

Abstract
Dopamine D4 receptors (D4R) are localized in the globus pallidus (GP), but their function remains unknown. In contrast, dopamine D2 receptor activation hyperpolarizes medium spiny neurons projecting from the striatum to the GP and inhibits GABA release. However, using slice preparations from D2R-deficient [D2 knock-out (D2KO)] mice, we found that dopamine inhibited GABAA-receptor-mediated currents in GP neurons. The paired-pulse ratio was statistically unchanged after dopamine application but was significantly elevated in D2KO wild-type littermates (WT). Furthermore, in D2KO mice, outward currents elicited by iontophoretically applied GABA were suppressed by dopamine. Dopamine (30 μm) decreased the amplitude of miniature IPSCs in both WT and D2KO mice, but the decrease in the frequency was observed only in the former but not significantly in the latter. Dopamine-induced suppression of IPSCs was blocked by selective D4R antagonists (clozapine or 3-[4-(4-iodophenyl)piperazin-1-yl]methyl-1H-pyrrolo[2,3-b]pyridine trihydrochloride), and a D4R-selective agonist N-[[4-(2-cyanophenyl)-1-piperazinyl]methyl]-3-methyl-benzamide reversibly and dose-dependently suppressed IPSCs, whereas agonists [SKF38,393 ((+/-)-1-phenyl-2,3,4,5-tetrahydro-(1H)-3-benzazepine-7,8-diol hydrochloride) or (+)-(4aR,10bR)-3,4,4a,10b-tetrahydro-4-propyl-2H,5H-[1]benzopyrano[4,3-b]-1,4-oxazin-9-ol] or antagonists [SCH23,390 (R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride) or sulpiride] of other receptor subtypes had little effect. In GP neurons from D4R-deficient mice, dopamine-induced inhibition of GABAergic outward currents was undetectable. D4R activation suppressed the activity of protein kinase A in GP neurons, resulting in a decrease in the amplitude of GABAergic IPSCs. These findings showed that postsynaptic activation of D4R on the GP neurons reduces GABAergic currents through the suppression of PKA activity.
Keywords: motor, basal ganglia, dopamine, GABA, globus pallidus, Parkinson's disease
Introduction
The rodent globus pallidus (GP) is equivalent to the external segment of the primate globus pallidus (GPe). The GP receives GABAergic inputs from striatal medium spiny (MS) neurons and seems to play a pivotal role in the function of basal ganglia. A model of the functional organization of the basal ganglia suggests that a loss of nigrostriatal dopamine (DA) neurons in Parkinson's disease (PD) leads to overactivity of the “indirect” MS neurons containing DA D2 receptors (D2R) that project from the striatum to the GPe (Albin et al., 1989; DeLong, 1990; Chesselet and Delfs, 1996). Therefore, the systemic administration of DA receptor agonists may increase the activity of GP (or GPe) neurons in vivo because of a decrease in GABA transmission in the GP (Nakanishi et al., 1985; DeLong, 1990; Hooper et al., 1997). However, DA D3 and D4 receptors are located in the striatum in addition to D2R, and the GP itself receives dopaminergic fibers from collaterals of the nigrostriatal pathway (Lindvall and Bjorklund, 1979; Surmeier et al., 1996). In fact, adding DA to the GP induces an increase in GP activity by attenuating GABAergic transmission (Bergstrom and Walters, 1984), suggesting that activation of DA receptors located in the GP modulates GABAergic transmission. The activation of D2-like receptors, which are presumably located on the striatopallidal terminals, reduces GABA release both in vitro (Floran et al., 1997) and in vivo (Querejeta et al., 2001), and thus reduces GABAA receptor-mediated IPSCs at the striatopallidal synapses (Cooper and Stanford, 2001).
The effects of DA neurotransmission are mediated by five genetically defined receptors, but DA receptors are pharmacologically divided into D1-like and D2-like subfamilies (Seeman and Van Tol, 1994). The D1-like receptor family activates adenylate cyclase, and D1 and D5 subtypes have been cloned. The D2-like receptor family is composed of the D2, D3, and D4 subtypes and inhibits cAMP. Studies using reverse transcription (RT)-PCR have indicated that the MS neurons projecting exclusively to the GP express high levels of D2R mRNA and low or modest levels of D3, D4, and D5 receptor mRNAs (Surmeier et al., 1996). In contrast, in the GP the D4 DA receptors (D4R) are expressed at high levels on the somata of GP neurons (Mrzljak et al., 1996; Ariano et al., 1997; Mauger et al., 1998). Because the DA receptors are not precisely pharmacologically distinguishable (Seeman and Van Tol, 1994), mice deficient in the DA receptor subtype would be the most useful tools with which to examine the specific role of each DA receptor subtype. Using D2R- and D4R-deficient mice, we show here that D4R is involved with GP neurons in the DA-mediated postsynaptic suppression of GABAergic IPSCs. This represents an additional mechanism for the regulation of GABA transmission in the GP.
Materials and Methods
All experimental procedures using laboratory animals were approved by the Animal Care and Use Committee of the Tokyo Metropolitan Institute of Gerontology.
Gene knock-out mice. D2R-deficient [D2 knock-out (D2KO)] mice were derived as described previously (Yamaguchi et al., 1996). D4R-deficient (D4KO) mice were generated by gene targeting with mouse embryonic stem (ES) cells (Kobayashi et al., 1995a,b). Mice heterozygous for the mutation were bred with C57BL/6J mice for several generations. Control wild-type (WT) mice were littermates of the homozygous mutants.
Generation of D4KO mice. Phage clones containing the mouse D4R gene were isolated from a λFIXII genomic DNA library (Stratagene, La Jolla, CA). The targeting vector contained the 5′-homologous region, the human interleukin-2 receptor α-subunit (IL-2Rα) gene cassette, the neo gene cassette, the 3′-homologous region, and the diphtheria toxin A-fragment (DTA) gene cassette (Fig. 1 A). Gene targeting with mouse ES cells proceeded as described previously (Kobayashi et al., 1995b). Correctly targeted ES cells were identified by Southern blotting (Fig. 1B). The ES cells were microinjected into C57BL/6J blastocysts, and the resulting chimeras were crossed with the C57BL/6J strain to produce mice heterozygous for the targeted mutation. The IL-2Rα gene cassette was introduced into the mouse D4R locus for conditional ablation of cell types expressing the receptor by immunotoxin-mediated cell targeting (Kobayashi et al., 1995a), but conditional cell ablation was not performed in this study.
Figure 1.
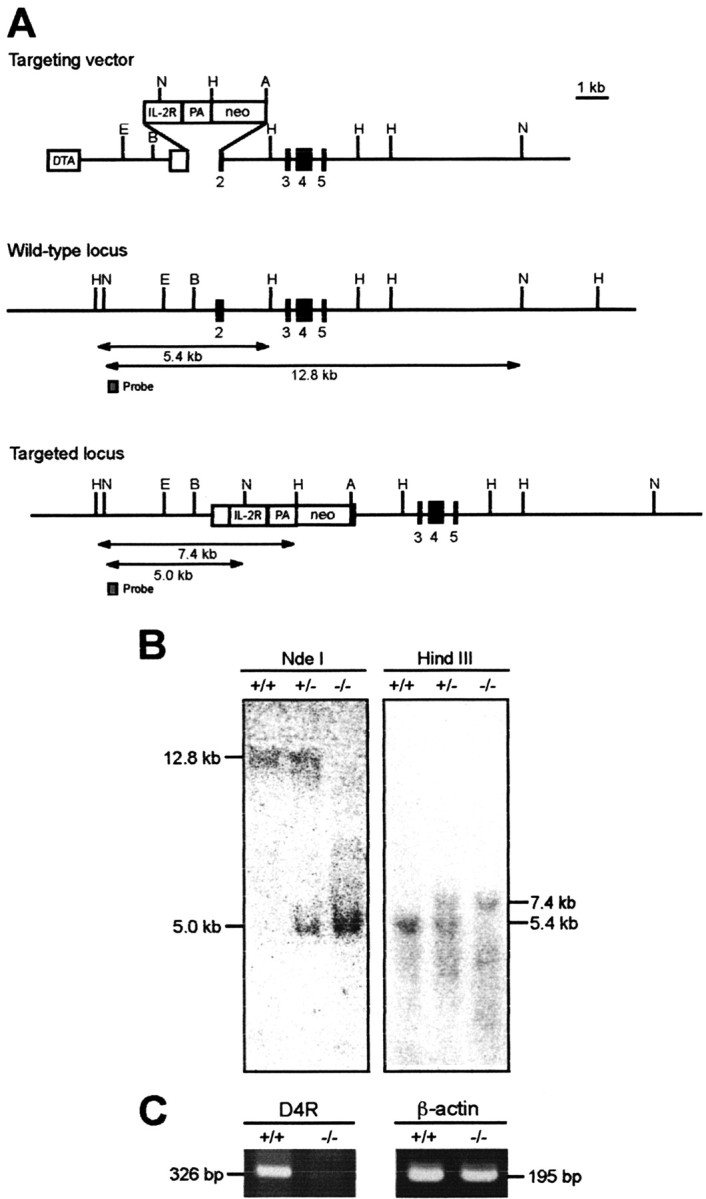
Gene targeting and RT-PCR analysis for generation of D4KO mice. A, Strategy for generation of D4KO mice. In the targeting vector, IL-2R gene and neo gene cassettes were introduced into exon 2 of mouse D4R gene. The DTA gene cassette was inserted at the 5′ end of the vector. Restriction enzyme fragments for Southern blots with probe A are indicated. A, AccI; B, BamHI; E, EcoRI; H, HindIII; N, NdeI; PA, polyadenylation signal. B, Genotyping of mice. Genomic DNA prepared from mouse tail tissues was digested with restriction enzymes and Southern blotted. After NdeI and HindIII digestion, the wild-type allele produced 12.8 and 5.4 kb bands, whereas the mutant produced 4.6 and 7.4 kb bands, respectively. C, The 326 bp band corresponding to the D4R mRNA was amplified in WT but not in D4KO mice.
The absence of D4R mRNA in the D4KO mice was confirmed by RT-PCR using total RNA (1 μg) from the GP. The primer set for D4R detection was 5′-CGGAGCGGGTTCAGCAGT-3′ (the nucleotides from -24 to -7 of mouse D4R cDNA) and 5′-CTCAGGAGCCACACGCCAC-3′ (the nucleotides from 284 to 302 of mouse D4R cDNA). The primer set for β-actin detection was 5′-AGAAGATCTGGCACCACACC-3′ (nucleotides from 329 to 348 of mouse β-actin cDNA) and 5′-GGACATACGGAGACCAGCATG-3′ (nucleotides from 504 to 524 of mouse β-actin cDNA). A 326 bp band corresponding to the D4R mRNA was amplified in WT but not in D4KO mice (Fig. 1C).
Electrophysiology. Whole brains from 10- to 20-d-old D2R- or D4R-deficient mice or their WT littermates under ether anesthesia were placed in ice-cold artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 3 KCl, 1 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 10 glucose, buffered to pH 7.4 with NaHCO3 (26 mm) saturated with 95% O2 and 5% CO2. Parasagittal slices (250 μm thick) cut using a DTK-1000 Microslicer (Dosaka, Kyoto, Japan) were continuously perfused with ACSF at a rate of 1-2 ml/min at 30°C for 1 hr. Whole-cell patch-clamp recordings were collected by an EPC9/2 amplifier (Heka Elektronik, Lambrecht/Pfalz, Germany), with infrared differential contrast visualization using an Olympus (Tokyo, Japan) BX50WI and a charge-coupled device camera (Hamamatsu, Shizouka, Japan). For current-clamp recordings, patch pipettes contained the following (in mm): 129 K-gluconate, 11 KCl, 2 MgCl2, 10 HEPES, 4 Na2-ATP, 0.3 GTP, and 0.5% biocytin (brought to pH 7.3 with KOH; osmolarity, 280 mOsm). For voltage-clamp recordings, patch pipettes (4-6 MΩ) contained the following (in mm): 124 Cs-methanesulfonate, 11 KCl, 2 MgCl2, 10 HEPES, 4 Na2-ATP, 0.3 GTP, 0.1 spermine, 5 N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide, and 0.5% biocytin (and were brought to 280 mOsm and pH 7.3 with CsOH). To record miniature IPSCs (mIPSCs), pipettes contained the following (in mm): 125 KCl, 10 NaCl, 1 CaCl2, 2 MgCl2, 10 BAPTA, 10 HEPES, 4 Na2-ATP, 0.3 GTP, and 0.5% biocytin (and were adjusted to pH 7.3 with KOH).
We evoked IPSCs by field stimulation of the striatum close to the GP at 0.1 Hz with a bipolar tungsten electrode (100-500 μsec, 100-800 μA; Microprobe, Potomac, MD) to obtain an ∼50% maximal response. To isolate GABAA-receptor-mediated currents, the NMDA receptor antagonist d(-)-2-amino-5-phosphonopentanoic acid (d-AP-5; 25 μm) and the AMPA receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; 10 μm) were applied to the ACSF. Voltage errors attributable to the liquid junction potential were subtracted off-line. To determine the effects of DA or DA receptor agonists on IPSCs, the averaged IPSC amplitudes of 12 traces during the peak response to the drug were compared with those obtained immediately before drug application.
We recorded mIPSCs at a potential of -60 mV in the presence of TTX (1 μm), CNQX (10 μm), d-AP-5 (25 μm), and strychnine (0.5 μm). The theoretical chloride reversal potential was ∼0 mV. Data were collected over continuous 10 min periods, acquired using PowerLab (AD Instruments, Castle Hill, Australia) and analyzed using Mini Analysis software (Synaptosoft, Decatur, GA).
Outward currents were evoked every 4 min by the iontophoretic application of GABA (1 mm) in the presence of the GABAB receptor antagonist 2-OH-saclofen (100 μm) and TTX (1 μm) in modified ACSF (2.4 mm MgCl2, 1.2 mm CaCl2) to suppress transmitter release (see Figs. 6 and 10). The modified ACSF was continuously perfused at a rate of 2-3 ml/min. To examine the effects of DA on outward currents, the averaged amplitudes of two traces in the presence of DA were compared with those obtained immediately before DA application. To investigate the effects of an inhibitor or an activator of PKA on IPSCs (see Fig. 11C,D), we plotted the averaged IPSC amplitudes of six consecutive traces.
Figure 6.
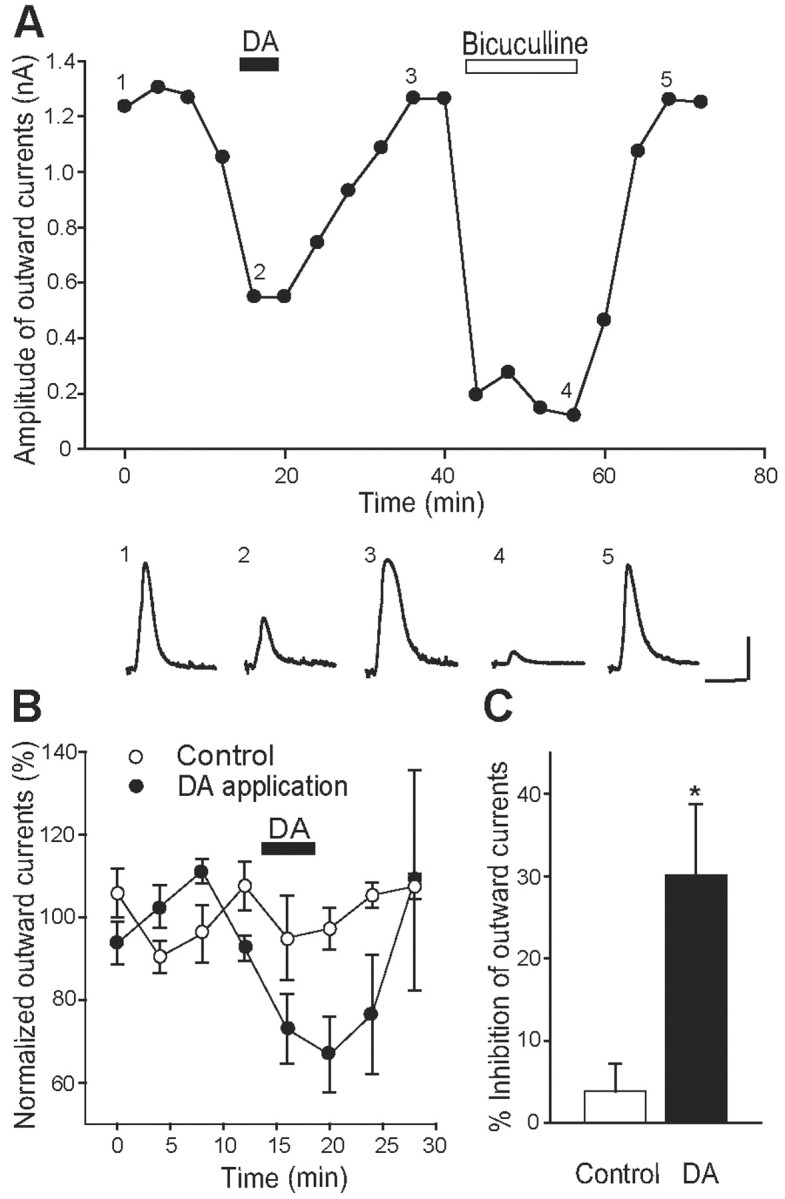
Effects of DA on outward currents elicited by exogenous GABA in D2KO mice. A, Effects of DA (10 μm; filled bar) and bicuculline (20 μm; open bar) on outward currents induced by iontophoretic application of GABA (1 mm) on GP neurons in the presence of TTX (1 μm) and 2-OH-saclofen (100 μm), a selective GABAB receptor antagonist. Evoked outward currents considerably fluctuated, but DA effectively and reversibly suppressed the currents. Each trace, taken at the indicated time point (1-5), represents outward current before, during, and after the application of DA or bicuculline. Calibration: 0.4 nA, 400 msec. B, Normalized outward currents were plotted with the application of DA for the period indicated by the bar (filled circles; n = 5) or with control vehicle (open circles; n = 5). C, Summarized histograms show the inhibitory effect of DA on the amplitude of outward currents. *p < 0.05; unpaired Student's t test.
Figure 10.

Dopamine-induced inhibition of GABAergic outward currents absent in GP neurons of D4KO mice. A, Current-voltage relationships of IPSCs recorded from WT (open triangles; n = 12) and D4KO (filled triangles; n = 10) mice. Current amplitudes were normalized to the value obtained at 0 mV. The inset indicates representative IPSCs at each holding potential from WT and D4KO mice. Calibration: 0.2 nA, 40 msec. B, Normalized outward currents recorded from GP neurons in acute slices prepared from D4R-deficient (n = 10) or littermate WT (n = 8) mice. The filled bar indicates application of DA (10 μm). C, Summarized histograms show effects of DA on the amplitude of outward currents recorded from D4KO and WT mice. *p < 0.05; unpaired Student's t test.
Figure 11.
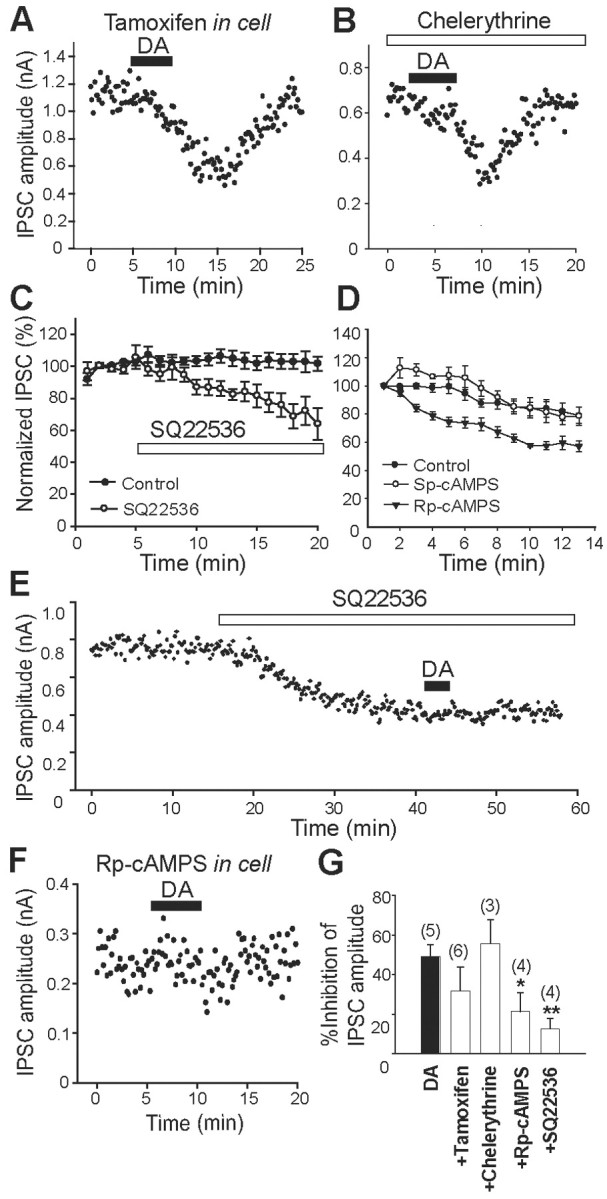
Dopamine-receptor-induced inhibition of IPSCs is mediated by PKA inhibition. A, B, Effects of tamoxifen (50 μm; A) and chelerythrine (5 μm; B), both of which are PKC inhibitors, on DA-receptor-induced inhibition of IPSCs in D2KO mice. Tamoxifen was included in a patch pipette, and chelerythrine was applied in bath solution. The filled bar indicates DA (30 μm) application in A, B, E, and F. C, Normalized IPSC amplitudes are averaged to plot the control (filled circles; n = 4) and the effects of SQ22,536 (100 μm; open circles; n = 6) for the period indicated by the open bar. D, Normalized IPSC amplitudes are plotted for the control (open circles; n = 5), Sp-cAMPS (filled circles; n = 5), and Rp-cAMPS (filled inverted triangles; n = 3) immediately after membrane rupture. Sp-cAMPS and Rp-cAMPS, an activator and an inhibitor of cAMP, respectively, were included in patch pipettes. E, Effect of the PKA inhibitor SQ22,536 (100 μm) on the amplitude of IPSCs and occlusion of DA receptor-induced inhibition. SQ22,536 was applied as a bath solution. F, Effect of Rp-cAMPS (1 mm), a PKA inhibitor, on DA-receptor-induced inhibition. Rp-cAMPS was included in patch pipettes. G, Summary of the percentage of inhibition of IPSC by DA in the absence (n = 5) or presence of tamoxifen (n = 6), chelerythrine (n = 3), Rp-cAMPS (n = 4), and SQ22,536 (n = 4). The numbers of experiments are shown in parentheses. *p < 0.05; **p < 0.01; unpaired Student's t test.
Signals were filtered at 5 kHz and digitized at 20 kHz with Pulse/PulseFit (Heka) running on a Macintosh computer (Apple Computers, Cupertino, CA). If series resistance was changed by >20%, the experiments were discarded. All values are expressed as means ± SEM. Statistical significance was assessed by unpaired or paired Student's t tests. Statistical differences in the cumulative frequency and amplitude of mIPSCs were analyzed using the Kolmogorov-Smirnov two-sample test (Synaptosoft).
Dopamine was dissolved in ACSF immediately before use, and sodium metabisulfite (50 μm) was always included to prevent oxidative degradation of DA. Rp-adenosine 3′,5′-cyclic monophosphorothioate triethylammonium (Rp-cAMPS) and Sp-adenosine 3′,5′-cyclic monophosphorothioate triethylammonium (Sp-cAMPS) were dissolved directly into patch solution, and tamoxifen was dissolved in methanol and added to the patch solution. Other drugs were stored as frozen stock solutions in distilled water and diluted 1000-fold into ACSF immediately before use, with the exception of 2-OH-saclofen, which was stored as a frozen 1000-fold stock solution in NaOH. We purchased TTX from Wako Chemicals (Osaka, Japan), 2-OH-saclofen and d-AP-5 from Tocris Cookson (Ballwin, MO), and all other drugs from Sigma/RBI (Poole, UK).
Histochemical procedures. Slices containing biocytin-filled cells were fixed in 0.1 m phosphate buffer (PB) containing 4% paraformaldehyde and 0.2% picric acid overnight at 4°C, rinsed in PB for 30 min, and incubated in PB containing 0.5% H2O2 for 30 min to suppress endogenous peroxidase activity. The slices were then incubated in 20% sucrose at 4°C overnight and stored frozen. Before use, the slices were washed with Tris-buffered saline containing 0.5% Triton X-100 and immersed in avidin-biotin-peroxidase complex (Vector Laboratories, Burlingame, CA) at a dilution of 1:100 for 4-6 hr at room temperature. The slices were rinsed and reacted with 3,3′-diaminobenzidine tetrahydrochloride (Kirkegaard & Perry, Gaithersburg, MD), and mounted on slides.
Results
Electrophysiological properties of mouse GP neurons
We visually identified and sampled GP neurons in brain slice preparations from mice by whole-cell recording under infrared differential contrast optics. According to other studies using guinea pigs (Nambu and Llinas, 1994, 1997) and rats (Kita and Kitai, 1991; Stanford and Cooper, 1999; Cooper and Stanford, 2000; Shindou et al., 2001), GP neurons can be classified into at least two major groups according to their electrophysiological properties. Type A (rat) or type II (guinea pig and rat) GP neurons were characterized by the presence of sag during hyperpolarizing current injection, which corresponded to a time- and voltage-dependent inward current (Ih) (Fig. 2A, left), and 26 of 38 of our mouse cells (68%) were of this type. The input resistance of these neurons was 399 ± 37 MΩ. Type B or type I neurons were identified in 12 of 38 cells (32%) by the absence of sag (Fig. 2A, right) and a lower input resistance (216 ± 23 MΩ; Student's t test; p = 0.003) than the type A or type II neurons. Other rare types such as type C or type III were not sampled during the initial phase of these experiments. Similar cell types were also found in the GP of the D2KO and the D4KO mouse, but the GP in each mutant and the physiological characterization of each cell type were not systematically determined in this study. Because the effects of DA or dopaminergic agents between the above cell types appeared identical, and because reliable cell typing in voltage-clamp experiments was practically impossible, we pooled the data from all neuronal cell types.
Figure 2.

Isolation of GABAA-receptor-mediated IPSCs in GP neurons. A, Superimposed voltage responses from type A (left) and B (right) neurons in response to 1000 msec hyperpolarizing and depolarizing currents. Calibration: 20 mV (voltage), 200 pA (current), 500 msec. B, Effect of bicuculline (20 μm), a GABAA receptor antagonist, on IPSCs at a holding potential of -10 mV in the presence of CNQX (10 μm) and d-AP-5 (25 μm). Traces 1-3, taken at indicated time points, are representative IPSCs in the WT mouse before, during, and after the application of bicuculline, respectively. Calibration: 500 pA, 10 msec. C, Current-voltage relationships of IPSCs recorded from D2KO (filled circles; n = 5) and WT (open circles; n = 5) mice. Current amplitudes were normalized to the value obtained at 0 mV and are expressed as means ± SEM here and in the following figures. The inset indicates representative IPSCs at each holding potential from a WT mouse. Calibration: 0.2 nA, 20 msec. D, Low-magnification micrographs of biocytin-filled GP neurons from WT (top) and D2KO (bottom) mice. Scale bar, 100 μm.
Identification of GABAA-receptor-mediated IPSCs on GP neurons of the D2R-deficient and the wild-type mouse
Synaptic currents were evoked by electrical stimulation of the dorsal striatum at a holding potential of -10 mV in the presence of CNQX (10 μm) and d-AP-5 (25 μm) to block glutamatergic components at a rate of 0.1 Hz (Fig. 2B). These currents were completely and reversibly suppressed by bicuculline (20 μm), a selective GABAA receptor antagonist, confirming that the currents were GABAA-receptor-mediated IPSCs. The results from acute slices prepared from D2R-deficient (D2KO mice, n = 4) and their littermate wild-type mice (n = 3) were the same. Current-voltage relationship curves (Fig. 2C) showed that the reversal potentials were approximately -51 mV (D2KO mice, -51.45 ± 1.09 mV; WT mice, -51.50 ± 2.51 mV; Student's t test; p = 0.986), which was close to the equilibrium potential for chloride calculated by the Nernst equation (ECl = -50 mV). Furthermore, we did not detect any difference in the kinetic properties of the IPSCs between the two (D2KO mice, n = 12 vs WT mice, n = 15; Student's t test; rise time, 20-80%, 1.09 ± 0.12 msec vs 1.04 ± 0.13 msec, p = 0.79; decay time constant, 12.31 ± 0.91 msec vs 13.55 ± 0.93 msec, p = 0.36). These results indicated that the physiological properties of GABAA receptor-mediated IPSCs were unchanged in D2KO mice. Subsequent staining with biocytin after recordings revealed no morphological differences between the GP neurons of D2KO and WT mice (Fig. 2D).
Origins of GABAergic IPSCs
At least one-third of GP neurons are thought to send their axons back to the striatum and have local axon collaterals in the GP (Kita and Kitai, 1991, 1994; Bevan et al., 1998; Kita et al., 1999; Kita and Kita, 2001). Therefore, electrical stimulation of the striatum would evoke both striatopallidal and pallidostriatal GABAergic fibers. Ogura and Kita (2000) presumed that small IPSCs with a latency of 7-10 msec are mediated by striatopallidal axons, and that large IPSCs with a latency of <4 msec might be derived from activation of the local collateral axons of the pallidostriatal neurons. Indeed, we occasionally obtained IPSCs that were composed of both short- and long-latency IPSCs. Figure 3 shows a typical result from a littermate of a D4KO mouse. The first short-latency IPSC was evoked as early as 80 μA of stimulus strength (filled circles). The second or third long-latency IPSCs (open triangles) started to appear at ≥150 μA, and their amplitude was smaller than the first. However, activating the two types of IPSCs separately was practically impossible, and the neurons that received multiple IPSCs as shown above were sampled only occasionally. The amplitude of the IPSCs we examined here (WT, D2KO, and D4KO mice) was approximately one-half of the maximal response to striatal stimulation, and their latency was usually <4 msec. Therefore, we considered that they contained both components, but that most of the IPSCs might be of short latency.
Figure 3.

Striatal stimulation induced short- and long-latency IPSCs in GP neurons. A, Sample traces of IPSCs to striatal stimulation with various stimulus intensities. This cell is from the WT littermate of a D4KO mouse. Filled circles indicate initial IPSCs; triangles indicate second and third IPSCs. Calibration: 10 msec, 100 pA. B, Plot of the IPSC amplitude against the stimulus intensity. Initial large IPSCs (filled circles) started to appear at a stimulus intensity of 80 μA; second and third IPSCs (triangles) appeared at 150 μA and were smaller. C, Plot of IPSC latency against stimulus intensity. The latency of initial IPSCs was 3.3 ± 0.1 msec, and that of the second and third IPSCs was 10.5 ± 0.7 msec.
Inhibition of IPSCs by DA in the D2KO mouse
Reports indicate that activation of D2R located on the terminals of MS neurons mediates presynaptic inhibition of IPSC, thereby causing increased GP neuron activity (Cooper and Stanford, 2001; Querejeta et al., 2001). To confirm these findings, we tested the effects of DA on IPSCs recorded from GP neurons. Bath application of DA (10 μm) for 5 min, together with sodium metabisulfite (50 μm), an antioxidant for DA, to acute slices prepared from WT mice reversibly inhibited the amplitude of IPSCs (Fig. 4A). However, DA continued to inhibit the amplitude of IPSCs reversibly, even in acute slices prepared from D2KO mice (Fig. 4B). These responses were evident in all neurons tested and were reversed on washing. Sodium metabisulfite alone had no effect on IPSCs (n = 12; data not shown). Figure 4C shows the IPSCs reduced by DA that were recorded from both types of mice (10 μm). The reduction was significantly larger in WT than in D2KO mice (WT mice, 53.26 ± 4.06%, n = 7; D2KO mice, 21.24 ± 4.97%, n = 5; p < 0.001; Student's t test). Resting membrane potentials or holding currents did not change during bath application of DA even at a concentration of 100 μm, and no difference was detected between the two. The difference in resting membrane potentials before and after DA application was -1.0 ± 1.354 mV (n = 4) in D2KO mice and -0.45 ± 0.38 mV (n = 9) in WT mice (p = 0.876; Student's t test). Figure 4D shows a comparison of the IPSC amplitude reduced by DA in both types of mice over a range of 1-30 μm. The concentration-response curves statistically and significantly differed in the range of 1-10 μm (WT vs D2KO, Student's t test: 1 μm, 16.05 ± 3.29 vs 0.12 ± 1.98%, p = 0.014; 3 μm, 41.69 ± 8.29 vs -2.41 ± 6.91%, p = 0.012; 10 μm, 53.26 ± 4.06 vs 21.24 ± 4.97%, p < 0.001) but at 30 μm (63.1 ± 7.68 vs 49.00 ± 6.18%; p = 0.174). The curve of WT mice could be fit by a summation of two Hill equations with IC50 values of 1.3 and 9.5 μm, respectively, whereas that of D2KO mice could be fit by a single Hill equation with an IC50 of 10.7 μm (Fig. 4D). This indicated a dual inhibition of IPSCs via activation of D2R and some other subtypes of DA receptors in striatopallidal synapses or synapses of the axon collaterals of the pallidostriatal neurons.
Figure 4.

Effects of DA on IPSCs. A, B, Effects of DA (10 μm; filled bars) on IPSCs recorded from WT (A) and D2KO (B) mice. Insets, Traces taken at indicated time points in A and B are representative IPSCs from acute slices from WT (1-3) and D2KO (4-6) mice before, during, and after the application of DA, respectively. Calibration: 0.4 nA, 20 msec. C, Normalized IPSC amplitudes were monitored before, during, and after the application of DA (10 μm) for the period indicated by the filled bar in acute slices from WT (open circles; n = 7) and D2KO (filled circles; n = 5) mice. D, Concentration-response relationships for DA-induced inhibition of IPSCs recorded from WT (open circles) and D2KO (filled circles) mice. The numbers of experiments are shown in parentheses. *p < 0.05; **p < 0.01; unpaired Student's t test.
Inhibition of GABAergic IPSCs by DA is postsynaptic in the D2KO mouse
We examined whether the DA-induced inhibition of GABAergic IPSCs in the D2KO mouse was mediated postsynaptically and/or presynaptically by testing the effects of DA on paired-pulse facilitation, which is commonly believed to be a mostly presynaptic mechanism (Zucker, 1989). A pair of IPSCs was elicited by two stimuli at an interval of 50 msec, and the paired-pulse ratio (PPR), which is the ratio of the amplitude of the second IPSC to the first, was monitored before and after DA application. Before DA application, the PPRs from the two types of mice did not differ (WT mice, 1.25 ± 0.05, n = 16; D2KO mice, 1.31 ± 0.05, n = 17; p = 0.456; Student's t test). However, as a result of DA (10 μm) application, PPRs increased in all seven experiments recorded from WT mice (Fig. 5A) and were relatively unaffected in D2KO mice (data not shown). In summary, the mean of the normalized PPR was significantly increased in WT mice after DA application (1.19 ± 0.06; p < 0.05; paired Student's t test; n = 7), whereas it was statistically unchanged in D2KO mice (1.03 ± 0.06; p = 0.625; paired Student's t test; n = 5) (Fig. 5B). These findings suggest that the activation of D2Rs inhibits IPSCs, probably via decreased GABA release in the mouse striatopallidal or pallidopallidal synapses. On the other hand, the inhibition in D2KO mice may be mainly postsynaptic. However, because several DA receptor subtypes have been identified in the striatal MS neurons (Surmeier et al., 1996), their contribution to the presynaptic inhibition should also be considered.
Figure 5.

Effects of DA on the PPR of IPSCs elicited by striatal stimulation. A, IPSCs elicited by two successive stimuli at an interval of 50 msec before and during application of DA (10 μm) obtained from WT mice. Each trace is the average of 12 consecutive traces. In the superimposed traces, initial IPSCs during DA application were normalized to those recorded under control conditions. Calibration: 0.5 nA, 20 msec. B, Averages of normalized PPRs obtained from WT (open circles; n = 7) and D2KO (filled circles; n = 5) mice. *p < 0.05; paired Student's t test.
To confirm the postsynaptic actions of DA, we investigated the effects of DA on outward currents elicited by iontophoretically applied GABA onto GP neurons prepared from D2KO mice. A pipette containing GABA (1 mm) was placed ∼20 μm from the soma of the recorded GP neuron. The modified ACSF with heightened Mg2+ (2.4 mm) and lowered Ca2+ (1.2 mm) containing TTX (0.5 μm) and 2-OH-saclofen (100 μm) to block transmitter release, sodium channels, and GABAB receptor-mediated currents, respectively, was continuously perfused at a rate of 2-3 ml/min. Thereafter, GABA was applied iontophoretically at an interval of 4 min to these neurons voltage-clamped at -10 mV. This yielded outward currents that substantially fluctuated, but Figure 6A shows that DA application (10 μm) for 5 min caused a reversible reduction in the amplitude of the outward currents. These currents were almost completely blocked by bicuculline (20 μm) but returned to the control level (Fig. 6A), confirming that they were mediated by the GABAA receptors. The ratio (percentage) of modulation of outward currents is plotted in the presence or absence of DA in Figure 6B. The outward currents were suppressed 30.16 ± 8.63% by DA, compared with 3.84 ± 3.40% by a vehicle (sodium metabisulfite), which was significantly lower than the value obtained with DA (p < 0.05; Student's t test) (Fig. 6C). These profiles supported the notion that some of the DA-induced inhibition of GABAergic IPSC in D2KO mice is postsynaptic.
DA decreases the amplitudes of mIPSCs in both D2KO and WT mice
If DA suppresses GABA release via the activation of presynaptic D2R and blocks GABAergic current via postsynaptic DA receptors other than D2R, the amplitudes of action potential-independent mIPSCs would be suppressed in both the D2KO and the WT mouse, whereas the frequency would be relatively unchanged in the D2KO mouse but suppressed in the WT mouse. To test this hypothesis, mIPSCs were recorded in chloride-loaded cells from WT and D2KO mice at a holding potential of -60 mV in the presence of TTX (1 μm), CNQX (10 μm), d-AP-5 (50 μm), and strychnine (0.5 μm). Under these conditions, the amplitudes of mIPSCs reached as high as 200 pA at a frequency of <10 Hz (Fig. 7A,C). The results showed that 30 μm DA significantly reduced the amplitudes of mIPSCs in all of six cells from WT mice to 87.1 ± 2.3% (WT: control, 44.1 ± 6.7 pA; after DA, 38.3 ± 5.7 pA; n = 6; p = 0.008) and in all of six cells from D2KO mice to 82.1 ± 5.7% (D2KO: control, 42.1 ± 14.8 pA; after DA, 35.1 ± 6.1 pA; n = 6; p = 0.013) (Fig. 7B,C, left). In contrast, DA caused insignificant and significant decreases in frequency in D2KO (control, 6.7 ± 1.6 Hz; after DA, 5.6 ± 1.7 Hz; n = 6; p = 0.276) and WT (control, 11.8 ± 3.0 Hz; after DA, 8.6 ± 2.3 Hz; n = 6; p = 0.033) mice, respectively. The amplitude and frequency between the two types of mice did not differ significantly (WT vs D2KO: amplitude, p = 0.825; frequency, p = 0.162), although the frequency of mIPSCs obtained from 10- to 11-d-old mice tended to be far smaller than that obtained from 13- to 15-d-old mice; this tendency appeared to affect the D2KO data. The amplitudes before DA application ranged from 5 to 280 pA, so the fact that DA further diminished the large number of small mIPSCs to the noise level, thereby reducing the apparent frequencies, should be considered. At any rate, these results indicate that although D3, D4, and D5 receptor mRNAs were also found in the striatal MS neurons (Surmeier et al., 1996), D2R is the major presynaptic receptor that suppresses the GABA release; in addition, it was found that postsynaptic activation of DA receptors other than D2R indeed causes IPSC suppression.
Figure 7.
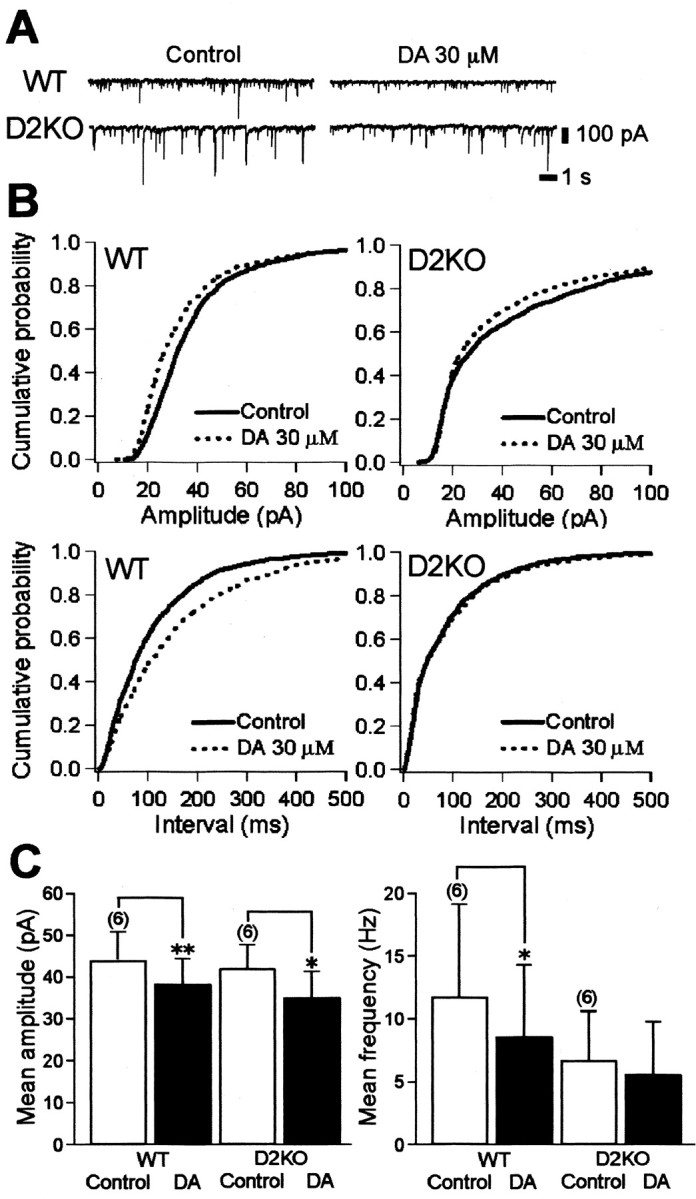
Effects of DA on mIPSCs in WT and D2KO mice. A, Continuous recordings (13 sec duration at a holding potential of -60 mV) of TTX-insensitive mIPSCs in WT and D2KO mice under control conditions and with 30 μm DA show decreased mIPSC amplitude with DA. Recordings were obtained using patch pipettes containing 125 mm chloride and 1 μm TTX, 10 μm CNQX, 50 μmd-AP-5, or 0.5 μm strychnine. B, Cumulative fraction histograms for the amplitude (top) and frequency (bottom) of mIPSCs in WT and a D2KO mice under control conditions and with DA in the same cells shown in A. Dopamine significantly decreased both the amplitude and the frequency of mIPSCs in the WT mouse but decreased the amplitude and not the frequency in the D2KO mouse. Kolmogorov-Smirnov two-sample test: amplitude, WT, p < 0.00001, D2KO, p = 0.0024; interevent interval, WT, p < 0.00001, D2KO, p = 0.1879. C, Normalized histograms of amplitudes and frequencies of mIPSCs before and after DA application. Dopamine significantly reduced amplitude in both the D2KO (p = 0.013) and WT (p = 0.008) mice. In contrast, the frequency of mIPSCs in the WT mouse significantly decreased after DA application (p = 0.033) but not significantly in the D2KO mouse (p = 0.276). *p < 0.05; **p < 0.01; paired Student's t test.
Postsynaptic inhibition of GABAergic IPSCs is mediated by D4R activation
We studied the pharmacological characteristics of postsynaptic IPSC inhibition in slices from D2KO mice. The DA receptor antagonists were continuously perfused throughout the experiments from at least 5 min before DA application. In the presence of R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH23,390) (10 μm), a D1/D5 receptor antagonist, or sulpiride (10 μm), a D2/D3 receptor antagonist, DA (30 μm) powerfully inhibited IPSCs, similar to the action evoked by DA alone (Fig. 8A,B,E). On the other hand, DA-induced inhibition was substantially blocked by clozapine (50 μm) or 3-[4-(4-iodophenyl)piperazin-1-yl]methyl-1H-pyrrolo[2,3-b]pyridine trihydrochloride (L750,667) (10 μm), both of which are selective D4R antagonists (Fig. 8C,D,E). The percentage of inhibition of IPSCs by DA in the absence or presence of these DA antagonists is summarized in Figure 8E. Dopamine had little effect on IPSCs of GP neurons pretreated with clozapine (11.30 ± 8.50%; n = 5; p < 0.01; Student's t test). This was significantly different from the effects of DA alone (49.00 ± 6.18%; n = 5), DA with SCH23,390 (56.64 ± 14.77%; n = 3; p = 0.594; Student's t test), or DA with sulpiride (45.09 ± 10.84%; n = 4; p = 0.750; Student's t test). In addition, L750,667 (10 and 50 μm) dose-dependently blocked DA-induced inhibition (10 μm, 22.09 ± 4.09%, n = 4, p < 0.05, Student's t test; 50 μm, 6.77 ± 3.90%, n = 3, p < 0.01, Student's t test). SCH23,390, sulpiride, clozapine, or L750,667 alone had no effect on the amplitude of IPSCs (data not shown).
Figure 8.
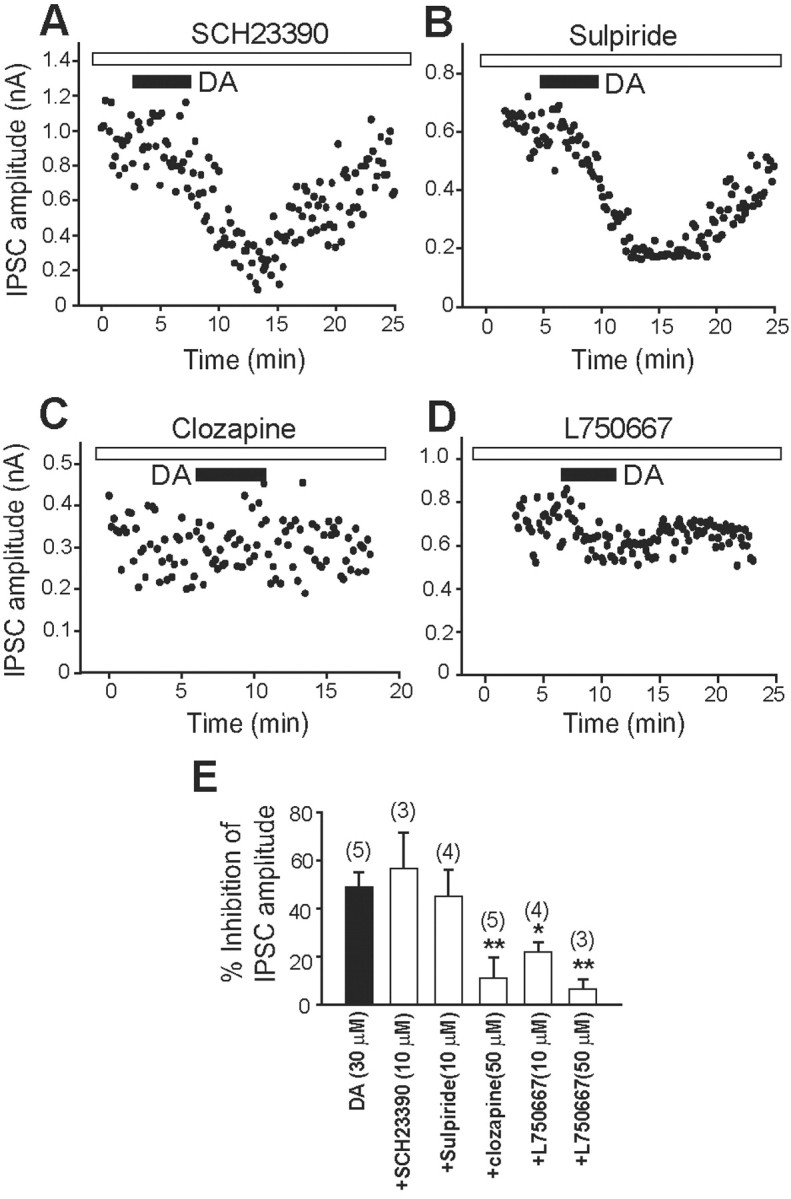
Pharmacological identification of a DA receptor subtype involved in IPSC inhibition in D2KO mice. A-D, Effects of the D1/D5 receptor antagonist SCH23,390 (10 μm; A), the D2/D3 receptor antagonist sulpiride (10 μm; B), and the D4 receptor antagonists clozapine (50 μm; C) and L750,667 (10 μm; D) on DA (30 μm)-induced inhibition of IPSCs. E, Summarized histograms show the effect of DA (30 μm) alone and of SCH23,390 (10 μm), sulpiride (10 μm), clozapine (50 μm), or L750,667 (10 and 50 μm) combined with DA (30 μm) on DA-receptor-induced IPSC inhibition. The numbers of experiments are shown in parentheses. *p < 0.05; **p < 0.01; unpaired Student's t test.
Furthermore, the selective D4R agonist N-[[4-(2-cyanophenyl)-1-piperazinyl]methyl]-3-benzazepine-7,8-diol hydrochloride (PD168,077) (3 μm) reversibly inhibited IPSCs (Fig. 9A), whereas the D1/D5 receptor agonist (+/-)-1-phenyl-2,3,4,5-tetrahydro-(1H)-3-benzazepine-7,8-diol hydrochloride (SKF38,393;30 μm) and the selective D3 receptor agonist (+)-4aR,10bR)-3,4,4a,10b-tetrahydro-4-propyl-2H,5H-[1]benzopyrano[4,3-b]-1,4-oxazin-9-ol (PD128,907) (1 μm) had little effect on IPSCs (Fig. 9B,C). Figure 9D shows the percentage of inhibition of IPSCs by these DA agonists. SKF38,393, PD128,907, or (+)-7-hydroxy-2-dipropylaminotetralin (7-OH-DPAT; 1 μm), another selective D3 receptor agonist, failed to mimic the DA-induced inhibition (8.67 ± 6.81%, n = 7, p < 0.01, Student's t test; 14.81 ± 7.47%, n = 5, p < 0.05; 15.14 ± 7.43%, n = 5, p < 0.05, respectively). In contrast, PD168,077 dose-dependently inhibited the IPSCs at concentrations of 0.3, 3, or 30 μm (22.79 ± 10.70%, n = 6; 40.60 ± 3.78, n = 5; 62.01 ± 11.79, n = 3, respectively). These pharmacological profiles indicate that D4R activation might be responsible for the postsynaptic inhibition of GABAergic IPSCs in D2KO mice.
Figure 9.

The selective D4R agonist PD168,077 dose-dependently inhibited IPSCs in D2KO mice. A-C, Effects of the selective D4 receptor agonist PD168,077 (3 μm; A), the D1/D5 receptor agonists SKF38,393 (30 μm; B), and the selective D3 receptor agonist PD128,907 (3 μm; C). D, Summarized histograms show inhibitory actions of DA (10 and 30 μm), SKF38,393 (30 μm), PD128,907 (1 μm), 7-OH-DPAT (1 μm; another selective D3 receptor antagonist), and PD168,077 (0.3, 3, and 30 μm) on IPSC amplitude. Only PD168,077 mimicked the DA effect. The numbers of experiments are shown in parentheses. *p < 0.05; **p < 0.01 (to the value obtained with 30 μm DA); unpaired Student's t test.
Absence of DA-induced inhibition of GABAergic currents in D4KO mouse
We confirmed D4R involvement in the dopaminergic postsynaptic inhibition of GABAergic currents in GP neurons using D4KO mice generated by disrupting exon 2 of the mouse D4R gene (Fig. 1). We also found at least two of the cell types in the D4KO mice described previously (Kita and Kitai, 1991, 1994; Nambu and Llinas, 1994, 1997; Cooper and Stanford, 2000; Shindou et al., 2001) (data not shown). The current-voltage relationship curves (Fig. 10A) showed that the reversal potentials of GABAergic IPSCs in both WT and D4KO mice were approximately -50 mV (D4KO mice, -52.1 ± 1.1 mV; WT mice, -53.2 ± 1.8 mV; p = 0.62; Student's t test), and the kinetic properties of the IPSCs did not differ between the two [D4KO (n = 8) vs WT (n = 10); Student's t test; rise time, 20-80%, 1.14 ± 0.18 msec vs 1.51 ± 0.27 msec, p = 0.58; decay time constant: 22.52 ± 0.37 msec vs 26.25 ± 3.96 msec, p = 0.41]. In addition, no morphological differences between the two were detectable.
To exclude the possible intervention of presynaptic D2-like receptors, we investigated the effect of DA on the GABAA-receptor-mediated outward currents elicited by iontophoretically applied GABA on GP neurons with a genetic deletion of D4R. Figure 10B shows that bath application of DA (10 μm) inhibited outward currents recorded on GP neurons of acute slices prepared from WT (n = 8) but not from D4KO (n = 10) mice. The normalized inhibition of outward currents by DA on GP neurons from both types of mice is shown in Figure 10C. The DA-induced inhibition of outward currents was 22.35 ± 6.98% in littermate WT mice, whereas that of D4KO mice was -2.31 ± 2.67%, which was significantly different from control (p < 0.05; Student's t test). These findings confirmed that DA-receptor-induced inhibition of GABA currents in GP neurons is dependent on the activation of postsynaptic D4R. A study of mIPSCs in the D4KO and the WT mouse is required to confirm these results further.
Inhibition of GABAA-receptor-mediated currents by D4R activation depends on decreased activity of protein kinase A
We investigated the signal transduction pathway that mediates the D4R-induced inhibition of GABAergic IPSCs. Other studies indicate that D4R activation suppresses cAMP synthesis in the mouse retina (Nir et al., 2002), in the rat cortical collecting duct (Li and Schafer, 1998), and in a mouse neuronal cell line (Yamaguchi et al., 1997). GABAA-receptor-mediated currents can be reduced by cAMP-dependent PKA (Porter et al., 1990) or PKC (Krishek et al., 1994). Therefore, we investigated the effect of selective PKC and PKA inhibitors on the DA-induced inhibition of GABAergic IPSCs in D2KO mice. Using these mice for this experiment did not exclude the contribution of other presynaptic D2-like receptors, but it at least minimized their contribution. Dopamine (30 μm) was applied 20-30 min after the start of the intracellular loading of tamoxifen (50 μm) (Fig. 11A) or the extracellular application of chelerythrine (5 μm) (Fig. 11B), both of which are selective PKC inhibitors. We found that neither of them had any effect. Figure 11G shows that DA inhibited IPSC by 31.69 ± 12.05% when pretreated with tamoxifen (n = 6; p = 0.261; Student's t test) and by 55.75 ± 12.16% with chelerythrine (n = 3; p = 0.561; Student's t test), neither of which were significantly smaller or larger than the effect of DA alone (49.00 ± 6.18%; n = 5). In contrast, 9-(tetrahydro-2-furanyl)-9H-purin-6-amine (SQ22,536) (100 μm), a membrane-permeable adenylyl cyclase inhibitor, significantly inhibited the IPSCs (64.01 ± 9.86% at 15 min; n = 6; p < 0.05; Student's t test), whereas the currents remained constant in its absence (101.72 ± 4.56% at 15 min; n = 4) (Fig. 11C,E). After reduction to a plateau by SQ22,536, DA failed to inhibit the IPSC further (Fig. 11E). In total, the DA-induced inhibition of IPSC in the presence of SQ22,536 was only 12.57 ± 5.33% (n = 4; p < 0.05; Student's t test) (Fig. 11G).
In addition, adding Rp-cAMPS (1 mm), an inhibitor of cAMP, to the patch pipette gradually decreased the IPSCs to a steady-state level 10 min after the membrane rupture (57.10 ± 3.76%; n = 3), which was significantly smaller than the effects of the control (79.02 ± 5.83%; n = 5) or Sp-cAMPS (78.44 ± 6.32%; n = 5), an activator of cAMP (Fig. 11D). Again, DA was ineffective. With Rp-cAMPS, DA inhibited the IPSC by 21.25 ± 9.54% (n = 4; p < 0.01; Student's t test), which was significantly smaller than the effects of DA alone (Fig. 11F,G). The decrease in IPSC by Rp-cAMPS (57.10 ± 3.76%, n = 3) was significantly different from the effect of Sp-cAMPS (78.44 ± 6.32%) or control (79.02 ± 5.83%; n = 5; Student's t test; p < 0.05). These results suggest that the inhibition of GABAA-receptor-mediated currents by D4R activation depends on PKA inhibition.
Discussion
In addition to the striatum, the internal and external pallidum, ventral pallidum, and subthalamic nucleus (STN) also receive significant dopaminergic input from the midbrain (Parent and Smith, 1987; Smith et al., 1989; Parent et al., 1990; Hassani et al., 1997; Smith and Kieval, 2000). In fact, several anatomical studies have identified D2R in the presynaptic terminals of striatopallidal MS neurons (Gerfen et al., 1990; Floran et al., 1997) and D4R in large neurons in the rodent GP and primate GPe (Mrzljak et al., 1996; Ariano et al., 1997; Mauger et al., 1998). The present study investigated how DA acts on the presynaptic D2R and postsynaptic D4R in the GP (GPe).
Using whole-cell patch-clamp recordings from visually identified GP neurons in rat slice preparations, Cooper and Stanford (2001) demonstrated that the presynaptic D2R activation on striatopallidal terminals reduces the release of GABA in the GP, whereas no subtle changes were detectable for the functional role of D4R in the GP. Similarly, we also found using mouse slice preparations that DA reduces GABAA IPSCs through presynaptic D2R activation in WT mice. However, the IPSC reduction by DA was biphasic, which was apparently mediated by the activation of both D2R and D4R in the GP. GABAA IPSCs recorded from GP neurons of D2KO mice were dose-dependently inhibited with an IC50 value similar to the higher value obtained in WT mice. Therefore, this inhibition appeared to be brought about by D4R activation. Indeed, the paired-pulse ratio of paired IPSCs evoked by electrical stimulation of the striatum increased after DA application in WT mice, whereas no significant change was elicited in D2KO mice. Outward currents elicited by bath application of GABA were suppressed by DA in D2KO mice. This was mimicked by D4R-selective agonists and dose-dependently blocked by D4R-selective antagonists. The study of mIPSCs in GP neurons also revealed that DA (30 μm) decreased the amplitude but not the frequency of the mIPSCs in D2KO mice, whereas it decreased both in WT mice. Moreover, our study using D4KO mice showed that GABAergic outward currents were not suppressed by DA, whereas a significant blockade of the outward currents was readily evoked in WT littermate GP neurons. This confirmed our notion that DA reduces GABAA IPSCs through postsynaptic D4R activation in the GP.
The discrepancy between a previous study (Cooper and Stanford, 2001) and ours concerning the role of D4R in the GP was probably not caused by species differences, because D4Rs have been found in large-sized GP neurons in both mice (Mauger et al., 1998) and rats (Ariano et al., 1997). Rather, it might have been caused at least partly by the difference in the concentration of DA that we used. Dopamine binds D2R with an affinity that is approximately fourfold higher than D4R (Seeman and Van Tol, 1994; Oak et al., 2000), which is consistent with our findings that DA inhibited GABAergic IPSCs through D2R and D4R at low and high concentration ranges, respectively. According to a study of the nucleus accumbens (Garris et al., 1994), a 1.6 mm concentration of DA released in the synaptic cleft was reduced by one-half at a distance of ∼10 μm through actions of dopamine transporters and diffusion, although the extracellular concentration of DA attained only 250 nm on a single electrical impulse. Therefore, a 30 μm concentration of DA may be within a physiological concentration in the GP. Dopamine would block the presynaptic D2R more readily than the postsynaptic D4R and suppress the release of GABA. Because the striatopallidal fibers bearing D2R might predominate in the GP, the reduction of the mIPSCs by D4R activation might have been masked when 3 μm DA was applied to WT mice. Alternatively, D4R might have been upregulated in the D2KO mice to compensate for the lack of presynaptic inhibition of GABA release, because the inhibition of IPSC amplitude in D2KO mice was not significantly different from that in WT mice at the high concentration of 30 μm. This is unlikely however, because the mIPSC amplitude also decreased in littermates of D2KO mice. Another possibility is that D4Rs are preferentially located in the pallidostriatal pathway. The latency of the evoked IPSCs examined here was within 4 msec. Thus, we considered that these were the main currents in the pallidostriatal pathway (Ogura and Kita, 2000). In fact, Cooper and Stanford (2001) reported that stimulation within the GP to minimize stimulation of striatopallidal fibers hardly affected the evoked IPSCs by bath application of 3 μm DA, whereas striatal stimulation activated both the striatopallidal and pallidostriatal fibers and significantly suppressed the frequency of the mIPSCs. Our observation that the mIPSCs of larger amplitudes seemed more suppressed fits with the fact that all pallidostriatal neurons have intrinsic collateral axons that form synapses on the somata and proximal dendrites of GP neurons and hence may elicit larger mIPSCs (Kita and Kitai, 1991, 1994; Bevan et al., 1998; Smith et al., 1998a). The effects of D4R activation on the mIPSCs were hardly detectable at 3 μm DA, because the pallidostriatal neurons consist of approximately one-third of the total neuronal population in the GP. Together, the findings of our study indicate that DA regulates GP activity by modulating GABAergic activity in two ways: suppression of GABA release via presynaptic D2R activation and reduction of the GABAergic current via postsynaptic D4R activation.
With respect to the signaling pathway, D4R activation decreases postsynaptic GABAA receptor function in the pyramidal neurons of the rat prefrontal cortex via regulation of the Yotiao-anchored PKA/protein phosphatase 1 signaling complex (Wang et al., 2002). Likewise, we found that PKA activation blocked and PKA inhibition occluded D4R modulation of GABAA receptor currents in D2KO mice. However, how the suppression of PKA activity actually reduces the efficacy of the GABAA receptor channel in the GP remains unclear. GABAA receptors are heteropentamers assembled predominantly from α1-α6, β1-β3, and γ2 subunits (Rabow et al., 1995), and their β3 subunit is one of the key substrates of PKA (McDonald et al., 1998). The β3 subunit of GABAA receptors plays an important role in assembly formation with another subunit (Ehya et al., 2003) and receptor localization or clustering (Connolly et al., 1996). In fact, GABA-evoked currents are obviously decreased in mice lacking GABAA receptor β3 subunit in vitro (Krasowski et al., 1998). These findings suggest that dephosphorylation of the β3 subunit by D4R activation causes the decreased efficacy of the GABAA receptor channel in the GP.
A model of the functional organization of the basal ganglia suggested that a loss of the nigrostriatal dopamine neurons would lead to overactivity of the striatopallidal GABAergic neurons and oscillation between the GPe and the STN (Albin et al., 1989; Alexander and Crutcher, 1990; DeLong, 1990; Chesselet and Delfs, 1996; Bevan et al., 2002). Therefore, the loss of postsynaptic inhibition of GABAergic IPSCs by D4R activation in the GP would enhance oscillation in PD. Moreover, multiple indirect pathways might function through the GPe, such as direct projections to output structures (the internal segment of the GP/substantia nigra pars reticulata) and to the reticular nucleus of the thalamus as well as reciprocal connections with the STN (Smith et al., 1998b). These studies indicate that the suppression of GABAergic activities in the GP significantly ameliorates PD symptoms. In fact, studies using parkinsonian model animals have shown increased GABA release within the GP (Robertson et al., 1990) and also shown that GABAA receptor antagonists applied directly into the GP significantly alleviate parkinsonian symptoms (Maneuf et al., 1994). Spontaneous movements in D2KO mice are significantly reduced, which is reminiscent of PD symptoms (Baik et al., 1995), and striatal MS neurons express long-term potentiation of EPSPs instead of the usual long-term depression in WT mice (Calabresi et al., 1997). The incidence of locomotion and rearing in D4R-deficient mice is also significantly reduced (Rubinstein et al., 1997), and long-term blockade of D4R with clozapine produces motor side effects such as a parkinsonian-like bradykinesia and mild akathisia, but no rigidity and rarely tremor (Gerlach and Peacock, 1994). Nigropallidal and nigrostriatal DA neurons arise from separate neuronal populations (Smith et al., 1989) and might be comparatively sparse in animal models of PD (Parent et al., 1990; Smith and Kieval, 2000). However, additional activation of D2R and D4R in the GP might cancel the outcome of overactivity of striatopallidal GABAergic neurons and suppress the occurrence of oscillatory activity between the GPe and STN.
Footnotes
This work was supported by Solution Oriented Research for Science and Technology at the Ministry of Education, Culture, Sports, Science and Technology of Japan (T.A.). We thank Drs. M. Katsuki and A. Aiba for donating the dopamine D2 knock-out mice and F. Nagumo for animal care.
Correspondence should be addressed to Dr. Toshihiko Aosaki, Tokyo Metropolitan Institute of Gerontology, Neural Circuits Dynamics Research Group, 35-2, Sakae-cho, Itabashi-ku, Tokyo 173-0015, Japan. E-mail: aosaki@tmig.or.jp.
R.-M. Shin's present address: Department of Psychiatry, McLean Hospital, Harvard Medical School, 115 Mill Street, Belmont, MA 02478.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2311662-•$15.00/0
References
- Albin RL, Young AB, Penney JB ( 1989) The functional anatomy of basal ganglia disorders. Trends Neurosci 12: 366-375. [DOI] [PubMed] [Google Scholar]
- Alexander GE, Crutcher MD ( 1990) Functional architecture of basal ganglia circuits: neural substrates of parallel processing. Trends Neurosci 13: 266-271. [DOI] [PubMed] [Google Scholar]
- Ariano MA, Wang J, Noblett KL, Larson ER, Sibley DR ( 1997) Cellular distribution of the rat D4 dopamine receptor protein in the CNS using anti-receptor antisera. Brain Res 752: 26-34. [DOI] [PubMed] [Google Scholar]
- Baik JH, Picetti R, Saiardi A, Thiriet G, Dierich A, Depaulis A, Le Meur M, Borrelli E ( 1995) Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature 377: 424-428. [DOI] [PubMed] [Google Scholar]
- Bergstrom DA, Walters JR ( 1984) Dopamine attenuates the effects of GABA on single unit activity in the globus pallidus. Brain Res 310: 23-33. [DOI] [PubMed] [Google Scholar]
- Bevan MD, Booth PA, Eaton SA, Bolam JP ( 1998) Selective innervation of neostriatal interneurons by a subclass of neuron in the globus pallidus of the rat. J Neurosci 18: 9438-9452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevan MD, Magill PJ, Terman D, Bolam JP, Wilson CJ ( 2002) Move to the rhythm: oscillations in the subthalamic nucleus-external globus pallidus network. Trends Neurosci 25: 525-531. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Saiardi A, Pisani A, Baik JH, Centonze D, Mercuri NB, Bernardi G, Borrelli E ( 1997) Abnormal synaptic plasticity in the striatum of mice lacking dopamine D2 receptors. J Neurosci 17: 4536-4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesselet MF, Delfs JM ( 1996) Basal ganglia and movement disorders: an update. Trends Neurosci 19: 417-422. [DOI] [PubMed] [Google Scholar]
- Connolly CN, Wooltorton JR, Smart TG, Moss SJ ( 1996) Subcellular localization of gamma-aminobutyric acid type A receptors is determined by receptor beta subunits. Proc Natl Acad Sci USA 93: 9899-9904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AJ, Stanford IM ( 2000) Electrophysiological and morphological characteristics of three subtypes of rat globus pallidus neurons in vitro. J Physiol (Lond) 527: 291-304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper AJ, Stanford IM ( 2001) Dopamine D2 receptor mediated presynaptic inhibition of striatopallidal GABA(A) IPSCs in vitro. Neuropharmacology 41: 62-71. [DOI] [PubMed] [Google Scholar]
- DeLong MR ( 1990) Primate models of movement disorders of basal ganglia origin. Trends Neurosci 13: 281-285. [DOI] [PubMed] [Google Scholar]
- Ehya N, Sarto I, Wabnegger L, Sieghart W ( 2003) Identification of an amino acid sequence within GABA(A) receptor beta3 subunits that is important for receptor assembly. J Neurochem 84: 127-135. [DOI] [PubMed] [Google Scholar]
- Floran B, Floran L, Sierra A, Aceves J ( 1997) D2 receptor-mediated inhibition of GABA release by endogenous dopamine in the rat globus pallidus. Neurosci Lett 237: 1-4. [DOI] [PubMed] [Google Scholar]
- Garris PA, Ciolkowski EL, Pastore P, Wightman RM ( 1994) Efflux of dopamine from the synaptic cleft in the nucleus accumbens of the rat brain. J Neurosci 14: 6084-6093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma Jr FJ, Sibley DR ( 1990) D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons. Science 250: 1429-1432. [DOI] [PubMed] [Google Scholar]
- Gerlach J, Peacock L ( 1994) Motor and mental side effects of clozapine. J Clin Psychiatry 55 [Suppl B]: 107-109. [PubMed] [Google Scholar]
- Hassani OK, Francois C, Yelnik J, Feger J ( 1997) Evidence for a dopaminergic innervation of the subthalamic nucleus in the rat. Brain Res 749: 88-94. [DOI] [PubMed] [Google Scholar]
- Hooper KC, Banks DA, Stordahl LJ, White IM, Rebec GV ( 1997) Quinpirole inhibits striatal and excites pallidal neurons in freely moving rats. Neurosci Lett 237: 69-72. [DOI] [PubMed] [Google Scholar]
- Kita H, Kitai ST ( 1991) Intracellular study of rat globus pallidus neurons: membrane properties and responses to neostriatal, subthalamic and nigral stimulation. Brain Res 564: 296-305. [DOI] [PubMed] [Google Scholar]
- Kita H, Kitai ST ( 1994) The morphology of globus pallidus projection neurons in the rat: an intracellular staining study. Brain Res 636: 308-319. [DOI] [PubMed] [Google Scholar]
- Kita H, Kita T ( 2001) Number, origins, and chemical types of rat pallidostriatal projection neurons. J Comp Neurol 437: 438-448. [DOI] [PubMed] [Google Scholar]
- Kita H, Tokuno H, Nambu A ( 1999) Monkey globus pallidus external segment neurons projecting to the neostriatum. NeuroReport 10: 1467-1472. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Morita S, Sawada H, Mizuguchi T, Yamada K, Nagatsu I, Fujita K, Kreitman RJ, Pastan I, Nagatsu T ( 1995a) Immunotoxin-mediated conditional disruption of specific neurons in transgenic mice. Proc Natl Acad Sci USA 92: 1132-1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Morita S, Sawada H, Mizuguchi T, Yamada K, Nagatsu I, Hata T, Watanabe Y, Fujita K, Nagatsu T ( 1995b) Targeted disruption of the tyrosine hydroxylase locus results in severe catecholamine depletion and perinatal lethality in mice. J Biol Chem 270: 27235-27243. [DOI] [PubMed] [Google Scholar]
- Krasowski MD, Rick CE, Harrison NL, Firestone LL, Homanics GE ( 1998) A deficit of functional GABA(A) receptors in neurons of beta 3 subunit knockout mice. Neurosci Lett 240: 81-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishek BJ, Xie X, Blackstone C, Huganir RL, Moss SJ, Smart TG ( 1994) Regulation of GABAA receptor function by protein kinase C phosphorylation. Neuron 12: 1081-1095. [DOI] [PubMed] [Google Scholar]
- Li L, Schafer JA ( 1998) Dopamine inhibits vasopressin-dependent cAMP production in the rat cortical collecting duct. Am J Physiol 275: F62-F67. [DOI] [PubMed] [Google Scholar]
- Lindvall O, Bjorklund A ( 1979) Dopaminergic innervation of the globus pallidus by collaterals from the nigrostriatal pathway. Brain Res 172: 169-173. [DOI] [PubMed] [Google Scholar]
- Maneuf YP, Mitchell IJ, Crossman AR, Brotchie JM ( 1994) On the role of enkephalin cotransmission in the GABAergic striatal efferents to the globus pallidus. Exp Neurol 125: 65-71. [DOI] [PubMed] [Google Scholar]
- Mauger C, Sivan B, Brockhaus M, Fuchs S, Civelli O, Monsma Jr F ( 1998) Development and characterization of antibodies directed against the mouse D4 dopamine receptor. Eur J Neurosci 10: 529-537. [DOI] [PubMed] [Google Scholar]
- McDonald BJ, Amato A, Connolly CN, Benke D, Moss SJ, Smart TG ( 1998) Adjacent phosphorylation sites on GABAA receptor beta subunits determine regulation by cAMP-dependent protein kinase. Nat Neurosci 1: 23-28. [DOI] [PubMed] [Google Scholar]
- Mrzljak L, Bergson C, Pappy M, Huff R, Levenson R, Goldman-Rakic PS ( 1996) Localization of dopamine D4 receptors in GABAergic neurons of the primate brain. Nature 381: 245-248. [DOI] [PubMed] [Google Scholar]
- Nakanishi H, Hori N, Katsuda N ( 1985) Neostriatal evoked inhibition and effects of dopamine on globus pallidal neurons in rat slice preparations. Brain Res 358: 282-286. [DOI] [PubMed] [Google Scholar]
- Nambu A, Llinas R ( 1994) Electrophysiology of globus pallidus neurons in vitro. J Neurophysiol 72: 1127-1139. [DOI] [PubMed] [Google Scholar]
- Nambu A, Llinas R ( 1997) Morphology of globus pallidus neurons: its correlation with electrophysiology in guinea pig brain slices. J Comp Neurol 377: 85-94. [PubMed] [Google Scholar]
- Nir I, Harrison JM, Haque R, Low MJ, Grandy DK, Rubinstein M, Iuvone PM ( 2002) Dysfunctional light-evoked regulation of cAMP in photoreceptors and abnormal retinal adaptation in mice lacking dopamine D4 receptors. J Neurosci 22: 2063-2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oak JN, Oldenhof J, Van Tol HH ( 2000) The dopamine D(4) receptor: one decade of research. Eur J Pharmacol 405: 303-327. [DOI] [PubMed] [Google Scholar]
- Ogura M, Kita H ( 2000) Dynorphin exerts both postsynaptic and presynaptic effects in the globus pallidus of the rat. J Neurophysiol 83: 3366-3376. [DOI] [PubMed] [Google Scholar]
- Parent A, Smith Y ( 1987) Differential dopaminergic innervation of the two pallidal segments in the squirrel monkey (Saimiri sciureus). Brain Res 426: 397-400. [DOI] [PubMed] [Google Scholar]
- Parent A, Lavoie B, Smith Y, Bedard P ( 1990) The dopaminergic nigropallidal projection in primates: distinct cellular origin and relative sparing in MPTP-treated monkeys. Adv Neurol 53: 111-116. [PubMed] [Google Scholar]
- Porter NM, Twyman RE, Uhler MD, Macdonald RL ( 1990) Cyclic AMP-dependent protein kinase decreases GABAA receptor current in mouse spinal neurons. Neuron 5: 789-796. [DOI] [PubMed] [Google Scholar]
- Querejeta E, Delgado A, Valdiosera R, Erlij D, Aceves J ( 2001) Intrapallidal D2 dopamine receptors control globus pallidus neuron activity in the rat. Neurosci Lett 300: 79-82. [DOI] [PubMed] [Google Scholar]
- Rabow LE, Russek SJ, Farb DH ( 1995) From ion currents to genomic analysis: recent advances in GABAA receptor research. Synapse 21: 189-274. [DOI] [PubMed] [Google Scholar]
- Robertson RG, Clarke CA, Boyce S, Sambrook MA, Crossman AR ( 1990) The role of striatopallidal neurones utilizing gamma-aminobutyric acid in the pathophysiology of MPTP-induced parkinsonism in the primate: evidence from [3H]flunitrazepam autoradiography. Brain Res 531: 95-104. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Phillips TJ, Bunzow JR, Falzone TL, Dziewczapolski G, Zhang G, Fang Y, Larson JL, McDougall JA, Chester JA, Saez C, Pugsley TA, Gershanik O, Low MJ, Grandy DK ( 1997) Mice lacking dopamine D4 receptors are supersensitive to ethanol, cocaine, and methamphetamine. Cell 90: 991-1001. [DOI] [PubMed] [Google Scholar]
- Seeman P, Van Tol HH ( 1994) Dopamine receptor pharmacology. Trends Pharmacol Sci 15: 264-270. [DOI] [PubMed] [Google Scholar]
- Shindou T, Mori A, Kase H, Ichimura M ( 2001) Adenosine A(2A) receptor enhances GABA(A)-mediated IPSCs in the rat globus pallidus. J Physiol (Lond) 532: 423-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith Y, Kieval JZ ( 2000) Anatomy of the dopamine system in the basal ganglia. Trends Neurosci 23: S28-S33. [DOI] [PubMed] [Google Scholar]
- Smith Y, Lavoie B, Dumas J, Parent A ( 1989) Evidence for a distinct nigropallidal dopaminergic projection in the squirrel monkey. Brain Res 482: 381-386. [DOI] [PubMed] [Google Scholar]
- Smith Y, Shink E, Sidibe M ( 1998a) Neuronal circuitry and synaptic connectivity of the basal ganglia. Neurosurg Clin N Am 9: 203-222. [PubMed] [Google Scholar]
- Smith Y, Bevan MD, Shink E, Bolam JP ( 1998b) Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience 86: 353-387. [DOI] [PubMed] [Google Scholar]
- Stanford IM, Cooper AJ ( 1999) Presynaptic μ and δ opioid receptor modulation of GABAA IPSCs in the rat globus pallidus in vitro J Neurosci 19: 4796-4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Song WJ, Yan Z ( 1996) Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci 16: 6579-6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhong P, Yan Z ( 2002) Dopamine D4 receptors modulate GABAergic signaling in pyramidal neurons of prefrontal cortex. J Neurosci 22: 9185-9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Aiba A, Nakamura K, Nakao K, Sakagami H, Goto K, Kondo H, Katsuki M ( 1996) Dopamine D2 receptor plays a critical role in cell proliferation and proopiomelanocortin expression in the pituitary. Genes Cells 1: 253-268. [DOI] [PubMed] [Google Scholar]
- Yamaguchi I, Harmon SK, Todd RD, O'Malley KL ( 1997) The rat D4 dopamine receptor couples to cone transducin (Gαt2) to inhibit forskolin-stimulated cAMP accumulation. J Biol Chem 272: 16599-16602. [DOI] [PubMed] [Google Scholar]
- Zucker RS ( 1989) Short-term synaptic plasticity. Annu Rev Neurosci 12: 13-31. [DOI] [PubMed] [Google Scholar]