rat mAb YL1/2 (Harlan Sera-Lab, Loughborough, UK); rabbit polyclonal anti-detyrosinated tubulin antibody (Gundersen et al., 1984) (a gift from Dr. G. Gundersen, Columbia University, New York, NY); anti-kinesin heavy chain mAb (Chemicon, Temecula, CA); anti-GFP mAb (Chemicon); anti-vimentin mAb V9 (Boehringer Mannheim, Indianapolis, IN); anti-MG160 mAb and rabbit polyclonal antibody (Gonatas et al., 1989) (gifts from Dr. N. K. Gonatas, University of Pennsylvania, Philadelphia, PA); and rabbit polyclonal anti-GFP antibodies (Molecular Probes).
For staining of mitochondria, MitoTracker Green (Molecular Probes) was added at a concentration of 400 nm and then incubated for 30 min before fixation as described above.
Results
Expression of human tau in primary astrocytes using adenovirus encoding the longest tau isoform
To develop a cell culture model of tau accumulation in primary astrocytes, we overexpressed tau by infecting rat astrocytes with T40-AV and determined the efficiency of infection using double indirect immunofluorescence analysis of tau and glial fibrillary acidic protein (GFAP), an IF protein that is a marker for astrocytes. At 4 hr after infection, 77 ± 4.3% of the cells were immunolabeled by both anti-tau and anti-GFAP antibodies, but, by 6 hr, >95% of the astrocytes were expressing the human tau T40 protein (data not shown). GFP-AV-infected cells showed almost the same efficiency (data not shown). To determine the level of tau protein expression, cell lysates from astrocytes at 0, 4, 8, and 24 hr after T40-AV infection were analyzed by quantitative Western blot analysis (Fig. 1A). The total amount of tau (based on the immunoreactivity of a phosphorylation-independent anti-tau antibody, i.e., 17026) increased with time, and, by 24 hr, the amount of tau expression was 10 and 4 times higher than those at 4 and 8 hr, respectively (Fig. 1A). The expression levels of tau achieved here are much higher than the levels estimated in Chinese hamster ovary (CHO) cells that stably expressed tau after transfection of human T40 (Vogelsberg-Ragaglia et al., 2000). Indeed, the expression levels of tau in the stably transfected CHO cells are almost identical to the astrocytes at 6 hr after infection (data not shown). Thus, the AV transfer system used in this study enabled a high expression level of tau in astrocytes. Although quantitative Western blotting using the phosphorylation-dependent tau antibodies PHF1 and 12E8 showed consistent increases of phosphorylated tau with longer infection times, these increases were almost proportional for total tau (17026) and non-phosphorylated tau (T1) (Fig. 1A), indicating that the fraction of phosphorylated tau remained unchanged during the time course of infection. Significantly, we also observed an increase in β-tubulin levels despite the fact that identical amounts of total protein were loaded.
Figure 1.

Quantitative Western blots of total tau and MT-bound and -unbound tau in cultured astrocytes infected with T40-AV. Western blot analyses of tau from astrocytes infected with T40-AV for different lengths of time: total tau (A) and MT-bound and -unbound tau (B) probed with primary antibodies against phosphorylation-independent (17026), phosphorylation-dependent (PHF1 and 12E8), and non-phosphorylation-dependent (T1) anti-tau and anti-β-tubulin (β-tub) antibodies, followed by [125I]-labeled secondary antibodies. Tau bound to MTs in the pellet (P) fractions comprised ∼65% of the total tau 8 hr after infection but it reduced to ∼35% at 24 hr, whereas tau in the pellet fractions increases twofold from 8 to 24 hr (B). Phosphorylated tau (PHF1 and 12E8) was mainly found in the supernatant (S) fractions (B).
To assess the binding of the overexpressed tau to MTs, we measured the ratio of MT bound and free tau in a MT-binding assay. At 4 hr after infection, the majority of the tau was recovered in the pellet fraction (P), suggesting that all the tau expressed at this time point was bound to MTs (Fig. 1B). By 8 hr, ∼65% of the total tau was recovered bound to MTs (Fig. 1B). However, at 24 hr, this bound fraction reduced to ∼35% (Fig. 1B). As described previously, phosphorylated tau showed a reduced ability to bind MTs, and our data at 24 hr confirmed that more phosphorylated tau was detected in supernatant (S) than in the pellet (the S/P tau ratio detected with PHF1 was 12:1 and with 12E8 was 3.4:1) than non-phosphorylated tau with T1 (S:P was 1.7:1) (Fig. 1B). Therefore, the relative abundance of phosphorylated tau in the supernatant fractions increased with time.
Tau overexpression alters the morphology of astrocytes by disrupting MT networks
To explore changes in the cytoskeletal organization and morphology of astrocytes overexpressing and accumulating tau, we analyzed the distribution of β-tubulin and tau by double-label indirect immunofluorescence. Non-infected cells showed classic MT networks without obvious tau staining (Fig. 2A). At 4 hr after infection, the mesh-like networks of MTs already showed slight disorganization in a subpopulation of cells expressing tau at higher levels than the other cells, although the majority of infected astrocytes showed no remarkable changes in MT organization (Fig. 2B). By 8 hr after infection, thick bundles of MTs replaced the normal MT networks in a significant number of cells, and not all of the tau staining was found to colocalize with MTs (Fig. 2C). At 16 hr, MT networks completely disappeared and long processes containing thick bundles of MTs were observed, whereas the AV-expressed tau uniformly filled the cytoplasm (Fig. 2D). These bundles, rather than radiating linearly from the cell body, occasionally encircled nuclei. Although similar findings were seen at 24 hr, tau-positive blobs that resemble tau aggregates in FTDs also were detected near the surface of the astrocytes (Fig. 2E, arrowheads), and this was associated with a dramatic reduction in the size of the astrocytes that inversely correlated with the levels of tau expression in individual cells (Fig. 2C-E, insets). At 4 and 8 hr after infection, there was a 25 and 50% reduction in cell size, respectively, but, by 16 and 24 hr after infection, there was an 80% decrease in the size of the astrocytes (Fig. 3). Significantly, despite the detection of tau structures that resemble aggregates, no SDS-insoluble tau protein was recovered biochemically.
Figure 2.

Changes in MTs and cell morphology in cultured astrocytes infected with T40-AV. Double-immunofluorescence staining for β-tubulin (red; Tub) and tau (green; 17026) was performed in astrocytes infected with T40-AV. Non-infected cells showed clear mesh-like MT networks but no tau staining (A), whereas a slight disturbance of the MT network was noticeable in some astrocytes just 4 hr after infection (B). Disruption of MT networks increased over time with abnormally thick bundles of MTs and free tau in a subset of cells at 8 hr after infection (C). Sixteen hours after infection, MT networks completely disappeared, and free tau (i.e., unbound to MTs) uniformly filled the cytoplasm (D). No additional changes in astrocyte morphology were detected 24 hr after infection, except for tau deposits near the cell periphery (E, arrowheads). Low-power photomicrographs (insets) clearly show alterations in the morphology of the astrocytes associated with increasing tau expression. Scale bars, 10 μm.
Figure 3.

Reduced viability and cell size of T40-AV-infected astrocytes. Astrocyte number and cell area after T40-AV infection were analyzed on the basis of image analysis of phase-contrast images using ImageQuant. Significant changes in astrocyte survival were not observed until 16 hr after T40-AV infection, but reductions in the area of astrocytes were seen as early as 4 hr after infection. Error bars represent SE. *p < 0.05; **p < 0.01.
We also monitored the survival of the astrocytes over time after T40-AV infection using phase-contrast microscopy. No cell death was observed during the first 8 hr of infection, but, by 16 and 24 hr, ∼75 and 50%, respectively, of the astrocytes remained attached to the plates and were viable on the basis of image analysis by ImageQuant software (Molecular Dynamics) (Fig. 3). Thus, all of the subsequent experiments were conducted with cells infected with T40-AV for 8 hr or less unless otherwise stated. Interestingly, there was no evidence of apoptosis on the basis of nuclear fragmentation, DNA laddering, and activation of caspase-3 when compared with astrocytes treated with staurosporine (Finkel, 2001), an obligatory “executor” of apoptosis as the positive control (Fig. 4).
Figure 4.

No obvious evidence of caspase-3 activation in T40-AV-infected astrocytes. Western blot analysis with a specific antibody to cleaved caspase-3 (Cell Signaling Technology, Beverly, MA). No clear band of cleaved caspase-3 (A) was observed in cell lysates from astrocytes infected with T40-AV for different lengths of time, unlike the astrocytes treated with staurosporin for 4 hr.
The accumulation of tau causes a specific reduction in the levels of detyrosinated Glu-MTs but increases the synthesis of total tubulin subunits
Previous studies have demonstrated that post-translational modifications of tubulin are markers of MT stability (for review, see MacRae, 1997). The formation of MT bundles in the T40-AV-infected astrocytes implies a production of hyperstabilized MTs by the overexpressed tau. To evaluate the effects of tau accumulation on MT dynamics, we assessed the different post-translational modified forms of tubulin in astrocytes. Quantitative Western blots revealed a time-dependent increase in the amounts of α- and β-tubulin, as well as Ac- and Tyr-tubulin concomitant with longer infection times (Fig. 5A,B). This suggests that increased tau expression caused tubulin subunits to accumulate. To determine whether or not this increase in tubulin levels was attributable to increased synthesis or reduced degradation of tubulin subunits, we performed metabolic labeling studies using radiolabeled [35S]methionine, followed by immunoprecipitation with an anti- β-tubulin antibody. These studies showed that the amounts of newly synthesized radiolabeled tubulin increased significantly with longer T40-AV infection times, suggesting that increased expression of tau in astrocytes led to de novo increase in tubulin synthesis (Fig. 5C). Because previous studies have shown that unpolymerized tubulin modulates the level of tubulin mRNA and that reduced levels of unassembled monomeric tubulin is a signal for increase tubulin synthesis (Cleveland et al., 1981), we assessed whether or not tau accumulation led to reduced tubulin monomers. To do this, we monitored directly the amount of soluble tubulin monomers and pelletable MTs, and we showed that increased tau expression decreased the levels of free monomeric tubulin in soluble fractions but accelerated MT polymerization by increasing the levels of assembled α-tubulins (Fig. 5D) and β-tubulins (Fig. 1B) in pellet fractions. Thus, the increased tubulin synthesis in response to tau accumulation is attributable to a decrease in unpolymerized monomeric tubulin subunits.
Figure 5.

Increase in acetylated and tyrosinated α- and β-tubulin subunits but selective reduction in Glu-tubulin in astrocytes accumulating tau. Quantitative Western blots of total cell lysates (A-C), supernatants (S), and cytoskeletal fractions (P) (D) probed with antibodies against several different post-translationally modified forms of tubulin subunits, followed by [125I]-labeled secondary antibodies. Synthesis of β-tubulin as monitored by [35S]methionine metabolic labeling of the T40-AV-infected astrocytes, followed by immunoprecipitation with anti-β-tubulin antibody (C) were performed. Quantitative analysis of Western blots showed increases of Ac-tubulin and Tyr-tubulin with time up to 180% of the initial levels 24 hr after infection, concomitant with the increases of α- and β-tubulins, whereas Glu-tubulin decreased with time down to 30% of the initial level at 24 hr (B). These alterations occurred primarily on assembled MTs rather than free tubulin subunits because the changes in the intensity of the protein bands in the cytoskeletal fractions (P) resembled the changes in the total cell lysates (A, D). The increase radiolabeled β-tubulin (C) with time correlated with the increased accumulation of total tubulin shown in A: Ac-tub, acetylated tubulin; Tyr-tub, tyrosinated tubulin; Glu-tub, detyrosinated tubulin. Statistical differences of the means, *p < 0.05; **p < 0.01.
Concomitant with an increase in tubulin synthesis, Ac- and Tyr-tubulin were also increased with time of infection (Fig. 5A,B). However, the level of detyrosinated Glu-tubulin is selectively reduced (70% reduction at 24 hr), whereas the levels of vimentin and GFAP remained unchanged (Fig. 5A,B). To further investigate the effects of tau accumulation on the localization of post-translationally modified MTs, indirect immunofluorescence staining was conducted using antibodies to Ac-, Glu-, and Tyr-tubulin (Fig. 6). In uninfected astrocytes, the distributions of stable Glu- and Ac-MTs appeared to be almost identical, but the overlay image showed incomplete colocalization with some segments of MTs that were exclusively positive for either Ac-tubulin or Glu-tubulin (Fig. 6A and inset). Tyr-tubulin also did not completely colocalize with Ac- and Glu-tubulin in control astrocytes (data not shown). In the T40-AV-infected astrocytes, Glu-tubulin immunoreactivity appeared to be selectively eliminated from cell periphery (Fig. 6B,C), whereas those of Ac-tubulin (Fig. 6B) and Tyr-tubulin (Fig. 6C) remained unchanged. Triple-immunofluorescence staining for Glu-tubulin, Tyr-tubulin, and tau clearly showed a remarkable loss of Glu-tubulin immunoreactivity at the periphery of the astrocytes accumulating tau. In contrast, Tyr-tubulin immunostaining remained colocalized with tau throughout the entire cell (Fig. 6C). These findings imply that tau accumulation led to the selective loss of Glu-MTs, particularly at the edge of the astrocytes.
Figure 6.
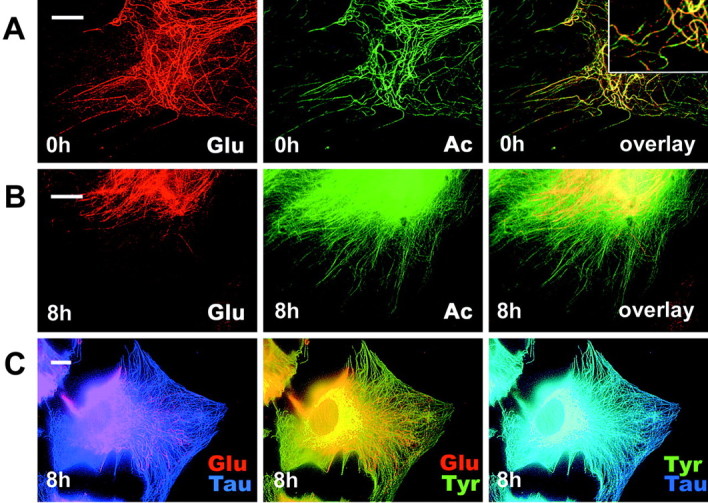
Glu-MTs are selectively eliminated from the cell periphery in astrocytes accumulating tau. Double- and triple-immunofluorescence staining with specific antibodies to post-translationally modified tubulin subunits was performed in astrocytes 8 hr after T40-AV infection. Although the distribution of the more stable Glu- (red) and Ac- (green) MTs appeared almost identical, the overlay picture showed that these two forms of modified tubulin did not completely colocalize with each other even in non-infected cells, but some segments of MTs were either Glu-MT-rich or Ac-MT-rich (A and inset). Eight hours after infection, Glu-MTs, but not Ac-MTs (B) or Tyr-MTs (C), disappeared selectively from the periphery of the astrocytes. Triple staining for Glu- (red) and Tyr- (green) MTs together with tau (blue) confirmed colocalization of tau with Tyr-MTs (C). Scale bars, 10 μm.
Tau accumulation disturbs interaction between kinesin and Glu-MTs
Because previous studies have shown that Glu-MTs bind to kinesin with high affinity (Liao and Gundersen, 1998), we asked whether or not a reduction in Glu-MTs in astrocytes accumulating tau affect the distribution and levels of kinesin. To accomplish this, we used an antibody (Pfister et al., 1989) against kinesin heavy chain (KHC) to assess the distribution of kinesin in astrocytes with and without tau accumulations. In non-infected astrocytes, KHC immunostaining showed vesicle-like and linear distributions of kinesin in close proximity to MTs throughout the entire cytoplasm (Fig. 7A). In contrast, in astrocytes infected with T40-AV, KHC immunostaining was found to be collapsed and clustered around the perinuclear area (Fig. 7, compare A, B). Thus, tau accumulation in astrocytes also caused a redistribution of kinesin. To evaluate whether or not this kinesin redistribution caused a change in the amount of kinesin associated with the cytoskeleton, we quantified the amount of KHC in the cytoskeletal fractions generated from astrocytes infected with T40-AV for different lengths of time. Significantly, we found a dramatic reduction of kinesin in the cytoekeletal fraction (Fig. 7C). Furthermore, this reduction appeared to be specific for kinesin because the levels of dynein associated with the cytoskeletal fractions remained unchanged (Fig. 7C). Thus, our data showed that tau accumulations in astrocytes not only cause a redistribution and reduction of Glu-MTs but also led to the redistribution and reduction of kinesin. Because the levels of dynein were not affected, we conclude that tau accumulations selectively reduced Glu-MTs levels, which directly or indirectly mitigate the disruption of Glu-MT and kinesin interactions, thereby resulting in a reduction of kinesin associated with the cytoskeleton.
Figure 7.

Kinesin is selectively reduced and redistributed in T40-AV-infected astrocytes. Double-label immunofluorescence of kinesin heavy chain and Glu-MTs before T40-AV infection (A) and of kinesin and tau 8 hr after T40-AV infection (B). Kinesin staining at high magnification showed vesicular or linear staining distributed over the entire cytoplasm extending to the edge of the cell. This distribution was not identical to that of MTs, although kinesin staining frequently existed in close proximity to MTs in the non-infected cells (A). T40-AV-infected astrocytes showed kinesin staining clustered near the nucleus (B). Western blot analysis of kinesin heavy chain and dynein in the cytoskeletal fractions (C) of T40-AV-infected astrocytes showed a decrease in kinesin with longer infection time, whereas the amount of dynein remained unchanged. Scale bar, 10 μm.
Tau accumulation alters distribution of organelles
Because kinesin is a motor protein responsible for plus-end-directed transport of organelles and vesicles (Hirokawa et al., 1991), the reduction and reorganization of kinesin as a consequence of reduced Glu-MTs in astrocytes accumulating tau could disrupt intracellular MT-dependent transport. Furthermore, several studies suggested that tau inhibits kinesin-dependent transport in other types of cultured cells (Ebneth et al., 1998; Trinczek et al., 1999; Stamer et al., 2002). Thus, we analyzed the trafficking of mitochondria and endoplasmic reticulum (ER) in astrocytes accumulating tau using MitoTracker as a marker for mitochondria and an antibody to calnexin to localize the ER. In control non-infected astrocytes, mitochondria were distributed in the entire cytoplasm of non-infected astrocytes (Fig. 8A), but mitochondria completely disappeared from the periphery of the cytoplasm and clustered around the perinuculear area in the tau-accumulating cells (Fig. 8B,C). Similar to mitochondria, calnexin, a molecular chaperone that resides in the ER membrane, also disappeared from the periphery and clustered in the vicinity of the nucleus of the tau-expressing astrocytes as well (Fig. 8F,G). ER-tracker labeling also showed reduced fraction of the cell area containing ER (data not shown). In contrast, dispersed mitochondria (Fig. 8D, green) and ER-associating calnexin (Fig. 8H, red) were also detected in control GFP-expressing astrocytes just like non-infected cells. The clustering of mitochondria and ER was observed 4 hr after infection, when a reduction of detyrosinated Glu-MTs and kinesin were seen without obvious abnormalities of MT networks. Thus, it is likely that dysfunction of plus-end-directed MT-dependent transport of organelles caused by reduced levels of Glu-MTs and kinesin emerged before the morphological disorganization of MTs.
Figure 8.

Redistribution of cellular organelles in T40-AV-infected astrocytes. Double-label immunofluorescence staining with MitoTracker (Mit) for mitochondria and β-tubulin (Tub) before (A) or after (B) 8 hr of T40-AV infection, for Mit and tau (C), as well as for Mit with GFP (D) 8 hr after infection. Double-label immunofluorescence staining of calnexin (Cal, E-H), a marker for ER, and MG160 (I-N), a marker for the GA, in astrocytes before (A, E, I, K) or after (B, C, F, G, J, L-N) infection with T40-AV or with GFP-AV (D, H). Mitochondria (green) and ER (green) spread over the whole cytoplasm and reached the distal ends of MTs in astrocytes before infection (A, E) and with GFP-AV infection (D, H), whereas the mitochondria and ER accumulated in the vicinity of the nucleus 8 hr after T40-AV infection (B, C, F, G), even in the cells showing preserved MT networks (B, F) and expressing small amounts of tau (C, G). However, the collapse of the ER did not lead to changes in the expression level of calnexin (O). D has a high red pseudocolor fluorescent signal attributable to the expression of GFP. The GA was clearly stained with the anti-MG160 antibody in non-infected astrocytes (I), in which the staining appeared in the anastomotic reticulum and extended to a considerable distance (K). GA staining distinctly decreased 8 hr after T40-AV infection (J), and fragmentation of the GA occurred, as shown in higher magnification (M). This fragmentation of the GA was observed as early as 4 hr after T40-AV infection (L). Double immunostaining for MG160 (red) and tau (17026; green) indicated the relationship between the loss of GA and the expression of tau (N): cells with higher expression levels of tau lost their GA (N, arrowheads), and cells with lower expression levels of tau had some GA remaining (N, arrow). Western blot analysis showed decreased expression of MG160 (O), which is consistent with the immunofluorescence staining. Mit, MitoTracker; Cal, calnexin; Tub, tubulin. Scale bars, 10 μm.
Tau accumulation induces fragmentation and loss of Golgi apparatus
Because the GA has a close functional association with the ER and dynamically interacts with MTs (Infante et al., 1999), the distribution and morphology of the GA was examined by immunocytochemistry with an mAb against the rat MG160, a 160 kDa sialoglycoprotein of medial cisternae of GA (Gonatas et al., 1989). In non-infected astrocytes, immunostaining for MG160 clearly showed a typical perinuclear network of the cisternae (Fig. 8I,K). However, 8 hr after T40-AV infection, MG160 immunostaining was dramatically diminished (Fig. 8J), and the remaining GA showed fragmentation and dispersion (Fig. 8M). Interestingly, disorganization of the reticular structure of GA also occurred at 4 hr (Fig. 8L), suggesting that the GA fragmentation is one of the early events caused by the accumulation of tau. A subset of the infected cells exhibited a markedly reduced area containing GA networks when compared with dispersed GA in the control cells (Fig. 8, compare cell with arrow in N with K). Double-immunofluorescence staining for MG160 and tau demonstrated that the cells with higher tau accumulation showed much more prominent loss of GA (Fig. 8N, arrowheads) than the cells with lower levels of tau accumulation (Fig. 8N, arrow). Notably, whereas the level of calnexin remained unchanged (Fig. 8O) despite the collapse of the ER (Fig. 8F,G), the amount of MG160 detected by Western blotting rapidly decreased 4 hr after T40-AV infection. Together, the GA showed a specific structural disintegration linked to the accumulation of tau that was different from other organelles. Thus, the impact of tau accumulation on the GA may be a significant and distinct process that is linked mechanistically to tau-induced astrocytic degeneration.
Tau accumulation alters distribution of IFs
Because Glu-MTs and kinesin have been implicated in maintaining the extended distribution of vimentin IFs (Gurland and Gundersen, 1995; Liao and Gundersen, 1998; Kreitzer et al., 1999) and because other studies also have indicated a collapse of IF networks by the overexpression of tau in vimentin-containing cells (Trinczek et al., 1999; Stamer et al., 2002), we investigated whether or not IF networks also were altered by tau accumulation in astrocytes. Immunofluorescence staining for vimentin (Fig. 9A-C, G) and GFAP (Fig. 9D-F, 9H) showed a collapse of the IF network in astrocytes even in those with a preserved MT organization (Fig. 9B,E) and with low levels of tau accumulation that aligned with MT networks (Fig. 9C,F). On the other hand, GFP-AV-infected astrocytes showed no alternations in the distribution of IFs (Fig. 9G,H). Interestingly, quantitative Western blot indicated that the levels of vimentin and GFAP remained unchanged after the T40-AV infection (Fig. 5A).
Figure 9.
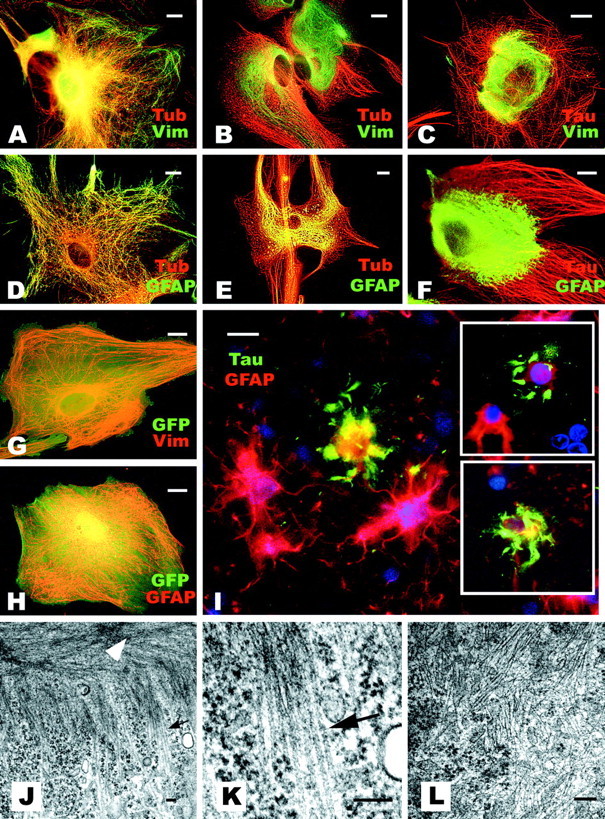
Disruption of IF network by tau accumulation in astrocytes recapitulate features of astrocytic pathology in human tauopathies. Double-label immunofluorescence staining for vimentin (Vim) and β-tubulin (Tub) before (A) and 8 hr after (B) T40-AV infection. Similar double labeling also was performed for GFAP and Tub before (D) and 8 hr after (E) T40-AV infection. Double labeling of tau (Tau) and Vim (C) or tau and GFAP (F) after T40-AV infection, as well as after GFP-AV infection (G, H). Note the collapse of the IF network and its accumulation on the vicinity of the nucleus 8 hr after T40-AV infection, even in the cells with preserved MT networks (B,E) with relatively low tau expression levels (C,F). A similar collapse of GFAP IF networks can be detected in a brain section from a patient with PSP immunostained with PHF1 and an anti-GFAP antibody (I). Note that the PHF1-positive astrocytes have lost their GFAP-positive long processes and exhibit a disrupted GFAP IF network in which there are abnormal tau accumulations (I, inset). Scale bars: A-I, 10 μm. Transmission EM shows the collapse of the IF network. Eight hours after T40-AV infection, low-power photomicrograph shows MT bundles in the cell periphery (J, arrow), and compacted IFs are seen in the vicinity of the nucleus (data not shown) but not in the peripheral area (J, white arrowhead). Higher magnification of MT bundles (K) and IFs (L) show exclusion of IFs in K and MTs in L, respectively. Scale bars: J-L, 100 nm.
Alterations of IFs in astrocytes resembling the above-mentioned findings were also revealed in human tauopathies by double-immunofluorescence staining with GFAP and PHF1 antibodies in brain sections from a PSP patient. Astrocytes with tau lesions lost GFAP immunoreactivity in their processes, in which tau resided as abnormal deposits (Fig. 9I and insets). These results imply a similar molecular mechanism of degeneration in cultured astrocytes accumulating tau in astrocytes of human tauopathies. It is also noteworthy that the collapse of IF networks in tauopathies may be caused by a dysfunction of IF transport rather than by the physical displacement of IFs in astrocytic processes by tau deposits, because the distribution of IFs was perturbed in astrocytes without unequivocal morphological evidence of tau aggregates (Fig. 9C,F).
The cytoskeletal alterations were also examined by transmission EM. Bundles of MTs were observed in peripheral areas of the cytoplasm with compact IFs localized close to the nucleus of the T40-AV-infected astrocytes (Fig. 9J, arrow and arrowheads, respectively). A high-power view clearly showed the fine structure of the MT bundles with an inter-MT distance of 10-30 nm (Fig. 9K), which is in agreement with the distance between MTs polymerized with tau in vitro (Hirokawa et al., 1988). Interestingly, almost no IFs were found in the peripheral area of the cytoplasm, in which MT bundles were localized (Fig. 9J). In contrast, abundant IFs were observed in the perinuculear area in the infected astrocytes, and they were randomly oriented with very few interspersed MTs (Fig. 9L). Thus, the loss of GFAP and/or vimentin from astrocytic processes and the abnormal deposition of tau in the distal processes of astrocytes detected in human tauopathies may be a consequence of Glu-MTs and kinesin reduction caused by tau accumulations.
Discussion
A cell culture model was developed to examine the pathogenic mechanisms of tau accumulation-induced astrocyte degeneration by overexpressing tau in primary rat astrocytes. We assessed the biological consequence of increased tau expression and determined whether this accumulation recapitulated features of astrocytic tau pathology in human FTDs, such as PSP and CBD. Our data showed that tau accumulation directly or indirectly led to the selective destruction of Glu-MTs and a concomitant decrease in kinesin in astrocytes. This reduction of Glu-MTs and kinesin caused the collapse of the IF network, disruption of kinesin-mediated anterograde organelle transport, GA fragmentation to culminate in the atrophy, and death of affected astrocytes. These alternations in T40-AV-infected astrocytes do not recapitulate events accompanying all types of cell death, because the selective destruction of Glu-MTs or GA fragmentation was not observed in astrocytes treated with other toxic agents, such as staurosporin. Significantly, the collapse of the IF network and the accumulation of tau at the cell periphery in our cultured astrocytes did recapitulate features of astrocytic plaques in CBD and PSP in which GFAP is found collapsed in the cell body associated with tau accumulations at their distal processes (Forman et al., 2002).
In addition to the selective reduction of the more stable Glu-MTs, the accumulation of tau in astrocytes caused other profound changes in MT dynamics, including the formation of abnormal MT bundles and increased MT assembly. These changes are likely attributable to increased levels of non-phosphorylated tau because this form of tau functions to promote MT assembly and stabilize MTs in the polymerized state. This increased promotion of MT assembly by tau also led to a de novo increase in tubulin biosynthesis. However, this is not surprising, because previously published studies have shown that the level of unpolymerized tubulin controls the synthesis of tubulin subunits by translational regulation of mRNA stability (Cleveland et al., 1981; Yen et al., 1988). The observed increase in tubulin biosynthesis is most likely attributable to the depletion of tubulin as a consequence of increased assembly of tubulin into MTs stimulated by high levels of tau. In addition to higher levels of α- and β-tubulin subunits with increasing tau levels, post-translationally modified forms of tubulin (with the exception of Glu-MTs) i.e., Ac- and Tyr-tubulin, also consistently increased with time in T40-AV-infected astrocytes. These results support the notion that the post-translational modification of tubulin occurs primarily when they are assembled into MTs rather than on the unassembled tubulin subunits.
A number of studies have shown that Glu- and Ac-MTs are found in the more stable pool of MTs and they are resistant to MT-disrupting agents compared with Tyr-MTs (Kreis, 1987). Thus, it is unclear why tau expression in astrocytes has opposite effects on Glu-MTs and Ac-MTs. At first glance, the reduction of Glu-MTs with increasing levels of tau could be interpreted as a result of more stabilized MTs induced by tau overexpression such that the more stable detyrosinated Glu-MTs are no longer needed. However, other more stable MTs, i.e., Ac-MTs, increased in proportion to both the total amount of MTs and the amount of tau, implying that the effect of tau on Glu-MTs is very selective and not simply attributable to more stabilized MTs. Interestingly, these differential effects of overexpressed tau on Glu-MTs and Ac-MTs in astrocytes were also noted in hippocampal neurons derived from tau knock-out mice wherein a reduction in Glu-MTs but not Ac-MTs was observed in the absence of tau (Rapoport et al., 2002). Furthermore, a dramatic reduction of Glu-MTs and to a lesser extent Ac-MTs has been observed in cultured human NT2N neurons treated with phosphatase inhibitors, such as okadaic acid, resulting in tau hyperphosphorylation and axonal degeneration (Merrick et al., 1997). Thus, tau levels and tau phsophorylation may play a specific role in regulating Glu-MT levels. Another explanation for the tau-induced disruption of Glu-MTs is the possibility that overexpressed tau could inhibit the activity of tubulin-carboxypeptidase (TCP), the enzyme responsible for detyrosination of tubulin (MacRae, 1997). Detyrosination and retyrosination of tubulins are regulated by TCP and tubulin-tyrosine ligase (TTL), respectively. TCP acts gradually and mainly on MTs, whereas the action of TTL is prompt and primarily on non-assembled tubulin subunits (Gundersen et al., 1987; MacRae, 1997; Contin et al., 1999). Disassembled Glu-tubulin can be rapidly retyrosinated into Tyr-tubulin by TTL, and newly assembled Tyr-MTs may not be detyrosinated because tau inhibits TCP. Finally, it is noteworthy that tau accumulations caused the selective reduction of Glu-MTs from the more dynamic distal portion of the MT network at the cell periphery but not the more stable proximal portion close to the nucleus. The reason for this is unknown, but the biological consequences of reduced Glu-MTs in this region of the cells has profound effects on the IF network and anterograde organelle transport, as demonstrated in our studies here, and we also provided evidence implicating kinesin as a downstream mediator of these changes.
Several studies have revealed unique interactions between Glu-MTs, kinesin, and IFs. For example, the recombinant head domain of the squid KHC binds to taxol-stabilized Glu-MTs with an affinity approximately threefold higher than the affinity of this KHC to Tyr-MTs (Liao et al., 1998). Double-immunofluorescence staining with anti-Glu-tubulin and anti-vimentin antibodies also demonstrated a preferential association between Glu-MTs and IFs (Prahlad et al., 1998; Kreitzer et al., 1999). Moreover, microinjection of kinesin antibodies into cultured cells induced a collapse of IFs (Gyoeva and Gelfand, 1991), and polyclonal anti-Glu-tubulin antibodies, but not anti-Tyr-tubulin IgG, caused a collapse of IFs in fibroblasts (Gurland et al., 1995). These and other findings indicate that Glu-MTs interact with kinesin and provide the “tracks” for kinesin-dependent transport. In addition, our data suggest that Glu-MTs and kinesin may be coregulated under specific conditions because tau accumulation in astrocytes induced a progressive reduction in Glu-MTs at cell periphery, which in turn caused a concomitant decrease in kinesin protein levels also at the cell periphery. The specific reduction of both Glu-MTs and kinesin at cell periphery of T40-AV-infected astrocytes most likely explains why kinesin mediated plus-end-directed transport to the cell periphery is compromised and why IF networks collapse around the nucleus.
This interpretation of our data are at odds with conclusions drawn from recent reports demonstrating that the overexpression of tau inhibits kinesin-dependent trafficking of vesicles as well as the collapse of IFs in CHO cells (Ebneth et al., 1998; Trinczek et al., 1999), N2a neuroblastoma cells, and cultured rat neurons (Stamer et al., 2002). In these studies, the authors suggest that the mechanism whereby tau exerts inhibitory effects on kinesin-dependent intracellular transport are attributable to the excessive binding of tau to MTs thereby reducing the attachment of kinesin to MTs. However, Glu-MTs were not studied, and only morphological, morphometric, and time-lapse experiments were performed without assessing the levels of kinesin and Glu-MTs in these tau-overexpressing systems. Another possible mechanism by which tau might inhibit binding of kinesin to MTs would be a direct blocking of the kinesin-binding sites (Hagiwara et al., 1994). This scenario seems unlikely because kinesin can bind to MTs saturated with tau, and C-terminal truncated tubulin can bind to kinesin but not to tau (Marya et al., 1994; Larcher et al., 1996), implying that the binding sites for kinesin and tau on MTs are distinct. Finally, because kinesin but not dynein binds to Glu-MTs, it is not surprising that the minus-end-directed transport remained unaffected or less affected by the accumulations of tau (Liao et al., 1998).
Previous studies in the lamprey have shown that the overexpression of human tau in neurons disrupts the cytoskeleton and alters the distribution of organelles (Hall et al., 2000). Here we showed that the accumulation of tau in T40-AV-infected astrocytes leads to the fragmentation of the GA, and similar disruptions of the GA have been reported in diverse neurodegenerative diseases, including Alzheimer's disease (Stieber et al., 1996), CBD (Sakurai et al., 2000), and amyotrophic lateral sclerosis (Gonatas et al., 1992; Mourelatos et al., 1993), as well as in transgenic mice expressing mutant Cu, Zn superoxide dismutase (Mourelatos et al., 1996) or tau with the V337M mutation (Tanemura et al., 2002). Therefore, morphological and functional changes of the GA might contribute to astrocytic degeneration in human neurodegenerative disorders. However, additional studies of the GA in tau-positive astrocytes in human tauopathies are required to clarify how perturbations of the GA contribute to astrocytic degeneration in these human disorders.
To summarize, the present study delineates for the first time a molecular mechanism for tau-induced neuropathology and degeneration in astrocytes. Specifically, the loss of Glu-MTs as a direct or indirect consequence of tau accumulation initiates a cascade of events that includes alterations of MT dynamics, a reduction in kinesin levels (particularly those associated with Glu-MTs at cell periphery) leading to impaired trafficking of organelles to the cell periphery, and the eventual collapse of the IF network, which, coupled to the fragmentation of the GA and the loss of Glu-MTs and kinesin, culminates in the atrophy and death of affected cells.
Footnotes
This work was supported by National Institute on Aging Grant AG 17586 (V.M.-Y.L., J.Q.T.). V.M.-Y.L. is the John H. Ware III professor of Alzheimer's research. We thank Dr. N. Gonatas for the Golgi-specific antibodies, Dr. V. Vogelsberg-Ragaglia for advice on T40 adenovirus, and Drs. M. Forman, B. Giasson, N. Gonatas, and M. Higuchi for critical reading and suggestion in the preparation of this manuscript.
Correspondence should be addressed to Dr. Virginia M.-Y. Lee, Maloney 3, Hospital of the University of Pennsylvania, 3600 Spruce Street, Philadelphia, PA 19104-4283. E-mail: vmylee@mail.med.upenn.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310662-10$15.00/0
References
- Binder LI, Frankfurter A, Rebhun LI ( 1985) The distribution of tau in the mammalian central nervous system. J Cell Biol 101: 1371-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botez G, Probst A, Ipsen S, Tolnay M ( 1999) Astrocytes expressing hyperphosphorylated tau protein without glial fibrillary tangles in argyophilic grain disease. Acta Neuropathol 98: 231-236. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E ( 1987) Argyrophilic grains: characteristic pathology of cerebral cortex in cases of adult onset dementia without Alzheimer changes. Neurosci Lett 76: 124-127. [DOI] [PubMed] [Google Scholar]
- Carmignoto G ( 2000) Reciprocal communication systems between astrocytes and neurones. Prog Neurobiol 62: 561-581. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Lopata MA, Sherline P, Kirschner MW ( 1981) Unpolymerized tubulin modulates the level of tubulin mRNAs. Cell 25: 537-546. [DOI] [PubMed] [Google Scholar]
- Contin MA, Sironi JJ, Barra HS, Arce CA ( 1999) Association of tubulin carboxypeptidase with microtubules in living cells. Biochem J 339: 463-471. [PMC free article] [PubMed] [Google Scholar]
- Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E ( 1998) Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer's disease. J Cell Biol 143: 777-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany MB, Dickson DW ( 1995) Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am J Pathol 146: 1388-1396. [PMC free article] [PubMed] [Google Scholar]
- Finkel E ( 2001) The mitochondrion: is it central to apoptosis? Science 292: 624-626. [DOI] [PubMed] [Google Scholar]
- Forman MS, Zhukareva V, Bergeron C, Chin SS-M, Grossman M, Clark C, Lee VM-Y, Trojanowski JQ ( 2002) Signature tau neuropathology in gray and white matter of corticobasal degeneration. Am J Pathol 160: 2045-2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG, Crowther RA, Chen SG, Parchi P, Tabaton M, Lanska DJ, Markesbery WR, Wilhelmsen KC, Dickson DW, Petersen RB, Gambetti P ( 1999) Tau gene mutation in familial progressive subcortical gliosis. Nat Med 5: 454-457. [DOI] [PubMed] [Google Scholar]
- Gonatas JO, Mezitis SG, Stieber A, Fleischer B, Gonatas NK ( 1989) MG-160. A novel sialoglycoprotein of the medial cisternae of the Golgi. J Biol Chem 264: 646-653. [PubMed] [Google Scholar]
- Gonatas NK, Stieber A, Mourelatos Z, Chen Y, Gonatas JO, Appel SH, Hays AP, Hickey WF, Hauw JJ ( 1992) Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis. Am J Pathol 140: 731-737. [PMC free article] [PubMed] [Google Scholar]
- Gundersen GG, Kalnoski MH, Bulinski JC ( 1984) Distinct populations of microtubules: tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell 38: 779-789. [DOI] [PubMed] [Google Scholar]
- Gundersen GG, Khawaja S, Bulinski JC ( 1987) Postpolymerization detyrosinatin of alpha-tubulin: a mechanism for subcellular differentiation of microtubules. J Cell Biol 106: 141-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurland G, Gundersen GG ( 1995) Stable detyrosinated microtubules function to localize vimentin intermediate filaments in fibroblast. J Cell Biol 131: 1275-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyoeva FK, Gelfand VI ( 1991) Coalignment of vimentin intermediate filaments with microtubules depends on kinesin. Nature 353: 445-448. [DOI] [PubMed] [Google Scholar]
- Hagiwara H, Yorifuji H, Sato-Yoshitake R, Hirokawa N ( 1994) Competition between motor molecules (kinesin and cytoplasmic dynein) and fibrous microtubule-associated proteins in binding to microtubules. J Biol Chem 269: 3581-3589. [PubMed] [Google Scholar]
- Hall GF, Chu B, Lee G, Yao J ( 2000) Human tau filaments induce microtubules and synapse loss in an in vivo model of neurofibrillary degenerative disease. J Cell Sci 113: 1373-1387. [DOI] [PubMed] [Google Scholar]
- Hauw JJ, Verny M, Delacre P, He Y, Duyckaerts C ( 1990) Constant neurofibrillary changes in the neocortex in progressive supranuclear palsy: basic differences with Alzheimer disease and aging. Neurosci Lett 119: 182-186. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Ishihara T, Zhang B, Hong M, Andreadis A, Trojanowski JQ, Lee VM-Y ( 2002) Transgenic mouse model of tauopathies with glia pathology and nervous system degeneration. Neuron 35: 433-446. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Shiomura Y, Okabe S ( 1988) Tau proteins: the molecular structure and mode of binding on microtubules. J Cell Biol 107: 1449-1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Sato-Yoshitake R, Kobayashi N, Pfister KK, Bloom GS, Brady ST ( 1991) Kinesin associates with anterogradely transported membranous organelles in vivo. J Cell Biol 114: 295-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante C, Ramos-Morales F, Fedriani C, Bornens M, Rios RM ( 1999) GMAP-210, A cis-Golgi network-associated protein, is a minus end microtubule-binding protein. J Cell Biol 145: 83-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM-Y ( 1999) Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 24: 751-762. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Orecchio LD, Binder L, Trojanowski JQ, Lee VM-Y, Lee G ( 1988) Epitopes that span the tau molecule are shared with paired helical filaments. Neuron 1: 817-825. [DOI] [PubMed] [Google Scholar]
- Kreis TE ( 1987) Microtubules containing detyrosinated tubulin are less dynamic. EMBO J 6: 2597-2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer G, Liao G, Gundersen GG ( 1999) Detyrosination of tubulin regulates the interaction of intermediate filaments with microtubules in vivo via a kinesin-dependent mechanism. Mol Biol Cell 10: 1105-1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larcher JC, Boucher D, Lazereg S, Gros F, Denoulet P ( 1996) Interaction of kinesin motor domains with α- and β-tubulin subunits at a tau-independent binding site. Regulation by polyglutamylation. J Biol Chem 271: 22117-22124. [DOI] [PubMed] [Google Scholar]
- Lee VM-Y, Goedert M, Trojanowski JQ ( 2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24: 1121-1159. [DOI] [PubMed] [Google Scholar]
- Liao G, Gundersen GG ( 1998) Kinesin is a candidate for cross-bridging microtubules and intermediate filaments. Selective binding of kinesin to detyrosinated tubulin and vimentin. J Biol Chem 273: 9797-9803. [DOI] [PubMed] [Google Scholar]
- MacRae TH ( 1997) Tubulin post-translational modifications—enzymes and their mechanisms of action. Eur J Biochem 244: 265-278. [DOI] [PubMed] [Google Scholar]
- Manthorpe M, Rudge JS, Varon S ( 1986) Astroclial cell contributions to neuronal survival and neuritic growth. In: Astrocytes (Fedoroff S, Vernadakis A, eds). Orlando, FL: Academic.
- Marya PK, Syed Z, Fraylich PE, Eagles PA ( 1994) Kinesin and tau bind to distinct sites on microtubules. J Cell Sci 107: 339-344. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J ( 1980) Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85: 890-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrick SE, Trojanowski JQ, Lee VM-Y ( 1997) Selective destruction of stable microtubules and axons by inhibitors of protein serine/threonine phosphatases in cultured human neurons (NT2N cells). J Neurosci 17: 5726-5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mourelatos Z, Yachnis A, Rorke L, Mikol L, Gonatas NK ( 1993) The Golgi apparatus of motor neurons in amyotrophic lateral sclerosis. Ann Neurol 33: 608-615. [DOI] [PubMed] [Google Scholar]
- Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC ( 1996) The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu, Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci USA 93: 5472-5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister KK, Wagner MC, Stenoien DL, Brady ST, Bloom GS ( 1989) Monoclonal antibodies to kinesin heavy and light chains stain vesicle-like structures, but not microtubules, in cultured cells. J Cell Biol 108: 1453-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prahlad V, Yoon M, Moir RD, Vale RD, Goldman RD ( 1998) Rapid movements of vimentin on microtubule tracks: kinesin-dependent assembly of intermediate filament networks. J Cell Biol 143: 159-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A ( 2002) Tau is essential to β-amyloid-induced neurotoxicity. Proc Natl Acad Sci USA 99: 6364-6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai A, Okamoto K, Fujita Y, Nakazato Y, Wakabayashi K, Takahashi H, Gonatas NK ( 2000) Fragmentation of the Golgi apparatus of the ballooned neurons in patients with corticobasal degeneration and Creutzfeldt-Jakob disease. Acta Neuropathol 100: 270-274. [DOI] [PubMed] [Google Scholar]
- Seubert P, Mawal-Dewan M, Barbour R, Jakes R, Goedert M, Johnson GV, Litersky JM, Schenk D, Lieberburg I, Trojanowski JQ, Lee V. M.-Y ( 1995) Detection of phosphorylated Ser262 in fetal tau, adult tau, and paired helical filament tau. J Biol Chem 270: 18917-18922. [DOI] [PubMed] [Google Scholar]
- Shin RW, Iwaki T, Kitamoto T, Tateishi J ( 1991) Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer's disease brain tissue. Lab Invest 64: 693-702. [PubMed] [Google Scholar]
- Spillantini MG, Goedert M ( 1998) Tau protein pathology in neurodegenerative diseases. Trends Neurosci 21: 428-433. [DOI] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM ( 2002) Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 156: 1051-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieber A, Mourelatos Z, Gonatas NK ( 1996) In Alzheimer's disease the Golgi apparatus of a population of neurons without neurofibrillary tangles is fragmented and atrophic. Am J Pathol 148: 415-426. [PMC free article] [PubMed] [Google Scholar]
- Tanemura K, Murayama M, Akagi T, Hashikawa T, Tominaga T, Ichikawa M, Yamaguchi H, Takashima A ( 2002) Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J Neurosci 22: 133-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinczek B, Ebneth A, Mandelkow EM, Mandelkow E ( 1999) Tau regulates the attachment/detachment but not the speed of motors in microtubule-dependent transport of single vesicles and organelles. J Cell Sci 112: 2355-2367. [DOI] [PubMed] [Google Scholar]
- Vogelsberg-Ragaglia V, Bruce J, Richter-Landsberg C, Zhang B, Hong M, Trojanowski JQ, Lee VM-Y ( 2000) Distinct FTDP-17 missense mutations in tau produce tau aggregates and other pathological phenotypes in transfected CHO cells. Mol Biol Cell 11: 4093-4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, McGeer PL, McGeer EG ( 1992) Appearance of paired nucleated, tau-positive glia in patients with progressive supranuclear palsy brain tissue. Neurosci Lett 135: 99-102. [DOI] [PubMed] [Google Scholar]
- Yen TJ, Gay DA, Pachter JS, Cleveland DW ( 1988) Autoregulated changes in stability of polyribosome-bound beta-tubulin mRNAs are specified by the first 13 translated nucleotides. Mol Cell Biol 8: 1224-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]