

Abstract
The presence of protein aggregates in the nervous system is associated with various pathological conditions, yet their contribution to disease mechanisms is poorly understood. One type of aggregate, the aggresome, accumulates misfolded proteins destined for degradation by the ubiquitin-proteasome pathway. Peripheral myelin protein 22 (PMP22) is a short-lived Schwann cell (SC) protein that forms aggresomes when the proteasome is inhibited or the protein is overexpressed. Duplication, deletion, or point mutations in PMP22 are associated with a host of demyelinating peripheral neuropathies, suggesting that, for normal SC cell function, the levels of PMP22 must be tightly regulated. Therefore, we speculate that mutant, misfolded PMP22 might overload the proteasome and promote aggresome formation. To test this, sciatic nerves of Trembler J (TrJ) neuropathy mice carrying a leucine-to-proline mutation in PMP22 were studied. In TrJ neuropathy nerves, PMP22 has an extended half-life and forms aggresome-like structures that are surrounded by molecular chaperones and lysosomes. On the basis of these characteristics, we hypothesized that PMP22 aggresomes are transitory, linking the proteasomal and lysosomal protein degradation pathways. Here we show that Schwann cells have the ability to eliminate aggresomes by a mechanism that is enhanced when autophagy is activated and is primarily prevented when autophagy is inhibited. This mechanism of aggresome clearance is not unique to peripheral glia, because L fibroblasts were also capable of removing aggresomes. Our results provide evidence for the involvement of the proteasome pathway in TrJ neuropathy and for the role of autophagy in the clearance of aggresomes.
Keywords: neuropathy, Schwann cells, myelin, protein aggregation, peripheral myelin protein 22, autophagy
Introduction
Intracellular protein aggregates are a common feature of numerous CNS and PNS disorders (Berke and Paulson, 2003). The mechanism by which aggregates are formed is not fully understood, but, in many conditions, it involves the misfolding of a protein into a state favoring its aggregation (Kopito and Ron, 2000; Sherman and Goldberg, 2001). A shared characteristic among cytoplasmic aggregates is the presence of molecular chaperones and components of the ubiquitin-proteasome pathway (Sherman and Goldberg, 2001; Garcia-Mata et al., 2002). One type of inclusion, the aggresome, is an assembly of protein aggregates that forms when the activity of the proteasome is inhibited (Johnston et al., 1998). A growing number of disease-associated proteins have been found to accumulate in aggresomes, including peripheral myelin protein 22 (PMP22) (Notterpek et al., 1999; Ryan et al., 2002), huntingtin (Waelter et al., 2001), parkin, and α-synuclein (Junn et al., 2002). The aggregation of these proteins is thought to be involved in the pathogenesis of peripheral neuropathies, Huntington's and Parkinson's diseases, respectively, and both protective and toxic roles have been proposed (Kopito, 2000).
PMP22 is a hydrophobic Schwann cell (SC) glycoprotein whose precise role in PNS myelin is unknown (Naef and Suter, 1998). Duplication of or point mutations within the PMP22 gene are associated with various demyelinating peripheral neuropathies, including Charcot-Marie-Tooth disease type 1A (CMT1A) (Lupski et al., 1991). One of these point mutations (Leu16Pro), carried by the Trembler J (TrJ) mouse, accurately reproduces the pathological findings of CMT1A nerves (Suter et al., 1992; Notterpek and Tolwani, 1999). Because in normal SCs the majority of the newly synthesized PMP22 is rapidly turned over, presumably by the proteasome (Pareek et al., 1997), we hypothesized that overproduction or misfolding of mutated PMP22 might overwhelm the protein degradative pathway. Indeed, in vitro studies indicate that overproduced wild-type (Wt) and mutated TrJ and Trembler (Tr) PMP22s form aggresomes when the proteasome is inhibited (Notterpek et al., 1999; Ryan et al., 2002). Comparison of the Tr (Gly150Asp) and TrJ mutations suggests that the tendency of mutant PMP22s to aggregate may correlate with disease severity (Tobler et al., 2002). Furthermore, an unbiased study of three non-naturally occurring PMP22 point mutations shows that the accumulation of mutant PMP22s in large perinuclear aggregates might be protective (Isaacs et al., 2002). Therefore, in vivo and in vitro studies support the involvement of PMP22 aggregates in the pathogenesis of CMT1A neuropathies. Nevertheless, the presence or the potential role of PMP22 aggregates in neuropathy nerves has not been examined.
Here we show that, in TrJ nerves, PMP22 has an extended half-life and accumulates in the perinuclear region of the SCs. These cytosolic PMP22 aggresome-like structures frequently associate with molecular chaperones, including heat shock cognate protein 70 (Hsc70), and recruit lysosomes, suggesting that these systems may be involved in the clearance of aggresomes. Indeed, SCs are able to eliminate PMP22 aggresomes by an autophagy-mediated mechanism. These studies provide additional evidence for the involvement of the proteasome in PMP22 neuropathies and support the idea that aggresomes are transitory structures linking the ubiquitin-proteasome and lysosomal pathways.
Materials and Methods
Mouse colonies. TrJ (The Jackson Laboratory, Bar Harbor, ME) and PMP22-deficient (Adlkofer et al., 1995) mouse breeding colonies were housed under specific pathogen-fee conditions at the McKnight Brain Institute animal facility. The use of animals for these studies has been approved by the University of Florida Institutional Animal Care and Use Committee. Genomic DNA was isolated from tail biopsies of younger than 10-d-old mice, and litters were genotyped by PCR followed by BanI enzymatic digestion (TrJ) or Southern blots (PMP22 deficient) (Adlkofer et al., 1995; Notterpek et al., 1997). For all experiments, unless otherwise specified, age-matched, 1-year-old heterozygous TrJ and Wt mice were used.
Metabolic labeling. Freshly isolated sciatic nerves from 18-d-old Wt, TrJ, and heterozygous PMP22-deficient (+/-) mice were metabolically labeled with 0.4 mCi/ml trans35S (ICN Biochemicals, Costa Mesa, CA) (Pareek et al., 1997). For the chase experiments, samples were incubated with an excess of cold methionine and cysteine for 0, 1, 2, 4, 6, 8, and 24 hr. Nerve pieces were then lysed for 45 min at 4°C with radioimmunoprecipitation buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% deoxycholate, 0.5% Nonidet P-40, and 0.1% SDS) supplemented with a mixture of protease inhibitors (Complete; Roche Products, Indianapolis, IN). PMP22 was immunoprecipitated with a rabbit polyclonal anti-PMP22 antibody, as described previously (Pareek et al., 1997). Treatment with endoglycosidase H (endo H) (New England Biolabs, Beverly, MA) was performed according to the instructions of the manufacturer. Samples were separated on 4-15% acrylamide gradient gels (Bio-Rad, Hercules, CA), and the gels were fixed and treated with AMPLIFY (Amersham Biosciences, Arlington Heights, IL). Dried gels were exposed to Kodak XAR film (Eastman Kodak, Rochester, NY) at -80°C. The half-lives of Wt and mutant PMP22s were determined quantitatively by densitometry using Scion Image software (Scion, Frederick, MD) in three separate experiments. The decay curve was plotted as the percentage of total trans35S-labeled PMP22 remaining at the different time intervals (± SEM).
Western blot analyses. The detergent partitioning procedure for aggresome characterization has been described previously (Johnston et al., 1998; Ryan et al., 2002). Briefly, frozen sciatic nerves were crushed under liquid nitrogen and lysed in immunoprecipitation buffer (IPB) (10 mm Tris-HCl, pH 7.5, 5 mm EDTA, 1% NP-40, 0.5% deoxycholate, and 150 mm NaCl) supplemented with protease inhibitors. The lysates were microcentrifuged, the supernatant was removed, and the insoluble material was incubated with 10 mm Tris-HCl and 1% SDS for 10 min. After the addition of 150 μl of IPB, the pellets were briefly sonicated and total protein was determined. Samples were separated on 12.5% SDS gels and transferred onto nitrocellulose membranes. Blots were blocked and incubated with the indicated primary antibodies. After incubation with anti-rabbit or anti-mouse horseradish peroxidase-conjugated secondary antibodies (Sigma, St. Louis, MO), membranes were reacted with an enhanced chemiluminescent substrate (PerkinElmer Life Sciences, Boston, MA). Films were digitally imaged using a GS-710 densitometer (Bio-Rad).
Primary antibodies. To detect PMP22 in the studied samples, rabbit polyclonal (for the mouse nerves) (Pareek et al., 1997) and mouse monoclonal (for rat SCs) (Chemicon, Temencula, CA) antibodies were used. Polyclonal rabbit anti-Gsa7 (glucose-induced autophagy 7 protein, an ubiquitin-activating enzyme-like molecule) (Dorn et al., 2001) and monoclonal rat FITC-conjugated anti-transferrin receptor (Chemicon) antibodies were used to label early autophagosomes and early endosomes, respectively. Protein chaperone antibodies used included αB-crystallin, calreticulin, heat shock protein 70 (Hsp70), and Hsc70 (all from Stressgen, Victoria, British Columbia, Canada). Antibodies against the 11S and 19S proteasome subunits (Affinity Research Products, Exeter, UK), the lysosome-associated membrane protein 1 (LAMP1) (clone 1D4B; Developmental Studies Hybridoma Bank, Iowa City, IA), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (loading control) (clone 1D4; EnCor Biotechnology, Alachua, FL), and ubiquitin (Dako, Carpinteria, CA) were obtained from the indicated commercial suppliers.
Immunostaining of teased nerve fibers. Sciatic nerves from 1-year-old Wt and TrJ mice were fixed in 4% paraformaldehyde and teased into single fibers (Arroyo et al., 1999). After drying, samples for aggresome detection were permeabilized with methanol (5 min, -20°C) or with 0.2% Triton X-100 (20 min, room temperature) for the transferrin receptor (endosome marker). Twenty percent normal goat serum in PBS was used to block nonspecific binding sites. Because of the high levels of endogenous mouse Igs in TrJ nerves, only rat origin monoclonal and rabbit polyclonal antibodies were used for the double-immunolabeling studies (Notterpek et al., 1997). After an overnight incubation at 4°C, bound primary antibodies were detected with goat anti-rabbit Texas Red and goat anti-rat FITC-conjugated Alexa dyes (Molecular Probes, Eugene, OR). Hoechst dye was included with the secondary antibodies. Coverslips were mounted using the Prolong Antifade kit (Molecular Probes), and images were acquired with a SPOT digital camera attached to a Nikon (Tokyo, Japan) Eclipse 1000 microscope or an Olympus Optical (Tokyo, Japan) MRC-1024 confocal microscope. Images were processed for printing by using Adobe Photoshop 5.0 (Adobe Systems, San Jose, CA).
Ultrastructural studies. In parallel with the biochemical studies, nerve samples were fixed in 2% paraformaldehyde and 1% glutaraldehyde in Tyrode's buffer, pH 7.4, for 60 min. The specimens were then treated with 2% OsO4 in 0.1 m sodium cacodylate, pH 7.5, dehydrated, and embedded in Taab. The samples were then sectioned, poststained with lead citrate and uranyl acetate, and examined on a Jeol (Peabody, MA) 100CX transmission electron microscope.
Aggresome formation and removal. Primary rat SC cultures were established and maintained as described previously (Notterpek et al., 1999). Mouse L fibroblasts were purchased from American Type Culture Collection (Rockville, MD) and maintained in DMEM with 10% fetal calf serum (HyClone, Logan, UT). SCs or L fibroblasts on glass coverslips were treated with 10 μm lactacystin (Calbiochem, La Jolla, CA) to inhibit the proteasome or dimethlysulfoxide (DMSO) (Sigma) as a control. After 16 hr incubation, a subset of lactacystin-treated cells was immediately processed for immunostaining to visualize aggresomes (Notterpek et al., 1999), whereas the rest were allowed to recover for 8 or 24 hr washout periods. During the 8 hr washout period, parallel samples were incubated with starvation medium (amino acid- and serum-free) to stimulate autophagy (Aplin et al., 1992) or 3-methyladenine (3-MA) (10 mm) (Sigma) to block autophagy (Dorn et al., 2001). After each paradigm, cells were immunolabeled to evaluate the presence of PMP22 aggresomes (Notterpek et al., 1999). Cell viability was monitored by Trypan blue exclusion after each pharmacological treatment (Stefanis et al., 2001). Bromodeoxyuridine (BrdU) incorporation during the washout periods was performed using a BrdU labeling and detection kit (Roche Products). Cells with PMP22 aggresomes were counted and expressed as percentage of total number of cells evaluated by nuclear staining with Hoechst 33258 (Molecular Probes). Statistically significant differences between the various paradigms were determined using a Student's t test.
Results
PMP22 accumulates in cytoplasmic aggregates in TrJ nerves
In normal nerves, the majority of newly synthesized Wt PMP22 is rapidly degraded from the endoplasmic reticulum (ER), likely attributable to inefficient folding (Pareek et al., 1997). In agreement, in proteasome-inhibited rat SCs, PMP22 accumulates in cytoplasmic, perinuclear, membrane-free structures termed aggresomes (Notterpek et al., 1999). Similarly, overexpressed Wt and mutated PMP22 also form aggresomes (Ryan et al., 2002). On the basis of these in vitro observations, we hypothesized that disease-associated mutations in PMP22 will further interfere with the correct folding of the newly synthesized protein and potentially saturate the proteasomal pathway, resulting in aggresome formation. To explore this idea, pulse-chase analyses in sciatic nerves from Wt, heterozygous PMP22-deficient (+/-), and TrJ mutant mice (Fig. 1A,B) were performed. In Wt mouse nerves, the newly synthesized PMP22 has a short half-life (∼45 min) and is mostly sensitive to endo H digestion (Fig. 1A,B) (Pareek et al., 1997). In comparison, the turnover rate of PMP22 in TrJ nerves is reduced by approximately eightfold, with nearly 75% of the newly synthesized protein remaining at the 2 hr chase time point (Fig. 1A,B). These data are in agreement with our previous findings in rat SCs, in which overexpressed TrJ PMP22 had an increased half-life compared with Wt PMP22 (Ryan et al., 2002). Extended chase time points reveal that, in TrJ nerves, the half-life of PMP22 is ∼4 hr (data not shown). Endoglycosidase treatments of the immunoprecipitated PMP22 at the early (1-2 hr) (Fig. 1A), as well as late, chase time points (4-8 hr) (data not shown) demonstrate that, in affected nerves, the majority of the newly synthesized endo H-sensitive PMP22 accumulates before the medial Golgi compartment (Fig. 1A). PMP22 from sciatic nerves of PMP22 +/- mice behaves as in Wt nerves (Fig. 1B), suggesting that the effects on PMP22 turnover were attributable to the presence of the TrJ allele rather than the absence of a Wt allele.
Figure 1.
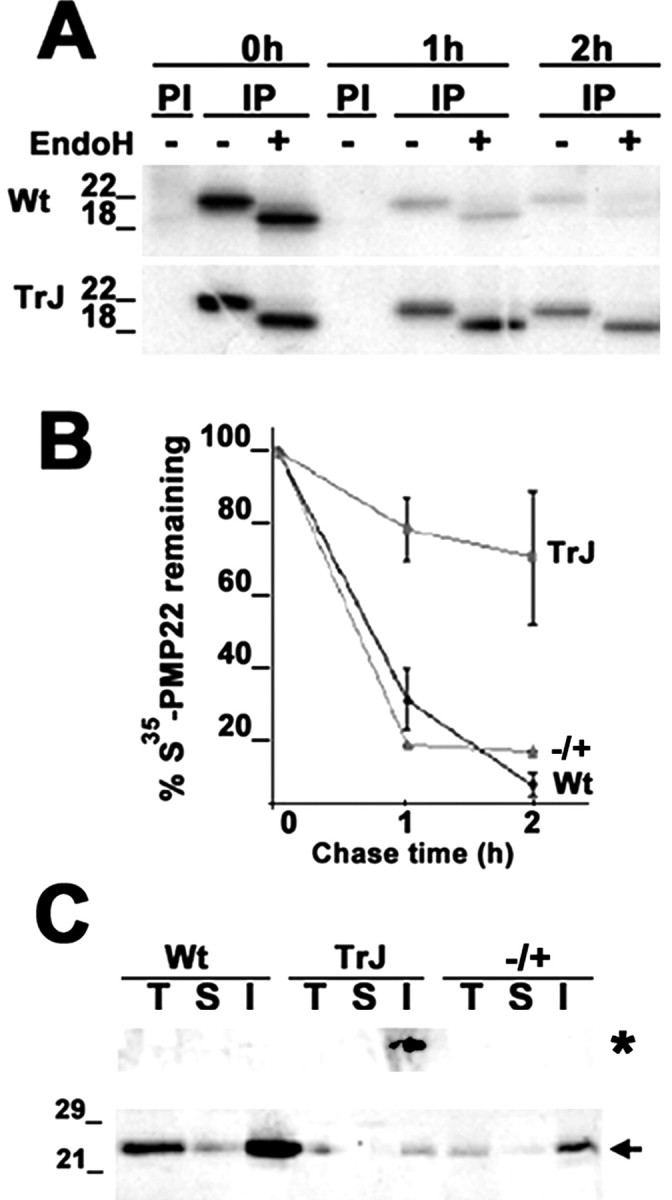
Accumulation of PMP22 in TrJ sciatic nerves. Sciatic nerves from Wt, heterozygous TrJ, and heterozygous PMP22-deficient (+/-; autoradiograph not shown) mice were metabolically labeled with trans35S, and PMP22 was immunoprecipitated from pulse (0 hr) and chase (1 and 2 hr) time points (A, B). PMP22 immunoprecipitates (IP) were incubated with (+) or without (-) endoH and separated on a 4-15% acrylamide gradient gel. PI, Preimmune. The decay curve was plotted as the percentage of total 35S-PMP22 remaining at the different chase intervals (n = 3; ±SEM) (B). Total (T), IPB-soluble (S), and IPB-insoluble (I) fractions (20 μg/lane) of nerve lysates were immunoblotted with a polyclonal anti-PMP22 antibody (C). The high molecular weight aggregates (asterisk, top of the gel) and the 22 kDa monomer (arrow) of PMP22 are indicated. Molecular mass is in kilodaltons.
The finding that the newly synthesized, endo H-sensitive PMP22 has an extended half-life in TrJ nerves (Fig. 1A) suggests that the protein may accumulate in aggresomes. To investigate the occurrence of PMP22 aggregates at the biochemical level, Wt and TrJ sciatic nerves were homogenized (T) and separated into detergent soluble (S) and detergent insoluble (I) fractions, followed by Western blot analyses (Fig. 1C) (Ryan et al., 2002). Because of demyelination in the TrJ nerves, the overall steady-state levels of the 22 kDa PMP22 are reduced (Fig. 1C, arrow) (Notterpek et al., 1997). Nevertheless, slow-migrating anti-PMP22 antibody reactive aggregates are detected at the top of the gel in the detergent-insoluble fraction of adult TrJ nerves but not in Wt or PMP22 +/- samples (Fig. 1C, asterisk). In addition, although most PMP22 is detergent insoluble in all three genotypes, this insolubility is further increased (approximately fourfold) in TrJ compared with Wt (Fig. 1C).
Compared with other cellular inclusions, a unique characteristic of aggresomes is that they are membrane free (Johnston et al., 1998). To visualize aggresomes at the subcellular level in TrJ neuropathy samples, sciatic nerves from control and affected mice were processed for transmission electron microscopy (Fig. 2). As described previously, the majority of axons are well myelinated in the adult Wt sciatic nerve (Fig. 2A) (Notterpek et al., 1997). In comparison, unmyelinated small caliber fibers, supernumerary SC profiles, and excessive extracellular matrix are common findings in TrJ samples (Fig. 2B) (Henry et al., 1983; Notterpek et al., 1997). A higher-magnification view of the boxed area in Figure 2B reveals that a fraction of TrJ SCs contains cytoplasmic, membrane-free granular protein aggregates (Fig. 2C, arrow). Similarly, amorphous protein aggregates are detected in a transverse section of a TrJ nerve fiber (Fig. 2D, arrow). These aggregates are distinct from previously described “lamellar debris” seen within areas of uncompacted myelin and the cytoplasm of some of TrJ SCs (Fig. 2D, arrowheads) (Henry et al., 1983) and resemble aggresomes composed of misfolded cystic fibrosis transmembrane regulator protein (Fig. 2C,D, arrows) (Johnston et al., 1998).
Figure 2.
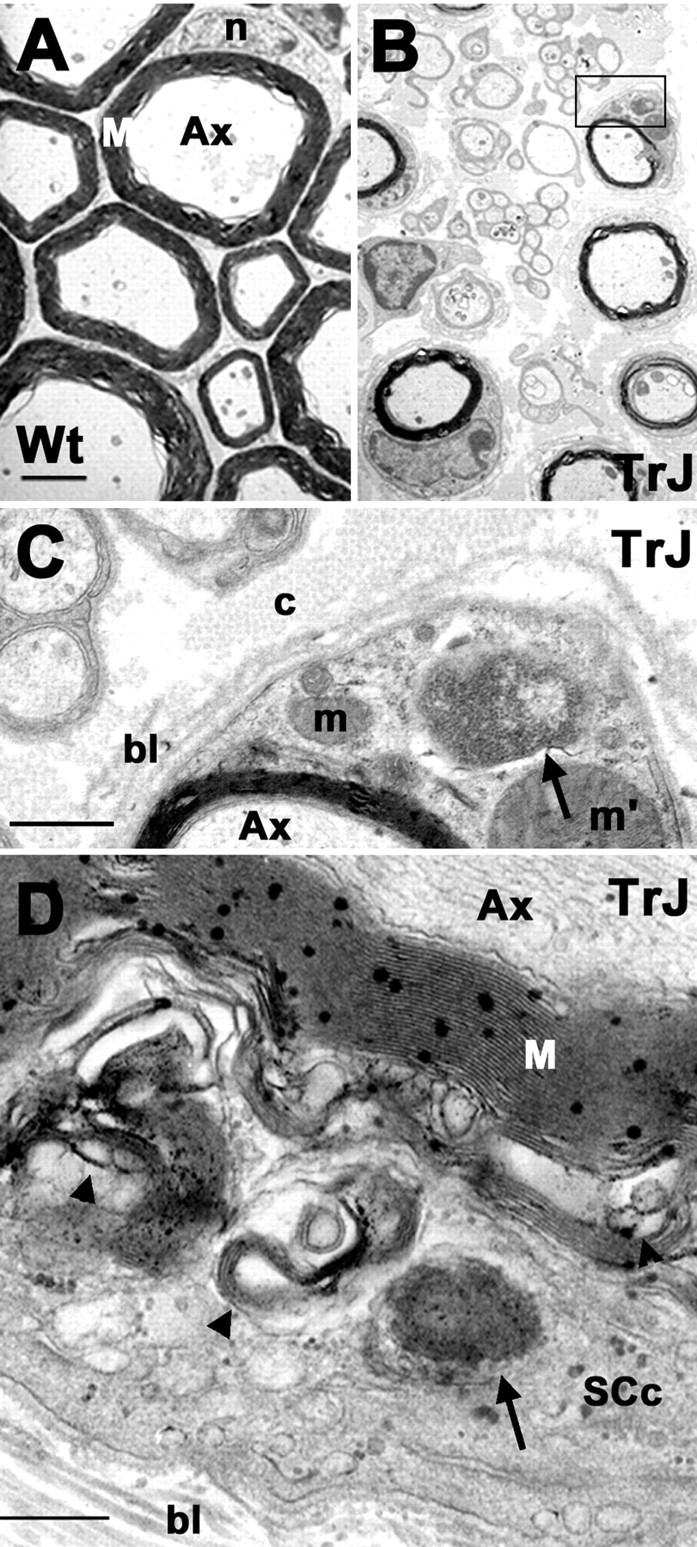
Aggresome-like structures are present in TrJ nerves. The ultrastructure of Wt (A) and TrJ (B-D) sciatic nerves are shown. A higher magnification of the area boxed in B is shown in C. Aggresome-like structures (C, D, arrows) and lamellar myelin debris (D, arrowhead) are visible in a transverse section of a TrJ nerve. Scale bars: A, B, 1 μm; C, 0.2 μm; D, 0.5 μm. Ax, Axon; bl, basal lamina; c, collagen; m, mitochondria; m′, enlarged mitochondria; M, myelin; n, nucleus; SCc, SC cytoplasm.
PMP22 aggregates recruit molecular chaperones
The formation of PMP22 aggresomes in vitro results in the upregulation of Hsps and their association with aggresomes (Ryan et al., 2002). To examine the levels and localization of Hsps in TrJ neuropathy nerves, teased fibers were processed for double immunolabeling with rabbit anti-PMP22 and rat anti-Hsc70 antibodies (Fig. 3). As described previously, in normal sciatic nerves, PMP22 is found in the compact portion of myelin and is absent from the nodes of Ranvier (Fig. 3A, inset) (Notterpek et al., 1997). In nerve fibers of TrJ mice, a fraction of PMP22 is detected in perinuclear aggregates (Fig. 3A, arrows) that are reminiscent of PMP22 aggresomes formed in vitro (Notterpek et al., 1999; Ryan et al., 2002). Less frequently, PMP22 can also be seen concentrated in distal, possibly membrane-associated structures (Fig. 3A, arrowheads). Compared with the normal nerve (Fig. 3B, inset), Hsc70-like immunoreactivity is readily detected in the TrJ sample (Fig. 3B), and Hsc70 seems to surround the perinuclear (Fig. 3B, arrows), but not the peripheral (Fig. 3B, arrowheads), PMP22-positive structures. The merged image of the PMP22- and Hsc70-stained panels shows that the two molecules do not exclusively overlap (Fig. 3C).
Figure 3.

Recruitment of cytosolic chaperones to PMP22 aggresomes. Teased sciatic nerves from Wt (A, B, insets) and TrJ (A-E) mice were double immunostained with anti-PMP22 (A, C-E, red) and anti-Hsc70 (B-D, green) or anti-transferrin receptor (E, green) antibodies. Nuclei are stained with Hoechst dye (A-C, E). In A-E, arrows indicate PMP22 aggresome-like structures, whereas arrowheads point at distal PMP22-positive structures. Scale bar: A-C, E, 10 μm. A confocal image of teased TrJ nerve fiber immunostained with anti-PMP22 (D, red) and anti-Hsc70 (D, green) antibodies is shown (D). A magnification (2×) of the perinuclear region of a TrJ SC (D, asterisk) is shown in the bottom left corner (D, box). Representative planes (D1-D4) of the composite image shown in D illustrate the spatial relationship between PMP22 and Hsc70. Distal PMP22-containing structures (E, arrowheads) costain with transferrin receptor (E, green) but not with Hsc70 (A-C). Total (T), IPB-soluble (S), and IPB-insoluble (I) fractions of Wt and TrJ sciatic nerve lysates (40 μg/lane) were analyzed by Western blots using anti-Hsp70, anti-αB-crystallin (αBc), anti-calreticulin (CRT), and anti-GAPDH (loading control) antibodies (F). Molecular mass is in kilodaltons.
To better define the relationship between PMP22-containing aggresome-like structures and Hsc70, teased fibers from TrJ mice were examined by confocal microscopy (Fig. 3D). In a composed image, Hsc70 is present in close proximity to the perinuclear PMP22 aggregate (Fig. 3D, arrows), as seen in the conventional light micrograph (Fig. 3A-C). Images collected at specific planes (Fig. 3D1-4), however, reveal that the PMP22-positive aggregates and Hsc70 do not strictly colocalize, but instead Hsc70 appears to surround the aggregates. Therefore, Hsc70 associates with PMP22 aggresomes in vitro (Ryan et al., 2002) and PMP22 aggresome-like structures in TrJ neuropathy nerves (Fig. 3). Similar to Hsc70, the small chaperone αB-crystallin is also recruited to these perinuclear PMP22 aggregates (data not shown). To better define the identity of the Hsc70-negative, PMP22-containing distal structures (Fig. 3A,C, arrowheads), TrJ nerve fibers were reacted with anti-PMP22 and anti-transferrin receptor antibodies (Fig. 3E). As the merged image reveals, a fraction of the PMP22-containing structures costains with the transferrin receptor antibody (Fig. 3E, arrowheads). Therefore, these structures most likely represent endocytosed myelin, which are also visible on the electron micrographs (Fig. 2D, arrowheads) and have been described previously (Henry et al., 1983; Notterpek et al., 1997).
To corroborate the observed prominent immunoreactivity of cytoplasmic chaperone antibodies in TrJ neuropathy samples, nerve lysates were analyzed by Western blots (Fig. 3F). Compared with Wt samples, the levels of Hsp70 (inducible form of Hsc70) and αB-crystallin are elevated in TrJ nerves (Fig. 3F). Furthermore, in agreement with our previous in vitro studies (Ryan et al., 2002), the detergent insolubility of these chaperones is increased, which likely reflects their association with PMP22 aggregates. In contrast to cytoplasmic chaperones, only a modest increase in the levels of the ER chaperones calreticulin (Fig. 3F) and calnexin (data not shown) were observed. The levels of GAPDH (protein loading control) remained primarily unaffected (Fig. 3F).
Alterations in protein degradation pathways
Lysosomes often surround PMP22 aggresomes formed in vitro, suggesting that the aggregates may serve as a staging ground for lysosomal degradation (Notterpek et al., 1999; Kopito, 2000). Coincidently, elevated lysosomal proteolysis has been implicated in the pathogenesis of TrJ neuropathy (Notterpek et al., 1997). To investigate the spatial relationship of in vivo PMP22 aggresomes with lysosomes, teased TrJ nerve fibers were double immunolabeled with anti-PMP22 and anti-LAMP1 antibodies (Fig. 4). As shown above, PMP22 accumulates in perinuclear regions in a fraction of TrJ SCs (Fig. 4A, arrows). Compared with Wt, the overall levels of LAMP1 are elevated in TrJ nerves (Notterpek et al., 1997), and LAMP1-like immunoreactivity is detected near the perinuclear PMP22 aggresome-like structures (Fig. 4B,C, arrows). When the images are merged and enlarged (Fig. 4C, inset), it becomes evident that the LAMP1-immunoreactive lysosomal vesicles are in close proximity to the PMP22 aggregates.
Figure 4.

Alterations in protein degradation pathways in TrJ nerves. Teased sciatic nerve fibers from TrJ mice were double immunostained for PMP22 (A, C, red) and LAMP1 (B, C, green). PMP22 aggresome-like structures (A, C, arrows) are surrounded by LAMP-1-immunoreactive lysosomes (B, C, arrows). An enlargement of a PMP22 aggresome-like structure and its spatial relationship with LAMP1 (C, asterisk) is shown (C, box). Total (T), IPB-soluble (S), and IPB-insoluble (I) fractions of Wt and TrJ nerve lysates (40 μg/lane) were analyzed for ubiquitin and the proteasomal subunits 11S and 19S (D). pUb, Polyubiquitinated substrates. Molecular mass is in kilodaltons.
Aggresomes assemble when the proteasome is impaired or overwhelmed, leading to the accumulation of polyubiquitinated protein substrates (Johnston et al., 1998). In agreement, elevated ubiquitin-like immunoreactivity (Ryan et al., 2002) and aggresome-like structures (Figs. 2, 3, 4) are detected in TrJ nerves. Western analysis of nerve lysates also revealed an accumulation of slow-migrating, polyubiquitinated protein complexes in affected nerves (Fig. 4D). Furthermore, a significant elevation in detergent insolubility of polyubiquitinated proteins is detected, likely reflecting their association with the aggregates (Fig. 4D). Polyubiquitination is the main targeting signal for degradation by the 26S proteasome, which is composed of a 20 S catalytic core and the regulatory complexes 11S or 19S (Hirsch and Ploegh, 2000). The levels of the 11S proteasome subunit were increased in TrJ nerves, whereas the levels of the 19S remained primarily unaltered (Fig. 4D). Because the maximal reaction velocity of the 20S proteasome is enhanced when bound to the 11S subunit (Ma et al., 1992), the elevated levels of 11S in TrJ nerves may represent an attempt of the cell to increase the degradation efficiency of newly synthesized misfolded proteins.
Implications of autophagy in the removal of TrJ aggresomes
The presence of lysosomes juxtaposed to the aggresomes (Fig. 4A-C) suggests that these degradative organelles may have a role in their removal. Because the primary avenue for the delivery of cytosolic proteins to lysosomes is autophagy (Dunn, 1990), it is probable that the aggresomes in TrJ SCs are eliminated by this mechanism. Autophagy is a vital process by which cytoplasmic cargo is engulfed by autophagosomes, which subsequently fuse with lysosomes to ensure degradation (Ohsumi, 2001). A morphological evidence of active autophagy is the presence of autophagosomes, which are characterized by being double-membrane bound (Dunn, 1990). Therefore, we investigated the occurrence of autophagosomes in TrJ sciatic nerves (Fig. 5). Various autophagic profiles are detected in the cytoplasm of TrJ SCs (Fig. 5A,B) but are noticeably absent from Wt SCs (Fig. 2A). A small autophagosome with the characteristic double-membrane border is visible (Fig. 5A, box). In the same SC cytoplasm, an amorphous electron-dense aggregate is found within an autophagosome (Fig. 5A, arrow). As autophagosomes mature, they fuse with lysosomes and give rise to the autolysosomes (Dunn, 1990). A late-stage autophagosome-autolysosome, with concentric arrays of membranes and electron-dense inclusions, is visible in the cytoplasm of a TrJ SC (Fig. 5B, arrow).
Figure 5.

Autophagosomes in TrJ nerves. Ultrastructural analyses of TrJ sciatic nerve cross-sections reveal the presence of autophagosomes (arrows) in SC cytoplasm (SCc) (A, B). An enlargement of a double-membrane autophagosome (asterisk) is shown (A, box). A late-stage autophagosome-autolysosome (arrow) and lamellar, myelin debris (arrowhead) are visible in the cytoplasm of a TrJ SC (B). Scale bar, 0.25 μm. Ax, Axon; M, myelin.
To obtain additional evidence for the involvement of autophagy in TrJ neuropathy, parallel nerve samples were reacted with a polyclonal anti-Gsa7 antibody (Fig. 6) (Dorn et al., 2001). Gsa7/Apg5 (autophagy-defective 5) is an enzyme required for two protein conjugation events that are essential for the formation of autophagosomes (Ohsumi, 2001). The first is the conjugation of Apg5 to Apg12, whereas the second is the amide linkage of MAP-LC3 (microtubule-associated protein-light chain) to the amino group of phosphatidyl-ethanolamine, located at the surface of autophagosomes. On Western blots, we detected an increase in the steady-state levels of Gsa7 in TrJ nerve lysates compared with Wt (Fig. 6A). The elevated levels of Gsa7 in the detergent-insoluble fraction of the TrJ nerve lysate could represent a transient association with the aggregates. Indeed, immunolabeling of teased TrJ nerve fibers with the Gsa7 antibody reveals bright Gsa7-like immunoreactivity in the perinuclear regions of affected SCs (Fig. 6B). In normal nerves, the levels of Gsa7 are low and the protein is diffuse (Fig. 6A, inset). The lysosomal marker LAMP1 associates with the majority of the Gsa7-positive vesicular structures (Fig. 6C,D), which likely represent early events in the fusion of autophagosomes and lysosomes. Together, these findings indicate that autophagy is activated in TrJ nerves, most likely to facilitate the removal of PMP22 aggresomes (see below).
Figure 6.
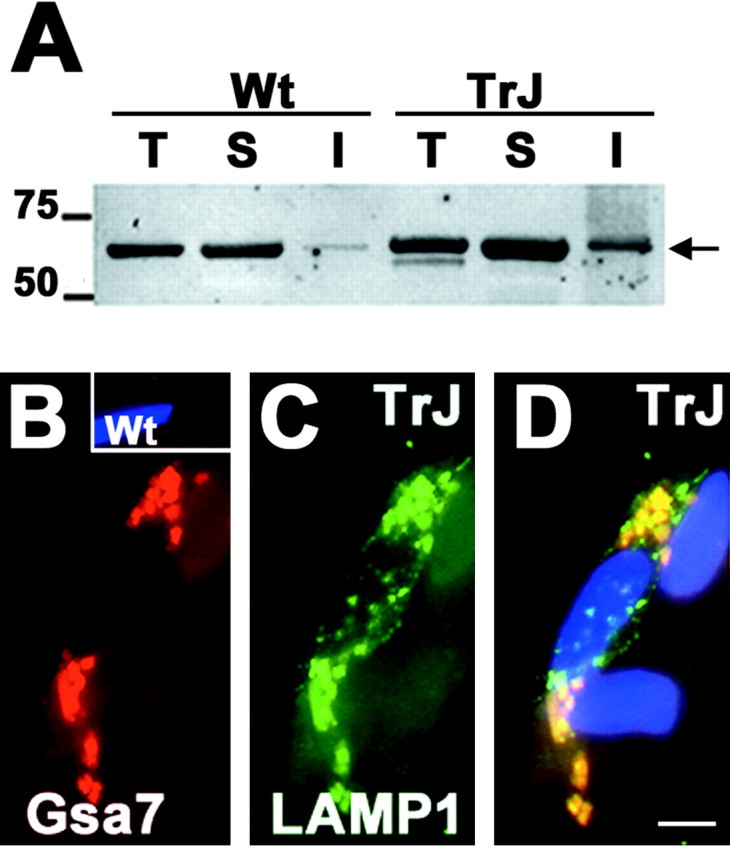
Autophagic constituents are present at perinuclear locations. Western blot analysis of a Gsa7 antibody on total (T), IPB-soluble (S), and IPB-insoluble (I) fractions of Wt and TrJ nerve lysates (40 μg/lane) (A). The levels of Gsa7 (in kilodaltons) are elevated in the IPB-insoluble fraction of the TrJ nerve. Molecular mass is in kilodaltons. Teased nerve fibers from Wt (B, inset) and TrJ (B-D) mice were double immunolabeled with the same polyclonal anti-Gsa7 (B, D, red) and a monoclonal anti-LAMP-1 (C, D, green) antibodies. Nuclei are visualized by Hoechst dye (D). Scale bar, 5 μm (D).
Aggresomes are reversible and can be cleared by autophagy
The consequences of aggresome formation are unknown, although it has been hypothesized they might result in cellular toxicity (Kopito, 2000). Nonetheless, the lack of abnormal nuclear profiles on Hoechst-stained, as well as ultrastructural, TrJ samples indicates that cell death is not a prominent feature of TrJ neuropathy (Notterpek et al., 1997). Furthermore, as shown above (Figs. 2, 3, 4), at a given time, only a subset of TrJ SCs contains aggresomes. Therefore, we hypothesized that aggresomes are transient structures, which under permissive conditions, can be removed by cells. To test this, normal rat SCs were treated with the proteasome inhibitor lactacystin for 16 hr, which results in aggresome formation in nearly all of the cells (Fig. 7A) (Notterpek et al., 1999). Subsequently, lactacystin was removed and the cells were allowed to recover in normal growth medium for 24 hr (Fig. 7B). After the 24 hr washout period, only ∼20% of the cells contained aggresomes (Fig. 7B,C). Using a BrdU incorporation assay during the washout period, cell proliferation was ruled out as a possible reason for the reduction in the number of aggresome-containing cells (data not shown). In addition, based on a Trypan blue viability assay (Stefanis et al., 2001), there was no significant difference in cell death between lactacystin-treated cells and those allowed to washout for 24 hr (data not shown). Thus, these results demonstrate for the first time that aggresomes are reversible structures once the conditions that favor their formation are eliminated.
Figure 7.
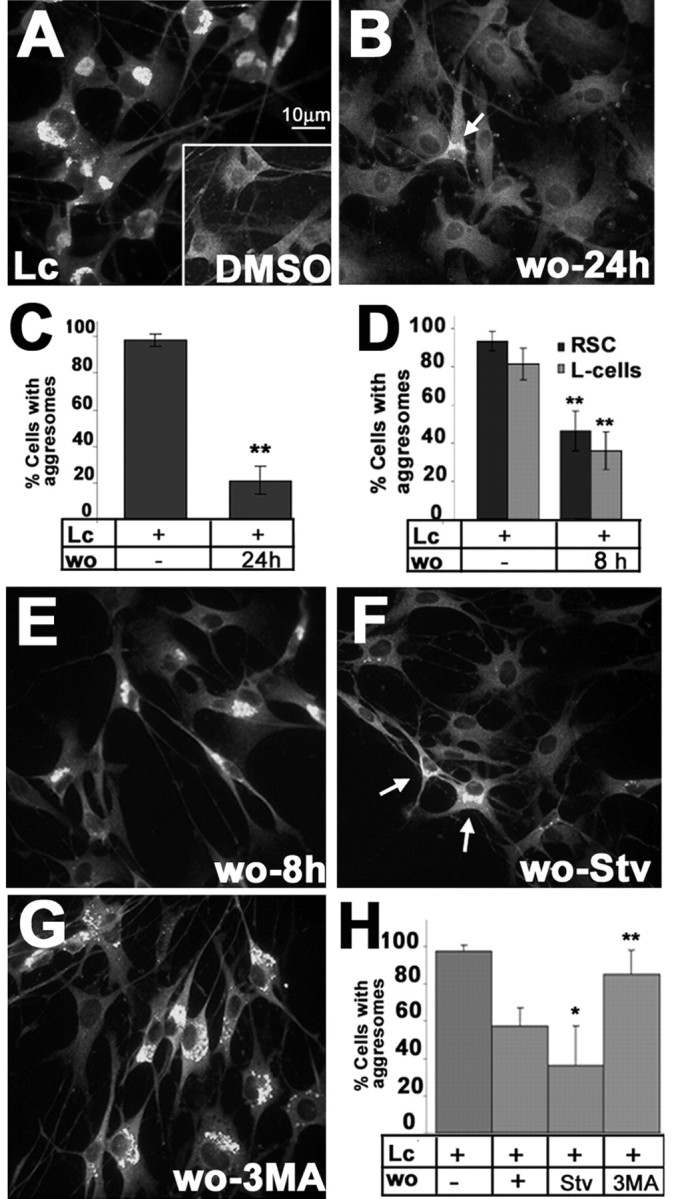
Reversibility of aggresomes in cultured SCs. SCs were treated with lactacystin (Lc) (A) or DMSO (A, inset) for 16 hr, after which the drug was removed and the cells were cultured for an additional 24 hr (B), followed by anti-PMP22 immunolabeling (A, B). An SC with a remaining PMP22 aggregate is visible (B, arrow). Scale bar, 10 μm. For each condition, cells with aggresomes were counted. The results from eight independent counts were graphed (C) (**p < 0.005). Rat SCs (RSC) and L cells were treated with Lc for 16 hr in parallel, and the percentage of cells with PMP22 aggresomes after an 8 hr washout was determined and graphed in D (**p < 0.005). RSCs treated with Lc (16 hr) (E-H) were subsequently incubated for 8 hr under the specified conditions: washout with normal media (E, wo-8 hr); starvation conditions (F, wo-Stv); or 3-methyladenine (G, wo-3MA). Arrows in F indicate two cells with remaining aggregates. Cells with aggresomes in eight independent fields were counted and graphed as a percentage of total (Hoechst dye) cells (H) (*p < 0.05; **p < 0.005).
SCs possess remarkable reparatory properties and are able to phagocytose and degrade large quantities of myelin proteins during Wallarian degeneration (Hirata and Kawabuchi, 2002). To investigate whether aggresome removal was unique to SCs, mouse L fibroblasts were subjected to the same experimental paradigm of lactacystin treatment and subsequent washout (Fig. 7D). After a 16 hr lactacystin treatment, over ∼80% of the SCs and L fibroblasts contained aggresomes (Fig. 7D). After an 8 hr washout period, only approximately one-half of these cells had aggresomes (Fig. 7D). These data indicate that L fibroblasts are able to eliminate aggresomes at approximately the same rate observed in SCs. Thus, this clearance event is not unique to peripheral glia.
The most likely mechanism by which cells can remove aggresomes is by their sequestration into autophagosomes for lysosomal degradation (Kopito, 2000). Indeed, our studies show that autophagy is an ongoing process in TrJ SCs (Figs. 5, 6). To determine whether autophagy is involved in the elimination of aggresomes, subsequent to the lactacystin treatment, pharmacologic modulators of autophagy were included in the washout medium. For these experiments, a washout period of 8 hr was chosen, at which time point ∼57% of the SCs still contain aggresomes (Fig. 7E). To stimulate autophagy during the 8 hr washout period, cells were incubated with amino acid-serum-deprived medium (Aplin et al., 1992), which resulted in a significant reduction in the number of cells with aggresomes (∼36%) (Fig. 7F,H). Conversely, when autophagy was inhibited with 3-MA (Dorn et al., 2001), the clearance of aggresomes was suppressed (Fig. 7G). The differences in the number of aggresome-containing cells between washout alone and autophagy modulators are statistically significant (Fig. 7H). There were no significant differences in cell death or cell proliferation between the cells undergoing the various 8 hr washout paradigms (data not shown). Together, these results indicate that autophagy has a substantial role in the removal of PMP22 aggresomes in SCs.
Discussion
The formation of aggresomes in neurons has been linked to several neurodegenerative disorders of the CNS (Waelter et al., 2001; Junn et al., 2002), but much less is known about protein aggregation in glial cells. PMP22 is a demyelinating peripheral neuropathy-associated SC protein that accumulates in aggresomes when the proteasome is impaired or the protein is misfolded (Notterpek et al., 1999; Ryan et al., 2002). In this report, we show that, in TrJ PMP22 neuropathy SCs, PMP22 forms perinuclear aggregates, which share characteristics of PMP22 aggresomes described in vitro (Notterpek et al., 1999; Isaacs et al., 2002; Ryan et al., 2002). Concurrently with the presence of aggresomes, TrJ nerves upregulate the lysosomal pathway (Notterpek et al., 1997) and contain autophagosomes, which are likely involved in the removal of the aggresomes. SCs in culture indeed have a remarkable capacity to clear PMP22 aggresomes by an autophagy-dependent mechanism. These results provide in vivo evidence for the involvement of aggresomes in PMP22 neuropathy nerves and for the role of autophagy in the removal of glial aggresomes.
If aggresomes are present in PMP22 neuropathy nerves, why have they not been described previously? One reason might be that, as our studies indicate, aggresomes are transitory and are only present in a fraction of the SCs at a given time. Review of the literature, however, suggests that aggresomes have been seen previously in PMP22 neuropathy specimens, but they were not identified as such. In the original studies of the Tr and TrJ mice, Henry et al. (1983) comment on the presence of non-lamellar electron-dense debris in the cytoplasm of some of the SCs. In nerve biopsies of neuropathy patients with PMP22 gene duplication, cytosolic accumulation of PMP22 was noted and confirmed by double immunolabeling with PMP22 and S100 antibodies (Nishimura et al., 1996). Studies of nerve biopsies from patients with PMP22 point mutations also revealed the retention of PMP22 in the SC cytoplasm with only partial overlap with the ER marker Bip (Hanemann et al., 2000). In agreement, whereas TrJ PMP22 has a prolonged association with the ER chaperone calnexin, likely as an attempt of correct folding, the unfolded protein response is not activated in TrJ nerves (Dickson et al., 2002). This finding points to a post-ER accumulation for the mutated PMP22. Indeed, because PMP22 is destined for degradation by the proteasome (Pareek et al., 1997; Notterpek et al., 1999), when the capacity of the proteasome is overwhelmed the protein will accumulate in the cytoplasm (Berke and Paulson, 2003).
Aggresome formation is thought to be a protective response to the accumulation of abnormal, likely misfolded proteins (Kopito, 2000; Garcia-Mata et al., 2002). Aggresomes form near the centrosome, a site enriched in proteasomal components and cell stress chaperones (Wigley et al., 1999). These aggresomes then sequester misfolded proteins in a central location, thereby increasing the efficiency of their degradation (Kopito, 2000). Indeed, preventing aggresome formation results in reduced turnover of expanded polyglutamine repeats and exacerbates cell death (Taylor et al., 2003). Similarly, the presence of large aggregates-aggresomes correlates with a less severe phenotype in three genetically engineered PMP22 mutants (Isaacs et al., 2002). Because aggresomes can further inhibit protein degradation via the proteasome, the extended presence of aggregated protein near the centrosomes is likely to induce cellular dysregulation and cell death (Bence et al., 2001).
Whereas SCs and L fibroblasts can effectively clear aggresomes under permissive conditions, it is unclear whether CNS glia and/or neurons share this ability. For example, although parkin and α-synuclein are widely expressed in the CNS and PNS, including SCs (Hase et al., 2002; Mori et al., 2002), the presence of these proteins in aggregates is associated with the selective loss of dopaminergic neurons in the substantia nigra without apparent peripheral nerve dysfunction (Giasson and Lee, 2001; Tanaka et al., 2001). Thus, it is possible that SCs, but not striatal neurons, use autophagy to clear these aggresomes. Nonetheless, it was shown previously that activation of autophagy reduced the presence of polyglutamine aggregates and ameliorated cell death in PC12 cells (Ravikumar et al., 2002). Therefore, this pathway could be pharmacologically enhanced in neurons in an attempt to reduce the toxicity of these inclusions. Alternatively, dopaminergic neurons may use autophagy, but the levels of parkin and α-synuclein may differ among cell types.
Morphological and biochemical studies indicate that autophagy is an active pathway in 1-year-old TrJ mouse neuropathy nerves. Furthermore, the in vitro pharmacologic experiments show that SCs effectively use this pathway to remove aggregates. Why then does this pathway fail to prevent the disease progression in PMP22 mutant mice? We believe that, in aged animals, this pathway might be less efficient, allowing it to become saturated. An age-related decline in macroautophagy and chaperone-mediated autophagy has been suggested previously (Cuervo and Dice, 2000; Ward, 2002). Given the progressive nature of peripheral neuropathies, and other protein aggregation disorders, it is possible that aggresomes are not prominent during the initial stages of the disease because they are continuously disposed of through autophagy. However, over time, the autophagy-lysosomal pathway gets saturated, resulting in the accumulation of protein aggregates that may become toxic if not eliminated. The prevalence of aggresomes with disease progression could explain the gain-of-function behavior of some PMP22 point mutations (Adlkofer et al., 1997; Gabreels-Festen and Wetering, 1999).
Although autophagy plays a central role in the removal of aggresomes in SCs, our studies suggest that this is not the only mechanism involved. For example, when autophagy was inhibited during the 8 hr washout period, compared with proteasome-inhibited controls, ∼15% of the cells still did not contain any type of aggregates (p = 0.01). These data indicate that either the autophagy blocking agent 3-MA was not entirely effective or autophagy is aided by other cellular events. In support of the latter, we found that the aggregates remaining during the 3-MA washout lost their initial compactness and appeared more diffuse (Fig. 7, compare A, G). This suggests that there is a step preceding autophagy that is responsible for the initial breakdown of the aggresome into less compact aggregates. Because cytosolic chaperones play an important role in the disassembly of molecular aggregates and accelerate the refolding of insoluble molecules (Cummings et al., 1998; Wang and Spector, 2000; Sherman and Goldberg, 2001), it is likely that chaperones are involved in the initial fragmentation of aggresomes. In agreement, cytoplasmic chaperones are recruited to PMP22 aggresomes both in vitro (Ryan et al., 2002) and in vivo (Fig. 3).
PMP22 is an excellent example of a protein that accumulates in aggregates, which can interfere with the normal functioning of the cell. Although cell death is not a prominent feature of PMP22-associated neuropathies, other protein aggregates are known to result in toxicity (Kopito 2000; Waelter et al., 2001). Whether enhancing the clearance of protein aggregates in different models of neurodegenerative disease will slow or block disease progression is an important question that awaits an answer. The finding that aggresome clearance can be enhanced by the stimulation of autophagy identifies a promising therapeutic approach to eliminate protein aggregates and avoid the potential toxic gain-of-function of these structures.
Footnotes
These studies were supported by the Muscular Dystrophy Association and National Institutes of Health-National Institute of Neurological Disorders and Stroke (L.N.) and the National Institutes of Health-National Cancer Institute (W.A.D.). We thank Julie Oakley and the Electron Microscopy Core Facility for technical assistance with these studies. We also thank Dr. Gerry Shaw for critical reading of this manuscript and members of the Notterpek laboratory for helpful discussions.
Correspondence should be addressed to Dr. Lucia Notterpek, Department of Neuroscience, McKnight Brain Institute, 100 Newell Drive, Box 100244, Gainesville, FL 32610-0244. E-mail: notterp@ufbi.ufl.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310672-09$15.00/0
References
- Adlkofer K, Martini R, Aguzzi A, Zielasek J, Toyka KV, Suter U ( 1995) Hypermyelination and demyelinating peripheral neuropathy in Pmp22- deficient mice. Nat Genet 11: 274-280. [DOI] [PubMed] [Google Scholar]
- Adlkofer K, Naef R, Suter U ( 1997) Analysis of compound heterozygous mice reveals the Trembler mutation can behave as a gain-of-function. J Neurosci Res 49: 671-680. [DOI] [PubMed] [Google Scholar]
- Aplin A, Jasionowski T, Tuttle DL, Lenk SE, Dunn Jr WA ( 1992) Cytoskeletal elements are required for the formation and maturation of autophagic vacuoles. J Cell Physiol 152: 458-466. [DOI] [PubMed] [Google Scholar]
- Arroyo EJ, Xu YT, Zhou L, Messing A, Peles E, Chiu SY, Scherer SS ( 1999) Myelinating Schwann cells determine the internodal localization of Kv1.1, Kv1.2, Kvbeta2, and Caspr. J Neurocytol 28: 333-347. [DOI] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, Kopito RR ( 2001) Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292: 1552-1555. [DOI] [PubMed] [Google Scholar]
- Berke SJ, Paulson HL ( 2003) Protein aggregation and the ubiquitin proteasome pathway: gaining the UPPer hand on neurodegeneration. Curr Opin Genet Dev 13: 253-261. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF ( 2000) Age related decline in chaperone-mediated autophagy. J Biol Chem 275: 31505-31513. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, Orr HT, Beaudet AL, Zoghbi HY ( 1998) Chaperone suppression of aggregation altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet 19: 148-154. [DOI] [PubMed] [Google Scholar]
- Dickson KM, Bergeron JJ, Shames I, Colby J, Nguyen DT, Chevet E, Thomas DY, Snipes GJ ( 2002) Association of calnexin with mutant peripheral myelin protein-22 ex vivo: a basis for “gain-of-function” ER diseases. Proc Natl Acad Sci USA 99: 9852-9857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn BR, Dunn Jr WA, Progulske-Fox A ( 2001) Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect Immun 69: 5698-5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn Jr WA ( 1990) Studies on the mechanisms of autophagy: formation of the autophagic vacuole. J Cell Biol 110: 1923-1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabreels-Festen A, Wetering RV ( 1999) Human nerve pathology caused by different mutational mechanisms of the PMP22 gene. Ann NY Acad Sci 883: 336-343. [PubMed] [Google Scholar]
- Garcia-Mata R, Gao YS, Sztul E ( 2002) Hassles with taking out the garbage: aggravating aggresomes. Traffic 3: 388-396. [DOI] [PubMed] [Google Scholar]
- Giasson BI, Lee VM ( 2001) Parkin and the molecular pathways of Parkinson's disease. Neuron 31: 885-888. [DOI] [PubMed] [Google Scholar]
- Hanemann CO, D'Urso D, Gabreels-Festen AA, Muller HW ( 2000) Mutation-dependent alternation in cellular distribution of peripheral myelin protein 22 in nerve biopsies from Charcot-Marie-Tooth type 1A. Brain 123: 1001-1006. [DOI] [PubMed] [Google Scholar]
- Hase A, Yamada H, Arai K, Sunada Y, Shimizu T, Matsumura K ( 2002) Characterization of parkin in bovine peripheral nerve. Brain Res 930: 143-149. [DOI] [PubMed] [Google Scholar]
- Henry EW, Cowen JS, Sidman RL ( 1983) Comparison of Trembler and Trembler-J mouse phenotypes: varying severity of peripheral hypomyelination. J Neuropathol Exp Neurol 42: 688-706. [DOI] [PubMed] [Google Scholar]
- Hirata K, Kawabuchi M ( 2002) Myelin phagocytosis by macrophages and nonmacrophages during Wallerian degeneration. Microsc Res Tech 57: 541-547. [DOI] [PubMed] [Google Scholar]
- Hirsch C, Ploegh HL ( 2000) Intracellular targeting of the proteasome. Trends Cell Biol 10: 268-272. [DOI] [PubMed] [Google Scholar]
- Isaacs A, Jeans A, Oliver P, Vizor L, Brown S, Hunter A, Davies K ( 2002) Identification of a new Pmp22 mouse mutant and trafficking analysis of a Pmp22 allelic series suggesting that protein aggregates may be protective in Pmp22-associated peripheral neuropathy. Mol Cell Neurosci 1: 114-125. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, Kopito RR ( 1998) Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143: 1883-1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junn E, Lee SS, Suhr UT, Mouradian MM ( 2002) Parkin accumulation in aggresomes due to proteasome impairment. J Biol Chem 277: 47870-47877. [DOI] [PubMed] [Google Scholar]
- Kopito RR ( 2000) Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol 10: 524-530. [DOI] [PubMed] [Google Scholar]
- Kopito RR, Ron D ( 2000) Conformational disease. Nat Cell Biol 2: E207-E209. [DOI] [PubMed] [Google Scholar]
- Lupski JR, de Oca-Luna RM, Slaugenhaupt S, Pentao L, Guzzetta V, Trask BJ, Saucedo-Cardenas O, Barker DF, Killian JM, Garcia CA ( 1991) DNA duplication associated with Charcot-Marie-Tooth disease type 1A. Cell 66: 219-232. [DOI] [PubMed] [Google Scholar]
- Ma CP, Slaughter CA, DeMartino GN ( 1992) Identification, purification, and characterization of a protein activator (PA28) of the 20 S proteasome (macropain). J Biol Chem 267: 10515-10523. [PubMed] [Google Scholar]
- Mori F, Hayashi S, Yamagishi S, Yoshimoto M, Yagihashi S, Takahashi H, Wakabayashi K ( 2002) Pick's disease: alpha- and beta-synuclein-immunoreactive Pick bodies in the dentate gyrus. Acta Neuropathol (Berl) 104: 455-461. [DOI] [PubMed] [Google Scholar]
- Naef R, Suter U ( 1998) Many facets of the peripheral myelin protein PMP22 in myelination and disease. Microsc Res Tech 41: 359-371. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Yoshikawa H, Fujimura H, Sakoda S, Yanagihara T ( 1996) Accumulation of peripheral myelin protein 22 in onion bulbs and Schwann cells of biopsied nerves from patients with Charcot-Marie-Tooth disease type 1A. Acta Neuropathol 92: 454-460. [DOI] [PubMed] [Google Scholar]
- Notterpek L, Tolwani RJ ( 1999) Experimental models of peripheral neuropathies. Lab Anim Sci 49: 588-599. [PubMed] [Google Scholar]
- Notterpek L, Shooter EM, Snipes GJ ( 1997) Upregulation of the endosomal-lysosomal pathway in the trembler-J neuropathy. J Neurosci 17: 4190-4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notterpek L, Ryan MC, Tobler AR, Shooter EM ( 1999) PMP22 accumulation in aggresomes: implications for CMT1A pathology. Neurobiol Dis 6: 450-460. [DOI] [PubMed] [Google Scholar]
- Ohsumi Y ( 2001) Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2: 211-216. [DOI] [PubMed] [Google Scholar]
- Pareek S, Notterpek L, Snipes GJ, Naef R, Sossin W, Laliberte J, Iacampo S, Suter U, Shooter EM, Murphy RA ( 1997) Neurons promote the translocation of peripheral myelin protein 22 into myelin. J Neurosci 17: 7754-7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, Duden R, Rubinsztein DC ( 2002) Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 11: 1107-1117. [DOI] [PubMed] [Google Scholar]
- Ryan MC, Shooter EM, Notterpek L ( 2002) Aggresome formation in neuropathy models based on peripheral myelin protein 22 mutations. Neurobiol Dis 10: 109-118. [DOI] [PubMed] [Google Scholar]
- Sherman MY, Goldberg AL ( 2001) Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron 29: 15-32. [DOI] [PubMed] [Google Scholar]
- Stefanis L, Larsen EL, Rideout HJ, Sulzer D, Greene LA ( 2001) Expression of A53T mutant but not wild-type α-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci 21: 9549-9560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter U, Moskow JJ, Welcher AA, Snipes GJ, Kosaras B, Sidman RL, Buchberg AM, Shooter EM ( 1992) A leucine-to-proline mutation in the putative first transmembrane domain of the 22-kDa peripheral myelin protein in the trembler-J mouse. Proc Natl Acad Sci USA 89: 4382-4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Suzuki T, Chiba T, Shimura H, Hattori N, Mizuno Y ( 2001) Parkin is linked to the ubiquitin pathway. J Mol Med 79: 482-494. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, Fischbeck KH ( 2003) Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet 7: 749-757. [DOI] [PubMed] [Google Scholar]
- Tobler AR, Liu N, Mueller L, Shooter EM ( 2002) Differential aggregation of the Trembler and Trembler J mutants of peripheral myelin protein 22. Proc Natl Acad Sci USA 99: 483-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE ( 2001) Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell 12: 1393-1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Spector A ( 2000) α-Crystallin prevents irreversible protein denaturation and acts cooperatively with other heat shock proteins to renature the stabilized partially denatured protein in an ATP-dependent manner. Eur J Biochem 267: 4705-4712. [DOI] [PubMed] [Google Scholar]
- Ward WF ( 2002) Protein degradation in the aging organism. Prog Mol Subcell Biol 29: 35-42. [DOI] [PubMed] [Google Scholar]
- Wigley WC, Fabunmi RP, Lee MG, Marino CR, Muallem S, DeMartino GN, Thomas PJ ( 1999) Dynamic association of proteasomal machinery with the centrosome. J Cell Biol 145: 481-490. [DOI] [PMC free article] [PubMed] [Google Scholar]