

Abstract
The accumulation of amyloid-β into insoluble plaques is a characteristic feature of Alzheimer's disease. Neuronal morphology is distorted by plaques: rather than being essentially straight, they are substantially more curved than those in control tissue, their trajectories become altered, and they are frequently distended or swollen, presumably affecting synaptic transmission. Clearance of plaques by administration of antibodies to amyloid-β is a promising therapeutic approach to the treatment of Alzheimer's disease, leading to stabilization of dementia by an unknown cellular mechanism. The effect of plaque clearance on plaque-induced neuronal alterations has not been studied previously. Here we show that both plaques and neuritic lesions are reversible in a strikingly short period of time after administration of a single dose of amyloid-β antibody. Amyloid clearance and recovery of normal neuronal geometries were observed as early as 4 d and lasted at least 32 d after a single treatment. These results demonstrate that, once plaques are cleared, neuronal morphology is self-correcting and that passive antibody treatment has the potential to reverse neuronal damage caused by Alzheimer's disease and, hence, directly impact cognitive decline. Moreover, the rapid normalization of neuritic dystrophy suggests an unexpected degree of plasticity in the adult nervous system.
Keywords: Alzheimer, immunotherapy, antibody, plasticity, neurodegeneration, amyloid
Introduction
Alzheimer's disease (AD) is characterized in postmortem analysis by the presence of neurofibrillary tangles and senile plaques. The amyloid-β (Aβ) peptide, a cleavage product of the amyloid precursor protein (APP), is the primary component of senile plaques (Selkoe, 1996). The bulk of genetic, histological, and biochemical studies support the amyloid hypothesis, which proposes that aberrant accumulation of amyloid-β is the cause of AD. Transgenic mouse models that overexpress mutant human APP develop senile plaques as they age and show signs of behavioral impairment, also supporting the amyloid hypothesis (Games et al., 1995; Hsiao et al., 1996). Although the mouse models do not show the marked neuronal loss that is found in late-stage AD in humans (Irizarry et al., 1997), they do exhibit similar pathophysiological alterations in neuronal morphology associated with plaque deposition in the brain. These alterations include bulbous swelling, curvy trajectories, and accumulation of neurofilament-associated inclusions in neurites that are in close proximity to specific subtypes of plaques (Irizarry et al., 1997; Schwab et al., 1997; Knowles et al., 1998, 1999; Masliah et al., 2001). We proposed that these local alterations in neurite morphology may be a source of cortical desynchronization, whereby neuronal signaling is distorted as the processes traverse through a volume of brain with sporadic, local disruptions in transmission (Knowles et al., 1999). The sum of these disruptions likely contributes to the cognitive impairment observed in AD.
The current study builds on previous work using anti-amyloid antibodies as a means of clearing existing amyloid deposits in older transgenic mice (Bacskai et al., 2001, 2002). These mice show behavioral impairment and display the full range of neuritic pathology associated with dense-core senile plaques (Games et al., 1995; Irizarry et al., 1997). Our work examines the neuritic pathology in antibody-treated mice to address whether removal of amyloid plaques is sufficient to restore normal neuronal morphology in the aged brain. We demonstrate that curvy, distorted neurites are indeed “straightened” in a remarkably short period of time and remain straightened for a surprisingly long period of time after a single treatment. The rapid normalization of neuritic morphology offers unexpected hope that an effective anti-amyloid therapy can stabilize the illness and perhaps even reverse the impairment. Likewise, these results demonstrate a startling degree of neuronal plasticity in an aged, adult animal that would not have been predicted.
Materials and Methods
Mice were anesthetized with Avertin (1.3% 2,2,2-tribromoethanol, 0.8% tert-pentylalcohol; 250 mg/kg). A small circular portion of the skull (∼7 mm diameter) was removed, and the dura was gently peeled back from the cortical surface. Fifteen microliters of antibody [1 mg/ml, anti-amyloid-β (10D5) or anti-tau (16B5)] were applied to the surface of the brain (Bacskai et al., 2001), after which a circular glass coverslip was affixed over the craniotomy with dental cement. After 4-32 d, mice were anesthetized and perfused intracardially with 4% paraformaldehyde, and their brains were extracted. Brains were sliced into 40-μm-thick coronal sections, which were then immunostained with SMI-32 (1:1000 in normal goat serum), Cy3, a fluorescent anti-mouse IgG, and fluorescein-labeled 3D6, at 1:500 in normal goat serum. 10D5 and 3D6 antibodies colocalize to amyloid-β deposits noncompetitively (Bacskai et al., 2002). Other sections were stained with 13G8 anti-APP at 1:800 in normal goat serum and visualized with horseradish peroxidase-linked secondary antibodies and diaminobenzadine. Sections were mounted onto slides, dehydrated, and coverslipped. Ratios of curvature, SMI-32 neuronal density, amyloid burden, and neuritic plaque count were made using a fluorescent microscope with BIOQUANT software (BIOQUANT Image Analysis, Nashville, TN). A 40× objective was used to view neurites. Field size was 48,896 μm2. The craniotomy borders were clearly delineated anatomically. Field selection of craniotomy and remote sites followed systematic random sampling rules. Ten to 18 fields per animal were sampled in all conditions, and all layers of cortex were sampled. The location of the fields was determined by the craniotomy, which was the same for all animals.
Data analysis was done using SAS (SAS Institute, Cary, NC). Significance tests were performed using an a priori significance level of p < 0.01, and post hoc test were performed using an a priori significance level of p < 0.05. In all figures, the error bars represent the errors among sections. The principal source for variability in our study was the within-animal variability. Thus, it was possible to achieve sufficient statistics and power using a relatively small number of animals. This was tested in a preliminary ANOVA, in which animals were used as independent variables nested within condition. No significant differences were found between animals (within conditions). A post hoc test for homogeneity of variances showed that the principal source of variability was within animals and that it was possible to combine results over animals.
Results
We used transgenic mice overexpressing mutant human APP (V717F, PDAPP mice), which develop amyloid-β plaques, gliosis, and neuritic dystrophy with increasing age (Games et al., 1995; Irizarry et al., 1997). Nontransgenic C57BL/6J mice aged 18-23 months were used as controls. The average age of the transgenic mice was 21 months (range of 19-23 months), at which time the average amyloid burden measured with 10D5 in untreated tissue was 10.2%. Anti-Aβ antibodies were topically applied to a discrete portion of the cortex after preparing a craniotomy, as described previously (Bacskai et al., 2001, 2002). Because under these circumstances diffusion of the antibody is limited, areas outside the application region serve as controls.
An example of amyloid-β clearance is shown in Figure 1, in which cortical tissue was treated once with 10D5, an anti-Aβ antibody (Hyman et al., 1992), and then perfused after a 32 d survival period. Figure 1 shows immunostaining with direct-labeled 3D6 anti-amyloid-β antibody [which has a distinct epitope from 10D5 (Bacskai et al., 2002)]. Cortical tissue in an area remote from the craniotomy, i.e., tissue that did not receive direct 10D5 application (Fig. 1a), is unchanged compared with untreated tissue or tissue treated with sham antibody (anti-human tau that does not cross-react with mouse tissue). Figure 1b shows staining in an area of cortex directly under the cranial window, and thus received 10D5 antibody, from the same animal. The amyloid-β burden is markedly reduced. Figure 1, c and d, shows similar treatment with the anti-tau antibody 16B5, in which the amyloid-β levels in the remote and craniotomy sites are similar to that in Figure 1a. Statistical analysis of quantitative measures of amyloid burden of the entire sample confirmed these results (Fig. 1e). We found that the amyloid-β burden was significantly reduced only in areas treated with 10D5 (2 × 2 ANOVA; df = 3, 96; F = 15.35; p < 0.01; Tukey's post hoc test; p < 0.05). These results demonstrate that one dose of the anti-amyloid antibody 10D5 was sufficient to significantly reduce the amyloid protein burden in treated cortical tissue for up to 32 d after treatment.
Figure 1.

Application of anti-amyloid antibody 10D5 clears amyloid-β plaques. Amyloid-β plaque clearance occurred only in cortical tissue treated with 10D5. Amyloid protein was immunostained with fluorescent 3D6. a, Untreated area of cortex in animal treated with 10D5. Plaques are clearly visible (arrow). Scale bar, 50 μm. b, Plaques are absent in treated area from the same animal. c, Untreated area of cortex in animal treated with human anti-tau antibody 16B5. d, Treated area of cortex from same animal as in c. e, Comparison of amyloid burden measured with 3D6 antibody under different experimental conditions. The values are the means; error bars indicate SD.
Because a major effect of plaques is an increase in neuritic curvature in their immediate vicinity (Knowles et al., 1999; Le et al., 2001), we tested whether clearing plaques would allow plaque-induced changes in neuronal morphology to recover. We immunostained neurofilaments in coronal sections using SMI-32 (Campbell and Morrison, 1989). We quantified neuritic curvature by dividing the straight-line length of neuritic processes by the curvilinear length of processes (Knowles et al., 1999; D'Amore et al., 2003).
Figure 2 shows the effect of 10D5 antibody treatment on neuritic curvature 32 d after application. Amyloid-β (top row) and stained neurites (middle row) are shown for areas remote from the cranial window (a, c) and, for areas below it, those directly receiving anti-amyloid-β antibody application (b, d). The two channels are merged in the bottom row. The relationship between the prominently curved neurites and the dense-core plaque is clearly seen. The amyloid-β in the treated areas is almost completely gone. The SMI-32-immunoreactive processes in the cleared area (Fig. 2d) are significantly straighter than those in the remote area (Fig. 2c).
Figure 2.
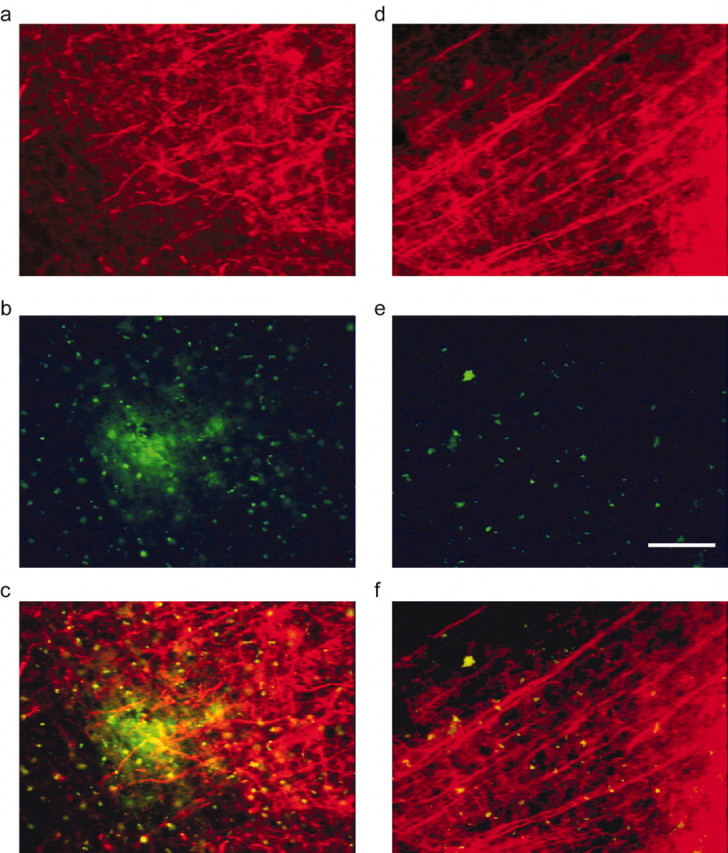
Amyloid-β clearance restores the morphology of neurites. a, Untreated area of cortex from 10D5-treated animal, immunostained with Cy-3-SMI-32. b, Plaques are clearly visible with 3D6 immunostaining. c, Overlay of a and b showing both plaques and dystrophic neurites. d, Treated area of cortex in same animal as a. Neurites are noticably straighter. e, Plaques are absent in treated 3D6 immunostained area, corrected for autofluorescence. f, Overlay of d and e. Scale bar, 50 μm.
The results for the entire sample are shown in Figure 3, top, in which the average curvature ratio histograms were calculated for remote and 10D5-treated brain areas, as well as corresponding areas receiving irrelevant (anti-human tau) antibody application. Neurites in the 10D5-treated areas qualitatively appear normal. Quantitative analysis supports this conclusion. The histogram of the 10D5-treated areas has a significantly greater curvature ratio than do the other three conditions, which do not differ significantly from each other (ANOVA; df = 2, 1715; F = 122.74; p < 0.01; Tukey's post hoc test; p < 0.05). Because of the high degree of skew in the histograms, the median and median absolute deviation were chosen as measures of central tendency and dispersion. These are shown in Figure 3, bottom. The neuritic curvature in the areas receiving 10D5 was reduced to values no different from those in control (nontransgenic) mice (Le et al., 2001; D'Amore et al., 2003). The results are summarized in Table 1.
Figure 3.
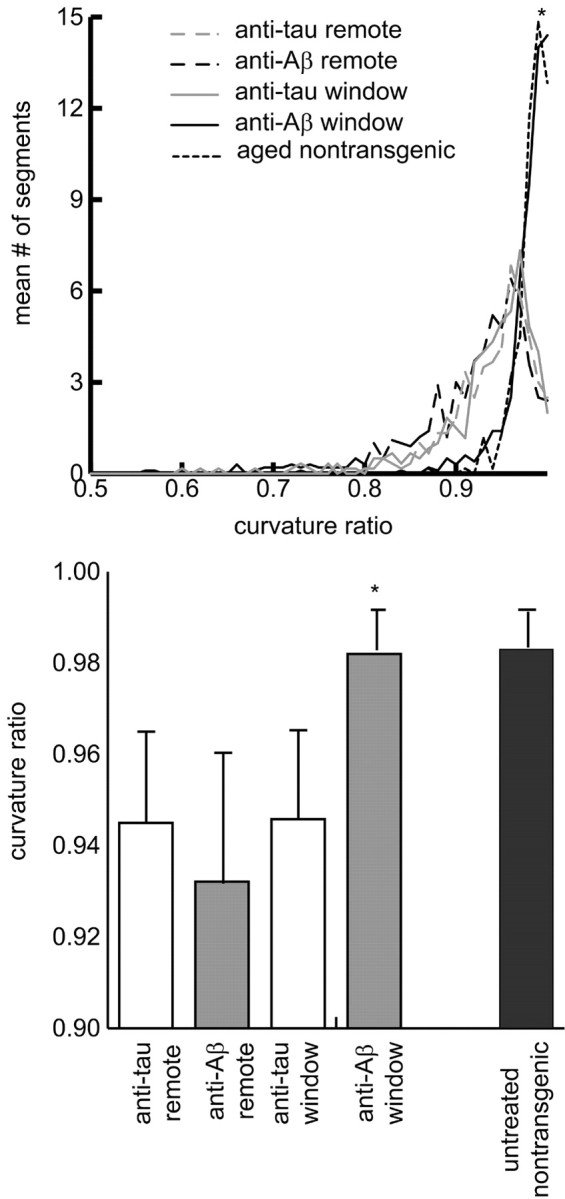
Comparison of curvature ratios from different experimental conditions. Top, Mean curvature ratio histogram for each experimental condition. Bottom, Comparison of curvature ratios for the different experimental conditions. The values are the medians ± median absolute deviations across animals for each condition.
Table 1.
Amyloid burden, curvature ratios, and neuritic densities measured under craniotomies and remote sites
|
|
Control antibody remote |
Control antibody window |
Anti-Aβ antibody remote |
Anti-Aβ antibody window |
|---|---|---|---|---|
| Amyloid burden | 10.78 ± 4.83 | 12.74 ± 9.42 | 9.57 ± 5.85 | 1.93 ± 1.61* |
| (n = 6) | (n = 6) | (n = 10) | (n = 10) | |
| Curvature ratio (all time points) | 0.945 ± 0.026 | 0.946 ± 0.024 | 0.932 ± 0.034 | 0.982 ± 0.010* |
| (n = 6) | (n = 6) | (n = 10) | (n = 10) | |
| 4 d curvature ratio | 0.947 ± 0.05 | 0.950 ± 0.04 | 0.925 ± 0.07 | 0.981 ± 0.04* |
| (n = 2) | (n = 2) | (n = 4) | (n = 4) | |
| 32 d curvature ratio | 0.943 ± 0.06 | 0.941 ± 0.06 | 0.935 ± 0.06 | 0.982 ± 0.02* |
| (n = 3) | (n = 3) | (n = 4) | (n = 4) | |
| SMI-32 density | 66.00 ± 3.52 | 66.65 ± 3.36 | 65.61 ± 3.10 | 66.03 ± 4.44 |
|
|
(n = 6)
|
(n = 6)
|
(n = 10)
|
(n = 10)
|
Curvature ratio for untreated nontransgenic neurites, 0.983 ± 0.009. *p≤0.01, significantly different. Amyloid burden and SMI-32 density are means ± SDs, and curvature ratios are medians ± median absolute deviations. n denotes number of animals.
This experiment showed that a remarkable degree of plasticity occurred in the aged adult mouse within 32 d. To refine the time course over which recovery of neurite morphology occurs, we repeated the experiment, waiting various periods between 4 and 30 d. The results are summarized in Table 1. Surprisingly, neurite morphology has returned to normal within 4 d of treatment and remains so for the duration of the experiment. Within any condition, neuritic curvature did not differ between 4 and 32 d (ANOVA; df = 7, 1510; F = 6.24; NS). For both 4 and 32 d treated animals, the neurites in the 10D5-treated area were significantly straighter. These results indicate that a single dose of antibody not only reduces amyloid-β plaque burden but can reduce neuritic curvature to values equivalent to those in age-matched nontransgenic animals within a few days of treatment and that the treatment itself is long lasting.
To test whether application of 10D5 selectively killed neurons whose morphology was distorted by plaques rather than reducing neuritic curvature (i.e., only neurons with relatively straight neurites survived antibody application), we measured neuritic density in SMI-32-stained tissue. No change in neurite density was observed within the treated area (ANOVA; df = 2, 136; F = 0.73; NS), suggesting that selective loss of dystrophic neurites could not explain the normalization.
Neuritic plaques are a subset of plaques with grossly distorted, bulbous processes that immunostain for APP (Joachim et al., 1991). In PDAPP mice, they represent only a small proportion of total plaques. To test the effects of anti-amyloid-β antibody on these most severe morphological changes, we stained sections with 13G8 against APP, indicative of neuritic plaques, and assessed the number of neuritic plaques in treated and nontreated areas. We found no significant difference in neuritic plaque count between areas of cortex treated with 10D5 and those left untreated (ANOVA; df = 2, 116; F = 2.8; NS), although the total number of neuritic plaques was fairly small in all circumstances. This result indicates that neuritic plaques are most resistant to antibody treatment and may require repeated treatments or higher concentrations for effective clearance.
Discussion
In both human and APP transgenic mouse tissue, there are marked morphological alterations in neurites that are most severe in the immediate vicinity of dense-core senile plaques (Knowles et al., 1998). Neurites that normally have straight trajectories become distorted and “curvy” (Le et al., 2001). Repeated local curviness may contribute to a desynchronization of neuronal signaling that alters synaptic transmission to a degree that is manifested in measurable cognitive impairment (Knowles et al., 1999). Vaccination against amyloid-β is a promising therapeutic approach for prevention and removal of senile plaques. Recent data suggest that the decline in cognitive performance is arrested in patients that received the vaccine (Hock et al., 2003), but the mechanism of this effect is unknown. There are currently no published reports linking functional disruption with the structural deficits observed in this study; however, at least one theoretical study predicts functional consequences of structural pathology (Stepanyants et al., 2002).
The current study used topical application of anti-amyloid-β to the surface of the cortex in aged transgenic mice that resulted in rapid and effective clearance of amyloid-β deposits within a limited radius of diffusion from the site. The local clearance of amyloid-β had a striking, local effect on neuritic morphology. Neurites were straightened to an extent found in nontransgenic mouse tissue, without a change in neurite density.
Our study is the first to measure the consequences to neurons of clearing amyloid-β plaques. Our results imply that (1) neurite abnormalities are directly related to Aβ deposition and (2) neurites appear to have a self-correcting mechanism by which they are able to rapidly restore their morphology to normal after plaques have been eliminated.
What is striking in the experiments presented here is both the rapid recovery of abnormal neurites and the long-lived effects of passive immunization on amyloid-β deposits. It is interesting to compare the current results with those from a recent human case study of a patient who had suffered meningoencephalitis after active immunization with amyloid-β protein (Nicoll et al., 2003). First, 20 months after the final immunization, the autopsy revealed clearance of amyloid-β deposits, albeit incomplete, suggesting a long-lived effect. In the patient, however, tau-positive plaque-associated dystrophic neurites were also cleared, contrasting with the negligible effect on neuritic plaques within 1 month after a single dose of antibody in the mice. It will be interesting to see whether the dystrophic neurites associated with the dense-core subset of plaques in the mice are self-correcting after prolonged amyloid clearance.
The mechanism by which neurites are restored to a straight morphology is unclear. It may be that standard ultrastructural and membrane turnover processes in these neurons are sufficiently rapid to account for the straightening, or it could be that upregulation of these processes is caused by the removal of amyloid-β. Alternatively, it could be that plaques simply exert mechanical force on neurites, and, after clearance, the processes simply “snap back” to their original position. Additional analysis of tissue with reference to the role of the microtubule protein tau may offer insight into how neurites are able to achieve a straight geometry after the clearance of plaques. In any case, our findings present significant new evidence for structural plasticity in these neurons: a previously unknown stable reversibility of some forms of neuritic dystrophy in the brains of transgenic mice attributable to the clearance of amyloid-β plaques. Although recent data suggest that structural plasticity in the adult cortex occurs with regard to movement of dendritic spines (Trachtenberg et al., 2002), the gross changes in morphology observed here over just a few days are unexpected, especially in aged animals. Alzheimer's disease has been proposed to be a failure of neuronal plasticity (Mesulam, 2000), and a recent theoretical study has shown that the structural changes most likely to affect memory would take place at the spatial scale most likely to be affected by plaques (Stepanyants et al., 2002). Clearance of plaques through the administration of antibody may thus provide a means by which neuritic damage suffered by Alzheimer's disease patients may be reversed. These findings offer a plausible mechanism for the reported stabilization of cognitive deficits observed in Alzheimer's patients who had received an anti-amyloid-β vaccine (Hock et al., 2003).
Footnotes
This work was supported by National Institutes of Health Grants AG08487, AG15453, AG020570, and EB00768, a Pioneer Award from the Alzheimer Association, and the Walters Family Foundation. We thank Drs. Dale Schenk and Dora Games at Elan Pharmaceuticals for providing transgenic mice and antibodies.
Correspondence should be addressed to Dr. Bradley T. Hyman, Department of Neurology/Alzheimer Unit, Massachusetts General Hospital, 114 Sixteenth Street (CAGN 2009), Charlestown, MA 02129. E-mail: bhyman@partners.org.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310879-05$15.00/0
References
- Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT ( 2001) Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med 7: 369-372. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT ( 2002) Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci 22: 7873-7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MJ, Morrison JH ( 1989) Monoclonal antibody to neurofilament protein (SMI-32) labels a subpopulation of pyramidal neurons in the human and monkey neocortex. J Comp Neurol 282: 191-205. [DOI] [PubMed] [Google Scholar]
- D'Amore JD, Kajdasz ST, McLellan ME, Bacskai BJ, Stern EA, Hyman BT ( 2003) In vivo multiphoton imaging of a transgenic mouse model of Alzheimer disease reveals marked thioflavine-S-associated alterations in neurite trajectories. J Neuropathol Exp Neurol 62: 137-145. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagoplan S, Johnson-Wood K, Kahn K, Lee M, Leibowitz P, Lieberburg I, Little S, Masliah E, McConlogue L, et al. ( 1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373: 523-527. [DOI] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Müller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer M, Umbricht D, deq Uervain D, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch R ( 2003) Antibodies against β-amyloid slow cognitive decline in Alzheimer's disease. Neuron 38: 547-554. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G ( 1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99-102. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Tanzi RE, Marzloff K, Barbour R, Schenk D ( 1992) Kunitz protease inhibitor-containing amyloid beta protein precursor immunoreactivity in Alzheimer's disease. J Neuropathol Exp Neurol 51: 76-83. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT ( 1997) Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci 17: 7053-7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachim C, Games D, Morris J, Ward P, Frenkel D, Selkoe D ( 1991) Antibodies to non-beta regions of the beta-amyloid precursor protein detect a subset of senile plaques. Am J Pathol 138: 373-384. [PMC free article] [PubMed] [Google Scholar]
- Knowles RB, Gomez-Isla T, Hyman BT ( 1998) Abeta associated neuropil changes: correlation with neuronal loss and dementia. J Neuropathol Exp Neurol 57: 1122-1130. [DOI] [PubMed] [Google Scholar]
- Knowles RB, Wyart C, Buldyrev SV, Cruz L, Urbanc B, Hasselmo ME, Stanley HE, Hyman BT ( 1999) Plaque-induced neurite abnormalities: implications for disruption of neural networks in Alzheimer's disease. Proc Natl Acad Sci USA 96: 5274-5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le R, Cruz L, Urbanc B, Knowles RB, Hsiao-Ashe K, Duff K, Irizarry MC, Stanley HE, Hyman BT ( 2001) Plaque-induced abnormalities in neurite geometry in transgenic models of Alzheimer disease: implications for neural system disruption. J Neuropathol Exp Neurol 60: 753-758. [DOI] [PubMed] [Google Scholar]
- Masliah E, Sisk A, Mallory M, Games D ( 2001) Neurofibrillary pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. J Neuropathol Exp Neurol 60: 357-368. [DOI] [PubMed] [Google Scholar]
- Mesulam MM ( 2000) A plasticity-based theory of the pathogenesis of Alzheimer's disease. Ann NY Acad Sci 924: 42-52. [DOI] [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO ( 2003) Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med 9: 448-452. [DOI] [PubMed] [Google Scholar]
- Schwab C, Steele JC, McGeer PL ( 1997) Dystrophic neurites are associated with early stage extracellular neurofibrillary tangles in the parkinsonismdementia complex of Guam. Acta Neuropathologica 94: 486-492. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ ( 1996) Amyloid beta-protein and the genetics of Alzheimer's disease. J Biol Chem 271: 18295-18298. [DOI] [PubMed] [Google Scholar]
- Stepanyants A, Hof PR, Chklovskii DB ( 2002) Geometry and structural plasticity of synaptic connectivity. Neuron 34: 275-288. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K ( 2002) Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420: 788-794. [DOI] [PubMed] [Google Scholar]