

Abstract
We studied the effects of phosphorylation by protein kinase A (PKA) on GABAA receptors (α1β1γ2L andα1β3γ2L) transiently expressed in HEK 293T cells. Under conditions favorable for PKA activation, currents obtained using whole-cell patch clamp of lifted cells displayed increased rate and extent of the fast phases of desensitization, decreased rate of current deactivation after GABA removal, and prolongation of brief IPSC-like currents. Mutation of serine residues (β1 S409, β3 S407, β3 S408) revealed that only β1 S409 and β3 S408 were critical for the modulatory effect of PKA on GABAA receptor currents. Additionally, repeated pulse inhibition was increased in receptors after mutation of the critical serine to glutamate and decreased when the serine was mutated to alanine. These data demonstrate that PKA phosphorylation modulated GABAA receptor currents by increasing fast phases of macroscopic desensitization and suggest a role for PKA in regulating GABAergic IPSC duration.
Keywords: protein kinase A, chloride ion channel, GABAA receptor channels, rapid application, patch clamp, mutagenesis
Introduction
Both ligand- and voltage-gated ion channels have been shown to be modulated by phosphorylation (Levitan, 1999). The GABAA receptor is a ligand-gated chloride ion channel composed of five subunit subtypes from seven families (α1-6, β1-3, γ1-3, δ1, ϵ1, π1, θ1) (Macdonald and Olsen, 1994; Olsen and Macdonald, 2002). All β subtypes contain a conserved site in the cytoplasmic loop between M3 and M4 transmembrane domains that is a consensus phosphorylation site for multiple kinases including protein kinase A (PKA), PKC, protein kinase G, and calcium calmodulin kinase II. Moreover, purified GABAA receptors were phosphorylated by exogenously applied PKA (Kirkness et al., 1989; Browning et al., 1990; Moss et al., 1992b). Specifically, fusion proteins containing the β subtype cytoplasmic loops were phosphorylated by PKA (Moss et al., 1992a; McDonald and Moss, 1997), whereas PKA did not phosphorylate similar α1 and γ2L subunit fusion proteins (Moss et al., 1992a). However, β subtype-containing receptors expressed in human embryonic kidney 293 (HEK293) cells were phosphorylated when β3, but not β2, subtype was transfected (McDonald et al., 1998). Furthermore, a kinase anchoring protein, 79/150, associated with β1 and β3, but not β2, subtypes in cultured cortical neurons (Brandon et al., 2003).
Although GABAA receptors containing β subtypes are phosphorylated by PKA, the physiological effect of the phosphorylation has been controversial. Several studies reported that PKA phosphorylation reduced GABAA receptor current (Chen et al., 1990; Porter et al., 1990; Moss et al., 1992b; Gyenes et al., 1994), whereas others reported that PKA enhanced GABAA receptor current (Cheun and Yeh, 1992; Angelotti et al., 1993; Feigenspan and Bormann, 1994; Cheun and Yeh, 1996; Kapur and Macdonald, 1996; Jones and Westbrook, 1997; Poisbeau et al., 1999). The different results reported in these studies are likely attributable to differences in techniques used to apply GABA, modulate phosphorylation, and control basal phosphorylation. To attempt to determine how acute PKA phosphorylation affects GABAA receptor current, we used rapid application of GABA to transiently transfected HEK293T cells expressing rat α1β1γ2L or α1β3γ2L receptors. To minimize effects of cytoskeletal disruption and obtain rapid solution exchange, we used a “lifted-cell” technique; entire cells were lifted from the bottom of the dish before GABA and modulator application. We controlled phosphorylation using activators (cAMP, forskolin), phosphatase [protein phosphatase 1 (PP1)], specific inhibitory peptide (PKI), culture conditions, and β subtype mutation. We focused on the role of the serines in β1 (S409) and β3 (S407, S408) subtypes shown to be phosphorylated by PKA (Moss et al., 1992a,b; McDonald and Moss, 1997; McDonald et al., 1998).
Our results suggest that intracellular conditions in HEK293T cells promoted basal phosphorylation of GABAA receptors and that S409 in β1 and S408 in β3 subtypes were critical residues that regulated GABAA receptor desensitization and deactivation. Promoting PKA phosphorylation of β subtypes increased fast phases of desensitization, slowed deactivation, prolonged brief IPSP-like GABAA receptor currents, and increased repeated pulse inhibition. These observations demonstrate the importance of GABAA receptor phosphorylation by PKA in shaping GABAA receptor currents and suggest a role for phosphorylation in regulation of GABAAergic IPSC duration.
Materials and Methods
Cell culture. HEK293T cells were grown in DMEM (Invitrogen, Carlsbad, CA) with 10% FBS (Invitrogen) and 100 IU/ml each of penicillin and streptomycin (Invitrogen) at 37°C in 5% CO2/95% air, except for serum starvation experiments in which FBS-free media were used 24 hr before recording to reduce basal levels of phosphorylation (see Results). One to 2 d before transfection, cells were passaged with 0.25% trypsin/0.2% EDTA solution and plated (200,000-400,000 cells/dish) on a 60 mm culture dish (Corning Glass Works, Corning, NY).
Transient transfection of HEK293T cells. Cells attached to the plastic surface of a 60 mm culture dish were cotransfected with pCMVrα1, pCMVrβ1 or pCMVrβ3, and pCMVrγ2L plasmids using a modified calcium phosphate precipitation method. pHook-1 (Invitrogen) was cotransfected with the GABAA receptor subunits at one-half the concentration of the subunits as a marker of positively transfected cells (Greenfield et al., 1997). The plasmid constructs were added in a 1:1:1:0.5 ratio. Because it has been suggested that differing amounts of γ2 plasmid may result in differential expression of αβγ and αβ GABAA receptors and altered current kinetic properties, we transfected some cells with 10 times the amount of γ2L subunit cDNA relative to the amount of α1 and β3 subunit cDNA. Experiments in which GABAA receptor α1, β3, and γ2L plasmid ratios were 1:1:10 exhibited current kinetic properties similar to those transfected in a 1:1:1 ratio (data not shown). Furthermore, currents from cells transfected with a 1:1:1 αβγ ratio were insensitive to 30 μm Zn2+; substantial inhibition by Zn2+ would be expected if the currents contained a substantial mix of αβγ and αβ GABAA receptors, as has been suggested (Boileau et al., 2002).
For the data presented in this study, GABAA receptor α1, β3, and γ2L plasmids and the pHook plasmid were transfected in a 1:1:1:0.5 ratio, maintaining the total amount of DNA added at 14 μg per 60 mm culture dish. Four to 5 hr after the addition of DNA, the cells were shocked for 30 sec with 15% glycerol in 2-[bis(2-hydroxyethyl)amino]ethanesulfonic acid-buffered saline. Approximately 24 hr after transfection, cells were trypsinized and collected by centrifugation at 400 × g (2000 rpm) for 10 min. Cells were then rotated in a 37°C incubator for 30-40 min to allow the surface-expressed pHook-1 coded antibody (sFv) to bind to magnetic hapten-coated beads. The marker-positive cells were then isolated using a magnetic stand and plated on 35 mm culture dishes. For serum-starved experiments, media in 35 mm dishes were serum free for 24-32 hr before recording. Electrophysiological analysis was performed 24-32 hr after plating on 35 mm dishes.
Mutagenesis. In some experiments, mutated β subunits were transfected in place of wild-type β subunits at the same relative concentrations. β Subunits were mutated using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA). Serine 409 in the β1 subunit and serines 407 and 408 in the β3 subunit were mutated to either alanine or glutamate. β1(S409A), β1(S409E), β3(S408A,S409A), β3(S408E,S409E), β3(S408A,S409E), and β3(S408E,S409A) mutants were generated. These subunits have the following flanking sequence in the area of the mutation (mutated serines underlined): β1-396, HGVPGKGRIRRRA SQLKVKI 415; β3-395, SIPHKKTHLRRR SSQLKIKI 414.
Recording solutions. Before recording, the culture dish solution was exchanged by three 2 ml washings with external recording solution containing the following (in mm): 142 NaCl, 8 KCl, 6 MgCl2, 1 CaCl2, 10 glucose, and 10 HEPES, pH 7.4; osmolarity, 320-325 mOsm). The intrapipette solution contained (in mm): 153 KCl, 1 MgCl2, 5 EGTA, 10 HEPES, and 2 Mg2+ ATP, pH 7.3; osmolarity, 298-312 mOsm). This combination of external and intrapipette solutions produced a chloride equilibrium potential (ECl) of ∼0 mV. Recording solutions were backfilled into pipettes for lifted cell (1-1.5 MΩ) and patch (5-10 MΩ) experiments, which were pulled from borosilicate capillary glass (World Precision Instruments, Sarasota, FL) on a P-87 Flaming Brown or P-2000 laser puller (Sutter Instrument Co., San Rafael, CA).
Electrophysiological recording. GABAA receptor currents were recorded using a lifted whole-cell patch-clamp technique. Currents were recorded only 42-52 hr after transfection to reduce variability observed when cells are recorded for multiple days after transfection. Small (15-20 μm; 8-20 pF) HEK293T cells with no more than two visible processes and no processes longer than the cell diameter were selected to minimize exchange time with the lifted cell technique. We did not record from cells larger than 20 μm, because cells with a capacitance >40 pF showed a decreased percentage contribution of the fastest phase of decay to the overall decay during macroscopic desensitization. Furthermore, we monitored the cell morphology carefully throughout the recording. If excessively rapid solution flow or bubbles caused the cell appearance to change such that the cell was pulling away from the electrode and positioned mostly behind the electrode tip, the cells were not used because of the degradation of space clamp and increase in access resistance that consistently occurs when the cell is markedly deformed in this manner. Signals were processed using a List EPC-7 amplifier (List Electronics, Darmstadt, Germany) or 200B amplifier (Axon Instruments, Union City, CA) in voltage-clamp mode. Signals were acquired simultaneously on a WR7400 chart recorder (Graphtec, Irvine, CA), on a videocassette recorder (Toshiba M-250), and on computer. Current amplitudes were measured on computer using the pClamp 8.2 software package (Axon Instruments). All cells were voltage clamped at -15 mV during recordings. With the range of current amplitudes (400-3000 pA) observed in this study, we saw no significant difference in current decay kinetics correlated with peak current size as has been reported previously (Bianchi and Macdonald, 2002). Most patch-clamp amplifiers (including the ones used in this study), although able to compensate for resting series resistance errors, are not able to dynamically compensate for series resistance errors introduced when channels open and the ratio of pipette to cell resistance increases dramatically (Axon Instruments, 1999). Furthermore, resting series resistance compensation did not affect peak current levels or I-V relationships of GABAA receptors transfected in HEK293T cells (Bianchi and Macdonald, 2002). Finally, voltage-step protocols are sensitive to resting series resistance errors, because the voltage step occurs before the channels open; however, no voltage-step protocols were used during any of these experiments. Because the most significant series resistance error by far was the error during channel opening and not the resting error, we chose not to perform resting series resistance compensation.
GABA and drug application system. GABA and kinase modulator application was achieved using a modified SF-77B Perfusion Fast-Step application system (Warner Instrument Corp., Hamden, CT) (Hinkle et al., 2003). The application system provided for simultaneous flow of all solutions to which the cells were exposed through three parallel glass square barrels. All step protocols began with a cell positioned in the flow of external bath solution from which the multibarreled array was repositioned such that the unmoved cell and electrode were now exposed to a drug (e.g., GABA). GABA (1 mm) was applied in all experiments, whereas forskolin (10 μm) or dideoxyforskolin (10 μm) were preapplied in some experiments. The drug application was initiated by an analog pulse triggered by the pClamp 8.2 software that caused the motor of the Warner Fast-Step to reposition the multibarrel array from one barrel to another (e.g., external solution to GABA). We added preapplication steps to the protocol for modulator experiments that enabled us to preapply a membrane-permeable modulator and rapidly wash in bath solution before GABA application. We modified the standard Warner Fast-Step configuration (Bianchi et al., 2002) to obtain rise times (10-90%) of 300-400 μsec with open-tip electrodes stepped from standard bath solution to 90% bath solution. We heated the glass with a Bunsen burner to reduce the square barrel cross-sectional area as well as to reduce the septum thickness of the glass barrels. This modification allowed us to increase flow and decrease the step distance (large step distances often result in poor exchange times), both of which contributed to decreased exchange time relative to the standard Warner setup. Additionally, we improved exchange time by increasing the stability of the unit by securing all tubing to the micromanipulator and using a long post holder (Newport, Irvine, CA) to stabilize the stepper motor unit. We repeatedly tested the open-tip electrode exchange time throughout the day to ensure that solution exchange time remained consistent for all cells from which we recorded.
Kinase activation. To assess the effect of PKA on GABAA receptor current, PKA was activated by either intracellular cAMP (300 μm) applied via the recording pipette or extracellular forskolin (10 μm; 7β-acetoxy-1α,6β,9α-trihydroxy-8,13-epoxy-labd-14-en-11-one) applied for 5 min (Sigma, St. Louis, MO). The inactive analog of forskolin, dideoxyforskolin (10 μm; 7β-acetoxy-6β-hydroxy-8,13-epoxy-labd-14-en-11-one), was applied extracellularly as a control for nonkinase-specific forskolin effects. PKA activity was blocked using intracellular application of PKI (50 μg/ml; Sigma). β Subunit phosphorylation sites were mutated using the QuikChange Site-Directed Mutagenesis kit (Stratagene) to either block receptor phosphorylation entirely with alanine substitutions of the identified serines or to mimic phosphorylation with glutamate substitutions of the serines.
Analysis of rapid application currents. Lifted cell patch data were low-pass filtered at 2 kHz, digitized at 20 kHz, and analyzed with the pClamp 8.2 software. Current decay was fit for each application duration of each cell studied. The desensitization and deactivation time courses were fit to one to three exponential functions using the Levenberg-Marquardt least squares method with the form ΣAn e-t/τn ± C, where n is the number of exponential components, t is time, A is relative amplitude of a given component at time = 0, τ is the time constant for a given component, and C is a constant to account for residual (nondesensitized) current. After initial fitting to a single component, additional components were added only if they significantly improved the fit compared with the previous exponential function(s), as determined by an F test on the sum of squared residuals. Currents elicited by 400 msec GABA applications were all fitted best with two exponential functions for desensitization. Six-second GABA-induced currents were fitted best with three exponential functions in all data analyzed here. Deactivation for 6 sec, 400 msec, and 5 msec GABA applications were all fitted best with two exponential functions. For 6 sec, 400 msec, and 5 msec GABA applications, deactivation time constants were weighted [(A1*τ1)/(A1 ± A2)*100 ± (A2*τ2)/(A1 ± A2)*100], where τ1 was the fast decay component, τ2 was the slow decay component, and A1 and A2 were the corresponding relative proportion of fast and slow components at peak current (time = 0). For all three application durations, the weighted time constant was designated τD. Extent of desensitization was defined as (peak current - current at time of GABA offset)/(peak current). All numerical and bar graph data were expressed as mean ± SEM. All measures of statistical significance were comparisons between wild-type control and the experimental condition being examined. In the case of forskolin and dideoxyforskolin experiments, the control was defined as the GABA-evoked current recorded before the modulator, and the experimental group was the GABA-evoked current recorded after modulator application. Statistical significance was assessed using an unpaired Student's t test: *p < 0.05, **p < 0.01, or **p < 0.001.
Macroscopic current modeling. Macroscopic currents were simulated using the Berkley Madonna 8.0.1 software package. The model used in simulations was based on a model that was developed using both single channel and rapid application data from α1β3γ2L GABAA receptors (Haas and Macdonald, 1999). Wild-type currents were simulated using the rate constants from the previously published model. The time constants listed here (see Fig. 9B) were corrected from the original publication that had a typographical error in which the rate constants (a1c, a2c, a3c, b1c, b2c, and b3c) for entry into distal closed states were transposed with the opening (a1o, a2o, a3o, b1o, b2o, and b3o) rates from the distal closed states. The rate constants were iteratively changed to simulate experimental data. Once a modified model was constructed to simulate experimental data, it was tested with multiple application durations and repeated pulse application to verify that the modified rate constants generated currents that were consistent with all of the acquired data.
Figure 9.

Simulated currents approximated experimentally observed changes in current time course when rate of entry into desensitization was increased. A, Wild-type current time courses were simulated using the Haas and Macdonald (1999) model. B, The time constants listed here were corrected from the original publication that had a typographical error in which the rate constants (a1c, a2c, a3c, b1c, b2c, and b3c) for entry into distal closed states were transposed with the opening (a1o, a2o, a3o, b1o, b2o, and b3o) rates from the distal closed states. Units for all rate constants were (s-1), except for koff (M-1 s-1). Currents with decreased rate and extent of rapid macroscopic desensitization and accelerated macroscopic deactivation observed under conditions unfavorable to receptor phosphorylation were simulated by increasing both fast (df) and intermediate (di) entry rates into microscopic desensitized states in by 50% or by increasing the unbinding rate (koff) by 5000%. No significant change was seen when the entry rate (ds) into the slowest microscopic desensitization state (Ds) was increased or decreased. (C, D) Decreasing df and di resulted in a simulated current time course qualitatively identical to experimental data for 6 sec, 400 msec, and 5 msec application of agonist, as did increasing koff (data not shown). E, Changing both koff and df and di resulted in simulated data with faster deactivation with 5 msec agonist applications. However, the reduction seen with increased koff was much greater than observed experimentally, whereas decreasing df and di by 50% approximated the prolongation in brief IPSC-like current well. F, When df and di were decreased 50%, the degree of repeated pulse inhibition was similar to that observed under dephosphorylating experimental conditions. A dashed line shows the peak current level of the second simulated agonist pulse using the unaltered model (w.t.) to aid comparison with the other simulations. Increasing koff resulted in simulated currents with a nearly complete loss of repeated pulse depression that was not observed experimentally. w.t., Wild type.
Results
Intracellular cAMP modestly increased desensitization of GABAA receptor currents under standard culture conditions
When GABA (1 mm) was applied rapidly for 6 sec to cells expressing α1β1γ2L or α1β3γ2L GABAA receptors, rapidly desensitizing (i.e., current decay in the persistent presence of agonist) currents were recorded (Fig. 1A,B, control internal), as described previously (Haas and Macdonald, 1999; Bianchi et al., 2001). Control α1β1γ2L and α1β3γ2L currents desensitized similarly with three exponential components to a “constant” current (supplemental Table A; available at www.jneurosci.org). No significant difference in any kinetic parameter was seen between α1β1γ2L and α1β3γ2L receptor currents (unpaired Student's t test). The addition of cAMP (300 μm) to the intracellular recording pipette solution resulted in a modest increase in the rate and extent of desensitization for both β1 and β3 subunit subtype-containing receptors (Fig. 1A,B, cAMP internal). In general, all three desensitization time constants were reduced slightly, relative amplitudes were increased, and the residual constant current was reduced by cAMP treatment. Although these data suggested that intracellular cAMP stimulated phosphorylation of an intracellular protein that increased the rate and extent of desensitization of GABAA receptor currents, the observed effects of cAMP were quite modest and not significant (supplemental Table A). This raised the possibility that intracellular proteins, including the GABAA receptors, might be constitutively phosphorylated in our expression system, thus occluding the effect of PKA activation.
Figure 1.
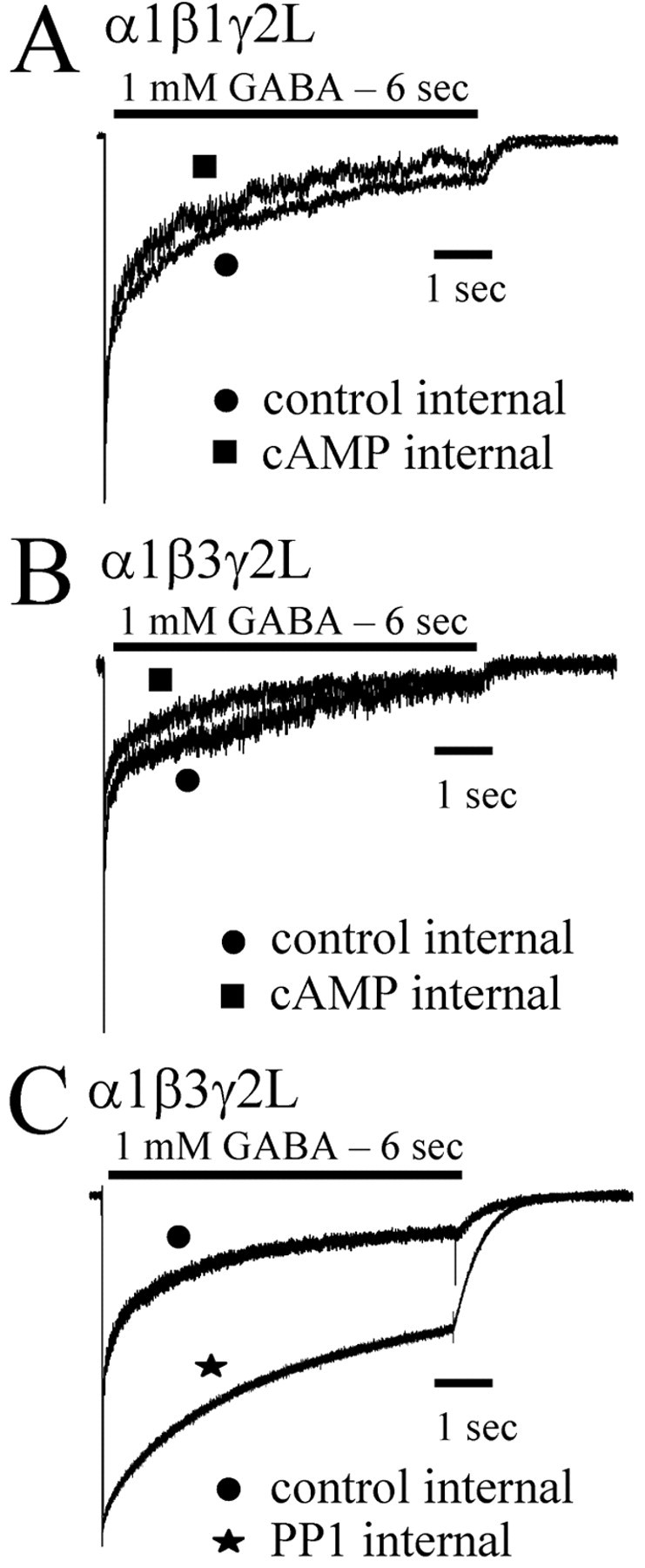
PKA modulation of α1β1γ2L and α1β3γ2L currents. GABA (1 mm) was rapidly applied for 6 sec to lifted HEK293T cells expressing α1β1γ2L (A) or α1β3γ2L (B, C) receptors. Data shown in all panels were normalized to peak and show GABA applications initiated 5 min after rupturing the patch and forming a whole-cell recording. Cells were then lifted from the surface of the dish and placed in the flow of bath solution from the multibarreled array of the rapid application system. Computer-initiated steps from the bath solution to the GABA solution elicited the recorded current. In separate populations of transfected cells, cAMP (300 μm) was included in the intracellular recording pipette along with the standard intracellular solution used in controls (A, B). cAMP caused a modest increase in the rate and extent of desensitization. C, When PP1 was included in the intracellular pipette, the rate and extent of desensitization were decreased.
PP1 catalytic subunit decreased desensitization of GABAA receptor currents
To explore the possibility that there was significant basal phosphorylation of proteins in HEK293T cells, we included PP1 catalytic subunit (200 U/μl) in the intracellular recording pipette to dephosphorylate phosphorylated serine or threonine residues. Intracellular PP1 caused a substantial decrease in the rate and extent of desensitization of α1β3γ2L GABAA receptor current (Fig. 1C). The current response to a 6 sec application of GABA in the presence of intracellular PP1 was fit to three exponential functions (supplemental Table A). Intracellular PP1 significantly increased the briefest time constant (τ1 = 21.6 ± 4.2 msec) compared with control (τ1 = 10.1 ± 2.0 msec). In addition, the relative contributions of each of the briefest two time constants (A1 = 5.3 ± 3.7%; A2 = 6.7 ± 1.1%) were decreased compared with control contributions (A1 = 23.5 ± 2.9%; A2 = 19.1 ± 3.2%). The extent of desensitization as indicated inversely by the constant term (C = 45.2 ± 4.6%) was significantly decreased compared with the control extent (C = 18.2 ± 2.6%). The reduction of desensitization and acceleration of deactivation by PP1 suggested that critical residues that influenced GABAA receptor kinetic properties were constitutively phosphorylated.
GABAA receptor currents recorded from serum-starved cells had decreased desensitization and accelerated deactivation
To explore the effects of PKA activation on GABAA receptor currents, we expressed GABAA receptors under conditions with reduced basal phosphorylation. In vitro PKA activity was reported to be downregulated in the absence of BSA (Vargas et al., 1999). PKC activity (Okuda et al., 2001) and endogenous Ca2+ channels (Berjukow et al., 1996) have been demonstrated to be upregulated by FBS in HEK293 cells. Furthermore, PKA activity in HEK293 cells has been indirectly shown to be downregulated by withdrawal of serum (Virdee et al., 2000). To determine whether the effects of cAMP on GABAA receptor currents were occluded by basal phosphorylation, we incubated the cells in serum-free media for 24 hr (serum starved) to reduce basal PKA activity before recording. Because of the extensive desensitization of currents produced by 6 sec GABA applications, shorter (400 msec) GABA applications were used. Both α1β1γ2L and α1β3γ2L GABAA receptor currents evoked by 400 msec GABA applications desensitized biexponentially to a constant current (supplemental Table B) (Haas and Macdonald, 1999; Bianchi et al., 2002). α1β3γ2L GABAA receptor currents recorded from serum-starved cells differed from those recorded from serum-fed cells (Fig. 2A). Currents from serum-starved cells had decreased the rate and extent of desensitization and accelerated deactivation (supplemental Table B). Control α1β3γ2L currents (serum-fed conditions) desensitized with two exponential components (τ1 = 10.5 ± 0.8 msec; A1 = 23.2 ± 2.5%; τ2 = 285 ± 22.4 msec; A2 = 29.2 ± 1.9%) to a residual current (C = 40.9 ± 1.8%) (supplemental Table B). With serum starvation, the briefest desensitization time constant (τ1) was increased to 17.3 ± 3.4 msec, and its relative contribution was decreased to 16.6 ± 2.2% (supplemental Table B). The slower desensitization component was unaltered, but the residual current was increased to 51.4 ± 2.1%. Thus, currents recorded from serum-starved cells had slower and less extensive desensitization because of a reduction in the rate and extent of fast desensitization.
Figure 2.

Modulation of α1β3γ2L currents by culture conditions and PKI. GABA (1 mm) was applied for 400 msec to lifted HEK293T cells expressing α1β3γ2L receptors. Serum-fed cells were cultured under standard cell conditions using DMEM with 10% FBS before recording. Serum-starved cells were cultured for 24 hr before recording in DMEM with no added FBS. In this and subsequent figures, filled symbols were used for currents from serum-fed cells, and open symbols were used for currents from serum-starved cells. Currents were normalized to peak for both panels. A, Rapid application currents elicited from serum-fed and serum-starved cells are overlaid for comparison. Desensitization was decreased, and deactivation was accelerated in serum-starved cells. B, PKI (50 μg/ml) was included in the intracellular recording pipette of some lifted cells, which were grown under standard serum-fed culture conditions. PKI reduced desensitization and accelerated deactivation in a manner similar to serum-starved cells.
Deactivation was defined as the time course of current decay after offset of GABA application and has been shown to be substantially different for different GABAA receptor subunit combinations (Haas and Macdonald, 1999). GABAA receptor currents deactivate with two time constants after 400 msec applications of GABA. Deactivation of α1β3γ2L currents was fit by two exponential functions and was reported as a weighted sum (τD) of the two time constants obtained for the fits. α1β3γ2L currents deactivated more rapidly when recorded from serum-starved (τD = 117 ± 14.6 msec) cells than from serum-fed (τD = 205 ± 28.2 msec) cells.
Enhancement of desensitization and slowing of deactivation of GABAA receptor currents from serum-fed cells was attributable to PKA phosphorylation
To determine whether the enhanced desensitization and accelerated deactivation of α1β3γ2L currents from serum-fed cells was caused by PKA phosphorylation, PKI was included in the intracellular recording pipette (50 μg/ml), and recordings were made from cells that were cultured normally (serum fed). When PKI was included in the recording pipette, the extent and rate of desensitization was decreased, and the rate of deactivation was increased (Fig. 2B). Desensitization was again fit with two exponential functions for cells containing PKI in the recording pipette (supplemental Table B). The changes produced by PKI were similar to those obtained with serum starvation. The briefer time constant (τ1) was increased to 13.2 ± 2.6 msec, and its relative amplitude (A1) was decreased to 18.2 ± 4.8%, but the changes were not statistically significant (supplemental Table B). The longer time constant (τ2 = 270 ± 52.6 msec) was not significantly different, nor was its contribution (A2 = 28.7 ± 1.5%) to peak amplitude. However, as with serum starvation, the current remaining at GABA offset was significantly increased with PKI (C = 53.1 ± 4.1%) compared with control (C = 40.9 ± 1.8%) (supplemental Table B). The currents recorded in the presence of PKI had an increased deactivation rate (Fig. 2B) similar to the effect of serum starvation (Fig. 2A). The mean deactivation rate (τD) was reduced by PKI from 205 ± 28.2 to 112 ± 11.9 msec (supplemental Table B).
These data were consistent with the premise that basal levels of PKA-mediated phosphorylation were high in HEK293T cells and that GABAA receptors were significantly phosphorylated when grown under serum-fed conditions. The effect of PKA phosphorylation appeared to involve increased desensitization and slowed deactivation because of a selective enhancement of the fast component of desensitization.
Activation of PKA by forskolin reduced desensitization and accelerated deactivation of GABAA receptor currents
All of the previous data were obtained from separate populations of cells. To determine directly whether GABAA receptor currents were acutely modulated by PKA phosphorylation, we used a multibarrelled array (see Materials and Methods) to preincubate with a PKA activator (forskolin) after obtaining control currents from the same cell. We used the membrane-permeable PKA activator forskolin because it has been shown to activate PKA and increase PKA-mediated phosphorylation of GABAA receptors in HEK293 cells (Moss et al., 1992b). Cells in which membrane-permeable activators and analogs were used were all serum starved to minimize basal phosphorylation, thus maximizing effects of the activator that would otherwise be difficult to detect under serum-fed cell culture conditions. Cells treated with forskolin were preincubated with forskolin (10 μm) for 5 min, washed in external bath solution for 1 sec, and then exposed to GABA (1 mm) for 400 msec. The wash in external solution was to eliminate any extracellular effects of forskolin on GABAA receptor currents. To keep wash times as brief as possible, we chose to use a 1 sec wash time, because longer washes (5 and 10 sec) did not alter the postwash current (data not shown). Forskolin treatment increased the rate and extent of desensitization and slowed deactivation (Fig. 3A). Forskolin decreased the fast desensitization (τ1) from 17.3 ± 3.4 to 7.1 ± 1.1 msec and increased its relative amplitude from 16.6 ± 2.2 to 39.0 ± 5.9% (supplemental Table B). The slower time constant and relative amplitude were not affected, but the residual constant component was reduced from 51.4 ± 2.1 to 30.7 ± 7.2%. Forskolin also slowed deactivation (Fig. 3A). The weighted deactivation time constant (τD) was significantly increased from 117 ± 14.6 to 188 ± 8.7 msec. No significant changes in peak current were seen over the brief period of recording we examined in this study. The inactive analog of forskolin (dideoxyforskolin) did not alter GABAA receptor current desensitization or deactivation (Fig. 3B).
Figure 3.

Modulation of α1β3γ2L currents by forskolin. GABA was applied to lifted HEK293T cells expressing α1β3γ2L GABA receptors. A, GABA (1 mm) was applied for 400 msec for all traces. After formation of a gigaohm seal, each cell was lifted from the bottom of the dish, and GABA was applied as an initial control (pre-forskolin). After the control GABA application, the lifted cell was incubated in forskolin (10 μm) for 5 min and was then washed for 1 sec in external bath solution before another 400 msec GABA application (post-forskolin). The extent of current desensitization was greater after incubation in forskolin (left). The right panel shows the same deactivation shown on the left, except the amount of current at the time of GABA offset is normalized to aid comparison. Deactivation was accelerated after forskolin application. B, No change in desensitization or deactivation was seen after dideoxyforskolin application. C, When the forskolin paradigm in A was repeated with PKI (50 μg/ml) in the intracellular recording pipette, no changes in desensitization or deactivation were observed.
To determine whether the forskolin effect was attributable to PKA phosphorylation, we repeated the forskolin treatment experiments but included PKI (50 μg/ml) in the intracellular recording pipette. In the presence of intracellular PKI, forskolin had no effect on current desensitization or deactivation (Fig. 3C). These results demonstrated that PKA modulated GABAA receptor currents by enhancing the fast component of desensitization.
Modification of desensitization and deactivation of α1β1γ2L currents by PKA was caused by phosphorylation of GABAA receptor β1 subunit serine 409
Although the previous results demonstrated that PKA modulated GABAA receptor currents, they could not distinguish between PKA phosphorylation of a receptor-coupled protein and direct receptor phosphorylation. To determine whether the effect of PKA was because of GABAA receptor phosphorylation, the β1 subtype serine 409 phosphorylation site was mutated to an alanine (S409A) to prevent phosphorylation of that serine by PKA. GABA (1 mm) was applied for both 400 msec and 6 sec durations. The briefer 400 msec applications allowed better resolution of deactivation and fast components of desensitization (Fig. 4A), whereas the longer 6 sec applications allowed resolution of the third phase of desensitization (Fig. 4B). All currents recorded from receptors containing mutated subunits were performed using normal (serum fed) culture conditions. Qualitatively, α1β1γ2L receptors containing the mutated β1 subtype [β1(S409A) receptors] had decreased the extent of desensitization and accelerated deactivation, similar to α1β1γ2L currents recorded from serum-starved cells (Fig. 4A,B).
Figure 4.

Mutation of the β1 subtype serine 409 to alanine decreased desensitization and accelerated deactivation. Wild-type α1β1γ2L receptor currents were compared with α1β1(S409A)γ2L receptor currents. A, Currents evoked by a 400 msec application of GABA (1 mm) to α1β1γ2L and α1β1(S409A)γ2L receptors were normalized to peak current. α1β1(S409A)γ2L receptor currents had decreased desensitization and accelerated deactivation compared with currents from α1β1γ2L receptors. B, Currents evoked by a 6 sec application of GABA (1 mm) to α1β1γ2L and α1β1(S409A)γ2L receptors were normalized to peak current. α1β1(S409A)γ2L receptor currents had decreased desensitization and accelerated deactivation compared with currents from α1β1γ2L receptors. Desensitization of α1β1(S409A)γ2L receptor currents was greater than desensitization observed with α1β1γ2L receptor currents evoked by 400 msec and 6 sec GABA application. Deactivation after time of GABA offset was accelerated with α1β1(S409A)γ2L receptor currents compared with α1β1γ2L receptor currents for evoked by 400 msec and 6 sec GABA application.
Desensitization of α1β1γ2L receptor currents elicited with 400 msec applications of GABA were fit to two exponential functions and a constant. Desensitization of β1(S409A) receptor currents elicited with 400 msec applications of GABA were also fit to two exponential functions and a constant (supplemental Table B). When compared with wild-type current desensitization, the briefest β1(S409A) receptor desensitization time constant (τ1 = 12.6 ± 2.0 msec) was longer than wild type (τ1 = 8.7 ± 0.9 msec) (supplemental Table B). The β1(S409A) receptor slower time constant (τ2) was not significantly different than wild type. Furthermore, the relative contribution of both exponential components (A1 = 5.7 ± 1.1%; A2 = 18.2 ± 4.8%) of desensitization were reduced for the β1(S409A) receptor compared with wild-type receptors (A1 = 21.8 ± 2.3%; A2 = 31.4 ± 3.5%), whereas the contribution of the constant term was increased in β1(S409A) receptors (C = 74.4 ± 5.2%) versus wild type (C = 46.8 ± 4.9%). The deactivation time courses of 400 msec currents were fit to two exponential functions and converted to a weighted time constant (τD). The β1(S409A) receptor deactivation (τD = 90.9 ± 13.3 msec) was faster than that of wild-type currents (τD = 158 ± 14.3 msec).
Similar results were obtained with 6 sec GABA applications (Fig. 4B). Desensitization of β1 receptor currents elicited with 6 sec GABA applications were fit to three exponential functions and a constant (supplemental Table A). Deactivation for this and all 6 sec applications were fit with two exponential functions and reported as weighted deactivation (τD). When compared with α1β1γ2L receptor currents, the briefest β1(S409A) receptor time constant (τ1 = 25.6 ± 7.3 msec) was significantly longer than wild type (τ1 = 10.3 ± 2.5 msec) (supplemental Table A). Furthermore, the relative contribution (A1 = 9.6 ± 1.5%) of the briefest time constant (τ1) to desensitization was reduced for the β1(S409A) receptor compared with wild type (A1 = 22.8 ± 4.3%), whereas the contribution of the constant term (C = 34.9 ± 5.5%) was increased in β1(S409A) receptor compared with wild type (C = 19.3 ± 5.3%). The intermediate (τ2) and longest (τ3) time constants and relative amplitudes (A2 and A3) were not changed by the β1(S409A) mutation. Mutant receptor current deactivation was faster (τD = 119 ± 9.9 msec) than wild-type current deactivation (τD = 219 ± 57.6 msec). These data suggested that PKA phosphorylation affected GABAA receptor current kinetic properties by direct receptor phosphorylation.
Modification of desensitization and deactivation of α1β3γ2L currents by PKA was attributable to phosphorylation of β3 subunit serine 408 but not serine 407
In contrast to the β1 subunit, β3 subunits contain two adjacent amino acids (S407 and S408) that are phosphorylated by PKA (McDonald et al., 1998). Because both amino acids are phosphorylated by PKA, we mutated both amino acids to either alanine (β3AA), to block receptor phosphorylation, or glutamate (β3EE), to mimic a tonically phosphorylated state. β3AA receptor currents evoked by 400 msec GABA applications desensitized biexponentially and had a larger brief time constant and faster deactivation (τ1 = 18.3 ± 3.6 msec; τD = 145 ± 14.3 msec) than wild-type receptor currents (τ1 = 10.5 ± 0.8 msec; τD = 205 ± 28.2 msec) (Fig. 5; supplemental Table B). Additionally, the β3AA receptor fast component contributed less to the desensitization (A1 = 12.9 ± 2.6%) and had a greater proportion of residual current (C = 53.6 ± 4.6%) than wild-type receptor currents (A1 = 23.2 ± 2.5%; C = 40.9 ± 1.8%). Neither the longer time constant (τ2) nor its contribution (A2) to peak current was altered in β3AA receptors (supplemental Table B). No significant differences were found between β3EE and wild-type receptors for any parameter for currents evoked by 400 msec GABA applications (supplemental Table B).
Figure 5.
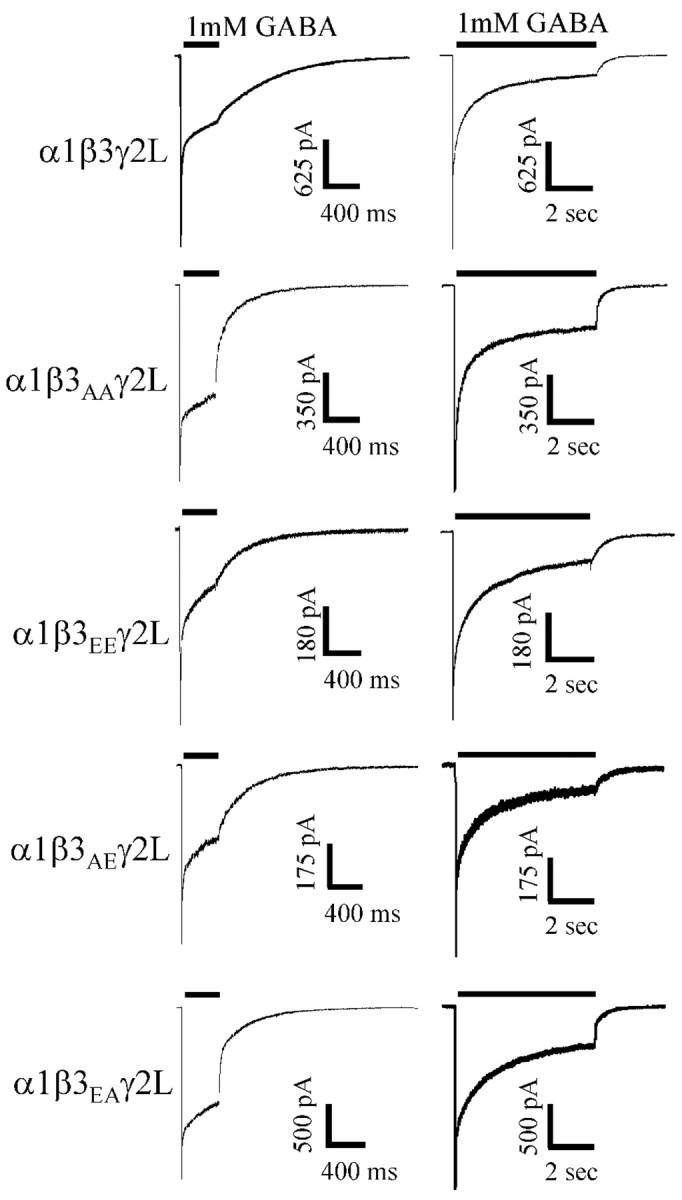
Mutation of β3 (S407A,S408A) decreased desensitization and accelerated deactivation, whereas β3(S407E, S408E)-containing receptors did not differ from wild type. Wild-type α1β3γ2L receptors were compared with α1β3(S407A,S408A)γ2L (β3AA receptors), α1β3(S407E,S408A)γ2L (β3EA), α1β3(S407A,S408E)γ2L (β3AE), and α1β3(S407E,S408E)γ2L (β3EE receptors) mutant receptors. Currents were normalized to peak. Initially, we compared β3AA receptors and β3EE receptors with wild type. A 400 msec GABA (1 mm) application to β3AA and β3EA receptors resulted in a decreased desensitization and accelerated deactivation compared with wild-type, β3EE, and β3AE GABA receptors. A 6 sec GABA (1 mm) application to β3AA and β3EA receptors resulted in an decreased desensitization and accelerated deactivation compared with wild-type β3EE, and β3AE GABA receptors.
As in previous experiments, three exponential functions were used to fit the desensitization in response to a 6 sec GABA application for α1β3γ2L wild-type receptor currents, β3AA receptor currents, and β3EE receptor currents (supplemental Table A). β3AA receptor currents had a larger brief desensitization time constant (τ1 = 34.4 ± 2.5 msec) and decreased contribution of that exponential to the overall desensitization (A1 = 11.5 ± 1.7%) compared with wild-type receptors (τ1 = 10.1 ± 2.0 msec; A1 = 23.5 ± 2.9%). There was an increase in the relative amplitude of current at GABA offset for a 6 sec application to β3AA receptors (C = 31.7 ± 2.7%) compared with wild-type receptors (C = 18.2 ± 2.6%). The deactivation of β3AA receptors (τD = 162 ± 12.4 msec) was accelerated compared with wild type (τD = 244 ± 43.7 msec). Finally, the intermediate (τ2) and slowest time constant (τ3) of the β3AA receptor were not significantly different than wild type, nor were the percentage contributions of these time constants (A2 and A3) significantly different (supplemental Table A). No significant kinetic differences between wild-type and β3EE receptor currents were found for 6 sec application durations.
Although both β3 phosphorylation sites are likely phosphorylated in HEK293 cells under our standard culture conditions (McDonald et al., 1998), we sought to determine whether S407, S408, or both were critical for PKA-induced changes in the current time course. To this end, we generated two β3 mutations [(S407A,S408E) and (S407E,S408A)] in which one phosphorylated serine was mutated to alanine and the other to glutamate to mimic phosphorylation at one site and to block it at the other site. These experiments used mutant or wild-type β3 subunits cotransfected with α1 and γ2L subunits. With 400 msec GABA applications, α1β3(S407E, S408A)γ2L (β3EA receptor) receptor current time courses were similar to those of β3AA receptor current time courses, having an increased brief desensitization time constant (τ1 = 26.7 ± 2.4 msec), a decreased contribution of the fast desensitization component to peak amplitude (A1 = 16.0 ± 2.9%), an increased residual (C = 56.9 ± 2.2%) contribution, and an accelerated deactivation (τD = 119 ± 10.3 msec) (Fig. 5; supplemental Table B) compared with wild type (above). The β3EA receptor long time constant (τ2) and relative contribution (A2) of that time constant to peak were not significantly different than wild type (supplemental Table B).
Although qualitatively similar to α1β3γ2L receptor currents (Fig. 5), β3AE receptor currents were quantitatively different with 400 msec GABA applications. The β3EA receptor brief time constant was decreased (τ1 = 6.8 ± 1.9 msec), and the slower time constant contributed less to the current (A2 = 16.9 ± 5.4%) than in wild-type receptor currents (Fig. 5). However, we were unable to resolve these or any other significant differences between β3AE receptors and wild-type receptors with 6 sec applications (Fig. 5; supplemental Table A). Six-second GABA applications to β3EA receptors resulted in differences similar to those seen for β3AA receptors. The briefest β3EA receptor time constant was longer (τ1 = 24.7 ± 0.8 msec) and contributed less to the peak amplitude of the current (A1 = 14.1 ± 1.6%) than for wild-type currents (Fig. 5; supplemental Table A). Additionally, the β3EA receptor residual current (C = 34.7 ± 1.5%) was larger than wild-type current (C = 18.2 ± 2.6%), and deactivation was faster (τD = 173 ± 3.4 msec) than wild-type deactivation (τD = 244 ± 43.7 msec) (Fig. 5). Finally, neither β3EA receptor intermediate (τ2) and long (τ3) time constants nor the relative contribution of these time constants (A2 and A3) to the peak current were significantly different than wild-type receptors.
To summarize, β3 mutant receptors in which S408 was mutated to alanine (β3AA and β3EA receptors) had current time courses that were similar to those of the serum-starved, PKI-treated, and β1(S409A) receptors, and to each other. The key macroscopic kinetic parameters that changed significantly for these groups were τ1 (increased), A1 (decreased), C (increased), and τD (decreased) (Fig. 6A,B). β3EE currents were not statistically different from wild-type currents in any measured kinetic parameter. β3AE currents were very similar to wild-type currents, but with 400 msec GABA application, the briefest time constant (τ1) was smaller (i.e., the opposite direction of the shift in β3 S408 alanine mutants) and the relative contribution (A2) of the slow time constant (τ2) was decreased.
Figure 6.

Altering the β phosphorylation site primarily altered τ1, A1, C, and τD. GABA (1 mm) was applied to lifted cells for 6 sec (A) or 400 msec (B). Selected kinetic parameters (τ1, A1, C, and τD) were expressed as a mean percentage of the value of the appropriate β subunit control. Control was defined as 100% and is indicated by a dotted line. In the case of forskolin, the percentage was calculated using the mean of the paired controls recorded before forskolin application. Only groups with significant differences from control were displayed. Groups transfected with α1β3γ2L GABAA receptors include PP1, serum starved (SS), PKI treated (β3I), forskolin treated (F), β3(S407A, S408A), and β3(S407E, S408A). One group transfected with α1β1γ2L GABAA receptors is the β1(S409A) mutant (β1A). All data displayed were significantly different from control, except for the A1 parameter for PP1 (A) and τ1 and A1 for β3I (supplemental Tables A and B).
Mutation of β3 subunit serine 408 altered brief “IPSC-like” GABAA receptor currents
Because brief applications of GABA mimic the kinetic behavior of IPSCs (Jones and Westbrook, 1995), we compared brief β3AA receptor currents to α1β3γ2L receptor and β3EE receptor currents to explore how unphosphorylatable GABAA receptors might differ from GABAA receptors mimicking full phosphorylation in response to very brief (5 msec) GABA (1 mm) applications (Fig. 7A,B). The time courses of deactivation after 5 msec GABA applications were fit with two exponential functions. Deactivation after a 5 msec application of GABA (1 mm) was accelerated in β3AA receptors compared with wild type, whereas β3EE receptors had slowed deactivation similar to wild-type receptors (supplemental Table C). Deactivation was also accelerated in β3AA for both 400 msec and 6 sec applications (Fig. 7C). The overall weighted time constant (τD) for 5 msec applications was significantly different for β3AA (τD = 46 ± 3.3 msec) and β3EA (τD = 49 ± 3.1 msec) receptors compared with control receptors (τD = 66 msec). When individual time constants and relative contributions were analyzed, only the longer time constant (τ2) was significantly different for any group (supplemental Table C). β3AA (τ2 = 132 ± 11 msec) and β3EA (τ2 = 135 ± 8.0 msec) time constant were decreased compared with wild type (τ2 = 184 ± 13 msec). Although the weighted deactivation for β3EE (τD = 95 ± 17 msec) and β3AE (τD = 81 ± 8.1 msec) receptor deactivation was slower than deactivation of wild-type receptors, neither the weighted deactivation nor individual time constants and relative contributions reached statistical significance. Based on these observations, one would expect IPSCs evoked from GABAA receptors containing phosphorylated β3 subunits to be prolonged.
Figure 7.
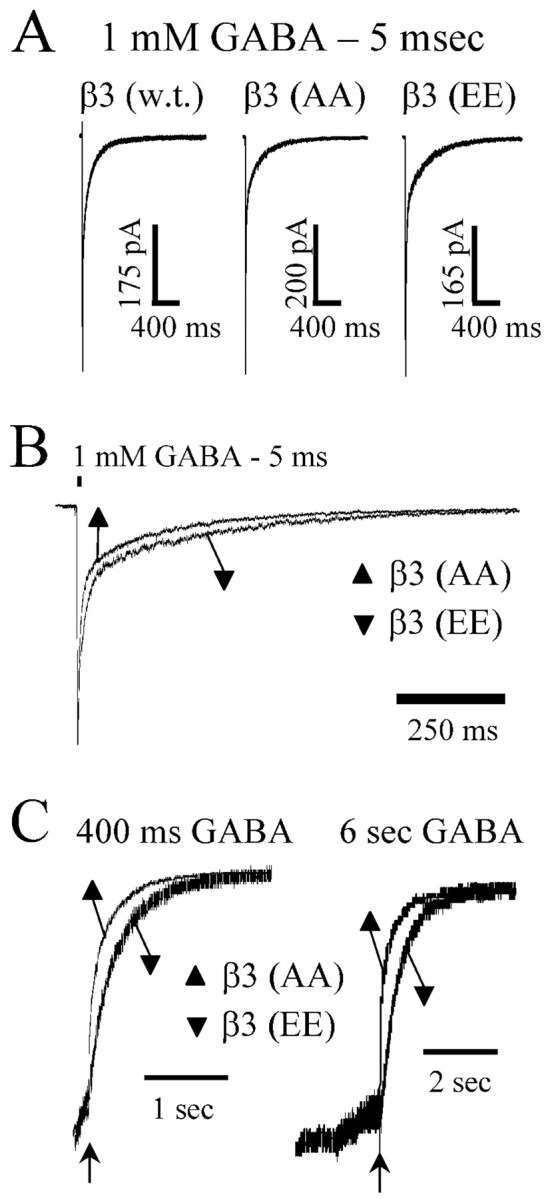
Brief (5 msec) GABA application response durations were modulated by mutation of β3 subtype serine 407 and 408 to alanine. GABA (1 mm) was applied to excised macropatches for 5 msec. A, Representative data traces are displayed for α1β3γ2L, α1β3(S407A,S408A)γ2L (β3AA), and α1β3(S407A,S408A)γ2L (β3EE) receptors. B, Data from A are normalized to peak and overlaid for comparison showing faster deactivation for β3AA receptors and slower deactivation for β3EE receptors compared with wild-type receptors. Deactivation was accelerated in β3AA receptor currents compared with β3EE and wild-type receptor currents. C, Representative data showing current deactivation after 400 msec and 6 sec GABA application. For purposes of comparison, the level of current at offset of GABA (arrow) is normalized, and most current in the presence of GABA has been excluded from the display. Deactivation was accelerated in β3AA receptor currents compared with β3EE receptor currents.
Mutation of β3 subunit serine 408 modulated repeated pulse inhibition of brief IPSC-like GABAA receptor currents
Repeated pulse protocols have been used to examine changes in microscopic desensitization in GABAA receptors (Jones and Westbrook, 1995; Bianchi and Macdonald, 2002). We performed repeated pulse experiments during which GABA (1 mm) was applied for 5 msec twice at varying interapplication intervals. The peak in response to the second GABA application was normalized (peak second current/peak initial current × 100) and displayed as a percentage of the peak of that was generated by the first GABA application. Peak currents in response to the second application of GABA for β3AA (66.9 ± 3.7%) and β3EA (69.3 ± 3.5%) receptors at 100 msec interapplication intervals were larger than wild-type currents (46.2 ± 6.0%) (Fig. 8B,C). The second pulse with β3EE (47.6 ± 0.5%) or β3AE (44.0 ± 2.0%) receptors were not significantly different than wild type at any interpulse interval (Fig. 8A,C). These results suggest that phosphorylated receptors spend more time in desensitized states than dephosphorylated receptors.
Figure 8.

Brief (5 msec) GABA repeated pulse inhibition was decreased by of β3 subtype serine 408 to alanine.GABA (1 mm) was applied for 5 msec twice in succession to excised macropatches at varying interpulse intervals (100 msec, 300 msec, 1 sec, 2 sec). Shown are representative data traces for α1β3(S407A,S408E)γ2L (β3AE) receptors (A) and α1β3(S407E,S408A)γ2L (β3EA) receptors (B). Each panel of representative data are four overlaid raw data traces aligned such that the initial currents were superimposed and each subsequent GABA current represented the second pulse of repeated pulse protocols. Each second pulse represented a separate trial on the same macropatch. C, Repeated pulse inhibition was measured for α1β3γ2L (β3), α1β3(S407A,S408A)γ2L (β3AA), α1β3(S407E,S408E)γ2L (β3EE), α1β3(S407A,S408E)γ2L (β3AE), and α1β3(S407E,S408A)γ2L (β3EA) receptors and was displayed as the percentage of second pulse peak to initial pulse peak (amplitudesecond current/amplitudeinitial current × 100). Repeated pulse inhibition was decreased for β3AA (66.9%) and β3EA (69.3%) receptors at a 100 msec interpulse interval compared with wild-type (46.2%), β3EE (47.6%), and β3AE (44.0%) receptors.
Simulated macroscopic currents closely approximated experimental data when entry into desensitized states was decreased
We used 6 sec, 400 msec, and 5 msec agonist application durations in simulating the time course of the GABAA currents using a model based on both single channel and macropatch rapid application data (Fig. 9A,B) (Haas and Macdonald, 1999). Macroscopic rapid application currents were simulated using the Berkley Madonna 8.0.1 software package. Because much of the data shown here were from experiments using lifted cells rather than macropatches, the simulated data are qualitatively the same, yet quantitatively different in some respects. When recording from lifted cells, the fast phase of desensitization is somewhat underrepresented in lifted-cell currents compared with excised macropatch currents, because of slower rise times (Bianchi and Macdonald, 2002). Consequently, when comparing lifted-cell data to simulated data, we fitted the simulated current by filtering the peak of the simulated data at 250 Hz to approximate the amount of effective peak filtering introduced by the lifted cell technique without additional external filtering. The time courses (or shapes) of the simulated currents (Fig. 9C-E, w.t.) were quantitatively similar (Table 1; supplemental Table C) to those of wild-type currents (Figs. 4, 5, 6; supplemental Tables A-C) recorded in this study. Because conditions favorable to phosphorylation resulted in current time courses that were similar to control time courses, the time course of currents simulated with the Haas and Macdonald (1999) time constants also closely approximated β3 EE and β3 AE receptor currents (Fig. 9C-E; Table 1; supplemental Table C).
Table 1.
Desensitization and deactivation time constants for simulated currents (400 msec)
|
Simulation group |
τ1 |
A1 |
τ2 |
A2 |
τ3 |
A3 |
C |
τd |
|---|---|---|---|---|---|---|---|---|
| 6 sec agonist | ||||||||
| Unaltered model | 9.3 | 24 | 353 | 21 | 1240 | 38 | 16 | 269 |
| Decreased df and di | 15.1 | 16 | 395 | 18 | 1378 | 43 | 23 | 198 |
| Increased koff | 15.4 | 23 | 361 | 20 | 1276 | 39 | 17 | 79 |
| 400 msec agonist | ||||||||
| Unaltered model | 9.5 | 28 | 333 | 33 | 37 | 236 | ||
| Decreased df and di | 13 | 16 | 382 | 36 | 47 | 184 | ||
| Increased koff
|
13
|
22
|
341
|
37
|
|
|
40
|
34
|
All data were simulated using the Haas and Macdonald model (1999). Although effects on τ1 and A1 were similar with both model perturbations (decreased df and di, increased koff), only the change in df and di resulted in a similar change in C and τd as was seen experimentally with serum starvation, PKI, and mutations [β1 (S409A), β3AA, and β3EA].
Because the significant differences we observed experimentally were between currents recorded under conditions favorable for or simulating phosphorylation (i.e., wild-type currents recorded from serum-fed cells, β3AE currents, or β3EE currents) and currents recorded under conditions unfavorable to receptor phosphorylation [i.e., wild-type currents recorded from serum-starved cells, β1(S409A), β3AA, and β3EA currents], we altered rate constants in the Haas and Macdonald (1999) model, which were presumably based on phosphorylated receptor currents, to generate simulated currents that had time courses similar to those observed experimentally for low phosphorylation conditions (Figs. 2A, 5). Increasing the opening rates (β1, β2, β3) (Fig. 9B) either individually or all together to all open states (O1, O2, and O3) (Fig. 9A) decreased the magnitude of macroscopic desensitization but did not accelerate deactivation for simulated currents having 6 sec, 400 msec, or 5 msec agonist application durations (data not shown). Similarly, decreasing the closing rates (α1, α2, α3) (Fig. 9B) also decreased macroscopic desensitization without accelerating deactivation (data not shown). Increasing the GABA unbinding rate (koff) (Fig. 9B) by 5000% (koff = 8500) or decreasing desensitization rates (df, di) (Fig. 9B) by 50% (df = 48 and di = 4) (Figs. 9C, D) both decreased macroscopic desensitization and accelerated deactivation for 6 sec, 400 msec and 5 msec applications (Table 1). It was necessary to increase both df and di to simulate “dephosphorylated” currents, because decreasing df alone produced simulated currents without an increased residual at time of agonist offset (column C in tables) and decreasing di alone did not alter deactivation. Decreasing ds (entry rate into the longest-lasting desensitization state, Ds) to 0 or increasing ds 3 orders of magnitude either alone or in addition to decreases in df and di did not significantly affect simulated brief (5 msec) or intermediate (400 msec) current traces. Currents simulated by increasing koff or decreasing df and di were qualitatively similar to those obtained experimentally under dephosphorylating conditions. The change in macroscopic desensitization was very similar for both sets of simulated currents (Table 1; supplemental Table C); however, the weighted deactivation time constant τD with increased koff was reduced 70-98% relative to the τD from the unaltered model, whereas the decrease in di and df only resulted in a 22-26% reduction in the τD.
Because increasing GABA unbinding and decreasing entry into fast and intermediate microscopic desensitized states both qualitatively reproduced the alterations in current time course recorded under conditions favoring dephosphorylation (i.e., macroscopically, both reduced desensitization and accelerated deactivation), we attempted to determine whether one or both of these changes were sufficient to explain the effect of dephosphorylation. One approach was to compare the effects of changing unbinding rate and decreasing entry into brief and intermediate microscopic desensitization states on repeated pulse inhibition. One would expect both changes to decrease repeated pulse inhibition; however, one would not necessarily predict that the amount of loss of repeated pulse inhibition would be the same in both cases. We simulated currents obtained with repeated pulse experiments (100 msec interpulse interval) and found that the increase in unbinding rate (koff = 8500) that was necessary to simulate changes in macroscopic desensitization and deactivation produced by 6 sec and 400 msec GABA application durations experimentally resulted in virtually no repeated pulse inhibition (repeated pulse = 97% of control) (Fig. 9E). When the desensitization rate constants were sufficiently decreased to simulate changes in macroscopic desensitization and deactivation produced by 6 sec and 400 msec GABA applications, the amount of repeated pulse inhibition (repeated pulse = 77% of control) (Fig. 9E) was similar to that obtained experimentally (repeated pulse = 69.3% of control) (Fig. 8C) (i.e., repeated pulse inhibition was decreased, but not eliminated). Entry into slow desensitized states has been shown to decrease peak current response with multiple repetitive stimulation (Bianchi and Macdonald, 2002). However, increasing the rate of entry (ds) into the slowest desensitization state (Ds) produced <1% change in peak current, a single repeated pulse with an interpulse interval of 100 msec. Furthermore, simulated 5 msec currents obtained (Fig. 9F; supplemental Table C) using an appropriately decreased unbinding rate constant had a macroscopic deactivation rate that was faster than the more modest macroscopic acceleration of desensitization observed experimentally (Fig. 7A,B; supplemental Table C). When entry into microscopic desensitized states was decreased, the time course of simulated 5 msec currents closely approximated the experimental 5 msec currents obtained under “dephosphorylated” conditions. Our simulations suggested that the effect of dephosphorylation could be explained by decreased entry into fast and intermediate microscopic desensitization states and the effect of phosphorylation by increased entry into fast and intermediate microscopic desensitized states. Although an effect on unbinding was not required to explain our data, we cannot completely rule out a minor effect on unbinding in addition to the altered desensitization. It should be noted that despite construction of an alternative model, Jones and Westbrook (1995) also came to the conclusion that increased repeated pulse inhibition was indicative of increased occupancy of desensitized states. When taken together, these simulations support the conclusion that PKA-mediated phosphorylation alters macroscopic currents by increasing entry into fast and intermediate desensitized states.
Discussion
It has become clear that phosphorylation has multiple, controversial physiological effects on GABAA receptor function (Brandon et al., 2002). Exogenously applied cAMP decreased GABAA receptor current over time in HEK293 cells transiently transfected with rat α1, β1, and γ2L subunits (Moss et al., 1992b). In contrast, larger α1β1γ2S GABAA receptor peak currents were observed in L929 mouse fibroblast cell lines constitutively expressing high amounts of catalytic subunit of PKA (Cα 12 cells) compared with cell lines expressing intermediate (L929 cells) or low (RAB 10 cells) amounts of active PKA (Angelotti et al., 1993). In cultured hippocampal neurons, IPSC decay was shortened, and macroscopic desensitization of GABA-evoked currents was increased in the rate and extent with calcineurin inhibition and no exogenous kinase (Jones and Westbrook, 1997). However, Jones and Westbrook (1997) did not see prolonged deactivation with phosphorylation, as was observed here. CA1 hippocampal neurons were reported to be tonically phosphorylated by PKA, which caused a decrease in miniature IPSC (mIPSC) amplitude and an increase in the proportion of mIPSCs with a double exponential decay (Poisbeau et al., 1999).
Many studies have investigated changes in GABAA receptor function associated with phosphorylation. However, the results obtained were very dependent on the hypotheses being tested and the methodology used to test them. For example, many studies used peak current as a functional assay of GABAA receptor function (Kano and Konnerth, 1992; Moss et al., 1992b; Angelotti et al., 1993; Robello et al., 1993; Cheun and Yeh, 1996; McDonald et al., 1998). However, measurements of peak current alone do not permit an accurate assessment of changes in desensitization and deactivation, which may have a significant effect on inhibition. Furthermore, true peak currents were often missed by the slow (>50 msec 10-90% rise time) application systems that were available. Additionally, membrane-permeable PKA activators were frequently coapplied with GABA, which did not allow investigators to distinguish between direct effects of the activator on GABAA receptor currents and effects attributable to PKA activation (Schwartz et al., 1991; Cheun and Yeh, 1992; Robello et al., 1993). Some investigators have reported changes that occur slowly (increasing effects over 10-30 min) (McDonald et al., 1998), whereas we have observed the full effect of PKA-induced change in desensitization in as little as 3 min using intracellular cAMP and extracellular forskolin. It is likely that PKA phosphorylation modulates GABAA receptor currents somewhat differently when activated acutely and chronically. Because the modulation of GABAA receptor currents is more complicated than a simple increase or decrease in peak, we have examined GABAA receptor currents more rigorously by analyzing transient kinetic properties observed with rapid application of GABA to lifted cells. Because receptor subtypes other than the ones we transfected were not present in our preparation, changes in current shape and time course were attributable to functional changes in the receptor not altered trafficking of different subunit combinations.
Phosphorylation of β1 and β3 GABAA receptor subunits increased desensitization and decreased deactivation
Rapid GABA application to lifted cells revealed an increase in the rate and extent of desensitization and slowed deactivation under serum-fed culture conditions or with β subunit serine to glutamate mutations that mimic receptor phosphorylation. When cells were serum starved or transfected with β subunit serine to alanine mutations of the critical serine [β1(S409), β3(S408)] that mimic receptor dephosphorylation, both the rate and extent of desensitization were decreased, and the rate of deactivation was increased. Mutation of β3(S407) had no effect on desensitization or deactivation, supporting the conclusion that the phosphorylation state of S408 in β3 is sufficient to modulate of kinetics by β subunit phosphorylation. McDonald et al. (1998) have reported that the site we have identified in β3 as crucial for affecting receptor kinetics causes a decreased peak current over time. Although their application system was inadequate to rigorously assess kinetic changes, their β3 mutant sample records show changes in desensitization similar to ours, even under control conditions. Whereas increased desensitization is consistent with a previous study (Jones and Westbrook, 1997) of hippocampal neurons and endogenous kinases, we observed prolonged deactivation and Jones and Westbrook did not. This may be because of different receptor subunit combinations present in these neurons or direct receptor effects of calcineurin inhibitors, which could not be assessed with the drug application systems available at that time.
Our results are consistent with findings that constitutive levels of kinase activity are present in both cultured neurons (Brandon et al., 2000) and HEK293 cells (Moss et al., 1992b; McDonald et al., 1998), which result in basal phosphorylation of GABAA receptor β subunits. Although the previous studies in HEK293 cells only showed moderate levels of receptor phosphorylation under basal conditions, our results suggest that those levels are sufficient for near maximal PKA modulation of GABAA receptor kinetic properties. However, because the phosphorylation site we have identified is also phosphorylated by PKC and we observed a smaller change in τ1 with PKI experiments than with mutant experiments, we cannot rule out the possibility that part of the effect of serum starvation was attributable to a reduction of other constitutive kinase activities. Another explanation of the PKI data are that the amount of PKI and its incubation time were not sufficient to fully block basal PKA activity, but we cannot conclude that based on our data. If another kinase was responsible for the basal level of GABAA receptor phosphorylation, it likely was not PKC, because the intracellular recording solution contained a calcium-chelating agent (EGTA). Furthermore, these data underscore the importance of cell culture conditions and intracellular environment in determining receptor function for any experimental paradigm. Because other investigators have found changes in peak GABAA receptor currents over the course of 30 min when adding PKA activators to an already moderately phosphorylated background, it is possible that very high levels of PKA could affect receptor trafficking and play a more important role in longer-term modification of receptor and synapse function, whereas more moderate levels are responsible for more rapid modification of GABAA receptor kinetic properties, which could exert a faster and finer control over receptor function.
Phosphorylation of β3 subunit serine 408 prolonged brief IPSC-like currents
Mutation of the β3 subtype serine 408 to glutamate to simulate phosphorylation conferred a deactivation rate similar to that of wild-type receptors. These findings are contrary to a report that promotion of phosphorylation by inhibition of the phosphatase calcineurin (protein phosphatase 2B) shortened IPSCs and GABA-evoked currents in excised patches from hippocampal neurons (Jones and Westbrook, 1997). Because the GABAA receptor subunit composition was not known for the neuronal cells and calcineurin may serve to dephosphorylate residues other than the specific ones we mutated, the effects observed with calcineurin were likely a result of phosphorylation of other residues on the GABAA receptor, direct inhibitor effects, differing GABAA receptor subunit composition, or phosphorylation of different intracellular proteins that interact with GABAA receptors. It is more difficult to compare directly the kinetic properties reported by Poisbeau et al.(1999) to our results, because they recorded from neurons at physiological temperature compared with our recombinant experiments with slower kinetics at room temperature. Nevertheless, our findings are consistent with their finding of an increase in the proportion of CA1 mIPSCs with a double exponential decay. Because we have found a prolongation of the slower deactivation time constant in recombinant cells, it is possible that the increase in double exponentially decaying mIPSCs was the result of an increased ability to resolve the two time constants as the second time constant was prolonged. The fact that with biexponential decays one time constant was shorter and the other longer than the single exponential decay time constant allowed for the possibility that the single exponentially decaying time constant was actually two time constants that were too close in value to be resolved at physiological temperature, as opposed to our experiments at room temperature. In our system, we likely always see both time constants of decay because room temperature recordings slow the kinetics of deactivation sufficiently to permit consistent resolution of both time constants.
PKA-induced alteration in macroscopic kinetics was attributable to increased microscopic desensitization
Data presented here indicate that PKA phosphorylation of GABAA receptors causes an increase in fast macroscopic desensitization and a slowing of macroscopic deactivation. This effect would prolong IPSPs, as we are reporting (Jones and Westbrook, 1995; Haas and Macdonald, 1999). Based on longer GABA application experiments (400 msec and 6 sec) as well as brief (5 msec) and repeated pulse experiments, we propose that PKA causes more rapid entry into the fast and intermediate microscopic desensitized states compared with unphosphorylated receptors. We have identified S409 in β1 and S408 in β3 as critical phosphorylated residues, which mediate the effect on GABAA receptor function. These findings demonstrate the importance of GABAA receptor phosphorylation to its functional characteristics and may have implications for GABAA receptors function when kinase activity is altered in disease states (Tehrani and Barnes, 1995; Sik et al., 2000; Yechikhov et al., 2001).
Footnotes
This work was supported by National Institutes of Health Grant 5R01 NS037825. We thank Matt Bianchi for assistance with the kinetic modeling.
Correspondence should be addressed to Dr. Robert L. Macdonald, Vanderbilt University Medical Center, 6140 Medical Research Building III, 465 21st Avenue South, Nashville, TN 37232-8552. E-mail: robert.macdonald@vanderbilt.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2311698-•$15.00/0
References
- Angelotti TP, Uhler MD, Macdonald RL ( 1993) Enhancement of recombinant gamma-aminobutyric acid type A receptor currents by chronic activation of cAMP-dependent protein kinase. Mol Pharmacol 44: 1202-1210. [PubMed] [Google Scholar]
- Axon Instruments ( 1999) Principles of operation. In: Axopatch 200B patch clamp theory and operation, pp 75-101. Union City, CA: Axon Instruments.
- Berjukow S, Doring F, Froschmayr M, Grabner M, Glossmann H, Hering S ( 1996) Endogenous calcium channels in human embryonic kidney (HEK293) cells. Br J Pharmacol 118: 748-754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi MT, Macdonald RL ( 2002) Slow phases of GABAA receptor desensitization: structural determinants and possible relevance for synaptic function. J Physiol (Lond) 544: 3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi MT, Haas KF, Macdonald RL ( 2001) Structural determinants of fast desensitization and desensitization-deactivation coupling in GABAA receptors. J Neurosci 21: 1127-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi MT, Song L, Zhang H, Macdonald RL ( 2002) Two different mechanisms of disinhibition produced by GABAA receptor mutations linked to epilepsy in humans. J Neurosci 22: 5321-5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau AJ, Baur R, Sharkey LM, Sigel E, Czajkowski C ( 2002) The relative amount of cRNA coding for gamma2 subunits affects stimulation by benzodiazepines in GABAA receptors expressed in Xenopus oocytes. Neuropharmacology 43: 695-700. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Delmas P, Kittler JT, McDonald BJ, Sieghart W, Brown DA, Smart TG, Moss SJ ( 2000) GABAA receptor phosphorylation and functional modulation in cortical neurons by a protein kinase C-dependent pathway. J Biol Chem 275: 38856-38862. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Jovanovic JN, Moss SJ ( 2002) Multiple roles of protein kinases in the modulation of GABAA receptor function and cell surface expression. Pharmacol Ther 94: 113-122. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Jovanovic JN, Colledge M, Kittler JT, Brandon JM, Scott JD, Moss SJ ( 2003) A-kinase anchoring protein 79/150 facilitates the phosphorylation of GABAA receptors by cAMP-dependent protein kinase via selective interaction with receptor beta subunits. Mol Cell Neurosci 22: 87-97. [DOI] [PubMed] [Google Scholar]
- Browning MD, Bureau M, Dudek EM, Olsen RW ( 1990) Protein kinase C and cAMP-dependent protein kinase phosphorylate the beta subunit of the purified gamma-aminobutyric acid A receptor. Proc Natl Acad Sci USA 87: 1315-1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QX, Stelzer A, Kay AR, Wong RK ( 1990) GABAA receptor function is regulated by phosphorylation in acutely dissociated guinea-pig hippocampal neurones. J Physiol (Lond) 420: 207-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheun JE, Yeh HH ( 1992) Modulation of GABAA receptor-activated current by norepinephrine in cerebellar Purkinje cells. Neuroscience 51: 951-960. [DOI] [PubMed] [Google Scholar]
- Cheun JE, Yeh HH ( 1996) Noradrenergic potentiation of cerebellar Purkinje cell responses to GABA: cyclic AMP as intracellular intermediary. Neuroscience 74: 835-844. [DOI] [PubMed] [Google Scholar]
- Feigenspan A, Bormann J ( 1994) Facilitation of GABAergic signaling in the retina by receptors stimulating adenylate cyclase. Proc Natl Acad Sci USA 91: 10893-10897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield Jr LJ, Sun F, Neelands TR, Burgard EC, Donnelly JL, Macdonald RL ( 1997) Expression of functional GABAA receptors in transfected L929 cells isolated by immunomagnetic bead separation. Neuropharmacology 36: 63-73. [DOI] [PubMed] [Google Scholar]
- Gyenes M, Wang Q, Gibbs TT, Farb DH ( 1994) Phosphorylation factors control neurotransmitter and neuromodulator actions at the gamma-aminobutyric acid type A receptor. Mol Pharmacol 46: 542-549. [PubMed] [Google Scholar]
- Haas KF, Macdonald RL ( 1999) GABAA receptor subunit gamma2 and delta subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol (Lond) 514: 27-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle DJ, Bianchi MT, Macdonald RL ( 2003) Modifications of a commercial perfusion system for use in ultrafast solution exchange during patch clamp recording. Biotechniques 35: 472-476. [PubMed] [Google Scholar]
- Jones MV, Westbrook GL ( 1995) Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron 15: 181-191. [DOI] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL ( 1997) Shaping of IPSCs by endogenous calcineurin activity. J Neurosci 17: 7626-7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Konnerth A ( 1992) Potentiation of GABA-mediated currents by cAMP-dependent protein kinase. NeuroReport 3: 563-566. [DOI] [PubMed] [Google Scholar]
- Kapur J, Macdonald RL ( 1996) Cyclic AMP-dependent protein kinase enhances hippocampal dentate granule cell GABAA receptor currents. J Neurophysiol 76: 2626-2634. [DOI] [PubMed] [Google Scholar]
- Kirkness EF, Bovenkerk CF, Ueda T, Turner AJ ( 1989) Phosphorylation of gamma-aminobutyrate (GABA)/benzodiazepine receptors by cyclic AMP-dependent protein kinase. Biochem J 259: 613-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitan IB ( 1999) Modulation of ion channels by protein phosphorylation. How the brain works. Adv Second Messenger Phosphoprotein Res 33: 3-22. [DOI] [PubMed] [Google Scholar]
- Macdonald RL, Olsen RW ( 1994) GABAA receptor channels. Annu Rev Neurosci 17: 569-602. [DOI] [PubMed] [Google Scholar]
- McDonald BJ, Moss SJ ( 1997) Conserved phosphorylation of the intracellular domains of GABAA receptor beta2 and beta3 subunits by cAMP-dependent protein kinase, cGMP-dependent protein kinase protein kinase C and Ca2+/calmodulin type II-dependent protein kinase. Neuropharmacology 36: 1377-1385. [DOI] [PubMed] [Google Scholar]
- McDonald BJ, Amato A, Connolly CN, Benke D, Moss SJ, Smart TG ( 1998) Adjacent phosphorylation sites on GABAA receptor beta subunits determine regulation by cAMP-dependent protein kinase. Nat Neurosci 1: 23-28. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Doherty CA, Huganir RL ( 1992a) Identification of the cAMP-dependent protein kinase and protein kinase C phosphorylation sites within the major intracellular domains of the beta 1, gamma 2S, and gamma 2L subunits of the gamma-aminobutyric acid type A receptor. J Biol Chem 267: 14470-14476. [PubMed] [Google Scholar]
- Moss SJ, Smart TG, Blackstone CD, Huganir RL ( 1992b) Functional modulation of GABAA receptors by cAMP-dependent protein phosphorylation. Science 257: 661-665. [DOI] [PubMed] [Google Scholar]
- Okuda H, Saitoh K, Hirai S, Iwai K, Takaki Y, Baba M, Minato N, Ohno S, Shuin T ( 2001) The von Hippel-Lindau tumor suppressor protein mediates ubiquitination of activated atypical protein kinase C. J Biol Chem 276: 43611-43617. [DOI] [PubMed] [Google Scholar]
- Olsen RW, Macdonald RL ( 2002) GABAA receptor complex: structure and function. In: Glutamate and GABA receptors and transporters: structure, function, and pharmacology (Egebjerg J, Schousboe A, Krogsgaard-Larsen P, eds), pp 203-235. London: Taylor and Francis.
- Poisbeau P, Cheney MC, Browning MD, Mody I ( 1999) Modulation of synaptic GABAA receptor function by PKA and PKC in adult hippocampal neurons. J Neurosci 19: 674-683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter NM, Twyman RE, Uhler MD, Macdonald RL ( 1990) Cyclic AMP-dependent protein kinase decreases GABAA receptor current in mouse spinal neurons. Neuron 5: 789-796. [DOI] [PubMed] [Google Scholar]
- Robello M, Amico C, Cupello A ( 1993) Regulation of GABAA receptor in cerebellar granule cells in culture: differential involvement of kinase activities. Neuroscience 53: 131-138. [DOI] [PubMed] [Google Scholar]
- Schwartz RD, Heuschneider G, Edgar PP, Cohn JA ( 1991) cAMP analogs inhibit gamma-aminobutyric acid-gated chloride flux and activate protein kinase A in brain synaptoneurosomes. Mol Pharmacol 39: 370-375. [PubMed] [Google Scholar]
- Sik A, Gulacsi A, Lai Y, Doyle WK, Pacia S, Mody I, Freund TF ( 2000) Localization of the A kinase anchoring protein AKAP79 in the human hippocampus. Eur J Neurosci 12: 1155-1164. [DOI] [PubMed] [Google Scholar]
- Tehrani MH, Barnes Jr EM ( 1995) Reduced function of gamma-aminobutyric acidA receptors in tottering mouse brain: role of cAMP-dependent protein kinase. Epilepsy Res 22: 13-21. [DOI] [PubMed] [Google Scholar]
- Vargas G, Yeh TY, Blumenthal DK, Lucero MT ( 1999) Common components of patch-clamp internal recording solutions can significantly affect protein kinase A activity. Brain Res 828: 169-173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virdee K, Parone PA, Tolkovsky AM ( 2000) Phosphorylation of the pro-apoptotic protein BAD on serine 155, a novel site, contributes to cell survival. Curr Biol 10: 1151-1154. [DOI] [PubMed] [Google Scholar]
- Yechikhov S, Morenkov E, Chulanova T, Godukhin O, Shchipakina T ( 2001) Involvement of cAMP- and Ca2+/calmodulin-dependent neuronal protein phosphorylation in mechanisms underlying genetic predisposition to audiogenic seizures in rats. Epilepsy Res 46: 15-25. [DOI] [PubMed] [Google Scholar]