

Abstract
The amygdala plays a role in learning and memory processes that involve an emotional component. However, neural structures that regulate these amygdala-dependent processes are unknown. Previous studies indicate that regulation of affect may be imposed by the prefrontal cortex (PFC) and its efferents to the amygdala. The presentation of conditioned affective stimuli enhances activity of neurons in the lateral nucleus of the amygdala (LAT), which is thought to drive conditioned affective responses. Moreover, plasticity of LAT neuronal responses to stimuli during the course of conditioning is believed to underlie affective learning. This study examines the role of the PFC in the regulation of affective behaviors by evaluating how the PFC affects LAT neuronal plasticity and activity that is evoked by previously conditioned stimuli. In vivo intracellular recordings were performed from the LAT of anesthetized rats during pavlovian conditioning and during the presentation of stimuli that were conditioned in the awake rat before recording. Train stimulation of the PFC suppressed LAT neuronal activity that was evoked by both previously conditioned and neutral stimuli. In addition, PFC stimulation blocked LAT neuronal plasticity associated with an affective conditioning procedure. These results indicate that the PFC has the potential to regulate affective processes by inhibition of the LAT. Patients with disruptions of the PFC-LAT interaction often display an inability to regulate affective responses. This may be attributable to the loss of PFC-imposed inhibition of the emotional response to a stimulus but may also include the formation or diminished extinction of inappropriate associations.
Keywords: amygdala, prefrontal cortex, plasticity, pavlovian conditioning, in vivo, intracellular electrophysiology, inhibition, Te3, odor, affect
Introduction
Abnormal interactions between the prefrontal cortex (PFC) and amygdala have been implicated in depression (Soares and Mann, 1997; Van Elst et al., 1999, 2001; Drevets, 2000; Sheline et al., 2001; Beyer and Krishnan, 2002; Bowley et al., 2002; Davidson et al., 2002; Siegle et al., 2002; Tekin and Cummings, 2002; Whalen et al., 2002), schizophrenia (Pakkenberg, 1992; Lawrie and Abukmeil, 1998; Benes and Berretta, 2001; Rajarethinam et al., 2001; Wible et al., 2001; Ananth et al., 2002; Anderson et al., 2002; Chance et al., 2002; Gur et al., 2002; Lawrie et al., 2002; Selemon et al., 2002; Taylor et al., 2002; Volk and Lewis, 2002; Perlstein et al., 2003), and other psychiatric disorders (Coplan and Lydiard, 1998; Ninan, 1999; Strakowski et al., 2000; Davidson, 2002). Patients with frontal lobe deficits often display inadequate regulation of affective behaviors (Damasio et al., 1994; Paradiso et al., 1999; Anderson et al., 2000; Kim and Choi-Kwon, 2000). The affective symptoms of many psychiatric disorders are consistent with disrupted regulation of the amygdala by PFC, resulting in disinhibited emotional behavior. Several imaging studies in humans have demonstrated that both activation of the PFC and higher cognitive processing are reciprocally related to amygdala activity and amygdala-dependent fear conditioning (Hariri et al., 2000, 2003; Liberzon et al., 2000; Beauregard et al., 2001; Ochsner et al., 2002; Schaefer et al., 2002; Carter et al., 2003). It has been reported in studies of nonhuman animals that lesion or inactivation of the medial PFC (mPFC) facilitates affective behaviors, whereas stimulation of the PFC suppresses similar behaviors (al Maskati and Zbrozyna, 1989; Jaskiw and Weinberger, 1990; Zbrozyna and Westwood, 1991, 1993; Morgan and LeDoux, 1995; Dias et al., 1996; Jinks and McGregor, 1997; Vouimba et al., 2000; Sacchetti et al., 2002). Thus, it appears that the PFC, and in particular the medial PFC (infralimbic and prelimbic cortices), plays a role in regulation of affective behaviors, some of which are mediated by the basolateral amygdala complex (BLA) (lateral, basal, and accessory basal nuclei). Our previous data from rats demonstrate that the mPFC afferents suppress BLA output, perhaps providing a mechanism for mPFC regulation of BLA-dependent affective behaviors (Rosenkranz and Grace, 2001).
The lateral nucleus (LAT) of the BLA may play a dual role in the acquisition and expression of some affective behaviors. Lesion or inactivation of the LAT blocks the formation of affective learning (Wilensky et al., 1999; Goosens and Maren, 2001; Nader et al., 2001; Bauer et al., 2002; Tazumi and Okaichi, 2002; but see Cahill et al., 2000), and during acquisition of BLA-dependent learning, the LAT is a site of neuronal plasticity (Ben-Ari and Le Gal la Salle, 1972; Rogan and LeDoux, 1995; Quirk et al., 1997; Poremba and Gabriel, 1999; Collins and Pare, 2000; Blair et al., 2001). Lesions of the LAT also disrupt the expression of some of these same affective behaviors (Kim et al., 1993; Cousens and Otto, 1998; Heldt et al., 2000; Wallace and Rosen, 2001), and LAT neurons display enhanced activity correlated with affective responses to conditioned sensory stimuli (Maren, 2000; Pare and Collins, 2000; Repa et al., 2001). However, it is not always appropriate to express affective behaviors, especially to behaviorally benign stimuli, and this suppression may be a function of the mPFC. Evidence indicates that plasticity of LAT neuronal responses is a substrate of affective learning, and the enhanced response of LAT neurons to conditioned sensory stimuli is a substrate of expression of BLA-dependent affective behaviors. Therefore, we assessed the contribution of the mPFC to learning and expression by examining how mPFC afferent activation alters LAT plasticity and LAT neuronal responses to conditioned sensory stimuli compared with inputs from a sensory association cortical region (Te3). Thus, this study uses in vivo intracellular recordings from neurons of the LATs of anesthetized rats to test the hypothesis that mPFC stimulation will suppress LAT plasticity that occurs during an affective conditioning procedure as well as LAT neuronal responses to previously conditioned sensory stimuli.
Materials and Methods
Animal preparation. All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Male Sprague Dawley rats (250-350 gm) were housed in pairs using a 12 hr light/dark cycle in a temperature-controlled environment. Food and water were available ad libitum. Rats were anesthetized with an initial intraperitoneal injection of 400 mg/kg 8% chloral hydrate. Supplemental anesthesia (8% chloral hydrate) was delivered via a lateral tail vein catheter as necessary to maintain suppression of hindlimb withdrawal reflex. Anesthesia level was also verified by pupil constriction and the lack of an eyeblink reflex. Rats were placed on a temperature-controlled heating pad (Heater VL-20F; Fintronics, Orange, CT), and their core temperature was monitored with a rectal thermometer (Precision Thermometer 4600; Yellow Springs Instruments, Yellow Springs, CO) and maintained at ∼37°C. The rats were placed in a stereotaxic device (David Kopf Instruments, Tujunga, CA) that was modified to allow contained odor delivery to the rat's nose. An empirically measured constant flow rate of 1.5 l/min of air was delivered from control vials or vials of odorant (anise or almond) across the rat's nose and then traveled to a container of odor-scavenging activated charcoal. On the basis of the air flow rate and the volume of tubing that separated the odorant source from the rat's nose (4.6 ml), it is estimated that odorants reached the rat's nose within 0.2 sec of switching the air flow path (using remotely controlled solenoid valves; Bio-Chem Valves, Boonton, NJ). Incisions were made in the scalp to expose the skull. Burr holes were drilled, and the dura was removed overlying the LAT and mPFC or Te3. Coordinates were determined using a stereotaxic atlas (Paxinos and Watson, 1998) as follows: LAT, -5.0 mm lateral, -3.3 mm caudal from bregma; mPFC, -0.3 lateral, 2.2 mm rostral from bregma; Te3, -6.0 lateral, +2.2 rostral from interaural. A concentric bipolar stimulating electrode was lowered into the mPFC or Te3. Experiments started no sooner than 45 min after completion of surgery.
Intracellular recordings. Electrodes composed of borosilicate glass tubing (1.5 mm outer diameter, 0.84 mm inner diameter; World Precision Instruments, Sarasota, FL) were pulled with a Flaming-Brown micropipette puller (model P-80/PC; Sutter Instruments, Novato, CA). Electrodes were filled with 2% biocytin in 3 m potassium acetate (Sigma, St. Louis, MO). Impedances were measured in situ and ranged between 45 and 75 MΩ. Electrodes were lowered slowly to the LAT via a hydraulic micromanipulator (model 640; David Kopf Instruments). A stimulator (Grass S88; Grass Instruments, Quincy, MA) in series with an intracellular recording amplifier (IR-283; Neuro Data Instruments, New York, NY) and a stimulus isolation unit (PSIU6; Grass Instruments) was used to deliver DC to the electrode. Electrode potentials were monitored visually with an oscilloscope (V134; Hitachi, Tokyo, Japan) and a multimeter (179A TRMS multimeter; Keithley, Cleveland, OH) and audially with a Grass AM5 audio monitor (Grass Instruments). Data were fed from the amplifier to a personal computer, monitored using custom-designed software (Neuroscope), and stored on a hard disk for analysis off-line. Data were also digitized (Neuro-corder DR-484; Neuro Data Instruments) and saved on videotapes using a videocassette recorder (Panasonic AG-1280; Matsushita Electric Industrial, Osaka, Japan). At the completion of experiments, neurons were hyperpolarized with negative current to below spike threshold, and positive DC pulses (0.1-1.0 nA, 250 msec, 2 Hz) were used to eject biocytin from the electrode. Rats were perfused transcardially with 0.85% cold saline followed by 4% paraformaldehyde in 0.1 m phosphate buffer. Brains were removed and stored in perfusion fixative and then cryoprotected in 25% sucrose and 0.1 m phosphate buffer solution. Brains were then sliced on a freezing microtome into 60- to 80-μm-thick sections and labeled for biocytin with the Vectastain Elite ABC peroxidase kit (Vector Laboratories, Burlingame, CA) using nickel-enhanced DAB as the chromogen. Sections were counterstained with a custom mixture of cresyl violet and neutral red stains, and stimulating electrode and recording electrode placements were identified as described previously (Rosenkranz and Grace, 2001). A cell was determined to lie within the LAT after localization of a biocytin-filled neuron or by determination of the recording site from the visible electrode track and the experimental records of the dorsoventral coordinates of the cell relative to the dorsal and ventral surfaces of the brain.
Electrical stimulation. Stimulation of the mPFC was performed during three sets of recordings. The mPFC was stimulated in trains at 15-20 Hz, 0.1-0.4 mA, 0.3 msec single-pulse duration. The train duration depended on the neuronal activity of interest. When the effects of the mPFC on spontaneous postsynaptic events were examined, stimulation lasted 5 sec. When determining the effects of mPFC stimulation on odor-evoked potentials or plasticity of odor-evoked potentials, the train lasted 10 sec, which was equal to the duration of the odor-footshock presentation.
Te3 stimulation was performed during two sets of recordings, and parameters were equivalent to mPFC stimulation. The stimulation lasted 5 sec when the effects of the Te3 on spontaneous postsynaptic events were examined and 10 sec when determining the effects of the Te3 on plasticity of odor-evoked potentials.
Pavlovian conditioning in awake rats. A pavlovian conditioning procedure was performed by pairing of an odor with footshock (Rescorla, 1988; Otto et al., 1997; Paschall and Davis, 2002). Rats (n = 8) were taken from their home cages and placed in a fan-ventilated activity chamber. After a 30 min habituation phase, each of two odors (almond and anise) were presented alternately to the chamber at 2 min intervals, two times each to maximize similarity to the conditioning protocol in anesthetized rats (see below). Subsequently, one odor was paired with footshock (0.1-0.5 mA, 3 sec, delivered via the grid floor of the activity chamber), whereas the other odor was presented in the absence of footshock, in interleaved trials at 2 min intervals, three times each. After another 20 min, rats were brought back to their home cages. After 24-48 hr, rats were anesthetized and prepared for electrophysiological recordings. After stable recordings from a neuron, both the paired [conditioned stimulus (CS+)] and nonpaired (CS-) odors were presented to the rats at 60 sec intervals, at least two times each.
A separate set of 10 rats was trained using the protocol described above or with a slightly modified protocol in which four CS+-CS- trials were given and the duration of CS- exposure per trial was 8 min. Twenty-four hours after training, the rats were placed in the training apparatus, and exploratory behavior, grooming, and freezing were scored during presentation of nonpaired-paired-nonpaired odor block of extinction (i.e., no shock) trials with a duration of 8 min each. Exploratory behavior was scored using the following scale: 0, no locomotion; 1, ambulation across the side of the testing chamber; 2, ambulation across two sides; 3, exploration of the full perimeter of the testing chamber; and 4, exploration of the center and perimeter. Grooming and freezing or other conditioned changes in posture were scored as present or absent during a CS exposure. Grooming consisted of paw washing, head washing, body-directed licking and biting, and pulling on fur (body washing), occurring singly, in bouts, or in sequences in which the target or the specific behavior was variable, to exclude potential stereotyped behavior. Exploratory activity scores during presentation of the paired and nonpaired odors (averaged across trials before and after the CS+) were compared with a Wilcoxon signed rank test. Other behaviors were analyzed using a binomial test in which the expected proportion (0-1.0) of values (yes or no) was defined as the proportion of values in the CS- condition.
Pavlovian conditioning in anesthetized rats. In a separate group of anesthetized rats, a pavlovian conditioning procedure was performed by pairing of an odor with footshock while recording intracellularly from a neuron. Footshock was delivered via two 28 gauge needles inserted beneath the skin of the lateral surface of the foot contralateral to the neuronal recordings. Footshock current intensity was controlled by a Grass S88 stimulation unit (Grass Instruments) in series with a stimulus isolation unit (PSIU 88; Grass Instruments). The conditioning procedure occurred as follows: Each odor (anise or almond) was presented two times for 10 sec each, with a 60 sec delay between presentations. One odor was chosen to be paired with the footshock. Paired odor selection was counterbalanced. This chosen odor was paired with the footshock (4 sec, 2-5 mA, 20 Hz, 0.2 msec duration pulses) such that the footshock was presented 5 sec after initiation of the odor delivery. The footshock intensity chosen was dependent on the level of depolarization achieved in the neuron by the footshock. Typically, an intensity of ∼4 mA was used, evoking a response that was subthreshold to spike generation. This pairing was performed five times at 60 sec intervals. After these pairings, each odor was again presented at least twice, 10 sec each, at 60 sec intervals. This procedure verified that the neuron was capable of displaying pavlovian conditioning-related plasticity.
To examine the effects of afferent activation on this neuronal plasticity, the above procedure was replicated while still recording from the same neuron with two modifications: the previously unpaired odor was now paired with footshock while the previously paired odor was not, and during this odor-footshock pairing, either the Te3 or the mPFC was stimulated (as described above) (see Fig. 1) at an intensity that evoked an ∼1-3 mV postsynaptic potential at resting membrane potentials (Vrest).
Figure 1.

Neuron locations and electrode placements. A, Stimulation electrodes were placed in the mPFC (infralimbic and prelimbic cortices; filled circles) and Te3 (filled triangles). Stimulation of the mPFC and Te3 alters the activity of neurons in the LAT (open circles and open triangles, respectively). B, The conditioning procedure in anesthetized rats included the pairing of a 10 sec odor presentation with a 4 sec footshock. When noted, the mPFC or Te3 was stimulated during the odor-footshock pairings. C, Morphology of biocytin-filled LAT neurons is consistent with projection neurons (20× magnification). D, The biocytin-filled neuron (in C) displays electrophysiological characteristics consistent with LAT projection neurons, including adapting firing pattern (1), input resistance (2; 47 MΩ), and membrane potential (3; -74 mV) with spontaneous PSPs.
Intracellular data collection. Data collection typically began 5-10 min after neuronal impalement to allow time for membrane stabilization. After collection of several minutes of baseline data, DC pulses were injected into the neurons (±0.02-1.2 nA, 250-350 msec, 0.2-0.4 Hz). Only hyperpolarizing DC pulses were used to determine input resistance, and only the linear portion of the plot was included for this analysis. Additionally, the input resistance was determined only from traces that displayed no co-occurring membrane fluctuations or postsynaptic potentials. Mean resting membrane potential and SD were determined from 30 sec sampling periods, and action potentials were eliminated from this analysis. Action potential threshold was measured at the onset of the rapid transition in membrane potential at the initiation of the spike. The amplitude of the action potential was the voltage difference between the threshold and the action potential peak. The duration of action potentials was measured as the time between action potential initiation at spike threshold and the return to baseline preceding the afterhyperpolarization, if present. The membrane potential calculated during intracellular recording was adjusted on the basis of the potential obtained after withdrawal of the electrode from the neuron after the recording.
Odor-evoked postsynaptic potentials (PSPs) were measured from LAT neurons before, during, and after conditioning, as well as in neurons from naive control rats that did not undergo any form of conditioning. The area under the odor-evoked PSP was calculated. Because the neurons displayed spontaneously occurring PSPs (sPSPs), odor-evoked responses were determined by comparing them with baseline activity. Thus, the area under the PSP was analyzed by measuring the first 5 sec of the area of the odor-evoked PSP using the resting membrane potential as the baseline and subtracting out the area of spontaneously occurring PSPs of the 5 sec immediately preceding odor presentation. Only the first 5 sec were used, because footshock was presented 5 sec into the odor during the conditioning protocol. Using only the first 5 sec of the odor presentation for analysis facilitated comparison of odor-evoked responses during conditioning with responses obtained before and after the conditioning protocol.
Data analysis. To determine the type of interaction between sPSPs and afferent-evoked PSPs, the area under these events was quantified. If the interaction between spontaneous and afferent-evoked PSPs is linear, their predicted mathematical summation should approximate the empirically measured area under PSPs during afferent train stimulation. Based on the average area of single afferent-evoked PSPs and the average baseline area of sPSPs, a predicted area under the PSPs during afferent stimulation can be estimated using the following algorithm: mean area of 5 sec sPSP + [mean evoked single PSP area × frequency of stimulation (15 or 20 Hz) × duration of stimulation (5 sec)].
To analyze differences in the odor-evoked PSP attributable to the conditioning protocol in anesthetized rats, the areas under the odor-evoked PSPs were compared before and after conditioning for each odor. Additionally, the difference between the paired and nonpaired odor was analyzed before and after conditioning.
To analyze differences in odor-evoked PSPs recorded from anesthetized rats attributable to the conditioning procedure in awake animals, the paired odor was compared with the nonpaired odor as well as with novel odors in naive rats. Additional analysis included comparison of the odor-evoked PSP in the presence and absence of afferent stimulation. For analysis of electrophysiological data, repeated-measures ANOVA was used as appropriate; otherwise, t tests were used. When post hoc analysis was performed, Bonferroni corrections were used.
Neurons were not included in this study if the resting membrane potential was less polarized than -65 mV, if their action potentials did not overshoot 0 mV, if the measured input resistance was <20 MΩ, or if they were found to lie outside the LAT.
Results
A total of 63 neurons that fit electrophysiological and morphological criteria for projection neurons (Washburn and Moises, 1992; Rainnie et al., 1993; Faber et al., 2001), and were located in the LAT were used for this study. Neurons displayed an average action potential duration of 2.0 ± 0.03 msec, an action potential amplitude of 69.9 ± 0.4 mV (from threshold), an input resistance of 42.7 ± 1.2 MΩ, and a resting membrane potential of -77.1 ± 0.8 mV. Biocytin-filled neurons that were recovered for anatomical analysis (n = 24) (Fig. 1) typically displayed an identifiable primary dendrite and were covered with spines. Within the LAT, neurons were mostly recorded from the dorsolateral and ventrolateral divisions (Fig. 1).
Train stimulation of afferents
In 37 neurons from 18 naive rats, the effects of mPFC or Te3 stimulation on spontaneous PSPs were examined to determine how these afferents interact with other synaptic inputs. Train stimulation of the mPFC resulted in qualitatively different effects than stimulation of the Te3, regardless of stimulation intensity or frequency. At membrane potentials shifted to depolarized values with constant DC injection (approximately -55 to -65 mV, resulting in an ∼4 Hz action potential firing rate), train stimulation of the mPFC resulted in hyperpolarization and in a cessation of spike firing (for analysis, 15 Hz stimulation was used, similar to frequencies used below; baseline firing frequency, 4.6 ± 0.8 Hz; firing frequency during mPFC stimulation, 0 Hz; p < 0.05; df = 11; paired t test), whereas train stimulation of the Te3 resulted in additional depolarization and increased spike firing (Fig. 2A) (baseline firing frequency, 4.1 ± 1.2 Hz; firing frequency during Te3 stimulation, 8.9 ± 2.0 Hz; p < 0.05; df = 11; paired t test). At Vrest, train stimulation of the Te3 evoked trains of PSPs that summated in a near-linear or supralinear manner with sPSPs (Fig. 3A). Thus, when Te3-evoked PSPs overlapped with sPSPs, the area under the co-occurring events was close to the linear sum of the predicted Te3-evoked PSP with the mean area under baseline sPSPs (Fig. 3A) (+157% change from baseline; baseline area under sPSPs, 25,496.6 ± 2317.7 mV/msec, area under PSPs during Te3 stimulation, 40,199.2 ± 4291.9 mV/msec; p < 0.05; df = 15; paired t test; predicted area, 39,017.3 ± 3424.4 mV/msec; single Te3 PSP area, 176.0 ± 21 mV/msec). In contrast, in most instances, train stimulation of the mPFC suppressed sPSPs below baseline, as reflected in a reduction in the SD of the mean membrane potential during mPFC stimulation (Fig. 3B) (baseline SD, 2.93 ± 0.37 mV; stimulation SD, 1.18 ± 0.09 mV; p < 0.05; df = 20; paired t test; compare with the Te3 baseline SD, 3.32 ± 0.35 mV; Te3 stimulation, 3.57 ± 0.40 mV; p > 0.05; df = 15). This is consistent with sublinear summation of sPSPs with mPFC-evoked trains of PSPs (Fig. 3D) (-49% change from baseline; baseline area under sPSPs, 23,348.0 ± 4170.9 mV/msec; area under PSPs during mPFC stimulation, 11,448.1 ± 1400.4 mV/msec; p < 0.05; df = 20; paired t test; single mPFC PSP, 152.4 ± 13.9 mV/msec), so that not only was there a lack of linear summation, but mPFC stimulation suppressed ongoing activity (Fig. 3D). If linear summation occurs between sPSPs and mPFC-evoked PSPs, a value of 34,613.7 ± 4212.0 mV/msec was expected for a 5 sec period during mPFC stimulation, whereas the actual measured area was 11,448.1 ± 1400.4 mV/msec, which was markedly sublinear to the predicted value (Fig. 3D) (for a 5-sec-period average mPFC-evoked PSP area, 152.4 ± 13.9 mV/msec; average baseline PSP area, 23,348.0 ± 4170.9 mV/msec). Spontaneous activity returned to baseline levels rapidly after cessation of afferent stimulation. Stimulation of the mPFC evokes chloride-mediated inhibitory PSPs (Rosenkranz and Grace, 2002a). However, the observed sublinear summation is not simply attributable to hyperpolarizing IPSPs evoked by mPFC stimulation subtracting from excitatory sPSPs, because at Vrest, mPFC-evoked PSPs were typically in the depolarizing direction (Vrest was more hyperpolarized than the chloride reversal potential). But to further verify this, the summation of mPFC-evoked PSPs with sPSPs was examined at even more hyperpolarized membrane potentials (greater than -80 mV), and sublinear summation was still observed (data not shown). Furthermore, this sublinear summation is not attributable to a reduction in the amplitude of the mPFC-evoked PSPs during the train, because the amplitude of evoked PSPs remained fairly constant. The magnitude of the sublinearity was greater than could be accounted for by even a complete reduction of mPFC-evoked PSP amplitude during the train and was even less than the mean area of sPSPs during an equivalent time epoch, indicating suppression of spontaneous PSPs. Nor is the greater summation of Te3 inputs attributable to longer decay times than mPFC-evoked PSPs (Fig. 3C), because the duration of Te3- and mPFC-evoked PSPs was similar at Vrest at stimulation intensities used for this study. Therefore, it is apparent that mPFC stimulation caused a suppression of spontaneous PSPs independent of the synaptic events evoked during the stimulation itself.
Figure 2.

Stimulation of the mPFC suppresses spike firing. Te3 and mPFC stimulation exert opposite actions on membrane potential and action potential firing when the membrane is depolarized. A, Te3 train stimulation evokes membrane depolarization and spike firing (part of the voltage response, indicated by the solid horizontal bar, is expanded in bottom traces). B, Conversely, mPFC stimulation results in membrane hyperpolarization and cessation of spike firing.
Figure 3.

Stimulation of the mPFC suppresses sPSPs. A, Train stimulation of Te3 afferents evokes PSPs that summate with sPSPs. Stimulation is indicated by the horizontal bar in the top trace, which is expanded in the bottom traces. Note the occurrence of sPSPs during Te3 stimulation (arrows demark the start and end of stimulation). B, The lack of suppression of sPSPs by Te3 inputs can be demonstrated by the unchanged SD of the mean membrane potential and the linear summation of the area under the PSP (A, inset). C, PSPs of similar amplitudes evoked by mPFC and Te3 stimulation display similar decay times. D, When the mPFC is stimulated in trains, no summation is observed, and sPSPs in LAT neurons are suppressed. The expanded bottom traces demonstrate the abrupt cessation of sPSPs at the onset of mPFC stimulation (first arrow). This can be quantified as a reduction in the SD of the mean membrane potential (B) and a sublinear summation (D, inset). Stimulus artifacts have been removed from most traces for clarity in this and subsequent figures. *p < 0.05; t test (significant difference between the mPFC and the Te3); +p < 0.05; paired t test (significant difference between stimulation and baseline).
MPFC regulation of odor-evoked PSPs
In 17 neurons from seven naive rats, discrete 10 sec presentation of an odor resulted in a noticeable barrage of PSPs. These PSPs were time-locked to odor presentation and occasionally evoked action potential firing. Train stimulation of mPFC suppressed the response of LAT neurons to odors (Fig. 4) (at Vrest, baseline odor-evoked PSP, 2321 ± 744 mV/msec; mPFC plus odor, -601 ± 183 mV/msec; p < 0.05; df = 16; paired t test); only neurons displaying obvious odor-evoked responses were used for this analysis (greater than ∼15% change from baseline; neurons displaying smaller responses were not included) to ensure that mPFC interactions with odors were examined and not only interactions with sPSPs, which were analyzed above (smaller odor-evoked responses were also inhibited by mPFC stimulation, but it is difficult to distinguish some smaller odor-evoked responses from normal variability of spontaneous PSP activity).
Figure 4.

Stimulation of the mPFC suppresses responses to novel odors. Presentation of odors results in PSPs in many LAT neurons (A, top; odor was presented at the gray bar in all traces). Train stimulation of the mPFC (15 Hz, 0.2 mA, indicated by arrows) during the odor presentation suppresses the PSPs evoked by this odor (bottom traces; each set of traces represents two overlays of neuronal activity immediately before and during odor presentation). B, The mPFC stimulation significantly suppresses odor-evoked PSPs (*p < 0.05; paired t test), as quantified by the area under the PSP during baseline odor presentation and during odor presentation with mPFC stimulation.
To examine whether the mPFC may play a role in suppression of the amygdalar response to both previously conditioned and nonconditioned odors, a differential conditioning paradigm was implemented in eight awake animals, and the effects of mPFC stimulation on odor-evoked responses were examined in anesthetized animals while performing in vivo intracellular recordings. Odors that were paired with a footshock in awake animals evoked a greater response than nonpaired control odors when LAT neurons were recorded 24-48 hr after the conditioning procedure, as well as when compared with novel odors in naive anesthetized rats (Fig. 5A,B, top traces) (mean paired odor, 4195 ± 716 mV/msec, n = 14 neurons; mean nonpaired odor, 1142 ± 851 mV/msec, p < 0.05 compared with paired odors, n = 14 neurons; mean naive odor, 842 ± 577 mV/msec, p < 0.05 compared with paired odors; n = 23 neurons; t test; in these cases, the odor-evoked responses of every LAT neuron tested were included in analysis, not only those neurons that displayed a large response, as above). However, train stimulation of the mPFC suppressed the response of LAT neurons to all odors to a similar degree, regardless of whether the odor was previously paired with a footshock (Fig. 5A,B, bottom traces, C) (mean baseline, 4195 ± 716 mV/msec; mPFC, -526 ± 98 mV/msec; p < 0.05; df = 14; paired t test), explicitly nonpaired (mean baseline, 1142 ± 851 mV/msec; mPFC, -338 ± 164 mV/msec; p < 0.05; df = 14; paired t test), or novel (see above). Thus, it appears that inputs from the mPFC can suppress LAT responses driven by conditioned or nonconditioned odors and thereby perhaps suppress affective responses to both neutral stimuli and stimuli endowed with affective salience.
Figure 5.

Stimulation of the mPFC suppresses responses to paired odors. Pairing of odors with footshock in awake rats results in an enhanced response of LAT neurons to the paired odors (A, top traces) recorded in anesthetized rats compared with nonpaired odors (B, top traces). Stimulation of the mPFC (15 Hz, 0.1 mA) during the odor presentation suppresses the response of this LAT neuron to both paired (A, bottom) and nonpaired (B, bottom) odors. Traces are overlays of the voltage response of a single LAT neuron to consecutive odor presentations (solid horizontal bar). C, As a group, paired odors evoke a greater response than nonpaired odors, and mPFC stimulation suppresses both odors to a similar extent compared with baseline odor-evoked responses, quantified as the area under the PSP. *p < 0.05; t test (paired odor compared with nonpaired odor); +p < 0.05; t test (baseline response to odor compared with response to odor in the presence of mPFC stimulation). D, The training procedure also resulted in differential behavioral responses to footshock-paired and nonpaired odors. These rats display a suppression of exploration in the presence of the footshock-paired odor (CS+) compared with the nonpaired odor (CS-; +p < 0.02; Wilcoxon signed rank test), as well as a suppression of grooming (*p < 0.001; binomial test); some rats displayed freezing (*p < 0.05; binomial test).
To ensure that the odor paired with footshock was endowed with affective salience, the behavior of a separate group of 10 rats that underwent the training procedure but were not anesthetized for electrophysiology was examined (Fig. 5D). All animals showed a suppression of ongoing exploratory behavior or grooming after the onset of the paired odor (CS+). Four animals showed obvious freezing within 20 sec after the onset of the CS+ that was similar to what had been displayed during shock on the previous day. These behaviors were not displayed during the nonpaired odor (CS-) before CS+ exposure. Thus, exploratory behavior during the CS+ exposure was significantly decreased relative to CS- exposure (CS-, 2.0 ± 0.19; CS+, 0.7 ± 0.40; p < 0.02; CS+-CS- negative ranks, 8 of 10; Wilcoxon Z, 2.43; Wilcoxon test). Grooming was completely suppressed during the CS+, with 8 of 10 rats displaying grooming during CS- and 0 of 10 rats displaying grooming during CS+ (p < 0.0001; binomial test). Although exploratory and grooming behavior was suppressed in the presence of the CS+, true freezing behavior (including postural tension and not just lack of movement) was seen in four animals, and it never occurred in the presence of the CS- (p < 0.02; binomial test).
mPFC regulation of LAT neuronal plasticity
In a different set of eight rats, the modification of plasticity by the mPFC was examined. Neurons of the LAT display plasticity during the course of an affective conditioning procedure (Fig. 1) even when the odor and footshock pairings occur in anesthetized rats (Rosenkranz and Grace, 2002b). However, the response to the odor paired with footshock under anesthesia (2074 ± 226) (Fig. 6) is not as robust as the response to the odor paired with footshock in awake animals (4195 ± 716; p < 0.05; df = 20; t test) (Fig. 6), although the prepairing baseline response to odors (928 ± 178) is similar to the response to the nonpaired control odor presented to awake restrained (1142 ± 851; NS; df = 20; t test). Nonetheless, in the anesthetized rat, an odor that evoked a minimal response under baseline conditions will evoke a greater response from the same LAT neurons after this odor has been paired with footshock (Fig. 7) (odor A baseline, 928 ± 178 mV/msec; postpairing, 2074 ± 226 mV/msec; p < 0.05; df = 7; paired t test). The non-footshock-paired odor does not evoke enhanced responses from LAT neurons after the conditioning procedure (Fig. 7) (odor B baseline, 899 ± 167 mV/msec; odor B postpairing of odor A, 916 ± 210 mV/msec; p > 0.05; paired t test; however, odor B is significantly different from odor A; p < 0.05; t test). Not only is the response to this odor enhanced compared with baseline values, it is also enhanced relative to a control, nonpaired odor. A previous study in awake rats demonstrated that separate groups of neurons in the dorsal subdivision of the LAT display differences in the duration of plasticity and the onset of responses to stimuli (Repa et al., 2001). In the current study, there were no obvious systematic differences in the plasticity of odor-evoked responses between neurons of the dorsal or other subdivisions of the LAT. However, it is unclear whether the presence of anesthesia may have obscured some of the subtle differences between the subregions of the LAT in awake rats. Thus, similar to our previous study, the plasticity of the odor-evoked response is specific for a footshock-paired odor and not for nonpaired odors. However, repeating the conditioning procedure with train stimulation of the mPFC while the odor is paired with footshock blocks this form of plasticity (Fig. 7) (postpairing B odor A, 2209 ± 275 mV/msec; odor B, 834 ± 199 mV/msec; p < 0.05; df = 7; t test; odor A is significantly different from odor B, and odor B postpairing of odor B is not significantly changed). Train stimulation of the mPFC reduces the usually observed enhancement of a footshock-paired odor, although it has no long-lasting effect on a previously conditioned odor. In addition, there was no dorsoventral gradient in the effectiveness of infralimbic and prelimbic prefrontal cortical stimulation during plasticity. The lack of enhancement of the odor-evoked response when it is paired with mPFC stimulation is not attributable to a general lack of plasticity in that particular neuron, because that same cell displays plasticity in response to odors paired with footshock in the absence of afferent stimulation. In addition, stimulation of the mPFC does not permanently disrupt plasticity, because in a few separate instances, odor and footshock were paired with mPFC stimulation first, and then a second odor was paired with footshock alone. The plasticity in the initial pairings was blocked by mPFC stimulation, although plasticity was seen after the second pairings in the absence of mPFC stimulation (baseline response to odor A, 847 ± 411 mV/msec; postpairing odor A with footshock and mPFC stimulation, 811 ± 371 mV/msec; odor B baseline, 732 ± 359 mV/msec; postpairing odor B with footshock, 2198 ± 571 mV/msec; n = 3). Thus, the mPFC-induced disruption of plasticity is not a generalized effect on that neuron, because plasticity of the response to the non-mPFC-paired odor can still occur. However, it is still not apparent whether pairing of mPFC stimulation with a specific odor will disrupt subsequent conditioning to this odor in the absence of mPFC stimulation. Our results show that in rats that were conditioned when awake, a small number of mPFC-CS+ pairings were insufficient to disrupt LAT neuronal responses to the CS+ in subsequent trials (see above), suggesting that the effects of mPFC pairings with the odor might not affect subsequent conditioning to that odor.
Figure 6.
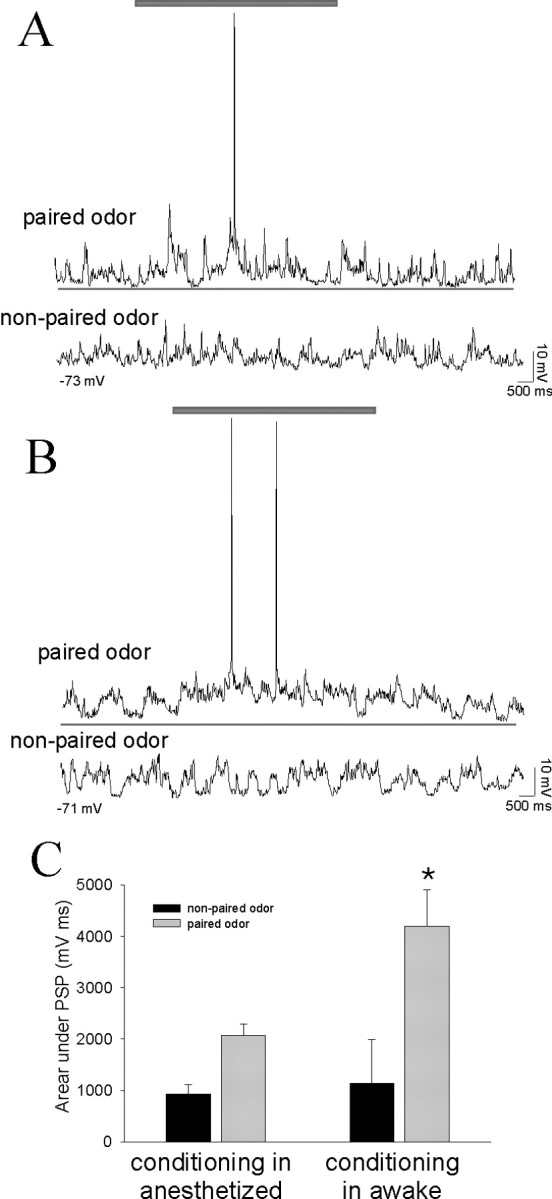
BLA neurons exhibit significantly greater responses to odors that were conditioned previously in the awake rat compared with the response to odors conditioned concurrently with recordings under anesthesia. A, After an odor has been paired with footshock in an anesthetized rat, a noticeable response to the paired odor develops (top trace; gray bar above traces denotes duration of odor presentation in all traces), whereas the nonpaired odor evokes no obvious response from this LAT neuron (bottom trace). B, When the odor has been paired with footshock in awake rats, the paired odor evokes a robust response from LAT neurons (top trace; the thin line beneath traces is used to facilitate comparison between the response to odors that were conditioned in awake and anesthetized rats) recorded from anesthetized rats 24 hr after the conditioning procedure. The nonpaired odor does not evoke an obvious response (bottom trace). C, The activity of LAT neurons, measured as the area under the odor-evoked PSPs, is significantly greater in response to odors conditioned in awake rats compared with odors conditioned in anesthetized rats (*p < 0.05; t test). The response to nonpaired odors is not significantly different.
Figure 7.

Stimulation of the mPFC suppresses the plasticity of LAT neurons. A, In anesthetized rats, an initially minimal response of a LAT neuron to an odor (1; presentation indicated by the gray bar) is enhanced after pairing of that odor with footshock (2, odor A, arrow), whereas the nonpaired odor does not display enhancement (2, odor B). Additional plasticity is blocked by stimulation of the mPFC during the odor-footshock pairings (3, odor B), whereas the previous enhancement of odor A is not reversed (3, odor A; compare arrows in the odor A and odor B traces in A3). B, The pairing procedure enhances a footshock-paired odor but not the nonpaired odor, and mPFC stimulation suppresses this plasticity (*p < 0.05 compared with baseline; +p < 0.05 compared with other odor), as quantified by the area under the PSP. C, Stimulation of the Te3, however, does not suppress this form of plasticity. Similar to the example above, this neuron does not display a noticeable response to odors (presented at bar, top traces). However, pairing of odor A with footshock results in an enhanced response to odor A (middle traces, arrow) but not odor B. Unlike stimulation of the mPFC, stimulation of the Te3 during the conditioning phase does not suppress the plasticity, as reflected by the enhanced response to odor B (the odor paired with footshock and Te3 stimulation). D, When quantified by the area under the PSP, as a group, it can be seen that the response of LAT neurons to paired odors is enhanced after the odor is paired with footshock. Furthermore, the enhancement of footshock-paired odors is not disrupted by Te3 train stimulation.
To rule out the possibility that this mPFC-induced disruption of plasticity is attributable to nonspecific actions of afferent activation or to time-dependent factors, this experiment was repeated in six other rats in which the Te3 was stimulated instead of the mPFC. Train stimulation of the Te3 did not suppress this form of neuronal plasticity, and in fact appears to enhance it (Fig. 7) (baseline response to odor, 725 ± 300 mV/msec; after footshock and Te3 stimulation pairing, 2735 ± 672 mV/msec; p < 0.05; df = 5; paired t test).
Discussion
The PFC is thought to play a role in the regulation of affect. Our data demonstrate that the mPFC may regulate two aspects of affective behavior, learning and expression, via interactions with the LAT. In accord with this, the mPFC inputs to the LAT suppress responses of LAT neurons to previously conditioned affective stimuli as well as their plasticity. This inhibition by the mPFC inputs may underlie the regulation of affective expression and affective learning, respectively.
Inputs from the mPFC not only disrupt the response to odors but also suppress spontaneous PSPs. The mechanisms for the differential actions of the mPFC and Te3 on postsynaptic activity and by extension on plasticity likely arise in part by the different nature of the PSPs they evoke. Te3 stimulation primarily evokes an EPSP, whereas mPFC stimulation evokes a disynaptic IPSP (Rosenkranz and Grace, 2002a). The summation of mPFC-evoked PSPs with spontaneous PSPs is markedly sublinear, although the mPFC-evoked PSPs are also in the depolarizing direction at Vrest. There are several potential mechanisms that may account for this, including differential GABAB-mediated components, differential activation of voltage-dependent conductances that suppress PSPs, differential location of inputs along the somatodendritic axis or compartments relative to sPSPs resulting in different degrees of shunting, and/or the fact that mPFC-evoked IPSPs may clamp the membrane potential close to the reversal potential of the mixed GABAA-GABAB events. Whatever the cause of the differential interactions of Te3 and mPFC inputs, the mPFC-induced suppression of the postsynaptic depolarization caused by odor-footshock pairing is likely the mechanism for mPFC-induced inhibition of the plasticity of LAT neurons, which depends on the summation of inputs in a voltage-dependent manner (Rosenkranz and Grace, 2002b). In contrast, stimulation of the Te3 had a small enhancing effect on LAT neuronal plasticity. This is likely attributable to the above-noted summation of Te3 inputs with other synaptic inputs, enhancing the net depolarization of the postsynaptic membrane during odor-footshock pairings. Te3 inputs convey polysensory information to the LAT. Stimulation of the Te3 may therefore be functionally similar to the presence of an object to which affective salience may be attached.
The opposing actions of mPFC and Te3 inputs on LAT activity and plasticity indicate a fundamentally distinct role for these inputs in modulating affective behaviors. A near-linear summation of Te3 afferent input and spontaneous events brings the LAT neurons closer to spike threshold and enhances spike firing. The sublinear and suppressive actions of mPFC inputs on spontaneous activity reduce spike firing and the ability of PSPs to evoke spike firing. The IPSPs evoked during stimulation of the mPFC at the stimulation intensities used in this study were of a small amplitude. Even when stimulus intensities were used that evoked a PSP of <1 mV, stimulation was associated with a suppression of LAT PSPs. This is an indication that IPSPs in the LAT may not function only by membrane hyperpolarization but also predominantly as a large non-voltage-dependent conductance, which reduces the ionic driving force for nearby synapses as well as shunts membrane conductance.
The plasticity observed is not dependent on postsynaptic somatic action potentials, implying that if this plasticity is dependent on calcium influx during the combined odor-footshock-evoked depolarization, ample calcium accumulates via flux through NMDA channels, opening of voltage-gated channels, or calcium-induced calcium release; higher-threshold calcium channels appear not to be involved. However, spike firing may contribute to conditioning in awake animals, perhaps resulting in stronger associations. Thus, the response of LAT neurons to odors conditioned in awake rats was more than double the response to odors conditioned in anesthetized rats. Furthermore, the conditioning of awake rats resulted in clear behavioral correlates. Yet rats that underwent conditioning while anesthetized and testing 1 d after conditioning displayed weak or no differential conditioned behavioral responses to the odors (J. A. Rosenkranz and A. A. Grace, unpublished data). Although this may be accounted for by an anesthesia-induced disruption of consolidation, it may also be attributable to less potent initial conditioning. It is still unclear under what circumstances the mPFC may exert actions analogous to those observed here, because lesions of the mPFC have little or no effect on basic pavlovian conditioning (Gewirtz et al., 1997). However, the lack of an effect of mPFC lesions does not preclude its involvement, which may be better observed during chemical or electrical stimulation of the mPFC. In addition, a recent study using temporary inactivation of the mPFC demonstrates effects not observed in lesion studies (Sacchetti et al., 2002), and other studies demonstrate changes in mPFC neuronal firing during conditioning (Maxwell et al., 1994; Garcia et al., 1999). The mPFC may play a role in conditioning in more complex situations in which ambiguities exist, such as delays, or when competing contingencies are present. Consistent with this, studies have demonstrated that the mPFC is involved in extinction or recovery of affective behaviors (Eichenbaum et al., 1974; Quirk et al., 2000; Milad and Quirk, 2002; Myers and Davis, 2002).
The inputs from the mPFC suppress both conditioned and nonconditioned neuronal responses to odors to a similar degree. However, the lack of a differential suppression of paired and nonpaired odors by the mPFC does not necessarily imply that these odors are equally regulated by the mPFC. Affective associations may be stored in regions other than the LAT, and the mPFC may exert a differential regulation over neural representations of these odors in those regions. It is also possible that in awake animals, the more diverse or differential patterns of mPFC neuronal activity in response to paired and nonpaired stimuli are not perfectly mimicked by the bulk fiber stimulation used here. The result would be more selective and circumscribed effects of the mPFC on LAT neuronal responses to various stimuli.
There was a clear differential behavioral response of the rats to the CS+ and CS- when measured with exploratory activity and grooming behavior. This conditioned response measured in the awake rat is reflected by an increased electrophysiological response of LAT neurons and demonstrates an electrophysiological correlate of a behavioral affective response profile. However, freezing behavior was seen less than expected. Cessation of appetitive behavior is considered to be a sensitive measure of the aversiveness of a stimulus. The high rate of suppression of grooming and exploratory activity versus the low rate of stereotypical freezing indicates that the level of conditioned fear in the rats was not maximal. The less than maximal magnitude of conditioned freezing behaviors while the activity of LAT neurons in response to conditioned odors was quite robust indicates that the LAT neuronal plasticity may occur before the clear emergence of some conditioned behaviors, and that amygdalar neurons encode an aversive US-CS association at a relatively early stage of conditioning.
Train stimulation of the mPFC is likely to exert actions outside of the LAT that may indirectly influence the LAT activity. Nevertheless, although this possibility cannot be ruled out with this type of study, several pieces of data indicate that the primary inhibitory action exerted by train stimulation of the mPFC is attributable to PFC afferents to the LAT. Thus, there are few extrinsic inhibitory inputs to the LAT. MPFC-evoked excitation of these extrinsic inhibitory inputs would likely involve a disynaptic circuit, the latency of which would be inconsistent with the effects observed here (Rosenkranz and Grace, 2001, 2002a), whereas interneurons of the LAT are potently excited by mPFC stimulation with a time course that is consistent with the IPSPs recorded from LAT projection neurons (Rosenkranz and Grace, 2001, 2002a). However, to date, most anatomical studies demonstrate that the glutamatergic input from the mPFC to the BLA complex synapses predominantly with spines (Brinley-Reed et al., 1995; Smith et al., 2000), which are found mostly on pyramidal-like neurons and not interneurons. It is possible that the small percentage of mPFC inputs that synapse with interneurons is sufficient to drive their activity, because interneurons tend to be more electrotonically compact, with higher input resistances (Chitwood et al., 1999), allowing a greater response than pyramidal-like neurons to a smaller input. Indeed, such an anatomical arrangement is present in the striatum, in which a small number of GABAergic interneurons provide feedforward inhibition to a vast array of projection neurons (Koos and Tepper, 1999). Nevertheless, the functional impact of glutamatergic mPFC inputs on pyramidal neuron spines is unclear, because they seldom contribute to spike firing. One possibility is that they play a role in local dendritic plasticity. Alternatively, if there are situations in which the GABAergic inputs to the pyramidal neurons are reduced, such as may occur when BLA interneurons are inhibited by other interneurons (Muller et al., 2003), the mPFC may then be capable of driving spike firing of BLA projection neurons.
From a functional standpoint, regulation of LAT neuronal responses to conditioned and nonconditioned odors may reflect a role for the mPFC in maintaining an appropriate magnitude and timing of affective responses. Furthermore, the regulation of LAT neuronal plasticity may reflect a role for the mPFC in formation of associations. Disruptions of the frontal cortex, such as those that may occur after lesions and drug abuse, or the hypofrontal responses of schizophrenia, are often associated with abnormalities in the regulation of emotional behavior. Our current data suggest that loss of frontal regulation not only results in inadequate attenuation of the response to an affective stimulus but may also lead to more pervasive problems associated with the inability to regulate amygdalar plasticity. One potential result is the formation of abnormal associations, a problem that has been observed in schizophrenia patients (Kapur, 2003) or in the inability to maintain extinction behaviors that have been learned to counter inappropriate associations. Furthermore, supranormal activity of the PFC may lead to over-regulation of affective behavior and the inability to learn appropriate associations. In agreement with this, ictal events of frontal lobe epilepsy are sometimes associated with behavioral inhibition, the opposite of temporal lobe epilepsy (Shulman, 2000; Helmstaedter, 2001).
Note added in proof. In the time since this manuscript has been reviewed, a paper (Quirk et al., J Neurosci 2003 23:8800) appeared in which the authors propose that PFC-evoked inhibition of LAT neurons may be attributable to the antidromic activation of LAT projection neuron collaterals that excite LAT inhibitory interneurons, and not attributable to direct PFC-evoked activation of LAT interneurons, as we have suggested. The argument is based on the finding that only a few PFC terminals contact interneurons. However, it is known from other systems that, in the case of interneurons, few synaptic contacts are required to drive spike firing. Furthermore, as has been described previously (Rosenkranz and Grace, 2001, 2002a), PFC stimulation was found to evoke EPSPs and spikes in LAT interneurons at a shorter latency than PFC-evoked antidromic spikes or PSPs evoked in LAT projection neurons. Therefore, the suggestion of antidromic activation of LAT neurons is not consistent with either the anatomical data or with our previously published data.
Footnotes
This work was supported by National Institutes of Health Grants MH57440, DA15408 (A.A.G.), and MH12533 (J.A.R.). We thank Christy Smolak, Nicole MacMurdo, and Brian Lowry for their technical assistance and Dr. Mauricio Delgado for useful discussion.
Correspondence should be addressed to Anthony A. Grace, 446 Crawford Hall, Department of Neuroscience, University of Pittsburgh, Pittsburgh, PA 15260. E-mail: grace@bns.pitt.edu.
J. A. Rosenkranz's present address: Department of Neuroscience, Baylor College of Medicine, Houston, TX 77030.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2311054-11$15.00/0
References
- al Maskati HA, Zbrozyna AW ( 1989) Stimulation in prefrontal cortex area inhibits cardiovascular and motor components of the defense reaction in rats. J Auton Nerv Syst 28: 117-125. [DOI] [PubMed] [Google Scholar]
- Ananth H, Popescu I, Critchley HD, Good CD, Frackowiak RS, Dolan RJ ( 2002) Cortical and subcortical gray matter abnormalities in schizophrenia determined through structural magnetic resonance imaging with optimized volumetric voxel-based morphometry. Am J Psychiatry 159: 1497-1505. [DOI] [PubMed] [Google Scholar]
- Anderson JE, Wible CG, McCarley RW, Jakab M, Kasai K, Shenton ME ( 2002) An MRI study of temporal lobe abnormalities and negative symptoms in chronic schizophrenia. Schizophr Res 58: 123-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SW, Damasio H, Tranel D, Damasio AR ( 2000) Long-term sequelae of prefrontal cortex damage acquired in early childhood. Dev Neuropsychol 18: 281-296. [DOI] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE ( 2002) NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci 22: 5239-5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauregard M, Levesque J, Bourgouin P ( 2001) Neural correlates of conscious self-regulation of emotion. J Neurosci 21: RC165(1-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Le Gal la Salle G ( 1972) Plasticity at unitary level. II. Modifications during sensory-sensory association procedures. Electroencephalogr Clin Neurophysiol 32: 667-679. [DOI] [PubMed] [Google Scholar]
- Benes FM, Berretta S ( 2001) GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 25: 1-27. [DOI] [PubMed] [Google Scholar]
- Beyer JL, Krishnan KR ( 2002) Volumetric brain imaging findings in mood disorders. Bipolar Disord 4: 89-104. [DOI] [PubMed] [Google Scholar]
- Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE ( 2001) Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn Mem 8: 229-242. [DOI] [PubMed] [Google Scholar]
- Bowley MP, Drevets WC, Ongur D, Price JL ( 2002) Low glial numbers in the amygdala in major depressive disorder. Biol Psychiatry 52: 404-412. [DOI] [PubMed] [Google Scholar]
- Brinley-Reed M, Mascagni F, McDonald AJ ( 1995) Synaptology of prefrontal cortical projections to the basolateral amygdala: an electron microscopic study in the rat. Neurosci Lett 202: 45-48. [DOI] [PubMed] [Google Scholar]
- Cahill L, Vazdarjanova A, Setlow B ( 2000) The basolateral amygdala complex is involved with, but is not necessary for, rapid acquisition of Pavlovian “fear conditioning.” Eur J Neurosci 12: 3044-3050. [DOI] [PubMed] [Google Scholar]
- Carter RM, Hofstotter C, Tsuchiya N, Koch C ( 2003) Working memory and fear conditioning. Proc Natl Acad Sci USA 100: 1399-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chance SA, Esiri MM, Crow TJ ( 2002) Amygdala volume in schizophrenia: post-mortem study and review of magnetic resonance imaging findings. Br J Psychiatry 180: 331-338. [DOI] [PubMed] [Google Scholar]
- Chitwood RA, Hubbard A, Jaffe DB ( 1999) Passive electrotonic properties of rat hippocampal CA3 interneurones. J Physiol (Lond) 515: 743-756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins DR, Pare D ( 2000) Differential fear conditioning induces reciprocal changes in the sensory responses of lateral amygdala neurons to the CS(+) and CS(-). Learn Mem 7: 97-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coplan JD, Lydiard RB ( 1998) Brain circuits in panic disorder. Biol Psychiatry 44: 1264-1276. [DOI] [PubMed] [Google Scholar]
- Cousens G, Otto T ( 1998) Both pre- and posttraining excitotoxic lesions of the basolateral amygdala abolish the expression of olfactory and contextual fear conditioning. Behav Neurosci 112: 1092-1103. [DOI] [PubMed] [Google Scholar]
- Damasio H, Grabowski T, Frank R, Galaburda AM, Damasio AR ( 1994) The return of Phineas Gage: clues about the brain from the skull of a famous patient. Science 264: 1102-1105. [DOI] [PubMed] [Google Scholar]
- Davidson RJ ( 2002) Anxiety and affective style: role of prefrontal cortex and amygdala. Biol Psychiatry 51: 68-80. [DOI] [PubMed] [Google Scholar]
- Davidson RJ, Pizzagalli D, Nitschke JB, Putnam K ( 2002) Depression: perspectives from affective neuroscience. Annu Rev Psychol 53: 545-574. [DOI] [PubMed] [Google Scholar]
- Dias R, Robbins TW, Roberts AC ( 1996) Dissociation in prefrontal cortex of affective and attentional shifts. Nature 380: 69-72. [DOI] [PubMed] [Google Scholar]
- Drevets WC ( 2000) Functional anatomical abnormalities in limbic and prefrontal cortical structures in major depression. Prog Brain Res 126: 413-431. [DOI] [PubMed] [Google Scholar]
- Eichenbaum H, Potter H, Papsdorf J, Butter CM ( 1974) Effects of frontal cortex lesions on differentiation and extinction of the classically conditioned nictitating membrane response in rabbits. J Comp Physiol Psychol 86: 179-186. [DOI] [PubMed] [Google Scholar]
- Faber ES, Callister RJ, Sah P ( 2001) Morphological and electrophysiological properties of principal neurons in the rat lateral amygdala in vitro J Neurophysiol 85: 714-723. [DOI] [PubMed] [Google Scholar]
- Garcia R, Vouimba RM, Baudry M, Thompson RF ( 1999) The amygdala modulates prefrontal cortex activity relative to conditioned fear. Nature 402: 294-296. [DOI] [PubMed] [Google Scholar]
- Gewirtz JC, Falls WA, Davis M ( 1997) Normal conditioned inhibition and extinction of freezing and fear-potentiated startle following electrolytic lesions of medial prefrontal cortex in rats. Behav Neurosci 111: 712-726. [DOI] [PubMed] [Google Scholar]
- Goosens KA, Maren S ( 2001) Contextual and auditory fear conditioning are mediated by the lateral, basal, and central amygdaloid nuclei in rats. Learn Mem 8: 148-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur RE, McGrath C, Chan RM, Schroeder L, Turner T, Turetsky BI, Kohler C, Alsop D, Maldji J, Ragland JD, Gur RC ( 2002) An fMRI study of facial emotion processing in patients with schizophrenia. Am J Psychiatry 159: 1992-1999. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Bookheimer SY, Mazziotta JC ( 2000) Modulating emotional responses: effects of a neocortical network on the limbic system. NeuroReport 11: 43-48. [DOI] [PubMed] [Google Scholar]
- Hariri AR, Mattay VS, Tessitore A, Fera F, Weinberger DR ( 2003) Neocortical modulation of the amygdala responses to fearful stimuli. Biol Psychiatry 53: 494-501. [DOI] [PubMed] [Google Scholar]
- Heldt S, Sundin V, Willott JF, Falls WA ( 2000) Posttraining lesions of the amygdala interfere with fear-potentiated startle to both visual and auditory conditioned stimuli in C57BL/6J mice. Behav Neurosci 114: 749-579. [PubMed] [Google Scholar]
- Helmstaedter C ( 2001) Behavioral aspects of frontal lobe epilepsy. Epilepsy Behav 2: 384-395. [DOI] [PubMed] [Google Scholar]
- Jaskiw GE, Weinberger DR ( 1990) Ibotenic acid lesions of the medial prefrontal cortex potentiate FG-7142-induced attenuation of exploratory activity in the rat. Pharmacol Biochem Behav 36: 695-697. [DOI] [PubMed] [Google Scholar]
- Jinks AL, McGregor IS ( 1997) Modulation of anxiety-related behaviours following lesions of the prelimbic or infralimbic cortex in the rat. Brain Res 772: 181-190. [DOI] [PubMed] [Google Scholar]
- Kapur S ( 2003) Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry 160: 13-23. [DOI] [PubMed] [Google Scholar]
- Kim JS, Choi-Kwon S ( 2000) Poststroke depression and emotional incontinence: correlation with lesion location. Neurology 54: 1805-1810. [DOI] [PubMed] [Google Scholar]
- Kim M, Campeau S, Falls WA, Davis M ( 1993) Infusion of the non-NMDA receptor antagonist CNQX into the amygdala blocks the expression of fear-potentiated startle. Behav Neural Biol 59: 5-8. [DOI] [PubMed] [Google Scholar]
- Koos T, Tepper JM ( 1999) Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat Neurosci 2: 467-472. [DOI] [PubMed] [Google Scholar]
- Lawrie SM, Abukmeil SS ( 1998) Brain abnormality in schizophrenia. A systematic and quantitative review of volumetric magnetic resonance imaging studies. Br J Psychiatry 172: 110-120. [DOI] [PubMed] [Google Scholar]
- Lawrie SM, Buechel C, Whalley HC, Frith CD, Friston KJ, Johnstone EC ( 2002) Reduced frontotemporal functional connectivity in schizophrenia associated with auditory hallucinations. Biol Psychiatry 51: 1008-1011. [DOI] [PubMed] [Google Scholar]
- Liberzon I, Taylor SF, Fig LM, Decker LR, Koeppe RA, Minoshima S ( 2000) Limbic activation and psychophysiologic responses to aversive visual stimuli. Neuropsychopharmacology 23: 508-516. [DOI] [PubMed] [Google Scholar]
- Maren S ( 2000) Auditory fear conditioning increases CS-elicited spike firing in lateral amygdala neurons even after extensive overtraining. Eur J Neurosci 12: 4047-4054. [DOI] [PubMed] [Google Scholar]
- Maxwell B, Powell DA, Buchanan SL ( 1994) Multiple- and single-unit activity in area 32 (prelimbic region) of the medial prefrontal cortex during Pavlovian heart rate conditioning in rabbits. Cereb Cortex 4: 230-246. [DOI] [PubMed] [Google Scholar]
- Milad MR, Quirk GJ ( 2002) Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 420: 70-74. [DOI] [PubMed] [Google Scholar]
- Morgan MA, LeDoux JE ( 1995) Differential contribution of dorsal and ventral medial prefrontal cortex to the acquisition and extinction of conditioned fear in rats. Behav Neurosci 109: 681-688. [DOI] [PubMed] [Google Scholar]
- Muller JF, Mascagni F, McDonald AJ ( 2003) Synaptic connections of distinct interneuronal subpopulations in the rat basolateral amygdalar nucleus. J Comp Neurol 456: 217-236. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M ( 2002) Behavioral and neural analysis of extinction. Neuron 36: 567-584. [DOI] [PubMed] [Google Scholar]
- Nader K, Majidishad P, Amorapanth P, LeDoux JE ( 2001) Damage to the lateral and central, but not other, amygdaloid nuclei prevents the acquisition of auditory fear conditioning. Learn Mem 8: 156-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninan PT ( 1999) The functional anatomy, neurochemistry, and pharmacology of anxiety. J Clin Psychiatry 60 [Suppl 22]: 12-17. [PubMed] [Google Scholar]
- Ochsner KN, Bunge SA, Gross JJ, Gabrieli JD ( 2002) Rethinking feelings: an FMRI study of the cognitive regulation of emotion. J Cogn Neurosci 14: 1215-1229. [DOI] [PubMed] [Google Scholar]
- Otto T, Cousens G, Rajewski K ( 1997) Odor-guided fear conditioning in rats: 1. Acquisition, retention, and latent inhibition. Behav Neurosci 111: 1257-1264. [PubMed] [Google Scholar]
- Pakkenberg B ( 1992) Stereological quantitation of human brains from normal and schizophrenic individuals. Acta Neurol Scand Suppl 137: 20-33. [DOI] [PubMed] [Google Scholar]
- Paradiso S, Chemerinski E, Yazici KM, Tartaro A, Robinson RG ( 1999) Frontal lobe syndrome reassessed: comparison of patients with lateral or medial frontal brain damage. J Neurol Neurosurg Psychiatry 67: 664-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pare D, Collins DR ( 2000) Neuronal correlates of fear in the lateral amygdala: multiple extracellular recordings in conscious cats. J Neurosci 20: 2701-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschall GY, Davis M ( 2002) Olfactory-mediated fear-potentiated startle. Behav Neurosci 116: 4-12. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C ( 1998) The rat brain in stereotaxic coordinates. San Diego: Academic.
- Perlstein WM, Dixit NK, Carter CS, Noll DC, Cohen JD ( 2003) Prefrontal cortex dysfunction mediates deficits in working memory and prepotent responding in schizophrenia. Biol Psychiatry 53: 25-38. [DOI] [PubMed] [Google Scholar]
- Poremba A, Gabriel M ( 1999) Amygdala neurons mediate acquisition but not maintenance of instrumental avoidance behavior in rabbits. J Neurosci 19: 9635-9641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Armony JL, LeDoux JE ( 1997) Fear conditioning enhances different temporal components of tone-evoked spike trains in auditory cortex and lateral amygdala. Neuron 19: 613-624. [DOI] [PubMed] [Google Scholar]
- Quirk GJ, Russo GK, Barron JL, Lebron K ( 2000) The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci 20: 6225-6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainnie DG, Asprodini EK, Shinnick-Gallagher P ( 1993) Intracellular recordings from morphologically identified neurons of the basolateral amygdala. J Neurophysiol 69: 1350-1362. [DOI] [PubMed] [Google Scholar]
- Rajarethinam R, DeQuardo JR, Miedler J, Arndt S, Kirbat R, Brunberg JA, Tandon R ( 2001) Hippocampus and amygdala in schizophrenia: assessment of the relationship of neuroanatomy to psychopathology. Psychiatry Res 108: 79-87. [DOI] [PubMed] [Google Scholar]
- Repa JC, Muller J, Apergis J, Desrochers TM, Zhou Y, LeDoux JE ( 2001) Two different lateral amygdala cell populations contribute to the initiation and storage of memory. Nat Neurosci 4: 724-731. [DOI] [PubMed] [Google Scholar]
- Rescorla RA ( 1988) Behavioral studies of Pavlovian conditioning. Annu Rev Neurosci 11: 329-352. [DOI] [PubMed] [Google Scholar]
- Rogan MT, LeDoux JE ( 1995) LTP is accompanied by commensurate enhancement of auditory-evoked responses in a fear conditioning circuit. Neuron 15: 127-136. [DOI] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA ( 2001) Dopamine attenuates prefrontal cortical suppression of sensory inputs to the basolateral amygdala of rats. J Neurosci 21: 4090-4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA ( 2002a) Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo J Neurosci 22: 324-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz JA, Grace AA ( 2002b) Dopamine-mediated modulation of odour-evoked amygdala potentials during pavlovian conditioning. Nature 417: 282-287. [DOI] [PubMed] [Google Scholar]
- Sacchetti B, Baldi E, Lorenzini CA, Bucherelli C ( 2002) Differential contribution of some cortical sites to the formation of memory traces supporting fear conditioning. Exp Brain Res 146: 223-232. [DOI] [PubMed] [Google Scholar]
- Schaefer SM, Jackson DC, Davidson RJ, Aguirre GK, Kimberg DY, Thompson-Schill SL ( 2002) Modulation of amygdalar activity by the conscious regulation of negative emotion. J Cogn Neurosci 14: 913-921. [DOI] [PubMed] [Google Scholar]
- Selemon LD, Kleinman JE, Herman MM, Goldman-Rakic PS ( 2002) Smaller frontal gray matter volume in postmortem schizophrenic brains. Am J Psychiatry 59: 1983-1991. [DOI] [PubMed] [Google Scholar]
- Sheline YI, Barch DM, Donnelly JM, Ollinger JM, Snyder AZ, Mintun MA ( 2001) Increased amygdala response to masked emotional faces in depressed subjects resolves with antidepressant treatment: an fMRI study. Biol Psychiatry 50: 651-658. [DOI] [PubMed] [Google Scholar]
- Shulman MB ( 2000) The frontal lobes, epilepsy, and behavior. Epilepsy Behav 1: 384-395. [DOI] [PubMed] [Google Scholar]
- Siegle GJ, Steinhauer SR, Thase ME, Stenger VA, Carter CS ( 2002) Can't shake that feeling: event-related fMRI assessment of sustained amygdala activity in response to emotional information in depressed individuals. Biol Psychiatry 51: 693-707. [DOI] [PubMed] [Google Scholar]
- Smith Y, Pare JF, Pare D ( 2000) Differential innervation of parvalbumin-immunoreactive interneurons of the basolateral amygdaloid complex by cortical and intrinsic inputs. J Comp Neurol 416: 496-508. [PubMed] [Google Scholar]
- Soares JC, Mann JJ ( 1997) The anatomy of mood disorders-review of structural neuroimaging studies. Biol Psychiatry 41: 86-106. [DOI] [PubMed] [Google Scholar]
- Strakowski SM, DelBello MP, Adler C, Cecil DM, Sax KW ( 2000) Neuroimaging in bipolar disorder. Bipolar Disord 2: 148-164. [DOI] [PubMed] [Google Scholar]
- Taylor SF, Liberzon I, Decker LR, Koeppe RA ( 2002) A functional anatomic study of emotion in schizophrenia. Schizophr Res 58: 159-172. [DOI] [PubMed] [Google Scholar]
- Tazumi T, Okaichi H ( 2002) Effect of lesions in the lateral nucleus of the amygdala on fear conditioning using auditory and visual conditioned stimuli in rats. Neurosci Res 43: 163-170. [DOI] [PubMed] [Google Scholar]
- Tekin S, Cummings JL ( 2002) Frontal-subcortical neuronal circuits and clinical neuropsychiatry: an update. J Psychosom Res 53: 647-654. [DOI] [PubMed] [Google Scholar]
- Van Elst LT, Woermann FG, Lemieux L, Trimble MR ( 1999) Amygdala enlargement in dysthymia-a volumetric study of patients with temporal lobe epilepsy. Biol Psychiatry 46: 1614-1623. [DOI] [PubMed] [Google Scholar]
- Van Elst LT, Ebert D, Trimble MR ( 2001) Hippocampus and amygdala pathology in depression. Am J Psychiatry 158: 652-653. [DOI] [PubMed] [Google Scholar]
- Volk DW, Lewis DA ( 2002) Impaired prefrontal inhibition in schizophrenia: relevance for cognitive dysfunction. Physiol Behav 77: 501-505. [DOI] [PubMed] [Google Scholar]
- Vouimba RM, Garcia R, Baudry M, Thompson RF ( 2000) Potentiation of conditioned freezing following dorsomedial prefrontal cortex lesions does not interfere with fear reduction in mice. Behav Neurosci 114: 720-724. [DOI] [PubMed] [Google Scholar]
- Wallace KJ, Rosen JB ( 2001) Neurotoxic lesions of the lateral nucleus of the amygdala decrease conditioned fear but not unconditioned fear of a predator odor: comparison with electrolytic lesions. J Neurosci 21: 3619-3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MS, Moises HC ( 1992) Electrophysiological and morphological properties of rat basolateral amygdaloid neurons in vitro J Neurosci 12: 4066-4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen PJ, Shin LM, Somerville LH, McLean AA, Kim H ( 2002) Functional neuroimaging studies of the amygdala in depression. Semin Clin Neuropsychiatry 7: 234-242. [DOI] [PubMed] [Google Scholar]
- Wible CG, Anderson J, Shenton ME, Kricun A, Hirayasu Y, Tanaka S, Levitt JJ, O'Donnell BF, Kikinis R, Jolesz FA, McCarley RW ( 2001) Prefrontal cortex, negative symptoms, and schizophrenia: an MRI study. Psychiatr Res 108: 65-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilensky AE, Schafe GE, LeDoux JE ( 1999) Functional inactivation of the amygdala before but not after auditory fear conditioning prevents memory formation. J Neurosci 19: RC48(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zbrozyna AW, Westwood DM ( 1991) Stimulation in prefrontal cortex inhibits conditioned increase in blood pressure and avoidance bar pressing in rats. Physiol Behav 49: 705-708. [DOI] [PubMed] [Google Scholar]
- Zbrozyna AW, Westwood DM ( 1993) Inhibitory control of the defense-aggression reaction including its cardiovascular components. Acta Neurobiol Exp 53: 209-213. [PubMed] [Google Scholar]