

Abstract
Molecules comprising the extracellular matrix (ECM), and the family of matrix metalloproteinases (MMPs) that regulate them, perform essential functions during neuroplasticity in both developing and adult nervous systems, including substrate guidance during neuritogenesis and the establishment of boundaries for axonal terminal fields. MMP proteolysis of ECM molecules may perform a permissive or inductive role in fiber remodeling and synaptogenesis initiated by deafferentation. This study examined functional and structural effects of MMP inhibition during the early phases of deafferentation-induced sprouting, characterizing components of the degeneration/proliferation cycle that may be dependent on MMP activity. Adult rats received unilateral lesions of the entorhinal cortex to induce collateral sprouting of the crossed temporodentate fiber pathway. This was followed by intraventricular infusion of the MMP inhibitor FN-439 (2.9 mg/kg) or saline vehicle. After 7 d postlesion, rats underwent in vivo electrophysiological recording or histological processing for electron microscopic analysis. Lesioned rats receiving vehicle exhibited normal sprouting and synaptogenesis, with the emergence of the capacity for long-term potentiation (LTP) within the sprouting pathway, and the successful clearance of degenerating terminals with subsequent synaptic proliferation. In contrast, lesioned rats receiving the MMP inhibitor failed to develop the capacity for LTP and showed persistent cellular debris. Current source density analysis also revealed an FN-439-induced disruption of the current sink, normally localized to the middle region of the granule cell dendrites, corresponding to the terminal field of the crossed temporodentate fibers. These results establish a role for MMP-dependent processes in the deafferentation/sprouting cycle.
Keywords: deafferentation, collateral sprouting, synaptogenesis, matrix metalloproteinase, extracellular matrix, synaptic plasticity
Introduction
After brain injuries that elicit cell death or deafferentation, surviving neurons undergo reactive synaptogenesis to reestablish connectivity with postsynaptic sites vacated by degenerating terminals (for review, see Cotman et al., 1981; Steward, 1994; Deller and Frotscher, 1997). Experimental models suggest this process may play a role in functional recovery (Loesche and Steward, 1977; Ramirez and Stein, 1984; Reeves and Steward, 1986; Reeves and Smith, 1987; Erb and Povlishock, 1991). Recent findings revealed roles for the extracellular matrix (ECM) in neuronal remodeling in development and during injury-induced structural reorganization (Lander, 1993; Choi, 1994; Wang et al., 2000). Proteolytic modification of ECM molecules, mediated by matrix metalloproteinases (MMPs), is posited to prepare denervated neuropil for synaptogenesis and influence the spatial distribution of the growth response.
The dentate gyrus is well suited to examine the MMP role in injury-induced sprouting, having afferents that terminate in segregated laminae, facilitating the interpretation of synaptic reorganization (for review, see Frotscher, 1988; Amaral and Witter, 1995). Unilateral entorhinal cortex (EC) lesions, in adults rats, massively deafferent ipsilateral dentate granule neurons, which lose ∼90% of synapses on their distal dendrites (Matthews et al., 1976a, b). One of the surviving afferent systems induced to sprout originates in the EC contralateral to the lesion. This crossed temporodentate pathway (CTD) is normally small relative to the ipsilateral entorhinodentate projection. The two systems, arising from common cortical neurons (Steward, 1976), share overlapping terminal fields (Wyss, 1981) and establish ultrastructurally similar synapses (Davis et al., 1988). Because of these similarities, prior research focused on whether CTD sprouting contributes to functional recovery. It was recognized that the CTD acquired the capability for long-term potentiation (LTP) after sprouting (Wilson 1981; Wilson et al., 1981; Reeves and Steward, 1986), and some behavioral indices correlated with the time course of sprouting (Reeves and Smith, 1987; Ramirez et al., 1996). The role of ECM molecules and MMPs in the deafferented neuropil remains uncertain; however, gelatin zymography showed elevated MMP-2 activity after unilateral EC lesions (Phillips and Reeves, 2001).
The present study examined the effects of MMP suppression during the initial phases of synaptogenesis in the dentate gyrus after unilateral EC lesion. It was hypothesized that reactive CTD fibers normally undergo growth in an ECM milieu of upregulated MMP activity and that pharmacological inhibition of MMP activity would alter the functional and structural properties of the reactive growth response. Electrophysiological measurements were selected that reflect, at least in part, the functioning and spatial distribution of presynaptic terminals: LTP, paired-pulse plasticity, and current source density (CSD) analysis. In the current study, MMP activity was inhibited with FN-439, a commercially available peptidyl hyroxamic acid developed for its action on human MMPs (Odake et al., 1994). This compound is water soluble and retains its inhibitory activity in the presence of neutral proteinases, making it a good choice for the modulation of MMP activity in vivo (Kigasawa et al., 1995; Anan et al., 1996). We applied FN-439 at 30 min postlesion, followed by electrophysiological and electron microscopic analysis at 7 d survival.
Materials and Methods
The procedures for this study followed all national guidelines for the care and use of laboratory animals, and the experimental protocol was approved by the Medical College of Virginia Animal Research Committee. All efforts were made to minimize animal suffering and to reduce the number of animals used. Twenty-four Sprague Dawley rats (Hilltop Lab Animals, Scottsdale, PA), weighing 250-300 gm, were used in these experiments. Animals were housed in individual cages in a temperature-controlled (22°C) and humidity-controlled (50% relative) animal facility on a 12 hr light/dark cycle. Rat chow and water were available ad libitum.
Unilateral entorhinal cortical lesion. Under inhalation anesthesia (70% N2O, 30% O2, and 2% isoflurane), rats were placed in a stereotaxic frame. During all surgical procedures, body temperature was maintained at 37°C. An area of skull was removed to expose the EC of one hemisphere (right side). A Teflon-insulated wire electrode was angled at 10° from perpendicular, and current passed (1.5 mA for 40 sec) at a total of nine stereotaxic sites: 1.5 mm anterior to the transverse sinus; 3, 4, and 5 mm lateral to midline, at 2, 4, and 6 mm ventral to the brain surface. The skull opening was filled with gelfoam, the scalp was sutured, and the animals were given an i.m. injection of Wycillin.
Intraventricular FN-439 injection. FN-439 (MMP inhibitor 1; Calbiochem, La Jolla, CA) is a synthetic peptide hydroxamate with the capacity to chelate Zn2+, thereby blocking the active site of metalloproteinases (Odake et al., 1994; Kigasawa et al., 1995). The drug exhibits specificity for interstitial collagenase, MMP-9, and MMP-3, with IC50 values of 1.0 μm, 30 μm, and 150 μm, respectively. Given that EC lesion models induce both MMP-3 and -9 gene expression and that FN-439 has been used in vivo to block these MMPs, we first sought to establish whether our pharmacological paradigm would affect functional activity of MMP-3 and -9. Using a commercial fluorescent substrate assay for MMP-3 activity, we determined percentage inhibition of purified MMP-3 over a range of FN-439 doses (75, 150, and 750 μm), inclusive of the published IC50 concentration. Our results showed 39.7, 51.6, and 69.3% inhibition, respectively, confirming 150 μm as the FN-439 IC50 value for MMP-3 and identifying 750 μm as a reasonable dosage for significant MMP-3 inhibition. Subsequent in vitro zymography experiments showed that exposure of EC lesion hippocampal preparations to 150 μm FN-439 blocked up to 98.8% of the MMP-9 gelatinolytic activity relative to matched untreated samples. Based on these findings, we chose to target both MMP-3 and -9 in vivo using the 720 μm dose.
Intracerebral ventricular injection of drug or saline vehicle was achieved by a modification of the method of Buki et al. (1999). At 30 min after entorhinal lesions, rats were placed in a stereotaxic frame under gas inhalation anesthesia (70% N2O, 30% O2, and 2% isoflurane), and a burr hole was prepared in the skull to permit direct access to the lateral ventricle of the right hemisphere. A pediatric spinal needle was inserted through the brain tissue to tap the CSF-filled space of the ventricle. Approximately 0.05 cc of CSF was withdrawn to confirm target site, and FN-439 (7.2 mm stock in sterile saline; total injected volume, 0.1 cc) was infused slowly using a Harvard Mechanical Syringe Pump [(Harvard Apparatus, Holliston, MA) (over a 30 min period, in eight steps; 2 min interstep interval; final effective ventricular concentration of drug, 720 μm)]. Pupillary reflexes were monitored during infusion to assess any significant change in intracranial pressure. At completion of the infusion, the spinal needle was left in place for 2-3 min before retraction and surgical site closure. In a subset of FN-439- and saline-infused EC lesion cases, hippocampal MMP-9 lytic activity was tested with gelatin zymography. In vivo delivery of FN-439 inhibited 83.6% of the MMP-9 functional activity observed in saline vehicle controls (data not shown), confirming that our paradigm achieved significant MMP inhibition.
Electrophysiological recording procedures. At 7 d postlesion, rats were reanesthetized with urethane (1.5 gm/kg, i.p.), and a concentric bipolar-stimulating electrode (SNE-100; tip separation, 0.5 mm; Rhodes Medical Instruments, Woodland Hills, CA) was positioned stereotaxically into the unlesioned (left) entorhinal cortex (8.0 mm posterior to bregma; 4.2 mm lateral; initial depth, 2.5 mm). An extracellular recording electrode (micropipette filled with 150 mm NaCl; impedance, 2-6 MΩ) was lowered into the dentate gyrus (2.5 mm posterior to bregma; 1.5-1.7 mm lateral to midline) of the right hemisphere. Stimulation pulses consisted of constant current stimulus-isolated square wave pulses of 150 μsec duration, with variable pulse amplitudes (ranging between 25 and 400 μA). Potentials were amplified (bandpass = DC to 10 kHz), digitized at 25 kHz, and stored on magnetic media for offline analysis. The extracellular population EPSP [referred to here as field EPSP (fEPSP)] for each evoked response was measured as the waveform slope between 1.5 and 2.0 sec after the onset of the fEPSP. In waveforms also having a negative-going population spike, the spike amplitude was measured from the peak negativity to a tangent line drawn between the preceding and following positive peaks.
The recording electrode was initially lowered to the dorsal blade of the dentate stratum granulosum, ∼3.0 mm below the cortical surface. This electrode position was confirmed by a shift in the polarity of the fEPSP from a negative wave (corresponding to the dentate stratum moleculare) to a positive wave (corresponding to the cell body layer). An input/output (I/O) function was then generated for each animal by varying stimulus intensity from threshold to that evoking maximum response, in 10 equal increments. The stimulus intensity that evoked a fEPSP response of ∼80% of maximum, as determined in the I/O function, was used for subsequent current source density and LTP testing.
CSD analysis. The recording electrode was raised, in 25 μm increments, from the granule cell body layer to the hippocampal fissure. The electrode was paused for at least 3 min at each 25 μm “stop point” along the electrode track, and four responses were evoked at a rate of 1/30 sec. The uniform cytoarchitecture of the dentate gyrus (segregation of afferents into laminae, stereotypic dendritic orientation) allows the CSD computation to simplify to one spatial dimension (Richardson et al., 1987; White et al., 1990; Johnston and Wu, 1995). Specifically, the extracellular membrane current density, im(t), is estimated as the second spatial derivative of the field potential and is computed using a three spatial point formula:
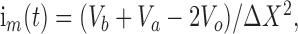 |
where Va, Vo, and Vb are consecutive recording sites along the dendritic axis and X is distance in micrometers (here 25 μm). The significance of changes in the distribution of current sinks and sources, observed among the experimental conditions, was evaluated using a mixed-model ANOVA, with distance along the granule dendritic axis as the within-subjects factor.
Paired-pulse plasticity analysis. Before paired-pulse testing and LTP trials, the recording electrode was repositioned into the cell body layer (dorsal stratum granulosum) and a stable positive-going fEPSP evoked potential was verified. Stimulation parameters were used that have been shown previously to elicit paired-pulse facilitation (PPF) in the hippocampus (Lomo, 1971; Steward et al., 1976; Zucker, 1989; Leung and Fu, 1994). In the PPF procedure, two identical stimulus pulses, subthreshold for target cell discharges and separated by <100 msec, usually evoke nonidentical responses; augmentation of the second response is considered to be mediated by residual Ca2+ loaded into presynaptic terminals during the first response (Leung and Fu, 1994). Paired-pulse plasticity was assessed at stimulus pair intervals ranging from 20 to 1000 msec, using the ratio of the second fEPSP slope to the first (fEPSP2/fEPSP1), and group differences in this ratio were evaluated with ANOVA.
LTP analysis. During LTP testing, field potentials were collected continuously at a rate of 1/30 sec, beginning 10 min before high-frequency stimulus trains (“tetanus”) and ending at 60 min after tetanus. The current level used for tetanic stimulation and for low-frequency test pulses was determined during initial I/O function testing, as noted above. The high-frequency conditioning stimulation consisted of eight trains, of four pulses each, at 400 Hz, for a total of 32 conditioning pulses. The intertrain interval was 200 msec. Tetanus-induced changes in mean fEPSP slope and mean population spike amplitude were evaluated with ANOVA and the Bonferroni post hoc test.
Electron microscopic methods. Seven days postlesion, rats were anesthetized with a lethal dose of sodium pentobarbital and killed by cardiac pressure perfusion of mixed aldehyde fixative (4% paraformaldehyde and 0.2% glutaraldehyde in 0.1 m Na phosphate buffer). Before aldehyde infusion, the vascular bed was flushed with 0.9% saline, and after in situ fixation, brains were removed and stored in the same fixative overnight at 4°C. As in previous ultrastructural studies (Yaghmai and Povlishock, 1992; Phillips et al., 1994), 40 μm serial coronal vibratome sections were cut containing bilateral hippocampi. Sections underwent osmication and were flat embedded on plastic slides sandwiched between two glass slides. After the plastic had cured to acceptable hardness, the glass slides were removed and sample regions of mid-dorsal hippocampus containing the CA1 and dentate gyrus were excised from the plastic-embedded material. These regions were mounted, and a series of thick and thin sections were cut on the ultramicrotome. The thin sections were collected on membrane-coated slotted grids, and the tissues were observed with a Zeiss electron microscope. Within each subregion of the dentate molecular layer (inner, middle, and outer zones), several sites were photographed at 3,000-20,000× magnification.
Results
This study evaluated the effects of MMP inhibition at 7 d postlesion to observe treatment effects during the process of active synaptogenesis. Sixteen rats were used to evaluate the effect of MMP inhibition on electrophysiological properties of the sprouting CTD fibers: unlesioned control rats (n = 4), EC-lesioned rats given vehicle (saline, intracerebral ventricular; n = 6), and EC-lesioned rats given FN-439 (intracerebral ventricular; n = 6). In addition, a second group of eight rats was used to evaluate the effect of MMP inhibition on the structural elements underlying sprouting and synaptogenesis: EC-lesioned rats given FN-439 (intracerebral ventricular; n = 4) and EC-lesioned rats given vehicle (saline, intracerebral ventricular; n = 4). Because the sprouting response does not approach asymptotic levels until ∼14 d postlesion (Matthews et al., 1976b; Steward and Vinsant, 1983; Steward, 1991), it was anticipated that only moderate increases in evoked potential amplitude would be observed. Consistent with this expectation, the maximum fEPSP peak amplitude was 3.51 ± 0.72 mV (all results reported as mean ± SEM) in unlesioned control rats, 4.62 ± 0.56 mV in vehicle-treated rats measured at 7 d postlesion, and 4.85 ± 1.15 mV in FN-439-treated rats recorded at 7 d postlesion. These group differences were not significant (F(2,13) = 0.55; p = 0.590). However, numerous studies have determined that measurement of the initial fEPSP slope provides a more stable estimate of synaptic efficacy than the fEPSP peak amplitude, which may be affected by embedded population spikes and polysynaptic influences (Wilson, 1981; Wilson et al., 1981; White et al., 1990; Reeves et al., 1995, 1997). Similar to the fEPSP peak amplitude, the baseline (pre-LTP) fEPSP slopes did not significantly differ among experimental groups (F(2,12) = 2.63; p = 0.113), but, as detailed below, the measurement of fEPSP slopes revealed significant group and treatment differences in functional plasticity of LTP and paired-pulse responses.
Long-term potentiation
Two types of analyses were used to evaluate group differences in LTP: (1) fEPSP slopes and population spike amplitudes were compared before and after tetanization to quantify the extent and significance of LTP within each group; and (2) the magnitude of potentiation was compared between groups, which directly addressed the effect of MMP inhibition. Consistent with prior investigations of the small unsprouted CTD system (Wilson, 1981; Wilson et al., 1981; Reeves and Steward, 1986), unlesioned control rats did not exhibit significant potentiation. In the current study, only the vehicle-treated lesioned rats demonstrated significant LTP of the fEPSP slopes, increasing from a pretetanus baseline of 0.87 ± 0.07 mV/msec to a post-tetanus level of 1.06 ± 0.07 mV/msec (F(5,1) = 25.13; p = 0.004) (Table 1). In contrast, fEPSP slopes in EC-lesioned rats receiving FN-439 treatment were not significantly changed by the tetanic stimulation (F(5,1) = 0.35; p = 0.579) (Table 1). Example baseline and post-tetanus averaged evoked potentials are shown in Figure 1, and the average tetanus-induced percentage increase in fEPSP slopes is shown in Figure 1D. In EC-lesioned rats receiving vehicle administration, the high-frequency conditioning trains produced an average 22.01 ± 5.10% increase in fEPSP slope. This was significantly different from the group of EC-lesioned rats receiving FN-439 treatments, in which post-tetanic fEPSP slopes were essentially unchanged (reduced by an average of 1.03 ± 5.78%) from pretetanus levels (F(1,11) = 8.94; p = 0.014) (Fig. 1D).
Table 1.
Evoked potential measurements (mean ± SEM) averaged for baseline (pretetanus) and for 10 - 60 min post-tetanus
|
|
|
fEPSP slope |
Population spike amplitude |
||||
|---|---|---|---|---|---|---|---|
| Experimental condition
|
n
|
Pretetanus (mV/msec)
|
Post-tetanus (mV/msec)
|
Pretetanus (mV)
|
Post-tetanus (mV)
|
||
| Unlesioned control | 4 | 0.81 ± 0.23 | 0.83 ± 0.22 | 0.56 ± 0.29 | 0.68 ± 0.36 | ||
| Lesioned, vehicle treated | 6 | 0.87 ± 0.07 | 1.06 ± 0.07** | 0.31 ± 0.06 | 0.51 ± 0.13* | ||
| Lesioned, FN-439 treated
|
6
|
0.93 ± 0.13
|
0.90 ± 0.11
|
0.24 ± 0.07
|
0.26 ± 0.09
|
||
*p < 0.05 and **p < 0.01, comparing post-tetanus to pretetanus within each experimental condition (repeated measures ANOVA).
Figure 1.

Effects of MMP inhibition on LTP in the crossed temporodentate system. A-C, Example field potentials evoked through the crossed perforant path. Pretetanus (top trace) and post-tetanus (bottom trace) waveforms are shown for an unlesioned control rat (A), a lesioned rat given intracerebral ventricular vehicle (B), and a lesioned rat given intracerebral ventricular FN-439 (C). Arrows indicate time of stimulus artifact. D, Group averages of tetanus-induced increases, expressed as percentage of pretetanus baseline, in fEPSP initial slope (left) and in population spike amplitude (right). *p < 0.05.
Inhibition of MMP activity also reduced potentiation of the population spike, as measured at 7 d postlesion. Population spikes evoked through the sparse CTD system in normal unlesioned rats were variable in waveform and did not exhibit significant changes induced by high-frequency tetanic stimulation. However, population spike amplitudes in vehicle-treated lesioned rats were significantly potentiated from a baseline of 0.31 ± 0.06 mV to a post-tetanus level of 0.51 ± 0.13 mV (F(5,1) = 8.52; p = 0.033) (Table 1). In comparison, pretetanus population spikes in FN-439-treated lesioned rats measured 0.24 ± 0.07 mV, which was not different from the post-tetanus mean of 0.26 ± 0.09 mV (F(5,1) = 0.46; p = 0.529). Figure 1D shows that in the group of lesioned rats given vehicle injections, high-frequency tetanus induced a 64.68 ± 16.34% increase in population spike amplitude that was significantly greater than the 1.21 ± 13.04% increase observed in FN-439-treated rats (F(1,10) = 9.219; p = 0.013) and greater than the 7.09 ± 10.72% increase measured in unlesioned control rats (F(1,8) = 6.78; p = 0.031).
Paired-pulse plasticity
This investigation used paired-pulse stimulation parameters that have been shown, in other fiber systems, to elicit facilitation of the second fEPSP relative to the first. Thus, current intensities that are subthreshold for population spikes, in the first response of the pair, may generate an augmented second fEPSP, attributed to residual Ca2+ loaded into presynaptic terminals during the first response (Zucker, 1989; Leung and Fu, 1994). However, the paired-pulse plasticity observed in this study was, in many cases, a paired-pulse depression in which the second fEPSP was smaller than the first. In unlesioned control rats, the ratio of the second fEPSP to the first [(fEPSP2/fEPSP1) × 100] was consistently below 100%, and this depression was significant for interpulse intervals (IPIs) of 250 and 500 msec (Fig. 2B). In vehicle-treated lesioned rats, a biphasic pattern of paired-pulse plasticity was noted, with relative paired-pulse facilitation observed at IPI <75 msec and depression of the second response at longer IPI values. However, for this group of rats, fEPSP2 was significantly different from fEPSP1 only for the paired-pulse depression noted at IPI = 500 msec (Fig. 2B). Importantly, the fEPSP2/fEPSP1 ratios for the vehicle-treated group, at IPI = 50 msec, were significantly elevated compared with unlesioned control rats (F(1,8) = 5.82; p = 0.042) and compared with lesioned rats treated with FN-439 (F(1,10) = 10.72; p = 0.008) (Fig. 2B). The paired-pulse plasticity responses observed for the FN-439-treated group were similar to those measured in unlesioned control rats, with significant depression (fEPSP2/fEPSP1 < 100%) seen at IPI values of 40, 50, 100, and 1000 msec (Fig. 2B).
Figure 2.
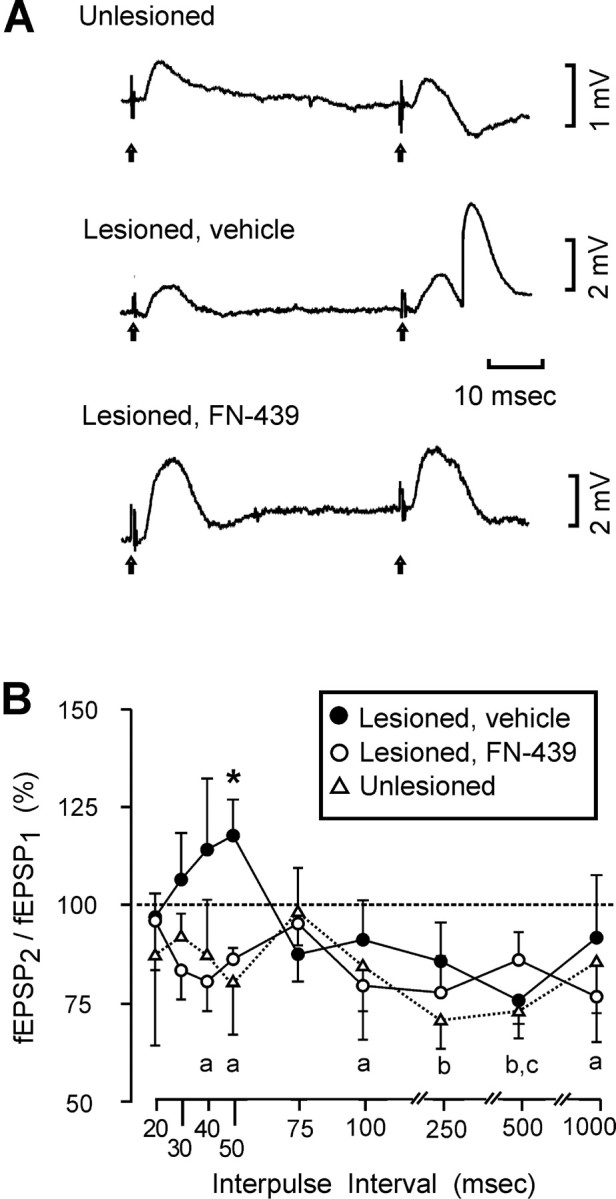
Effects of MMP inhibition on evoked paired-pulse responses. A, Field potentials evoked with identical paired stimulus pulses, with interpulse interval (50 msec) for a control rat, a lesioned rat given intracerebral ventricular vehicle, and a lesioned rat given intracerebral ventricular FN-439. Note that this stimulation protocol induced a population spike in the second response only for the vehicle-treated lesioned animal. B, Average ratio of the second fEPSP to the first (fEPSP2/fEPSP1 × 100), for all experimental groups, using interpulse intervals from 20 to 1000 msec. Note that the predominate pattern for the CTD pathway was a paired-pulse depression, with fEPSP2 significantly below fEPSP1 in the following conditions: a, lesioned, FN-439-treated rats (IPI = 40, 50, 100, and 1000 msec); b, unlesioned control rats (IPI = 250 and 500 msec); c, lesioned, vehicle-treated rats (IPI = 500 msec). Also note that for IPI <75 msec, a relative paired-pulse facilitation was observed for the lesioned rats given intracerebral ventricular vehicle. For this group, the fEPSP2/fEPSP1 ratio was significantly elevated above that for the control rats (*p < 0.05) and above that for the lesioned rats given FN-439 (*p < 0.01).
CSD analysis
CSD analysis also compared unlesioned control and EC-lesioned rats with and without pharmacological MMP inhibition. Figure 3 shows the CSD analysis of the CTD fiber system in the dentate molecular layer of an unlesioned control rat, with the raw field potentials shown in Figure 3B and the computed current sources in Figure 3C (also duplicated in a shaded version in Fig. 3D). Consistent with the known pattern of afferent termination (CTD fibers innervate the middle portion of the dentate molecular layer), the largest current sink was found in the middle molecular layer and the largest current source in the cell body region. Figure 4 shows examples of CSD profiles for control and for lesioned rats given intracerebral ventricular vehicle and FN-439. To facilitate group comparisons, the areas under the CSD curves were integrated and the resulting average sinks and sources were plotted (bottom panel Fig. 4), corresponding to eight equidistant bands along the granule cell dendritic axis. Vehicle-treated lesioned rats exhibited a CSD pattern similar to controls, with a prominent current sink occupying approximately the middle 100 μm in the molecular layer. In contrast, lesioned rats given intracerebral ventricular FN-439 demonstrated an aberrant distribution of sinks and sources, with a relatively narrow current sink in a portion of the middle dendritic zone. The statistical analysis of the averaged sinks and sources showed that one of the bands in the FN-439-treated group differed from that in the control (F(1,5) = 18.55; p = 0.008) and the vehicle-treated (F(1,5) = 22.46; p = 0.005) groups.
Figure 3.

CSD analysis of crossed temporodentate fiber system in unlesioned control rat. A, Distance scale along granule cell dendritic axis in dentate molecular layer. B, Field potentials evoked at 25 μm intervals. C, Computed CSD, with positive-going sinks and negative-going sources. D, Shaded CSD (from C) to emphasize major sinks and sources.
Figure 4.

Average CSD results in control group and in lesioned groups with and without MMP inhibition. Curves in each column show the average current sources for each group. Bars at bottom show areas under CSD curves averaged for each rat in each group. Note that the control group and vehicle-treated lesioned group show similar current sink in middle molecular layer, contrasting with a reduced sink in lesioned group given intracerebral ventricular FN-439. Statistical analysis of the averaged CSD showed that one stratum in the FN-439-treated group significantly differed from that in the control group (*p < 0.01) and the vehicle-treated group (*p < 0.01).
Structural effects of FN-439 treatment
The effect of MMP inhibition on the structural components underlying reactive synaptogenesis was assessed through two qualitative comparisons at 7 d postlesion. The first directly compared the morphology of ipsilateral dentate molecular layers, the target site of entorhinal lesion-induced plasticity, within the FN-439- and vehicle-treated cases. A second compared the ipsilateral effect of FN-439 with drug effect in the nondeafferented contralateral dentate molecular layer, an intra-animal control that would identify any nonspecific neurotoxic effects. When ipsilateral molecular layers were compared between treated and vehicle groups, the previously reported 7 d pattern of rapid sprouting and synaptogenesis was not observed. Rather, the acute FN-439 exposure seemed to interfere with the removal of the majority of degenerated presynaptic terminals, which has been reported previously to occur over the first 4 d postlesion. At 7 d survival, persistent sites of degenerative terminals were visible throughout the deafferented molecular layer of animals subjected to MMP inhibition (Fig. 5A,D), whereas the normal evolution of reactive synaptogenesis was observed in the lesioned cases receiving the vehicle treatment (Fig. 5B,E). No evidence of nonspecific pathological effects of FN-439 on the dentate neuropil was seen in the contralateral, nonlesioned hippocampus of the drug-treated animals (Fig. 5C,F).
Figure 5.
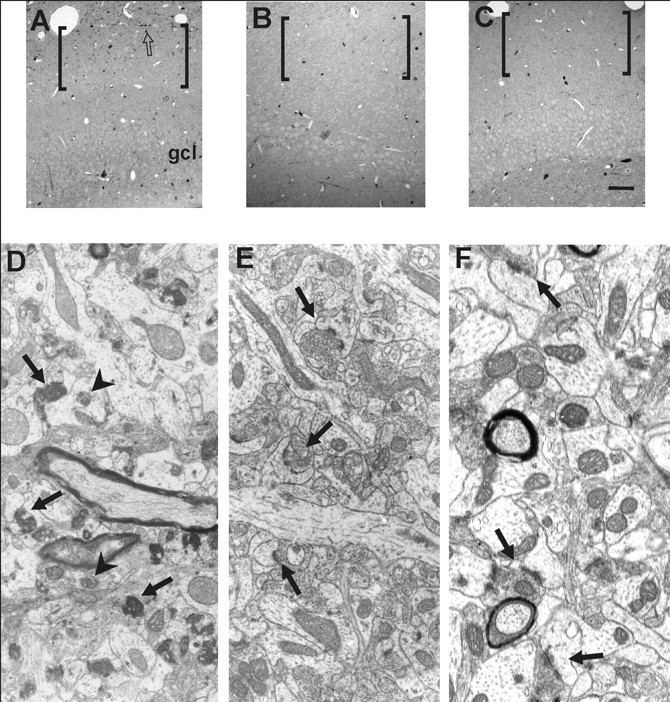
The structural effects of acute MMP inhibition with FN-439 on the dentate gyrus molecular layer at 7 d after UEC lesion. In A-C, thick (1 μm) plastic sections stained with toluidine blue show the cytoarchitecture of the dentate molecular layer (brackets) and adjacent granule cell bodies (gcl). In A, the ipsilateral denervated molecular layer of FN-439-treated lesion cases bore clear sites of degenerative debris (bracketed area; example of debris aggregate shown at open arrow), suggestive of the abnormal progress of reactive synaptogenesis. By contrast, the ipsilateral side of the vehicle-treated animals (B) shows little evidence of degenerative debris within the denervated neuropil. Similarly, the molecular layer within contralateral control in FN-439-treated cases exhibited no degenerative pathology (C). At the ultrastructural level (D-F), the dentate molecular layer morphology supports the observations shown in A-C. The effects of acute FN-439 treatment on molecular layer ultrastructure are profound (D). Numerous profiles of degenerative presynaptic terminals (arrows) remain visible. Few intact synapses can be seen, and dendrites appear distended, with disorganized cytoarchitecture and damaged mitochondria (arrowheads). In contrast, the ipsilateral side of cases receiving vehicle (E) exhibit successful synaptic reorganization, with multiple dendritic spines and synaptic profiles visible (arrows). Dendritic processes contain intact cytoskeletal substructure and normal profiles of mitochondria. Notably, the contralateral, nondeafferented neuropil of FN-439-treated animals shows intact synaptic architecture (see arrows in F). Scale bar (in C) A-C, 50 μm; magnification D, E, F, 20,000×.
Discussion
This study examined the degree to which inhibiting MMPs altered functional and structural properties of a fiber system undergoing deafferentation-induced sprouting in the adult nervous system. In vehicle-treated lesioned rats, the CTD fiber system exhibited a reactive sprouting response consistent with prior reports in this area, demonstrating normal clearance of degenerating terminals while revealing typical patterns of reactive synaptogenesis along with the acquisition of the capacity for LTP. In contrast, the deafferented neuropil in lesioned rats given FN-439 exhibited persistent degenerative debris and failed to develop the capacity for LTP. CSD profiles suggested that the normal dense current sink in the middle molecular layer was disrupted by MMP inhibition during the reactive sprouting process. Taken together, these results are consistent with a supportive role for MMPs in reactive synaptogenesis.
The MMP inhibitor interfered with the emergence of the capacity for LTP in the sprouting CTD, a functional correlate of the sprouting response that was documented systematically in a number of previous studies (Wilson, 1981; Wilson et al., 1981; Reeves and Steward, 1986). The strength of LTP observed at this relatively early stage in sprouting (7 d) was predictably modest (22% in fEPSP slopes). However, the significant suppression of this capacity by a single intraventricular FN-439 injection demonstrates that this survival interval was adequate to demonstrate the MMP dependence of this form of plasticity. The degree of the observed structural recovery in the dentate molecular layer was consistent with the observed changes in electrophysiology after MMP inhibition, because those sites that failed to exhibit LTP revealed persistent pathology and a paucity of synaptic reorganization.
Treatment of EC-lesioned rats with the MMP inhibitor also altered the evoked responses of the sprouting pathway to paired-pulse stimulation. The pattern of paired-pulse synaptic plasticity seen in this study was predominantly one of paired-pulse depression: the second fEPSP was reduced at all interpulse intervals for unlesioned control rats and for lesioned rats treated with the MMP inhibitor. Compared with these groups, rats with sprouting CTD systems (lesioned rats given only saline vehicle) exhibited a facilitation of the second fEPSP at an interpulse interval of 50 msec, but this group as well showed mainly paired-pulse depression for pulse intervals over 75 msec. Although stimulus currents in this study were selected to be subthreshold for granule cell population spikes, measurement stability in the sprouting commissural system often requires a relatively higher level of stimulation, conceivably activating feedforward inhibition, thereby contributing to the observed tendency toward paired-pulse depression. Although paired-pulse plasticity has been described extensively in the ipsilateral perforant pathway system (Lomo, 1971; DiScenna and Teyler, 1994; Burdette and Gilbert, 1995), only limited reports of similar measurements in the CTD system exist (Steward et al., 1976), conducting measurements at 30+ d postlesion and reporting that sprouting in the CTD augmented paired-pulse facilitation. It is unclear why the current results showed a greater tendency toward a depressive pattern in paired-pulse responses, however, these two studies both agree that the reinnervation process did shift this form of short-term plasticity in a facilitatory direction (Fig. 2 B).
CSD analysis in the present study indicated that MMP inhibition during the initial sprouting period altered the distribution of current sinks and sources in the dentate molecular layer. In normal rats and in lesioned rats not treated with the MMP inhibitor, a distinct current sink was observed in the middle molecular layer, analogous to the sink associated with the terminal region of the ipsilateral entorhinodentate system (Desmond and Levy, 1983; White et al., 1990; Leung et al., 1995; Canning and Leung, 1997). In contrast, the FN-439-treated group exhibited an altered sink/source pattern with a reduction in the middle dendritic sink. It is probable that this result reflects an interference in the putative boundary-forming and guidance functions of the MMPs and their ECM substrate molecules. The branching pattern of regenerating neurons, for example, has been demonstrated to be affected by the presence of chondroitin sulfate proteoglycan (CSPG) in the neuropil substrate (Davies et al., 1999), and, in the context of the current experiment, it is possible that pharmacological MMP inhibition altered the pattern of spatial arborization of the sprouting CTD fibers. Such a difference in spatial distribution of afferents after MMP inhibition is consistent with our previous observation (Phillips and Reeves, 2001) that the ECMs collagen IV, CSPG, and tenascin are all selectively elevated within the deafferented dendritic subregions when brain injuries involved entorhinal lesion.
There is growing evidence that common molecular mechanisms may be active in the development of neuron-substrate contacts and in use-dependent synaptic plasticity in the adult. Thus, diverse experimental approaches have shown that manipulation of ECM molecules, having documented developmental roles, also impairs LTP. Knock-out mice lacking heparin-binding growth-associated molecule (Amet et al., 2001) or tenascin-R (Bukalo et al., 2001; Chang et al., 2001) exhibit altered LTP, as do brain slice experiments showing an inhibition of LTP induction after bath application of neuropsin, an ECM serine protease (Komai et al., 2000), or of heparitinase (Lauri et al., 1999). A particularly relevant link between brain development and plasticity is the description of regional and cell-specific expression of MMPs and their endogenous inhibitors (tissue inhibitors of metalloproteinases) during cerebellar ontogenesis and in the adult (Vaillant et al., 1999). Vaillant et al. (1999) described the actions of some proteases in degrading receptors and cell adhesion molecules to eliminate synapses or ECM molecules, thus exposing cryptic adhesion sites for new synapses. These are properties expected of molecules regulating the remodeling of denervated neuropil. It is noteworthy that MMPs are also implicated in the pathogenesis of multiple sclerosis and Alzheimer's disease and may mediate angiogenesis in glioma cells (Yong et al., 1998; Lukes et al., 1999; Fillmore et al., 2001).
Changes in hippocampal ECM and regulatory enzyme expression are concurrent with multiple injury-induced processes. By the first postinjury day, glial hypertrophy (for review, see Deller et al., 1997) is coupled with upregulation of c-fos and c-jun (Phillips and Belardo, 1994; Haas et al., 1997) and neurotrophin mRNA (Forster et al., 1997). Within this same period, increased MMP expression and lysis of synapse-associated ECM has been documented using the kainate-induced model of hippocampal excitotoxicity (Yuan et al., 2002). Specifically, acute elevation of ADAMTS (a disintegrin and metalloprotease with thrombospondin motifs) mRNA correlated with both lysis of the CSPG, brevican, and reduction of synapse density in the dentate molecular layer. Such results support the concept that acute MMP inhibition may target ECM mechanisms critical to removal of degenerative synapses. During the first week after unilateral EC lesion, synapse proliferation is marked by increased protein synthesis within postsynaptic compartments (Phillips and Steward, 1990) and astrocytic biosynthesis of ECM and neurotrophins (Gage et al., 1988; Lee et al., 1997). Consistent with activation of ADAMTS proteinases after seizure-induced injury, our previous unilateral EC lesion results show that MMP expression and functional activity are spatially and temporally correlated with ECM enhancement during reactive synaptogenesis (Phillips and Reeves, 2001). ECMs within the denervated dentate neuropil may provide spatial guidance for collateral sprouting, an idea supported by the fact that tenascin-C and the CSPG neurocan are also spatially and temporally correlated with the pattern of dendritic reinnervation (Deller et al., 1997; Haas et al., 1999). Specifically, CTD terminal growth is restricted to the outer and commissural/associational projections to the inner molecular layer, with no translaminar sprouting observed across ECM boundaries (Deller et al., 1996).
Because MMPs can degrade essentially all components of the ECM when they are activated (Fujimura et al., 1999), MMPs may act to break down matrix constituents to enable efficient phagocytic clearance of degenerating fibers and terminals. Indeed, in the current study, rats treated with the MMP inhibitor exhibited persistent degenerative profiles, consistent with a prephagocytic MMP role. Beyond the initial degenerative phase, MMP regulation is likely to provide guidance during the proliferation of new synapses. Several observations support such a guidance function. For example, in the process of axonal extension, MMPs have been localized at the growth cone tips, permitting attachment/detachment between the neurons and the matrix substratum (Monard, 1988), and oligodendrocytes use MMPs to extend their processes (Uhm et al., 1998).
In summary, brain injuries that involve significant cell death or axonal injury will necessarily lead to diffuse or focal deafferentation (Erb and Povlishock, 1991), and the recovery of function after such injuries may depend, in part, on the ECM/MMP-directed repopulation of presynaptic input to deafferented target neurons. It is not necessary to assume that new synapses contribute to recovery by replicating the function of the degenerating terminals. Instead, collateral sprouting may restore the balance of excitation/inhibition in target neurons or reestablish the supply of trophic factors to denervated cells. Moreover, there is evidence that synaptogenesis interacts with other components of brain injury, most notably the excessive neuroexcitation resulting from posttraumatic elevations in excitatory neurotransmitter release (Reeves et al., 1997; Phillips and Reeves, 2001) and that ECM/MMP expression can vary with these interactions. It is conceivable that future progress in this area will impact the selection and sequence of postinjury pharmacological interventions, which may be chosen to facilitate useful synaptogenesis or to suppress instances of synaptogenesis that may be maladaptive.
Footnotes
This work was supported by National Institutes of Health Grants NS44372 and NS20193. We thank Susan Walker, Tom Coburn, Lesley Harris, and Raiford Black for technical assistance and Dr. Helen Fillmore for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Thomas M. Reeves, Department of Anatomy and Neurobiology, 1217 East Marshall Street, Room 740, Medical College of Virginia, Box 980709, Virginia Commonwealth University, Richmond, VA 23298. E-mail: tmreeves@hsc.vcu.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310182-08$15.00/0
References
- Amaral DG, Witter MP ( 1995) The hippocampal formation. In: The rat nervous system, Ed 2 (Paxinos G, ed), pp 443-494. New York: Academic.
- Amet LE, Lauri SE, Hienola A, Croll SD, Lu Y, Levorse JM, Prabhakaran B, Taira T, Rauvala H, Vogt TF ( 2001) Enhanced hippocampal long-term potentiation in mice lacking heparin-binding growth-associated molecule. Mol Cell Neurosci 17: 1014-1024. [DOI] [PubMed] [Google Scholar]
- Anan H, Matsumoto A, Hamachi T, Yoshimine Y, Morita Y, Maeda K ( 1996) Effects of a combination of an antibacterial agent (ofloxacin) and a collagenase inhibitor (FN-439) on the healing of rat periapical lesions. J Endod 22: 668-673. [DOI] [PubMed] [Google Scholar]
- Bukalo O, Schachner M, Dityatev A ( 2001) Modification of extracellular matrix by enzymatic removal of chondroitin sulfate and by lack of tenascin-R differentially affects several forms of synaptic plasticity in the hippocampus. Neuroscience 104: 359-369. [DOI] [PubMed] [Google Scholar]
- Buki A, Okonkwo DO, Povlishock JT ( 1999) Postinjury cyclosporin A administration limits damage and disconnection in traumatic brain injury. J Neurotrauma 16: 511-521. [DOI] [PubMed] [Google Scholar]
- Burdette LJ, Gilbert ME ( 1995) Stimulus parameters affecting paired-pulse depression of dentate granule cell field potentials. I. Stimulus intensity. Brain Res 680: 53-62. [DOI] [PubMed] [Google Scholar]
- Canning KJ, Leung LS ( 1997) Lateral entorhinal, perirhinal, and amygdalaentorhinal transition projections to hippocampal CA1 and dentate gyrus in the rat: a current source density study. Hippocampus 7: 643-655. [DOI] [PubMed] [Google Scholar]
- Chang HP, Ma YL, Wan FJ, Tsai LY, Lindberg FP, Lee EH ( 2001) Functional blocking of integrin-associated protein impairs memory retention and decreases glutamate release from the hippocampus. Neuroscience 102: 289-296. [DOI] [PubMed] [Google Scholar]
- Choi BH ( 1994) Role of the basement membrane in neurogenesis and repair of injury in the central nervous system. Microsc Res Tech 28: 193-203. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Nieto-Sampedro M, Harris EW ( 1981) Synapse replacement in the nervous system of adult vertebrates. Physiol Rev 61: 684-784. [DOI] [PubMed] [Google Scholar]
- Davies SJA, Goucher DA, Doller C, Silver J ( 1999) Robust regeneration of adult sensory axons in degenerating white matter of the adult rat spinal cord. J Neurosci 19: 5810-5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis L, Vinsant SL, Steward O ( 1988) Ultrastructural characterization of the synapses of the crossed temporodentate pathway in rats. J Comp Neurol 267: 190-202. [DOI] [PubMed] [Google Scholar]
- Deller T, Frotscher M ( 1997) Lesion-induced plasticity of central neurons: sprouting of single fibres in the rat hippocampus after unilateral entorhinal cortex lesion. Prog Neurobiol 53: 687-727. [DOI] [PubMed] [Google Scholar]
- Deller T, Nitsch R, Frotscher M ( 1996) Layer-specific sprouting of commissural fibers to the rat fascia dentata after unilateral entorhinal cortex lesion: a Phaseolus vulgaris leucoagglutinin tracing study. Neuroscience 71: 651-660. [DOI] [PubMed] [Google Scholar]
- Deller T, Haas CA, Naumann T, Joester A, Faissner A, Frotscher M ( 1997) Upregulation of astrocyte-derived tenascin-C correlates with neurite outgrowth in the rat dentate gyrus after unilateral entorhinal cortex lesion. Neuroscience 81: 829-846. [DOI] [PubMed] [Google Scholar]
- Desmond NL, Levy WB ( 1983) Synaptic correlates of associative potentiation/depression: an ultrastructural study in the hippocampus. Brain Res 265: 21-30. [DOI] [PubMed] [Google Scholar]
- DiScenna PG, Teyler TJ ( 1994) Development of inhibitory and excitatory synaptic transmission in the rat dentate gyrus. Hippocampus 4: 569-576. [DOI] [PubMed] [Google Scholar]
- Erb DE, Povlishock JT ( 1991) Neuroplasticity following traumatic brain injury: a study of GABAergic terminal loss and recovery in the cat dorsal lateral vestibular nucleus. Exp Brain Res 83: 253-267. [DOI] [PubMed] [Google Scholar]
- Fillmore HL, VanMeter TE, Broaddus WC ( 2001) Membrane-type matrix metalloproteinases (MT-MMPs): expression and function during glioma invasion. J Neuro-oncol 53: 187-202. [DOI] [PubMed] [Google Scholar]
- Forster E, Naumann T, Deller T, Straube A, Nitsch R, Frotscher M ( 1997) Cholinergic sprouting in the rat fascia dentata after entorhinal lesions is not linked to early changes in neurotrophin messenger RNA expression. Neuroscience 80: 731-739. [DOI] [PubMed] [Google Scholar]
- Frotscher M ( 1988) Neuronal elements in the hippocampus and their synaptic connections. Adv Anat Embryol 111: 2-17. [Google Scholar]
- Fujimura M, Gasche Y, Morita-Fujimura Y, Massengale J, Kawase M, Chan PH ( 1999) Early appearance of activated matrix metalloproteinase-9 and blood-brain barrier disruption in mice after focal cerebral ischemia and reperfusion. Brain Res 842: 92-100. [DOI] [PubMed] [Google Scholar]
- Gage FH, Olejniczak P, Armstrong DM ( 1988) Astrocytes are important for sprouting in the septohippocampal circuit. Exp Neurol 102: 2-13. [DOI] [PubMed] [Google Scholar]
- Haas CA, Deller T, Frotscher M ( 1997) Basal expression, subcellular distribution, and upregulation of the protooncogene c-JUN in the rat dentate gyrus after unilateral entorhinal cortex lesion. Neuroscience 81: 33-45. [DOI] [PubMed] [Google Scholar]
- Haas CA, Rauch U, Thon N, Merten T, Deller T ( 1999) Entorhinal cortex lesion in adult rats induces the expression of the neuronal chondroitin sulfate proteoglycan neurocan in reactive astrocytes. J Neurosci 19: 9953-9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D, Wu SM-S ( 1995) Foundations of cellular neurophysiology. Cambridge, MA: MIT.
- Kigasawa K, Murata H, Morita Y, Odake S, Suda E, Shimizu I, Morikawa T, Nagai Y ( 1995) Inhibition of corneal ulceration by tetrapeptidyl hydroxamic acid. Jpn J Opthalmol 39: 35-42. [PubMed] [Google Scholar]
- Komai S, Matsuyama T, Matsumoto K, Kato K, Kobayashi M, Imamura K, Yoshida S, Ugawa S, Shiosaka S ( 2000) Neuropsin regulates an early phase of schaffer-collateral long-term potentiation in the murine hippocampus. Eur J Neurosci 12: 1479-1486. [DOI] [PubMed] [Google Scholar]
- Lander AD ( 1993) Proteoglycans in the nervous system. Curr Opin Neurobiol 3: 716-723. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Kaukinen S, Kinnunen T, Yilnen A, Imai S, Kaila K, Taira T, Rauvala H ( 1999) Regulatory role and molecular interactions of a cell-surface heparan sulfate proteoglycan (N-syndecan) in hippocampal long-term potentiation. J Neurosci 19: 1226-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M-Y, Hofmann H-D, Kirsch M ( 1997) Differential regulation of CNTF and CNTF receptor alpha expression in astrocytes and neurons of the fascia dentata following entorhinal cortex lesion. J Neurosci 17: 1137-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung LS, Fu X-W ( 1994) Factors affecting paired-pulse facilitation in hippocampal CA1 neruons in vitro Brain Res 650: 75-84. [DOI] [PubMed] [Google Scholar]
- Leung LS, Roth L, Canning KJ ( 1995) Entorhinal inputs to hippocampal CA1 and dentate gyrus in the rat: a current-source-density study. J Neurophysiol 73: 2392-2403. [DOI] [PubMed] [Google Scholar]
- Loesche J, Steward O ( 1977) Behavioral correlates of denervation and reinnervation of the hippocampal formation of the rat: recovery of alternation performance following unilateral entorhinal cortex lesions. Brain Res Bull 2: 31-39. [DOI] [PubMed] [Google Scholar]
- Lomo T ( 1971) Potentiation of monosynaptic EPSPs in the perforant path dentate granule cell synapse. Exp Brain Res 12: 46-63. [PubMed] [Google Scholar]
- Lukes A, Mun-Bryce S, Lukes M, Rosenberg GA ( 1999) Extracellular matrix degradation by metalloproteinases and central nervous system diseases. Mol Neurobiol 19: 267-284. [DOI] [PubMed] [Google Scholar]
- Matthews DA, Cotman CW, Lynch G ( 1976a) An electron microscopic study of lesion-induced synaptogenesis in the dentate gyrus of the adult rat. I. Magnitude and time course of degeneration. Brain Res 115: 1-21. [DOI] [PubMed] [Google Scholar]
- Matthews DA, Cotman CW, Lynch G ( 1976b) An electron microscopic study of lesion-induced synaptogenesis in the dentate gyrus of the adult rat. II. Reappearance of morphologically normal synaptic contacts. Brain Res 115: 23-41. [DOI] [PubMed] [Google Scholar]
- Monard D ( 1988) Cell-derived proteases and protease inhibitors as regulators of neurite outgrowth. Trends Neurosci 11: 541-544. [DOI] [PubMed] [Google Scholar]
- Odake S, Morita Y, Morikawa T, Yoshida N, Hori H, Nagai Y ( 1994) Inhibition of metalloproteinases by peptidyl hydroxiamic acids. Biochem Biophys Res Commun 199: 1442-1446. [DOI] [PubMed] [Google Scholar]
- Phillips LL, Belardo ET ( 1994) Increase of c-fos and ras oncoproteins in the denervated neuropil of the rat dentate gyrus. Neuroscience 58: 503-514. [DOI] [PubMed] [Google Scholar]
- Phillips LL, Reeves TM ( 2001) Interactive pathology following traumatic brain injury modifies hippocampal plasticity. Restor Neurol Neurosci 19: 213-235. [PubMed] [Google Scholar]
- Phillips LL, Steward O ( 1990) Increases in mRNA for cytoskeletal proteins in the denervated neuropil of the dentate gyrus: an in situ hybridization study using riboprobes for beta-actin and beta-tubulin. Mol Brain Res 8: 249-257. [DOI] [PubMed] [Google Scholar]
- Phillips LL, Lyeth BG, Hamm RJ, Povlishock JT ( 1994) Combined fluid percussion brain injury and entorhinal cortical lesion: a model for assessing the interaction between neuroexcitation and deafferentation. J Neurotrauma 11: 641-656. [DOI] [PubMed] [Google Scholar]
- Ramirez JJ, Stein DG ( 1984) Sparing and recovery of spatial alternation performance after entorhinal cortex lesions in rats. Behav Brain Res 13: 53-61. [DOI] [PubMed] [Google Scholar]
- Ramirez JJ, McQuilkin M, Carrigan T, MacDonald K, Kelly MS ( 1996) Progressive enthorhinal cortex lesions accelerate hippocampal sprouting and spare spatial memory in rats. Proc Natl Acad Sci USA 93: 15512-15517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves TM, Smith DC ( 1987) Reinnervation of the dentate gyrus and recovery of alternation behavior following entorhinal cortical lesions. Behav Neurosci 101: 179-186. [DOI] [PubMed] [Google Scholar]
- Reeves TM, Steward O ( 1986) Emergence of the capacity for LTP during reinnervation of the dentate gyrus: evidence that abnormally shaped spines can mediate LTP. Exp Brain Res 65: 167-175. [DOI] [PubMed] [Google Scholar]
- Reeves TM, Lyeth BG, Povlishock JT ( 1995) Long-term potentiation deficits and excitability changes following traumatic brain injury. Exp Brain Res 106: 248-256. [DOI] [PubMed] [Google Scholar]
- Reeves TM, Zhu J, Povlishock JT, Phillips LL ( 1997) The effect of combined fluid percussion and entorhinal cortical lesions on long-term potentiation. Neuroscience 77: 431-444. [DOI] [PubMed] [Google Scholar]
- Richardson TL, Turner RW, Miller JJ ( 1987) Action-potential discharge in hippocampal CA1 pyramidal neurons: current source-density analysis. J Neurophysiol 58: 981-996. [DOI] [PubMed] [Google Scholar]
- Steward O ( 1976) Reinnervation of the dentate gyrus by homologous afferents following entorhinal cortex lesion in adult rats. Science 194: 426-428. [DOI] [PubMed] [Google Scholar]
- Steward O ( 1991) Synapse replacement on cortical neurons following denervation. Cereb Cortex 9: 81-132. [Google Scholar]
- Steward O ( 1994) Reorganization of neuronal circuitry following central nervous system trauma: naturally occurring processes and opportunities for therapeutic intervention. In: The neurobiology of central nervous system trauma (Salzman SK, Faden AI, eds), pp 266-287. New York: Oxford UP.
- Steward O, Vinsant SL ( 1983) The process of reinnervation in the dentate gyrus of the adult rat: a quantitative electron microscopic analysis of terminal proliferation and reactive synaptogenesis. J Comp Neurol 214: 370-386. [Google Scholar]
- Steward O, White CW, Cotman CW, Lynch G ( 1976) Potentiation of excitatory synaptic transmission in the normal and in the reinnervated dentate gyrus of the rat. Exp Brain Res 26: 423-441. [DOI] [PubMed] [Google Scholar]
- Uhm JH, Dooley NP, Oh LY, Yong VW ( 1998) Oligodendrocytes utilize a matrix metalloproteinase, MMP-9, to extend processes along an astrocyte extracellular matrix. Glia 22: 53-63. [DOI] [PubMed] [Google Scholar]
- Vaillant C, Didier-Bazes M, Hutter A, Belin M-F, Thomasset N ( 1999) Spatiotemporal expression patterns of metalloproteinases and their inhibitors in the postnatal developing rat cerebellum. J Neurosci 19: 4994-5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz M, Dixon CE, Fini ME, Lo EH ( 2000) Effects of matrix metalloproteinase-9 gene knockout on morphological and motor outcomes after traumatic brain injury. J Neurosci 20: 7037-7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White G, Levy WB, Steward O ( 1990) Spatial overlap between populations of synapses determines the extent of their associative interaction during the induction of long-term potentiation and depression. J Neurophysiol 64: 1186-1198. [DOI] [PubMed] [Google Scholar]
- Wilson RC ( 1981) Changes in translation of synaptic excitation to dentate granule cell discharge accompanying long-term potentiation. I. Differences between normal and reinnervated dentate gyrus. J Neurophysiol 46: 324-338. [DOI] [PubMed] [Google Scholar]
- Wilson RC, Levy WB, Steward O ( 1981) Changes in translation of synaptic excitation to dentate granule cell discharge accompanying long-term potentiation. II. An evaluation of mechanisms utilizing dentate gyrus dually innervated by surviving ipsilateral and sprouted crossed temporodentate inputs. J Neurophysiol 46: 339-355. [DOI] [PubMed] [Google Scholar]
- Wyss JM ( 1981) An autoradiographic study of the efferent connections of the entorhinal cortex in the rat. J Comp Neurol 199: 495-512. [DOI] [PubMed] [Google Scholar]
- Yaghmai A, Povlishock JT ( 1992) Traumatically induced reactive change as visualized through the use of monoclonal antibodies targeted to neuro-filament subunits. J Neuropathol Exp Neurol 51: 158-176. [DOI] [PubMed] [Google Scholar]
- Yong VW, Krekoski CA, Forsyth PA, Bell R, Edwards DR ( 1998) Matrix metalloproteinases and disease of the CNS. Trends Neurosci 21: 75-80. [DOI] [PubMed] [Google Scholar]
- Yuan W, Matthews RT, Sandy JD, Gottschall PE ( 2002) Association between protease-specific proteolytic cleavage of brevican and synaptic loss in the dentate gyrus of kainate-treated rats. Neuroscience 114: 1091-1101. [DOI] [PubMed] [Google Scholar]
- Zucker RS ( 1989) Short-term synaptic plasticity. Annu Rev Neurosci 12: 13-31. [DOI] [PubMed] [Google Scholar]