

Abstract
Positive allosteric regulation of glutamate AMPA receptors involves conformational changes that can attenuate receptor desensitization and enhance ion flux through the channel pore. Many allosteric modulators (e.g., cyclothiazide and aniracetam) preferentially affect the flip (i) or flop (o) alternatively spliced isoform of AMPA receptors, implicating residues in the flip-flop domain as critical determinants of splice variant sensitivity. Indeed, previous mutational analyses have demonstrated that the differential sensitivity to cyclothiazide and aniracetam depends on a single amino acid, Ser (flip) and Asn (flop), suggesting that this residue may be solely responsible for differences in modulation of AMPA receptor isoforms. The present studies tested this hypothesis by investigating the molecular determinants of modulation of AMPA receptor splice variants by a structurally distinct compound, LY404187, which displays strikingly different and opposing kinetics of allosteric regulation characterized by a time-dependent enhancement in potentiation of homomeric GluR1-GluR4i and a time-dependent reduction in potentiation of GluR1-GluR4o. Site-directed mutagenesis of residues in the flip-flop domain of GluR2 revealed that, although exchange of Asn775 for Ser in GluR2o was sufficient to confer the GluR2i phenotype of potentiation, the corresponding mutation, Ser775Asn, in GluR2i did not impart the GluR2o response. In fact, the GluR2o kinetics of modulation depended on a novel set of substitutions in GluR2i, including Thr765Asn, Pro766Ala, and Val779Leu in combination with Ser775Asn. Collectively, these results show that, unlike cyclothiazide and aniracetam, the residues that confer splice variant differences in modulation by LY404187 are not identical and indicate that allosteric regulation of AMPA receptors can arise from multiple molecular determinants.
Keywords: aniracetam, biarylpropylsulfonamide, cyclothiazide, flip, flop, alternative splicing
Introduction
The AMPA subtype of ionotropic glutamate receptors mediates the majority of rapid excitatory synaptic transmission in the brain. These receptors are tetrameric complexes assembled from subunits encoded by four different genes termed GluR1-GluR4 (or GluRA-GluRD) in various stoichiometries, giving rise to functional homomeric and heteromeric receptors (Hollmann et al., 1989; Keinanen et al., 1990). Additional diversity comes from alternative splicing of RNA for each subunit in a region that encodes a 38 amino acid sequence in the extracellular domain between M3 and M4. These two splice variants named flip (i) and flop (o) deviate in sequence by less than 10 amino acids and display different rates of receptor desensitization that contribute to the heterogeneity of AMPA receptor synaptic responses in brain (Sommer et al., 1990; Dingledine et al., 1999). Flip and flop isoforms also display preferential sensitivity to allosteric regulation by distinct classes of compounds, including benzothiadiazines (e.g., cyclothiazide), biarylpropylsulfonamides (e.g., LY404187), and pyrrolidinones (e.g., aniracetam). The functional consequence of these positive modulators is to amplify excitatory synaptic transmission in part by reducing receptor desensitization (Dingledine et al., 1999). Significantly, an accumulating body of evidence has demonstrated that positive modulators can enhance performance of animals in models of cognitive function (Pontecorvo and Evans, 1985; Staubli et al., 1994b), as well as improve memory encoding in humans (Ingvar et al., 1997), suggesting that allosteric regulation AMPA receptors may be a novel therapeutic approach for cognitive disorders (Yamada, 2000; Quirk and Nisenbaum, 2002).
The preferential sensitivity of flip and flop receptors to allosteric regulation has focused efforts to elucidate the molecular determinants of these differences on the flip-flop domain. Indeed, mutagenic studies have demonstrated that a single residue, Ser750 in GluR1i and Asn750 in GluR1o, governs the greater sensitivity of GluR1i to cyclothiazide and aniracetam, respectively (Partin et al., 1995, 1996). The critical nature of this residue was further established in recent crystallography studies of cyclothiazide in the ligand-bind core (LBC) of GluR2o containing this Asn to Ser mutation. Cyclothiazide was proposed to attenuate desensitization by stabilizing the intradimer interface between two adjacent ligand-binding cores, in part through binding to this Ser residue. In addition, when this GluR2o-Ser crystal was superimposed with that of native GluR2o containing an Asn at this position, it was observed that tight binding of cyclothiazide would be precluded (Sun et al., 2002). Collectively, these results suggest that this single residue is responsible for conferring the sensitivity of flip and flop isoforms to allosteric regulation.
Recent studies with the biarylpropylsulfonamide compound LY404187 showed that human GluR2i were preferentially sensitive to modulation compared with GluR2o (Ornstein et al., 2000; Miu et al., 2001). The present studies have further demonstrated that the potency of LY404187 is greater for flip splice variants of all recombinant GluR1-GluR4, as well as revealed striking differences in the kinetics of allosteric regulation of the two isoforms for each subunit (see Fig. 1). To determine whether the Ser/Asn residue is responsible for these splice variant differences, we used site-directed mutagenesis to exchange residues in the flip-flop domain of GluR2i/o. Our results show that, unlike cyclothiazide, the molecular substrates for allosteric regulation by LY404187 of AMPA receptor splice variants are not identical and do not depend solely on the Ser/Asn residue.
Figure 1.
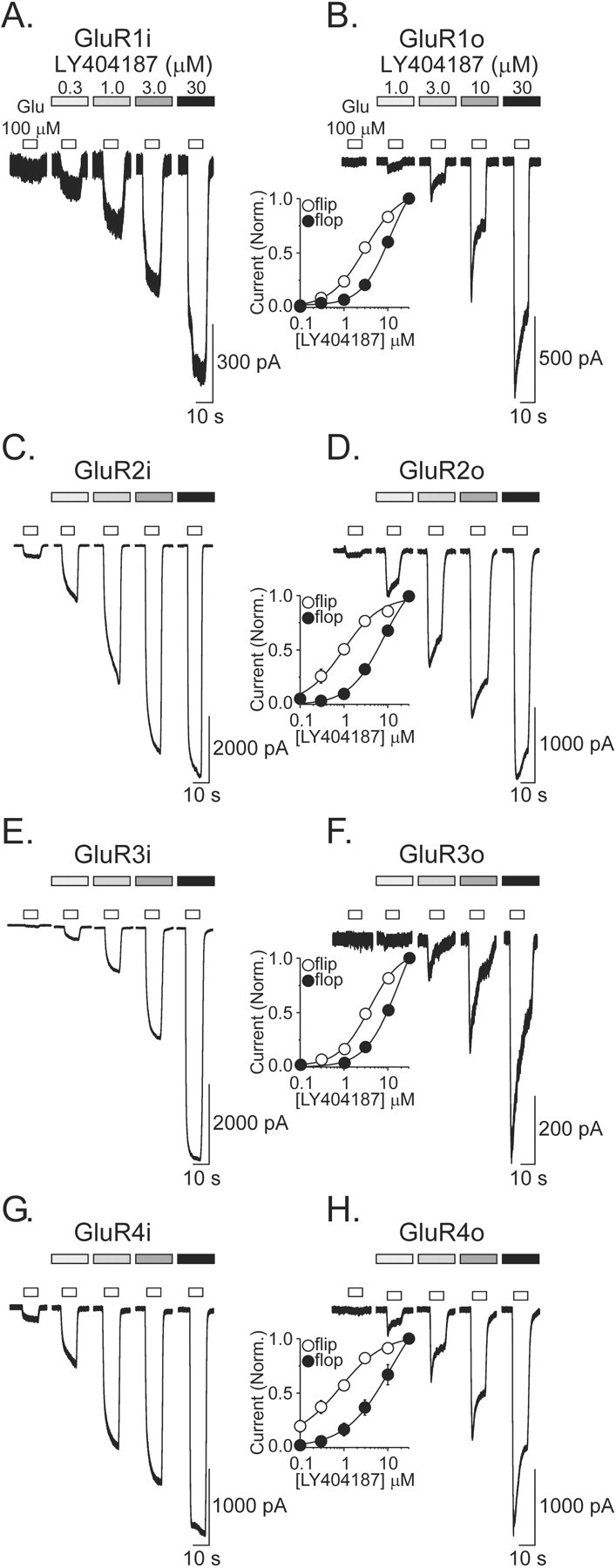
Concentration-response profile for LY404187 potentiation of homomeric GluR1-GluR4 flip and flop receptor-mediated currents. A-H, The concentration-response profile for LY404187 (0.03-30 μm) potentiation of 10 sec glutamate (100 μm; Glu) applications from cells stably expressing human homomeric GluR1i (A), GluR1o (B), GluR2i (C), GluR2o (D), GluR3i (E), GluR3o (F), GluR4i (G), or GluR4o (H). Insets, Plot of the average relative degree of potentiation normalized to the maximal response to 100 μm glutamte in the presence of 30 μm LY404187. LY404187 is more potent at homomeric flip receptors than flop receptors. Points represent the mean ± SEM; n = 2-11.
Materials and Methods
Molecular biology. Stable cell lines of HEK 293 cells transfected with cDNA encoding human GluR1-GluR4 flip or GluR1, GluR2, and GluR4 flop subunits were kindly provided by Dr. Keith R. Jarvie (Allelix Biopharmaceuticals, Mississauga, Ontario, Canada). These cells lines were established as reported previously (Fletcher et al., 1995). HEK 293 cells were transfected with the mammalian expression vectors pcDNA3.1 or pRc/CMV containing a GluR subunit gene. Transfected cells were selected on the basis of G418 resistance, and expression of genes was confirmed by reverse transcription-PCR. The stable GluR3o cell line was established in a similar manner using AV12 cells.
For the mutagenesis and rapid perfusion studies, GluR2i and GluR2o were first subcloned into the mammalian expression vector pcDNA5/FRT (catalog #V6010-20; Invitrogen, Carlsbad, CA). Native GluR2 subunits are modified in the pore region (M2) so that a glutamine residue (Q; CGA) is edited to an arginine (R; CGG). However, these edited subunits have a significantly lower single channel conductance (Swanson et al., 1997). Therefore, in the present studies, the unedited, or Q, form of the GluR2 subunit was used. Stable cell lines were then generated using the Flp-In System (catalog #K6010-02; Invitrogen, Carlsbad, CA). Cell lines were established on the basis of Hygromycin B (100 μg/ml; catalog #10687010; Invitrogen, Carlsbad, CA) resistance.
Site-directed mutagenesis was performed with the QuikChange mutagenesis kit (Stratagene, La Jolla, CA) using complementary primers. Presence of the desired mutant was confirmed by direct sequencing. To verify the absence of new, unwanted substitutions, the complete coding region was sequenced as well. Amino acids are numbered according to the GenBank sequence (accession number NP_000817).
Transient expression of recombinant AMPA receptors in HEK 293 cells. Flp-In-293 HEK cells (catalog #R750-07; Invitrogen, Carlsbad, CA) were maintained in DMEM with high glucose, l-glutamine, and pyridoxine hydrochloride, without sodium pyruvate (catalog #11965-092; Invitrogen, Gaithersburg, MD), 10% fetal bovine serum (catalog #16000-044; Invitrogen, Gaithersburg, MD), and 100 μg/ml Zeocin (catalog #45-0430; Invitrogen, Carlsbad, CA) in 5% CO2 at 37°C. For transfections, cultures were plated at a density of 10 × 105 cells/ml. After 12-18 hr, the plasmid containing the gene of interest was transfected using the standard Fugene 6 transfection reagent protocol (catalog #1-814-443; Roche Diagnostics, Indianapolis, IN). For transiently transfected cells, the pTracer-SV40 plasmid (catalog #V870-01; Invitrogen, Carlsbad, CA) that encodes for green fluorescent protein was cotransfected, permitting identification of transfected cells. The pTracer-SV40 plasmid was present at a ratio of 1:4 of the total DNA (2.5-4.0 μg/ml). Cells were replated onto collagen-coated coverslips (catalog #C-8919; Sigma, St. Louis, MO) 12 hr after the transfection.
Electrophysiological recordings from HEK 293 cells. The whole-cell variant of the patch-clamp technique was used for recording current from stably and transiently transfected HEK 293 cells. Recordings were performed 1-3 d after plating for stably transfected cells and 30-54 hr after transient transfections. Recording electrodes were pulled from borosilicate capillary tubing (Corning 7052; World Precision Instruments, Sarasota, FL) using a multistage puller (model P-97; Sutter Instruments, Novato CA). The electrodes were fire polished using a microforge (model MF-830; Narishige, Tokyo, Japan) before use. The internal electrode filling solution contained (in mm): 165.0 N-methyl-d-glucamine, 4.0 MgCl2, 40.0 HEPES, 3.5 BAPTA, 3.6 Na2ATP, and 0.35 GTP (pH was adjusted to 7.2 with KOH, and osmolarity was adjusted to 280 ± 3 mOsm/l with phosphocreatine). The extracellular solution contained (in mm): 120.0 Na isethionate, 5.0 BaCl2, 1.0 MgCl2, 20.0 CsCl, and 10.0 HEPES (pH was adjusted to 7.4 with 1.0 m NaOH, and osmolarity was adjusted to 300 ± 3 mOsm/l with glucose.
Before recording, the transfected HEK cells were placed into a 50 mm transparent plastic Petri dish that was mounted onto the stage of an inverted microscope. After placing the recording electrode into the extracellular solution bath, offset potentials were corrected, and electrode resistances ranged between 2 and 7 MΩ. Voltage-clamp recordings were made using an Axon Instruments (Union City, CA) 200B amplifier. The membrane potential of cells was held at -80 mV unless stated otherwise. Currents were digitized and monitored with pClamp software version 8.0 (Axon Instruments) running on a Pentium personal computer. A small amount of constant positive pressure (2-3 cm H2O) was applied to the electrodes as they were advanced through the bath. After achieving the whole-cell configuration, series resistance was compensated (70-85%) and monitored periodically.
Application of drugs was accomplished using one of two methods. For concentration-response profiles, drugs were delivered using a 16-barrel pipette array made from small-diameter (∼600 μm) glass capillary tubing. Electronic valves controlled the gravity-induced flow of each solution from a 10 ml syringe to an individual barrel. The pipette array was positioned 100-200 μm from the cell before seal formation. The solutions from the drug array were changed (∼100 msec) by altering the array position with a DC actuator (Newport, Irvine, CA). For rapid perfusion experiments, a theta tube constructed from double-barrel borosilicate glass tubing (catalog #TGC200-4; Warner Instruments, Hamden, CT) was used to deliver solutions to individual cells. The theta tubing was pulled so that the final diameter of each barrel was ∼125 μm, and the septum between barrels was ∼80 μm. The theta tube position was shifted using a piezoelectric actuator (Burleigh Instruments, Fishers, NY) having a charging time of 0.3 msec. Onset-to-offset time of junction potential measurements (0.1 and 1.0 m NaCl) was ∼700 μsec. A four-to-one manifold was attached to the input of each barrel of the theta tubing, permitting multiple solutions to be applied to individual cells. Solutions were delivered to the theta tubing using the gravity-induced flow method described above. All experiments were conducted at room temperature.
pClamp software version 8.0 (Axon Instruments), SigmaPlot 2002 for Windows version 8.02 (SPSS, Chicago, IL), and Prism version 3.02 (GraphPad Software, San Diego, CA) were used in the analysis of the data. Using the Prism software, significant differences between groups were determined using an ANOVA followed by Tukey's multiple post hoc comparison tests. The F value and p value from each ANOVA are presented in the corresponding figure legends. When significant differences between groups were observed, the p values from the Tukey's post hoc comparisons are presented in the text.
Chemicals.l-Glutamic acid (catalog #0218) and cyclothiazide (catalog #0713) were obtained from Tocris Cookson (Ballwin, MO). LY404187 was synthesized at Eli Lilly and Company. Concentrated stock solutions of 40 mm l-glutamic acid were prepared by dissolving in 1 equivalent NaOH. Concentrated stock solutions of cyclothiazide and LY404187 were prepared by dissolving in 100% dimethylsulfoxide. On the day of recording, solutions were diluted to the desired concentrations in the extracellular bathing solution.
Results
Differences in potency and kinetics of allosteric modulation by LY404187 on recombinant homomeric AMPA receptors
Previous studies have demonstrated that human homomeric GluR2i are more sensitive to allosteric regulation by LY404187 than GluR2o (Miu et al., 2001). In addition, the kinetics of potentiation of GluR4i by LY404187 is distinct from that for cyclothiazide. Specifically, cyclothiazide rapidly enhances the desensitized current of GluR4i to steady-state level, whereas potentiation by LY404187 displays a unique time-dependent increase in potentiation such that a steady-state level is rarely achieved during even prolonged applications (Miu et al., 2001). A similar response has been described for 4-[2-(phenylsulfonylamino)ethylthio]-2,6-difluoro-phenoxyacetamide and GluR3o (Sekiguchi et al., 1997, 2001). Initial studies investigated the extent to which the greater potency at flip receptors and the distinct time-dependent kinetics of allosteric regulation by LY404187 was detected for all of the human homomeric recombinant AMPA receptor subunits. Using whole-cell patch-clamp recording, the effects of glutamate (100 μm, 10 sec duration; holding potential, -80 mV) in the absence and presence of increasing concentrations of LY404187 (0.03-30 μm) on the desensitized current of human homomeric AMPA receptors were measured. Concentrations of LY404187 >30 μm were not tested because of solubility limitations of the compound. LY404187 potentiated glutamate-evoked responses in all cell lines in a concentration-dependent manner. For homomeric flip receptors (GluR1-GluR4i), potentiation was characterized by a continuous increase in the glutamate-evoked current such that a steady-state level was not achieved during the 10 sec pulse (Fig. 1A,C,E,G). Although the rates of the time-dependent increase in current varied among flip receptors, the effect was apparent in each subunit in response to all concentrations of LY404187. In marked contrast, potentiation of homomeric flop receptors (GluR1-GluR4o) by LY404187 was characterized by an initial enhancement of the desensitized current followed by a subsequent decay in the response. As for flip receptors, the time-dependent decay of LY404187 potentiation of flop receptors was evident in each subunit and at all concentrations tested (Fig. 1B,D,F,H). Because potentiation never reached a steady-state value for any subunit-splice variant combination, potency values were estimated by measuring the maximal response during the 10 sec pulse for each concentration of LY404187 and normalizing to the response in the presence of 30 μm of the compound. Examination of the concentration-response profiles shows that LY404187 was more potent at all flip receptors than flop receptors (EC50 values: GluR1i, 3.0 ± 0.3 μm, n = 6 vs GluR1o, 10.2 ± 1.3 μm, n = 11; GluR2i, 1.1 ± 0.2 μm, n = 6 vs GluR2o, 10.1 ± 3.7 μm, n = 9; GluR3i, 3.6 ± 0.4 μm, n = 8 vs GluR3o, 29.1 ± 9.8 μm, n = 6; GluR4i, 1.0 ± 0.1 μm, n = 7 vs GluR4o, 11.5 ± 4.2 μm, n = 8; mean ± SEM). Collectively, these results demonstrate unique differences in the kinetics of allosteric modulation by LY404187 of AMPA receptor splice variants.
To better characterize the differences in kinetics of modulation of flip and flop receptors, rapid perfusion studies on GluR2i or GluR2o were conducted. Whole-cell responses to rapid application of glutamate (1 mm, 1.5 sec duration; holding potential, -80 mV) in the absence and presence of LY404187 (0.3-3.0 μm) were measured. Similar to the results described above, LY404187 produced a time-dependent increase in potentiation at GluR2i and a time-dependent decrease in the potentiation at GluR2o (Fig. 2). The degree of potentiation of the AMPA receptor splice variants was quantified by measuring the current level at the end of the 1.5 sec pulse and expressing this value as a percentage of the amplitude of the initial desensitized current. Operationally, these values were used to describe the percentage change in potentiation of GluR2i and GluR2o responses. Results showed that the percentage change in potentiation of GluR2i was dose dependent, with the percentage change in potentiation at 3.0 μm being significantly greater than that for 0.3 μm (0.3 μmLY404187, 157.7 ± 6.7%, n = 21; 1.0 μmLY404187, 184.4 ± 11.0%, n = 14; 3.0 μmLY404187, 211.8 ± 13.4%, n = 13; F(2,45) = 7.8; p < 0.001). In contrast, the kinetics of potentiation of GluR2o was not dose dependent because the current at the end of the 1.5 sec pulse was ∼65% of the initial desensitized current for all concentrations of LY404187 tested (0.3 μmLY404187, 61.7 ± 10.0%, n = 14; 1.0 μmLY404187, 65.0 ± 3.9%, n = 13; 3.0 μmLY404187, 73.1 ± 6.5%, n = 9). These results confirm the splice variant differences in kinetics of allosteric modulation by LY404187.
Figure 2.
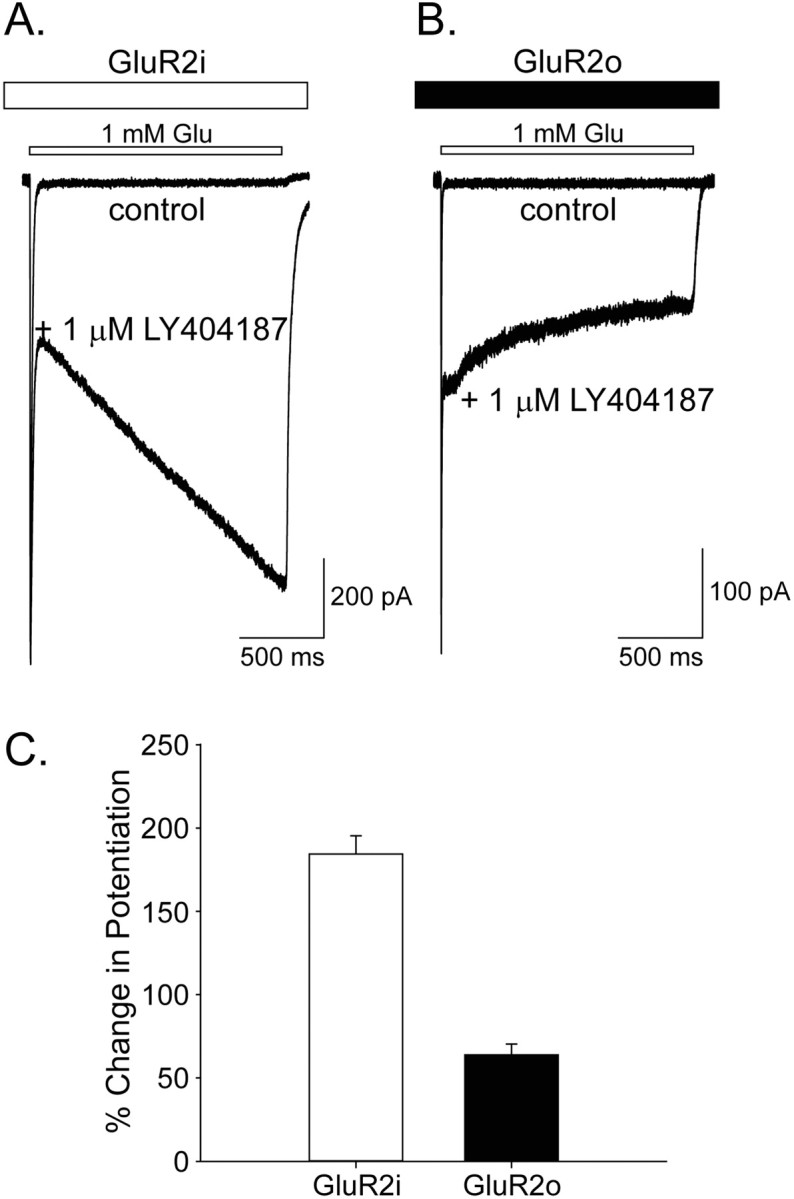
Effects of LY404187 on wild-type GluR2i and GluR2o. A, B, Whole-cell responses to rapid glutamate (1 mm) applications in the absence and presence of 1 μmLY404187 from HEK 293 cells expressing either wild-type GluR2i (A) or GluR2o (B). C, Plot of the percentage change in potentiation in the presence of LY404187 for wild-type homomeric GluR2i and GluR2o. The percentage change in potentiation was determined by expressing the amount of current at the end of the 1.5 sec rapid glutamate application as a percentage of the amount of current at the initial desensitization. Error bars represent mean ± SEM; n = 8-14.
Molecular determinants of allosteric regulation by LY404187 of flip receptors
Because the distinct kinetics of potentiation by LY404187 depended on isoform and not subunit, the mutational strategy focused on residue exchanges within the flip-flop module and specifically concentrated on GluR2 subunits. As originally described by Sommer et al. (1990), alignment of the flip and flop region for all four genes reveals that only a few residues differ between the alternatively spliced domains (Fig. 3). These residues are located in three different regions and are referred to in this paper as regions 1, 2, and 3 (according to Partin et al., 1995). Region 1 is at the N-terminal border of the alternatively spliced domain and consists of a Thr765-Pro766 in GluR2i and Asn765-Ala766 in GluR2o subunits. Region 2 is 10 amino acids downstream and contains a Ser775 in GluR2i and an Asn775 in GluR2o subunits. Region 3 is located at the C-terminal boundary of the flip-flop domain and comprises four residues, Lys797-Asp798-Ser799-Gly800 in GluR2i and Gly797-Gly798-Gly799-Asp800 in GluR2o subunits. Figure 3 shows an alignment of the flip-flop domain for GluR1-GluR4 subunits with the three regions identified. In addition to these three regions, there are several additional residues that differ between flip and flop isoforms. However, these other differences are fairly conservative and/or are not conserved in all flip and all flop receptors and have not been shown previously to contribute to allosteric regulation of AMPA receptors. In this paper, the three regions in GluR2i and GluR2o will be identified as a subscripted “i” or “o” in the mutated receptors. For example, exchange of region 2 from GluR2i into GluR2o will be indicated as GluR2oo,i,o. Wild-type GluR2i and GluR2o will have no subscripted letters.
Figure 3.

Sequence alignment of the flip and flop domains of GluR1-GluR4 subunits. The three regions of heterogeneity between the flip and flop splice variants (regions 1, 2, and 3) are labeled. Asterisks indicate two other positions that were exchanged in the GluR2i subunit with the residue in the equivalent position in the GluR2o subunit. In subsequent figures, an open rectangle will refer to the flip domain, and a dark rectangle will refer to the flop domain. Exchanges will then be highlighted in the appropriate color.
Initial studies focused on identification of the structural determinants for allosteric regulation of flip receptors. For these experiments, the three regions in the flip-flop module of GluR2o were individually exchanged for the corresponding regions in GluR2i. The importance of region 2 (Ser775 in flip and Asn775 in flop) for determining the nature of modulation by cyclothiazide of flip and flop receptors suggested that the identity of this residue also may confer the distinct kinetic phenotypes of potentiation by LY404187 of the two isoforms. After transient transfection into HEK 293 cells, all of the recombinant mutated GluR2o were functional, as evidenced by the response to application of glutamate (1 mm) and modulation by LY404187 (Fig. 4). As predicted from studies of cyclothiazide, exchange of region 2 (N775S) completely reversed the kinetics of potentiation so that the percentage change in potentiation for GluR2oo,i,o (207.0 ± 14.0%; n = 12) was statistically equivalent to wild-type GluR2i (184.4 ± 11.0%; n = 14; p > 0.05) (Fig. 4B,C). Substitution of region 1 (N765T, A766P) also significantly altered the kinetics of potentiation of GluR2o (p < 0.05) but did not restore the flip receptor phenotype. Instead, the percentage change in potentiation for GluR2oi,o,o (110.5 ± 6.3%; n = 6) was intermediate to that for wild-type GluR2i or GluR2o (Fig. 4A,C). In contrast, exchange of region 3 (G797K, G798D, G799S, D800G) failed to alter the kinetics of potentiation (percentage change in potentiation: GluR2oo,o,i, 77.0 ± 4.4%; n = 15) (Fig. 4C). These results indicate that exchange of a single residue, Asn775 for Ser, in GluR2o is sufficient to impart GluR2i kinetic phenotype of allosteric modulation by LY404187.
Figure 4.
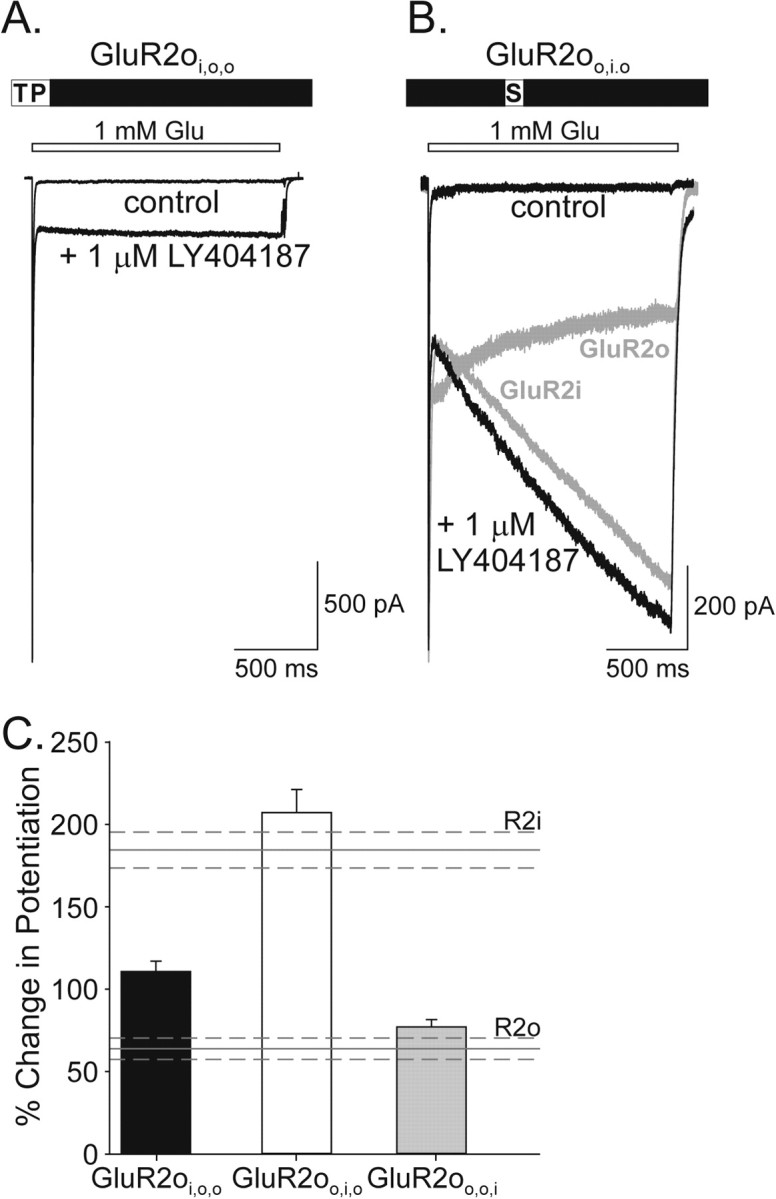
Exchange of a single residue in GluR2o can produce the GluR2i kinetic response to allosteric regulation by LY404187. A, B, Whole-cell responses to 1.5 sec glutamate (1 mm) applications in the absence and presence of 1 μmLY404187 from HEK 293 cells expressing GluR2oi,o,o (A) or GluR2oo,i,o (B). In B, the responses of GluR2i and GluR2o are superimposed in light gray for comparison. C, Plot of the percentage change in potentiation in the presence of LY404187 for the different mutated GluR2o. The percentage change in potentiation for GluR2i (R2i) and GluR2o (R2o) is indicated for comparison. Error bars represent mean ± SEM; n = 6-15 (F(4,55) = 52.57; p < 0.0001).
Molecular determinants of allosteric regulation by LY404187 of flop receptors
A similar mutational strategy was used to determine the residues that were critical for conferring the flop receptor phenotype of potentiation. In contrast to the dependence of the GluR2i kinetic phenotype on the presence of Ser in region 2, exchange of Ser775 for Asn in GluR2i did not alter the percentage change in potentiation from that displayed by wild-type GluR2i (GluR2i, 184.4 ± 11.0%, n = 14; GluR2ii,o,i, 176.5 ± 4.9%, n = 27) (Fig. 5A,B). Subsequent experiments showed that exchange of region 3 (K797G, D798G, S799G, G800D) also was not capable of reversing the kinetics of potentiation (GluR2ii,i,o, 199.0 ± 6.0%; n = 9) (Fig. 5B). Likewise, the substitution of region 1 (T765N, P766A) into GluR2i did not reverse the kinetics of potentiation by LY404187. However, a significant increase in the percentage change in potentiation was observed for GluR2io,i,i compared with wild-type GluR2i (GluR2io,i,i, 255.4 ± 13.2%; n = 10; p < 0.001) (Fig. 5B). This effect required substitution of both amino acids in region 1, because the exchange of either residue alone did not significantly increase the percentage change in potentiation over wild-type GluR2i (GluR2iT765N, 208.9 ± 12.0%, n = 8; GluR2iP766A, 177.5 ± 10.3%, n = 9). Collectively, these results indicate that, unlike the kinetics of potentiation of flip receptors, the phenotype of flop receptor potentiation does not depend solely on the identity of the residue (i.e., Asn775) in region 2 or on any individual region of the flip-flop domain.
Figure 5.
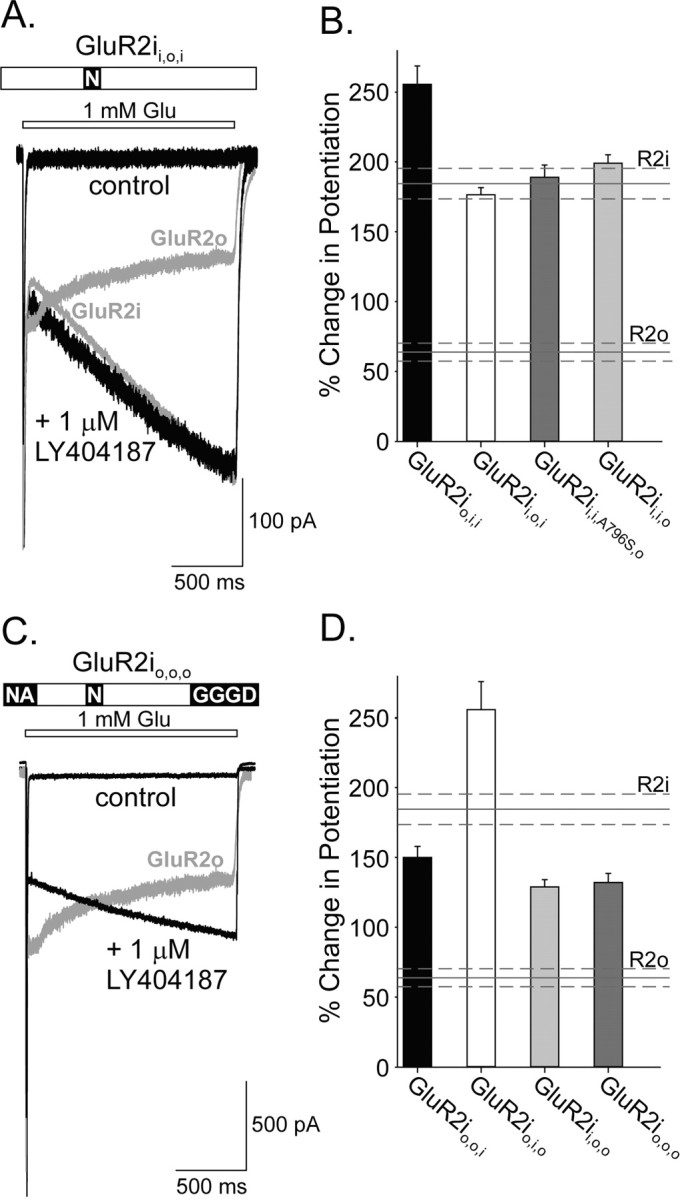
Exchange of any single region alone or in combination in GluR2i cannot produce the GluR2o kinetic response to allosteric regulation by LY404187. A, C, Whole-cell responses to 1.5 sec glutamate (1 mm) applications in the absence and presence of 1 μmLY404187 from HEK 293 cells expressing GluR2ii,o,i (A) or GluR2io,o,o (C) receptors. The responses of GluR2i or GluR2o are superimposed in light gray for comparison. B, D, Plot of the percentage change in potentiation in the presence of LY404187 for mutated receptors. B, Because the amino acid sequences of the GluR2 isoforms differ by one residue at the N-terminal border of region 3 (Ala796 in GluR2i and Ser796 in GluR2o; Fig. 3), an additional mutation of region 3 was made including this residue. Results showed that this mutation also did not alter the potentiation by LY40187 from that for GluR2i (GluR2ii,i,A796S,o, 188.9 ± 8.8%; n = 10; for other exchanges, see Results). D, Combination substitutions of regions failed to confer GluR2o potentiation (GluR2io,o,i, 150.1 ± 7.8%, n = 18; GluR2ii,o,o, 128.9 ± 5.2%, n = 6; GluR2io,i,o, 255.6 ± 20.2%, n = 7; for GluR2io,o,o, see Results). The percentage change in potentiation for GluR2i (R2i) and GluR2o (R2o) are indicated for comparison. Error bars represent mean ± SEM; n = 9-27 (F(5,77) = 53.22; p < 0.0001).
The failure of exchanges of individual regions in GluR2i to restore the GluR2o kinetics of potentiation implied that multiple regions contributed to manifestation of the GluR2o response to LY404187. To test this hypothesis, the mutational strategy was expanded to evaluate the effects of combinations of regional substitutions in GluR2i. Results showed that none of these combinations of mutations gave rise to the GluR2o kinetics of modulation. Surprisingly, even when all three regions of the GluR2i were exchanged for the corresponding regions in GluR2o, the kinetics of potentiation still displayed a positive percentage change in potentiation, albeit at a reduced level (GluR2io,o,o, 132.0 ± 6.5%; n = 17; p < 0.001) (Fig. 5C,D). Together, these results indicate that these three regions alone are not sufficient to confer the flop kinetic phenotype of allosteric modulation by LY404187.
Besides the three regions originally described by Partin et al. (1995), there is a relatively conservative difference in the amino acid sequence of flip and flop isoforms four residues downstream from region 2 (Fig. 3). For the GluR2 subunit, this residue is Val779 in the flip variant and Leu779 in the flop variant. Initially, the single exchange of Val779 for Leu in GluR2i receptors was conducted to determine whether this residue alone was sufficient to confer the flop kinetics of potentiation. Although the GluR2iV779L mutation reduced the percentage change in potentiation, the response was still different from wild-type GluR2o modulation (GluR2iV779L, 144.3 ± 13.8%; n = 11; p < 0.001) (Fig. 6A,C). Therefore, exchanges of Val779 to Leu with individual regions alone and in combinations were performed. Results showed that substitution of region 1, region 2, and Val779Leu in the GluR2i was necessary and sufficient to confer the GluR2o kinetic phenotype of modulation to LY404187 (GluR2io,o,V779L,i, 79.4 ± 3.3%; n = 15) (Fig. 6B,C). Interestingly, for each of the other single region and combination substitutions that included Val779Leu, the percentage change in potentiation was reduced compared with each of these mutations without this residue exchanged, further indicating the critical nature of this residue in the expression of the flop kinetic phenotype of allosteric modulation (Fig. 7C).
Figure 6.
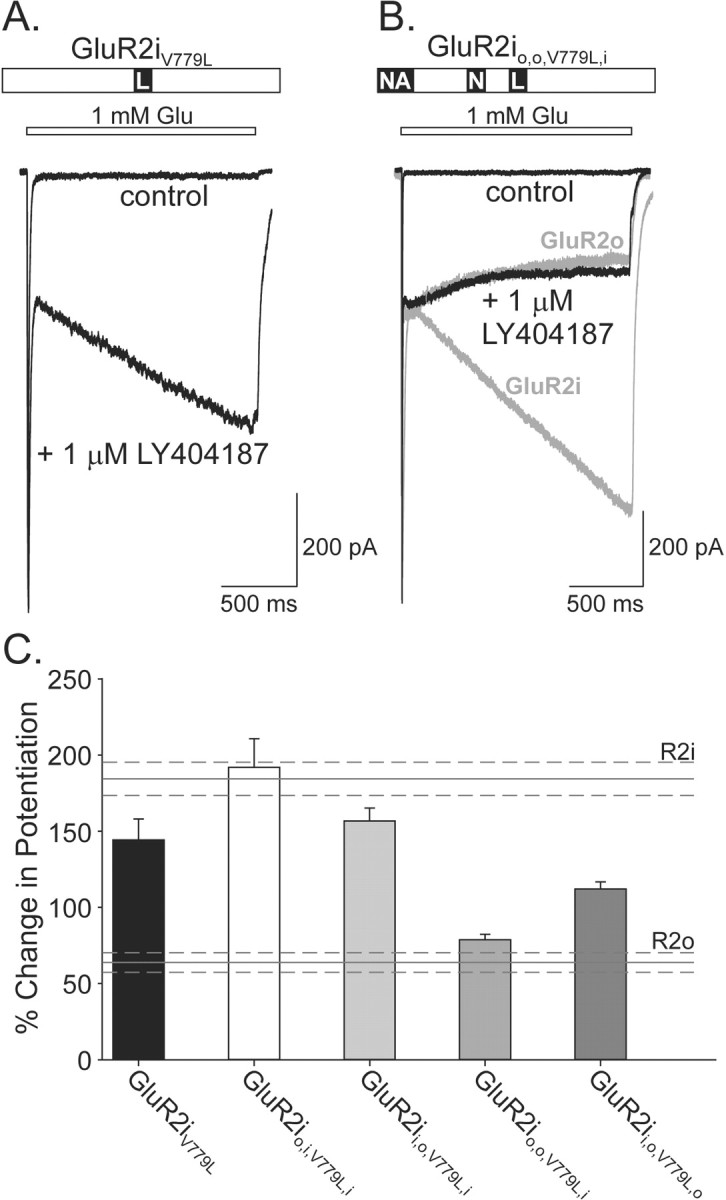
The GluR2o kinetic response to allosteric regulation by LY404187 requires the exchange of four residues in GluR2i. A, B, Whole-cell responses to 1.5 sec glutamate (1 mm) applications in the absence and presence of 1 μmLY404187 from HEK 293 cells expressing GluR2iV779L (A) or GluR2io,o,V779L,i (B). In B, the responses of GluR2i or GluR2o are superimposed in light gray for comparison. C, Plot of the percentage change in potentiation in the presence of LY404187 for mutated receptors (GluR2io,i,V779L,i, 192.0 ± 18.8%, n = 10; GluR2ii,o,V779L,i, 156.8 ± 8.4%, n = 14; GluR2ii,o,V779L,o, 112.0 ± 4.8%, n = 20; for other exchanges, see Results). The percentage change in potentiation for GluR2i (R2i) and GluR2o (R2o) wild-type receptors are indicated for comparison. Error bars represent mean ± SEM; n = 10-20 (F(6,90) = 28.62; p < 0.0001).
Figure 7.

The same residues confer differences in the kinetics and potency of allosteric modulation by LY404187. The concentration-response profile for LY404187 (0.1-30 μm) potentiation of 10 sec glutamate (100 μm) applications from HEK 293 cells expressing GluR2i, GluR2o, GluR2io,o,V779L,i, or GluR2oo,i,o were acquired. Because all of the mutated receptors were expressed in Flp-In -293 cells (catalog #R750-07; Invitrogen, Carlsbad, CA), for these potency studies, all of the receptors compared were expressed in these cells. The plot of the average EC50 for these four receptors is shown. Error bars represent the mean ± SEM; n = 10-20 (F(3,81) = 8.54; p < 0.0001).
The residue immediately adjacent to region 1 (residue 764 in human GluR2) can be edited from Arg to a Gly in GluR2-GluR4 subunits and has been shown to influence the rate of desensitization (Lomeli et al., 1994). In the present studies, the edited forms (Gly764) for both GluR2i and GluR2o were used, suggesting that this residue should not contribute to the different kinetic phenotypes of allosteric regulation of the two isoforms. However, given the influence of region 1 on potentiation by LY404187, the effect of exchanging Gly764 for Arg in GluR2i was examined. As expected, this exchange failed to alter percentage potentiation by LY404187 (GluR2iG764R, 174.6 ± 4.9%; n = 14).
Potency of LY404187 potentiation
LY404187 also displayed splice variant differences in potency at homomeric GluR1-GluR4, with the flip isoforms of each subunit exhibiting greater sensitivity to the compound (Fig. 1). To determine whether these differences in potency were conferred by the same residues as those required for the differential kinetics of modulation, the potency of LY404187 at GluR2oo,i,o and GluR2io,o,V779L,i were compared with the potency at wild-type GluR2i and GluR2o. Results showed that the potency of LY404187 for GluR2oo,i,o (EC50 of 2.1 ± 0.2 μm; n = 17) was equivalent to that for GluR2i (EC50 of 1.4 ± 0.1 μm; n = 19) (Fig. 7). Likewise, the potency for GluR2io,o,V779L,i (EC50 of 5.1 ± 0.8 μm; n = 20) was equivalent to GluR2o (EC50 of 5.4 ± 0.9 μm; n = 29) (Fig. 7) Therefore, the residues responsible for conferring differences in kinetics of potentiation by LY404187 for AMPA receptor splice variant are also sufficient for conferring differences in potency.
Role of region 2 in allosteric regulation by cyclothiazide of flip and flop receptors
The present results indicate that the expression of GluR2i kinetic phenotype of potentiation by LY404187 depends on the presence of Ser775 in region 2 but that expression of the GluR2o kinetic phenotype cannot be similarly conferred solely by substitution of Asn775 in this region. In contrast, previous studies have shown that the preferential sensitivity to cyclothiazide of rat homomeric GluR1i versus GluR1o depends entirely on the identity of the amino acid residue in region 2 (Ser750 in flip and Asn750 in flop) (Partin et al., 1995). Together, these data suggest that the structural determinants for allosteric regulation of flip and flop AMPA receptors by LY404187 and cyclothiazide are different. To explore this possibility, the effects of mutating region 2 in GluR2i and GluR2o on potentiation by cyclothiazide were evaluated. As described previously for GluR1 (Partin et al., 1995), attenuation of desensitization by a maximal concentration of cyclothiazide (100 μm) was markedly greater for GluR2i than GluR2o (Fig. 8A,B), such that the percentage change in potentiation was 104.4 ± 3.5% (n = 9) and 6.0 ± 1.3% (n = 5), respectively. After mutation of region 2 (Asn775Ser) in GluR2o, cyclothiazide (100 μm) also completely suppressed desensitization so that the percentage change in potentiation (105.4 ± 4.6%; n = 5) (Fig. 8D,E) was indistinguishable from that for GluR2i. Similar to responses in GluR1 (Partin et al., 1995), but unlike the lack of effect on LY404187 potentiation (Fig. 5A), mutation of region 2 (Ser775Asn) in GluR2i significantly reduced the activity of cyclothiazide (GluR2ii,o,i percentage change in potentiation, 42.6 ± 3.9%; n = 9; p < 0.001) compared with effects on wild-type GluR2i (Fig. 8C). However, the degree of potentiation by cyclothiazide of GluR2ii,o,i also was significantly greater than that for wild-type GluR2o (p < 0.001). Subsequent mutations revealed that exchange of region 1 and 2 and Val779Leu in GluR2i reduced the magnitude of potentiation (GluR2io,o,V779L,i percentage change in potentiation, 10.3 ± 1.8%; n = 10) to levels equivalent to those for GluR2o. These data demonstrate subtle differences in the mechanism of cyclothiazide modulation of rat GluR1 and human GluR2, suggesting either species and/or subunit variations. More importantly, these results indicate that the differential sensitivity of flip and flop receptors to cyclothiazide depends on region 2 to a greater extent than modulation of these receptor isoforms by LY404187.
Figure 8.
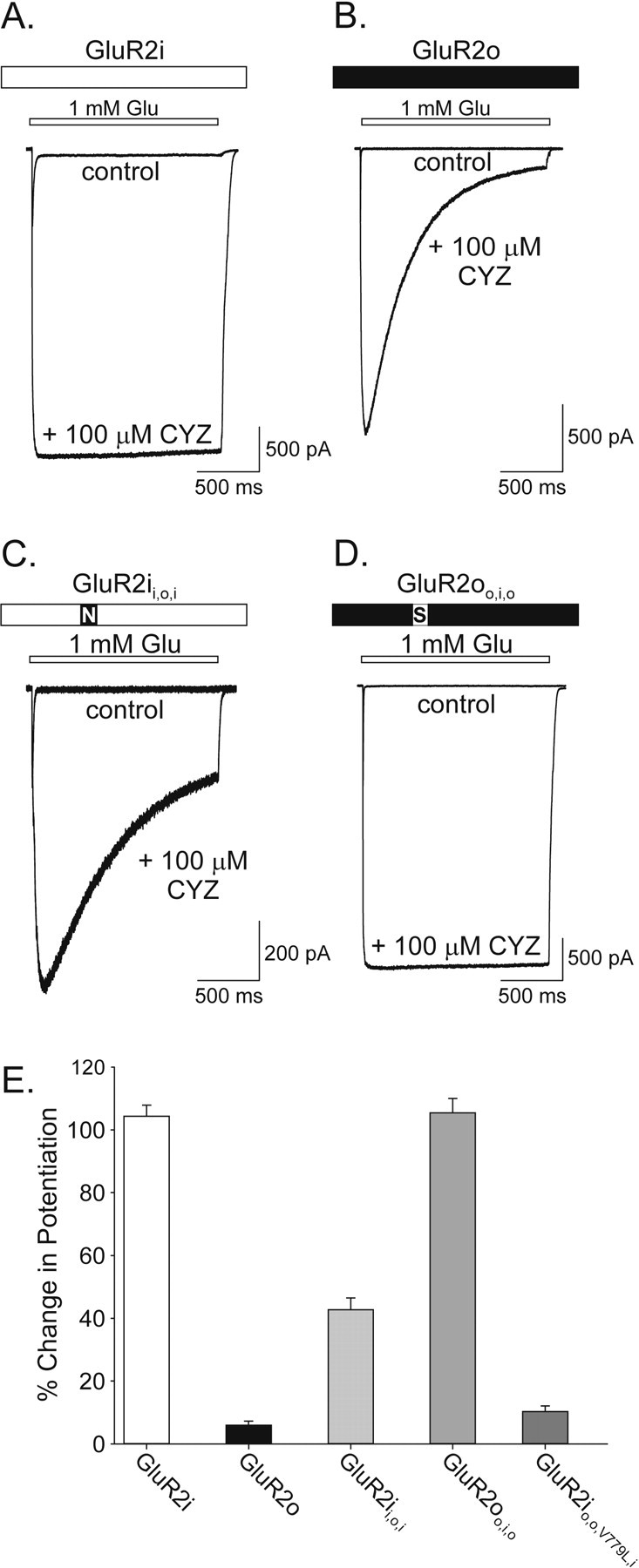
Activity of cyclothiazide on wild-type and mutant GluR2i and GluR2o. A-D, Whole-cell responses to 1.5 sec glutamate (1 mm) applications in the absence and presence of 100 μm cyclothiazide (CYZ) from HEK 293 cells expressing GluR2i (A), GluR2o (B), GluR2ii,o,i (C), or GluR2oo,i,o (D). E, Plot of the percentage change in potentiation in the presence of 100 μm cyclothiazide. The percentage change in potentiation was determined by expressing the current at the end of a rapid 1.5 sec glutamate (1 mm) application as a percentage of the initial current. Error bars represent mean ± SEM; n = 5-19 (F(4,45) = 241.7; p < 0.0001).
Further evidence supporting a greater dependence on region 2 for allosteric regulation of AMPA receptors by cyclothiazide than LY404187 comes from additional mutational studies. Previous research has demonstrated that exchange of Ser750 for Gln (corresponding residue in kainate receptors) in rat GluR1i eliminates potentiation by cyclothiazide (Partin et al., 1995, 1996). Consistent with these findings, mutation of the corresponding amino acid, Ser776, in human GluR4i subunits to Gln nearly abolishes potentiation by cyclothiazide. However, the same mutation only partially reduces the activity and binding of LY395153 (another biarylpropylsulfonamide compound that is an allosteric modulator of AMPA receptors), suggesting that binding to the flip-flop domain may occur at overlapping but distinct sites from the cyclothiazide binding site (Quirk et al., 2002). In the present studies, the relative dependence of cyclothiazide and LY404187 modulation on region 2 was evaluated in wild-type (Ser775) and mutant (Ser775Gln) human GluR2i. Both cyclothiazide (1-300 μm) and LY404187 (0.1-30 μm) enhanced the glutamate-evoked desensitized current of GluR2i in a concentration-dependent manner (cyclothiazide EC50 of 7.6 ± 0.9 μm, n = 10; LY404187 EC50 of 1.4 ± 0.1 μm, n = 19) (Fig. 9A,B). Maximal concentrations of cyclothiazide (100 μm) and LY404187 (30 μm) potentiated GluR2i responses by 42.4 ± 8.7-fold and 156.1 ± 31.6-fold, respectively. Consistent with previous results (Partin et al., 1995; Quirk et al., 2002), exchange of a Ser775 for Gln in GluR2i nearly eliminated potentiation by cyclothiazide (100 μm), such that the response was only 1.2 ± 0.01-fold (n = 8) greater than that for glutamate alone (Fig. 9B). In contrast, potentiation by LY404187 was less sensitive to the Ser775 to Gln mutation, with the maximal potentiation ∼40% of the potentiation found in wild-type GluR2i. The amino acid exchange also shifted the LY404187 concentration-response curve to the right (GluR2i EC50 of 1.4 ± 0.1 μm, n = 19; GluR2iS775Q EC50 of 10 ± 1.6 μm, n = 7). These results indicate that the activity of LY404187 is more tolerant than cyclothiazide of a Gln in position 775 and suggest that allosteric regulation of AMPA receptors can be conferred by multiple molecular mechanisms that may or may not depend solely on the identity of the amino acid residue in region 2.
Figure 9.
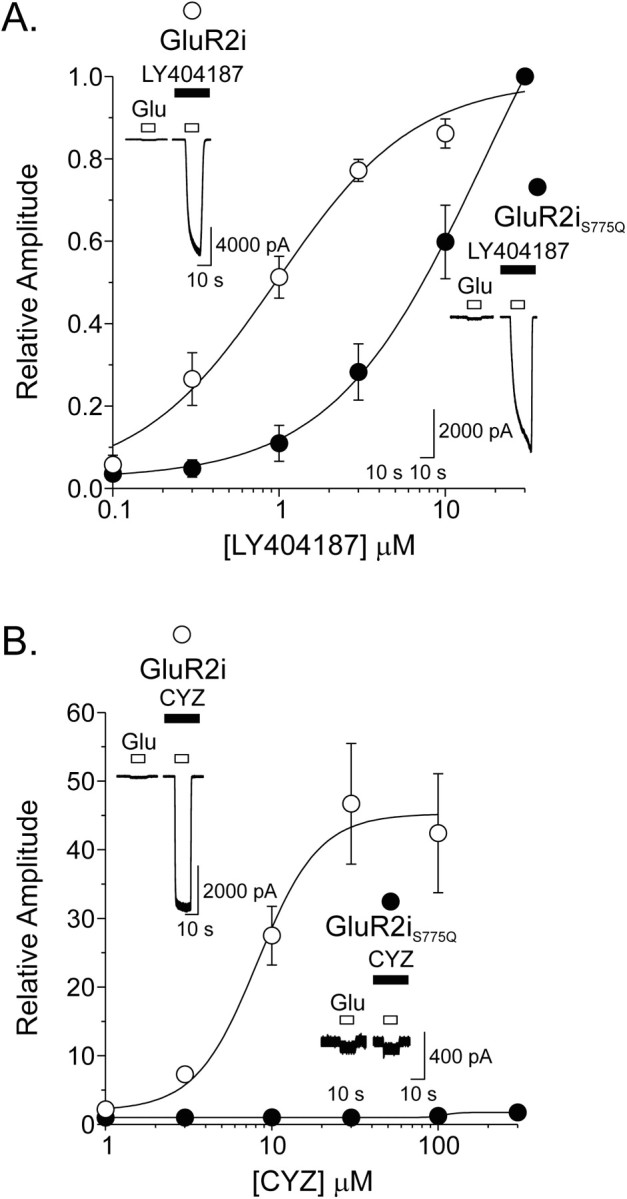
LY404187 and cyclothiazide are differentially sensitive to mutation of region 2 in GluR2i. A, Plot of the average relative degree of potentiation by LY404187 (0.1-30 μm) normalized to the maximal response to the application of 100 μm glutamate in the presence of 30 μmLY404187 for wild-type (S775) GluR2i (open circles) and mutant (S775Q) GluR2i (filled circles). Insets, The responses of wild-type GluR2i (top) and mutant GluR2iS775Q (bottom) to glutamate alone and in the presence of 30 μm LY404187. B, Plot of the average relative degree of potentiation by cyclothiazide (CYZ; 1-300 μm) normalized to the response to 100 μm glutamate alone for wild-type GluR2i (open circles) and mutant GluR2iS775Q (filled circles). Insets, The responses of wild-type GluR2i (top) and mutant GluR2iS775Q (bottom) to glutamate alone and in the presence of 300 μm cyclothiazide. Points represent mean ± SEM; n = 8-14.
Discussion
Novel molecular determinants of allosteric modulation of AMPA receptor splice variants
Allosteric modulation of receptor activity involves conformational change(s) of a receptor protein that is transmitted either to the orthosteric (agonist) site and/or directly to effector mechanisms (Christopoulos, 2002). Examples of both endogenous and exogenous allosteric modulators of ligand-gated ion channels have been identified, including glycine for the NMDA receptor (endogenous) and benzodiazepines (exogenous) for the GABAA receptor (Kardos and Nyikos, 2001). Cyclothiazide was among the first modulators of AMPA receptors to be discovered and subsequently shown to preferentially affect the activity of flip and flop isoforms, suggesting that the structural basis for its differential activity arose from differences in this extracellular domain. Indeed, previous studies demonstrated that the Ser/Asn residue in region 2 confers the differential action of cyclothiazide (Partin et al., 1995), a finding supported by the present results (Fig. 8). Similarly, the preferential flop activity of aniracetam is sensitive to exchange of region 2 (Partin et al., 1996), suggesting that this residue may be the sole determinant of sensitivity of AMPA receptor splice variants to allosteric modulation. To test this hypothesis, the present study investigated the molecular determinants for the differential sensitivity of AMPA receptor isoforms to modulation by LY404187, initially focusing on region 2. Mutational analyses revealed that, similar to cyclothiazide, exchange of region 2 (Asn775Ser) in GluR2o was necessary and sufficient to confer the GluR2i kinetic phenotype of allosteric modulation by LY404187. Surprisingly, however, the corresponding mutation in GluR2i failed to impart the GluR2o kinetics of potentiation. Moreover, exchange of none of the previously identified regions alone or in combination was sufficient to confer the GluR2o response to LY404187. Results showed that the GluR2o kinetics of modulation depended on a novel set of substitutions in GluR2i, including Val779Leu in combination with regions 1 and 2. The difference in potency of the receptor isoforms to LY404187 also was dependent on these same residues. These results demonstrate that, unlike cyclothiazide and aniracetam, the structural determinants for allosteric regulation of AMPA receptor splice variants by LY404187 are not identical. More specifically, although the amino acid identity of region 2 is necessary, it is not sufficient to impart differences in the splice variant responses to modulation by LY404187, indicating that allosteric regulation of AMPA receptors can arise from multiple molecular mechanisms.
Structural implications for LY404187 modulation
Insights into the structural determinants of LY404187 modulation of AMPA receptor splice variants may be inferred from recent crystallography studies that have investigated the binding of cyclothiazide to the glutamate LBC for a GluR2o possessing a single Ser for Asn point mutation in region 2 (i.e., LBC for GluR2oo,i,o) (Sun et al., 2002). The LBC contained the extracellular glutamate binding region and most of the flip-flop domain, including region 1, region 2, and the Val/Leu site (Fig. 3). Evidence from functional studies with cyclothiazide on the full-length receptor would indicate that this ligand-binding core most closely reflects a flip receptor (Partin et al., 1995). In the crystal structure, two cyclothiazide molecules were shown to link two adjacent ligand-binding cores, in part through formation of a hydrogen bond with Ser in region 2 of the flip-flop domain (Sun et al., 2002). At first glance, the observation that both cyclothiazide and LY404187 produce the flip splice variant phenotype of potentiation in the GluR2oo,i,o suggests that LY404187 may bind to the receptor in an identical manner. However, closer inspection of the modulation by the two compounds shows clear differences in their kinetics of potentiation, suggesting that their binding domains might be distinct. In addition, the present results demonstrated that substitution of Gln for Ser775 in GluR2i altered modulation by these two compounds in a markedly different manner, such that potentiation by cyclothiazide was eliminated, whereas modulation by LY404187 was only attenuated. In a similar manner, both the functional activity and binding of LY395153, a structural analog of LY404187, were partially blocked by the Gln exchange for Ser (Quirk et al., 2002). These results indicate that, although the binding domains of cyclothiazide and LY404187 in GluR2i may be overlapping and involve a close interaction with Ser in region 2 of the flip-flop module, they are most likely not identical. In a broader context, previous studies have suggested that other classes of positive modulators, including pyrollidinones (e.g., aniracetam) and benzamides (e.g., CX516 and CX546) display differing degrees of dependence on region 2 for their functional effects (Partin et al., 1996; Arai et al., 2000, 2002). Together, these data suggest that allosteric regulation of AMPA receptors may arise from multiple binding motifs that produce distinct conformational changes and may account for differences in kinetics of potentiation.
A novel finding of the present studies was the discovery that the flop kinetic phenotype of potentiation by LY404187 depended on the identity of amino acids in regions 1 and 2, as well as Leu779. In the crystal structure, region 2 is located on α helix J and binds directly to cyclothiazide, whereas region 1 is positioned at the beginning of this α helix but is rotated away from cyclothiazide so that a direct interaction is not possible (Sun et al., 2002). Exchange of region 1 in either GluR2i or GluR2o influenced the activity of LY404187, suggesting a direct interaction with region 1. Alternatively, the influence of region 1 on LY404187 activity could be indirect. For example, the identity of region 1 could determine the degree of rotation of α helix J and consequently influence the location of region 2 and its interaction with LY404187.
The Val/Leu779 residue has been disregarded in previous studies of flip and flop receptors because of the relatively conservative difference between these amino acids. In the crystal structure, Leu779 is located in the linker region between helix J and helix K with its side chain positioned away from the dimer interface, making an interaction between this residue and LY404187 unclear (Sun et al., 2002). However, helix K is proposed to point toward the plasma membrane, and therefore it is possible that the Leu779 residue contributes to transduction of LY404187 binding into movements that influence channel gating. Alternatively, Leu779 may affect the degree of rotation of helix J and thus the location of region 2, as described above for region 1. Ultimately, crystallographic studies of LY404187 bound to the native GluR2i and GluR2o ligand-binding cores will be critical for elucidating the contribution of region 1 and the Val/Leu779 residue to allosteric modulation.
Therapeutic potential of positive allosteric modulators of AMPA receptors
Considerable evidence has indicated that dysfunction of glutamatergic signaling in the CNS may contribute to cognitive deficits associated with a variety of neurological and psychiatric disorders (Yamada, 2000). Several therapeutic approaches designed to enhance glutamatergic signaling are being pursued, one of which involves augmentation of AMPA receptor function using positive allosteric modulators. Support for the therapeutic potential of AMPA receptor modulators has come from preclinical studies demonstrating that modulators, including biarylpropylsulfonamides, improve performance of animals on a variety of cognitive tasks that require multiple types of mnemonic processes (Staubli et al., 1994a; Hampson et al., 1998; Quirk and Nisenbaum, 2002). More importantly, several clinical studies have demonstrated that allosteric modulators can enhance cognitive function in both healthy and aged subjects, as well as in patients with Parkinson's disease and schizophrenia (Dimond et al., 1979; Oepen et al., 1985; Ingvar et al., 1997). Collectively, these data support the possibility that positive modulation of AMPA receptors may be a novel therapeutic approach for cognitive deficits in a variety of disorders, particularly those that are associated with reduced glutamatergic signaling.
Given the promising therapeutic value of allosteric modulators of AMPA receptors, there is great interest in developing compounds that selectively modulate flip and flop isoforms to ascertain their relative contribution to cognitive enhancement. Toward this end, the present functional studies have identified amino acids that are critical for conferring differences in allosteric modulation of these splice variants. Future crystallographic analyses will provide complementary information on the specific involvement of these residues in the binding of modulators to flip and flop receptors. In combination with these structural and functional data, molecular modeling techniques could be used to design novel compounds for which specific interactions are strengthened to yield truly selective molecules for AMPA receptor splice variants.
Footnotes
Correspondence should be addressed to Dr. Eric S. Nisenbaum, Eli Lilly and Company, Lilly Corporate Center, Indianapolis, IN 46285. E-mail: esn@lilly.com.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310953-10$15.00/0
References
- Arai AC, Kessler M, Rogers G, Lynch G ( 2000) Effects of the potent ampakine CX614 on hippocampal and recombinant AMPA receptors: interactions with cyclothiazide and GYKI 52466. Mol Pharmacol 58: 802-813. [DOI] [PubMed] [Google Scholar]
- Arai AC, Xia YF, Rogers G, Lynch G, Kessler M ( 2002) Benzamide-type AMPA receptor modulators form two subfamilies with distinct modes of action. J Pharmacol Exp Ther 303: 1075-1085. [DOI] [PubMed] [Google Scholar]
- Christopoulos A ( 2002) Allosteric binding sites on cell-surface receptors: novel targets for drug discovery. Nat Rev Drug Discov 1: 198-210. [DOI] [PubMed] [Google Scholar]
- Dimond SJ, Scammell RE, Pryce IG, Huws D, Gray C ( 1979) Some effects of piracetam (UCB 6215 Nootropyl) on chronic schizophrenia. Psychopharmacology 64: 341-348. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF ( 1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7-61. [PubMed] [Google Scholar]
- Fletcher E, Nutt S, Hoo K, Elliott C, Korczak B, McWhinnie E, Kamboj R ( 1995) Cloning, expression and pharmacological characterization of a human glutamate receptor: hGluR4. Receptors Channels 3: 21-31. [PubMed] [Google Scholar]
- Hampson RE, Rogers G, Lynch G, Deadwyler SA ( 1998) Facilitative effects of the ampakine CX516 on short-term memory in rats: correlations with hippocampal neuronal activity. J Neurosci 18: 2748-2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, O'Shea-Greenfield A, Rogers SW, Heinemann S ( 1989) Cloning and functional expression of a member of the glutamate receptor family. Nature 342: 643-648. [DOI] [PubMed] [Google Scholar]
- Ingvar M, Ambros-Ingerson J, Davis M, Granger R, Kessler M, Rogers GA, Schehr RS, Lynch G ( 1997) Enhancement by an ampakine of memory encoding in humans. Exp Neurol 146: 553-559. [DOI] [PubMed] [Google Scholar]
- Kardos J, Nyikos L ( 2001) Universality of receptor channel responses. Trends Pharmacol Sci 22: 642-645. [DOI] [PubMed] [Google Scholar]
- Keinanen K, Wisden W, Sommer B, Werner P, Herb A, Verdoorn TA, Sakmann B, Seeburg PH ( 1990) A family of AMPA-selective glutamate receptors. Science 249: 556-560. [DOI] [PubMed] [Google Scholar]
- Lomeli H, Mosbacher J, Melcher T, Hoger T, Geiger JR, Kuner T, Monyer H, Higuchi M, Bach A, Seeburg PH ( 1994) Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science 266: 1709-1713. [DOI] [PubMed] [Google Scholar]
- Miu P, Jarvie KR, Radhakrishnan V, Gates MR, Ogden A, Ornstein PL, Zarrinmayeh H, Ho K, Peters D, Grabell J, Gupta A, Zimmerman DM, Bleakman D ( 2001) Novel AMPA receptor potentiators LY392098 and LY404187: effects on recombinant human AMPA receptors in vitro. Neuropharmacology 40: 976-983. [DOI] [PubMed] [Google Scholar]
- Oepen G, Eisele K, Thoden U, Birg W ( 1985) Piracetam improves visuomotor and cognitive deficits in early Parkinsonism—a pilot study. Pharmacopsychiatry 18: 343-346. [DOI] [PubMed] [Google Scholar]
- Ornstein PL, Zimmerman DM, Arnold MB, Bleisch TJ, Cantrell B, Simon R, Zarrinmayeh H, Baker SR, Gates M, Tizzano JP, Bleakman D, Mandelzys A, Jarvie KR, Ho D, Deverill M, Kamboj RK ( 2000) Biarylpropylsulfonamides as novel, potent potentiators of 2-amino-3-(5-methyl-3-hydroxyisoxazol-4-yl)-propanoic acid (AMPA) receptors. J. Med Chem 43: 4354-4358. [DOI] [PubMed] [Google Scholar]
- Partin KM, Bowie D, Mayer ML ( 1995) Structural determinants of allosteric regulation in alternatively spliced AMPA receptors. Neuron 14: 833-843. [DOI] [PubMed] [Google Scholar]
- Partin KM, Fleck MW, Mayer ML ( 1996) AMPA receptor flip/flop mutants affecting deactivation, desensitization, and modulation by cyclothiazide, aniracetam, and thiocyanate. J Neurosci 16: 6634-6647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontecorvo MJ, Evans HL ( 1985) Effects of aniracetam on delayed matching-to-sample performance of monkeys and pigeons. Pharmacol Biochem Behav 22: 745-752. [DOI] [PubMed] [Google Scholar]
- Quirk J, Nisenbaum E ( 2002) LY404187: a novel positive allosteric modulator of AMPA receptors. CNS Drug Rev 8: 255-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk JC, Linden AM, Strakhova M, Yu H, Skolnick P, Nisenbaum ES ( 2002) A single residue contributes sensitivity of allosteric modulation of AMPA receptors by LY395153. Eur J Pharmacol 454: 125-129. [DOI] [PubMed] [Google Scholar]
- Sekiguchi M, Fleck MW, Mayer ML, Takeo J, Chiba Y, Yamashita S, Wada K ( 1997) A novel allosteric potentiator of AMPA receptors: 4-[2-(phenylsulfonylamino)ethylthio]-2, 6-difluoro-phenoxyacetamide. J Neurosci 17: 5760-5771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi M, Yamada K, Jin J, Hachitanda M, Murata Y, Namura S, Kamichi S, Kimura I, Wada K ( 2001) The AMPA receptor allosteric potentiator PEPA ameliorates post-ischemic memory impairment. NeuroReport 12: 2947-2950. [DOI] [PubMed] [Google Scholar]
- Sommer B, Keinanen K, Verdoorn TA, Wisden W, Burnashev N, Herb A, Kohler M, Takagi T, Sakmann B, Seeburg PH ( 1990) Flip and flop: a cell-specific functional switch in glutamate-operated channels of the CNS. Science 249: 1580-1585. [DOI] [PubMed] [Google Scholar]
- Staubli U, Rogers G, Lynch G ( 1994a) Facilitation of glutamate receptors enhances memory. Proc Natl Acad Sci USA 91: 777-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staubli U, Perez Y, Xu F, Rogers G, Ingvar M, Stone-Elander S, Lynch G ( 1994b) Centrally active modulators of glutamate receptors facilitate the induction of long-term potentiation in vivo. Proc Natl Acad Sci USA 91: 11158-11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Olson R, Horning M, Armstrong N, Mayer M, Gouaux E ( 2002) Mechanism of glutamate receptor desensitization. Nature 417: 245-253. [DOI] [PubMed] [Google Scholar]
- Swanson GT, Kamboj SK, Cull-Candy SG ( 1997) Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci 17: 58-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KA ( 2000) Therapeutic potential of positive AMPA receptor modulators in the treatment of neurological disease. Expert Opin Investig Drugs 9: 765-778. [DOI] [PubMed] [Google Scholar]