

Abstract
When acutely dissociated small-diameter dorsal root ganglion (DRG) neurons were stimulated with repeated current injections or prolonged application of capsaicin, their action potential firing quickly adapted. Because TTX-resistant (TTX-R) sodium current in these presumptive nociceptors generates a large fraction of depolarizing current during the action potential, we examined the possible role of inactivation of TTX-R sodium channels in producing adaptation. Under voltage clamp, TTX-R current elicited by short depolarizations showed strong use dependence at frequencies as low as 1 Hz, although recovery from fast inactivation was complete in ∼10-30 msec. This use-dependent reduction was the result of the entry of TTX-R sodium channels into slow inactivated states. Slow inactivation was more effectively produced by steady depolarization than by cycling channels through open states. Slow inactivation was steeply voltage dependent, with a Boltzmann slope factor of 5 mV, a midpoint near -45 mV (5 sec conditioning pulses), and completeness of ∼93% positive to -20 mV. The time constant for entry (∼200 msec) was independent of voltage from -20 mV to +60 mV, whereas recovery kinetics were moderately voltage dependent (time constant, ∼1.5 sec at -60 mV and ∼0.5 sec at -100 mV). Using a prerecorded current-clamp response to capsaicin as a voltage-clamp command waveform, we found that adaptation of firing occurred with a time course similar to that of development of slow inactivation. Thus, slow inactivation of the TTX-R sodium current limits the duration of small DRG cell firing in response to maintained stimuli and may contribute to cross desensitization between chemical and electrical stimuli.
Keywords: action potential, dorsal root ganglion, excitability, nociceptor, sodium channel, spike-frequency adaptation, tetrodotoxin
Introduction
In response to maintained mechanical, heat, or capsaicin stimulation, nociceptive fibers respond with action potential firing that shows prominent adaptation over several seconds (Handwerker et al., 1987; Gallar et al., 1993; Andrew and Greenspan, 1999; Slugg et al., 2000). In some cases, excitation by one stimulus can reduce the response of the nociceptor to other stimuli (Petsche et al., 1983; LaMotte et al., 1992; Gallar et al., 1993; Peng et al., 2003), suggesting that processes downstream to receptor desensitization are involved. Adaptation of firing frequency is often accompanied by a reduction in conduction velocity (Thalhammer et al., 1994; Gee et al., 1996; Serra et al., 1999), implying that intrinsic membrane excitability is depressed.
In neurons in general, several mechanisms produce adaptation of action potential firing during maintained depolarizing stimuli, including activation of calcium-activated potassium current (Madison and Nicoll, 1984; Weinreich and Wonderlin, 1987; Scholz et al., 1998b), the potassium current IM (Wang and McKinnon, 1995), and hyperpolarizing current from enhanced Na/K-ATPase activity (French, 1989). Another mechanism is a progressive reduction in available sodium conductance through a process of “slow inactivation” that develops and reverses with time constants of hundreds of milliseconds to seconds, much slower than normal fast inactivation (for review, see Vilin and Ruben, 2001). Fleidervish et al. (1996) showed that a reduction in available sodium current caused by slow inactivation contributes to adaptation of firing in neocortical pyramidal neurons. Slow inactivation of sodium current has also been proposed to underlie time-dependent changes in the excitability of hippocampal CA1 dendrites (Colbert et al., 1997; Jung et al., 1997; Mickus et al., 1999), hypoglossal motoneurons (Powers et al., 1999), salamander retinal ganglion neurons (Kim and Rieke, 2003), and subthalamic nucleus neurons (Do and Bean, 2003).
Nociceptive neurons differ from most other neurons in expressing voltage-gated sodium current resistant to TTX (for review, see McCleskey and Gold, 1999; Waxman et al., 1999; Baker and Wood, 2001), generally in combination with TTX-sensitive (TTX-S) sodium channels. Expression of the channels mediating TTX-resistant (TTX-R) current, NaV1.8 and NaV1.9, has been implicated in mediating the sensation of pain (Akopian et al., 1999; Porreca et al., 1999; Yoshimura et al., 2001; Laird et al., 2002). The kinetic properties of TTX-R channels differ from those of TTX-S channels in ways that are likely to be significant for their functional roles. In particular, TTX-R current decays more slowly during a step depolarization (fast inactivation) than do TTX-S channels and recovers from inactivation more quickly. However, TTX-R sodium current in sensory neurons also shows prominent slow inactivation (Ogata and Tatebayashi, 1992; Rush et al., 1998; Fazan et al., 2001), which can be sufficiently pronounced to produce substantial use-dependent reduction of TTX-R current in response to low-frequency stimulation (Rush et al., 1998; Scholz et al., 1998b). Similarly, NaV1.8 channels heterologously expressed in Xenopus oocytes show prominent slow inactivation (Vijayaragavan et al., 2001). We examined the kinetics and voltage dependence of the slow inactivation of TTX-R channels in small dorsal root ganglion (DRG) neurons and tested whether it can account for the adaptation of firing during maintained stimulation of the cells.
Materials and Methods
Cell preparation. Dissociated DRG neurons were prepared as described previously (Blair and Bean, 2002). DRGs were removed from Long-Evans rats (postnatal day 14-18), cut in half, and treated for 20 min at 37°C with 20 U/ml papain (Worthington Biochemical, Lakewood, NJ) and 5 mm dl-cysteine in a Ca2+, Mg2+-free (CMF) HBSS containing the following (in mm): 136.9 NaCl, 5.4 KCl, 0.34 Na2HPO4, 0.44 KH2PO4, 5.55 glucose, 5 HEPES, and 0.005% phenol red, pH 7.4. Ganglia were then treated for 20 min at 37°C with 3 mg/ml collagenase (type I; Sigma-Aldrich, St. Louis, MO) and 4 mg/ml dispase II (Boehringer Mannheim, Indianapolis, IN) in CMF HBSS. Cells were dispersed by trituration with a fire-polished glass Pasteur pipette in a solution of Leibovitz's L-15 medium (Invitrogen, San Diego, CA) supplemented with 10% fetal calf serum, 5 mm HEPES, 50 U/ml penicillin, 50 μg/ml streptomycin, 2 mml-glutamine, and 100 ng/ml NGF (Invitrogen), and then plated on glass coverslips treated with 200 μg/ml poly-d-lysine. Cells were incubated in the supplemented L-15 solution at 33°C (room air) for 2-4 hr, after which they were stored at 4°C and used within 48 hr.
General electrophysiology. Whole-cell voltage- and current-clamp recordings were made from DRG cells using an Axopatch 200B Amplifier (Axon Instruments, Union City, CA), with electrodes pulled from Corning 7052 glass (A-M systems, Sequim, WA) that had resistances of 2-6 MΩ when filled with K-methanesulfonate or N-methyl-d-glucamine (NMDG)-aspartate internal solutions. Pipette tips were wrapped with thin strips of parafilm to reduce capacitance. Seals were formed in Tyrode's solution containing the following (in mm): 150 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 glucose, and 10 HEPES, pH 7.4 (with NaOH). Series resistance was compensated 85-95%. Solutions were applied after lifting the cell in front of an array of quartz fiber flow pipes. Cells with less than -500 pA TTX-R sodium current during a test step from -100 mV to 0 mV were not included in the analysis. Experiments were conducted within 25 min after the establishment of a whole-cell configuration to help minimize the time-dependent rundown in TTX-R sodium currents (Schild and Kunze, 1997). Cell diameters were measured from images captured to computer from a CCD camera (Hitachi, Woodbury, NY) using a video acquisition card (Scion, Frederick, MD) with a resolution of 0.4 μm. All experiments were performed at room temperature (20-24°C).
Data acquisition and analysis. Currents and voltages were controlled and sampled using a Digidata 1321A interface and pClamp 8 software (Axon Instruments). Signals were filtered at 5-10 kHz (-3 dB, four pole Bessel) and digitized at 10-100 μsec. Analysis was performed using Igor Pro (version 4.06; Wavemetrics, Lake Oswego, OR) with DataAccess (Bruxton, Seattle, WA) used to import pClamp files. Currents were corrected for leak and residual uncompensated capacity currents by scaling averaged responses to inverted voltage steps that were 0.1-0.5 in amplitude.
Current-clamp experiments. The K-aspartate internal solution used in current-clamp recordings contained the following (in mm): 140 K-aspartate, 13.5 NaCl, 1.6 MgCl2, 0.09 EGTA, 9 HEPES, 4 MgATP, 14 Tris-creatine PO4, and 0.3 Tris-GTP, pH 7.4 (with KOH). The external solution was Tyrode's solution. The prerecorded capsaicin response used as a voltage-clamp waveform (see Figs. 1 A,8, 9, 10) was recorded with a K-methanesulfonate internal solution containing the following (in mm): 140 K-methanesulfonate, 13.5 NaCl, 1.6 MgCl2, 0.09 EGTA, 0.9 glucose, 9 HEPES, 4 MgATP, 14 Tris-creatine PO4, and 0.3 Tris-GTP, pH 7.4 (with KOH). No differences were noted between recordings made with K-aspartate and K-methanesulfonate internal solutions. All recordings were made using the fast current-clamp mode of the Axopatch 200B.
Figure 1.
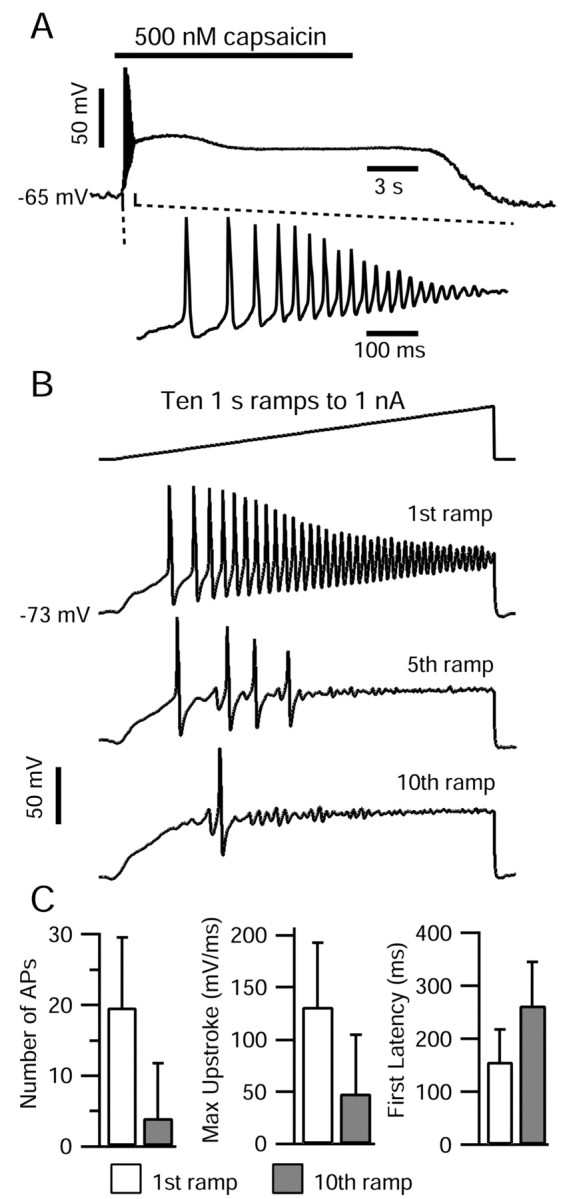
Current-clamp responses of DRG cells to capsaicin application and to repeated current ramp injections. A, Top, Application of 500 nm capsaicin (bar) elicited a brief period of action potential firing, after which the cell remained depolarized at -27 mV. A, Bottom, 800 msec of the same capsaicin response on an expanded time base. The internal solution contained the following (in mm): 140 K-methanesulfonate, 13.5 NaCl, 1.6 MgCl2, 0.09 EGTA, 9 HEPES, 4 MgATP, 14 Tris-creatine PO4, and 0.3 Tris-GTP, pH 7.4 (with KOH). The external solution was Tyrode's solution. B, Ten 1 sec current ramps to 1 nA were injected at 0.5 Hz. The internal solution contained the following (in mm): 140 K-aspartate, 13.5 NaCl, 1.6 MgCl2, 0.09 EGTA, 9 HEPES, 4 MgATP, 14 Tris-creatine PO4, and 0.3 Tris-GTP, pH 7.4 (with KOH). The external solution was Tyrode's solution. C, Comparison of action potential firing elicited during first current ramp (open bars) and 10th current ramp (gray bars) applied at 0.5 Hz. The total number of action potentials (peaks >0 mV), the maximal upstroke of the first action potential, and the latency from the current ramp onset to the first action potential are shown. Error bars indicate SD.
Figure 8.

TTX-S and TTX-R sodium currents flowing during the voltage waveform elicited by capsaicin application. Top, Voltage command taken from current-clamp response to 500 nm capsaicin (Fig. 1 A). Middle, TTX-R sodium currents flowing in response to capsaicin response waveform. To improve voltage control, currents were recorded in an external solution with reduced sodium containing the following (in mm): 50 NaCl, 100 TEA-Cl, 4 CsCl, 2 CaCl2, 2 MgCl2, 0.03 CdCl2, 10 glucose, and 10 HEPES, pH 7.4 (with TEA-OH). Outward currents occur when spike height exceeds sodium reversal potential in reduced sodium (∼31 mV). Bottom, TTX-S sodium current flowing during the capsaicin response waveform, determined as the current sensitive to block by 300 nm TTX in an external solution containing the following (in mm): 50 NaCl, 100 TEA-Cl, 2 BaCl2, 0.3 CdCl2, 10 glucose, and 10 HEPES, pH 7.4 (with TEA-OH) (different cell from top). The internal solution for both recordings contained the following (in mm): 126 NMDG, 120 aspartate, 15 NaCl, 1.8 MgCl2, 9 EGTA, and 9 HEPES, pH 7.4 (with 7 mm CsOH).
Figure 9.
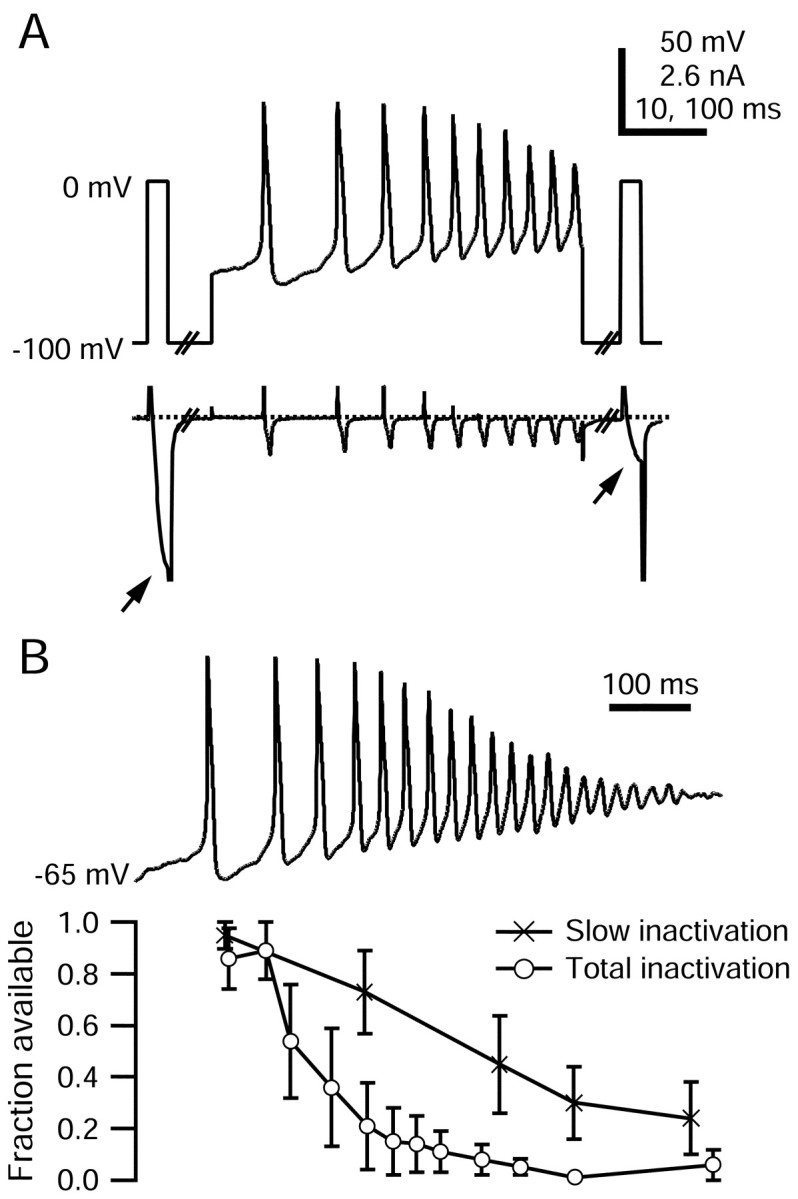
Inactivation of TTX-R sodium current during capsaicin response. A, Voltage protocol and TTX-R sodium currents evoked during a single sweep to measure slow inactivation elicited by an ∼490 msec segment of the capsaicin response. TTX-R sodium currents during 2.5 msec test steps to 0 mV (arrows) are shown on an expanded time scale. A 30 msec step to -100 mV preceded the second test pulse to remove fast inactivation. Large inward tail currents flowing during repolarizations to -100 mV have been truncated. Persistent sodium current between spikes is compressed because of the vertical scale. The internal solution contained the following (in mm): 130 NMDG, 120 aspartate, 15 NaCl, 1.8 MgCl2, 9 EGTA, 9 HEPES, 4 MgATP, 14 Tris-creatine PO4, and 0.3 Tris-GTP, pH 7.4 (with 7 mm CsOH). The external solution contained the following (in mm): 50 NaCl, 100 TEA-Cl, 4 CsCl, 2 CaCl2, 2 MgCl2, 0.03 CdCl2, 10 glucose, and 10 HEPES, pH 7.4 (with TEA-OH). B, Fraction of TTX-R sodium current available (second test step current normalized to the first) plotted as a function of time during the capsaicin response. Total inactivation (open circles) was measured with test steps directly to 0 mV; slow inactivation (crosses) was measured with test steps preceded by hyperpolarizing pulses to remove fast inactivation. Error bars indicate SD.
Figure 10.

Recovery from fast inactivation during interspike interval in the capsaicin response waveform. A, Top, Voltage protocol used to measure TTX-R sodium channel recovery from fast inactivation in the interval between the third and fourth action potentials (outlined in B). For comparison, the TTX-R sodium current available after removal of all fast inactivation by a 30 msec step to -100 mV applied near the peak of the AHP is shown at right (69% of initial current). Dotted lines show zero current level. Large inward tail currents flowing during repolarizations to -100 mV have been truncated, as have outward currents during the action potential upstroke and the test step onset. The internal solution contained the following (in mm): 130 NMDG, 120 aspartate, 15 NaCl, 1.8 MgCl2, 9 EGTA, 9 HEPES, 4 MgATP, 14 Tris-creatine PO4, and 0.3 Tris-GTP, pH 7.4 (with 7 mm CsOH). The external solution contained the following (in mm): 50 NaCl, 100 TEA-Cl, 4 CsCl, 2 CaCl2, 2 MgCl2, 0.03 CdCl2, 10 glucose, and 10 HEPES, pH 7.4 (with TEA-OH). B, Average fraction available TTX-R sodium current is shown throughout the capsaicin response. Available current ∼5 msec after a spike (open squares) and later in interspike interval (filled circles). Error bars indicate SD. Lines connect values during the same interspike interval. Reduction in TTX-R current availability caused by slow inactivation is shown in crosses (error bars omitted).
Cells recorded in current clamp were assayed for the expression of the hyperpolarization-activated cation current Ih and the transient potassium current IA, and for response to the application of 500 nm capsaicin. Ih was measured in voltage clamp as the inward current activated at the completion of a 500 msec step from -60 to -130 mV. IA was measured as the peak outward current flowing within 60 msec after the return to -60 mV after the 500 msec step to -130 mV. Sensitivity to capsaicin was tested in either current clamp or voltage clamp. Cells were classified as capsaicin-sensitive if in current clamp the application of 500 nm capsaicin (prepared daily from stock) generated a depolarization, or if in voltage clamp the inward current at a holding potential of -70 mV increased; in both cases the effect of capsaicin reversed when the cell was returned to control Tyrode's solution.
The majority of DRG cells from which we recorded had properties consistent with nociceptors. Cells had small diameters (27.9 ± 3.2 μm; n = 25); action potentials elicited by 0.5 msec current injections had prominent “shoulders” during the repolarization, long durations (average, 4.2 ± 1.9 msec at half-maximal amplitude; n = 26), and long duration afterhyperpolarizations (AHPs) that decayed by 80% within 315 ± 177 msec (n = 24; in six cells the AHP had not decayed by 80% within the 550 msec recorded duration; a value of 550 msec was used for statistics; two other cells were followed by afterdepolarizations). Twenty of 25 cells responded to 500 nm capsaicin, added in current or voltage clamp. Ih expression was generally low, with an average density of 2.2 ± 3.1 pA/pF (n = 26). The expression of IA was more variable, with 19 of 26 cells expressing <5 pA/pF, whereas the remaining seven cells expressed up to 59 pA/pF (overall average, 11 ± 17 pA/pF; n = 26). According to the classification system of Cardenas et al. (1995) and Petruska et al. (2000, 2002), the majority of cells would correspond to types 1 and 2, whereas the few cells with larger Ih would possibly correspond to type 7; all of these types are believed to correspond with types of nociceptors.
Voltage-clamp experiments. We isolated the TTX-R sodium current using an NMDG-aspartate internal solution containing the following (in mm): 126 or 130 NMDG, 120 aspartate, 15 NaCl, 1.8 MgCl2, 9 EGTA, 9 HEPES, 4 MgATP, 14 Tris-creatine PO4, and 0.3 Tris-GTP, pH 7.4 (with 7 mm CsOH). The external solution contained the following (in mm): 50 NaCl, 100 TEA-Cl, 4 CsCl, 2 CaCl2, 2 MgCl2, 0.03 CdCl2, 10 glucose, and 10 HEPES, pH 7.4 (with NaOH) with 300 nm TTX. The low concentration of CdCl2 minimizes the direct block of TTX-R sodium channels (Ikeda and Schofield, 1987; Roy and Narahashi, 1992; Kuo et al., 2002). Calcium channels are >95% blocked by 30 μm Cd2+ at 0 mV, leaving test steps used to assay TTX-R sodium current availability free of calcium current (Swandulla and Armstrong, 1989).
TTX-S sodium current was isolated as the current that was blocked by the addition of 300 nm TTX, a concentration that fully blocks TTX-S sodium channels while sparing TTX-R sodium channels (Roy and Narahashi, 1992; Ogata and Tatebayashi, 1993). TTX was applied in an external solution containing the following (in mm): 50 NaCl, 100 TEA-Cl, 2 BaCl2, 0.3 CdCl2, 10 glucose, and 10 HEPES, pH 7.4 (with NaOH).
Voltage-clamp protocols. In experiments examining the recovery of TTX-R sodium channels from fast inactivation and the details of TTX-R sodium channel slow inactivation, protocols were designed to minimize the accumulation of use dependence. The holding potential was -100 mV; test steps applied to assay available current were brief, typically 2-4 msec; intersweep intervals were 10-45 sec.
All voltages have been corrected for the measured liquid junction potentials between internal solutions and Tyrode's solution present when zeroing pipette current. The K-aspartate solution had a liquid junction potential of -10 mV, and the NMDG-aspartate had a liquid junction potential of -4 mV (both measured using a flowing 3 m KCl bridge, as described by Neher (1992). The capsaicin-evoked waveform used in Figures 8, 9, 10 was first adjusted by -8 mV to account for the liquid junction potential present when recorded with the K-methanesulfonate internal solution, and then adjusted by +4 mV to account for the NMDG-aspartate internal solution used in voltage-clamp experiments.
Drugs. TTX was from Calbiochem (La Jolla, CA). All other chemicals were from Sigma-Aldrich.
Statistical analysis. Data are presented as means ± SD.
Results
Firing adaptation in small DRG neurons
Figure 1 shows a typical record of action potential firing caused by the application of 500 nm capsaicin to a small-diameter DRG neuron recorded in current clamp. Action potential firing began within ∼200 msec. However, firing lasted for >1 sec, although the cell remained depolarized (between -15 and -27 mV) for the full duration (∼15 sec) that capsaicin was present. The peaks of the first action potentials were quite positive (more than +40 mV), but after the first four or five spikes, the peaks steadily decreased until damped oscillations remained. After ∼1 sec, the oscillations ceased.
Of the 16 DRG neurons tested in current clamp with 500 nm capsaicin, seven responded with an initial burst of between 2 and 11 action potentials, after which they remained depolarized at an average membrane potential of -29 ± 17 mV. Six other cells depolarized to an average potential of -43 ± 7 mV after capsaicin application but did not fire any action potentials. In all capsaicin-sensitive cells, depolarization began quickly, within 5 sec of capsaicin application. Three tested cells did not respond to capsaicin. A brief period of action potential firing followed by maintained depolarization in response to capsaicin matches previous reports of recordings from dissociated DRG cells (Urban and Dray, 1993; Baumann et al., 1996; Lopshire and Nicol, 1997) and other sensory neurons (Liu et al., 2001).
The adaptation of spike firing during capsaicin stimulation is obviously not entirely attributable to desensitization of the receptors for capsaicin, because many cells remained depolarized well beyond threshold (approximately -40 mV) after spike firing had ceased. One element underlying the cessation of firing might be a reduction of sodium current produced by capsaicin (Su et al., 1999), which is evidently mediated at least in part by second-messenger pathways (Liu et al., 2001). Another possible element is the inactivation of sodium current during the maintained depolarization. To distinguish these, we examined adaptation during depolarizations resulting from injected current. The responses of DRG neurons to ramp injections of depolarizing current were qualitatively similar to responses to prolonged capsaicin application: injection of 1 sec ramps from 0 to 1 nA elicited a period of action potential firing that adapted within ∼1 sec (Fig. 1B). The number of action potentials in response to such a current ramp ranged from 8 to 36 (average, 19.6 ± 10.0; n = 7), and the peak of the first action potential reached an average of 48 ± 4 mV. As the current ramp progressed, action potential amplitude steadily decreased until in five of seven cells the membrane potential displayed a series of damped oscillations with peak amplitudes of 10-20 mV. Action potentials at the end of current ramps in the two remaining cells had peaks >0 mV.
When 10 1 sec current ramps were injected with a 1 sec interval separating them (i.e., at 0.5 Hz), the firing response during each ramp progressively declined. The number of action potentials during the 10th ramp was reduced to 19 ± 18% (n = 7) of the number during the first ramp (Fig. 1C). The decrease in firing was accompanied by an increase in the latency to the first action potential, from 145 ± 84 to 263 ± 108 msec (n = 7) and a decrease in the maximal upstroke velocity, dV/dtmax, of the first action potential from 132 ± 68 to 48 ± 35 mV/msec (n = 7). Damped oscillations were also abolished by the 10th current ramp injection. These trends were observed to a lesser degree when 1 sec current ramps were separated by 9 sec (i.e., 0.1 Hz).
Use dependence of the TTX-R sodium current
The reduction in nociceptor cell firing during repetitive current ramp injections might result from the use-dependent reduction of TTX-R sodium current observed in voltage clamp (Roy and Narahashi, 1992; Schild and Kunze, 1997; Rush et al., 1998; Scholz et al., 1998a; Fazan et al., 2001; D'Alcantara et al., 2002). TTX-R sodium current is expressed in the majority of small DRGs and is important in the generation of action potentials in response to depolarizing current injection (Caffrey et al., 1992; Scholz et al., 1998a; Blair and Bean, 2002). When we recorded TTX-R sodium current in voltage clamp under conditions minimizing the contribution of other currents (300 nm TTX to block TTX-S sodium channels, internal NMDG+ and external TEA+ and Cs+ to block potassium channels, and external Cd2+ to block calcium channels) the TTX-R sodium current was strongly use dependent (Fig. 2). During a step to 0 mV from -80 mV holding potential, the TTX-R sodium current activated, reached a peak within ∼2 msec, and then inactivated until the sodium current remaining after 30 msec was 6 ± 3% (n = 19) of the initial peak.
Figure 2.
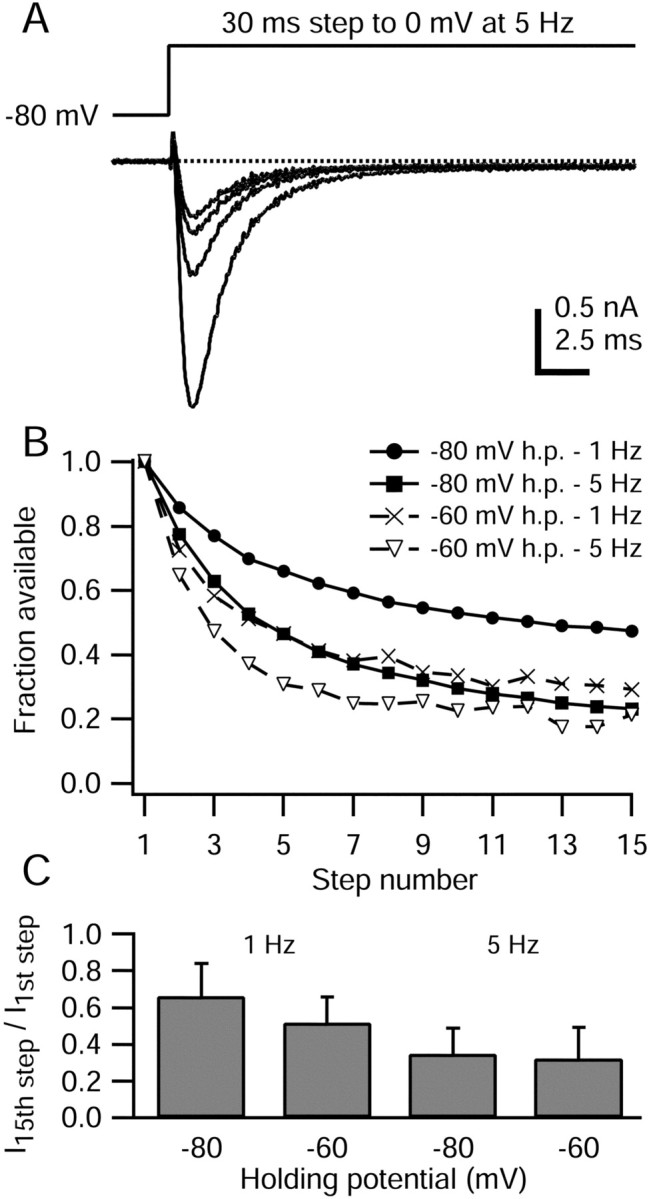
Use dependence of TTX-R sodium current. A, TTX-R sodium current elicited during 30 msec steps to 0 mV applied at 5 Hz, from a holding potential of -80 mV. For clarity, only the 1st, 5th, 10th, and 15th sweeps are shown. Dotted line indicates zero current level. At the onset of the voltage step, 200 μsec were blanked to remove residual uncompensated capacity current. The internal solution contained the following (in mm): 130 NMDG, 120 aspartate, 15 NaCl, 1.8 MgCl2, 9 EGTA, 9 HEPES, 4 MgATP, 14 Tris-creatine PO4, and 0.3 Tris-GTP, pH 7.4 (with 7 mm CsOH). The external solution contained the following (in mm): 50 NaCl, 100 TEA-Cl, 4 CsCl, 2 CaCl2, 2 MgCl2, 0.03 CdCl2, 10 glucose, and 10 HEPES, pH 7.4 (with TEA-OH). B, Peak TTX-R sodium currents from the cell in A were normalized to the initial value and plotted against the step number. Results for holding potentials of -60 and -80 mV and stimulation rates of 1 and 5 Hz are also shown for this cell. C, Average decline in TTX-R sodium current amplitude over 15 steps for holding potentials of -60 and -80 mV and stimulation rates of 1 and 5 Hz. Error bars indicate SD.
When 15 30 msec step depolarizations (from -80 to 0 mV) were applied repetitively at 5 Hz, the amplitude of the evoked TTX-R sodium current progressively decreased until the current during the 15th step was 23% of the initial peak (Fig. 2). Overall, when TTX-R sodium current was evoked from -80 mV holding potential at 5 Hz, the average peak amplitude during the 15th step to 0 mV was 35 ± 15% (n = 11) (Fig. 2C) of the initial peak.
The use-dependent decrease of the TTX-R sodium current was also present when the stimulation frequency was 1 Hz, such that 15 30 msec steps to 0 mV (-80 mV holding potential) reduced the TTX-R sodium current to 66 ± 18% (n = 14) of the initial value. Use dependence was also substantial when the holding potential was -60 mV rather than -80 mV. In this case, after 15 30 msec steps to 0 mV at 1 Hz, the TTX-R current was reduced to 52 ± 14% (n = 15) after 15 steps, whereas at 5 Hz the current was reduced to 32 ± 17% (n = 8) of the initial value.
Although previous studies have reported use dependence of TTX-R current in DRG neurons, the degree of use dependence has varied widely, and in some cases there was much less use dependence at 1 Hz (Roy and Narahashi; 1992; Gold and Thut, 2001) than the 35-50% reduction that we saw. The reasons for the differences are unclear, but one possibility is the use of different internal solutions. Most previous investigations have used cesium as the main internal cation, whereas in most of our experiments we used NMDG. In some experiments we used potassium as the main internal cation and saw a similar amount of use dependence as with NMDG (see Fig. 11). The degree of use dependence can be affected by test pulse length (see Fig. 4) and by holding potential (Fig. 2C), which have both varied somewhat among different studies. There could also be differences in the particular population of neurons chosen for study, and Rush et al. (1998) showed dramatic differences in the degree of use dependence among different cells even with identical solutions and protocols.
Figure 11.

TTX-R sodium current use dependence elicited by action potential waveforms. A, top, Single action potentials recorded from two DRG neurons applied as voltage commands at 1 Hz, from a holding potential of -60 or -70 mV. Solid line, Short-duration action potential recorded with internal solution containing the following (in mm): 140 K-methanesulfonate, 13.5 NaCl, 1.6 MgCl2, 0.09 EGTA, 9 HEPES, 0.9 glucose, 14 Tris-creatine PO4, 4 MgATP, and 0.3 Tris-GTP, pH 7.4. Dashed line, Long-duration action potential recorded with the same internal solution as above, with 140 mm K-aspartate replacing K-methanesulfonate. In both cases, the external solution was Tyrode's solution. A, Bottom, Peak inward current elicited during action potential waveforms (applied in the same cell), was normalized to the initial value and plotted against sweep number. Filled triangles, The degree of TTX-R sodium current use dependence in this cell during 30 msec steps to 0 mV, applied at 1 Hz from a holding potential of -60 mV. The internal solution contained the following (in mm): 140 K-aspartate, 13.5 NaCl, 1.6 MgCl2, 0.09 EGTA, 9 HEPES, 14 Tris-creatine PO4, 4 MgATP, and 0.3 Tris-GTP, pH 7.4. The external solution contained the following (in mm): 150 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES, 10 glucose, and 5 TEA-Cl, pH 7.4 with 300 nm TTX.
Figure 4.
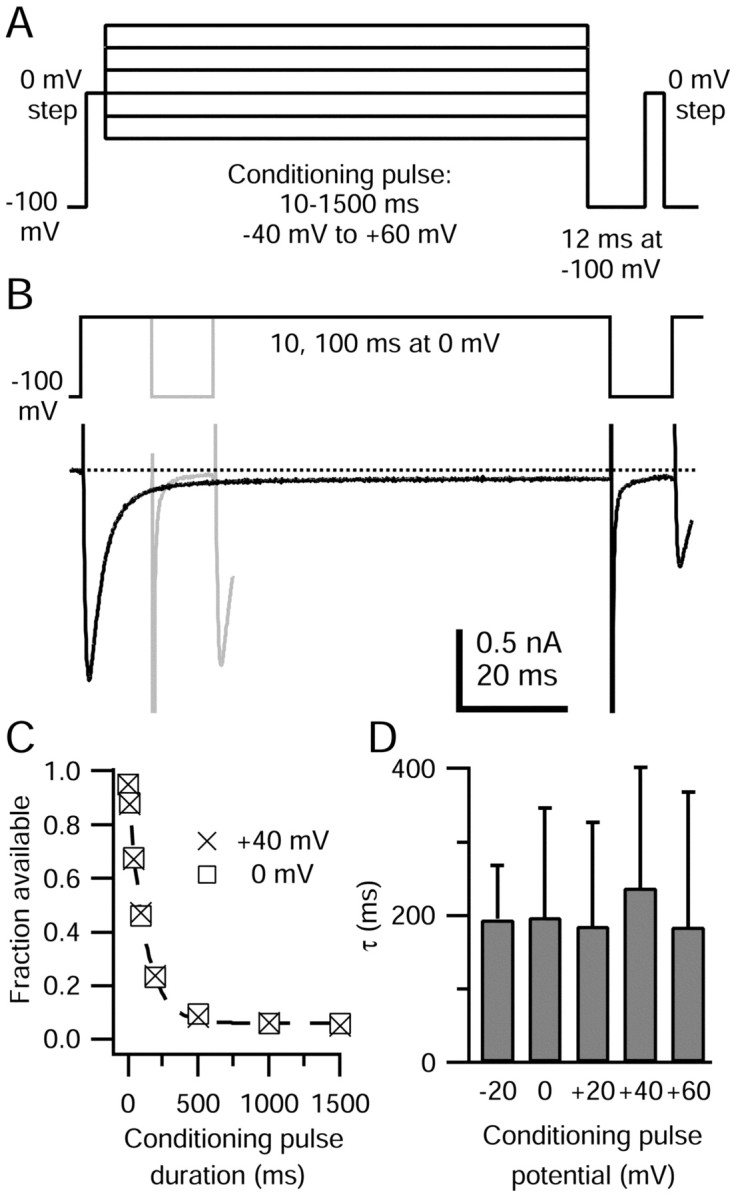
Rate of TTX-R sodium channel entry into slow inactivated states. A, Protocol for measuring the rate of entry into slow inactivated states. Test steps to 0 mV were separated by conditioning pulses of 10-1500 msec at potentials from -40 to +60 mV. To isolate the contribution of slow inactivation, fast inactivation was removed during a 12 msec step to -100 mV preceding the second test step. B, TTX-R sodium currents during measurement of slow inactivation elicited by 10 msec (gray) and 100 msec (black) conditioning pulses at 0 mV. Approximately 5% of TTX-R channels were slow inactivated after the 10 msec pulse to 0 mV, whereas ∼50% were slow inactivated after the 100 msec pulse. (Conditioning pulse durations do not include the initial 4 msec step to 0 mV.) The fast inward tail current during the repolarization to -100 mV is probably carried by a mixture of sodium ions flowing through TTX-R channels and calcium ions flowing through calcium channels; the block of calcium channels by 30 μm is partially relieved at strongly hyperpolarized potentials. The internal solution contained the following (in mm): 130 NMDG, 120 aspartate, 15 NaCl, 1.8 MgCl2, 9 EGTA, 9 HEPES, 4 MgATP, 14 Tris-creatine PO4, and 0.3 Tris-GTP, pH 7.4 (with 7 mm CsOH). The external solution contained the following (in mm): 50 NaCl, 100 TEA-Cl, 4 CsCl, 2 CaCl2, 2 MgCl2, 0.03 CdCl2, 10 glucose, and 10 HEPES, pH 7.4 (with TEA-OH). C, Fraction of available TTX-R sodium current (second test step current normalized to the first) plotted against the duration of the conditioning pulse at 0 and +40 mV (same cell). The dashed line shows the monoexponential fit to the data for 0 mV conditioning potential, with a time constant of 111 msec. D, Average time constants for TTX-R sodium channel entry to slow inactivation at conditioning potentials between -20 and +60 mV. At most, two conditioning potentials were tested in a single cell. Error bars indicate SD.
TTX-R sodium channel recovery from fast inactivation
Because fast inactivation of TTX-R sodium channels is nearly complete at the end of the 30 msec test step to 0 mV, the use dependence shown in Figure 2 could result from incomplete recovery from fast inactivation during the 200 or 1000 msec intersweep intervals. Therefore, we measured the rates of TTX-R sodium channel recovery from fast inactivation (Fig. 3). Cells were depolarized to 0 mV for 30 msec to inactivate TTX-R sodium channels, followed by recovery periods at potentials from -40 to -120 mV, applied for durations of 0.5-200 msec. A second step to 0 mV was applied to assay available TTX-R sodium current. Recovery could be fitted with a monoexponential curve, with rate constants of 1.75 ± 0.42 msec-1 at -120 mV (n = 7), 0.82 ± 0.26 msec-1 at -100 mV (n = 9), 0.42 ± 0.11 msec-1 at -80 mV (n = 8), 0.14 ± 0.05 msec-1 at -60 mV (n = 9), and 0.038 ± 0.03 msec-1 at -40 mV (n = 4). In fitting the recovery at -40 mV, we ignored the 3-5 msec delay before recovery started, which likely reflects the requirement that channels deactivate before recovery from inactivation can begin, as in TTX-S sodium channels from hippocampal CA1 neurons (Kuo and Bean, 1994a). The rapid recovery from inactivation of TTX-R sodium channels fits with previous reports (Elliott and Elliott, 1993; Schild and Kunze, 1997; Scholz et al., 1998a; Fazan et al., 2001; but see Ogata and Tatebayashi, 1993).
Figure 3.

TTX-R sodium channels recover rapidly from fast inactivation. A, Top, Voltage protocol to measure recovery from fast inactivation. A 30 msec step to 0 mV is followed by varying durations at the recovery potential (here, -60 mV), before a second test step to 0 mV to assay available TTX-R sodium current. A, Bottom, TTX-R sodium currents recorded during the above protocol with recovery durations of 1, 3, 10, 25, and 50 msec. The internal solution contained the following (in mm): 130 NMDG, 120 aspartate, 15 NaCl, 1.8 MgCl2, 9 EGTA, 9 HEPES, 4 MgATP, 14 Tris-creatine PO4, and 0.3 Tris-GTP, pH 7.4 (with 7 mm CsOH). The external solution contained the following (in mm): 50 NaCl, 100 TEA-Cl, 4 CsCl, 2 CaCl2, 2 MgCl2, 0.03 CdCl2, 10 glucose, and 10 HEPES, pH 7.4 (with TEA-OH). B, TTX-R sodium current (cell in A), during the second test step normalized to the current during the initial step, is plotted against the recovery period duration (symbols). Dashed lines show monoexponential fits to the data, with indicated time constants. C, Average TTX-R sodium current recovery rates plotted against voltage. Error bars indicate SD; absent error bars are smaller than symbols.
Although recovery from fast inactivation after a 30 msec step to 0 mV was rapid and reached a steady state within 10-30 msec, it was incomplete. As noted previously (Elliott and Elliott, 1993), ∼10% of the current recovered with much slower kinetics: after 50 msec, recovery was 89 ± 6% complete at -100 mV (n = 9) and 89 ± 9% complete at -80 mV (n = 8). Recovery in 50 msec was slightly more complete at -120 mV (96 ± 4%, n = 3) but less complete at -60 mV (78 ± 16%, n = 9). The incomplete recovery of TTX-R sodium channels after 50 msec, when the initial recovery process had plateaued, suggests that a small fraction of channels entered inactivated states from which recovery was slower.
Slow inactivation of the TTX-R sodium current
During long depolarizations lasting seconds, voltage-gated TTX-sensitive sodium channels enter slow inactivated states from which recovery can take several seconds (Narahashi, 1964; Rudy, 1978). If accumulation of TTX-R sodium channels in slow inactivated states caused the use dependence shown in Figure 3, then slow inactivation entry rates must be rapid enough for an appreciable fraction of channels to enter during a 30 msec step to 0 mV. Furthermore, the recovery of TTX-R sodium channels from slow inactivated states must be incomplete after 200 or 1000 msec at -60 or -80 mV, the interpulse intervals and potentials during the use-dependence experiments.
To determine the extent of slow inactivation elicited by short-duration depolarizations, we measured the entry of TTX-R sodium channels into slow inactivated states using 10-1500 msec conditioning pulses from -40 to +60 mV, separating two 4 msec test steps to 0 mV (Fig. 4A). The conditioning pulse was followed by a 12 msec step to -100 mV, during which recovery from fast inactivation should be complete (∼10 time constants for recovery). Any reduction in current during the second test step should reflect slow inactivation generated during the conditioning pulse.
An example of TTX-R sodium current evoked during this protocol is shown in Figure 4B. TTX-R sodium current activated during the step to 0 mV, here consisting of the initial 4 msec test step, immediately followed by the 10 msec conditioning pulse at 0 mV. After the hyperpolarizing pulse, the TTX-R sodium current during the second test step to 0 mV was ∼5% smaller than during the initial pulse, indicating that ∼5% of TTX-R channels were slow inactivated during the conditioning pulse. When the conditioning pulse was lengthened to 100 msec at 0 mV, ∼50% of TTX-R sodium channels were slow inactivated.
The kinetics of TTX-R sodium channel entry into slow inactivated states are shown in Figure 4C. The available TTX-R sodium current after conditioning pulses to 0 and 40 mV (normalized to the initial test step TTX-R sodium current) decreased as channels rapidly entered slow inactivated states. The data were fitted with monoexponential functions, which in this cell had time constants of 111 msec for entry at 0 mV and 116 msec for entry at 40 mV.
Time constants for entry of TTX-R sodium channels into slow inactivated states at potentials between -20 and +60 mV, determined in 38 DRG cells, ranged from ∼55 to ∼550 msec. Entry time constants were not voltage dependent over this range: average time constants were 195 ± 73 msec at -20 mV (n = 6), 197 ± 148 msec at 0 mV (n = 15), 186 ± 141 msec at +20 mV (n = 9), 238 ± 163 msec at +40 mV (n = 9), and 184 ± 184 msec at +60 mV (n = 8). The large variability occurred between different cells, because when slow inactivation entry time constants were measured at two conditioning pulse potentials in a single cell, they differed by only 7 ± 5% (n = 9).
The entry of TTX-R sodium channels into slow inactivated states was a monoexponential process at potentials from -20 mV to +60 mV, but the entry at -40 mV was generally biphasic. The fast component of entry had an average time constant of 56 ± 20 msec (n = 8), similar to the fastest slow inactivation entry time constants observed at more depolarized potentials, and consisted of 34 ± 20% of the entry process. The slower entry phase had time constants ranging from ∼460 msec to ∼3.6 sec (average, 1414 ± 1004 msec; n = 8) and accounted for 66 ± 20% of the total.
Entry from open states versus fast inactivated states
Some TTX-S sodium channels appear to enter slow inactivated states more rapidly from open states than from fast inactivated states, generating use-dependent decreases similar to that shown in Figure 2 (Rudy, 1981; Jung et al., 1997; Mickus et al., 1999). To test whether the slow inactivation of TTX-R channels is produced most effectively from the open state, we compared the extent of TTX-R channel slow inactivation by conditioning protocols designed to maximize the time spent in the open state (trains of many short pulses) or the inactivated state (a few long pulses). Figure 5A illustrates the different conditioning trains. In each case, a series of steps to 0 mV were given such that the total duration at 0 mV was 500 msec. TTX-R sodium current reached a peak 1.7 ± 0.3 msec (n = 9) after the onset of a step to 0 mV (Fig. 5B). Thus, with 1 and 2 msec steps, there should be little occupancy of the fast inactivated state during the pulse, with channels primarily in the open state. In contrast, with 50 msec steps, TTX-R current decayed to 10% within ∼10 msec and to 5 ± 2% (n = 7) after 50 msec, so that channels spend the majority of time in the fast inactivated state during the 50 msec pulses.
Figure 5.
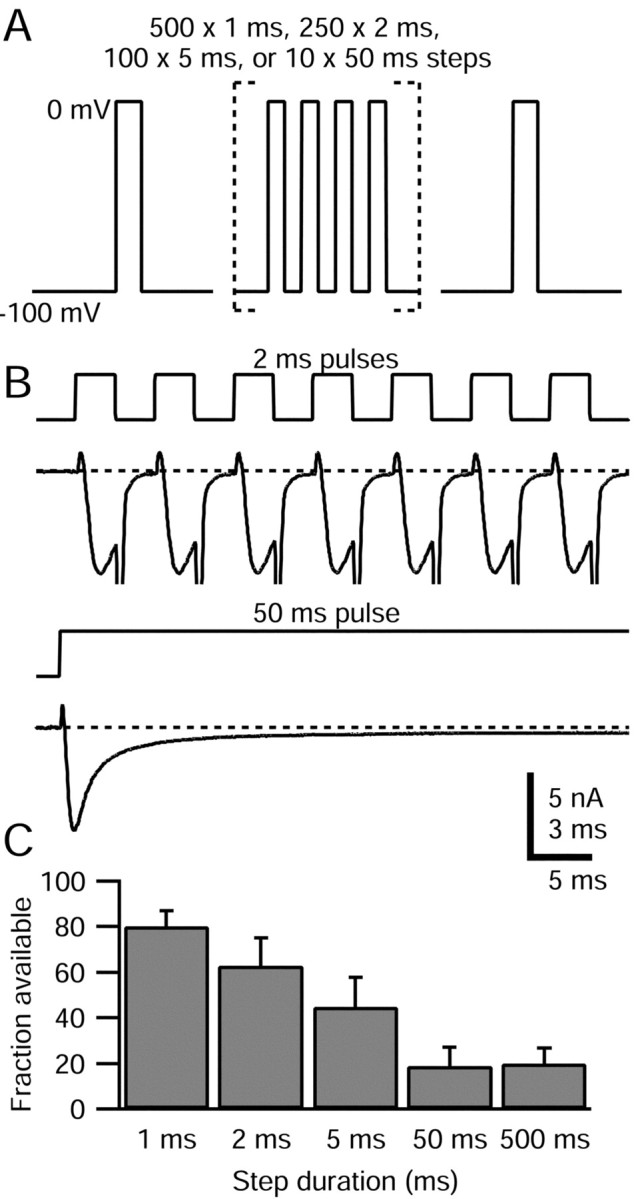
TTX-R sodium channel slow inactivation elicited by steps of varying duration. A, Schematic of voltage protocol: a train of 1, 2, 5, and 50 msec pulses to 0 mV was applied, with a total duration of 500 msec spent at 0 mV in each case. A test step to 0 mV after the train (preceded by 15 msec at -100 mV) assayed TTX-R sodium channel slow inactivation. The interpulse interval was 2 msec for the 1 and 2 msec pulse trains and 5 msec for the 5 and 50 msec pulse trains. B, Comparison of TTX-R sodium currents flowing during 2 and 50 msec pulses. The first seven pulses during the 2 msec train and the first 50 msec pulse are shown. Large inward tail currents during repolarizations were truncated. The dotted line indicates zero current level. Note different time scales: bar corresponds to 3 msec of 2 msec pulse train and 5 msec of 50 msec pulse train. The internal solution contained the following (in mm): 130 NMDG, 120 aspartate, 15 NaCl, 1.8 MgCl2, 9 EGTA, 9 HEPES, 14 Tris-creatine PO4, 4 MgATP, and 0.3 Tris-GTP, pH 7.4. The external solution contained the following (in mm): 50 NaCl, 100 TEA-Cl, 4 CsCl, 2 CaCl2, 2 MgCl2, 0.03 CdCl2, 10 glucose, and 10 HEPES, pH 7.4 (with TEA-OH). C, Average available TTX-R sodium current (second test step current normalized to test step current applied before pulse train) for different pulse durations. Error bars indicate SD.
Slow inactivation of TTX-R sodium channels was generated more effectively by fewer long-duration pulses, in which channels are mostly in the fast inactivated state, than by numerous short pulses, in which channels are mostly in the open state (Fig. 5C). TTX-R channels were 63 ± 12% (n = 4) available after 250 2 msec pulses to 0 mV, but only 19 ± 8% (n = 7) available after 10 50 msec pulses and 21 ± 7% (n = 9) available after a single 500 msec pulse. This result shows that it is not necessary for channels to cycle through the open state to enter slow inactivation. The results are consistent with TTX-R channels entering slow inactivation more rapidly from the fast inactivated state than the open state, but it is not possible to conclude this with certainty, because the protocols with multiple short pulses incorporate periods at -100 mV during which there could be recovery from slow inactivation.
TTX-R sodium channel recovery from slow inactivation
We measured the recovery of TTX-R sodium channels from slow inactivated states by eliciting slow inactivation with a single 500 msec conditioning pulse to +40 mV (Fig. 6). After this conditioning pulse, the membrane potential was stepped to the recovery potential (-60, -80, or -100 mV) for a total of 4 sec. From the recovery voltage, 3 msec test steps to 0 mV were given periodically to measure available TTX-R sodium current (during recovery at -60 and -80 mV, test steps were preceded by 15 msec at -100 mV to ensure recovery from fast inactivation). This protocol should accurately measure the recovery from slow inactivation, because test steps are brief enough that they will not generate significant slow inactivation themselves.
Figure 6.

Kinetics of recovery from slow inactivation. A, Slow inactivation was produced by a 500 msec conditioning pulse to 40 mV. Recovery was assayed by periodic 3 msec steps to 0 mV, preceded by 15 msec at -100 mV to recover fast inactivation. Current was normalized to that elicited by an initial test step to 0 mV. Dashed lines show single exponential fits to the data, with indicated time constants. B, Average time constants of recovery from slow inactivation at -60, -80, and -100 mV. Recovery duration includes the 15 msec periods at -100 mV used to remove fast inactivation. Error bars indicate SD.
The 500 msec conditioning pulse to +40 mV drove between 66 and 77% of TTX-R sodium channels into slow inactivated states, as measured after the first recovery duration of 50 msec (Fig. 6A). Recovery of TTX-R sodium channels from slow inactivation was monoexponential, with time constants dependent on the recovery potential. Recovery was most rapid at -100 mV, with an average time constant of 0.5 ± 0.2 sec (n = 15), slower at -80 mV, with an average time constant of 1.2 ± 0.7 sec (n = 11), and slowest at -60 mV, with an average time constant of 1.5 ± 0.7 sec (n = 12). The extent of recovery after 4 sec at the recovery potential was 96 ± 2% (n = 15) at -100 mV, 83 ± 8% (n = 11) at -80 mV, and 62 ± 12% (n = 11) at -60 mV. The incomplete recovery after nearly three time constants suggests the existence of an even slower component of recovery, similar to previous reports for TTX-R sodium channels (Ogata and Tatebayashi, 1992; Rush et al., 1998; Fazan et al., 2001).
The relatively rapid rate of TTX-R sodium channel entry into slow inactivated states, together with the relative slowness of recovery, accounts for the strong use dependence we observed when stimulating cells with short depolarizations at 1 and 5 Hz. During each 30 msec step to 0 mV, ∼3-30% of TTX-R sodium channels enter slow inactivated states. With recovery from slow inactivation proceeding with time constants of >1 sec at -60 and -80 mV, the TTX-R sodium current will not completely recover during the interpulse intervals of 200 and 1000 msec.
Voltage dependence of TTX-R sodium channel slow inactivation
To determine the steady-state voltage dependence of TTX-R sodium channel slow inactivation, we used 5 sec conditioning pulses (Fig. 7). The normalized available TTX-R sodium current was plotted against conditioning pulse potential and could be fitted by a Boltzmann function with an average midpoint of -44 ± 5 mV and a slope factor of 5 ± 1 mV (n = 11). Thus, the slow inactivation of TTX-R sodium channels was quite voltage-dependent, going from completely available to completely slow inactivated over a range of 40 mV. The maximal steady-state fraction of TTX-R sodium channels that were slow inactivated after 5 sec conditioning pulses was 93 ± 3% (n = 11), saturating at potentials from -20 to +60 mV.
Figure 7.
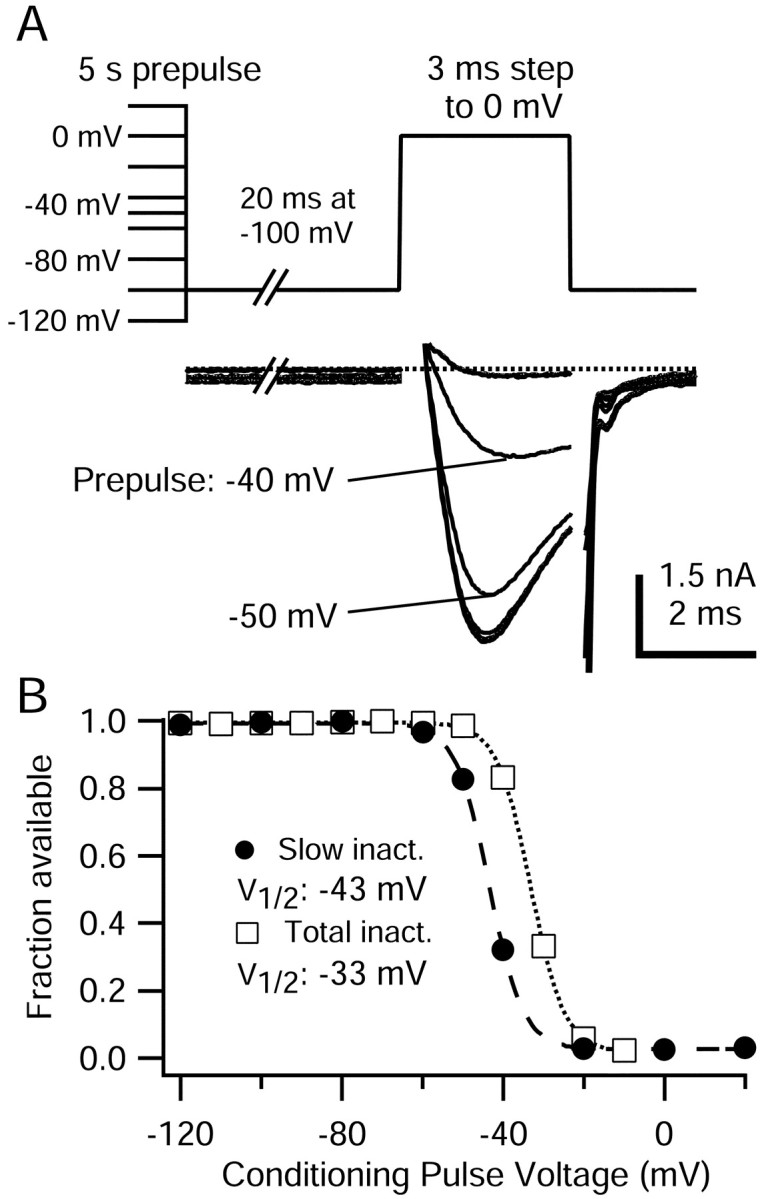
Voltage dependence of TTX-R sodium channel slow inactivation. A, Top, Protocol for measuring steady-state voltage dependence of slow inactivation. Conditioning pulses of 5 sec to potentials between -120 and +20 mV were followed by a 20 msec step to -100 mV to remove fast inactivation and then a test step to 0 mV. A, Bottom, Representative TTX-R sodium currents elicited during 0 mV test step. To remove residual uncompensated capacity current, 200 μsec of current traces at the leading and falling edges of the voltage step were blanked. The internal solution contained the following (in mm): 130 NMDG, 120 aspartate, 15 NaCl, 1.8 MgCl2, 9 EGTA, 9 HEPES, 4 MgATP, 14 Tris-creatine PO4, and 0.3 Tris-GTP, pH 7.4 (with 7 mm CsOH). The external solution contained the following (in mm): 50 NaCl, 100 TEA-Cl, 4 CsCl, 2 CaCl2, 2 MgCl2, 0.03 CdCl2, 10 glucose, and 10 HEPES, pH 7.4 (with TEA-OH). B, TTX-R sodium currents during 0 mV step from cell in B were normalized to their maximum and plotted against conditioning pulse voltage. Filled circles, Test pulse current after 5 sec conditioning pulses and 20 msec at -100 mV to remove fast inactivation. Open squares, Test pulse current after 500 msec conditioning pulses, and no removal of fast inactivation. This measurement includes the effects of slow inactivation, and is therefore labeled “total inactivation.” Lines show the best fit to the Boltzmann equation, I/Imax = 1/(1 + exp[(V - V1/2)/k]), where V is the conditioning pulse potential, V1/2 is the half-maximal voltage in millivolts, and k is the slope factor in millivolts. Slow inactivation, V1/2 = -43 mV, k = 4.2 mV. Total inactivation, V1/2 = -33 mV, k = 4.4 mV.
We compared the voltage dependence of slow inactivation with that of fast inactivation, measured in the same cells using 500 msec prepulses from -120 to -10 mV; this had a midpoint of -34 ± 5 mV (n = 11), or ∼10 mV depolarized relative to slow inactivation. The hyperpolarized position of the slow inactivation curve is similar to that seen in skeletal muscle TTX-S sodium channels (Ruff et al., 1988; Featherstone et al., 1996) but distinct from sodium current in heart cells (Richmond et al., 1998), neocortical neurons (Fleidervish et al., 1996), and hippocampal neurons (Kuo and Bean, 1994b), all of which have slow inactivation curves with midpoints more depolarized than fast inactivation curves. Because the voltage dependence of slow inactivation is hyperpolarized relative to fast inactivation, the inactivation measured with 500 msec prepulses probably includes significant influence of slow inactivation and is shifted hyperpolarized relative to a hypothetical curve of “pure” fast inactivation.
When a 10 sec prepulse was used instead of a 5 sec prepulse, the midpoint of the TTX-R sodium current slow inactivation curve was shifted hyperpolarized by ∼4 mV (midpoint, -48 ± 5 mV; n = 4). Further lengthening the prepulse to 60 sec again shifted the midpoint hyperpolarized, to an average midpoint of -51 ± 6 mV (n = 3). The maximal fraction of slow inactivated TTX-R sodium channels increased to 99-100% after 60 sec conditioning pulses. These results are similar to those obtained previously for TTX-R channels in rat DRG neurons by Ogata and Tatebayashi (1992) and Rush et al. (1998) and by Fazan et al. (2001) for nodose ganglion cells and suggest the existence of multiple slow inactivated states, similar to “ultra-slow” inactivation that TTX-S sodium channels enter during very long depolarizations (Fox, 1976; Ellerkmann et al., 2001).
TTX-R sodium current slow inactivation elicited by physiological waveforms
Because TTX-R sodium channels enter slow inactivated states rapidly and with modest depolarizations, physiological waveforms, including those elicited by capsaicin application (Fig. 1A), may elicit substantial slow inactivation of the TTX-R sodium current. If so, the resulting long-lasting reduction in sodium current would reduce the ability of nociceptors to generate action potentials both during and after strong stimuli. In addition, the more hyperpolarized position of slow inactivation compared with fast inactivation means that for small depolarizations, channels may actually enter slow inactivated states before fast inactivated states.
To examine directly the contribution of slow inactivation of TTX-R sodium current to the control of nociceptor excitability, we used as the command waveform in voltage-clamp experiments a previously recorded current-clamp response to 500 nm capsaicin. In particular, we used the ∼800 msec fragment of the initial capsaicin evoked response shown in Figure 1A, interrupted at various points with test steps to assay the available TTX-R sodium current.
TTX-R and TTX-S sodium currents during prerecorded capsaicin response
When the capsaicin-evoked waveform was applied to cells in voltage clamp under ionic conditions designed to minimize the contribution of other currents, the TTX-R sodium current activated soon after the initial step from the holding potential of -100 to -64 mV, and continued to flow throughout the entire waveform, including during damped oscillations (Fig. 8). As expected, the TTX-R current shows phasic behavior during the spikes, activating and then inactivating. However, in addition there is a smaller component of steady TTX-R current, most evident at the end of the waveform as steady current of approximately -150 pA. This may represent a small “persistent” component of TTX-R current, such as has been proposed to arise from NaV1.9 channels (Cummins et al., 1999; Rugiero et al., 2003).
We also recorded TTX-S sodium current during the capsaicin-evoked waveform by determining the current blocked by the addition of 300 nm TTX. TTX-S sodium current also began flowing early in the response, during the interval leading up to the first action potential. The maximal TTX-S sodium current amplitude occurred during the upstroke of the first action potential, and it was almost fully inactivated by the third action potential. Because fast inactivation of TTX-S sodium current is nearly complete at the peak of a single action potential (∼80%; Blair and Bean, 2002), the reduction during subsequent action potentials likely results from incomplete recovery during interspike intervals. This experiment shows that both TTX-S and TTX-R currents contribute to the first two spikes of the capsaicin response, but subsequent spikes are nearly completely attributable to TTX-R current.
Slow inactivation during the capsaicin response
Is the loss of firing during the capsaicin response caused by inactivation of TTX-R channels? To assay the availability of TTX-R sodium channels during the capsaicin-evoked waveform directly, we applied short test steps to 0 mV at varying points throughout the response. The test pulse was either given directly (to measure total inactivation) or preceded by a 30 msec step to -100 mV (to remove fast inactivation and measure slow inactivation).
Figure 9A shows an example in which slow inactivation 490 msec into the capsaicin-evoked waveform, after 10 spikes, was measured. Approximately 70% of TTX-R sodium channels were slow inactivated at this point, approximately halfway through the capsaicin-evoked waveform.
Total inactivation of TTX-R sodium channels accumulated throughout the capsaicin-evoked waveform; it was ∼50% after two spikes and ∼98% at the end of the waveform (Fig. 9B, open symbols). The nearly complete inactivation of TTX-R current during the waveform is nicely consistent with the failure of spike generation at the end of the waveform. Slow inactivation was somewhat less complete than total inactivation at all points during the capsaicin-evoked waveform. However, slow inactivation was 76 ± 14% complete (n = 7) at the end of the waveform. This is very similar to maximal slow inactivation induced by 500 msec steps to 0 mV (74 ± 15%), determined in four of the same cells.
The time points in Figure 9B measuring total availability of TTX-R sodium current occurred soon after the completion of the previous spike. However, because TTX-R sodium channels recover from fast inactivation quite quickly (Fig. 3), the availability of TTX-R channels could change substantially during the interspike interval. Therefore, we measured the recovery of TTX-R sodium channels from fast inactivation during intervals between action potentials by comparing availability immediately after a spike and just before the next spike (Fig. 10). In fact, there was substantial recovery from fast inactivation during the interspike intervals. The total availability of TTX-R sodium channels increased from ∼20% after the third spike in the capsaicin-evoked waveform to ∼51% just before the fourth spike. The extent of TTX-R channel recovery from fast inactivation during subsequent interspike intervals progressively decreased (Fig. 10B). For example, between the 4th and 5th action potentials, availability recovered from 14 ± 1 to 31 ± 10%, and between the 9th and 10th action potentials, spikes availability increased only slightly, from 5 ± 4 to 7 ± 4% (n = 5). The reduced recovery of TTX-R sodium current availability between later spikes is primarily attributable to the progressive development of slow inactivation, which would not be expected to recover significantly between spikes. In addition, recovery from fast inactivation is reduced between later spikes because interspike intervals are shorter, and interspike intervals are more depolarized.
Is the slow inactivation during the capsaicin-evoked waveform more attributable to the spikes themselves or to the steady depolarization? To address the contribution of spikes to the generation of slow inactivation, we tested the ability of action potential waveforms to induce slow inactivation. We applied action potentials as command waveforms in voltage clamp (Fig. 11) and examined the use-dependent decrease in TTX-R sodium current flowing during the upstroke when the spike waveform was given at 1 Hz. An action potential with a relatively short duration (2.9 msec at half-maximal amplitude) delivered at 1 Hz was not very effective at eliciting slow inactivation, with 91 ± 4% of TTX-R channels still available after 15 sweeps (n = 7). We also tested a spike waveform with a longer duration (5.4 msec at half-maximal amplitude). This waveform drove a somewhat larger fraction of TTX-R channels into slow inactivation, with 82 ± 9% available after 15 spikes (n = 7). However, both action potential waveforms were much less effective than a 30 msec step to 0 mV (from -60 mV holding potential), which in these same cells reduced the available TTX-R sodium current to 53 ± 9% (n = 7). These results suggest that action potential waveforms are not especially effective in producing slow inactivation and fit with the results comparing short versus long conditioning steps (Fig. 5). These results suggest that slow inactivation during the capsaicin response is generated mainly by the underlying depolarization rather than the spikes.
Discussion
These results suggest that slow inactivation of TTX-R sodium current plays a major role in controlling adaptation of action potential firing in nociceptive sensory neurons. TTX-R sodium channels readily enter slow inactivated states at physiological voltages. Using a prerecorded response to capsaicin as a voltage command, we found that at the point at which the action potential firing ceased (after ∼1 sec of depolarization), slow inactivation of TTX-R sodium channels was ∼75% complete and total inactivation was ∼98% complete. This is consistent with slow inactivation of TTX-R channels being primarily responsible for quick adaptation of firing in response to capsaicin application and suggests that firing would adapt quickly even without the effect of capsaicin depressing sodium current through second messenger pathways (Su et al., 1999; Liu et al., 2001). The latter effect might produce more prolonged adaptation than would occur from slow inactivation alone.
Slow inactivation appears to be a ubiquitous feature of voltage-dependent sodium channels, including all types of TTX-R and TTX-S channels examined so far. However, there are a number of significant differences in the properties of slow inactivation observed among various types of channels. In general, compared with TTX-S channels, slow inactivation of TTX-R channels is more complete at voltages that are likely to be reached with physiological stimuli. Thus, we found that slow inactivation reached ∼95% completion with 5 sec depolarization to -20 mV and nearly 100% completion with 60 sec depolarization to -30 mV. The nearly complete development of slow inactivation of TTX-R channels for moderate depolarizations, which agrees well with previous observations (Ogata and Tatebayashi, 1992; Rush et al., 1998; Fazan et al., 2001), is in contrast to maximal steady-state slow inactivation for TTX-S channels of ∼50% in various central neurons (Kuo and Bean, 1994b; Fleidervish et al., 1996; Do and Bean, 2003) using similar protocols.
For neuronal TTX-S channels, the voltage dependence of slow inactivation has a midpoint considerably depolarized to that of fast inactivation (Kuo and Bean, 1994b; Fleidervish et al., 1996; Fazan et al., 2001). In contrast, for TTX-R channels, the midpoint for slow inactivation is considerably hyperpolarized compared with that for fast inactivation. Thus, in our experiments slow inactivation determined even with relatively short conditioning pulses (5 sec) had a midpoint of -44 mV, compared with a midpoint of -34 mV for fast inactivation. For TTX-S sodium channels, steady depolarizations first produce fast inactivation and then slow inactivation, whereas for TTX-R channels, slow inactivation occurs before fast inactivation, at least for small, steady depolarizations to near threshold voltages. In fact, because of this, it is not easy to determine the voltage dependence of fast inactivation by itself, and it likely has a midpoint even more depolarized than -34 mV.
For TTX-S channels, the fast inactivation process determines the extent and time course of inactivation both for inactivation during spikes and also for subthreshold depolarizations lasting hundreds of milliseconds. For TTX-R channels, fast inactivation dominates during spikes, but it is slow inactivation that regulates the extent of inactivation for smaller steady depolarizations, such as those produced by stimulatory ligands. In addition, although most nociceptors have both TTX-S and TTX-R currents, TTX-S current is completely inactivated much earlier during the waveform produced by application of capsaicin (Fig. 8), so that it is inactivation of TTX-R current that limits the ability of the cell to spike later in the application. Thus, slow inactivation of TTX-R channels should play a large role in determining the extent and time course of accommodation and adaptation during maintained stimulation. Previous studies have shown that calcium-activated potassium current also contributes to adaptation in sensory neurons (Weinreich and Wonderlin, 1987; Scholz et al., 1998b). Calcium-activated potassium current should depend more on spiking than on steady depolarization and may serve a complementary role to slow inactivation of TTX-R channels.
For TTX-R channels, slow inactivation is reached much more effectively by a single steady depolarization than by repeated shorter depolarizations (Fig. 5). The ineffectiveness of short depolarizations, even when given at frequencies as high as 250 Hz, contrasts strongly with slow inactivation of neuronal TTX-S channels, which display pronounced slow inactivation with short-duration depolarizations during stimulation at 1-20 Hz, (Rudy, 1981; Colbert et al., 1997; Jung et al., 1997; Mickus et al., 1999). For these TTX-S channels, slow inactivation is evidently promoted by rapid cycling through the open state, whereas this does not appear to be true for TTX-R channels. These results suggest that slow inactivations of TTX-S and TTX-R sodium channels are likely to have quite different physiological roles in neurons that rely on each channel type for spike generation. Because of the rapid transition of TTX-S channels from open states to slow inactivated states, action potentials effectively drive TTX-S channels into slow inactivation, thus limiting cell firing depending on the number of previous spikes. Slow inactivation of TTX-R sodium channels should instead depend less on the number of spikes fired and more on the overall duration of the stimulus-induced depolarization. For TTX-R sodium channels, depolarizations below action potential threshold (approximately -40 mV) can elicit substantial slow inactivation (∼50%) in the complete absence of spikes.
The relative ineffectiveness of repeated short depolarizations seen for TTX-R channels is also characteristic of skeletal muscle sodium channels (Ong et al., 2000), which resemble neuronal TTX-R channels in having the voltage dependence of slow inactivation hyperpolarized relative to that of fast inactivation. However, skeletal muscle sodium channels enter slow inactivation far more slowly, with time constants of seconds to minutes (Ruff et al., 1988), than we found for TTX-R channels.
Adaptation properties in peripheral terminals of low-threshold mechanoreceptors match those in the soma (Harper, 1991). If nociceptors are similar in this regard, and slow inactivation of TTX-R sodium channels occurs in terminals as it does in the soma, it likely contributes to the rapid adaptation of firing seen with in vivo recordings during application of painful stimuli. C-fiber nociceptor responses to sustained heat and mechanical stimulation consist of firing that adapts over 3-10 sec to final frequencies of ∼1 Hz and less (Handwerker et al., 1987; Gallar et al., 1993; Slugg et al., 2000). TTX-R sodium channels were ∼75% slow inactivated at the completion of the 800 msec capsaicin-evoked response, and by extending the response duration, the steady-state value of slow inactivation (95%) would have been reached. The slower progression of adaptation during physiological receptive field stimulation suggests that the stimulation used generated less slow inactivation of TTX-R sodium channels than the direct application of 500 nm capsaicin.
Slow inactivation of TTX-R sodium current could contribute to the fatigue seen in nociceptors during repeated stimulation (Handwerker et al., 1987; Gallar et al., 1993; Slugg et al., 2000; Peng et al., 2003). Peng et al. (2003) showed that the application of heat or mechanical stimulus to a C-fiber nociceptor receptive field, or electrical stimulation of the axon, reduced the number of action potentials elicited during a subsequent heat stimulus. Full recovery from fatigue required 20-60 sec. This is longer than the time course of recovery from slow inactivation that we observed, but we used relatively short conditioning stimuli (5 sec). With longer conditioning stimuli, Rush et al. (1998) observed time constants for recovery from slow inactivation of up to 170 sec. Reduction in TTX-R current attributable to slow inactivation can account well for fatigue occurring across stimulation types.
The properties of slow inactivation suggest that maintained repetitive firing should be most effectively stimulated by steady depolarizations that are beyond threshold (approximately -40 mV) but not too far beyond, because slow inactivation is complete at approximately -20 mV (Fig. 7) and would reduce responses to very strong stimuli. Slow inactivation will tend to set a ceiling for the maximal response of nociceptors independently of the threshold. Inflammatory mediators such as prostaglandins E2 and serotonin increase nociceptor excitability through an increase in TTX-R sodium current and a decrease in potassium current (Gold et al., 1996; Cardenas et al., 1997; Nicol et al., 1997). Sensitization causes a decrease in the threshold stimulus generating a response, but limitation of firing by TTX-R channel slow inactivation may help explain why the response of nociceptors to very strong stimuli does not necessarily increase after sensitization (Andrew and Greenspan, 1999; Chen et al., 1999; Levy and Strassman, 2002).
TTX-R sodium channels constitute a logical target for new anti-nociceptive drugs, because they are confined to sensory neurons and help mediate pain signaling (McCleskey and Gold, 1999; Waxman et al., 1999; Baker and Wood, 2001). In general, clinically useful drug molecules that block ion channels show state-dependent binding. At least for TTX-S channels, slow inactivation seems to involve conformational changes distinct from fast inactivation (Vedantham and Cannon, 1998; Ong et al., 2000). Thus, in principle it should be possible to develop drug molecules that bind selectively to slow inactivated states. Drugs that bind to slow inactivated states of TTX-R channels sufficiently tightly should block firing of nociceptors by enhancing slow inactivation at resting potentials or during mild stimulation. Thus, in screening candidate drugs for block of TTX-R channels, it would make sense to use voltage protocols designed to enhance the occupancy of slow inactivated states.
Footnotes
This work was supported by National Institutes of Health Grants HL35034, NS36855, and NS38312. N.T.B. was supported by the Stuart H. Q. and Victoria Quan Fellowship.
Correspondence should be addressed to Dr. Bruce P. Bean, Department of Neurobiology, Harvard Medical School, 220 Longwood Avenue, Boston, MA 02115. E-mail: bruce_bean@hms.harvard.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310338-13$15.00/0
References
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN (1999) The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci 2:541-548. [DOI] [PubMed]
- Andrew D, Greenspan JD (1999) Mechanical and heat sensitization of cutaneous nociceptors after peripheral inflammation in the rat. J Neurophysiol 82:2649-2656. [DOI] [PubMed]
- Baker MD, Wood JN (2001) Involvement of Na+ channels in pain pathways. Trends Pharmacol Sci 22:27-31. [DOI] [PubMed]
- Baumann TK, Burchiel KJ, Ingram SL, Martenson ME (1996) Responses of adult human dorsal root ganglion neurons in culture to capsaicin and low pH. Pain 65:31-38. [DOI] [PubMed]
- Blair NT, Bean BP (2002) Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci 22:10277-10290. [DOI] [PMC free article] [PubMed]
- Caffrey JM, Eng DL, Black JA, Waxman SG, Kocsis JD (1992) Three types of sodium channels in adult rat dorsal root ganglion neurons. Brain Res 592:283-297. [DOI] [PubMed]
- Cardenas CG, Del Mar LP, Scroggs RS (1995) Variation in serotonergic inhibition of calcium channel currents in four types of rat sensory neurons differentiated by membrane properties. J Neurophysiol 74:1870-1879. [DOI] [PubMed]
- Cardenas CG, Del Mar LP, Cooper BY, Scroggs RS (1997) 5HT4 receptors couple positively to tetrodotoxin-insensitive sodium channels in a subpopulation of capsaicin-sensitive rat sensory neurons. J Neurosci 17:7181-7189. [DOI] [PMC free article] [PubMed]
- Chen X, Tanner K, Levine JD (1999) Mechanical sensitization of cutaneous C-fiber nociceptors by prostaglandin E2 in the rat. Neurosci Lett 267:105-108. [DOI] [PubMed]
- Colbert CM, Magee JC, Hoffman DA, Johnston D (1997) Slow recovery from inactivation of Na+ channels underlies the activity-dependent attenuation of dendritic action potentials in hippocampal CA1 pyramidal neurons. J Neurosci 17:6512-6521. [DOI] [PMC free article] [PubMed]
- Cummins TR, Dib-Hajj SD, Black JA, Akopian AN, Wood JN, Waxman SG (1999) A novel persistent tetrodotoxin-resistant sodium current in SNS-null and wild-type small primary sensory neurons. J Neurosci 19:RC43(1-6). [DOI] [PMC free article] [PubMed]
- D'Alcantara P, Cardenas LM, Swillens S, Scroggs RS (2002) Reduced transition between open and inactivated channel states underlies 5HT increased I(Na+) in rat nociceptors. Biophys J 83:5-21. [DOI] [PMC free article] [PubMed]
- Do MT, Bean BP (2003) Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron 39:109-120. [DOI] [PubMed]
- Ellerkmann RK, Riazanski V, Elger CE, Urban BW, Beck H (2001) Slow recovery from inactivation regulates the availability of voltage-dependent Na(+) channels in hippocampal granule cells, hilar neurons and basket cells. J Physiol (Lond) 532:385-397. [DOI] [PMC free article] [PubMed]
- Elliott AA, Elliott JR (1993) Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia. J Physiol (Lond) 463:39-56. [DOI] [PMC free article] [PubMed]
- Fazan Jr R, Whiteis CA, Chapleau MW, Abboud FM, Bielefeldt K (2001) Slow inactivation of sodium currents in the rat nodose neurons. Auton Neurosci 87:209-216. [DOI] [PubMed]
- Featherstone DE, Richmond JE, Ruben PC (1996) Interaction between fast and slow inactivation in Skm1 sodium channels. Biophys J 71:3098-3109. [DOI] [PMC free article] [PubMed]
- Fleidervish IA, Friedman A, Gutnick MJ (1996) Slow inactivation of Na+ current and slow cumulative spike adaptation in mouse and guinea-pig neocortical neurones in slices. J Physiol (Lond) 493:83-97. [DOI] [PMC free article] [PubMed]
- Fox JM (1976) Ultra-slow inactivation of the ionic currents through the membrane of myelinated nerve. Biochim Biophys Acta 426:232-244. [DOI] [PubMed]
- French AS (1989) Ouabain selectively affects the slow component of sensory adaptation in an insect mechanoreceptor. Brain Res 504:112-114. [DOI] [PubMed]
- Gallar J, Pozo MA, Tuckett RP, Belmonte C (1993) Response of sensory units with unmyelinated fibres to mechanical, thermal and chemical stimulation of the cat's cornea. J Physiol (Lond) 468:609-622. [DOI] [PMC free article] [PubMed]
- Gee MD, Lynn B, Cotsell B (1996) Activity-dependent slowing of conduction velocity provides a method for identifying different functional classes of C-fibre in the rat saphenous nerve. Neuroscience 73:667-675. [DOI] [PubMed]
- Gold MS, Thut PD (2001) Lithium increases the potency of lidocaine-induced block of voltage gated Na+ in rat sensory neurons in vitro. J Pharmacol Exp Ther 299:705-711. [PubMed]
- Gold MS, Reichling DB, Shuster MJ, Levine JD (1996) Hyperalgesic agents increase a tetrodotoxin-resistant Na+ current in nociceptors. Proc Natl Acad Sci USA 93:1108-1112. [DOI] [PMC free article] [PubMed]
- Handwerker HO, Anton F, Reeh PW (1987) Discharge patterns of afferent cutaneous nerve fibers from the rat's tail during prolonged noxious mechanical stimulation. Exp Brain Res 65:493-504. [DOI] [PubMed]
- Harper AA (1991) Similarities between some properties of the soma and sensory receptors of primary afferent neurones. Exp Physiol 76:369-377. [DOI] [PubMed]
- Ikeda SR, Schofield GG (1987) Tetrodotoxin-resistant sodium current of rat nodose neurones: monovalent cation selectivity and divalent cation block. J Physiol (Lond) 389:255-270. [DOI] [PMC free article] [PubMed]
- Jung HY, Mickus T, Spruston N (1997) Prolonged sodium channel inactivation contributes to dendritic action potential attenuation in hippocampal pyramidal neurons. J Neurosci 17:6639-6646. [DOI] [PMC free article] [PubMed]
- Kim KJ, Rieke F (2003) Slow Na+ inactivation and variance adaptation in salamander retinal ganglion cells. J Neurosci 23:1506-1516. [DOI] [PMC free article] [PubMed]
- Kuo CC, Bean BP (1994a) Na+ channels must deactivate to recover from inactivation. Neuron 12:819-829. [DOI] [PubMed]
- Kuo CC, Bean BP (1994b) Slow binding of phenytoin to inactivated sodium channels in rat hippocampal neurons. Mol Pharmacol 46:716-725. [PubMed]
- Kuo CC, Lin TJ, Hsieh CP (2002) Effect of Na+ flow on Cd2+ block of tetrodotoxin-resistant Na+ channels. J Gen Physiol 120:159-172. [DOI] [PMC free article] [PubMed]
- Laird JM, Souslova V, Wood JN, Cervero F (2002) Deficits in visceral pain and referred hyperalgesia in Nav1.8 (SNS/PN3)-null mice. J Neurosci 22:8352-8356. [DOI] [PMC free article] [PubMed]
- LaMotte RH, Lundberg LE, Torebjork HE (1992) Pain, hyperalgesia and activity in nociceptive C units in humans after intradermal injection of capsaicin. J Physiol (Lond) 448:749-764. [DOI] [PMC free article] [PubMed]
- Levy D, Strassman AM (2002) Distinct sensitizing effects of the cAMP-PKA second messenger cascade on rat dural mechanonociceptors. J Physiol (Lond) 538:483-493. [DOI] [PMC free article] [PubMed]
- Liu L, Oortgiesen M, Li L, Simon SA (2001) Capsaicin inhibits activation of voltage-gated sodium currents in capsaicin-sensitive trigeminal ganglion neurons. J Neurophysiol 85:745-758. [DOI] [PubMed]
- Lopshire JC, Nicol GD (1997) Activation and recovery of the PGE2-mediated sensitization of the capsaicin response in rat sensory neurons. J Neurophysiol 78:3154 -3164. [DOI] [PubMed]
- Madison DV, Nicoll RA (1984) Control of the repetitive discharge of rat CA 1 pyramidal neurones in vitro. J Physiol (Lond) 354:319-331. [DOI] [PMC free article] [PubMed]
- McCleskey EW, Gold MS (1999) Ion channels of nociception. Annu Rev Physiol 61:835-856. [DOI] [PubMed]
- Mickus T, Jung HY, Spruston N (1999) Slow sodium channel inactivation in CA1 pyramidal cells. Ann NY Acad Sci 868:97-101. [DOI] [PubMed]
- Narahashi T (1964) Restoration of action potential by anodal polarization in lobster giant axons. J Cell Comp Physiol 64:73-96. [DOI] [PubMed]
- Neher E (1992) Correction for liquid junction potentials in patch clamp experiments. In: Methods in enzymology: ion channels (Rudy B, Iverson LE, eds), pp 123-131. San Diego: Academic. [DOI] [PubMed]
- Nicol GD, Vasko MR, Evans AR (1997) Prostaglandins suppress an outward potassium current in embryonic rat sensory neurons. J Neurophysiol 77:167-176. [DOI] [PubMed]
- Ogata N, Tatebayashi H (1992) Slow inactivation of tetrodotoxin-insensitive Na+ channels in neurons of rat dorsal root ganglia. J Membr Biol 129:71-80. [DOI] [PubMed]
- Ogata N, Tatebayashi H (1993) Kinetic analysis of two types of Na+ channels in rat dorsal root ganglia. J Physiol (Lond) 466:9-37. [PMC free article] [PubMed]
- Ong BH, Tomaselli GF, Balser JR (2000) A structural rearrangement in the sodium channel pore linked to slow inactivation and use dependence. J Gen Physiol 116:653-662. [DOI] [PMC free article] [PubMed]
- Peng YB, Ringkamp M, Meyer RA, Campbell JN (2003) Fatigue and paradoxical enhancement of heat response in C-fiber nociceptors from cross-modal excitation. J Neurosci 23:4766-4774. [DOI] [PMC free article] [PubMed]
- Petruska JC, Napaporn J, Johnson RD, Gu JG, Cooper BY (2000) Subclassified acutely dissociated cells of rat DRG: histochemistry and patterns of capsaicin-, proton-, and ATP-activated currents. J Neurophysiol 84:2365-2379. [DOI] [PubMed]
- Petruska JC, Napaporn J, Johnson RD, Cooper BY (2002) Chemical responsiveness and histochemical phenotype of electrophysiologically classified cells of the adult rat dorsal root ganglion. Neuroscience 115:15-30. [DOI] [PubMed]
- Petsche U, Fleischer E, Lembeck F, Handwerker HO (1983) The effect of capsaicin application to a peripheral nerve on impulse conduction in functionally identified afferent nerve fibres. Brain Res 265:233-240. [DOI] [PubMed]
- Porreca F, Lai J, Bian D, Wegert S, Ossipov MH, Eglen RM, Kassotakis L, Novakovic S, Rabert DK, Sangameswaran L, Hunter JC (1999) A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc Natl Acad Sci USA 96:7640-7644. [DOI] [PMC free article] [PubMed]
- Powers RK, Sawczuk A, Musick JR, Binder MD (1999) Multiple mechanisms of spike-frequency adaptation in motoneurones. J Physiol (Paris) 93:101-114. [DOI] [PubMed]
- Richmond JE, Featherstone DE, Hartmann HA, Ruben PC (1998) Slow inactivation in human cardiac sodium channels. Biophys J 74:2945-2952. [DOI] [PMC free article] [PubMed]
- Roy ML, Narahashi T (1992) Differential properties of tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels in rat dorsal root ganglion neurons. J Neurosci 12:2104-2111. [DOI] [PMC free article] [PubMed]
- Rudy B (1978) Slow inactivation of the sodium conductance in squid giant axons: pronase resistance. J Physiol (Lond) 283:1-21. [DOI] [PMC free article] [PubMed]
- Rudy B (1981) Inactivation in Myxicola giant axons responsible for slow and accumulative adaptation phenomena. J Physiol (Lond) 312:531-549. [DOI] [PMC free article] [PubMed]
- Ruff RL, Simoncini L, Stuhmer W (1988) Slow sodium channel inactivation in mammalian muscle: a possible role in regulating excitability. Muscle Nerve 11:502-510. [DOI] [PubMed]
- Rugiero F, Mistry M, Sage D, Black JA, Waxman SG, Crest M, Clerc N, Delmas P, Gola M (2003) Selective expression of a persistent tetrodotoxin-resistant Na+ current and NaV1.9 subunit in myenteric sensory neurons. J Neurosci 23:2715-2725. [DOI] [PMC free article] [PubMed]
- Rush AM, Brau ME, Elliott AA, Elliott JR (1998) Electrophysiological properties of sodium current subtypes in small cells from adult rat dorsal root ganglia. J Physiol (Lond) 511:771-789. [DOI] [PMC free article] [PubMed]
- Schild JH, Kunze DL (1997) Experimental and modeling study of Na+ current heterogeneity in rat nodose neurons and its impact on neuronal discharge. J Neurophysiol 78:3198-3209. [DOI] [PubMed]
- Scholz A, Kuboyama N, Hempelmann G, Vogel W (1998a) Complex blockade of TTX-resistant Na+ currents by lidocaine and bupivacaine reduce firing frequency in DRG neurons. J Neurophysiol 79:1746-1754. [DOI] [PubMed]
- Scholz A, Gruss M, Vogel W (1998b) Properties and functions of calcium-activated K+ channels in small neurones of rat dorsal root ganglion studied in a thin slice preparation. J Physiol (Lond) 513:55-69. [DOI] [PMC free article] [PubMed]
- Serra J, Campero M, Ochoa J, Bostock H (1999) Activity-dependent slowing of conduction differentiates functional subtypes of C fibres innervating human skin. J Physiol (Lond) 515:799-811. [DOI] [PMC free article] [PubMed]
- Slugg RM, Meyer RA, Campbell JN (2000) Response of cutaneous A- and C-fiber nociceptors in the monkey to controlled-force stimuli. J Neurophysiol 83:2179-2191. [DOI] [PubMed]
- Su X, Wachtel RE, Gebhart GF (1999) Capsaicin sensitivity and voltage-gated sodium currents in colon sensory neurons from rat dorsal root ganglia. Am J Physiol 277:G1180-G1188. [DOI] [PubMed]
- Swandulla D, Armstrong CM (1989) Calcium channel block by cadmium in chicken sensory neurons. Proc Natl Acad Sci USA 86:1736-1740. [DOI] [PMC free article] [PubMed]
- Thalhammer JG, Raymond SA, Popitz-Bergez FA, Strichartz GR (1994) Modality-dependent modulation of conduction by impulse activity in functionally characterized single cutaneous afferents in the rat. Somatosens Mot Res 11:243-257. [DOI] [PubMed]
- Urban L, Dray A (1993) Actions of capsaicin on mouse dorsal root ganglion cells in vitro. Neurosci Lett 157:187-190. [DOI] [PubMed]
- Vedantham V, Cannon SC (1998) Slow inactivation does not affect movement of the fast inactivation gate in voltage-gated Na+ channels. J Gen Physiol 111:83-93. [DOI] [PMC free article] [PubMed]
- Vijayaragavan K, O'Leary ME, Chahine M (2001) Gating properties of Nav1.7 and Nav1.8 peripheral nerve sodium channels. J Neurosci 21:7909-7918. [DOI] [PMC free article] [PubMed]
- Vilin YY, Ruben PC (2001) Slow inactivation in voltage-gated sodium channels: molecular substrates and contributions to channelopathies. Cell Biochem Biophys 35:171-190. [DOI] [PubMed]
- Wang HS, McKinnon D (1995) Potassium currents in rat prevertebral and paravertebral sympathetic neurones: control of firing properties. J Physiol (Lond) 485:319-335. [DOI] [PMC free article] [PubMed]
- Waxman SG, Dib-Hajj S, Cummins TR, Black JA (1999) Sodium channels and pain. Proc Natl Acad Sci USA 96:7635-7639. [DOI] [PMC free article] [PubMed]
- Weinreich D, Wonderlin WF (1987) Inhibition of calcium-dependent spike after-hyperpolarization increases excitability of rabbit visceral sensory neurones. J Physiol (Lond) 394:415-427. [DOI] [PMC free article] [PubMed]
- Yoshimura N, Seki S, Novakovic SD, Tzoumaka E, Erickson VL, Erickson KA, Chancellor MB, de Groat WC (2001) The involvement of the tetrodotoxin-resistant sodium channel NaV1.8 (PN3/SNS) in a rat model of visceral pain. J Neurosci 21:8690-8696. [DOI] [PMC free article] [PubMed]