

Abstract
3-Hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase inhibitors, or statins, reduce the incidence of strokes and reduce infarct volume after cerebral ischemia in mice. Excitoxicity caused by overstimulation of glutamate receptors is a major cause of neuronal death after an ischemic brain insult. Experiments presented here explored whether statins protect cultured neurons from excitotoxic death caused by the glutamate receptor agonist NMDA. Treatment with statins preserved NMDA receptor-expressing cortical neurons and potently and substantially reduced lactate dehydrogenase release caused by exposure of embryonic mouse neocortical cultures to NMDA. The rank order of neuroprotective potency was rosuvastatin = simvastatin > atorvastatin = mevastatin > pravastatin, which is similar to the known rank order of potency for inhibition of the HMG-CoA reductase enzyme. Resistance of cultures to NMDA excitotoxicity developed after several days of statin exposure. Neuroprotection by rosuvastatin was coincident with a decrease in cell sterols and occurred with a similar potency as inhibition of cholesterol biosynthesis. Neuroprotection was substantially attenuated by cotreatment with either mevalonate or cholesterol and was mimicked by acute treatment with the cholesterol-extracting agent β-cyclodextrin, suggesting that neuroprotection was mediated by depletion of a cellular pool of cholesterol because of the inhibition of cholesterol biosynthesis. These results suggest the possibility that, in addition to effects on cerebrovascular function, statins have the potential to render cortical neurons more resistant to NMDA-induced excitotoxic death as a result of changes in cell cholesterol homeostasis.
Keywords: HMG-CoA reductase, statin, cholesterol, NMDA, excitotoxicity, neuron
Introduction
3-Hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase inhibitors (statins) are potent cholesterol-lowering drugs. Several clinical studies indicate that statins reduce the incidence of stroke in some patients (Scandinavian Simvastatin Survival Study Group, 1994; Shepherd et al., 1995; Sacks et al., 1996; The Long-Term Intervention with Pravastatin in Ischemic Disease (LIPID) Study Group, 1998; Warshafsky et al., 1999; Heart Protection Study Collaborative Group, 2002). Retrospective case-control studies further suggest that the prevalence of Alzheimer's disease and vascular dementia is reduced among people taking statins (Jick et al., 2000; Wolozin et al., 2000). One interpretation of these findings is that statins may exert a broad neuroprotective effect.
Pretreatment with statins reduces infarct volume and provides protection from neurological deficits in mouse models of cerebral ischemia (Endres et al., 1998; Laufs et al., 2000, 2002; Amin-Hanjani et al., 2001). Studies in these models indicate that statins provide protection from ischemic brain damage by cerebrovascular mechanisms such as increased expression and activity of endothelial nitric oxide synthase (NOS) in endothelial cells and increased cerebral blood flow (Endres et al., 1998). In addition, several clinical studies have documented improved endothelial function during statin therapy, and human arterial blood flow is improved even in response to a single high dose of atorvastatin (Laufs et al., 2001). Thus, cerebrovascular mechanisms of action probably account, in part, for the reduced incidence of stroke in patients taking statins.
The current study describes another possible mechanism by which statins might directly protect neurons from ischemia-induced neurodegeneration by reducing neuronal cholesterol. Two recent clinical studies reported that some statins alter CSF markers of brain cholesterol homeostasis. For example, treatment with a high dose of simvastatin reduced the CSF level of the cholesterol biosynthetic intermediate lathosterol and the brain cholesterol-efflux metabolite 24-OH-cholesterol (Simons et al., 2002), and hypercholesterolemic patients treated with statins exhibited significantly lower CSF concentrations of both cholesterol and lathosterol (Fassbender et al., 2002). If these changes correspond to a decrease in neuronal membrane cholesterol, then some statins may alter membrane-delimited processes that mediate ischemic neuronal death such as NMDA receptor (NMDAR)-mediated excitotoxicity.
Excitotoxicity mediated by glutamate-gated ion channels is a well documented form of neuronal death caused by brain ischemia (Sattler and Tymianski, 2001). The NMDAR subtype of glutamate receptors probably plays the major role because of its high calcium permeability. Reducing neuronal membrane cholesterol could have profound effects on NMDAR-mediated processes. For example, NMDA excitotoxicity and ischemic neurodegeneration is mediated, in part, by a tri-partite protein complex (Aarts et al., 2002) that resides in sterol-rich neuronal membrane microdomains known as lipid rafts (Perez and Bredt, 1998). Reducing cholesterol in cultured cells has been shown to alter the function and composition of several lipid-raft-associated neuronal protein complexes (Martens et al., 2000; Ehehalt et al., 2003). Furthermore, treatment with statins alters the function of lipid-raft-associated protein complexes in cultured neurons (Simons et al., 1998). These observations lead to the hypothesis that reducing neuronal cholesterol by inhibiting brain HMG-CoA reductase protects neurons from NMDAR-mediated excitotoxicity. The results presented here describe experiments designed to test this hypothesis using cultured neurons.
Materials and Methods
Drugs. Simvastatin, atorvastatin, and pravastatin were isolated from commercially available tablets using standard chemical extraction and purification techniques; simvastatin sodium salt was prepared by alkaline hydrolysis of the lactone form. The purity of active ingredient was determined to be: simvastatin lactone (97.4%); atorvastatin (81.4%); pravastatin (87.1%). All analytical data were consistent with the known structures of these drugs, and in no case were any significant organic impurities detected, the remainder of the weight being water, solvent, and minor inorganic impurities. The neuroprotective potencies of simvastatin lactone and simvastatin sodium salt were identical (data not shown). Rosuvastatin calcium salt was synthesized at AstraZeneca Pharmaceuticals (Wilmington, DE). Mevastatin (compactin), mevalonate, cholesterol, NMDA, and β-cyclodextrin were obtained from Sigma (St. Louis, MO). Drug stocks were prepared in DMSO at 1000-fold greater than experimental concentrations, unless indicated otherwise.
Cortical culture. Fetal mouse neocortical cells were cultured on a glial feeder layer. Cortices were harvested from E16 Swiss-Webster mouse embryos in ice-cold dissecting media [HBSS plus (in mm): 28 glucose, 10 sucrose, and 4.2 sodium bicarbonate]. To dissociate cortices into single cells, they were minced in 1 ml of dissecting media, incubated in 0.09% trypsin in media stock (MS) [Eagle's MEM with Earle's salts plus (in mm): 22 glucose and 29 sodium bicarbonate] for 30 min at 37°C, centrifuged at 2,500 rpm for 5 min, resuspended in 1 ml of MS, 5% FBS, 5% horse serum (HS), and triturated with glass pipettes. To prepare a glial feeder layer, cell suspensions similarly prepared from 2 d postnatal mouse cortices were plated at 0.5 cortices/plate in 24-well tissue-culture plates that had been coated overnight with poly-d-lysine and laminin. These cultures produced an astrocyte monolayer on which cells prepared from E16 cortices were plated 2 weeks later at a density of 350,000 cells/well. After 6 d in culture, plating media were replaced with growth media containing 10% HS and 10 μm cytosine arabinoside (Ara-C); 2 d later, the culture media were again exchanged with fresh growth media lacking Ara-C. Typically, statins were added to the culture by media exchange on days 9 and 12. In some experiments, mevalonate (1 mm in PBS) and cholesterol (10 mm in ethanol) were added on the same days.
NMDA-induced lactate dehydrogenase release. Neuronal death was elicited 14-15 d after plating of embryonic cultures. Cultures were first washed twice with HEPES-buffered control salt solution (HCSS) (in mm: 120 NaCl, 5.4 KCl, 0.8 MgCl2, 1.8 CaCl2, 15 glucose, 20 HEPES acid, 10 NaOH, and 0.5% phenol red, pH 7.4) to remove all drugs and then exposed to 300 μm NMDA and 10 μm glycine for 10 min in HCSS. Cultures were then washed three times and incubated overnight in MS. After 24 hr, a media sample was collected to determine lactate dehydrogenase (LDH) activity. LDH activity was determined by measuring the linear rate of consumption of NADH absorption (340 nm) during the reduction of pyruvate to lactate using a spectrophotometer. Neuronal death was calculated from LDH activity by determining the percentage of the total NMDA-releasable pool that was measured for a given test condition using the following equation: neuronal death = [(A-Basal)/(Max-Basal)] × 100, in which A was the mean LDH activity measured in media samples from four wells subjected to the test condition, Max was the total NMDA-releasable pool of LDH activity observed in four wells treated for 24 hr with 300 μm NMDA, and Basal was the LDH activity from four identically treated wells that were not exposed to NMDA. These controls were run on every plate in each culture and averaged across multiple plates derived from the same culture. In all but Figure 1 (a representative single experiment), results are the mean of experiments conducted in three different cultures. In the concentration-response and time course experiments shown in Figure 2, consistent drug effects were observed, but the neuronal death caused by a 10 min exposure to NMDA varied somewhat between cultures. To calculate the effects of the drug independent of the variability in the effect of transient exposure to NMDA in the cultures used in these experiments, 10 min NMDA-induced neuronal death observed in drug-treated wells was normalized to 10 min NMDA-induced neuronal death observed in nondrug-treated wells in the same culture. These data are, therefore, reported as percentages of NMDA-induced neuronal death. In this system, NMDA-induced decreases in neuronal cell counts correlate with NMDA-induced increases in LDH release, and both are prevented by the noncompetitive NMDAR antagonist MK-801 (data not shown).
Figure 1.

Simvastatin protects cultured mouse cortical neurons from NMDA excitotoxicity. Nine-day-old cortical cultures were treated with 100 nm simvastatin for 6 d or left untreated before a 10 min exposure to 300 μm NMDA. A, fluorescence photomicrographs (200×) of NMDAR-1 immunocytochemically labeled cultures show that NMDAR-expressing neurons were preserved in cultures pretreated with simvastatin before NMDA exposure. B, LDH release, calculated as a percentage of the total NMDA-releasable pool of LDH, shows that NMDA-induced LDH release from the same cultures was prevented by simvastatin pretreatment.
Figure 2.

Statin-mediated neuroprotection is concentration and time dependent. A, Statins potently protect neurons from NMDA excitotoxicity, consistent with inhibition of HMG-CoA reductase. Cortical cultures were treated with various concentrations of the indicated statins for 6 d before NMDA exposure. *, p < 0.05 vs rosuvastatin; ‡, p < 0.01 vs rosuvastatin. B, Statin-induced neuroprotection developed over several days of pretreatment. Cortical cultures were exposed to either 100 nm simvastatin or 300 nm mevastatin for the indicated number of days before NMDA exposure.
Immunocytochemistry. Cultures were washed with PBS for 5 min, fixed with 4% paraformaldehyde in PBS for 20 min, washed twice with PBS, incubated with ice-cold methanol for 10 min, washed twice with PBS, then blocked with 10% donkey serum in PBS for 1-2 hr. Cultures were then incubated overnight in 1:50 rabbit NMDAR subtype 1 (NMDAR-1) antibody (Chemicon, Temecula, CA) in blocking solution at room temperature, washed three times with PBS, incubated for 1 hr in 1:100 Cy3-conjugated anti-rabbit IgG secondary antibody (Jackson Immunoresearch, West Grove, PA) in blocking solution at room temperature, and washed three times with PBS. Digital photomicrographs were obtained using an inverted Olympus (Tokyo, Japan) fluorescence microscope.
Sterol Measurements. Cortical cultures treated with drugs as described above were saponified in ethanolic KOH and extracted with hexane. Extracts were derivatized to sterol esters using pyridine/acetic anhydride and analyzed by gas chromotography-mass spectroscopy. Cell culture sterols were identified and quantified by comparing retention times, spectra, and peak heights to those obtained with known amounts of authentic standards.
Electrophysiology. Both electrophysiology and calcium-imaging experiments were performed at room temperature on cultures grown on individual glass coverslips otherwise as described above. Before experiments, cultures were thoroughly washed free from drug-containing cell culture media using the relevant superfusion solutions. Currents were recorded using the whole-cell patch-clamp technique. Borosilicate glass pipettes with resistances of 5-7 MΩ were filled with the following solution (in mm): 135 CsCl, 10 EGTA, 10 HEPES, 1 MgCl2, and 4 Mg-ATP, pH 7.4, with CsOH and adjusted to 305 mOsm using sucrose. The superfusion solution contained the following (in mm): 135 NaCl, 5 KCl, 2 CaCl2, 5 HEPES, 11 glucose, and 0.01 glycine, pH 7.3, with NaOH and adjusted to 315 mOsm using sucrose. Drugs were applied using a linear array of gravity-fed, glass-lined tubes connected to solenoid valves under digital control (BME Systems, Baltimore MD). Whole-cell capacitance (Cm = integral of capacitive transient) was calculated by fitting currents recorded during a -1 mV, 10 msec step. Membrane currents were amplified with an Axopatch 2A amplifier (Axon Instruments, Foster City, CA), low-pass filtered at 10 kHz, digitized at 2 kHz, and stored as computer files using P-Clamp software (Axon Instruments) and a TL-1 interface (Scientific Solutions, Solon, OH). Cell membrane potential was held at -60 mV. NMDA (100 μm) was applied for 1.6 sec every 45 sec after break-in. Peak current amplitudes were calculated by subtracting NMDA-evoked currents obtained in the presence of 100 μm D(-)-2-amino-5-phosphonopentanoic acid. A variable, brief, and modest run-down in peak NMDA current amplitude was observed. Reported peak amplitudes were derived from traces obtained after currents had stabilized.
Calcium Imaging. Calcium microfluorimetry experiments were conducted using Axon Imaging Workbench 2.2 and a Sensicam CCD camera (PCO CCD Imaging, Kelheim, Germany) attached to a Nikon Eclipse TE300 inverted fluorescence microscope. Cultures were incubated with the acetoxy-methyl-ester of the fluorescent calcium-indicator dye fura-2 (Molecular Probes, Eugene, OR) at 37oC for 45 min, rinsed, then maintained in the dark at room temperature for an additional 15 min before use. Coverslips were attached with vacuum grease to a coverslip holder (Warner Instruments) and superfused for the remainder of the experiment with the following solution (in mm): 135 NaCl, 5 KCl, 2 CaCl2, 5 HEPES, and 11 glucose, pH 7.3; osmolarity adjusted to 315 mosM with sucrose and 200 nm TTX. The excitation exposure interval was adjusted to 3-10 msec to obtain fluorescence that was dim but distinct during excitation at 340 nm and less than half-maximal during excitation at 380 nm. Intraneuronal calcium concentration is proportional to the ratio (R) of the fura-2 fluorescence emission intensity measurements obtained when the dye is excited at 340 or 380 nm excitation wavelengths (F340/F380). Ratio images were collected at 5 sec intervals to determine baseline fluorescence, then images were collected at 1 sec intervals for ∼1 min during superfusion with 300 μm NMDA and 10 μm glycine. After the peak response was obtained, the sampling interval was increased to 5 sec. Baseline and peak values were determined for each cell from individual traces off-line. Cells that were unresponsive or exhibited either an unstable baseline or an oscillating response to NMDA were excluded from analysis. For statistical comparisons, the peak NMDA-induced increase in intraneuronal calcium concentration (ΔR) was measured in ∼20 neurons per microscope field and was averaged to generate a single value for each coverslip.
Statistics and pharmacology. Concentration-response curves were fitted using a four-parameter, sigmoidal concentration-response equation of the following form: y = Bottom + [(Top - Bottom)/(1 + 10(logIC50-[drug]*n))]. Statistical significance was determined using one of three tests. For electrophysiological and calcium-imaging studies, in which comparisons were made between mean values calculated for the two treatment groups, Student's t test was performed. For all other experiments, in which the mean values of multiple treatment groups were compared, an ANOVA was conducted to determine whether significant differences existed between the means. If the ANOVA reached significance (p < 0.05), then either a Dunnett's test (all means compared with a control) or a Bonferonni test (comparisons between individual means) was used.
Results
Simvastatin preserves NMDAR-1 immunoreactivity and reduces LDH release from NMDA-treated cortical cultures
Dissociated embryonic mouse neocortices were plated on a 2-week-old glial feeder layer derived from neonatal mice. Nine days later, cultures were treated with 100 nm of the HMG-CoA reductase inhibitor simvastatin. After 6 d of exposure to simvastatin, cultures were washed and exposed to 300 μm NMDA for 10 min. To determine the survival of cortical neurons under these conditions, both NMDAR-1 immunofluorescence microscopy and LDH release measurements were conducted 24 hr later in the identical simvastatin-treated and control cultures. As shown in Figure 1, 6 d of pretreatment with 100 nm simvastatin protected cortical neurons from NMDA-induced death, as indicated by the preservation of NMDAR-1 immunoreactive neurons, and a concomitant decrease in NMDA-induced LDH release in simvastatin-treated cultures.
In the representative experiment shown in Figure 1, exposure to 300 μm NMDA for 10 min elicited release of 69 ± 16% of maximal NMDA-induced LDH release, which occurred after a 24 hr exposure to 300 μm NMDA, and is subsequently referred to as “total LDH.” Basal LDH release was defined as that measured in cultures subjected to the identical wash protocol but without exposure to NMDA, which was 1.1 ± 0.9% of the maximum. Pretreatment with 100 nm simvastatin, a maximally neuroprotective concentration of simvastatin, reduced NMDA-induced LDH release to 10 ± 4% of total LDH (p < 0.01). Simvastatin produced a modest, and nonsignificant, decrease in basal LDH release (data not shown).
Concentration and time dependence of neuroprotection by statins
Under identical experimental conditions, the neuroprotective concentration-response relationship was determined for five HMG-CoA reductase inhibitors. As shown in Figure 2, all five drugs exhibited concentration-dependent and potent inhibition of NMDA-induced excitotoxicity. The drugs produced equivalent and nearly complete maximal neuroprotection but differed in their neuroprotective potency. The rank order of neuroprotective potency was rosuvastatin = simvastatin > atorvastatin = mevastatin > pravastatin. The IC50 values and the 95% confidence intervals of the IC50 values for inhibition of NMDA-induced neuronal death were 7 (5-10), 9 (6-13), 54 (32-91), 61 (50-74), and 363 (171-771) nm, for rosuvastatin, simvastatin, mevastatin, atorvastatin, and pravastatin, respectively (Fig. 2A). The potency of rosuvastatin and simvastatin was not statistically different, but mevastatin (p < 0.05), atorvastatin (p < 0.05), and pravastatin (p < 0.01) were less potent than rosuvastatin and simvastatin, and pravastatin was less potent than either atorvastatin or mevastatin (p < 0.05 in both cases). The time course of neuroprotection was determined by treating cultures with either 100 nm simvastatin or 300 nm mevastatin for 1-6 d before NMDA exposure on day 15 in culture. As shown in Figure 2B, most of the neuroprotection afforded by either drug developed between 2 and 4 d of pretreatment.
Cortical culture sterol analysis
To determine whether statins protect neurons from excitotoxicity by inhibiting cholesterol biosynthesis, the effect of rosuvastatin treatment on cortical culture sterol levels was measured. Treatment with rosuvastatin reduced cortical culture cholesterol and Δ-8-cholestenol and decreased the ratio of Δ-8-cholestenol to cholesterol. Δ-8-Cholestenol levels decreased to a minimum after 3 d of drug treatment (data not shown). Cholesterol was decreased by 29% from 6.6 ± 0.2 μg/well in vehicle-treated cultures to 4.7 ± 0.1 μg/well in cultures treated for 3 d with 30 nm rosuvastatin (p < 0.05). In the same cultures, Δ-8-cholestenol was decreased by 72% from 119 ± 17 to 33 ± 8 ng/well (p < 0.01). Δ-8-Cholestenol is an intermediate sterol in the biosynthesis of cholesterol, so a decrease in the ratio of Δ-8-cholestenol to cholesterol indicates that rosuvastatin inhibited sterol biosynthesis in cortical cultures. Rosuvastatin reduced the ratio of cortical culture Δ-8-cholestenol to cholesterol by more than threefold after 3 d. As shown in Figure 3, rosuvastatin decreased the ratio of Δ-8-cholestenol to cholesterol in a concentration-dependent manner with an IC50 value of 5 nm (95% confidence interval, 4-10 nm).
Figure 3.

Rosuvastatin potently inhibits cortical culture cholesterol biosynthesis. Δ-8-Cholestenol and cholesterol were measured in cortical cultures treated for 3 d with the indicated concentrations of rosuvastatin. *, p < 0.05; ‡, p < 0.01 vs vehicle-treated cultures.
Reversal of neuroprotection by mevalonate or cholesterol
The product of HMG-CoA reductase, mevalonate, is a biosynthetic intermediate in several biochemical pathways, including cholesterol biosynthesis. To determine whether depletion of mevalonate and subsequent depletion of cholesterol was involved in the prevention of NMDA excitotoxicity, cultures were cotreated with simvastatin alone or with simvastatin in combination with either 1 mm mevalonate or 50 μm cholesterol. As shown in Figure 4 (left), cotreatment with either mevalonate or cholesterol prevented the neuroprotective action of simvastatin. In simvastatin-treated cultures, NMDA-induced LDH release was reduced from 71 ± 2 to 6 ± 2% of total LDH (p < 0.001). In cultures exposed to simvastatin and either mevalonate or cholesterol, the extent of NMDA-induced LDH release was 43 ± 5 and 91 ± 7% of total LDH release, respectively, significantly more than that observed in cultures treated with simvastatin alone (p < 0.001). Cotreatment with cholesterol not only prevented the protective effect of simvastatin but increased NMDA-induced LDH release above that observed in control cultures (91 ± 7% compared with 71 ± 2% of total LDH; p < 0.05). These data suggested that manipulation of cell cholesterol levels could directly alter NMDA toxicity independent of HMG-CoA reductase inhibition.
Figure 4.

Resistance to excitotoxicity was prevented by coincubation with either mevalonate or cholesterol. In the experiment shown on the left side of the bar graph, before NMDA exposure, cortical cultures were left untreated, treated for 6 d with 100 nm simvastatin alone, or treated with both simvastatin and either 1 mm mevalonate or 50 μm cholesterol. NMDA-induced LDH release after cotreatment with simvastatin and either mevalonate or cholesterol was increased compared with NMDA-induced LDH release after treatment with simvastatin alone. In the experiment shown on the right side of the bar graph, cultures were treated with vehicle or cholesterol for 6 d before NMDA exposure. Cholesterol treatment increased NMDA-induced LDH release. §, p < 0.001; ‡, p < 0.01.
This possibility was explored in two subsequent experiments. First, the effect of chronic cholesterol loading on NMDA-induced excitotoxicity was measured. As shown in Figure 4 (right), pretreatment of cultures for 6 d with 50 μm cholesterol increased NMDA-induced LDH release to 82 ± 4% of total LDH release, a significant increase over that observed in vehicle-treated cultures (67 ± 3%; p < 0.01). This experiment suggests that loading cultures with cholesterol increases the excitotoxic effect of NMDA.
The cyclic glucose oligomer methyl-β-cyclodextrin (βCD) has been used to selectively deplete cell membranes of cholesterol, while leaving membrane phospholipid content intact (Kilsdonk et al., 1995; Klein et al., 1995; Simons et al., 1998; Martens et al., 2000). βCD was used to investigate the effect of acute cholesterol depletion and repletion on the excitotoxicity of NMDA in cortical cultures. As shown in Figure 5, NMDA-induced LDH release was reduced from 77 ± 2.2% in control cultures to 13 ± 0.2% of total LDH (p < 0.001) in cultures treated for 20 min with 1% βCD, followed by incubation in HCSS for 15 min before NMDA exposure. Treatment of cultures for 20 min with 1% βCD to deplete cholesterol, followed by a 15 min treatment with 2 mm cholesterol complexed with 2.5% βCD to replace cholesterol, restored NMDA-induced LDH release back to control levels (64 ± 8% of total LDH). This effect of cholesterol was concentration dependent, because restoring cholesterol levels by treatment with 0.6 mm cholesterol complexed with 0.7% βCD did not prevent the protective effect of cholesterol depletion by 1% βCD treatment. These data indicate that treatments that acutely deplete cell cholesterol produced a neuroprotective effect similar to that observed after chronic statin treatment.
Figure 5.

Specific depletion of membrane cholesterol with βCD prevents NMDA-induced excitotoxicity. Before NMDA exposure, cultures were left untreated or treated with 1% βCD for 20 min. βCD-treated cultures were then left untreated or treated for 15 min with the indicated dilutions of βCD/cholesterol. Cholesterol depletion prevented and cholesterol repletion restored NMDA excitotoxicity. §, p < 0.001.
NMDAR function
Whole-cell patch-clamp electrophysiological recordings and calcium microfluorimetry were used to investigate effects on NMDAR function caused by chronic exposure of mouse cortical cultures to simvastatin. As shown in Figure 6, treatment of cortical cultures for 4-6 d with simvastatin decreased NMDA-induced whole-cell currents and current density (data not shown) by 25% (mean ± SEM; untreated: 1947 ± 124 pA, 36 pA/pF, n = 33; simvastatin: 1479 ± 119pA, 27 pA/pF, n = 22; p < 0.01 for both whole-cell current and current density). In contrast, the magnitude of the 300 μm NMDA-induced increase in intraneuronal calcium concentration was not different in simvastatin-treated and control cultures (F340/F380; control: 0.330 ± 0.008, n = 22 coverslips, 331 cells; simvastatin: 0.319 ± 0.006, n = 21 coverslips, 371 cells). These data indicate that, although statin treatment slightly reduces NMDA-induced whole-cell current, this change is not reflected in reduced intraneuronal calcium concentrations.
Figure 6.
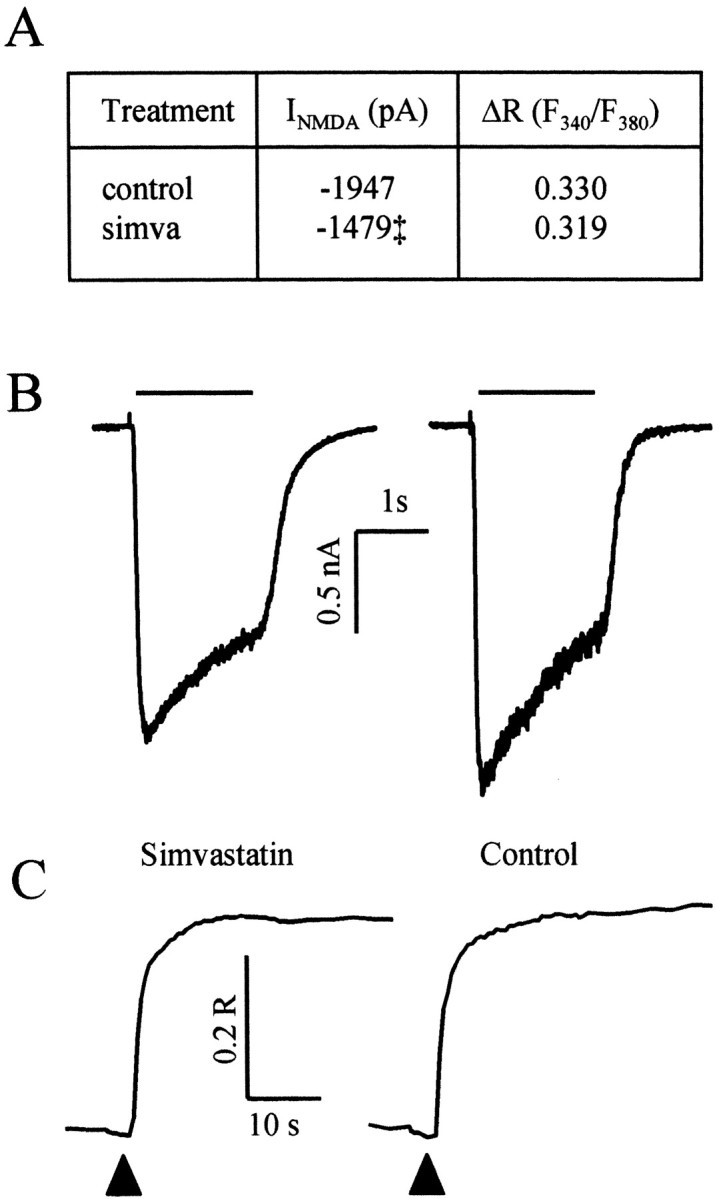
Simvastatin reduces NMDA-induced whole-cell currents but does not affect the NMDA-induced increase in intraneuronal calcium concentration. A, The table provides mean and SEM peak NMDA-induced whole-cell current (n = 22 cells for simvastatin; n = 33 cells for control; ‡, p < 0.01) and fura-2 ΔR (simvastatin, n = 331 cells from 22 coverslips; control, n = 371 cells from 21 coverslips) obtained in simvastatin-treated and control cultures. Mean data were obtained from experiments conducted in three cultures. B, Representative traces of NMDA-induced whole-cell currents. The horizontal bar indicates the application of 100 μm NMDA. C, Representative traces of NMDA-induced changes in the fura-2 fluorescence intensity ratio. The black triangles indicate the initiation of application of 300 μm NMDA.
Discussion
Here, HMG-CoA reductase inhibitors, or statins, are shown for the first time to elicit resistance of cultured neurons to NMDA-induced excitotoxic death, a potential neuroprotective action of statins distinct from their beneficial effects on cerebrovascular function.
The HMG-CoA reductase enzyme is rate limiting in cholesterol biosynthesis. The neuroprotective effect described here is attributable to inhibition of cholesterol biosynthesis and depletion of cholesterol or a metabolic product of cholesterol. The rank order of neuroprotective potency of statins is generally consistent with the rank order of potency reported for inhibition of either the purified HMG-CoA reductase enzyme or of cellular cholesterol biosynthesis. For example, the rank order potencies and IC50 values (as nanomolar concentrations) for inhibition of the purified catalytic domain of human HMG-CoA reductase were rosuvastatin (5.4) = atorvastatin (8.2) > simvastatin (11.2) > pravastatin (44.1) and for inhibition of cultured rat hepatocyte cholesterol synthesis were rosuvastatin (0.16) > atorvastatin (1.16) = simvastatin (2.74) = pravastatin (6.93) (McTaggart et al., 2001). The presence of specific uptake mechanisms increases the potency of rosuvastatin and pravastatin, but not simvastatin, by >100-fold in rat hepatocytes compared with fibroblasts. It is not known whether such uptake mechanisms are present in mouse cortical neurons.
Δ-8-Cholestenol is an intermediate in the biosynthesis of cholesterol and has a shorter half-life than cholesterol. Rosuvastatin decreased both cholesterol and Δ-8-cholestenol levels, and decreased the ratio of Δ-8-cholestenol to cholesterol in cortical cultures. These changes are consistent with depletion of cortical culture sterols after inhibition of HMG-CoA reductase. The observed decrease in the ratio of Δ-8-cholestenol to cholesterol indicates that rosuvastatin inhibited de novo cholesterol biosynthesis in cortical cultures. Treatment with rosuvastatin decreased culture cholesterol by inhibiting cholesterol biosynthesis. The time course and potency with which rosuvastatin reduced cell sterols closely matched the time course of mevastatin and simvastatin and the potency of rosuvastatin to protect cortical neurons from excitoxicity. Future studies should determine whether the rank order potency for inhibition of cholesterol biosynthesis matches the rank order potency for neuroprotection.
Statins inhibit cholesterol biosynthesis more potently than other mevalonate-dependent biochemical pathways. Isoprenylation of proteins, which is dependent on mevalonate biosynthesis, is inhibited by lovastatin at micromolar concentrations, 1000-fold higher than the concentration required for inhibition of cholesterol biosynthesis in the same cells (Sinensky et al., 1990). Furthermore, in cortical cultures, micromolar concentrations of statins are highly toxic (Tanaka et al., 2000), an effect that is prevented by cotreatment with the isoprenoid precursor geranyl-geranyl pyrophosphate. Similar observations were made in the cultures used in this study, and these observations suggest that the neuroprotective effect described here is not attributable to inhibition of isoprenoid biosynthesis. The neuroprotective potency of statins observed in this study is consistent with inhibition of cholesterol biosynthesis and less consistent with inhibition of other mevalonate-dependent biochemical pathways.
Resistance to excitotoxicity was substantially attenuated by coincubation with either mevalonate or cholesterol. Cotreatment with mevalonate restores cholesterol biosynthesis as well as other mevalonate-dependent biosynthetic pathways. The ability of cholesterol to reverse statin neuroprotection implies that inhibition of cholesterol synthesis and subsequent decrease in cell cholesterol content is essential to the statin neuroprotective mechanism (Michikawa and Yanagisawa, 1999).
Finally, two experiments suggest that manipulation of cell cholesterol without statins produced changes in the susceptibility of the cultures to NMDA-induced excitotoxicity. Treatment with cholesterol increased susceptibility to NMDA excitotoxicity. In addition, βCD, which extracts membrane cholesterol (Kilsdonk et al., 1995; Klein et al., 1995), reduced susceptibility to NMDA-induced excitotoxicity, an effect reversed by subsequent treatment with a cholesterol/βCD mixture, which restores extracted cell cholesterol (Klein et al., 1995). Taken together, these results indicate that decreased cholesterol content caused by inhibition of HMG-CoA reductase protects cultured cortical neurons from NMDA-induced excitotoxicity.
Two previous studies suggest that exposure of cultured neurons to statins triggers neuronal apoptosis (Michikawa and Yanagisawa, 1999;Tanaka et al., 2000). Tanaka et al. (2000) reported that simvastatin caused neurotoxicity with an EC50 value of 3 μm because of depletion of the isoprenoid ggPP and not because of depletion of intermediates in cholesterol biosynthesis. In cultures used in the present study, concentrations of simvastatin or mevastatin >3 μm also caused the death of cultured neurons, which was reversed by ggPP but not by fPP (data not shown), but the protection against NMDA excitotoxicity reported here was apparent at concentrations >300-fold less than those that cause toxicity. The potency of the neuroprotective effect of statins because of inhibition of cholesterol biosynthesis differentiates this effect from the cytotoxicity of statins because of inhibition of ggPP biosynthesis.
Michikawa and Yanagisawa (1999) also observed apoptosis of statin-treated cultured neurons and concluded that the toxicity of statins was caused by inhibition of cholesterol synthesis. These investigators used neuronal cultures 6 hr after plating and suggested that survival of newly plated neurons may strictly require cholesterol biosynthesis to support formation of new membranes.
The effect of reduced cortical culture cholesterol on NMDAR-mediated excitotoxic events is unknown. The modest decrease in NMDAR current observed in simvastatin-treated neurons may account partly for the observed neuroprotective efficacy of simvastatin, however the identical treatment decreased neuronal death by >90%. Under conditions used here, NMDAR-mediated excitotoxicity is largely calcium dependent, and the peak NMDA-induced intraneuronal calcium concentration in these cultures was unchanged by statin treatment. The cause of this discrepancy is unknown, but it could be attributable to either to insufficient sensitivity of the calcium measurements to detect decreased intraneuronal calcium, or to altered permeability of the cationic nonselective NMDAR pore such that the whole-cell current was reduced without reducing the resulting intraneuronal calcium load. Finally, NMDAR-1 immunocytochemistry revealed similar levels of NMDAR expression in statin-protected and untreated cultures. These data suggest that statins reduce susceptibility of cortical neurons to NMDA excitotoxicity by acting downstream of NMDAR function.
Depletion of cholesterol from plasma membrane microdomains may modulate key steps in the excitotoxic cascade downstream of NMDAR function. NMDAR-mediated calcium influx activates NOS (nNOS). nNOS-mediated production of nitric oxide and subsequent formation of reactive oxygen radicals is a cause of excitotoxic neuronal death, and inhibitors of nNOS protect cultured cortical neurons from excitotoxicity (Dawson et al., 1993). nNOS forms a tri-partite complex with the NMDAR and the postsynaptic density protein PSD-95 (Christopherson et al., 1999). nNOS and PSD-95 are found in sterol-rich plasma-membrane microdomains called lipid rafts (Perez and Bredt, 1998; Topinka and Bredt, 1998; Keshet et al., 1999), and depletion of membrane cholesterol alters localization, composition, and function of protein complexes associated with rafts (Martens et al., 2000; Ehehalt et al., 2003). Statins may alter the sterol content of lipid rafts, thereby uncoupling nNOS activation from NMDAR-mediated calcium flux. Antisense suppression of PSD-95 selectively uncouples NMDAR-induced calcium flux from nNOS activation and protects cultured neurons from NMDA excitotoxicity (Sattler et al., 1999), and peptides that dissociate NMDAR and PSD-95 prevent excitotoxic neuronal death and reduce infarct volume after cerebral ischemia (Aarts et al., 2002). Future studies should determine whether cholesterol depletion is neuroprotective because of changes in nNOS activation or NMDAR composition resulting from altered lipid raft sterol composition.
Laufs et al. (2000) have reported that 2 week pretreatment of mice with simvastatin, lovastatin, atorvastatin, or mevastatin reduces infarct size and neurological deficits caused by subsequent middle cerebral artery occlusion in mice (Endres et al., 1998; Amin-Hanjani et al., 2001). Recently, these investigators have described a more potent neuroprotective effect of rosuvastatin (Laufs et al., 2002). The proposed cerebrovascular neuroprotective mechanism of statins differs from the neuroprotective mechanism described here, in that cerebrovascular effects occurred without a change in plasma cholesterol and seemed to require reduced endothelial cell isoprenoids, not reduced cholesterol (Laufs and Liao, 1998; Laufs et al., 1998). Furthermore, whereas simvastatin can reduce brain cholesterol biosynthesis in vivo (Fassbender et al., 2001, 2002; Simons et al., 2002), this likely occurs at doses far exceeding those used by Endres et al. (1998) and could not occur independent of reduced plasma cholesterol. Additional experiments are needed to determine whether the neuroprotective effect described here occurs in vivo and whether cerebrovascular and central neuroprotective effects could be synergistic.
Clinically, it would be reasonable to postulate that vascular effects of statins might predominantly influence stroke incidence, whereas the neuroprotective mechanism described here might predominantly affect the severity of resultant neurological damage. Reliably distinguishing these, and which mechanism is responsible for which effect, is beyond the scope of data from currently available, large-scale clinical studies. Reduced incidence of stroke has been observed in middle-aged men treated with simvastatin (Scandinavian Simvastatin Survival Study Group, 1994) or pravastatin [The Long-Term Intervention with Pravastatin in Ischemic Disease (LIPID) Study Group, 1998], although pravastatin does not readily cross the blood-brain barrier. However, pravastatin did not reduce stroke incidence in elderly patients (Shepherd et al., 2002); this might be because of more advanced vascular disease in the elderly being less amenable to cerebrovascular protection. Detailed assessment of the extent and severity of neurological damage was not performed in these studies, making it difficult to judge whether the neuroprotection demonstrated here could have afforded any clinical benefit.
In conclusion, increased resistance of statin-treated cultured neurons to NMDAR-mediated excitotoxic death is attributable to reduced cell cholesterol, occurs in the absence of vascular elements, and represents an additional, plausible, “direct” neuroprotective mechanism.
Footnotes
We thank Charlotte Lott and My Linh Do for culture preparation and Drs. Fergus McTaggart, David Tuffin, and Adam Dudley for critical reading of this manuscript.
Correspondence should be addressed to Dr. Timothy Piser, Department of Neuroscience, AstraZeneca Pharmaceuticals, 1800 Concord Pike, Wilmington, DE 19850. E-mail: Timothy.Piser@AstraZeneca.com.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2311104-08$15.00/0
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M ( 2002) Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 298: 846-850. [DOI] [PubMed] [Google Scholar]
- Amin-Hanjani S, Stagliano NE, Yamada M, Huang PL, Liao JK, Moskowitz MA ( 2001) Mevastatin, an HMG-CoA reductase inhibitor, reduces stroke damage and upregulates endothelial nitric oxide synthase in mice. Stroke 32: 980-986. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Hillier BJ, Lim WA, Bredt DS ( 1999) PSD-95 assembles a ternary complex with the N-methyl-D-aspartic acid receptor and a bivalent neuronal NO synthase PDZ domain. J Biol Chem 274: 27467-27473. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, Bartley DA, Uhl GR, Snyder SH ( 1993) Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci 13: 2651-2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehehalt R, Keller P, Haass C, Thiele C, Simons K ( 2003) Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J Cell Biol 160: 113-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endres M, Laufs U, Huang Z, Nakamura T, Huang P, Moskowitz MA, Liao JK ( 1998) Stroke protection by 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase inhibitors mediated by endothelial nitric oxide synthase. Proc Natl Acad Sci USA 95: 8880-8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohann D, Keller P, Runz H, Kuhl S, Bertsch T, von Bergmann K, Hennerici M, Beyreuther K, Hartmann T ( 2001) Simvastatin strongly reduces levels of Alzheimer's disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci USA 98: 5856-5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassbender K, Stroick M, Bertsch T, Ragoschke A, Kuehl S, Walter S, Walter J, Brechtel K, Muehlhauser F, von Bergmann K, Lutjohann D ( 2002) Effects of statins on human cerebral cholesterol metabolism and secretion of Alzheimer amyloid peptide. Neurology 59: 1257-1258. [DOI] [PubMed] [Google Scholar]
- Heart Protection Study Collaborative Group ( 2002) MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 360: 7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA ( 2000) Statins and the risk of dementia. Lancet [Erratum (2001) 357:562] 356: 1627-1631. [DOI] [PubMed] [Google Scholar]
- Keshet GI, Ovadia H, Taraboulos A, Gabizon R ( 1999) Scrapie-infected mice and PrP knockout mice share abnormal localization and activity of neuronal nitric oxide synthase. J Neurochem 72: 1224-1231. [DOI] [PubMed] [Google Scholar]
- Kilsdonk EP, Yancey PG, Stoudt GW, Bangerter FW, Johnson WJ, Phillips MC, Rothblat GH ( 1995) Cellular cholesterol efflux mediated by cyclodextrins. J Biol Chem 270: 17250-17256. [DOI] [PubMed] [Google Scholar]
- Klein U, Gimpl G, Fahrenholz F ( 1995) Alteration of the myometrial plasma membrane cholesterol content with beta-cyclodextrin modulates the binding affinity of the oxytocin receptor. Biochemistry 34: 13784-13793. [DOI] [PubMed] [Google Scholar]
- Laufs U, Liao JK ( 1998) Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem 273: 24266-24271. [DOI] [PubMed] [Google Scholar]
- Laufs U, La FV, Plutzky J, Liao JK ( 1998) Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation 97: 1129-1135. [DOI] [PubMed] [Google Scholar]
- Laufs U, Gertz K, Huang P, Nickenig G, Bohm M, Dirnagl U, Endres M ( 2000) Atorvastatin upregulates type III nitric oxide synthase in thrombocytes, decreases platelet activation, and protects from cerebral ischemia in normocholesterolemic mice. Stroke 31: 2442-2449. [DOI] [PubMed] [Google Scholar]
- Laufs U, Wassmann S, Hilgers S, Ribaudo N, Bohm M, Nickenig G ( 2001) Rapid effects on vascular function after initiation and withdrawal of atorvastatin in healthy, normocholesterolemic men. Am J Cardiol 88: 1306-1307. [DOI] [PubMed] [Google Scholar]
- Laufs U, Gertz K, Dirnagl U, Bohm M, Nickenig G, Endres M ( 2002) Rosuvastatin, a new HMG-CoA reductase inhibitor, upregulates endothelial nitric oxide synthase and protects from ischemic stroke in mice. Brain Res 942: 23-30. [DOI] [PubMed] [Google Scholar]
- Martens JR, Navarro-Polanco R, Coppock EA, Nishiyama A, Parshley L, Grobaski TD, Tamkun MM ( 2000) Differential targeting of Shaker-like potassium channels to lipid rafts. J Biol Chem 275: 7443-7446. [DOI] [PubMed] [Google Scholar]
- McTaggart F, Buckett L, Davidson R, Holdgate G, McCormick A, Schneck D, Smith G, Warwick M ( 2001) Preclinical and clinical pharmacology of rosuvastatin, a new 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor. Am J Cardiol 87: 28B-32B. [DOI] [PubMed] [Google Scholar]
- Michikawa M, Yanagisawa K ( 1999) Inhibition of cholesterol production but not of nonsterol isoprenoid products induces neuronal cell death. J Neurochem 72: 2278-2285. [DOI] [PubMed] [Google Scholar]
- Perez AS, Bredt DS ( 1998) The N-terminal PDZ-containing region of postsynaptic density-95 mediates association with caveolar-like lipid domains. Neurosci Lett 258: 121-123. [DOI] [PubMed] [Google Scholar]
- Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, Brown L, Warnica JW, Arnold JM, Wun CC, Davis BR, Braunwald E ( 1996) The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med 335: 1001-1009. [DOI] [PubMed] [Google Scholar]
- Sattler R, Tymianski M ( 2001) Molecular mechanisms of glutamate receptor-mediated excitotoxic neuronal cell death. Mol Neurobiol 24: 107-129. [DOI] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M ( 1999) Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science 284: 1845-1848. [DOI] [PubMed] [Google Scholar]
- Scandinavian Simvastatin Survival Study Group ( 1994) Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet 344: 1383-1389. [PubMed] [Google Scholar]
- Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR, MacFarlane PW, McKillop JH, Packard CJ ( 1995) Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. West of Scotland Coronary Prevention Study Group. N Engl J Med 333: 1301-1307. [DOI] [PubMed] [Google Scholar]
- Shepherd J, Blauw GJ, Murphy MB, Bollen EL, Buckley BM, Cobbe SM, Ford I, Gaw A, Hyland M, Jukema JW, Kamper AM, MacFarlane PW, Meinders AE, Norrie J, Packard CJ, Perry IJ, Stott DJ, Sweeney BJ, Twomey C, Westendorp RG, PROSPER study group. Prospective Study of Pravastatin in the Elderly at Risk ( 2002) Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 360: 1623-1630. [DOI] [PubMed] [Google Scholar]
- Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K ( 1998) Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci USA 95: 6460-6464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M, Schwarzler F, Lutjohann D, von Bergmann K, Beyreuther K, Dichgans J, Wormstall H, Hartmann T, Schulz JB ( 2002) Treatment with simvastatin in normocholesterolemic patients with Alzheimer's disease: a 26-week randomized, placebo-controlled, double-blind trial. Ann Neurol 52: 346-350. [DOI] [PubMed] [Google Scholar]
- Sinensky M, Beck LA, Leonard S, Evans R ( 1990) Differential inhibitory effects of lovastatin on protein isoprenylation and sterol synthesis. J Biol Chem 265: 19937-19941. [PubMed] [Google Scholar]
- Tanaka T, Tatsuno I, Uchida D, Moroo I, Morio H, Nakamura S, Noguchi Y, Yasuda T, Kitagawa M, Saito Y, Hirai A ( 2000) Geranylgeranyl-pyrophosphate, an isoprenoid of mevalonate cascade, is a critical compound for rat primary cultured cortical neurons to protect the cell death induced by 3-hydroxy-3-methylglutaryl-CoA reductase inhibition. J Neurosci 20: 2852-2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group ( 1998) Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med 339: 1349-1357. [DOI] [PubMed] [Google Scholar]
- Topinka JR, Bredt DS ( 1998) N-terminal palmitoylation of PSD-95 regulates association with cell membranes and interaction with K+ channel Kv1.4. Neuron 20: 125-134. [DOI] [PubMed] [Google Scholar]
- Warshafsky S, Packard D, Marks SJ, Sachdeva N, Terashita DM, Kaufman G, Sang K, Deluca AJ, Peterson SJ, Frishman WH ( 1999) Efficacy of 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors for prevention of stroke. J Gen Intern Med 14: 763-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G ( 2000) Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol 57: 1439-1443. [DOI] [PubMed] [Google Scholar]