

Abstract
The number and shape of dendritic spines are influenced by activity and regulated by molecules that organize the actin cytoskeleton of spines. Cortactin is an F-actin binding protein and activator of the Arp2/3 actin nucleation machinery that also interacts with the postsynaptic density (PSD) protein Shank. Cortactin is concentrated in dendritic spines of cultured hippocampal neurons, and the N-terminal half of the protein containing the Arp2/3 and F-actin binding domains is necessary and sufficient for spine targeting. Knockdown of cortactin protein by short-interfering RNA (siRNA) results in depletion of dendritic spines in hippocampal neurons, whereas overexpression of cortactin causes elongation of spines. In response to synaptic stimulation and NMDA receptor activation, cortactin redistributes rapidly from spines to dendritic shaft, correlating with remodeling of the actin cytoskeleton, implicating cortactin in the activity-dependent regulation of spine morphogenesis.
Keywords: actin, hippocampal neuron, NMDA receptor, spine, live imaging, RNA interference
Introduction
Dendritic spines are small protrusions found on dendrites of principal neurons of mammalian brain. Serving as postsynaptic compartments for individual excitatory inputs, spines show rapid movements and shape changes that are influenced by synaptic activity (Fischer et al., 2000; Korkotian and Segal, 2001). The structural modifications of spines are believed to represent morphological correlates of synaptic plasticity. The form and motility of spines are determined mainly by the actin cytoskeleton (Matus, 2000). Understanding the molecular mechanisms that control the actin cytoskeleton of spines therefore should provide insights into the structural plasticity of this postsynaptic compartment.
Cortactin is an F-actin binding protein implicated in the stabilization and branching of actin filaments (Wu and Parsons, 1993; Weed et al., 2000; Uruno et al., 2001; Weaver et al., 2001). The F-actin binding site of cortactin has been localized to part of the tandem repeats in the central region of the protein (Weed et al., 2000). In addition, cortactin binds via its N-terminal acidic domain (NTA) to the Arp2/3 complex that functions in actin polymerization (Weed et al., 2000; Uruno et al., 2001). Cortactin is thought to bind F-actin and the Arp2/3 actin polymerization machinery simultaneously and thereby facilitate the nucleation of new actin branches at the sides of preexisting actin filaments (Uruno et al., 2001; Weaver et al., 2001). Cortactin also contains a C-terminal SH3 domain for which several binding partners have been identified (Katsube et al., 1998; Ohoka and Takai, 1998; Dudek et al., 2002; Kinley et al., 2003), including the Shank family of postsynaptic density (PSD) scaffold proteins (Du et al., 1998; Naisbitt et al., 1999) and dynamin (McNiven et al., 2000). The Shank-cortactin interaction offers a potential link between the postsynaptic NMDA receptor-PSD-95 complex and the actin cytoskeleton of the dendritic spine (Naisbitt et al., 1999).
In non-neural cells the cortactin translocates to sites of active actin remodeling in the cell periphery in response to growth factors, adhesion, bacterial invasion, or mechanical stress, all of which induce changes in the shape or motility of cells (Ozawa et al., 1995; Weed et al., 1998; Cantarelli et al., 2000; Lopez et al., 2001; Birukov et al., 2002; Di Ciano et al., 2002). This translocation requires the Arp2/3- and F-actin binding sites of cortactin (Weed et al., 2000; Di Ciano et al., 2002).
Here we show that cortactin is concentrated with F-actin in dendritic spines of cultured hippocampal neurons but is redistributed to the dendritic shaft in response to NMDA receptor activation. In neurons deficient in cortactin, spine density is diminished greatly, whereas overexpression of cortactin induces longitudinal growth of spines. Spine targeting of cortactin is mediated primarily by the tandem repeats, with a minor contribution from the NTA domain, and these same domains are also important for promoting spine elongation. These findings indicate a critical role for cortactin in the activity-dependent regulation of spine cytoskeleton.
Materials and Methods
Antibodies and drugs. The following antibodies have been described: rabbit antiserum Shank 56/e (Naisbitt et al., 1999), guinea pig Shank antiserum (Sala et al., 2003), guinea pig PSD-95 antiserum (Kim et al., 1995), and mouse monoclonal PSD-95 antibody K28/43 (gift from J. Trimmer, State University of New York, Stony Brook, NY). The following antibodies and fluorescent dyes were purchased from commercial sources: cortactin for immunostaining (H-191, Santa Cruz Biotechnology, Santa Cruz, CA); bassoon (StressGen, Sidney, Canada); hemagglutinin (HA; Y11, Santa Cruz Biotechnology); myc (9E10, Santa Cruz Biotechnology); monoclonal β-galactosidase (Promega, Madison, WI); rabbit β-galactosidase (Abcam, Cambridge, MA); GAD-6 (Developmental Studies Hybridoma Bank, Iowa City, IA); PV-28 (Swant, Bellinzona, Switzerland); MAP2 (HM-2, Sigma, St. Louis, MO); anti-mouse and anti-rabbit Alexa 488- and 543-conjugated secondary antibodies, propidium iodide (PI), Hoechst 33342, and rhodamine-phalloidin (Molecular Probes, Eugene, OR).
Pharmacological reagents were purchased from the following sources: AMPA, NMDA, CNQX, APV, TTX, bicuculline, and 4-AP (Sigma); l-glutamic acid (Research Biochemicals, Natick, MA); and jasplakinolide (Molecular Probes).
DNA constructs. All cortactin constructs were generated by PCR with the use of Pfu polymerase (Stratagene, La Jolla, CA) and primers with sequence extensions containing the restriction sites AscI and EcoRI, respectively. For the generation of HA-tagged cortactin deletion mutants the amplified DNA fragments were ligated into the AscI and EcoRI restriction sites of the expression vector pGW1-CMV (British Biotechnology, Oxford, UK) carrying an N-terminal HA tag. Inserts were verified by DNA sequencing. For all constructs except W22A and W22AΔtandem, murine wild-type cortactin was used as template (gift of J. Thomas Parson, University of Virginia, Charlottesville, VA). For W22A and W22AΔtandem the clone pJP20 was used as template (kind gift from Xi Zhan, American Red Cross, Rockville, MD). N-terminally tagged myc-cortactin, green fluorescent protein-cortactin (GFP-cortactin), and GFP-Shank1B (all in pGW1-CMV) were kind gifts from Carlo Sala (CNR Institute of Neuroscience, Milan, Italy). GFP-cortactin in Sindbis virus vector was generated by ligating GFP-cortactin into the XbaI and ApaI restriction sites of the pSinRep5 vector (gift of Sondra Schlesinger, Washington University, St. Louis, MO). For the generation of the short-interfering RNA construct pSUPERcort300 the following DNA oligonucleotides, 5′-gatccccgcactgctcacaagtggacttcaagagagtccacttgtgagcagtgctttttggaaa-3′ and 5′-agcttttccaaaaagcactgctcacaagtggactctcttgaagtccacttgtgagcagtgcggg-3′ (corresponding to nucleotides 300-318 of the rat and mouse cortactin sequence with the first nucleotide of the start codon counted as nucleotide 1), were annealed and subcloned into the HindIII and BglII sites of the pSUPER vector (OligoEngine Platform).
Sindbis virus generation. GFP-cortactin in pSinRep5 was linearized by using the restriction enzyme PacI and subsequentially transcribed by using the mMessage mMachine RNA transcription kit (Ambion, Austin, TX) according to the manufacturer's instruction. The virus RNA was electroporated into baby hamster kidney cells cultured in modified Eagle's medium (Invitrogen, San Diego, CA) supplemented with 20% fetal calf serum and nonessential amino acids (Invitrogen). After 2 d of incubation the culture medium containing the Sindbis virus particles was harvested and directly used for infection or stored at -80°C. The GFP Sindbis virus was a kind gift from Terunaga Nakagawa (Massachusetts Institute of Technology, Cambridge, MA).
Cell culture, transfection, immunostaining, and PI exclusion assay. Hippocampal neuron cultures were prepared from embryonic day 18-19 rat embryos as described previously (Brewer et al., 1993). Medium density cultures (∼150 cells mm-2) were plated on coverslips (Gallard-Schlessinger, New York, NY) or glass-bottom dishes (MatTek, Ashland, MA) coated with poly-d-lysine (30 μg/ml) and laminin (2.5 μg/ml). Cultures were grown in Neurobasal medium (Invitrogen) supplemented with B27 (Invitrogen), 0.5 mm glutamine, and 12.5 μm glutamate. For time-lapse studies 10 mm HEPES, pH 7.4, was added to the culture medium before the image acquisition. During the imaging period the neurons were kept in a humidified incubation chamber at 37°C and 5% CO2. COS-7 cells were maintained in DMEM supplemented with 10% calf serum and transfected with Lipofectamine (Invitrogen).
Neurons were transfected via the calcium phosphate method (Sala et al., 2001). For the short-interfering RNA (siRNA) inhibition experiments the neurons were transfected at 13 d in vitro (DIV13) and for the cortactin and deletion mutant overexpression experiments at DIV10. For the spine-targeting study (see Fig. 2) a DNA ratio of 1:10 of cortactin (or cortactin mutant) to β-galactosidase was used to obtain low expression levels of cortactin or cortactin mutants.
Figure 2.
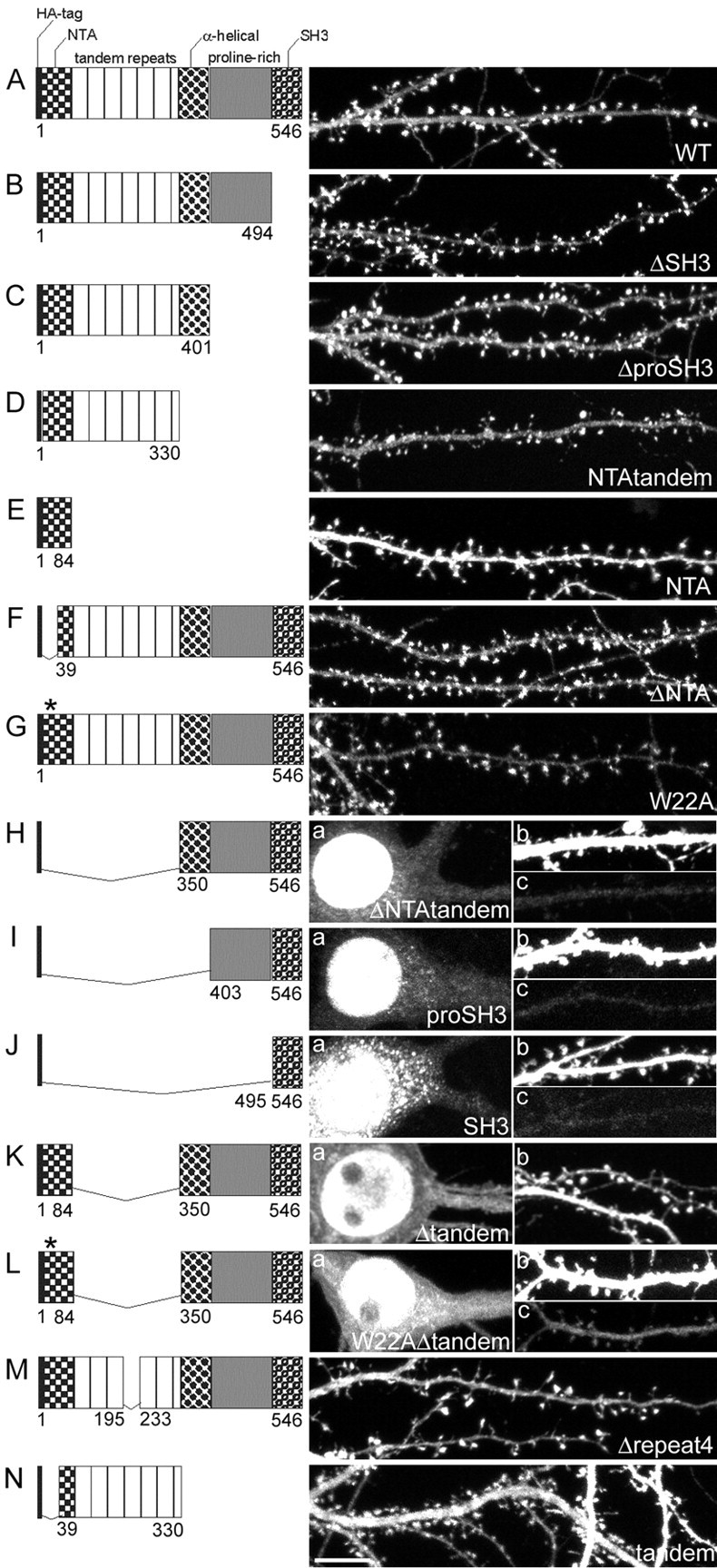
Domain requirements for spine targeting of cortactin. Micrographs show the distribution of transfected wild-type and mutant cortactin constructs (all HA-tagged) in hippocampal neurons (DIV20; 10 d after transfection). The domain structure of the transfected cortactin mutants is shown to the left of each image. The numbers indicate the first and last amino acid for each construct. An asterisk indicates the W22A point mutation. For neurons cotransfected with β-galactosidase and HA-tagged WT (A), ΔSH3 (B), ΔproSH3 (C), NTAtandem (D), ΔNTA (F), W22A (G), Δrepeat4 (M), or tandem (N), only the distribution of cortactin construct (HA staining) is shown. For neurons cotransfected with β-galactosidase and HA-tagged ΔNTAtandem (H), proSH3 (I), SH3 (J), Δtandem (K), and W22AΔtandem (L), the HA staining pattern is shown for the soma (a in H-L) and the dendrites (b in K and c in H-J, L). The outline of the same transfected dendrite (β-galactosidase staining) is shown in b in H-J and L. Scale bar, 10 μm.
For all immunostaining experiments except Shank staining the neurons were fixed in 4% formaldehyde/4% sucrose in PBS, pH 7.4, for 10 min at room temperature. For immunostaining of Shank the neurons were fixed either in methanol for 10 min at -20°C (see Fig. 5) or in 1% formaldehyde/4% sucrose in PBS, pH 7.4, for 3 min at room temperature, followed by methanol for 10 min at -20°C (see Fig. 3). Immunostaining was performed as described (Sala et al., 2000).
Figure 5.
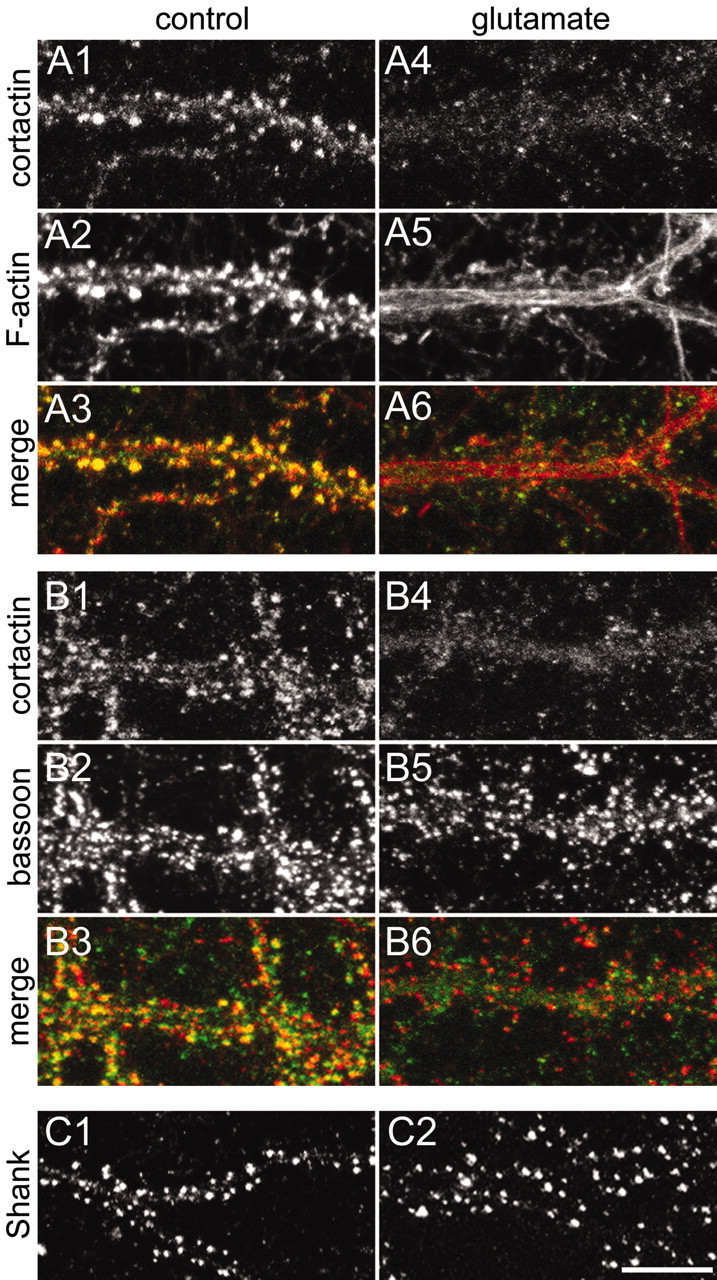
Glutamate-induced redistribution of cortactin from dendritic spines. All images were taken from hippocampal neurons at DIV20. A, Dendrites of a spiny neuron were double-stained for cortactin with antibody H-191 (A1, A4) and F-actin (A2, A5) in the absence of stimulation (A1-A3) or after stimulation with 100 μm glutamate for 15 min (A4-A6). Color merges are shown in A3 and A6. B, Dendrites of a spiny neuron were double-stained for cortactin with antibody H-191 (B1, B4) and bassoon (B2, B5) in the absence or presence of 100 μm glutamate for 15 min. Color merges are shown in B3 and B6. C, Dendrites of a spiny neuron were stained for Shank in the absence (C1) or presence (C2) of 100 μm glutamate for 15 min. Scale bar, 10 μm.
Figure 3.

Changes in spine morphology induced by cortactin mutants. All images were taken from hippocampal neurons at DIV20 after 10 d of transfection with β-galactosidase (A), HA-tagged WT (B), ΔSH3 (C), ΔproSH3 (D), or NTAtandem (E). The cumulative distribution of spine width (F) and length (G) in neurons transfected with the constructs indicated in A-E is shown. The key for the graph in F also applies to G. H, Dendrites of neurons overexpressing wild-type cortactin (WT; green) or β-galactosidase (β-gal; green) costained for Shank (red). Scale bar (in E): A-E, H, 10 μm.
For the PI exclusion assay the neurons were labeled with 10 μm Hoechst 33342 and 5 μg/ml PI for 15 min at 37°C and then fixed in 4% formaldehyde/4% sucrose in PBS, pH 7.4, for 10 min at room temperature.
Microscopy and quantitation. Immunofluorescence images were acquired with a LSM510 confocal microscope system (Zeiss, Oberkochen, Germany) and an Axioplan microscope (Zeiss), using a 63× oil immersion lens. Each image consisted of a z-stack of pictures taken at a depth interval of 0.5-0.8 μm and then projected into one image.
Time-lapse images were acquired by using Multi-time software (Zeiss) with a Pascal confocal system (Zeiss) and an Axiovert microscope (Zeiss), using a 100× oil immersion lens. Laser intensity of the argon laser at 488 nm was set to 0.1% for GFP-cortactin and 3% for GFP-Shank1B. Neurons with low expression levels of GFP-cortactin and GFP-Shank1B and relatively normal spine morphology were selected to be able to observe movements of the protein in response to stimulation.
Morphometric measurements were performed with the aid of MetaMorph Software (Universal Imaging, West Chester, PA) and was done in part by a third person who was completely unaware of the experimental conditions. Each experiment was repeated at least three times with independent neuronal preparations. Neurons for morphometric analysis were sampled randomly from each neuronal preparation.
For protrusion density (see Fig. 1) the number of protrusions (maximum length, 10 μm) was counted for 32-45 neurons (∼200 μm total dendritic length per neuron). For spine size (see Fig. 3) ∼4000 spines (17-23 neurons) were measured for each culture condition. The length of a spine from the base of the neck to the furthest point on the spine head and the maximal width of the spine head perpendicular to the long axis of the spine neck were measured. For each condition individual density measurements or spine dimensions were grouped first and averaged per neuron; means from multiple individual neurons then were averaged to obtain a mean and SEM for the population of neurons.
Figure 1.
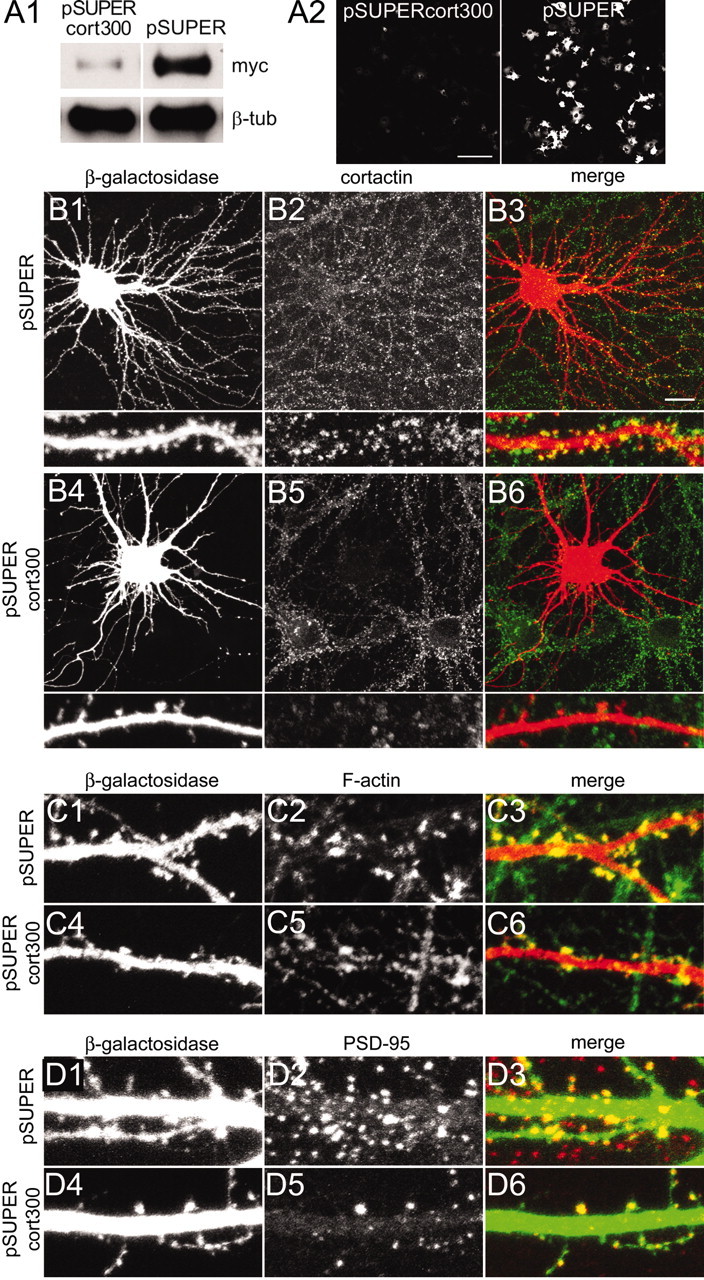
siRNA inhibition of cortactin causes loss of spines. A1, COS-7 cells transfected with myc-tagged murine cortactin plus pSUPERcort300 or pSUPER vector control were immunoblotted for myc and β-tubulin, as indicated. A2, GFP fluorescence in COS-7 cells cotransfected with GFP-tagged murine cortactin plus pSUPERcort300 or pSUPER. B-D, Hippocampal neurons at DIV16 cotransfected with β-galactosidase plus pSUPERcort300 or pSUPER were double-stained as indicated for cortactin (antibody H-191; B2, B5) and β-galactosidase (B1, B4); β-galactosidase (C1, C4) and rhodamine-phalloidin (C2, C5); β-galactosidase (D1, D4) and PSD-95 (D2, D5). Color merges of the double-labeled images are shown in B3, C3, D3 and B6, C6, D6 for each condition. Scale bars: A2, 200 μm; (in B3) B1-B6, 20 μm; for high-magnification micrographs in B1-B6, C, D, 5 μm.
For the PI exclusion assay 1096-1207 nuclei were analyzed from three different coverslips for each experimental condition. The ratio of PI to Hoechst 33342-positive nuclei was calculated for each coverslip and then averaged per coverslip to obtain the mean. Similar cell densities between experimental groups were confirmed by counting the number of Hoechst 33342-positive nuclei per microscopic view. Measurements are given as mean ± SEM, unless otherwise stated. Statistical significance was calculated by comparing the means from multiple neurons per experimental condition with the Mann-Whitney test, unless otherwise stated. Densitometric analysis of immunoblot bands was performed by using Quantity One V4.2.2 software (Bio-Rad, Hercules, CA).
Results
Loss of cortactin causes decrease in spine density
To investigate the role of cortactin in dendritic spine morphogenesis, we used siRNA to knock down specifically the expression of endogenous cortactin in relatively mature cultured hippocampal neurons (DIV16). Three different 19 nucleotide sequences specific for murine and rat cortactin mRNA were tested in plasmid (pSUPER)-based siRNA constructs. In heterologous cells only the sequence corresponding to nucleotides 300-318 of the rat and mouse cortactin sequence (pSUPERcort300) reduced the expression of cotransfected myc-tagged murine cortactin to 17.94 ± 6.84% of control levels (Fig. 1A1) and extinguished the fluorescence intensity of cotransfected GFP-cortactin (Fig. 1A2).
Hippocampal neurons transfected with pSUPERcort300 showed a strong decrease in the staining intensity for cortactin (Fig. 1), but no change in other cytoskeleton-associated proteins such as MAP2 (data not shown). The loss of cortactin immunoreactivity was most obvious in spines (Fig. 1B6), where cortactin is highly enriched in mature neurons (Fig. 1B3). Associated with the knockdown of cortactin, total protrusion density was reduced by 67% (from 3.77 ± 0.26 to 1.26 ± 0.08 protrusions per 10 μm dendrite length; p < 0.0001, Mann-Whitney test) in pSUPERcort300-transfected neurons. This result implies that cortactin is necessary for the formation or stability of spines. The remaining spines still were enriched for F-actin, as revealed by rhodamine-phalloidin labeling (Fig. 1C6), and they contained clusters of the PSD protein PSD-95 (Fig. 1D6). However, the density of F-actin and PSD-95 clusters on dendrites of cortactin-deficient neurons was diminished greatly, correlating with the loss of spines.
Domains of cortactin required for spine targeting
To address the molecular basis for the role of cortactin in spine morphogenesis, we first determined which domains of cortactin are required for its targeting to dendritic spines. Neurons were doubly transfected with a series of HA-tagged deletion mutants of cortactin together with β-galactosidase (β-galactosidase immunostaining was used to outline neuronal morphology) (Fig. 2). Overexpressed wild-type (WT) HA-cortactin accumulated specifically in spines (Fig. 2A). Progressive deletions of up to the C-terminal half of the protein, including the SH3 domain (ΔSH3, Fig. 2B), the proline-rich domain (ΔproSH3, Fig. 2C), and the α-helical region (NTAtandem, Fig. 2D), did not impair the targeting of cortactin to spines. Thus approximately the N-terminal half of cortactin (NTAtandem) is sufficient for concentration in spines, whereas interaction with Shank and other binding partners of the C terminus, such as dynamin, ZO-1, endothelial cell myosin light chain kinase, WASp-interacting protein (WIP), or CBP90 (Du et al., 1998; Katsube et al., 1998; Ohoka and Takai, 1998; Naisbitt et al., 1999; McNiven et al., 2000; Dudek et al., 2002; Kinley et al., 2003), are not essential.
The NTA alone, however, was distributed diffusely in dendrites with no enrichment in spines (Fig. 2E), indicating that the tandem repeats domain of cortactin is necessary for spine targeting. Deletion of the first 38 amino acids of the NTA domain (ΔNTA, Fig. 2F) or replacing tryptophan-22 by alanine (W22A, Fig. 2G), two mutations that abolish cortactin interaction with Arp2/3 (Uruno et al., 2001), did not prevent the cortactin enrichment in spines. The cortactin mutant that lacks the NTA and tandem repeats domain (ΔNTAtandem), however, was virtually absent from dendrites and spines (Fig. 2Hb,Hc) and instead accumulated in the nucleus (Fig. 2Ha). A similar nuclear localization was observed for the mutants containing only the proline-rich and SH3 domains (proSH3, Fig. 2I) or the SH3 domain alone (SH3, Fig. 2J). Taken together, the data indicate that the tandem repeats are important for cortactin localization in spines.
To support this conclusion, we generated a cortactin mutant with an internal deletion of the tandem repeats (Δtandem, Fig. 2K). This Δtandem mutant also mainly targeted to the nucleus (Fig. 2Ka), but some protein was detected in dendrites (Fig. 2Kb), where it showed only weak enrichment in spines. This result confirms the importance of the tandem repeats but suggests that other domains might contribute in minor manner to the spine targeting of cortactin. Because the NTA domain is the only difference between ΔNTAtandem (which is virtually absent from dendrites and spines) and Δtandem, this additional spine targeting determinant presumably resides in the NTA domain. Consistent with this idea, a “double” mutant lacking the tandem repeats and carrying the W22A point mutation in the NTA (W22AΔtandem) was targeted primarily to the nucleus (Fig. 2La), and very little could be detected in dendrites (Fig. 2Lc). We never observed any enrichment of W22AΔtandem in spines.
The fourth repeat within the tandem repeat domains has been shown to be essential for the binding of cortactin to F-actin (Weed et al., 2000). However, a cortactin mutant lacking repeat 4 (Δrepeat4) still accumulated in spines (Fig. 2M). Furthermore, a mutant containing only the tandem repeat region of cortactin (tandem, Fig. 2N) was distributed diffusely throughout the dendrites and spines, but with no enrichment in spines. We conclude that the tandem repeats domain is necessary, but not sufficient, for the targeting of cortactin to spines. In addition to the tandem repeats, the NTA domain seems to contribute a spine-targeting determinant.
Cortactin promotes longitudinal spine growth
In the spine targeting experiments we noticed that neurons expressing high levels of WT cortactin showed significantly longer spines (Fig. 3B) than control neurons expressing β-galactosidase alone (Fig. 3A). The increase in length appeared to be mainly attributable to increased length of the spine neck. Overexpression of the C-terminal deletion mutants ΔSH3, ΔproSH3, or NTAtandem (all of which target efficiently to spines) also resulted in spine elongation, similar in degree to WT overexpression (Fig. 3C-E,G, Table 1). The spine-elongating activity of the C-terminal deletion mutants correlates with the fact that the minimal cortactin fragment required for the promotion of actin polymerization comprises the NTA and tandem repeats domains (Weaver et al., 2001) and suggests that the elongated spines are a result of enhanced actin polymerization. In addition, however, overexpression of these C-terminal deletion mutants also prominently reduced the width of the spine head (Fig. 3F, Table 1). Neurons expressing the shortest C-terminal mutant (NTAtandem) had the thinnest protrusions, many of which resembled filopodia in that they lacked a spine head (Fig. 3E,F). These data suggest that domains in the C-terminal half of cortactin play a role in the morphogenesis of spines, particularly of the spine head. In addition to the difference from WT cortactin or control, we observed statistically significant differences with regard to spine width among several of the different C-terminal deletion mutants of cortactin (Table 1), suggesting a contribution of each of the C-terminal domains (α-helical, proline-rich, and SH3) to the formation of spine heads. The overexpression of WT cortactin did not cause an accumulation of endogenous Shank in spines (Fig. 3H), consistent with the observation that the interaction between Shank and cortactin is not required for the localization of the proteins to spines (Sala et al., 2001; this study).
Table 1.
Dendritic spine size in neurons overexpressing cortactin and cortactin mutants
|
Cortactin mutants |
Spine width (μm) |
Spine length (μm) |
|---|---|---|
|
|
Mean ± SD
|
Mean ± SD
|
| Control (β-gal) | 1.02 ± 0.08 | 1.78 ± 0.18 |
| WT | 0.94 ± 0.1 | 2.32 ± 0.5 |
| ΔSH3 | 0.86 ± 0.09 | 2.04 ± 0.26 |
| ΔproSH3 | 0.8 ± 0.1 | 2.06 ± 0.29 |
| NTAtandem
|
0.7 ± 0.12
|
2.29 ± 0.39
|
|
|
p value
|
p value
|
|---|---|---|
| β-gal : WT | 0.0014 | <0.0001 |
| β-gal : ΔSH3 | <0.0001 | 0.0013 |
| β-gal : ΔproSH3 | <0.0001 | 0.0024 |
| β-gal : NTAtandem | <0.0001 | <0.0001 |
| WT : ΔSH3 | 0.0047 | 0.0829 |
| WT : ΔproSH3 | 0.0003 | 0.1604 |
| WT : NTAtandem | <0.0001 | 0.8043 |
| ΔSH3 : ΔproSH3 | 0.1239 | 0.9635 |
| ΔSH3 : NTAtandem | <0.0001 | 0.0295 |
| ΔproSH3 : NTAtandem
|
0.0120
|
0.0610
|
Cortactin induces the formation of protrusions in aspiny neurons
Aspiny inhibitory interneurons comprise ∼5-10% of the neurons in our hippocampal cultures and can be identified by morphological criteria as well as molecular markers such as glutamic acid decarboxylase (GAD; Kunkel et al., 1986; Benson et al., 1994) and various calcium binding proteins, including parvalbumin (Sik et al., 1995; Lee et al., 2000). Additionally, these interneurons characteristically have elongated F-actin patches (data not shown) and a high density of bassoon puncta along their dendrites (Fig. 4A2). Staining with the cortactin antibody showed only weak and diffuse staining (Fig. 4A1, arrows), indicating that cortactin is expressed minimally in this type of neuron (for comparison, a dendrite of spiny neuron is indicated by an arrowhead in Fig. 4A1).
Figure 4.
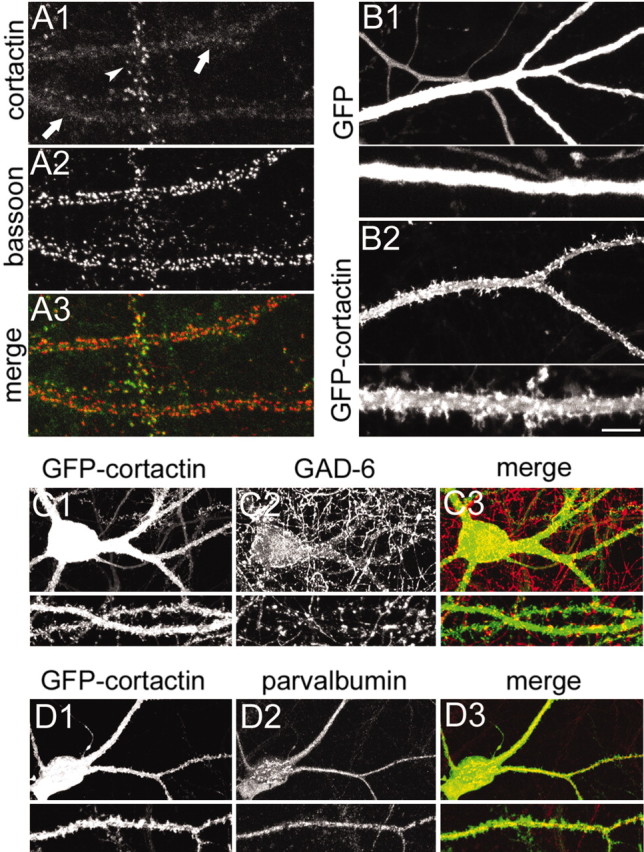
Effects of cortactin overexpression in aspiny interneurons. All images were taken from neurons at DIV20. A, Dendrites of an aspiny interneuron (arrow) and a spiny pyramidal neuron (arrowhead) costained for cortactin with antibody H-191 (A1) and bassoon (A2). A3, The color merge of the two images. B, Dendrites of interneurons infected with Sindbis virus expressing GFP (B1) or GFP-cortactin (B2) for 16-20 hr. Bottom panels show dendrites at higher magnification. C, Hippocampal neuron infected with Sindbis virus expressing GFP-cortactin (C1) and costained for GAD (C2), merged in C3. D, Hippocampal neuron infected with Sindbis virus expressing GFP-cortactin (D1) and costained for parvalbumin (D2), merged in D3. Scale bar (in B2): C1-C3, D1-D3, 20 μm; A1-A3, B1, B2, and high-magnification views of C, D, 10 μm; high-magnification views of B, 5 μm.
Interneurons infected with control Sindbis virus expressing GFP showed smooth dendrites with no protrusions (Fig. 4B1). However, interneurons (as defined by their typical dendritic morphology and GAD or parvalbumin immunoreactivity in the cell soma (Fig. 4C2,D2) infected with GFP-cortactin displayed numerous tiny protrusions resembling short filopodia emerging from the dendritic shaft (Fig. 4B2,C1,D1). Interestingly, GFP intensity in these protrusions was higher than in the dendrite, indicating that GFP-cortactin was enriched in these structures (Fig. 4B4). Thus overexpression of cortactin induces the outgrowth of atypical dendritic protrusions in neurons that normally do not form spines.
Cortactin is enriched in dendritic spines and translocates with glutamate stimulation
Spines undergo activity-dependent morphological changes that most likely are based on rearrangements of the underlying actin cytoskeleton (for review, see Matus, 2000; Hering and Sheng, 2001). Because cortactin binds to F-actin and affects actin polymerization, we asked whether cortactin changes distribution in response to synaptic activity. In basal conditions of mature hippocampal cultures, labeling with the cortactin antibody H-191 showed numerous immunoreactive puncta along the dendrites of spiny (presumptive pyramidal) neurons (Fig. 5A1; see also Fig. 1B). The punctate cortactin staining colocalized with the concentration of F-actin in dendritic spines, as revealed by double labeling with phalloidin (Fig. 5A2,A3). Cortactin puncta also showed overlap with the presynaptic protein bassoon (Fig. 5B), consistent with the postsynaptic enrichment of cortactin. We were unable to demonstrate a colocalization of cortactin and its binding partner Shank because of fixation incompatibility of the respective antibodies.
Bath application of glutamate (100 μm) for 15 min to cultured hippocampal neurons led to the disappearance of F-actin from spines and its reappearance as filamentous bundles within the dendritic shaft (Fig. 5A5), as reported previously (Halpain et al., 1998). The same glutamate treatment caused a redistribution of cortactin from dendritic spines to a diffuse pattern within dendrites (Fig. 5A4). Similar results also were obtained with lower concentrations of glutamate and with bicuculline treatment (see below), implying that cortactin redistribution is not attributable to excitotoxicity.
The disappearance of cortactin from spines was not attributable to the degradation of the protein, because immunoblot analysis showed unchanged levels of total cortactin in stimulated cultures (data not shown). In contrast to cortactin (Fig. 5B4), the immunostaining patterns of presynaptic protein bassoon and PSD protein Shank remained punctate after 15 min of glutamate stimulation (Fig. 5B5,C1,C2). Thus glutamate excitation induces the specific translocation of cortactin from postsynaptic sites in dendritic spines.
To investigate the dynamics of cortactin redistribution, we performed time-lapse imaging of GFP-tagged cortactin expressed by Sindbis virus vector in live hippocampal neurons. Exogenous GFP-cortactin accumulated in the heads of dendritic spines (Fig. 6A, 0 min; red pseudocolor represents high fluorescence intensity), similar to the distribution of endogenous cortactin and transfected HA-cortactin (Figs. 5A1, 2A). Diffuse GFP-cortactin signal also was observed at much lower intensity in the dendritic shaft. In unstimulated neurons GFP-cortactin showed little change in overall distribution (Fig. 6A, 0 and 6 min). However, glutamate stimulation induced a rapid and dramatic change in the localization of GFP-cortactin. GFP-cortactin fluorescence intensity decreased greatly in spines and increased strongly in the dendritic shaft, implying a redistribution of GFP-cortactin from spines to the shaft (Fig. 6B, 12-54 min). In some stimulated neurons vacuole-like areas could be seen that were relatively devoid of GFP-cortactin; these structures might reflect early excitotoxicity induced by glutamate (Bindokas and Miller, 1995; Hasbani et al., 2001). Concomitantly with GFP-cortactin redistribution, the morphology of spines changed, the majority becoming thinner and more elongated. The formation of new filopodia-like protrusions was observed, some of which grew from existing spine heads (Fig. 6A, arrows).
Figure 6.

Dynamics of cortactin in dendritic spines. Shown are time-lapse image series for GFP-cortactin or GFP-Shank in transfected neurons. Pixels saturated by high GFP fluorescence intensity are shown in red; pixels without fluorescence signal are shown in blue. Fluorescence intensities below saturation are shown in gray scale. Images were taken every 3 min for 1 hr (only every second image is shown; time stamps in minutes). Asterisks indicate the application of 100 μm glutamate ∼100 sec before the image acquisition. A, Dendrite of a spiny neuron (DIV20) 16-20 hr after infection with GFP-cortactin Sindbis virus. Arrows point to protrusions emerging from spine heads. B, Dendrites of a spiny neuron (DIV17) transfected with GFP-tagged Shank (GFP at N terminus of Shank). Scale bar, 10 μm.
GFP-tagged Shank1B also concentrated in dendritic spines but, in contrast to GFP-cortactin, showed little change in subcellular distribution after glutamate stimulation (Fig. 6B). In transfected neurons GFP-Shank1B was concentrated in spines with weak signal in dendritic shafts (Fig. 6B, 0 min), as reported before for HA-tagged Shank1B (Sala et al., 2001). In live imaging GFP-Shank1B clusters were localized stably to spines in unstimulated neurons (Fig. 6B, 0 and 6 min), and this distribution did not change after 100 μm glutamate application (Fig. 6B). These live-imaging data demonstrate that cortactin redistributes from spines within a few minutes of glutamate stimulation, whereas the localization of PSD proteins as represented by Shank remains grossly unchanged.
The direction of cortactin movement (from spines to shaft) contradicts a previous report from our lab in which glutamate stimulation increased cortactin staining at synaptic sites (Naisbitt et al., 1999). Although the culture conditions of those experiments and this study are similar, we cannot exclude that some subtle change in culture density or quality underlies this difference. We suspect, however, that the discrepancy might be attributable to the cortactin monoclonal antibody (4F11) used in Naisbitt et al. (1999), which we subsequently found recognizes a band around 45 kDa in addition to the 80/85 kDa cortactin doublet on immunoblots (H. H., unpublished observation). The cortactin antibody H-191 used in this study reacts only with the 80/85 kDa bands (H. H., unpublished observation). We believe that cortactin translocation from the spine to the dendrite is correct, because the immunostaining result is corroborated by direct visualization of translocation of GFP-cortactin in live neurons (Fig. 6).
NMDA receptor-dependent redistribution of cortactin
Which type of glutamate receptor mediates the glutamate-induced dispersal of cortactin? Hippocampal neurons stimulated for 15 min with 50 μm NMDA (in the presence of 30 μm CNQX and 1 μm TTX) showed cortactin redistribution from spines (Fig. 7A1) and rearrangement of F-actin (Fig. 7A2) similar to glutamate-stimulated cells. In contrast, specific stimulation of AMPA receptors with 100 μm AMPA (plus 100 μm APV and 1 μm TTX) caused no change in the staining pattern of cortactin (Fig. 7B1) or F-actin (Fig. 7B2). Moreover, the dispersal of cortactin by glutamate was blocked by the NMDA receptor antagonist APV (Fig. 7C). Taken together, these findings indicate that the translocation of cortactin is induced by the activation of NMDA receptors.
Figure 7.

NMDA receptor-dependent translocation of cortactin. All images were taken from hippocampal neurons at DIV20. A-E, Dendrites of spiny neurons double-stained for cortactin with antibody H-191 (A1-E1) and F-actin (A2-E2) after the application of 50 μm NMDA, 30 μm CNQX, and 1 μm TTX (A); 100 μm AMPA, 100 μm APV, and 1 μm TTX (B); 500 μm APV and 100 μm glutamate for 15 min (C); 50 μm bicuculline and 500 μm 4-AP for 1 hr (D); or 30 μm glutamate for 15 min (E). A3-E3, Color merges of double-labeled images. F, Time-lapse series of a dendrite of a spiny neuron infected with Sindbis virus expressing GFP-cortactin. Fluorescence intensity is represented as in Figure 6; time stamps are in minutes. Asterisks indicate the application of bicuculline and 4-AP ∼100 sec before the image was taken. G, Dendrites of spiny neurons stained with the cortactin antibody H-191 in the presence of 2 μm jasplakinolide (G1) or 2 μm jasplakinolide and 100 μm glutamate (G2). H, Dendrites of hippocampal neurons double-stained for cortactin with antibody H-191 and MAP2 after the application of 30 μm glutamate for 15 min (H1), 50 μm bicuculline and 500 μm 4-AP for 4 hr (H2), and control (H3). Scale bar (in D): A-E, G, 10 μm; F, 5 μm; H, 20 μm.
Bath application of glutamate or NMDA stimulates synaptic as well as extrasynaptic NMDA receptors (Hardingham et al., 2002) and can induce excitotoxicity in neurons (Rothman and Olney, 1995; Hardingham et al., 2002). We therefore stimulated hippocampal neurons with a lower concentration of glutamate (30 μm, 15 min). This treatment caused the dispersal of cortactin and the rearrangement of F-actin into dendritic bundles (Fig. 7E1-E3)in ∼95% of the neuronal population, similar to 100 μm glutamate. Furthermore, we assessed possible excitotoxic damage to neuronal dendrites by MAP2 staining (Fig. 7H). No obvious change in microtubule organization was observed in the majority of dendrites in which cortactin was dispersed after glutamate treatment (Fig. 7H1). Using the PI exclusion assay, which gives a more quantitative measurement of cellular damage, we found that the ratio of PI-positive nuclei to the total number of nuclei (as detected by Hoechst 33342 staining) was 0.38 ± 0.05 (mean ± SEM) after 24 hr of recovery from treatment with 30 μm glutamate for 15 min and was 0.17 ± 0.01 in untreated control neurons. The difference was statistically significant (p = 0.02, Student's t test), suggesting that an additional ∼20% of the neuronal population shows signs of cellular damage after glutamate treatment as compared with control neurons. However, this was substantially less than the percentage of neurons (∼95%) that shows a redistribution of cortactin and F-actin in response to 30 μm glutamate, indicating that the translocation of cortactin is not solely a result of excitotoxicity. The density of cells in control and glutamate treated cultures was similar after 24 hr of recovery (data not shown).
To test whether cortactin redistributes in response to more physiological NMDA receptor activation, we stimulated synaptic activity in hippocampal cultures with 50 μm bicuculline and 500 μm 4-AP (bic/4AP; Hardingham et al., 2002). Exposure of neurons to bic/4-AP for 15 min did not cause any obvious changes in the distribution of endogenous cortactin and F-actin (data not shown). However, after 1 hr in the presence of bic/4-AP approximately one-third of the neurons showed loss of cortactin and F-actin from spines (Fig. 7D1,H2) and the formation of F-actin fibers in dendrites (Fig. 7D2). Redistribution of cortactin in the majority of neurons was observed after 24 hr of bic/4-AP treatment (data not shown). Bic/4-AP had no excitotoxic effects on hippocampal neurons as revealed by MAP2 staining (Fig. 7H2)or the PI exclusion assay. The fraction of PI-positive nuclei in bic/4-AP-treated cultures was 0.19 ± 0.01 as compared with 0.17 ± 0.01 in control (p = 0.35, Student's t test).
Time-lapse imaging clearly confirmed the bic/4-AP-induced redistribution of GFP-cortactin from spines to dendritic shaft (Fig. 7F), although the time course of cortactin translocation was slower than with the bath application of glutamate. We never detected vacuolar swellings in dendrites of neurons treated with bic/4-AP. Thus cortactin mobilization occurs in response to synaptic stimulation in the absence of excitotoxicity.
Both cortactin and F-actin redistribute from spines in response to glutamate stimulation. Is there a possible causal relationship between these events? Treatment of hippocampal neurons with jasplakinolide (2 μm), a drug that stabilizes F-actin, did not affect the spine localization of cortactin in unstimulated conditions (Fig. 7G1) but prevented the dispersal of cortactin induced by glutamate (Fig. 7G2). These results suggest that glutamate-induced translocation of cortactin is secondary to the rearrangement of the actin cytoskeleton. The localization of F-actin could not be shown in this experiment, because phalloidin binds to the same site on actin filaments as jasplakinolide.
Discussion
Cortactin and spine morphogenesis
During neuronal development long thin filopodia protrude from dendrites and subsequently are replaced by shorter spines with well defined heads in more mature neurons. The emergence of filopodia and spines is presumably the result of the protrusive force of actin polymerization, which is regulated by multiple actin binding proteins (for review, see Cooper and Schafer, 2000). A variety of actin binding proteins, including neurabin/spinophilin, α-actinin, drebrin, synaptopodin, SPAR, and inositol 1,4,5-triphosphate 3-kinase A, has been localized in dendritic protrusions (Nakanishi et al., 1997; Wyszynski et al., 1997; Hayashi and Shirao, 1999; Deller et al., 2000; Pak et al., 2001; Schell et al., 2001), but their roles in spine morphogenesis are not clear. We find the actin binding protein cortactin to be enriched in spines. siRNA knockdown of cortactin resulted in a drastic reduction of spine density, and overexpression of cortactin caused an elongation of spines. Thus both gain- and loss-of-function experiments indicate a role for cortactin in spine morphogenesis.
How might cortactin act in promoting spine numbers? Cortactin has been shown to delay the debranching of aged actin networks (Weaver et al., 2001); thus it may be important for stabilizing the actin cytoskeleton in the spine. In addition, cortactin interacts with and facilitates the function of the Arp2/3 complex, which is involved in the growth of actin networks via branch nucleation. The longitudinal growth-promoting activity of cortactin was localized to the N-terminal half of the protein comprising the NTA and the tandem repeats domains. These are the domains that bind to the Arp2/3 complex and F-actin (Wu and Parsons, 1993; Weed et al., 2000; Uruno et al., 2001) and are essential for the activation of the Arp2/3 nucleation machinery by cortactin (Uruno et al., 2001; Weaver et al., 2001). Thus the spine-elongating activity of cortactin may be related to the promotion of actin branching via the Arp2/3 complex. Similarly, in non-neuronal cells the Arp2/3 complex is essential for the extension of lamellipodia (Bailly et al., 2001), where it colocalizes with cortactin (Weed et al., 2000).
In addition to spine elongation, mutants lacking the C-terminal region of cortactin resulted in thinner spines (shrinkage/loss of spine heads), implicating the C-terminal SH3, proline-rich, and α-helical domains in determination of spine structure. Exactly how these domains act in spine morphogenesis remains unclear. The biochemical significance of the α-helical domains is not known. The proline-rich region is phosphorylated by a variety of tyrosine and serine/threonine protein kinases (for review, see Weed and Parsons, 2001) and also may mediate protein-protein interactions. Of these C-terminal domains, the SH3 is best understood and binds to several proteins, including the Shank family of scaffold proteins in the PSD (Du et al., 1998; Naisbitt et al., 1999). Interestingly, Shank is known to play a role in the maturation and enlargement of spine heads (Sala et al., 2001). One could speculate, therefore, that the interaction between cortactin and Shank is involved in the establishment of spine head morphology. The functional significance of this interaction is probably not the localization of the two proteins, because spine targeting of Shank and cortactin is independent of the respective cortactin and Shank-binding sites (Sala et al., 2001; this study). The cortactin SH3 domain also binds to dynamin, which is implicated increasingly in membrane-actin cytoskeleton interactions and morphogenesis (Orth and McNiven, 2003), and to WIP, which functions in the regulation of the actin cytoskeleton (Kinley et al., 2003).
Activity-induced cortactin translocation in spines
In response to glutamate receptor stimulation, cortactin redistributed from dendritic spines, consistent with the idea that it might function in activity-dependent dynamic changes of the actin network. It is conceivable that glutamate receptor signaling leads to the dissociation of cortactin from the spine actin network, thereby facilitating the disassembly of actin filaments. However, arguing against this idea is that jasplakinolide inhibited the glutamate-induced dispersal of cortactin, suggesting that the loss of spine cortactin is secondary to actin depolymerization.
The stimulation of glutamate receptors has been demonstrated to slow down the turnover rate of spine actin (Star et al., 2002). Therefore, the translocation of cortactin from spines may function in the deactivation of the Arp2/3-based nucleation machinery, thereby decreasing the actin turnover rate in spines.
The translocation of cortactin depends on the activation of NMDA-type of glutamate receptors. Bath application of glutamate induced a much faster redistribution of GFP-cortactin than bicuculline/4-AP, possibly because of differential stimulation of extrasynaptic versus synaptic NMDA receptors, which activate distinct signaling pathways (Hardingham et al., 2002). Activation of total NMDA receptors induces prompt breakdown of the spine actin cytoskeleton (Halpain et al., 1998), which could explain the rapid loss of cortactin from spines after the application of glutamate. The stimulation of synaptic glutamate receptors, however, might activate a different set of downstream effectors that reorganize the spine actin cytoskeleton at a slower rate, leading to the gradual changes in spine structure that occur over a time scale of hours (Hosokawa et al., 1995; Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999). The slower mobilization of cortactin might reflect the slower kinetics of actin remodeling under these conditions. Alternatively, the rapid translocation of cortactin induced by bath-applied glutamate may reflect an excitotoxic response of hippocampal neurons (Park et al., 1996; Hasbani et al., 2001). We cannot rule out that excitotoxic events might contribute to the translocation of cortactin; however, the fact that nontoxic bicuculline/4-AP treatment had a similar effect implies that cortactin redistribution can occur in response to more physiological synaptic stimulation.
Rapid NMDA receptor-dependent redistribution from synaptic clusters also has been reported for the PSD protein Homer1c (Okabe et al., 2001). Homer family proteins are implicated in the structural organization of the PSD by bridging Shank to metabotropic glutamate receptors, IP3 receptors, and other proteins (Naisbitt et al., 1999; Tu et al., 1999; Xiao et al., 2000; Sala et al., 2001). Interestingly, both cortactin and Homer are Shank-binding proteins, yet the synaptic clustering of Shank itself is acutely unaffected by glutamate stimulation. Thus our findings point toward a relatively stable core within the PSD, comprising proteins such as PSD-95 (Okabe et al., 2001) and Shank, and a more dynamic periphery of interacting proteins such as cortactin and Homer that connect the PSD core to the actin cytoskeleton and signaling proteins.
Footnotes
M.S. is an Investigator of the Howard Hughes Medical Institute. The GAD-6 monoclonal antibody developed by David Gottlieb was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA 52242. We thank Sheila Rudolph-Correia for excellent technical assistance and the following for reagents: Sondra Schlesinger for the pSinRep5, J. Thomas Parson for murine cortactin cDNA, and Xi Zhan for clone pJP20. We are grateful to Carlo Sala and Terunaga Nakagawa for unpublished reagents.
Correspondence should be addressed to Morgan Sheng, Picower Center for Learning and Memory, Massachusetts Institute of Technology, 77 Massachusetts Avenue (E18-215), Cambridge, MA 02139. E-mail: msheng@mit.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2311759-11$15.00/0
References
- Bailly M, Ichetovkin I, Grant W, Zebda N, Machesky LM, Segall JE, Condeelis J ( 2001) The F-actin side binding activity of the Arp2/3 complex is essential for actin nucleation and lamellipod extension. Curr Biol 11: 620-625. [DOI] [PubMed] [Google Scholar]
- Benson DL, Watkins FH, Steward O, Banker G ( 1994) Characterization of GABAergic neurons in hippocampal cell cultures. J Neurocytol 23: 279-295. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Miller RJ ( 1995) Excitotoxic degeneration is initiated at non-random sites in cultured rat cerebellar neurons. J Neurosci 15: 6999-7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukov KG, Birukova AA, Dudek SM, Verin AD, Crow MT, Zhan X, DePaola N, Garcia JG ( 2002) Shear stress-mediated cytoskeletal remodeling and cortactin translocation in pulmonary endothelial cells. Am J Respir Cell Mol Biol 26: 453-464. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ ( 1993) Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res 35: 567-576. [DOI] [PubMed] [Google Scholar]
- Cantarelli VV, Takahashi A, Akeda Y, Nagayama K, Honda T ( 2000) Interaction of enteropathogenic or enterohemorrhagic Escherichia coli with HeLa cells results in translocation of cortactin to the bacterial adherence site. Infect Immun 68: 382-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA, Schafer DA ( 2000) Control of actin assembly and disassembly at filament ends. Curr Opin Cell Biol 12: 97-103. [DOI] [PubMed] [Google Scholar]
- Deller T, Merten T, Roth SU, Mundel P, Frotscher M ( 2000) Actin-associated protein synaptopodin in the rat hippocampal formation: localization in the spine neck and close association with the spine apparatus of principal neurons. J Comp Neurol 418: 164-181. [DOI] [PubMed] [Google Scholar]
- Di Ciano C, Nie Z, Szaszi K, Lewis A, Uruno T, Zhan X, Rotstein OD, Mak A, Kapus A ( 2002) Osmotic stress-induced remodeling of the cortical cytoskeleton. Am J Physiol Cell Physiol 283: C850-C865. [DOI] [PubMed] [Google Scholar]
- Du Y, Weed SA, Xiong WC, Marshall TD, Parsons JT ( 1998) Identification of a novel cortactin SH3 domain-binding protein and its localization to growth cones of cultured neurons. Mol Cell Biol 18: 5838-5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Birukov KG, Zhan X, Garcia JG ( 2002) Novel interaction of cortactin with endothelial cell myosin light chain kinase. Biochem Biophys Res Commun 298: 511-519. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T ( 1999) Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399: 66-70. [DOI] [PubMed] [Google Scholar]
- Fischer M, Kaech S, Wagner U, Brinkhaus H, Matus A ( 2000) Glutamate receptors regulate actin-based plasticity in dendritic spines. Nat Neurosci 3: 887-894. [DOI] [PubMed] [Google Scholar]
- Halpain S, Hipolito A, Saffer L ( 1998) Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J Neurosci 18: 9835-9844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H ( 2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5: 405-414. [DOI] [PubMed] [Google Scholar]
- Hasbani MJ, Schlief ML, Fisher DA, Goldberg MP ( 2001) Dendritic spines lost during glutamate receptor activation reemerge at original sites of synaptic contact. J Neurosci 21: 2393-2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Shirao T ( 1999) Change in the shape of dendritic spines caused by overexpression of drebrin in cultured cortical neurons. J Neurosci 19: 3918-3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering H, Sheng M ( 2001) Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci 2: 880-888. [DOI] [PubMed] [Google Scholar]
- Hosokawa T, Rusakov DA, Bliss TV, Fine A ( 1995) Repeated confocal imaging of individual dendritic spines in the living hippocampal slice: evidence for changes in length and orientation associated with chemically induced LTP. J Neurosci 15: 5560-5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsube T, Takahisa M, Ueda R, Hashimoto N, Kobayashi M, Togashi S ( 1998) Cortactin associates with the cell-cell junction protein ZO-1 in both Drosophila and mouse. J Biol Chem 273: 29672-29677. [DOI] [PubMed] [Google Scholar]
- Kim E, Niethammer M, Rothschild A, Jan YN, Sheng M ( 1995) Clustering of Shaker-type K+ channels by interaction with a family of membrane-associated guanylate kinases. Nature 378: 85-88. [DOI] [PubMed] [Google Scholar]
- Kinley AW, Weed SA, Weaver AM, Karginov AV, Bissonette E, Cooper JA, Parsons JT ( 2003) Cortactin interacts with WIP in regulating Arp2/3 activation and membrane protrusion. Curr Biol 13: 384-393. [DOI] [PubMed] [Google Scholar]
- Korkotian E, Segal M ( 2001) Regulation of dendritic spine motility in cultured hippocampal neurons. J Neurosci 21: 6115-6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel DD, Hendrickson AE, Wu JY, Schwartzkroin PA ( 1986) Glutamic acid decarboxylase (GAD) immunocytochemistry of developing rabbit hippocampus. J Neurosci 6: 541-552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Rosenmund C, Schwaller B, Neher E ( 2000) Differences in Ca2+ buffering properties between excitatory and inhibitory hippocampal neurons from the rat. J Physiol (Lond) 525[Pt 2]: 405-418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez I, Duprez V, Melle J, Dreyfus F, Levy-Toledano S, Fontenay-Roupie M ( 2001) Thrombopoietin stimulates cortactin translocation to the cytoskeleton independently of tyrosine phosphorylation. Biochem J 356: 875-881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K ( 1999) Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science 283: 1923-1927. [DOI] [PubMed] [Google Scholar]
- Matus A ( 2000) Actin-based plasticity in dendritic spines. Science 290: 754-758. [DOI] [PubMed] [Google Scholar]
- McNiven MA, Kim L, Krueger EW, Orth JD, Cao H, Wong TW ( 2000) Regulated interactions between dynamin and the actin-binding protein cortactin modulate cell shape. J Cell Biol 151: 187-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M ( 1999) Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23: 569-582. [DOI] [PubMed] [Google Scholar]
- Nakanishi H, Obaishi H, Satoh A, Wada M, Mandai K, Satoh K, Nishioka H, Matsuura Y, Mizoguchi A, Takai Y ( 1997) Neurabin: a novel neural tissue-specific actin filament-binding protein involved in neurite formation. J Cell Biol 139: 951-961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka Y, Takai Y ( 1998) Isolation and characterization of cortactin isoforms and a novel cortactin-binding protein, CBP90. Genes Cells 3: 603-612. [DOI] [PubMed] [Google Scholar]
- Okabe S, Urushido T, Konno D, Okado H, Sobue K ( 2001) Rapid redistribution of the postsynaptic density protein PSD-Zip45 (Homer 1c) and its differential regulation by NMDA receptors and calcium channels. J Neurosci 21: 9561-9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, McNiven MA ( 2003) Dynamin at the actin-membrane interface. Curr Opin Cell Biol 15: 31-39. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Kashiwada K, Takahashi M, Sobue K ( 1995) Translocation of cortactin (p80/85) to the actin-based cytoskeleton during thrombin receptor-mediated platelet activation. Exp Cell Res 221: 197-204. [DOI] [PubMed] [Google Scholar]
- Pak DT, Yang S, Rudolph-Correia S, Kim E, Sheng M ( 2001) Regulation of dendritic spine morphology by SPAR, a PSD-95-associated RapGAP. Neuron 31: 289-303. [DOI] [PubMed] [Google Scholar]
- Park JS, Bateman MC, Goldberg MP ( 1996) Rapid alterations in dendrite morphology during sublethal hypoxia or glutamate receptor activation. Neurobiol Dis 3: 215-227. [DOI] [PubMed] [Google Scholar]
- Rothman SM, Olney JW ( 1995) Excitotoxicity and the NMDA receptor—still lethal after eight years. Trends Neurosci 18: 57-58. [DOI] [PubMed] [Google Scholar]
- Sala C, Rudolph-Correia S, Sheng M ( 2000) Developmentally regulated NMDA receptor-dependent dephosphorylation of cAMP response element-binding protein (CREB) in hippocampal neurons. J Neurosci 20: 3529-3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Piech V, Wilson NR, Passafaro M, Liu G, Sheng M ( 2001) Regulation of dendritic spine morphology and synaptic function by Shank and Homer. Neuron 31: 115-130. [DOI] [PubMed] [Google Scholar]
- Sala C, Futai K, Yamamoto K, Worley PF, Hayashi Y, Sheng M ( 2003) Inhibition of dendritic spine morphogenesis and synaptic transmission by activity-inducible protein Homer1a. J Neurosci 23: 6327-6337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell MJ, Erneux C, Irvine RF ( 2001) Inositol 1,4,5-trisphosphate 3-kinase A associates with F-actin and dendritic spines via its N terminus. J Biol Chem 276: 37537-37546. [DOI] [PubMed] [Google Scholar]
- Sik A, Penttonen M, Ylinen A, Buzs↔ ki G ( 1995) Hippocampal CA1 interneurons: an in vivo intracellular labeling study. J Neurosci 15: 6651-6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Star EN, Kwiatkowski DJ, Murthy VN ( 2002) Rapid turnover of actin in dendritic spines and its regulation by activity. Nat Neurosci 5: 239-246. [DOI] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF ( 1999) Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron 23: 583-592. [DOI] [PubMed] [Google Scholar]
- Uruno T, Liu J, Zhang P, Fan Y, Egile C, Li R, Mueller SC, Zhan X ( 2001) Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat Cell Biol 3: 259-266. [DOI] [PubMed] [Google Scholar]
- Weaver AM, Karginov AV, Kinley AW, Weed SA, Li Y, Parsons JT, Cooper JA ( 2001) Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr Biol 11: 370-374. [DOI] [PubMed] [Google Scholar]
- Weed SA, Parsons JT ( 2001) Cortactin: coupling membrane dynamics to cortical actin assembly. Oncogene 20: 6418-6434. [DOI] [PubMed] [Google Scholar]
- Weed SA, Du Y, Parsons JT ( 1998) Translocation of cortactin to the cell periphery is mediated by the small GTPase Rac1. J Cell Sci 111[Pt 16]: 2433-2443. [DOI] [PubMed] [Google Scholar]
- Weed SA, Karginov AV, Schafer DA, Weaver AM, Kinley AW, Cooper JA, Parsons JT ( 2000) Cortactin localization to sites of actin assembly in lamellipodia requires interactions with F-actin and the Arp2/3 complex. J Cell Biol 151: 29-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Parsons JT ( 1993) Cortactin, an 80/85-kilodalton pp60src substrate, is a filamentous actin-binding protein enriched in the cell cortex. J Cell Biol 120: 1417-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M ( 1997) Competitive binding of α-actinin and calmodulin to the NMDA receptor. Nature 385: 439-442. [DOI] [PubMed] [Google Scholar]
- Xiao B, Tu JC, Worley PF ( 2000) Homer: a link between neural activity and glutamate receptor function. Curr Opin Neurobiol 10: 370-374. [DOI] [PubMed] [Google Scholar]