

Abstract
Neuromodulation is often thought to have a static, gain-setting function in neural circuits. Here we report a counter example: the neuromodulatory effect of a serotonergic neuron is dependent on the interval between its spikes and those of the neuron being modulated. The serotonergic dorsal swim interneurons (DSIs) are members of the escape swim central pattern generator (CPG) in the mollusk Tritonia diomedea. DSI spike trains heterosynaptically enhanced synaptic potentials evoked by another CPG neuron, ventral swim interneuron B (VSI-B), when VSI-B action potentials occurred within 10 sec of a DSI spike train; however, if VSI-B was stimulated 20-120 sec after DSI, then the amplitude of VSI-B synaptic potentials decreased. Consistent with this, VSI-B-evoked synaptic currents exhibited a temporally biphasic and bidirectional change in amplitude after DSI stimulation. Both the DSI-evoked enhancement and decrement were occluded by serotonin and blocked by the serotonin receptor antagonist methysergide, suggesting that both phases are mediated by serotonin. In most preparations, however, bath-applied serotonin caused only a sustained enhancement of VSI-B synaptic strength. The heterosynaptic modulation interacted with short-term homosynaptic plasticity: DSI-evoked depression was offset by VSI-B homosynaptic facilitation. This caused a complicated temporal pattern of neuromodulation when DSI and VSI-B were stimulated to fire in alternating bursts to mimic the natural motor pattern: DSI strongly enhanced summated VSI-B synaptic potentials and suppressed single synaptic potentials after the cessation of the artificial motor pattern. Thus, spike timing-dependent serotonergic neuromodulatory actions can impart temporal information that may be relevant to the operation of the CPG.
Keywords: biphasic, bidirectional, serotonin, opisthobranch mollusk, facilitation, synaptic depression, motor pattern
Introduction
Synaptic strength can be enhanced or diminished by various homosynaptic mechanisms that are dependent on the extent and timing of the presynaptic activity (Daniel et al., 1998; Malenka and Nicoll, 1999; Abbott and Nelson, 2000; Bennett, 2000; Bi and Poo, 2001; Zucker and Regehr, 2002). Synaptic strength also can be changed by heterosynaptic neuromodulatory actions, which are generally thought to be independent of the level or timing of activity in the target neurons (Powis and Bunn, 1995; Katz, 1999; Gu, 2002). Such neuromodulatory actions are important for learning and memory (Byrne and Kandel, 1996; Hammer, 1997; Hasselmo and Linster, 1999), sensory processing (Pasztor, 1989; Nagai et al., 1996; Mercer, 1999), and motor pattern generation (Marder and Calabrese, 1996; Calabrese, 1998; Kiehn and Katz, 1999; Marder and Bucher, 2001). Although some forms of homosynaptic plasticity exhibit spike-timing dependence (Abbott and Nelson, 2000; Bi and Poo, 2001; Sjöström and Nelson, 2002), neuromodulation is often thought to act tonically to cause a sustained change in neuronal and synaptic properties (Pearson, 1993). Here we report an example of a spike-timing-dependent neuromodulation that might play a functional role in a motor pattern-generating network.
The system that we studied is the escape swim network of the mollusk Tritonia diomedea (see Fig. 1). The central pattern generator (CPG) for the rhythmic escape swim behavior is composed of three cell types (see Fig. 1A,B): the dorsal swim interneurons (DSIs), the ventral swim interneurons A and B (VSI-A, VSI-B), and cerebral interneuron 2 (C2) (Getting, 1981, 1989). The swim motor pattern (see Fig. 1C) is produced by the interactions of these three CPG neurons, which convey the pattern of activity to efferent flexion neurons: the dorsal flexion neurons (DFNs) and the ventral flexion neurons (VFNs). The DSIs are serotonergic (Katz et al., 1994; McClellan et al., 1994; Fickbohm and Katz, 2000) and use serotonin (5-HT) to evoke both synaptic and neuromodulatory actions (Katz and Frost, 1995a). The previously described DSI neuromodulatory actions include heterosynaptic enhancement of the amount of neurotransmitter released by C2 (Katz et al., 1994; Katz and Frost, 1995b) and a decrease in C2 spike frequency adaptation (Katz and Frost, 1997). Thus, the DSIs function as neuromodulatory neurons intrinsic to the CPG circuit.
Figure 1.

The Tritonia swim network and swim motor program. A, A schematic diagram of the neural circuit underlying the swim motor pattern. All neurons have a contralateral homolog that is not represented. The shaded box indicates the neurons that comprise the CPG. Filled triangles represent excitatory synapses, and filled circles represent inhibitory synapses. Combinations of triangle and circle are multicomponent synapses. Dotted lines indicate that the connection is either polysynaptic or not determined. Tr1, Trigger neuron 1. The connectivity is derived from published work of Getting et al. (1980), Getting (1981), Hume and Getting (1982a), Frost and Katz (1996), and Frost et al. (2001). B, Schematic drawing of a dorsal view of the Tritonia brain showing the location of the swim CPG neurons and their axonal projections. DSI and C2 are located on the dorsal surface of the cerebral ganglion. VSI-B is located on the ventral side of the pleural ganglion. VFN is located on the dorsal side of the pedal ganglion. The VSI-B synapse on VFN and the DSI modulatory action are shown as a filled triangle and an arrow, respectively. C, An example of the swim motor pattern. Simultaneous intracellular recordings from three CPG neurons, C2, DSI, and VSI-B, and one efferent flexion neuron, VFN, are shown. The bursting pattern was elicited by stimulation of pedal nerve 3 by applying voltages pulses (10 V, 1 msec) at 10 Hz for 1 sec (arrow). DSI and VSI-B fire bursts of action potentials out of phase with one another. The cycle period varies from 7 to 12 sec.
In this study, we examined the neuromodulatory effect of DSI stimulation on the synaptic output of another essential component of the CPG, VSI-B (Getting, 1983, 1989). We found that activation of a DSI evoked either enhancement or decrement of VSI-B synaptic strength depending on the timing of VSI-B spikes with respect to DSI spikes. This timing-dependent heterosynaptic neuromodulation may play a role in operation of this CPG.
Some of these data have been published previously in abstract form (Sakurai and Katz, 2002).
Materials and Methods
Preparation. Tritonia diomedea were obtained from Living Elements (Vancouver, British Columbia, Canada). All experiments were performed on the isolated brain, which consisted of the fused cerebral and pleural ganglia and the pedal ganglia (see Fig. 1B). The brain was removed from the animal as described previously (Willows et al., 1973), placed immediately in a Sylgard-lined chamber (1 ml), and superfused with saline at 4°C. The composition of physiological saline was (in mm): 420 NaCl, 10 KCl, 10 CaCl2, 50 MgCl2, 11 d-glucose, and 10 HEPES, pH 7.6. The brain was pinned to the bottom of the experimental chamber, and the cell bodies were exposed by removing the connective tissue sheath from the surface of the ganglia as described previously (Willows et al., 1973; Katz and Frost, 1995a). Suction electrodes, made from polyethylene tubing, were placed on pedal nerves. The preparation then was left overnight superfused in saline at 8-10°C. All experiments were conducted at 10°C with chilled saline superfused at 2 ml/min.
5-HT (Sigma, St. Louis, MO) was dissolved in high divalent cation saline (see below) just before use at the final concentrations (1-100 μm). Methysergide (RBI, Natick, MA) was dissolved in dimethyl sulfoxide (Sigma) at 20 mm and diluted to 50 μm in the high divalent cation saline just before use. Drugs were bath-applied by switching the superfusion paths.
Intracellular recordings and stimulation. Neurons were identified by soma location, coloration, synaptic connectivity, and activity pattern at rest and during the swim motor program (Taghert and Willows, 1978; Getting et al., 1980; Getting, 1981, 1983). The soma of VSI-B is located on the ventral side of the pleural ganglion, whereas DSIs and C2 are located on the dorsal surface of the cerebral ganglion. VSI-B projects ipsilaterally, whereas C2 and DSIs project contralaterally to the pedal ganglion (see Fig. 1B) (Lynn-Bullock et al., 2002). To record from both DSI and VSI-B simultaneously, the cerebral commissure was twisted as described previously by Getting (1983). After the interneurons were identified, the bathing medium was switched to high divalent cation saline, which raises the threshold for spiking and reduces spontaneous neural firing. The composition of the high divalent cation saline was (in mm): 285 NaCl, 10 KCl, 25 CaCl2, 125 MgCl2, 11 d-glucose, and 10 HEPES, pH 7.6 (Katz et al., 1994).
Neurons were impaled with glass microelectrodes filled with 3 m potassium chloride or 4 m potassium acetate (resistance, 2-15 MΩ). In most EPSP and EPSC recordings, the postsynaptic neuron was impaled with two electrodes, one for passing current (2-5 MΩ) and the other (6-15 MΩ) for recording voltage. To elicit action potentials in a presynaptic neuron, 20 msec current pulses were injected through a single recording electrode via a balanced bridge circuit. Each current pulse evoked a single spike, allowing the spike frequency to be controlled precisely.
The recordings were digitized at >1 kHz with an analog-to-digital converter from Cambridge Electronic Design [(CED) Cambridge, UK]. Data acquisition and analysis were performed with Spike2 software (CED) and SigmaPlot (Jandel Scientific, San Rafael, CA). Statistical comparisons were made using paired t test or one-way ANOVA with Tukey test. In all cases, p < 0.05 was considered significant. Results are expressed as the mean ± SEM.
Experimental protocol. To examine the modulatory actions of DSI on VSI-B synapses, we chose to focus on the VSI-B synaptic connections to follower neurons in the pedal ganglion, including the VFNs (see Fig. 1A, B). VSI-B makes excitatory synapses onto these neurons, whereas DSIs make inhibitory synapses onto the same neurons (Hume and Getting, 1982a; Getting, 1983). We also recorded from other VSI-B follower cells that were not positively identified but may include class III neurons (Hume and Getting, 1982a), which receive excitatory input from DSIs (see Fig. 2B1,C1), and others that receive no apparent synaptic input from DSIs (see Fig. 11A). In this study, we refer to all VSI-B follower cells in the pedal ganglion that fire bursts of action potentials in the ventral flexion phase of the swim cycle as VFNs.
Figure 2.
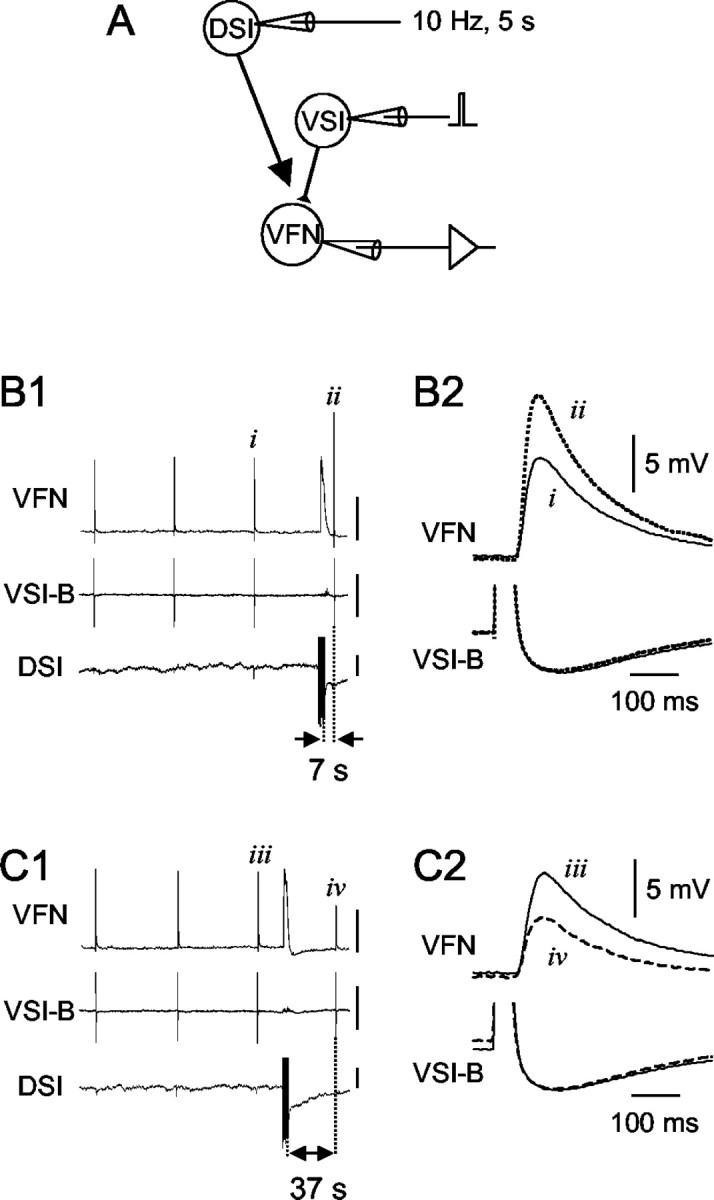
The effect of DSI stimulation on VSI-B-evoked EPSPs was dependent on the time between the DSI spike train and the VSI-B spike. A, A schematic diagram showing the experimental configuration. Intracellular microelectrodes were placed in the somata of DSI, VSI-B, and VFN. DSI and VSI-B were stimulated by injecting brief current pulses (7-10 nA, 20 msec). The membrane potential of VFN was set at approximately -70 mV by constantly injecting a hyperpolarizing current through another electrode to prevent action potential firing triggered by the EPSP. B1, Simultaneous intracellular recordings from VFN, VSI-B, and DSI show that DSI stimulation increased the amplitude of VSI-B-evoked EPSPs recorded in VFN when VSI-B was stimulated shortly after DSI. VSI-B was stimulated repeatedly to fire single action potentials at a 60 sec interval. After the third VSI-B action potential (i), DSI was stimulated to fire a train of action potentials (10 Hz, 5 sec) that ended 7 sec before the next VSI-B spike (ii). The amplitude of the EPSP in VFN was enhanced after DSI stimulation. In this recording, VFN showed a slow depolarization in response to a DSI spike train, which appeared to be the summated unitary EPSPs. The depolarized membrane potential in VFN recovered to the original potential within 5 sec. Action potentials in the VSI-B and DSI traces are truncated. Vertical calibration bars indicate 5 mV. B2, Expanded, superimposed waveforms of VSI-B-evoked EPSPs recorded in VFN (top trace) and VSI-B action potentials (bottom trace) before (i, solid trace) and 7 sec after (ii, dotted trace) the DSI stimulation show the synaptic enhancement. The action potentials in VSI-B are truncated to show the resting potential and spike afterhyperpolarization. C1, When the DSI spike train preceded VSI-B by 37 sec, then the amplitude of the VSI-B-evoked EPSP in VFN decreased. The stimulus protocol was the same as in B except for the timing of the DSI spike train. Vertical calibration bars indicate 5 mV. C2, The decreased amplitude of the EPSPs can be seen in superimposed waveforms evoked before (iii, solid trace) and 37 sec after (iv, dashed trace) the DSI stimulation. Resting potentials of VSI-B and DSI were -47 and -44 mV, respectively.
Figure 11.
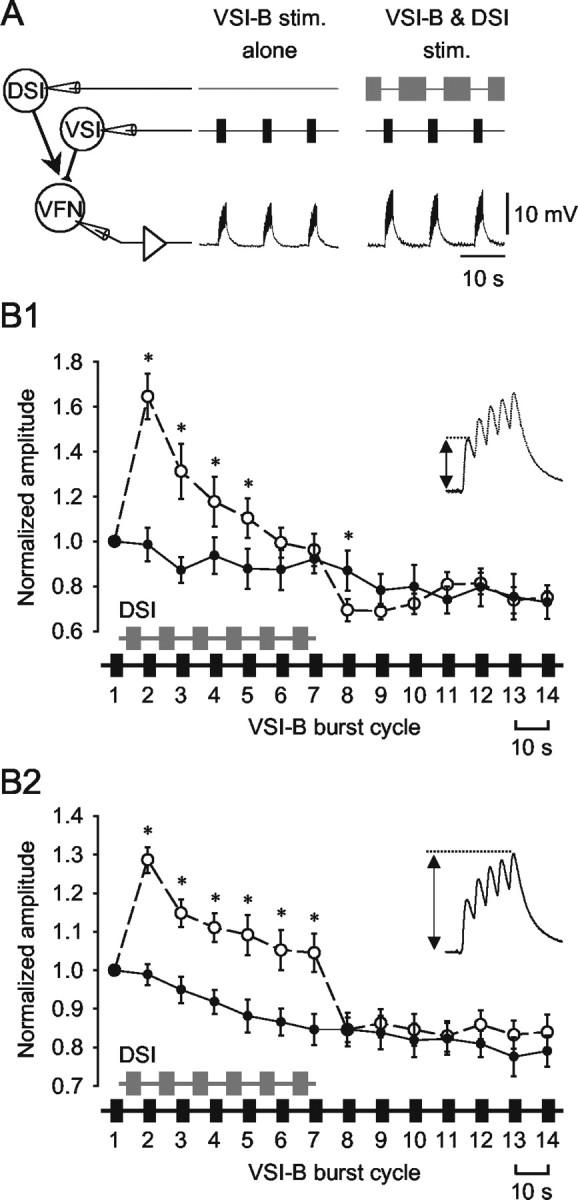
Stimulating DSI and VSI-B in a swim-like bursting pattern differentially modulated single and summated EPSPs. A, The configuration of intracellular stimulation to VSI-B and DSI and an intracellular recording of EPSPs in VFN. Bursts of 5-10 spikes were evoked in VSI-B every 10 sec at 5-10 Hz alone (left) or in alternation with DSI spike trains (5-10 Hz, 4-7 sec; right) mimicking the swim motor program. B, The normalized average amplitudes of the initial EPSP in each train (B1) and the summated fifth EPSP (B2) are plotted as a function of burst cycle number. When VSI-B was stimulated alone (solid circles), the amplitudes of both the initial EPSP (B1) and the summated EPSP (B2) gradually declined. When DSI bursts were interposed between VSI-B bursts for six cycles, the amplitudes of both the initial EPSP (B1) and the summated EPSP (B2) increased immediately (control, open circles). The initial EPSP declined gradually in amplitude despite continued DSI stimulation and by the seventh cycle was not significantly different from control (B1). In contrast, the summated EPSP decreased in amplitude after the first burst cycle but remained elevated for as long as DSI bursts were maintained (B2). After the cessation of DSI stimulation (the eighth cycle), the initial EPSP decreased to an amplitude that was significantly less than control, whereas the summated EPSP was not significantly different from control. Asterisks indicate significant difference from control (n = 7; p < 0.05; paired t test).
In most of the experiments, VSI-B was stimulated to fire single action potentials or short trains of action potentials at 5-10 Hz at regular intervals of 20-60 sec by injecting brief current pulses (5-20 nA, 20 msec). In some experiments, the intervals were varied from 60 sec up to 400 sec. To record VSI-B-evoked EPSPs, a follower cell of VSI-B was held hyperpolarized at -50 to -70 mV by a steady current injected though a bridge circuit or through another electrode to prevent action potential firing. For some experiments, the follower cell was voltage clamped at -50 to -60 mV with two electrodes to measure evoked synaptic currents. Single action potentials evoked in VSI-B at regular intervals produced a series of EPSPs with relatively constant amplitude in these follower cells (see Fig. 2B1,C1).
To determine the modulatory effects of DSI on VSI-B synaptic strength, VSI-B was stimulated four times at a regular interval (20-400 sec), and a DSI was stimulated to fire a train of action potentials at various times after the third VSI-B stimulus (see Fig. 2B1,C1). The DSI was stimulated at 5-10 Hz for 3-10 sec (precise stimulation parameters are noted in each experiment) by injecting current pulses (5-20 nA, 20 msec) into the soma or by applying voltage pulses (5-10 V, 1 msec) via a suction electrode to pedal nerve 5, which contains DSI axons (Lynn-Bullock et al., 2002). Unless noted otherwise, the interval between the DSI stimuli was always >2 min to prevent an accumulation of modulatory actions. The mean amplitude of three sequential EPSPs or EPSCs evoked by VSI-B before each DSI stimulation was used as a baseline for each trial; the amplitudes of all EPSPs or EPSCs were normalized to the baseline value for analysis. The effect of DSI stimulation on VSI-B synapses was assayed by comparing the normalized amplitudes of the EPSPs or EPSCs evoked before and after the DSI burst. The extent of homosynaptic facilitation of VSI-B-evoked EPSCs was quantified as the ratio of the fifth EPSC amplitude to the first EPSC amplitude.
Results
DSI modulated VSI-B-evoked synaptic potentials in a spike timing-dependent manner
The effect of DSI stimulation on synaptic potentials evoked by a VSI-B action potential was spike timing dependent. A spike train (10 Hz, 5 sec) was evoked in a DSI contralateral to VSI-B at varying times before a VSI-B spike and VSI-B-evoked synaptic potentials were recorded from a VFN in the pedal ganglion ipsilateral to the VSI-B soma (for soma locations and axon projection patterns, see Fig. 1B). The time between the DSI spike train and the VSI-B spike determined whether DSI caused the VSI-B-evoked EPSP to be enhanced or decreased (Fig. 2). For example, when VSI-B fired an action potential 7 sec after the end of the DSI spike train, then the VSI-B-evoked EPSP recorded in the VFN increased in amplitude (Fig. 2B). In contrast, when the spike from VSI-B occurred 37 sec after the end of a DSI spike train, then the EPSP decreased in amplitude (Fig. 2C).
The effect of DSI on VSI-B synaptic strength changed continuously over the course of 2 min after a DSI spike train (Fig. 3). A plot of normalized EPSP amplitudes versus the time after DSI spike train shows a biphasic-bidirectional change in the amplitude of VSI-B-evoked EPSPs: there was a smooth transition from enhancement to decrement (Fig. 3A). The plot was well fit by a double-exponential curve with decay time constants of 18.8 and 68.5 sec (see figure legend). The double-exponential fit suggests that the effect of DSI on VSI-B could be caused by two independent processes of opposite sign and different decay rates.
Figure 3.
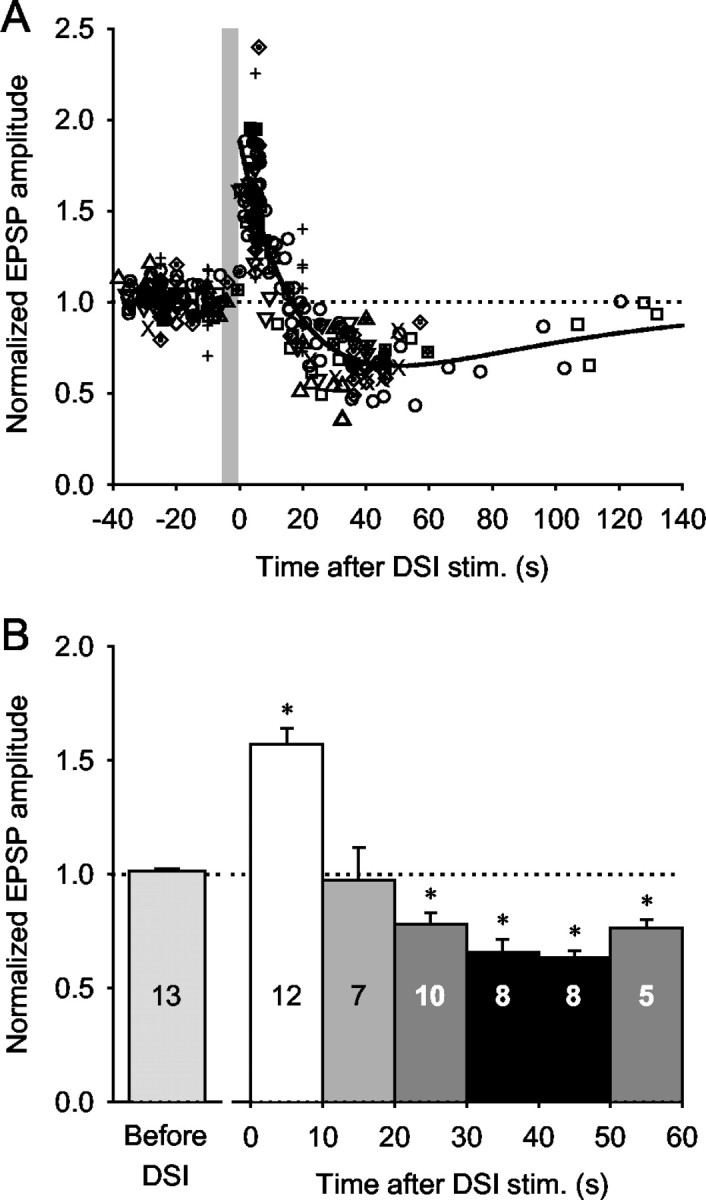
DSI stimulation caused a biphasic-bidirectional change in the amplitude of EPSPs evoked by VSI-B. A, The normalized amplitudes of VSI-B-evoked EPSPs were plotted as a function of time after the end of the DSI spike train. A DSI was stimulated to fire action potentials for 3-10 sec at 5-10 Hz (the gray vertical bar represents a 5 sec DSI spike train). Time 0 is the time of the last DSI spike. The peak amplitude of the VSI-B-evoked EPSPs occurred ∼5 sec after the DSI spike train. The decrement was maximal at ∼40 sec after the DSI spike train. Data from 13 preparations are plotted; each symbol represents data from a different preparation. The plot was fit by a double-exponential curve, f(t) = 1 + A × e-t/τ1 - e-t/τ2, where t is the time after the end of the DSI spike train, A = 1.89, τ1 = 18.8 sec, and τ2 = 68.5 sec. The EPSP amplitude was normalized to the average of the three EPSPs elicited before each DSI spike train (compare Fig. 2B1,C1). B, Bar graph showing averages of the normalized EPSP amplitudes at 10 sec intervals after DSI stimulation. Each bar shows mean ± SEM across preparations. The number of animals examined for each time period is indicated in each bar. There was a significant increase in EPSP amplitude when VSI-B was stimulated within 10 sec of DSI and a significant decrease when VSI-B was stimulated 20-60 sec after DSI (p < 0.001 by one-way ANOVA with Tukey test); however, there was no significant change during the transition period from enhancement to decrement (10-20 sec after DSI).
Normalized EPSP amplitudes were binned in 10 sec intervals to determine whether the changes in VSI-B synaptic strength were significant (Fig. 3B). The average increase within 10 sec of the DSI spike train was 56.9 ± 7.1%, which was a significant change (p < 0.001; one-way ANOVA with Tukey test). The maximum decrement occurred 40-50 sec after the DSI spike train (-36.7 ± 3.2%). The decrements for each of the 10 sec intervals between 20 and 60 sec after DSI were highly significant compared with pre-DSI controls (p < 0.001; one-way ANOVA with Tukey test); however, there was no significant change during the transition period from enhancement to depression (10-20 sec after DSI stimulation).
DSI stimulation evoked different temporal patterns of modulation depending on the timing of its spike train relative to VSI-B action potentials (Fig. 4). When a VSI-B was stimulated repeatedly at a regular interval (30 sec), it evoked an EPSP of consistent amplitude in VFN. Interposing a DSI spike train (10 Hz, 5 sec) 5 sec before a VSI-B spike caused an increase in the amplitude of the EPSP evoked by that spike (Fig. 4A). This synaptic enhancement was followed by a decrease in subsequent EPSPs, resulting in a temporally biphasic and bidirectional change in the EPSP amplitude (Fig. 4D, curve A); however, when the interval between the DSI spike train and the next VSI-B spike was 15 sec, then the first EPSP after the DSI spike train was unaffected, but subsequent VSI-B spikes produced decremented EPSPs (Fig. 4B,D, curve B). Finally, if the interval between the DSI spike train and the next VSI-B spike was 20 sec, then the next few EPSPs showed only a decrement (Fig. 4C,D, curve C). Thus, the time window during which DSI fired with respect to VSI-B critically determined the dynamics of subsequent VSI-B synaptic actions: it caused a biphasic change (Fig. 4A), an apparent delayed action (Fig. 4B), or a purely decrementing effect (Fig. 4C).
Figure 4.
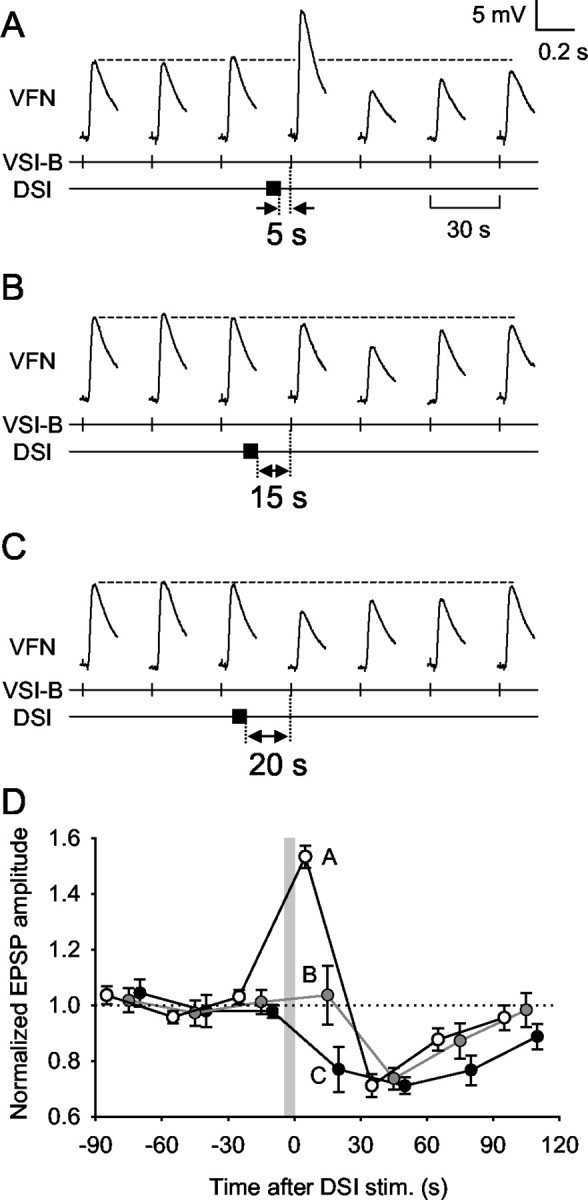
DSI stimulation caused different sequential neuromodulatory effects depending on the timing of its activation. A-C, VSI-B was stimulated to fire an action potential every 30 sec. A DSI was then stimulated to fire a single train of action potentials (10 Hz, 5 sec) that ended 5 sec (A), 15 sec (B), or 20 sec (C) before a VSI-B spike. D, Averaged data show the sequential effects of DSI stimulation on normalized VSI-B-evoked EPSPs recorded in VFNs. The gray vertical bar represents a 5 sec DSI spike train. When DSI was stimulated to fire 5 sec before a VSI-B spike, subsequent EPSPs showed an initial increase, followed by a decrease in amplitude (plot A, open circles; n = 7). When DSI was stimulated 15 sec before a VSI-B spike, there was no immediate effect, but subsequent EPSPs were decreased in amplitude (plot B, gray circles; n = 5). When DSI was stimulated 20 sec before a VSI-B spike, subsequent EPSPs were decreased in amplitude (plot C, filled circles; n = 4). The time course of recovery was slightly slower when the DSI stimulus was timed to produce only a decrement. In all cases, the EPSPs recovered to the original size after the decrement. Each plot shows a mean value (mean ± SEM) of pooled data obtained by repeated trials made in three preparations.
The DSI neuromodulatory action directly affected synaptic currents
We tested whether changes in postsynaptic input resistance in the VFN could account for the biphasic-bidirectional effect of DSI on VSI-B-evoked synaptic potentials. To mediate such changes, the postsynaptic input resistance would need to be higher during the period of synaptic enhancement and lower during the decrement; however, the input resistance of the VFN (five VFNs in three preparations) showed a transient decrease during and after (<10 sec) a DSI spike train, which was followed by a prolonged increase lasting from 10 sec to >2 min after the end of DSI stimulation (Fig. 5). Thus, although DSI stimulation caused a biphasic-bidirectional change in VFN input resistance, it was in a direction opposite to what would be needed to cause the observed change in synaptic strength. This effect on the postsynaptic membrane properties might function as a filter to gate synaptic inputs to the VFN (see Discussion).
Figure 5.

DSI stimulation produced a biphasic-bidirectional change in the input resistance of a VFN that was opposite to the observed change in synaptic strength. Two-electrode voltage clamp was performed on a VFN, which was held at -50 mV. Changes in the input resistance of the VFN were measured by recording membrane current responses evoked by repetitive command voltage pulses (amplitude, -40 mV, 1 sec duration) given at 0.5 Hz. Reciprocal values for the membrane current responses were normalized to the average before DSI stimulation and plotted against time after the DSI stimulation. The plot was fit by a double-exponential curve, f(t) = 1 + A × e-t/τ1 - B × e-t/τ2, where t is the time after the end of DSI stimulus train, A = 0.25, τ1 = 62.5 sec, B = 0.4, and τ2 = 15.9 sec.
The lack of involvement of postsynaptic membrane properties was further confirmed by voltage clamping the postsynaptic cell and recording the synaptic currents evoked by VSI-B (Fig. 6A). We found that DSI stimulation caused a biphasic-bidirectional change in synaptic currents evoked by VSI-B (Fig. 6B,C). As was observed with synaptic potentials, when VSI-B was stimulated to fire an action potential within 15 sec of a DSI spike train, the amplitude of the EPSC that it evoked in VFN increased (Fig. 6B1), whereas it decreased if VSI-B was stimulated 18 sec to 2 min after DSI (Fig. 6B2). The biphasic-bidirectional change in EPSC amplitude was very similar to that of EPSP amplitude, being well described by a double-exponential function (Fig. 6C). These results indicate that the DSI neuromodulatory action is a direct effect on the strength of the synapse, which can be caused either by altering presynaptic release from VSI-B or by changing the sensitivity of the postsynaptic receptors on the VFN.
Figure 6.
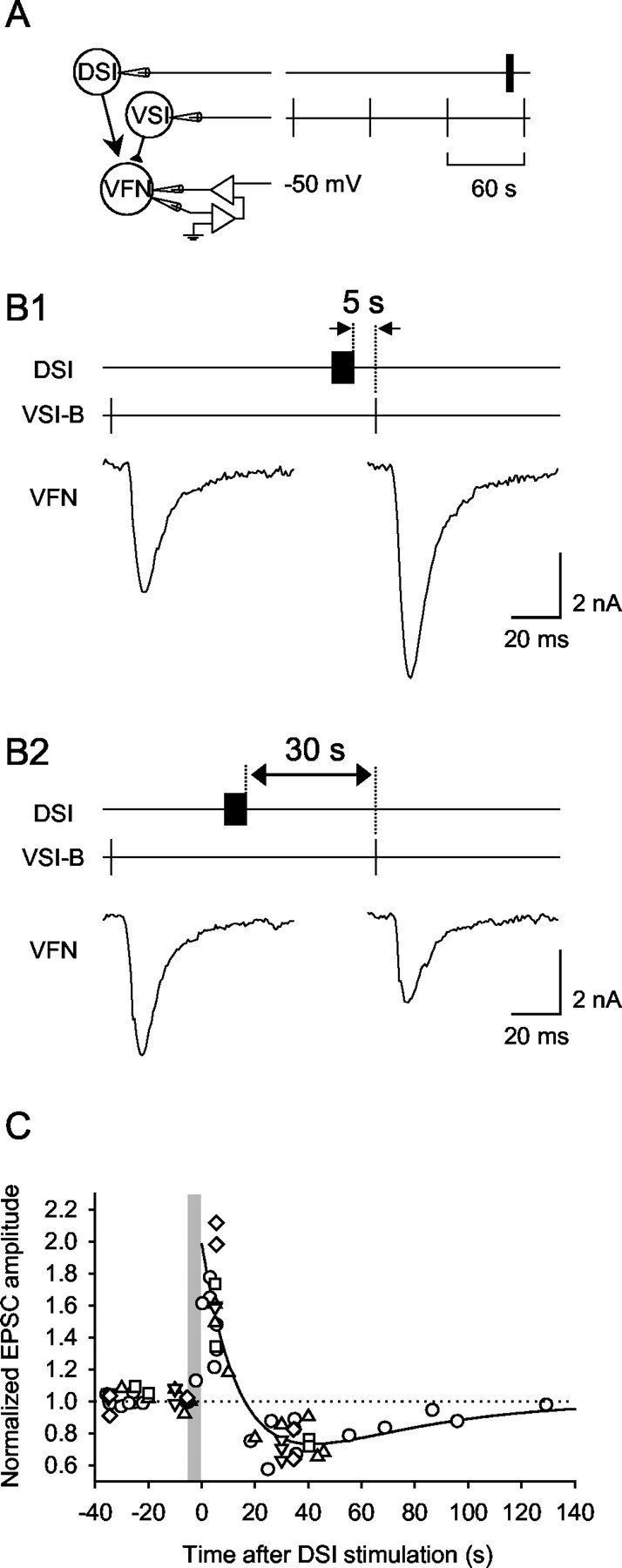
Synaptic currents evoked by VSI-B were modulated in a timing-dependent manner by DSI. A, Schematic diagram showing the experimental configuration. The membrane potential of the VFN soma was held at -50 mV by two-electrode voltage clamp. Intracellular electrodes were used to elicit action potentials in DSI and VSI-B. VSI-B was stimulated to fire an action potential every 60 sec. A DSI spike train (10 Hz, 5 sec) was elicited at varying times relative to VSI-B spikes. Changes in EPSC amplitude were normalized to the average of the three EPSCs before the DSI spike train. B, The effect of DSI on VSI-B-evoked EPSCs in VFN depended on the timing of the DSI spike train relative to VSI-B spikes. When DSI stimulation preceded VSI-B by 5 sec, then the amplitude of the following EPSC increased (B1). When the DSI spike train was evoked 30 sec before VSI-B spiking, then the amplitude of the EPSC decreased (B2). C, The normalized EPSC amplitudes plotted against time after the DSI stimulation show a biphasic-bidirectional change (n = 5). Each symbol represents a different animal. The plot was well fit by a double-exponential function: f(t) = 1 + A × e-t/τ1 - e-t/τ2, where A = 1.98, τ1 = 15.6 sec, and τ2 = 45.9 sec.
Serotonin mediates the DSI neuromodulatory actions
The DSIs are serotonergic (Katz et al., 1994; McClellan et al., 1994) and use 5-HT for both neurotransmission and neuromodulation of C2 synaptic strength (Katz et al., 1994; Katz and Frost, 1995a). We tested whether 5-HT mediates the biphasic-bidirectional effect of DSI stimulation on VSI-B synaptic strength.
Bath application of 5-HT had somewhat complicated effects on VSI-B synaptic strength (Fig. 7). In 13 of 14 preparations tested, bath-applied 5-HT (10-100 μm) did not mimic the biphasic-bidirectional action of DSI stimulation on VSI-B synaptic strength. In 11 of the 14 preparations, 5-HT caused only a sustained increase in the amplitude of VSI-B-evoked EPSPs or EPSCs recorded in VFN (Fig. 7A1). In 2 of the 14 preparations, 5-HT caused a sustained decrement in VSI-B synaptic strength (Fig. 7A2). In one preparation, bath-applied 5-HT (5.5 min) caused a somewhat biphasic-bidirectional change in VSI-B synaptic potentials, with an initial transient increase lasting for ∼2 min followed by a sustained decrease (Fig. 7A3).
Figure 7.
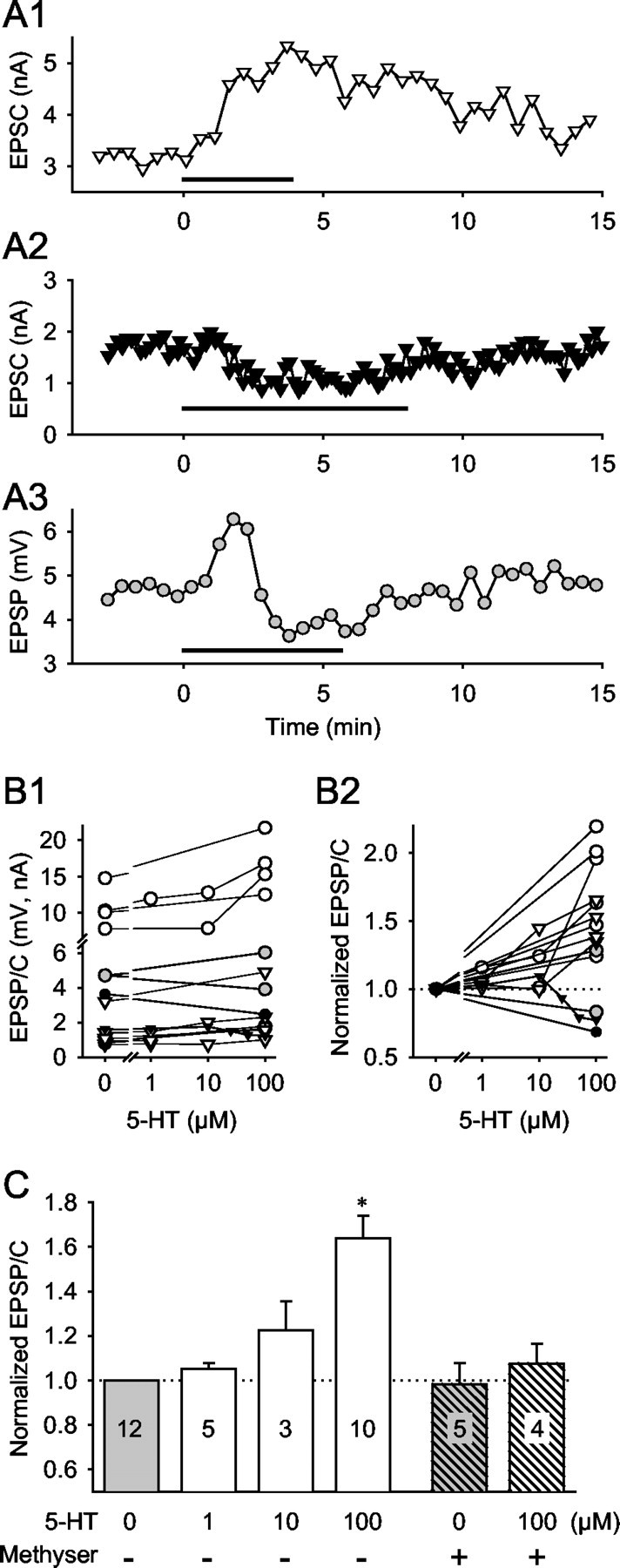
Effects of bath-applied serotonin on VSI-B synaptic strength. A, 5-HT (100 μm) caused three types of changes in the amplitude of the VSI-B-evoked EPSPs or EPSCs: a sustained increase (A1), a sustained decrease (A2), and a biphasic-bidirectional change with initial transient increase followed by a decrease (A3). The horizontal bar in each graph shows the duration of 5-HT bath application. B, Dose-responses to 5-HT for individual animals. The amplitudes (B1) and the normalized amplitudes (B2) of VSI-B-evoked EPSPs and EPSCs are plotted as a function of 5-HT concentration. Symbols represent the averaged amplitude of 10 EPSPs (millivolts, circles) or EPSCs (nanoamperes, triangles) before the onset of 5-HT application (0 μm) and in the presence of 5-HT (1, 10, or 100 μm). Open symbols indicate 5-HT-induced enhancement (n = 11), whereas filled symbols indicate the depression (n = 2). Gray circles represent a biphasic-bidirectional change (n = 1). C, Average dose-response relationship of the 5-HT-induced enhancement of VSI-B synaptic strength and the effect of methysergide. The presence or absence of methysergide (50 μm) is shown by + or -, respectively. 5-HT (100 μm) caused a significant increase in the amplitude of VSI-B-evoked EPSP and EPSCs (+64.1 ± 10.0%; n = 11; p < 0.05; paired t test). Methysergide (50 μm) by itself did not have a significant effect on the amplitude of VSI-B-evoked EPSP and EPSCs (p = 0.99; paired t test). In the presence of methysergide, 100 μm 5-HT caused no significant change (p = 0.46; paired t test).
The effect of 5-HT was dose dependent, but the direction of change was not (Fig. 7B,C). In four of five preparations, increasing the concentration of 5-HT increased the magnitude of the synaptic enhancement. In one preparation (Fig. 7B1,B2, filled triangles), 10 μm 5-HT caused a slight increase in synaptic strength, whereas higher concentrations caused a decrease in the amplitude of VSI-B-evoked EPSCs. The average responses of preparations that exhibited only enhancement are shown in Figure 7C. There was a trend toward increased synaptic strength with increased 5-HT concentration; however, only the effect of 100 μm 5-HT was significantly greater than control (p < 0.05; paired t test).
Although 5-HT generally did not mimic both phases of the DSI neuromodulatory action, it occluded both of them (Fig. 8). In normal saline, DSI stimulation evoked either enhancement or decrement depending on the timing of its spikes relative to the VSI-B spikes (n = 4; p < 0.05; paired t tests). In the presence of 100 μm 5-HT, DSI stimulation caused no significant change in VSI-B-evoked EPSP and EPSCs (n = 4; p = 0.18 and 0.22).
Figure 8.

Occlusion of DSI modulatory actions by bath-applied 5-HT. A, Superimposed waveforms of EPSPs evoked by VSI-B alone (solid traces) and those evoked 5 sec (dotted traces) and 35 sec (dashed traces) after DSI stimulation are shown. In normal saline (left), stimulation of DSI 5 sec before VSI-B increased the amplitude of the EPSP; stimulating DSI 35 sec before VSI-B decreased the EPSP amplitude. In 100 μm 5-HT (middle), the amplitude of the EPSP evoked by VSI-B alone increased; however, DSI stimulation caused little change in VSI-B-evoked EPSPs. The amplitude of the EPSP recovered during washout of 5-HT, and DSI-evoked enhancement-decrement was again observed (right). B, Bar graphs showing mean values of normalized amplitudes of the EPSPs evoked before DSI stimulation (gray), 0-10 sec after DSI stimulation (white), and 20-60 sec after DSI stimulation (black). In normal saline, there was a significant enhancement (+50.5 ± 16.5%) of VSI-B-evoked synaptic EPSPs when VSI-B was stimulated within 10 sec of a DSI train (n = 4; p < 0.05; paired t test) and a significant decrement (-11.2 ± 2.1%) when VSI-B was stimulated 20-60 sec after DSI (n = 4; p < 0.05; paired t test). In the presence of 100 μm 5-HT, there was no significant change in VSI-B-evoked EPSPs after DSI stimulation (p = 0.18 and 0.22; paired t test).
To further test whether 5-HT mediates both the enhancement and decrement of VSI-B synaptic strength, we tested the effect of the serotonin receptor antagonist methysergide on the DSI neuromodulatory actions. Bath application of methysergide (50 μm) had no effect on VSI-B-evoked EPSPs or EPSCs when applied alone (Figs. 7C, 9A), but it blocked the effect of 100 μm 5-HT (Fig. 7C). Methysergide reduced the extent of both the enhancement and decrement caused by DSI stimulation in all five preparations examined, such that DSI no longer caused a significant change in VSI-B synaptic strength (Fig. 9B). These results suggest that both phases of the DSI action were mediated by 5-HT.
Figure 9.

Blockade of DSI neuromodulatory actions by methysergide. A, Superimposed waveforms of the EPSPs evoked by VSI-B alone before DSI stimulation (solid traces) and those evoked 5 sec (dotted traces) and 35 sec (dashed traces) after DSI stimulation are shown. In normal saline (left), DSI stimulation caused either an enhancement or a decrement of VSI-B-evoked EPSPs depending on the timing of its firing relative to that of VSI-B. In the presence of methysergide (50 μm; middle), DSI stimulation caused little change in EPSP amplitude. The DSI-evoked enhancement-decrement did not recover after washing out for >1 hr (right). B, Average responses show that methysergide blocked both the enhancement and depression evoked by DSI stimulation. In normal saline, stimulation of DSI 10 sec before VSI-B caused a significant increase in synaptic strength (+48.2 ± 13.9%; n = 4; p < 0.05; paired t test) and a significant decrement (-24.6 ± 8.6%) when stimulated 20-60 sec before VSI-B (n = 4; p < 0.05; paired t test). In the presence of 50 μm methysergide, DSI stimulation had no significant effect on VSI-B synaptic strength in either time period (p = 0.23 and 0.18; paired t test).
The heterosynaptic DSI-evoked decrement is offset by homosynaptic facilitation
The preceding experiments focused on the effects of DSI stimulation on individual VSI-B-evoked EPSP and EPSCs; however, during the swim motor pattern, VSI-B fires action potentials at 5-20 Hz in each burst (Fig. 1B). Therefore, to better understand the effect that DSI would have on the output of VSI-B during a swim motor pattern, we examined the neuromodulatory actions of DSI on trains of EPSCs evoked by VSI-B (Fig. 10).
Figure 10.
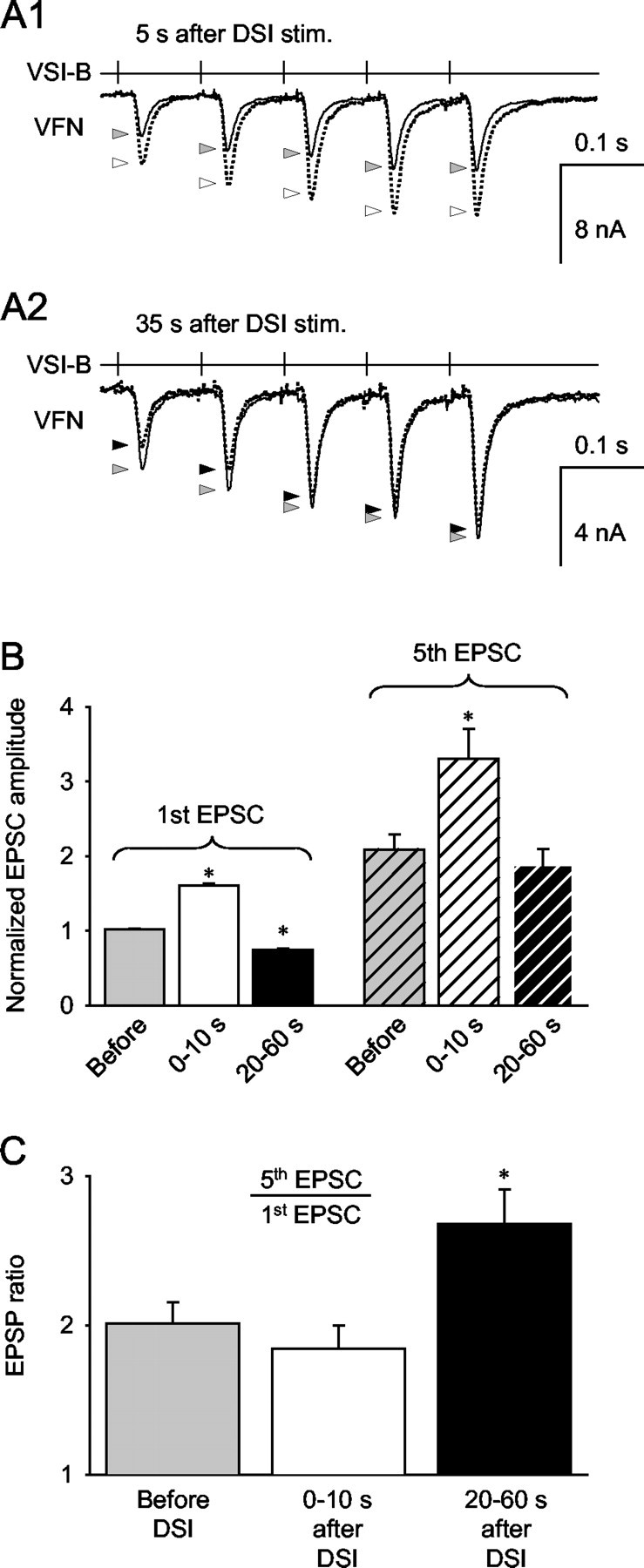
DSI modulatory actions interact with homosynaptic facilitation. A, Trains of five VSI-B-evoked EPSCs (10 Hz) recorded from VFN before DSI stimulation (solid lines) and 5 sec (A1) or 35 sec (A2) after DSI stimulation (dotted lines). The gray arrowheads point to the peak amplitudes of the unmodulated EPSCs. The white and black arrowheads indicate the peak of the EPSCs 5 and 35 sec after DSI stimulation, respectively. When VSI-B was stimulated 5 sec after DSI (A1), each EPSC increased in amplitude; however, when VSI-B was stimulated 35 sec after DSI (A2), there was a substantial decrease in the first two EPSPs and very little change in subsequent EPSPs. B, Averages of normalized responses show that the amplitude of the first EPSC (left) was significantly increased when VSI-B was stimulated within 10 sec of DSI (n = 5; p < 0.05; paired t test) and significantly decreased when stimulated 20-60 sec after DSI (n = 5; p < 0.05; paired t test). The fifth EPSC (right) of the train was larger than the first (left) because of to homosynaptic facilitation. When VSI-B was stimulated within 10 sec after a DSI spike train, the amplitude of the fifth EPSC was significantly larger than before DSI (n = 4; p < 0.05; paired t test); however, when VSI-B was stimulated 20-60 sec after DSI, the amplitude of the fifth EPSC in the train was not significantly different from the control fifth EPSC (n = 4; p = 0.35; paired t test). C, DSI-evoked changes in the ratio of fifth EPSC to first EPSC, an index of homosynaptic frequency facilitation of the VSI-B synapse. When VSI-B was stimulated 0-10 sec after DSI (white bar), there was no significant change in the ratio (n = 4; p = 0.06; paired t test); however, when VSI-B was stimulated 20-60 sec after DSI stimulation (black bar), there was a significant increase in the ratio (n = 4; p < 0.05; paired t test).
We found that when VSI-B was stimulated repeatedly to fire five action potentials at 10 Hz, the EPSCs recorded in VFN exhibited short-term homosynaptic facilitation (Fig. 10A1,A2, gray arrows). A DSI spike train that preceded the VSI-B train by 5 sec increased the amplitude of all five EPSCs (Fig. 10A1, white arrows). In contrast, when the DSI spike train preceded the VSI-B train by 35 sec, only the first few EPSCs evoked by VSI-B showed a substantial amplitude decrement; the fifth EPSC of the train was not strongly affected by the DSI stimulus (Fig. 10A2, black arrows). On average, the first EPSC of the train behaved in a similar manner to the individually evoked EPSCs: it increased in amplitude when the VSI-B spike train began within 10 sec of the DSI stimulus and decreased in amplitude when it began 20-60 sec after DSI (Fig. 10B, left). The fifth EPSC behaved differently. Although it also significantly increased in size when VSI-B was stimulated within 10 sec of DSI, its amplitude was not significantly changed by DSI stimuli that occurred 20-60 sec before the beginning of the VSI-B train (Fig. 10B, right). Thus, there was a differential effect of DSI stimulation on the first and fifth EPSCs evoked by VSI-B.
The differential effect of DSI stimulation on the first and fifth VSI-B EPSCs changed the extent of frequency-dependent homosynaptic facilitation (Fig. 10C). The EPSCs exhibited significantly more homosynaptic facilitation when VSI-B was stimulated 20-60 sec after DSI. Without DSI stimulation, the VSI-B-evoked EPSCs exhibited frequency-dependent facilitation, with the ratio of the fifth EPSC to the first EPSC being 2.17 ± 0.12 (n = 5). Stimulation of VSI-B within 10 sec of DSI produced little change in the ratio (2.00 ± 0.16; n = 5). In contrast, when the VSI-B spike train occurred 20-60 sec after the DSI stimulus, the ratio increased to 2.94 ± 0.14 (n = 5). Thus, the DSI-elicited decrement in VSI-B synaptic strength resulted in an apparent increase in VSI-B homosynaptic facilitation.
DSI dynamically shapes VSI-B synaptic strength during a swim-like bursting pattern
Our results showed that the effects of DSI on VSI-B synaptic strength depend on both the relative timing of VSI-B spikes with respect to DSI bursts and the firing frequency of VSI-B. During the swim motor pattern, VSI-B fires bursts of action potentials in rhythmic alternation with DSIs (Fig. 1B). Because a swim motor pattern consists of three to eight cycles with a periodicity of 7-12 sec, the effects of DSI might summate across cycles. To determine what effect DSI might have on VSI-B synaptic strength during the swim motor pattern, we measured VSI-B synaptic strength when VSI-B was stimulated to fire repeated bursts either alone or in alternation with DSI bursts, mimicking the motor pattern (Fig. 11A).
When VSI-B spike trains were evoked repeatedly at a constant interval without stimulating DSI, the amplitude of the initial EPSP of each burst declined gradually (Fig. 11B1, filled circles), indicating that the VSI-B synapse displays use-dependent synaptic depression. When DSI spike trains were interposed between the VSI-B bursts, however, the amplitude of the initial EPSP in each burst changed in a biphasic-bidirectional manner (Fig. 11B1, open circles). The amplitude became maximal after the initial DSI train and then decreased to a value close to the control amplitude by the seventh burst cycle. After termination of the 6th DSI train, the mean amplitude of the initial EPSP decreased to a value less than the comparable control amplitude (the 8th cycle in Fig. 11B1) and then recovered to the control amplitude by the 10th cycle. Thus, during a swim motor pattern, DSI activity would likely enhance the synaptic output of the initial VSI-B spikes in the motor pattern and then act to suppress the effects of individual VSI-B spikes that might occur after the cessation of the swim motor pattern.
The effect of DSI bursts on the total summated EPSP was different from the effect on the initial EPSP of the train (Fig. 11B2). As was true of the first EPSP in the train, the total summated EPSP also gradually decreased when VSI-B alone was stimulated repeatedly (Fig. 11B2, filled circles). When stimulated in alternation with DSI, however, the amplitude of the summated EPSPs showed an initial large increase in the first burst cycle, followed by a sustained lower level of enhancement (Fig. 11B2, open circles). The summated EPSP amplitude remained significantly larger than the control value throughout all of the artificial swim cycles (p < 0.05; paired t test). After the termination of the sixth DSI train, the amplitudes of the summated EPSPs returned to the control level without decrement (the eighth burst cycle). This difference between the effects on single EPSPs and summated bursts may have functional significance for controlling VSI-B synaptic strength during and shortly after a swim bout.
Discussion
Spike timing-dependent neuromodulatory actions
This study determined that the neuromodulatory actions of the serotonergic DSIs on the synaptic output of VSI-B are dependent on the timing of spikes in these two neurons. The spike timing dependence of DSI neuromodulatory actions resembles spike timing-dependent plasticity (STDP) described at cortical synapses that can lead to a bidirectional change in synaptic strength (Markram et al., 1997; Abbott and Nelson, 2000; Bi and Poo, 2001; Froemke and Dan, 2002); however, unlike STDP, the relative timing of the presynaptic and postsynaptic spikes is not important for the synaptic modulation reported in this study. Instead, the timing of heterosynaptic input (DSI) relative to the activation of the presynaptic neuron (VSI-B) determines the direction of change. Furthermore, STDP is a mechanism for long-term potentiation or long-term depression that can last for hours. In contrast, the synaptic modifications that we observed here were rather transient (< 15 sec for synaptic enhancement and <2 min for synaptic decrement) and resulted in a temporally biphasic-bidirectional change in VSI-B synaptic strength. Such neuromodulatory actions can provide temporal information as well as magnitude information relevant for the operation of a motor pattern generator circuit.
Biphasic-bidirectional neuromodulatory actions
There are few reports of temporal changes in neuromodulatory actions. This may be a consequence of the use of exogenous application of neuromodulatory substances rather than direct stimulation of modulatory neurons. Indeed, we found that exogenous application of 5-HT generally failed to mimic the biphasic nature of the DSI actions. Therefore, if this study had relied on 5-HT application alone, we would have reached the false conclusion that 5-HT only caused either a sustained synaptic enhancement or depression. The discrepancy may be attributable to the mode of 5-HT application: a slow, sustained increase in 5-HT concentration by superfusion may evoke different effects than a brief activation of the serotonergic neuron DSI. Differences in the concentration and time course of applied 5-HT can produce opposite modulatory effects in crayfish (Teshiba et al., 2001).
The biphasic-bidirectional neuromodulation could be caused by its action onto two different types of receptors (Mercer et al., 1991; Seamans et al., 2001). Multiphasic synaptic potentials, commonly seen in molluskan nervous systems (Shimahara and Tauc, 1975), can be mediated through actions at multiple receptor types. For example, in Tritonia the fast component of DSI-evoked synaptic potentials in DFN-A appears to be mediated by a 5-HT-gated conductance, whereas the slow component is mediated by G-protein-coupled receptors (Clemens and Katz, 2001). The types of 5-HT receptors that mediate the DSI actions onto VSI-B are currently unknown.
Differences in the distribution of the two receptor types could also underlie the biphasic nature of the DSI neuromodulatory actions (Skydsgaard and Hounsgaard, 1996; Burrell et al., 2001). For example, one type of receptor may be located near the DSI terminal, whereas the other type may be confined to sites that are distant from DSI synapses. In such a configuration, 5-HT released from DSIs would reach one site faster than the other and thus produce a biphasic response. When 5-HT was bath applied, these receptors could be activated at the same time, and two opposite responses could summate to produce a sustained response in one direction.
The biphasic-bidirectional response could result from the activation of multiple second messenger cascades. For example, in Aplysia, the neuropeptide FMRFamide acts on one receptor type to produce a biphasic-bidirectional change in membrane currents of the LFS neuron through a parallel activation of Na+ and K+ channels with different time courses (Belkin and Abrams, 1993). Similarly, glutamate evokes a biphasic-bidirectional change in MAP2 phosphorylation, presumably because of the kinetics of second messenger pathways (Quinlan and Halpain, 1996). In prefrontal cortex pyramidal neurons, 5-HT produces a bidirectional, although not biphasic, change in the GABAA receptor function depending on the background activity level of protein kinase A (Cai et al., 2002). 5-HT can also have effects on second messenger systems with different time courses leading to multiple phases of memory formation (Mauelshagen et al., 1998; Sutton and Carew, 2000). Further investigations are needed to elucidate the cellular mechanisms underlying the DSI spike timing-dependent neuromodulation of VSI-B synaptic strength.
Presynaptic and postsynaptic mechanisms may act as a selectivity filter
The results of voltage-clamp experiments suggest that the DSI modulation of VSI-B synaptic strength is not caused by changes in postsynaptic membrane properties (Figs. 5, 6). Furthermore, the increased extent of frequency facilitation during the DSI-evoked depression phase (Fig. 10) is consistent with a presynaptic locus, because low-output synapses tend to facilitate more than high-output synapses (Zucker and Regehr, 2002). A presynaptic site of action is also consistent with previous results that showed DSI increasing neurotransmitter release from the other CPG neuron, C2 (Katz and Frost, 1995b; Clemens and Katz, 2003); however, these results do not rule out the possibility of changes in the sensitivity of the postsynaptic receptors in VFN. A similar change in sensitivity of AMPA receptors has been shown to play a role in modulating inspiratory drive to hypoglossal motor neurons (Bocchiaro et al., 2003).
The voltage-clamp experiments also revealed that DSI evoked a biphasic-bidirectional change in the membrane resistance of VFN (Fig. 5). The change was in the opposite direction from what would be expected to cause an initial enhancement and subsequent decrement of VSI-B synaptic strength. The initial decrease in the postsynaptic resistance would shunt all synaptic inputs, whereas the later increase would enhance all synaptic inputs to VFN. Thus, the DSI-evoked changes in VFN input resistance counteract the changes in VSI-B synaptic strength and thus may function as a selectivity filter to enhance the contrast of VSI-B synapses with other synaptic inputs to VFN during the swim motor program.
Neuromodulatory signals can convey temporal information that affects CPG function
During the swim motor program, the pattern of changes in the VSI-B synaptic strength is probably determined by the temporal summation of the repetitive DSI neuromodulatory actions. Because VSI-B and DSIs fire bursts in alternation with a periodicity of 7-12 sec (Fig. 1C), the time window of 15 sec for the enhancement of VSI-B synaptic strength appears to be well tuned for augmenting the ventral phase of the swim motor program. As would be expected, the amplitude of the summated EPSPs in the artificial swim program showed a sustained enhancement when DSI bursts were interposed (Fig. 11B2).
The artificial swim motor program also revealed that there was the initial large enhancement of VSI-B synaptic strength after the first DSI burst (Fig. 11B1,B2). This may contribute to the high frequency of VFN spiking at the beginning of the swim program (Fig. 1C) (Hume and Getting, 1982b) and thus would be important for launching the animal during the initial strong ventral flexion. On the basis of simple summation of the DSI actions, it would be expected that a greater enhancement should occur during the third swim cycle (10-15 sec after the first DSI burst) in the artificial swim program because it would be within the enhancement period of the two preceding DSI bursts before the depression phase (compare Fig. 3A); however, we found that the enhanced EPSPs already started to decline by the third cycle, suggesting that the neuromodulatory actions do not exhibit linear summation. Further physiological and modeling studies are needed to fully understand the mechanisms underlying this biphasic-bidirectional modulation and how it contributes to motor pattern generation.
Shortly after the termination of the DSI bursts in the artificial swim program, the initial EPSP evoked by a VSI-B spike train was depressed (Fig. 11B1). The initial EPSP is equivalent to an EPSP evoked by a solitary spike because it exhibits neither homosynaptic facilitation nor temporal summation. Thus, there may be a suppression of spurious EPSPs evoked by VSI-B action potentials after the swim motor program. In addition to its role in the swim motor pattern, VSI-B stimulation decreases ciliary locomotion (Popescu and Frost, 2002). Thus, the suppression of VSI-B synapses after a swim would prevent this neuron from inhibiting crawling after the swim, a time when crawling is normally enhanced (Popescu and Frost, 2002).
In summary, the spike timing-dependent neuromodulation evoked by serotonergic interneurons may provide temporal information by causing a biphasic-bidirectional change in the strength of the synaptic output from the CPG circuit. Such neuromodulation may play a role in optimizing the synaptic strength for the patterned motor output, which would be relevant for controlling motor behavior. Such a mechanism might be common, although currently unexplored, in other modulatory systems.
Footnotes
This work was supported by National Institutes of Health Grant NS 35371 and funds from Georgia State University. We thank James Newcomb for valuable comments on this manuscript, Donald Edwards for many helpful discussions, and William Frost for advice on identifying the VSI-B neuron.
Correspondence should be addressed to Paul S. Katz, Department of Biology, Georgia State University, MSC 8L0389, 33 Gilmer Street Southeast, Unit 8, Atlanta, GA 30303-3088. E-mail: pkatz@gsu.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310745-11$15.00/0
References
- Abbott LF, Nelson SB ( 2000) Synaptic plasticity: taming the beast. Nat Neurosci [Suppl] 3: 1178-1183. [DOI] [PubMed] [Google Scholar]
- Belkin KJ, Abrams TW ( 1993) FMRFamide produces biphasic modulation of the LFS motor neurons in the neural circuit of the siphon withdrawal reflex of Aplysia by activating Na+ and K+ currents. J Neurosci 13: 5139-5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MR ( 2000) The concept of long term potentiation of transmission at synapses. Prog Neurobiol 60: 109-137. [DOI] [PubMed] [Google Scholar]
- Bi GQ, Poo MM ( 2001) Synaptic modification by correlated activity: Hebb's postulate revisited. Annu Rev Neurosci 24: 139-166. [DOI] [PubMed] [Google Scholar]
- Bocchiaro CM, Saywell SA, Feldman JL ( 2003) Dynamic modulation of inspiratory drive currents by protein kinase A and protein phosphatases in functionally active motoneurons. J Neurosci 23: 1099-1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell BD, Sahley CL, Muller KJ ( 2001) Non-associative learning and serotonin induce similar bi-directional changes in excitability of a neuron critical for learning in the medicinal leech. J Neurosci 21: 1401-1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne JH, Kandel ER ( 1996) Presynaptic facilitation revisited: state and time dependence. J Neurosci 16: 425-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Flores-Hernandez J, Feng J, Yan Z ( 2002) Activity-dependent bidirectional regulation of GABAA receptor channels by the 5-HT4 receptor-mediated signalling in rat prefrontal cortical pyramidal neurons. J Physiol (Lond) 540: 743-759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese RL ( 1998) Cellular, synaptic, network, and modulatory mechanisms involved in rhythm generation. Curr Opin Neurobiol 8: 710-717. [DOI] [PubMed] [Google Scholar]
- Clemens S, Katz PS ( 2001) Identified serotonergic neurons in the Tritonia swim CPG activate both ionotropic and metabotropic receptors. J Neurophysiol 85: 476-479. [DOI] [PubMed] [Google Scholar]
- Clemens S, Katz PS ( 2003) G Protein signaling in a neuronal network is necessary for rhythmic motor pattern production. J Neurophysiol 89: 762-772. [DOI] [PubMed] [Google Scholar]
- Daniel H, Levenes C, Crépel F ( 1998) Cellular mechanisms of cerebellar LTD. Trends Neurosci 21: 401-407. [DOI] [PubMed] [Google Scholar]
- Fickbohm DJ, Katz PS ( 2000) Paradoxical actions of the serotonin precursor 5-hydroxytryptophan on the activity of identified serotonergic neurons in a simple motor circuit. J Neurosci 20: 1622-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froemke RC, Dan Y ( 2002) Spike-timing-dependent synaptic modification induced by natural spike trains. Nature 416: 433-438. [DOI] [PubMed] [Google Scholar]
- Frost WN, Katz PS ( 1996) Single neuron control over a complex motor program. Proc Natl Acad Sci USA 93: 422-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost WN, Hoppe TA, Wang J, Tian LM ( 2001) Swim initiation neurons in Tritonia diomedea. Am Zool 41: 952-961. [Google Scholar]
- Getting PA ( 1981) Mechanisms of pattern generation underlying swimming in Tritonia. I. Neuronal network formed by monosynaptic connections. J Neurophysiol 46: 65-79. [DOI] [PubMed] [Google Scholar]
- Getting PA ( 1983) Mechanisms of pattern generation underlying swimming in Tritonia. III. Intrinsic and synaptic mechanisms for delayed excitation. J Neurophysiol 49: 1036-1050. [DOI] [PubMed] [Google Scholar]
- Getting PA ( 1989) A network oscillator underlying swimming in Tritonia. In: Neuronal and cellular oscillators (Jacklet JW, ed), pp 215-236. New York: Marcel Dekker.
- Getting PA, Lennard PR, Hume RI ( 1980) Central pattern generator mediating swimming in Tritonia. I. Identification and synaptic interactions. J Neurophysiol 44: 151-164. [DOI] [PubMed] [Google Scholar]
- Gu Q ( 2002) Neuromodulatory transmitter systems in the cortex and their role in cortical plasticity. Neuroscience 111: 815-835. [DOI] [PubMed] [Google Scholar]
- Hammer M ( 1997) The neural basis of associative reward learning in honeybees. Trends Neurosci 20: 245-252. [DOI] [PubMed] [Google Scholar]
- Hasselmo ME, Linster C ( 1999) Neuromodulation and memory function. In: Beyond neurotransmission: neuromodulation and its importance for information processing (Katz PS, ed), pp 318-348. Oxford: Oxford UP.
- Hume RI, Getting PA ( 1982a) Motor organization of Tritonia swimming. II. Synaptic drive to flexion neurons from premotor interneurons. J Neurophysiol 47: 75-90. [DOI] [PubMed] [Google Scholar]
- Hume RI, Getting PA ( 1982b) Motor organization of Tritonia swimming. III. Contribution of intrinsic membrane properties to flexion neuron burst formation. J Neurophysiol 47: 91-102. [DOI] [PubMed] [Google Scholar]
- Katz PS ( 1999) Beyond neurotransmission: neuromodulation and its importance for information processing. Oxford: Oxford UP.
- Katz PS, Frost WN ( 1995a) Intrinsic neuromodulation in the Tritonia swim CPG: serotonin mediates both neuromodulation and neurotransmission by the dorsal swim interneurons. J Neurophysiol 74: 2281-2294. [DOI] [PubMed] [Google Scholar]
- Katz PS, Frost WN ( 1995b) Intrinsic neuromodulation in the Tritonia swim CPG: the serotonergic dorsal swim interneurons act presynaptically to enhance transmitter release from interneuron C2. J Neurosci 15: 6035-6045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz PS, Frost WN ( 1997) Removal of spike frequency adaptation via neuromodulation intrinsic to the Tritonia escape swim central pattern generator. J Neurosci 17: 7703-7713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz PS, Getting PA, Frost WN ( 1994) Dynamic neuromodulation of synaptic strength intrinsic to a central pattern generator circuit. Nature 367: 729-731. [DOI] [PubMed] [Google Scholar]
- Kiehn O, Katz PS ( 1999) Making circuits dance: neuromodulation of motor systems. In: Beyond neurotransmission: neuromodulation and its importance for information processing (Katz PS, ed), pp 275-317. Oxford: Oxford UP.
- Lynn-Bullock CP, Clemens S, Sakurai A, Katz PS ( 2002) Functional anatomy of the escape swim CPG in Tritonia. Soc Neurosci Abstr 28: 67.25. [Google Scholar]
- Malenka RC, Nicoll RA ( 1999) Long-term potentiation—a decade of progress? Science 285: 1870-1874. [DOI] [PubMed] [Google Scholar]
- Marder E, Bucher D ( 2001) Central pattern generators and the control of rhythmic movements. Curr Biol 11: R986-R996. [DOI] [PubMed] [Google Scholar]
- Marder E, Calabrese RL ( 1996) Principles of rhythmic motor pattern generation. Physiol Rev 76: 687-717. [DOI] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Sakmann B ( 1997) Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275: 213-215. [DOI] [PubMed] [Google Scholar]
- Mauelshagen J, Sherff CM, Carew TJ ( 1998) Differential induction of long-term synaptic facilitation by spaced and massed applications of serotonin at sensory neuron synapses of Aplysia californica Learn Mem 5: 246-256. [PMC free article] [PubMed] [Google Scholar]
- McClellan AD, Brown GD, Getting PA ( 1994) Modulation of swimming in Tritonia: excitatory and inhibitory effects of serotonin. J Comp Physiol [A] 174: 257-266. [DOI] [PubMed] [Google Scholar]
- Mercer AR ( 1999) Changing the way we perceive things: sensory systems modulation. In: Beyond neurotransmission: neuromodulation and its importance for information processing (Katz PS, ed), pp 198-240. Oxford: Oxford UP.
- Mercer AR, Emptage NJ, Carew TJ ( 1991) Pharmacological dissociation of modulatory effects of serotonin in Aplysia sensory neurons. Science 254: 1811-1813. [DOI] [PubMed] [Google Scholar]
- Nagai T, Kim DJ, Delay RJ, Roper SD ( 1996) Neuromodulation of transduction and signal processing in the end organs of taste. Chem Senses 21: 353-365. [DOI] [PubMed] [Google Scholar]
- Pasztor VM ( 1989) Modulation of sensitivity in invertebrate sensory receptors. Semin Neurosci 1: 5-14. [Google Scholar]
- Pearson KG ( 1993) Common principles of motor control in vertebrates and invertebrates. Annu Rev Neurosci 16: 265-297. [DOI] [PubMed] [Google Scholar]
- Popescu IR, Frost WN ( 2002) Highly dissimilar behaviors mediated by a multifunctional network in the marine mollusk Tritonia diomedea. J Neurosci 22: 1985-1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powis, DA and Bunn, SJ (eds) ( 1995) Neurotransmitter release and its modulation: biochemical mechanisms, physiological function and clinical relevance. Cambridge, UK: Cambridge UP.
- Quinlan EM, Halpain S ( 1996) Postsynaptic mechanisms for bidirectional control of MAP2 phosphorylation by glutamate receptors. Neuron 16: 357-368. [DOI] [PubMed] [Google Scholar]
- Sakurai A, Katz PS ( 2002) Serotonergic interneurons in Tritonia evoke a biphasic neuromodulatory action. Soc Neurosci Abstr 28: 270.13. [Google Scholar]
- Seamans JK, Gorelova N, Durstewitz D, Yang CR ( 2001) Bidirectional dopamine modulation of GABAergic inhibition in prefrontal cortical pyramidal neurons. J Neurosci 21: 3628-3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimahara T, Tauc L ( 1975) Multiple interneuronal effects to the giant cells in Aplysia. J Physiol (Lond) 247: 299-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöström PJ, Nelson SB ( 2002) Spike timing, calcium signals and synaptic plasticity. Curr Opin Neurobiol 12: 305-314. [DOI] [PubMed] [Google Scholar]
- Skydsgaard M, Hounsgaard J ( 1996) Multiple actions of iontophoretically applied serotonin on motorneurones in the turtle spinal cord in vitro. Acta Physiol Scand 158: 301-310. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Carew TJ ( 2000) Parallel molecular pathways mediate expression of distinct forms of intermediate-term facilitation at tail sensory-motor synapses in Aplysia. Neuron 26: 219-231. [DOI] [PubMed] [Google Scholar]
- Taghert PH, Willows AOD ( 1978) Control of a fixed action pattern by single, central neurons in the marine mollusk, Tritonia diomedea. J Comp Physiol 123: 253-259. [Google Scholar]
- Teshiba T, Shamsian A, Yashar B, Yeh SR, Edwards DH, Krasne FB ( 2001) Dual and opposing modulatory effects of serotonin on crayfish lateral giant escape command neurons. J Neurosci 21: 4523-4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willows AOD, Dorsett DA, Hoyle G ( 1973) The neuronal basis of behavior in Tritonia I. Functional organization of the central nervous system. J Neurobiol 4: 207-237. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG ( 2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355-405. [DOI] [PubMed] [Google Scholar]