

Abstract
Planar cell polarity (PCP) regulates coordinated cellular polarity among neighboring cells to establish a polarity axis parallel to the plane of the tissue. Disruption in PCP results in a range of developmental anomalies and diseases. A key feature of PCP is the polarized and asymmetric localization of several membrane PCP proteins, which is essential to establish the polarity axis to orient cells coordinately. However, the machinery that regulates the asymmetric partition of PCP proteins remains largely unknown. In the present study, we show Van gogh-like 2 (Vangl2) in early and recycling endosomes as made evident by colocalization with diverse endosomal Rab proteins. Vangl2 biochemically interacts with adaptor protein-3 complex (AP-3). Using short hairpin RNA knockdown, we found that Vangl2 subcellular localization was modified in AP-3–depleted cells. Moreover, Vangl2 membrane localization within the cochlea is greatly reduced in AP-3–deficient mocha mice, which exhibit profound hearing loss. In inner ears from AP-3–deficient mocha mice, we observed PCP-dependent phenotypes, such as misorientation and deformation of hair cell stereociliary bundles and disorganization of hair cells characteristic of defects in convergent extension that is driven by PCP. These findings demonstrate a novel role of AP-3–mediated sorting mechanisms in regulating PCP proteins.
INTRODUCTION
Planar cell polarity (PCP) refers to the coordinated polarization of neighboring cells along an axis parallel to the plane of the tissue. PCP is critical for tissue development and function. Defects in PCP lead to a range of developmental anomalies and diseases, including cancers and developmental defects in the neural tube, the inner ear, heart, and kidney (Veeman et al., 2003; Guo et al., 2004; Park et al., 2006; Wang et al., 2006a,b). Many of these diseases are direct results of loss of coordinated polarity of cells or defective convergent extension (CE), which is driven by polarized cellular rearrangements under the regulation of PCP signaling (Wallingford and Harland, 2002; Wang et al., 2005; Axelrod, 2009).
The PCP pathway consists of a set of transmembrane proteins and their associated cytoplasmic proteins known as core PCP proteins, including Van gogh-like 1 and 2 (Vangl1 and Vangl2), Frizzled 3/6 (Fz3/Fz6), Dishevelled (Dvl), and Prickle (Pk) proteins (Axelrod, 2009; McNeill, 2010). These core PCP proteins are asymmetrically distributed within individual cells during PCP regulation. In the cochlea, the core PCP proteins are distributed in a polarized manner along the PCP axis in the terminally differentiated sensory epithelium known as the organ of Corti (Wang et al., 2006b; Montcouquiol et al., 2008). The core PCP proteins presumably form membrane-associated protein complexes at cellular boundaries to coordinate the polarity among neighboring cells (Guo et al., 2004; Wang et al., 2005; Montcouquiol et al., 2006; Chacon-Heszele et al., 2012). In mutant animals carrying a loss-of-function allele of Vangl2, Vangl2Lp/Lp (Kibar et al., 2001), core PCP proteins fail to correctly target the membrane in an asymmetrical manner, presumably underlying the loss of the coordinated orientation of sensory hair cells in those mutants (Curtin et al., 2003; Montcouquiol et al., 2003; Etheridge et al., 2008). Despite the apparent essential role of polarized localization of core PCP proteins, the central question of the role of intracellular trafficking in the selective sorting and localization of core PCP proteins in vertebrates (Merte et al., 2010; Wansleeben et al., 2010) remains unclear.
Proteins destined for plasma membrane domains are processed along the biosynthetic pathway and segregated into distinct subsets of transport carriers emanating from the trans-Golgi network (TGN). Coat proteins mediate protein sorting at the TGN. Among known coat proteins, adaptor protein complexes play a major role in cargo selection and sorting/transport to regulate intercellular transport (Bonifacino, 2014; Park and Guo, 2014). In particular, recent studies show a potential role for adaptor proteins in the trafficking of membrane PCP proteins, for example, AP-1 in exporting Vangl2 from the TGN in cultured cells and in targeting Fz during establishment of PCP in Drosophila wing (Guo et al., 2013; Carvajal-Gonzalez et al., 2015) . There are five known AP complexes in the vertebrates, AP-1, AP-2, AP-3, AP-4, and AP-5. Each AP is composed of two large subunits (α, β, γ, δ, ε, or ζ), one medium subunit (μ1–μ5), and one small subunit (σ1–σ5) (Nakatsu et al., 2014). Similar to small GTPase Rab proteins, AP complexes are localized to distinct but overlapping intracellular membranous compartments. AP-1, AP-3, and AP-4 are found at the TGN and endosomal membranes; AP-2 is localized to plasma membrane; and AP-5 is associated with late endosomes. Their localization denotes the trafficking events these APs regulate (Bonifacino, 2014; Park and Guo, 2014). AP complexes display specificity in membrane protein sorting by binding to selective sorting signals/motifs present in the cytoplasmic domain of membrane cargoes (Bonifacino and Traub, 2003; Bonifacino, 2014). Because adaptor selective localization and binding to sorting signals is required for the polarized partition of apical and basolateral proteins (Folsch, 2008), we asked whether adaptor-dependent sorting mechanisms segregate or regulate the subcellular localization of PCP membrane proteins along the polarity axis. We identified a novel interaction between AP-3 and Vangl2 in endosomal compartments and a requirement for AP-3 in Vangl2 membrane localization. We further took advantage of AP-3 mutant mice carrying a null mocha allele of the large subunit delta adaptin of AP-3, Ap3d1mh/mh, and identified PCP phenotypes in the organ of Corti that are associated with reduced membrane localization of Vangl2 and Fz3 and likely contribute to the auditory and vestibular dysfunctions we observed in the mutant mice. These results provide novel insight into the sorting pathways controlling PCP protein subcellular localization and reveal a novel role of the adaptor complex AP-3 in regulating PCP.
RESULTS
Vangl2 localizes to early and recycling endosomes
To explore PCP membrane protein–sorting endosomal pathways, we sought to identify cellular compartments where PCP proteins such as Vangl2 reside. We focused on adaptors and Rab proteins, which are small GTPases localizing to distinct membrane compartments within the endomembrane system (Schwartz et al., 2007; Stenmark, 2009; Pfeffer, 2013; Wandinger-Ness and Zerial, 2014). We also tested pharmacological agents capable of disrupting trafficking in an organelle-selective manner.
We first examined the distribution of Vangl2 in IMCD3 cells transiently expressing GFP-Vangl2 relative to red fluorescent protein (RFP)-tagged endosomal Rab proteins, Rab4, Rab5, Rab11, and Rab21, by time-lapse microscopy (Bhuin and Roy, 2014). Whole-cell colocalization was quantified using Pearson’s correlation coefficient (PCC), which quantifies how variations in two channels conform to each other irrespective of the gain (Newell-Litwa et al., 2007; Blumer et al., 2013; Tower-Gilchrist et al., 2014). Vangl2 partially colocalized with Rab5-positive and Rab21-positive early endosomes (Figure 1, C and L), and with recycling endosome markers Rab4a and Rab11a (Figure 1, F and I). The overlap between early endosomes and Vangl2 had a PCC of 0.38, while the PCC between Vangl2 and recycling endosome Rab4a or Rab11a was 0.79–0.8 (Figure 1M). Not only was Vangl2 seen in the endosomal compartments, but it also was observed on the cell surface (Figure 1, A, D, G, and J). The colocalization of Vangl2 with recycling endosome Rab protein was also confirmed by using an antibody that recognizes the endogenous Rab11a in IMCD3 cells (Supplemental Figure S1).
FIGURE 1:

Vangl2 association with endocytic compartments. (A–L) IMCD3 cells were cotransfected with GFP-Vangl2 (green) and RFP-Rab5 (B), Cherry-Rab4a (E), DsRed-Rab11a (H), or Cherry-Rab21 (K). After 48 h, cells were imaged using time-lapse microscopy and a series of consecutive images were captured. A single frame from each series is shown (A–L). The white boxes mark the regions that were magnified in the corner of each panel to illustrate the colocalization of Vangl2 with each Rab protein. (M) Colocalization was quantified by PCCs of Vangl2 with each Rab. N = 3 transfections; 25 cells analyzed per condition. (N) Movement of GFP-Vangl2 vesicles from Rab4-positive endosomes toward the plasma membrane. IMCD3 cells were transfected with GFP-Vangl2 and Cherry-Rab4, cultured for 24 h, and then imaged live. Consecutive images were captured, and stills from the series at different times are shown. Scale bar: 19 μm.
Recycling Rab GTPases regulate membrane targeting (Bastin and Heximer, 2013). We used live-cell microscopy to further explore whether Vangl2 trafficks through the Rab4-recycling endosome pathway en route to the plasma membrane. Still images from real-time microscopy illustrated several events of Vangl2 sorting from Rab4 vesicles to the cell periphery (Figure 1N). A Vangl2-positive vesicle could be seen fusing with Rab4 endosomes at time zero, followed by budding off from the Rab4 endosome and moving to the plasma membrane (Figure 1N and Supplemental Movie 1). These events were observed at several locations in all of the 25 cells examined.
Movie S1.
Vangl2 traffics from Rab4 positive endosomes. IMCD cells were transfected with GFP Vangl2 and cultured for 24 hours and live cell imaging was performed. A series of consecutive images were captured. Still frames from movie is presented (panel in F) at different times starting at time 0 and ending 315 seconds later. A vesicle containing GFP Vangl2 moves towards Rab4-positive endosomes (0 seconds) where it fuses (around 15 seconds, yellow). A vesicle containing Vangl2 buds from the Rab4-positive endosome and moves to plasma membrane (315 seconds). Scale bar, 19 μm.
To further examine whether Vangl2 is targeted to the plasma membrane via recycling endosomes, we employed a pharmacological approach using LY294002, a known phosphoinositide 3-kinase inhibitor that inhibits transferrin recycling in recycling endosomes (van Dam et al., 2002; Zhao and Keen, 2008) and in transporting cargoes to the cell membrane (Chen and Wang, 2001; Awwad et al., 2007), to block aspects of Vangl2 sorting. As expected, cells treated with LY294002 contained enlarged Rab4 vesicles (Supplemental Figure S2, A–H), consistent with a disruption in recycling endocytic processes. Vangl2 membrane localization was observed in cells transiently expressing GFP-Vangl2 in the presence of dimethyl sulfoxide (DMSO) (Supplemental Figure S2, A, C, E, and G). However, in the presence of LY294002, Vangl2 membrane localization was reduced and Vangl2 was trapped in punctate intracellular structures of apparently aberrant endosomes (Supplemental Figure S2, B, D, and F). Furthermore, the dominant-negative form of Rab4a caused a similar reduction of Vangl2 membrane localization (Supplemental Figure S2, F and G). These data suggested that Vangl2 cycles through Rab4a-positive endosomal compartments.
Vangl2 interacts with adaptor protein-3 complex (AP-3)
We examined the association of Vangl2 with a coat protein known to localize to endosomes for which a viable mouse null mutant exists. The delta subunit of the coat protein complex adaptor protein-3 complex (AP-3) is mutated in the mocha mouse (Ap3d1mh/mh) (Kantheti et al., 1998). AP-3 localizes to early and recycling endosomes in diverse cell types (Peden et al., 2004; Theos et al., 2005; Newell-Litwa et al., 2009). Following the finding that Vangl2 was present in endosomes and that a small fraction of Vangl2 colocalized with AP-3 (unpublished data), we tested whether AP-3 and Vangl2 proteins interact by coimmunoprecipitation (co-IP) (Figure 2). We detected a specific interaction between AP-3 and Vangl2 in cellular extracts (Figure 2A). The interaction between AP-3 and Vangl2 was also detected using brain extracts isolated from animals expressing Vangl2–green fluorescent protein (Vangl2-GFP) protein (Figure 2B). The specificity of the co-IP by the AP-3 delta antibody was confirmed by immunoblotting with transferrin receptor, whose presence was only observed in the input (Figure 2A) and is known not to be associated with AP-3 (Dell’Angelica et al., 1999; Newell-Litwa et al., 2009). Moreover, to exclude any spurious interactions between Vangl2 and AP-3 antibody–coated beads, we outcompeted the binding of the AP-3 adaptor to its antibody using the AP-3 immunogenic delta peptide (Figure 2, A and B).
FIGURE 2:
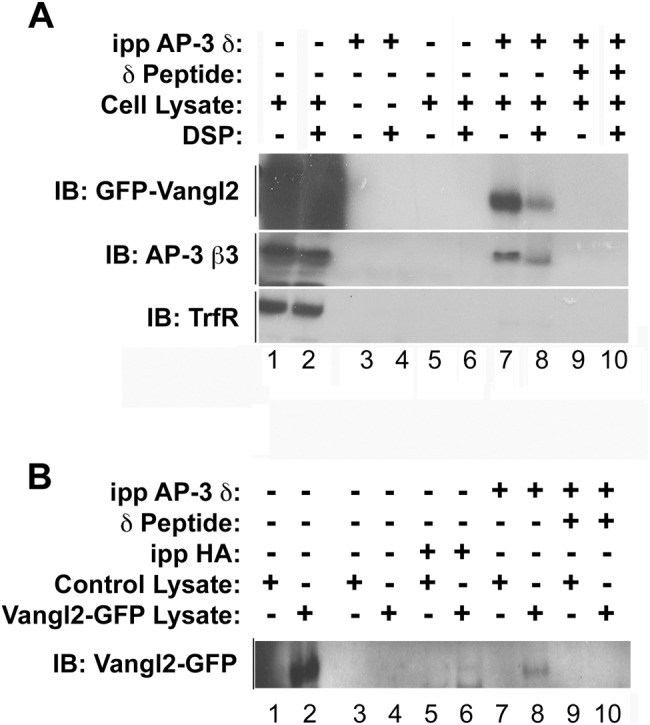
Vangl2 interacts with AP-3. (A) HEK293 cells were transiently transfected with GFP-Vangl2. Cell lysates were incubated at 4°C in the presence or absence of a cross-linker, DSP, and were immunoprecipitated with an anti–AP-3 SA4 delta antibody (A). Lanes 1 and 2 are input lanes (A,B). The resulting immune complexes were resolved by SDS–PAGE, and their compositions were assessed by immunoblot with antibodies against GFP, AP-3 beta, or transferrin receptor. Vangl2 was specifically pulled down by AP-3 antibody (lanes 7 and 8), and AP-3–specific peptide competition abolished the interaction between AP-3 and Vangl2 (lanes 9 and 10). (B) Brain protein extracts from neonatal control animals and animals expressing Vangl2-GFP were used for immunoprecipitations with an anti–AP-3 SA4 antibody. After incubation, the bound Vangl2 was detected by immunoblot using an anti-GFP antibody. AP-3 peptide competition abolished the interaction between AP-3 and Vangl2 (lanes 9 and 10). An antibody against HA served as an antibody control (lanes 5 and 6).
Depletion of the delta subunit of AP-3 causes loss of Vangl2 from the cell membrane
A functional interaction between AP-3 and Vangl2 predicts that Vangl2 subcellular distribution should be altered by AP-3 loss of function. Thus, we depleted AP-3 from cells using a short hairpin RNA (shRNA) against Ap3d1 (Figure 3 and Supplemental Figure S4) (Dong et al., 2005). We used nonpermeabilized cells to directly detect Vangl2-GFP fluorescence and Alexa Fluor–labeled wheat germ agglutinin (WGA) to mark the cell surface. GFP-Vangl2 localized to the cell surface in most scrambled shRNA-infected cells (Figure 3, A–F′). In contrast, Vangl2 accumulated in perinuclear structures in 95% of the cells transfected with AP-3 shRNA (Figure 3, D–F, F′, H, and I, and Supplemental Figure S4). AP-3 shRNA did not affect the expression of Vangl2 (Figure 3G). These findings show that AP-3 is required for the membrane localization of Vangl2 in cultured cells. In addition, the influence of the loss of AP-3 on Vangl2 membrane localization was observed in varying confluent culture conditions (Figure 3 and Supplemental Figure S4), suggesting that cell–cell contacts may not be a requirement for Vangl2 membrane localization mediated by AP-3 in cultured cells.
FIGURE 3:

Knockdown of AP-3δ redistributes Vangl2 from the plasma membrane. HEK293 cells were infected with lentiviral shRNA against nontargeting shRNA (scrambled) (A–C, C′) or with shRNA against AP-3δ subunit (D–F, F′) for 72 h before puromycin selection was started. Cells were then transfected with GFP-Vangl2 for 48 h; this was followed by a quick staining with WGA-594 to mark cell membranes. Vangl2 was not detected on the membrane and was observed in punctuate structures throughout the cell in most cells depleted of AP-3δ. C′ and F′ are large views of the boxed regions in C and F, respectively. The cultures were confluent for both scrambled and AP-3 shRNA conditions. Cultures with less confluency (Supplemental Figure S4) showed similar results. (G) Cell lysates were collected and subjected to Western blot analysis of protein lysates using anti–AP-3δ SA4 antibody, revealing a reduction of > 95% in AP-3 levels by shRNA against AP-3. (F) The percent of Vangl2 redistribution in AP-3–depleted cells was quantified (H). The numbers of cells with Vangl2 on the membrane were significantly lower than that of controls. One hundred cells were counted per condition in each experimental set, and the experiments were repeated three times. Vangl2 membrane localization was quantified by measuring Pearson’s coefficient of Vangl2 and WGA in shRNA Scrambled and shRNA AP-3 cells (I). N = 3; 50 cells analyzed. Scale bars: 19 µm. p < 0.002. Error bars represent SD.
Reduction of Vangl2 membrane localization in AP-3–deficient mice
Previous studies have reported postnatal hair cell loss in the organ of Corti in AP-3 delta 1 (Ap3d1)-deficient mocha animals (Lane and Deol, 1974; Rolfsen and Erway, 1984). However, it is not clear what the cellular defects are. Because AP-3 interacts with Vangl2 and is required for Vangl2 subcellular localization in cultured cells (Figures 3 and 4), we hypothesized that Vangl2 membrane localization may be disrupted and abnormal planar cell polarity may contribute to the overall defects observed in Ap3d1-deficient animals. To determine whether AP-3 has a role in Vangl2 trafficking in vivo, we bred animals carrying a Vangl2-GFP transgene (Qian et al., 2007) with mice carrying an AP-3 functional null allele, Ap3d1mh/mh (Kantheti et al., 1998) to obtain AP-3–deficient animals carrying the Vangl2-GFP transgene. The Vangl2-GFP transgenic animals carry two copies of the transgene that are under the control of Vangl2 regulatory elements and do not have any detectable cochlear or vestibular phenotypes (Qian et al., 2007). The subcellular localization of Vangl2-GFP is identical to that of the endogenous Vangl2 observed (Montcouquiol et al., 2006; Qian et al., 2007; Belotti et al., 2012), affording us a tool to examine Vangl2 localization in vivo.
FIGURE 4:

Disruption of Vangl2 membrane localization in AP-3–deficient cochlea. (A–F) Confocal images of cochlear whole mounts isolated at P0 from control mice (A, C, E) and AP-3–deficient mocha (Ap3d1mh/mh) mice (B, D, F) expressing Vangl2-GFP. Vangl2-GFP signal at the cellular junctions was much reduced in homozygous AP-3–deficient mocha mutants (B), in comparison to the littermate controls (A). The orthogonal images of the confocal scans of the control (C, E) and AP-3–deficient (D, F) littermates indicated by the white arrowhead (C, D) and by the white arrow (E, F) were shown to visualize the distribution of Vangl2 protein throughout the cells. The red and yellow arrows mark Vangl2 membrane enrichment in comparable regions in the control (A) and AP-3 mutant (B) organ of Corti. Scale bars: 10 μm. (G–J) Confocal images of cochlear whole mounts isolated at P0 from control mice (G, H) and AP-3–deficient mocha (Ap3d1mh/mh) mice (I, J) stained with antibodies against E-cadherin (blue) and Vangl2 (green) and phalloidin for F-actin (red). Note that Vangl2 asymmetric localization (indicated by arrows) was detected by the antibody against Vangl2, but the intensity of Vangl2 signaling in Ap3d1mh/mh mice was much reduced. The brackets mark the outer hair cell region, while the asterisks mark the inner hair cell region. E-cadherin showed a distinct boundary excluded from the inner hair cell region.
Membrane or membrane-associated PCP proteins normally show a polarized membrane localization at cellular junctions in the cochlear epithelium at postnatal day 0 (P0), and disruption of the membrane localization of Vangl2 and other PCP proteins results in misorientation of sensory hair cells and cellular patterning defects from abnormal CE (Kibar et al., 2001; Montcouquiol et al., 2003; Wang et al., 2005). As expected, Vangl2 appears on the apical cellular boundaries in control littermates (Figure 4, A, C, and E). In Ap3d1mh/mh animals, the plasma membrane levels of Vangl2 are drastically reduced across the length of the cochlear duct (Figure 4, B, D, and F), a phenotype reminiscent of the GFP-Vangl2 phenotypes observed in AP-3 down-regulated cells (Figure 3, D–F′).
We also performed antibody staining for Vangl2 and adherens junctional protein E-cadherin (Figure 4, G–J). We found a similar reduction of Vangl2 in AP-3–deficient mice (Figure 4, H and J) while the adherens junctional structure, revealed by staining for both E-cadherin and cortical F-actin associated with cellular junctional structures, appeared normal in AP-3–deficient mice (Figure 4, G and I). The loss or reduction of Vangl2 on the cell membrane in AP-3–deficient mice is likely to cause an alteration of PCP signaling, which will affect the polarized localization of other membrane or membrane-associated PCP proteins (Iliescu et al., 2011). Furthermore, interactions between membrane PCP protein complexes at the cellular boundaries reinforce the polarized distribution of these proteins (Axelrod, 2009). We tested the possibility of AP-3 loss having an effect on other membrane PCP proteins. We evaluated the distribution of Fz3 in the cochlea at P0 (Figure 5). In control cochleae, Fz3 asymmetric staining is apparent and readily detectable at the cellular boundaries on the medial side of the hair cell membrane (Wang et al., 2006a) (Figure 5, A–C). Confocal analysis of AP-3 mutant cochlea showed a reduction in Fz3 membrane localization (Figure 5, D–F). These results suggest that AP-3 is required for the membrane localization of Vangl2 at the apical cellular boundaries and that AP-3 deficiency influences Fz3 either directly or via the indirect effort of the loss of AP-3 on Vangl2 membrane targeting and PCP signaling.
FIGURE 5:

Distribution of Frizzled 3 in the organ of Corti of AP-3 deficient animals. (A–F) Immunostaining for Frizzled 3 (Fz3, red) in whole mounts of the organ of Corti at postnatal day 0 (P0) from basal regions of control (A–C) and Ap3d1mh/mh (D–F) cochleae. The level of Fz3 staining at the cell–cell junctional membranes is reduced in Ap3d1mh/mh mutants (D–F). Images in C and F are larger views of the boxed areas in A and D, respectively. The signal intensity of the boxed area (D) was enhanced (F) in order to illustrate the remaining junctional localizations of Fz3 in Ap3d1mh/mh mutants (arrowheads, F). Yellow arrows and asterisks (*) mark misoriented hair bundles and deformed hair bundles, respectively (E). IHC: inner hair cells; OHC1: outer hair cell row 1; OHC2: outer hair cell row 2; OHC3: outer hair cell row 3. (B, E). (G–J) Immunostaining for Arl13b (red), which visualizes the kinocilia, and F-actin (green) in whole mounts of the organ of Corti at postnatal day 0 (P0) from basal region of control (G, H) and Ap3d1mh/mh (I, J) cochleae. Images in H and J are larger views of the boxed areas in G and I, respectively. White arrowheads denote Fz3 localization. Note that the hair bundle morphology in Ap3d1mh/mh cochleae (I) appeared not as uniform as in control cochleae (G), and that the kinocilium remained at the vertex of the stereocilia bundle in normal and misoriented hair cells from Ap3d1mh/mh cochleae (I, J).
AP-3 mutants exhibit characteristic PCP defects in the cochlea and are deaf
A morphogenetic readout for epithelial PCP in the cochlea is the formation of a precisely polarized hair bundle on the apical surface of each hair cell and coordinated polarity of all hair cells across the entire cochlea (Kelly and Chen, 2009). We further explored the hypothesis that AP-3 deficiencies will have an effect on hair bundle morphology and orientation (Figure 6). In AP-3–deficient cochleae, there was a bundle misorientation phenotype as well as structural deformation of hair bundles where the hair bundles no longer formed a symmetric “V” shape (Figure 6, A–E). The hair bundles often appeared irregularly shaped, which was noticeable in the inner and outer third of hair cell rows in the organ of Corti of AP-3 mutants but not of control animals (Figures 5 and 6, A–C and E). The kinocilium remained near the cortex of the V-shaped stereociliary bundle even in misoriented hair cells (Figure 5, G and J). We measured the hair bundle orientation and plotted the cumulative probability of the hair bundle orientation. We found that the difference in angle distribution between control and AP-3–deficient animals is statistically significant using the Kolmogorov-Smirnov (KS) test (Figure 6D). Similarly, we performed statistical analysis for the malformation of hair bundles using Student’s t test and found that the difference between control and AP-3–deficient animals is significant (Figure 6E).
FIGURE 6:

Polarity and patterning defects in Ap3d1mh/mh organ of Corti. (A–C) Cochleae at P0 from Ap3d1+/mh (A) and Ap3d1mh/mh (B, C) animals were stained for actin (phalloidin). The asterisks mark irregular hair bundles. IHC, inner hair cells; OHC1, outer hair cell row 1; OHC2, outer hair cell row 2; OHC3, outer hair cell row 3. Scale bars: 10 μm. (D, E). Quantification of OHC3 stereociliary bundle orientation from basal and medial regions of cochleae (D) and malformation of the bundles (E). We measured the orientation of hair bundles from Ap3d1+/mh and Ap3d1mh/mh animals and plotted the distribution of the angles (D, insert) and the cumulative probability of the angles (D). We also quantified the percentage of hair cells from the basal region of Ap3d1+/mh and Ap3d1mh/mh animals with abnormally flattened or deformed bundles (E). The statistical significance of the cumulative probability of angle distribution was performed using KS test (D). The statistic significance of the percentage of hair bundle malformation was performed using Student’s t test (E). Data obtained from three animals per genotype. p < 0.0001 (bundle orientation); p < 0.02 (bundle morphology). (F) Ap3d1mh/mh animals have a smaller inner ear than control animals. The brackets mark the cochleae of inner ears in F.
Examination of the cochleae at the onset of hearing showed that the general structure of the cochlea is maintained in AP-3–deficient animals (Supplemental Figure S3). Hair cell loss was evident in older animals (Supplemental Figure S3), consistent with a previous report (Rolfsen and Erway, 1984). We found that the AP-3–deficient animals are profoundly deaf (Supplemental Figure S6A), potentially caused by both defects in hair bundles and loss of hair cells.
The vertebrate PCP pathway regulates CE, a type of polarized cellular rearrangement that drives the tissue to converge along one axis and extend along a perpendicular axis (Wallingford et al., 2001; Goto and Keller, 2002). In addition to regulating hair bundle morphology and orientation, essential vertebrate PCP genes also regulate CE in the terminally differentiating cochlea (Keller, 2002; Montcouquiol et al., 2003; Torban et al., 2004; Wang et al., 2005). Defects in PCP signaling and defective CE lead to cellular patterning defects in the cochlea, including a shortened cochlea with additional rows of sensory hair cells in a widened sensory epithelium, known as the organ of Corti (Montcouquiol and Kelley, 2003; Wang et al., 2005). In more than 60% of the Ap3d1mh/mh animals, we observed extra rows of hair cells (Figure 6C). In addition, the cochlear duct was shorter in some of the Ap3d1mh/mh cochleae compared with the control cochleae (Figure 6F).
Together, the data (Figure 4) indicate that AP-3 is required for membrane localization of Vangl2 at the cellular boundaries in the cochlea. The reduction of Vangl2 membrane localization in AP-3–deficient mice likely contributes to the abnormality in hair cell patterning and hair bundle polarity (Figure 6, B–E). The morphological abnormality observed in stereociliary bundles (Figure 6) may be caused by additional cargoes of AP-3 that are essential for the formation of stereociliary bundles. AP-3 is an essential gene for cochlear development, maturation, and function (Figure 6 and Supplemental Figures S3 and S4).
AP-3 mutants have characteristic PCP defects in the vestibular sensory organ
Ap3d1mh/mh mice exhibited head tilt and circling behavior (Jones et al., 2004). The underlying morphological and cellular defects, however, are not known. The tilted-head behavior in AP-3–deficient mice and the head bobbing observed in the Vangl2 loop-tail mutant animals (Strong and Hollander, 1949) are both consistent with vestibular abnormalities. We performed a number of behavioral tests to see whether the AP-3–deficient animals have additional behavior abnormalities indicative of a vestibular defect. We noticed that Ap3d1mh/mh mice performed poorly in walking assessments in comparison to littermate controls, while both Ap3d1+/mh animals and wild-type animals performed to the same normal capacity (Supplemental Figure S4B). In addition, Ap3d1mh/mh animals were unable to swim and had to be immediately rescued from drowning (Supplemental Figure S4C). These phenotypes are often associated with defects in the vestibule (Hoffman et al., 2006; Zhao et al., 2008).
Given the PCP defects in the cochlea in AP-3–deficient mice (Figure 6) and the vestibular behavioral abnormalities (Supplemental Figure S4), we performed morphological characterization of the vestibular organs from these animals to see whether there is similarly a PCP-related defect in the vestibule. In control animals, all the hair cells were oriented in the same direction in the posterior crista (Figure 7, B and D, arrows). In Ap3d1mh/mh animals, hair cells were oriented in various directions (Figure 7, B and D, arrows). Together, these data suggest that the loss of function of AP-3 leads to a PCP-dependent developmental defect in hair cell orientation in the posterior crista.
FIGURE 7:

PCP defects in the posterior crista from AP-3 mutant mice. Posterior crista from P0 control (A, B) and Ap3d1mh/mh (C, D) littermates were analyzed for hair cell orientation by staining with α-spectrin to visualize the position of the kinocilium (green). In Ap3d1mh/mh posterior crista, uniform hair cell orientation was disrupted and hair cells point in different directions (D, arrows) compared with the littermate control (B). The asterisks in D mark cells with spotty apical α-spectrin staining. Scale bars: 10 μm. (E) The misorientation of hair cells was quantified and plotted as described in Materials and Methods.
DISCUSSION
An essential feature of PCP signaling, the coordinated polarization of cells within the group, is achieved by membrane PCP protein complexes formed at cellular boundaries asymmetrically in a polarized manner to mediate the polarity coordination between neighboring cells (Peng and Axelrod, 2012; Singh and Mlodzik, 2012). Limited studies have suggested the requirement of specific trafficking of core PCP components for establishment of PCP (Yu et al., 2007; Mottola et al., 2010; Strutt et al., 2011). However, the network of trafficking machinery that contributes to the asymmetry distribution of membrane or membrane-associated PCP proteins is not clearly defined. Here, we showed that AP-3–dependent endosome traffic pathway is involved in membrane localization of Vangl2 in vitro and in vivo, supporting endosome traffic mechanisms in PCP regulation.
Vangl2 as a novel cargo of AP-3
Most membrane proteins are synthesized at the endoplasmic reticulum (ER), sorted by the COPII coating machinery into COPII vesicles, and exported from ER to the Golgi as the first trafficking event (Paczkowski et al., 2015). After passage through the Golgi complex, proteins are packaged into vesicles that bud off the TGN and are delivered either to endosomal compartments or to the cell membrane (Yap and Winckler, 2015).
Two previous studies established the interaction between Vangl2 and Sec24b, a component of the COPII coat. (Merte et al., 2010; Wansleeben et al., 2010). In this study, we examined the role of endosome trafficking mechanisms in Vangl2 membrane localization. AP complexes play crucial roles in the transport of proteins to distinct cellular compartments within the endomembrane system, including sorting and assembling cargoes into endocytic vesicles at the TGN for membrane protein trafficking, by interactions through tyrosine- and dileucine-based sorting signals in the cytoplasmic tails of cargo proteins (Badolato and Parolini, 2007; Park and Guo, 2014). The AP-3 coat complex has been shown to function on endosomes and aid in trafficking of membrane proteins from endosomes (Peden et al., 2004). A number of proteins have been identified as AP-3 cargoes, including TI-VAMP7 (Martinez-Arca et al., 2003), CLC-3 and ZnT3 (Salazar et al., 2004a, 2005a,b), phophatidylinositol-4-kinase type II α (Salazar et al., 2004a, 2005b; Craige et al., 2008), and tyrosinase (Theos et al., 2005; Di Pietro et al., 2006; Setty et al., 2007; Sitaram et al., 2012). Here, we propose that Vangl2 is a new AP-3 cargo. This model is supported by the coprecipitation of recombinant Vangl2 with endogenous AP-3, redistribution of Vangl2 away from the cell surface in AP-3–deplected cells in culture or in the cochlea from AP-3 mutant mice (Figures 2, 3, and 4), and phenotypes commonly associated with PCP mutants in AP-3–null cochleae and vestibules (Figures 5–7).
Roles of AP-3 in PCP protein membrane localization in vivo and in the formation and function of inner ear sensory organs
The mechanism of trafficking or sorting in the partition of PCP components has been explored by several recent studies. These studies identified a small repertoire of trafficking components such as Rabenosyn 5, Rab23, SEC24b, the adaptor complex AP-1, Arfaptin, and Arf1 (Mottola et al., 2010; Pataki et al., 2010; Wansleeben et al., 2010; Guo et al., 2013; Carvajal-Gonzalez et al., 2015). Disruption in the protein trafficking pathways regulated by Sec24b or AP-1 leads to several PCP mutant phenotypes in vivo, including inner ear defects and failure of Vangl2 to reach cell membrane (Merte et al., 2010; Wansleeben et al., 2010). In particular, the studies of the role of AP-1 in PCP protein trafficking in Drosophila and zebrafish revealed loss of polarized distribution of Fz and Vang when AP-1 function is compromised (Carvajal-Gonzalez et al., 2015). In this study, we found that membrane localization of Vangl2 is mediated by AP-3 endocytic pathways in cultured cells, as loss of AP-3 led to reduction of Vangl2 on the plasma membrane (Figure 3). We did note accumulation of Vangl2 into intracellular structures, and it would be important to further examine the destination of the missorted Vangl2. There a lack of overlap between Vangl2 with Rab4 and Rab5 in AP-3–depleted cells, which might suggest that Vangl2 is missorted away from the endosome (potentially into lysosomes) (unpublished data). Although we do not directly determine which pool of Vangl2 (newly synthesized or recycling) is being trafficked in this route, we provide evidence suggesting that the Rab4 endosome may play a role (Supplemental Figure S2). A study by Ma et al. (2018) demonstrates a distinct packaging of Vangl2 separate from Frizzled 6, events upstream of the endosomal pathway. Furthermore, the levels of Vangl2 on cellular junctions in the organ of Corti were greatly reduced in AP-3–deficient mice (Figure 4). This phenotype resembles another bona fide cargo, ZNT3, whose cellular content in neurons is reduced in AP-3 mocha animals (Kantheti et al., 1998; Salazar et al., 2004a,b). The results indicate AP-3 plays a role in Vangl2 membrane localization in vitro and in vivo.
Furthermore, we observed cochlear and vestibular morphological defects that are consistent with defective PCP signaling in AP-3–deficient mice (Figures 4–7) and may underlie the hearing and vestibular functional defects observed in such mice (Supplemental Figures S5 and S6). Previous studies show that Ap3d1mh/mh animals are deaf by testing hearing using the pinna response and report hair cell loss in older animals (Lane and Deol, 1974). In the vestibule, otoconial variations were seen in these animals (Rolfsen and Erway, 1984; Jones et al., 2004). In the current study, we found that the general structure of the cochlea is normal at onset of hearing, while the morphogenesis of the hair bundles and the patterning of the hair cells in the cochlea are affected in AP-3–deficient mice (Figure 6 and Supplemental Figure S3). The young AP-3–deficient animals are profoundly deaf, while littermates have normal hearing onset, as shown by auditory brainstem response (ABR) (Supplemental Figure S4). The PCP-like cellular defects likely contribute to the hearing defect in the cochlea. The loss of hair cells may be secondary to the loss of activity of hair cells, as hair cells remain in Vglut3 mutant mice that are deaf (Ruel et al., 2008; Akil et al., 2012). A hair cell polarity defect was also observed in the vestibule (Figure 7), which may contribute to the functional defects in the vestibule (Supplemental Figure S4). In addition to a CE and hair bundle polarity abnormality in AP-3–deficient animals, there are also apparent abnormalities in hair bundle morphology (Figures 5–7).
The apical enrichment of an F-actin–associated protein appears to be abnormal in AP-3 mutants (Figure 7). It is possible that additional cargoes of AP-3 could contribute to the abnormal bundle morphology and apical F-actin–associated protein enrichment. The published evidence suggests that apical-basal polarity can be established in cells lacking functional AP-3 complexes. Our results in neurons (Larimore et al., 2011) and the data by Junking et al. (2014) in epithelial cells indicate that markers such as cadherins retain their polarized apical–basal distribution despite clear indications of defective AP-3–dependent cargo sorting. We have also observed normal E-cadherin localization in the cochlea in AP-3 mutants (Figure 4). Moreover, ultrastructural studies of AP-3–null mouse kidney tubule cells indicate that there are no evident apical-basal polarity defects (Zhen et al., 1999). These reports are consistent with our observations that cellular junctional structures, such as E-cadherin adherens junctions and cortical F-actin, appear normal in AP-3–deficient cochlea (Figure 4 and Supplemental Figure S4). Additional cargoes that are affected in AP-3–deficient mutants may regulate actin dynamics in hair bundle morphogenesis and the apical actin network in the cochlea.
MATERIALS AND METHODS
Mouse strains and animal care
Animal care and usage was in accordance with U.S. National Institutes of Health (NIH, Bethesda, MD) guidelines and was approved by the Animal Care and Use Committee of Emory University under the IACUC protocol no.: DAR-2001963-082815BN. The transgenic, Vangl2-GFP animals were generated as previously reported (Qian et al., 2007). AP-3 mutant mice were purchased from Jackson Laboratories (stock no.: 000279, gr +/+ Ap3d1mh/mh dmh/J) and are referred to as the Ap3d1mh allele. The Ap3d1mh allele is a functional null allele of AP-3d1 (Kantheti et al., 1998).
Antibodies and other reagents
The following antibodies and dyes were used in this study: affinity-purified anti-VAMP7 (a gift from Andrew Peden, Cambridge Institute for Research, Cambridge, UK); polyclonal anti–phosphatidylinositol-4-kianse type II α (PI4KIIα) (Guo et al., 2003); monoclonal anti–AP-3 δ (SA4) (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA); polyclonal anti–AP-3 beta (cat. no. 13384-1-AP, Protein Tech Group, Chicago, IL); monoclonal anti–AP2 α-adaptin and anti-actin (cat. nos. A4325 and A5441, Sigma-Aldrich, St. Louis, MO); anti-hemagglutinin (HA; cat. no. A190108A, Bethyl Laboratories, Montgomery, TX); polyclonal anti-GFP (cat. no. 132002, Synaptic Systems, Goettingen, Germany); anti–α-spectrin (MAB1622, Chemicon); Rhodamine- or Alexa Fluor 488–conjugated phalloidin (Invitrogen); the antibody against Fz3 (a gift from J. Nathans, Johns Hopkins University, Baltimore, MD). Lipofectamine (cat. no. 11668-019) and puromycin (cat. no. 540411) were purchased from Invitrogen and Millipore, respectively. Anti-Vangl2 Clone 2G4 rat monoclonal antibody (gift from Jean-Paul Borg, INSERM-CNRS-Institut Paoli-Calmettes-Aix-Marseille Université, Marseille, France). Anti–E-cadherin monoclonal antibody was from BD Biosciences (cat. no. 610181, San Jose, CA). Anti-Rab11a rabbit antibody was from Cell Signaling (cat. no. 2413S, Danvers, MA). Anti-Rab4 and Rab5 antibodies were from Abcam (cat. nos. ab13252 and ab13253, Cambridge, MA). WGA, Alexa Fluor 594 conjugate was from ThermoFisher Scientific (cat. no. W11262, Grand Island, NY). LY294002 (cat. no. L9908-1MG) was from Sigma-Aldrich.
DNA constructs, oligonucleotides, and peptides
GFP-tagged Vangl2 (GFP-Vangl2) was obtained from Randy Schekman (University of California, Berkeley, CA). The pmCherry-C1-tagged Rab21 and pmCherry C2-Rab4aS22N were gifts from Elizabeth Sztul (University of Alabama at Birmingham, Birmingham, AL). pmCherry-C2-Rab4a wild-type was a gift from James Goldenring (Vanderbilt University Medical Center, Nashville, TN). DsRed-Rab11a wild-type (plasmid 12679) and mRFP-Rab5 wild-type plasmid (plasmid 14437) were from Addgene (Cambridge, MA) (Choudhury et al., 2002; Sharma et al., 2003; Vonderheit and Helenius, 2005). The oligonucleotides for AP-3δ (RHS4533-NM_003938) shRNA in a pLKO.1 vector for lentiviral infection were obtained from Open Biosystems (Huntsville, AL) and used to silence expression of human AP-3. The scramble control shRNA in pLKO.1 was obtained from Addgene (vector 1864). The peptide of AP-3δ SA4 (AQQVDIVTEEMPENALPSDEDDKDPNDPYRA) (Salazar et al., 2009) was purchased from Invitrogen (EvoQuest Team, Carlsbad, CA). The plasmid, μ2-HA-WT, was purchased from Addgene (plasmid 32752) (Nesterov et al., 1999).
Cell cultures, transfections, and lentiviral infections
Mouse IMCD3s were grown in DMEM/Ham’s F12 50/50 (cat. no. MT10092 CVRF, Invitrogen) mix with l-glutamine supplemented with 10% fetal bovine serum (cat. no. S11150, Atlanta Biologicals) and 100 U/ml penicillin/100 μg/ml streptomycin (cat. no. 15140122, Invitrogen) in a 37°C incubator with 5% CO2. PC12 cells were cultured as previously described (Newell-Litwa et al., 2007). Hek293T cells seeded in 35-mm plates were infected with 3 μl of lentivirus containing the shRNA constructs described earlier. After 24 h of being infected cells were incubated for up to 6 d in culture media supplemented with 4 μg/ml puromycin for selection as described in (Zlatic et al., 2011). Hek293 cells were then transfected with lipofectamine 2000 (Invitrogen) with 2 μg of DNA for 48 h.
Immunoprecipitations and Western blot analysis
Standard methods were used for protein extraction and Western blotting (Craige et al., 2008). Total protein concentrations were determined using the Bio-Rad protein assay (cat. no. 5000006). Proteins were separated by SDS–PAGE under reducing conditions and transferred onto a polyvinylidene difluoride (PVDF) membrane. Membranes were blocked in 5% dry milk and probed with specific primary antibodies and horseradish peroxidase–conjugated secondary antibodies.
Inner ear dissection, immunostaining, and imaging
Standard procedures were used for inner ear dissection, staining, and images (Jones and Chen, 2008). Inner ears were isolated with or without decalcification, fixed in 4% paraformaldehyde for 1 h either at room temperature or overnight at 4°C. The cochlear or vestibular epithelia were dissected and subjected to antibody staining as described previously (Wang et al., 2006a; Ren et al., 2013).
Time-lapse imaging was performed on a Nikon A1R confocal scope. Images were acquired for at least 15 s at 4 frames/s using NIS Elements (Nikon Instruments). Movies were then exported into Quicktime (Adobe Systems, San Jose, CA), and sequential still frames were taken from Quicktime movies. Localization and morphology images were captured using a Leica confocal imaging system. Further image analysis was done using Adobe Photoshop CS5.1 Extended, Image J (NIH), MetaMorph software (Sunnyvale, CA), or Volocity software programs. Confocal images were obtained using Nikon A1R, Olympus SzX12, Olympus FV1000/TIRF, or Zeiss LSM510.
For WGA, Alexa Fluor 594 labeling, cells were fixed in 4% paraformaldehyde for 15 min at room temperature; this was followed by three washes with Hank’s balanced salt solution (HBSS). Cells were incubated with 5 µg/ml WGA diluted in phosphate-buffered saline (PBS) for 10 min at room temperature. Cells were then washed with HBSS two times and with PBS. Cells were permeabilized with 0.1% Triton X-100 in PBS for 7 min. The coverslips were then washed with PBS-T (PBS containing 0.2% Tween-20), and nuclei were stained with Hoechst for 5 min and washed with PBS before cells were mounted on slides.
ABR tests
Tone-burst ABR is an objective measurement of hearing thresholds at specific frequencies to detect hearing impairment sensitively. ABR tests were performed as previously described (Ahmad et al., 2007; Chang et al., 2015). Briefly, sound stimuli of particular frequencies from 4 to 32 kHz generated by the BioSig software package (Tucker-Davis Technologies, Alachua, FL) were delivered to anesthetized mice, and ABR responses were recorded and analyzed as previously described (Ahmad et al., 2007).
Quantification of protein colocalization and morphology phenotype
We quantified colocalization using PCC, which quantifies how variations in two channels conform to each other irrespective of the gain. In-focus image planes were background subtracted with a 10-pixel rolling-ball algorithm in Fiji (ImageJ). A region of interest (ROI) immediately surrounding an individual cell was selected, and the Pearson coefficient was calculated by the Coloc 2 plug-in. Standard image rotation of 15° was used as a control to assess whether low colocalization values are just spurious channel overlap (Dunn et al., 2011). In all the analyses, the whole cell was included in the ROI.
For quantification of the number of Rab4a-enlarged endosomes in DMSO- or LY294002-treated cells, endosome size was measured based on exogenous cherry-Rab4a expression and endogenous EEA1 staining. Each segmented endosome was marked as an ROI using the image segmentation tool in ImageJ that defines endosomes based on signal intensity. Size was measured for each ROI/endosome (n = 30) for each cell (n = 25). Endosomes falling in the 0.5- to 4.5-µm range were considered small compared with larger endosomes with sizes from 5 µm and up.
For determination of the V-shaped hair bundle orientation in the cochlea, a line was drawn from the position of the kinocilium through the middle of the V-shaped stereocilia (bisecting line). We defined the angle of orientation as the angle formed between the bisecting line and the line parallel to the medial to lateral axis of the cochlear duct. Normally, this angle is close to 0°. Each row of hair cells was divided into three groups according to its position along the longitudinal axis of the cochlea (base, middle, and apex) owing to the presence of a differentiation gradient within the cochlea during development. Hair cells from the base and middle regions were included for quantification, and hair cells from comparable regions within the cochleae were used for comparison among different genotypes. Hair cells in the apex region of the cochlear duct are less developed and not included for quantification. At least 25 hair cells were quantified for each sample at each region of the cochlea and at least 3 animals per genotype were analyzed.
To quantify hair cell orientation in the posterior crista, we measured hair cell orientation throughout the length of the cristae with the use of alpha-Spectrin staining to highlight the position of the fonticulus, or the bare region devoid of F-actin and alpha-Spectrin. Orientation differences between neighboring cells were observed in four quadrants. Angle deviations of 30° from the axis perpendicular to the long axis of the crista were considered misoriented. The tissues may be stretched during mounting, and the long axis was adjusted to correct the twist caused by stretching. The cells in each quadrant with misorientation were counted and recorded. The data from 3 cristae per genotype were averaged and plotted.
Statistics
A KS comparison was used to test statistical significance of the cumulative probability of hair bundle angle distributions between control and mutant animals using the engine www.physics.csbsju.edu/stats/KS-test.n.plot_form.html or the software package StatPlus Mac Built 5.6.0pre/Universal (AnalystSoft). A p value < 0.05 is considered to be significant. For the percentage of malformed hair bundles in the control and mutant animals, the data are presented as average ± SD. Student’s t tests were used to determine statistical significance of data obtained from control and mutant animals. A p value < 0.05 is considered significant.
Supplementary Material
Acknowledgments
The pmCherry-C1-tagged Rab21 and pmCherry C2-Rab4aS22N were gifts from Elizabeth Sztul (University of Alabama at Birmingham, Birmingham, AL). pmCherry-C2-Rab4a wild-type was a gift from James Goldenring (Vanderbilt University Medical Center, Nashville, TN). Anti-Vangl2 Clone 2G4 rat monoclonal antibody was a gift from Jean-Paul Borg (INSERM-CNRS-Institut Paoli-Calmettes-Aix-Marseille Université, Marseille, France). This research project was supported by NIH grants ROI DC005213 to P.C., F32 DC0127 02 to C.T.-G., and GM077569 and CHOA for V.F. and S.A.Z. This research was also supported in part by the Emory University Integrated Cellular Imaging Microscopy Core.
Abbreviations used:
- ABR
auditory brainstem response
- AP-3
adaptor protein-3 complex
- CE
convergent extension
- co-IP
coimmunoprecipitation
- DMSO
dimethyl sulfoxide
- Dvl
Dishevelled
- ER
endoplasmic reticulum
- Fz3/Fz6
Frizzled 3/6
- GFP
green fluorescent protein
- HA
hemagglutinin
- KS
Kolmogorov-Smirnov
- NIH
National Institutes of Health
- P0
postnatal day 0
- PBS
phosphate-buffered saline
- PCC
Pearson’s correlation coefficient
- PCP
planar cell polarity
- Pk
Prickle
- RFP
red fluorescent protein
- ROI
region of interest
- shRNA
short hairpin RNA
- TGN
trans-Golgi network
- Vangl1 and Vangl2
Van gogh-like 1 and 2
- WGA
wheat germ agglutinin.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-08-0592) on July 3, 2019.
REFERENCES
- Ahmad S, Tang W, Chang Q, Qu Y, Hibshman J, Li Y, Sohl G, Willecke K, Chen P, Lin X. (2007). Restoration of connexin26 protein level in the cochlea completely rescues hearing in a mouse model of human connexin30-linked deafness. Proc Natl Acad Sci USA , 1337–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akil O, Seal RP, Burke K, Wang C, Alemi A, During M, Edwards RH, Lustig LR. (2012). Restoration of hearing in the VGLUT3 knockout mouse using virally mediated gene therapy. Neuron , 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awwad HO, Iyer V, Rosenfeld JL, Millman EE, Foster E, Moore RH, Knoll BJ. (2007). Inhibitors of phosphoinositide 3-kinase cause defects in the postendocytic sorting of beta2-adrenergic receptors. Exp Cell Res , 2586–2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelrod JD. (2009). Progress and challenges in understanding planar cell polarity signaling. Semin Cell Dev Biol , 964–971. [DOI] [PubMed] [Google Scholar]
- Badolato R, Parolini S. (2007). Novel insights from adaptor protein 3 complex deficiency. J Allergy Clin Immunol , 735–741; quiz 742–733. [DOI] [PubMed] [Google Scholar]
- Bastin G, Heximer SP. (2013). Rab family proteins regulate the endosomal trafficking and function of RGS4. J Biol Chem , 21836–21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotti E, Puvirajesinghe TM, Audebert S, Baudelet E, Camoin L, Pierres M, Lasvaux L, Ferracci G, Montcouquiol M, Borg JP. (2012). Molecular characterisation of endogenous Vangl2/Vangl1 heteromeric protein complexes. PLoS One , e46213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuin T, Roy JK. (2014). Rab proteins: the key regulators of intracellular vesicle transport. Exp Cell Res , 1–19. [DOI] [PubMed] [Google Scholar]
- Blumer J, Rey J, Dehmelt L, Mazel T, Wu YW, Bastiaens P, Goody RS, Itzen A. (2013). RabGEFs are a major determinant for specific Rab membrane targeting. J Cell Biol , 287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS. (2014). Adaptor proteins involved in polarized sorting. J Cell Biol , 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM. (2003). Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu Rev Biochem , 395–447. [DOI] [PubMed] [Google Scholar]
- Carvajal-Gonzalez JM, Balmer S, Mendoza M, Dussert A, Collu G, Roman AC, Weber U, Ciruna B, Mlodzik M. (2015). The clathrin adaptor AP-1 complex and Arf1 regulate planar cell polarity in vivo. Nat Commun , 6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacon-Heszele MF, Ren D, Reynolds AB, Chi F, Chen P. (2012). Regulation of cochlear convergent extension by the vertebrate planar cell polarity pathway is dependent on p120-catenin. Development , 968–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Wang J, Li Q, Kim Y, Zhou B, Wang Y, Li H, Lin X. (2015). Virally mediated Kcnq1 gene replacement therapy in the immature scala media restores hearing in a mouse model of human Jervell and Lange-Nielsen deafness syndrome. EMBO Mol Med , 1077–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wang Z. (2001). Regulation of epidermal growth factor receptor endocytosis by wortmannin through activation of Rab5 rather than inhibition of phosphatidylinositol 3-kinase. EMBO Rep , 842–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhury A, Dominguez M, Puri V, Sharma DK, Narita K, Wheatley CL, Marks DL, Pagano RE. (2002). Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J Clin Invest , 1541–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craige B, Salazar G, Faundez V. (2008). Phosphatidylinositol-4-kinase type II alpha contains an AP-3-sorting motif and a kinase domain that are both required for endosome traffic. Mol Biol Cell , 1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtin JA, Quint E, Tsipouri V, Arkell RM, Cattanach B, Copp AJ, Henderson DJ, Spurr N, Stanier P, Fisher EM, et al. (2003). Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr Biol , 1129–1133. [DOI] [PubMed] [Google Scholar]
- Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. (1999). Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta 3A subunit of the AP-3 adaptor. Mol Cell , 11–21. [DOI] [PubMed] [Google Scholar]
- Di Pietro SM, Falcon-Perez JM, Tenza D, Setty SR, Marks MS, Raposo G, Dell’Angelica EC. (2006). BLOC-1 interacts with BLOC-2 and the AP-3 complex to facilitate protein trafficking on endosomes. Mol Biol Cell , 4027–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Li H, Derdowski A, Ding L, Burnett A, Chen X, Peters TR, Dermody TS, Woodruff E, Wang JJ, et al. (2005). AP-3 directs the intracellular trafficking of HIV-1 Gag and plays a key role in particle assembly. Cell , 663–674. [DOI] [PubMed] [Google Scholar]
- Dunn KW, Kamocka MM, McDonald JH. (2011). A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol , C723–C742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etheridge SL, Ray S, Li S, Hamblet NS, Lijam N, Tsang M, Greer J, Kardos N, Wang J, Sussman DJ, et al. (2008). Murine dishevelled 3 functions in redundant pathways with dishevelled 1 and 2 in normal cardiac outflow tract, cochlea, and neural tube development. PLoS Genet , e1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folsch H. (2008). Regulation of membrane trafficking in polarized epithelial cells. Curr Opin Cell Biol , 208–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto T, Keller R. (2002). The planar cell polarity gene strabismus regulates convergence and extension and neural fold closure in Xenopus. Dev Biol , 165–181. [DOI] [PubMed] [Google Scholar]
- Guo J, Wenk MR, Pellegrini L, Onofri F, Benfenati F, De Camilli P. (2003). Phosphatidylinositol 4-kinase type IIα is responsible for the phosphatidylinositol 4-kinase activity associated with synaptic vesicles. Proc Natl Acad Sci USA , 3995–4000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo N, Hawkins C, Nathans J. (2004). Frizzled6 controls hair patterning in mice. Proc Natl Acad Sci USA , 9277–9281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Zanetti G, Schekman R. (2013). A novel GTP-binding protein-adaptor protein complex responsible for export of Vangl2 from the trans Golgi network. Elife , e00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LF, Ross MD, Varelas J, Jones SM, Jones TA. (2006). Afferent synapses are present in utricular hair cells from otoconia-deficient mice. Hear Res , 35–42. [DOI] [PubMed] [Google Scholar]
- Iliescu A, Gravel M, Horth C, Kibar Z, Gros P. (2011). Loss of membrane targeting of Vangl proteins causes neural tube defects. Biochemistry , 795–804. [DOI] [PubMed] [Google Scholar]
- Jones C, Chen P. (2008). Primary cilia in planar cell polarity regulation of the inner ear. Curr Top Dev Biol , 197–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones SM, Erway LC, Johnson KR, Yu H, Jones TA. (2004). Gravity receptor function in mice with graded otoconial deficiencies. Hear Res , 34–40. [DOI] [PubMed] [Google Scholar]
- Junking M, Sawasdee N, Duangtum N, Cheunsuchon B, Limjindaporn T, Yenchitsomanus PT. (2014). Role of adaptor proteins and clathrin in the trafficking of human kidney anion exchanger 1 (kAE1) to the cell surface. Traffic , 788–802. [DOI] [PubMed] [Google Scholar]
- Kantheti P, Qiao X, Diaz ME, Peden AA, Meyer GE, Carskadon SL, Kapfhamer D, Sufalko D, Robinson MS, Noebels JL, et al. (1998). Mutation in AP-3 delta in the mocha mouse links endosomal transport to storage deficiency in platelets, melanosomes, and synaptic vesicles. Neuron , 111–122. [DOI] [PubMed] [Google Scholar]
- Keller R. (2002). Shaping the vertebrate body plan by polarized embryonic cell movements. Science , 1950–1954. [DOI] [PubMed] [Google Scholar]
- Kelly MC, Chen P. (2009). Development of form and function in the mammalian cochlea. Curr Opin Neurobiol , 395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kibar Z, Vogan KJ, Groulx N, Justice MJ, Underhill DA, Gros P. (2001). Ltap, a mammalian homolog of Drosophila Strabismus/Van Gogh, is altered in the mouse neural tube mutant Loop-tail. Nat Genet , 251–255. [DOI] [PubMed] [Google Scholar]
- Lane PW, Deol MS. (1974). Mocha, a new coat color and behavior mutation on chromosome 10 of the mouse. J Hered , 362–364. [DOI] [PubMed] [Google Scholar]
- Larimore J, Tornieri K, Ryder PV, Gokhale A, Zlatic SA, Craige B, Lee JD, Talbot K, Pare JF, Smith Y, et al. (2011). The schizophrenia susceptibility factor dysbindin and its associated complex sort cargoes from cell bodies to the synapse. Mol Biol Cell , 4854–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T, Li B, Wang R, Lau PK, Huang Y, Jiang L, Schekman R, Guo Y. (2018). A mechanism for differential sorting of the planar cell polarity proteins Frizzled6 and Vangl2 at the trans-Golgi network. J Biol Chem , 8410–8427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Arca S, Rudge R, Vacca M, Raposo G, Camonis J, Proux-Gillardeaux V, Daviet L, Formstecher E, Hamburger A, Filippini F, et al. (2003). A dual mechanism controlling the localization and function of exocytic v-SNAREs. Proc Natl Acad Sci USA , 9011–9016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill H. (2010). Planar cell polarity: keeping hairs straight is not so simple. Cold Spring Harb Perspect Biol , a003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merte J, Jensen D, Wright K, Sarsfield S, Wang Y, Schekman R, Ginty DD. (2010). Sec24b selectively sorts Vangl2 to regulate planar cell polarity during neural tube closure. Nat Cell Biol , 41–46; sup pp 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montcouquiol M, Jones JM, Sans N. (2008). Detection of planar polarity proteins in mammalian cochlea. Methods Mol Biol , 207–219. [DOI] [PubMed] [Google Scholar]
- Montcouquiol M, Kelley MW. (2003). Planar and vertical signals control cellular differentiation and patterning in the mammalian cochlea. J Neurosci , 9469–9478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montcouquiol M, Rachel RA, Lanford PJ, Copeland NG, Jenkins NA, Kelley MW. (2003). Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature , 173–177. [DOI] [PubMed] [Google Scholar]
- Montcouquiol M, Sans N, Huss D, Kach J, Dickman JD, Forge A, Rachel RA, Copeland NG, Jenkins NA, Bogani D, et al. (2006). Asymmetric localization of Vangl2 and Fz3 indicate novel mechanisms for planar cell polarity in mammals. J Neurosci , 5265–5275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottola G, Classen AK, Gonzalez-Gaitan M, Eaton S, Zerial M. (2010). A novel function for the Rab5 effector Rabenosyn-5 in planar cell polarity. Development , 2353–2364. [DOI] [PubMed] [Google Scholar]
- Nakatsu F, Hase K, Ohno H. (2014). The role of the clathrin adaptor AP-1: polarized sorting and beyond. Membranes , 747–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesterov A, Carter RE, Sorkina T, Gill GN, Sorkin A. (1999). Inhibition of the receptor-binding function of clathrin adaptor protein AP-2 by dominant-negative mutant mu2 subunit and its effects on endocytosis. EMBO J , 2489–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell-Litwa K, Salazar G, Smith Y, Faundez V. (2009). Roles of BLOC-1 and adaptor protein-3 complexes in cargo sorting to synaptic vesicles. Mol Biol Cell , 1441–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell-Litwa K, Seong E, Burmeister M, Faundez V. (2007). Neuronal and non-neuronal functions of the AP-3 sorting machinery. J Cell Sci , 531–541. [DOI] [PubMed] [Google Scholar]
- Paczkowski JE, Richardson BC, Fromme JC. (2015). Cargo adaptors: structures illuminate mechanisms regulating vesicle biogenesis. Trends Cell Biol , 408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Guo X. (2014). Adaptor protein complexes and intracellular transport. Biosci Rep , e00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park TJ, Haigo SL, Wallingford JB. (2006). Ciliogenesis defects in embryos lacking inturned or fuzzy function are associated with failure of planar cell polarity and Hedgehog signaling. Nat Genet , 303–311. [DOI] [PubMed] [Google Scholar]
- Pataki C, Matusek T, Kurucz E, Ando I, Jenny A, Mihaly J. (2010). Drosophila Rab23 is involved in the regulation of the number and planar polarization of the adult cuticular hairs. Genetics , 1051–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peden AA, Oorschot V, Hesser BA, Austin CD, Scheller RH, Klumperman J. (2004). Localization of the AP-3 adaptor complex defines a novel endosomal exit site for lysosomal membrane proteins. J Cell Biol , 1065–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Axelrod JD. (2012). Asymmetric protein localization in planar cell polarity: mechanisms, puzzles, and challenges. Curr Top Dev Biol , 33–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer SR. (2013). Rab GTPase regulation of membrane identity. Curr Opin Cell Biol , 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian D, Jones C, Rzadzinska A, Mark S, Zhang X, Steel KP, Dai X, Chen P. (2007). Wnt5a functions in planar cell polarity regulation in mice. Dev Biol , 121–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren DD, Kelly M, Kim SM, Grimsley-Myers CM, Chi FL, Chen P. (2013). Testin interacts with vangl2 genetically to regulate inner ear sensory cell orientation and the normal development of the female reproductive tract in mice. Dev Dyn , 1454–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolfsen RM, Erway LC. (1984). Trace metals and otolith defects in mocha mice. J Hered , 159–162. [PubMed] [Google Scholar]
- Ruel J, Emery S, Nouvian R, Bersot T, Amilhon B, Van Rybroek JM, Rebillard G, Lenoir M, Eybalin M, Delprat B, et al. (2008). Impairment of SLC17A8 encoding vesicular glutamate transporter-3, VGLUT3, underlies nonsyndromic deafness DFNA25 and inner hair cell dysfunction in null mice. Am J Hum Genet , 278–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar G, Craige B, Love R, Kalman D, Faundez V. (2005a). Vglut1 and ZnT3 co-targeting mechanisms regulate vesicular zinc stores in PC12 cells. J Cell Sci , 1911–1921. [DOI] [PubMed] [Google Scholar]
- Salazar G, Craige B, Wainer BH, Guo J, De Camilli P, Faundez V. (2005b). Phosphatidylinositol-4-kinase type II alpha is a component of adaptor protein-3-derived vesicles. Mol Biol Cell , 3692–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar G, Love R, Styers ML, Werner E, Peden A, Rodriguez S, Gearing M, Wainer BH, Faundez V. (2004a). AP-3-dependent mechanisms control the targeting of a chloride channel (ClC-3) in neuronal and non-neuronal cells. J Biol Chem , 25430–25439. [DOI] [PubMed] [Google Scholar]
- Salazar G, Love R, Werner E, Doucette MM, Cheng S, Levey A, Faundez V. (2004b). The zinc transporter ZnT3 interacts with AP-3 and it is preferentially targeted to a distinct synaptic vesicle subpopulation. Mol Biol Cell , 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar G, Zlatic S, Craige B, Peden AA, Pohl J, Faundez V. (2009). Hermansky-Pudlak syndrome protein complexes associate with phosphatidylinositol 4-kinase type II alpha in neuronal and non-neuronal cells. J Biol Chem , 1790–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz SL, Cao C, Pylypenko O, Rak A, Wandinger-Ness A. (2007). Rab GTPases at a glance. J Cell Sci , 3905–3910. [DOI] [PubMed] [Google Scholar]
- Setty SR, Tenza D, Truschel ST, Chou E, Sviderskaya EV, Theos AC, Lamoreux ML, Di Pietro SM, Starcevic M, Bennett DC, et al. (2007). BLOC-1 is required for cargo-specific sorting from vacuolar early endosomes toward lysosome-related organelles. Mol Biol Cell , 768–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma DK, Choudhury A, Singh RD, Wheatley CL, Marks DL, Pagano RE. (2003). Glycosphingolipids internalized via caveolar-related endocytosis rapidly merge with the clathrin pathway in early endosomes and form microdomains for recycling. J Biol Chem , 7564–7572. [DOI] [PubMed] [Google Scholar]
- Singh J, Mlodzik M. (2012). Planar cell polarity signaling: coordination of cellular orientation across tissues. Wiley Interdiscip Rev Dev Biol , 479–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitaram A, Dennis MK, Chaudhuri R, De Jesus-Rojas W, Tenza D, Setty SR, Wood CS, Sviderskaya EV, Bennett DC, Raposo G, et al. (2012). Differential recognition of a dileucine-based sorting signal by AP-1 and AP-3 reveals a requirement for both BLOC-1 and AP-3 in delivery of OCA2 to melanosomes. Mol Biol Cell , 3178–3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenmark H. (2009). Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol , 513–525. [DOI] [PubMed] [Google Scholar]
- Strong LC, Hollander WF. (1949). Hereditary loop-tail in the house mouse—accompanied by imperforate vagina and with lethal craniorachischisis when homozygous. J Hered , 329–334. [Google Scholar]
- Strutt H, Warrington SJ, Strutt D. (2011). Dynamics of core planar polarity protein turnover and stable assembly into discrete membrane subdomains. Dev Cell , 511–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theos AC, Tenza D, Martina JA, Hurbain I, Peden AA, Sviderskaya EV, Stewart A, Robinson MS, Bennett DC, Cutler DF, et al. (2005). Functions of adaptor protein (AP)-3 and AP-1 in tyrosinase sorting from endosomes to melanosomes. Mol Biol Cell , 5356–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torban E, Kor C, Gros P. (2004). Van Gogh-like2 (Strabismus) and its role in planar cell polarity and convergent extension in vertebrates. Trends Genet , 570–577. [DOI] [PubMed] [Google Scholar]
- Tower-Gilchrist C, Styers ML, Yoder BK, Berbari NF, Sztul E. (2014). Monitoring endosomal trafficking of the g protein-coupled receptor somatostatin receptor 3. Methods Enzymol , 261–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dam EM, Ten Broeke T, Jansen K, Spijkers P, Stoorvogel W. (2002). Endocytosed transferrin receptors recycle via distinct dynamin and phosphatidylinositol 3-kinase-dependent pathways. J Biol Chem , 48876–48883. [DOI] [PubMed] [Google Scholar]
- Veeman MT, Axelrod JD, Moon RT. (2003). A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev Cell , 367–377. [DOI] [PubMed] [Google Scholar]
- Vonderheit A, Helenius A. (2005). Rab7 associates with early endosomes to mediate sorting and transport of Semliki Forest virus to late endosomes. PLoS Biol , e233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallingford JB, Harland RM. (2002). Neural tube closure requires Dishevelled-dependent convergent extension of the midline. Development , 5815–5825. [DOI] [PubMed] [Google Scholar]
- Wallingford JB, Vogeli KM, Harland RM. (2001). Regulation of convergent extension in Xenopus by Wnt5a and Frizzled-8 is independent of the canonical Wnt pathway. Int J Dev Biol , 225–227. [PubMed] [Google Scholar]
- Wandinger-Ness A, Zerial M. (2014). Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb Perspect Biol , a022616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Hamblet NS, Mark S, Dickinson ME, Brinkman BC, Segil N, Fraser SE, Chen P, Wallingford JB, Wynshaw-Boris A. (2006a). Dishevelled genes mediate a conserved mammalian PCP pathway to regulate convergent extension during neurulation. Development , 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Mark S, Zhang X, Qian D, Yoo SJ, Radde-Gallwitz K, Zhang Y, Lin X, Collazo A, Wynshaw-Boris A, et al. (2005). Regulation of polarized extension and planar cell polarity in the cochlea by the vertebrate PCP pathway. Nat Genet , 980–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Guo N, Nathans J. (2006b). The role of Frizzled3 and Frizzled6 in neural tube closure and in the planar polarity of inner-ear sensory hair cells. J Neurosci , 2147–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wansleeben C, Feitsma H, Montcouquiol M, Kroon C, Cuppen E, Meijlink F. (2010). Planar cell polarity defects and defective Vangl2 trafficking in mutants for the COPII gene Sec24b. Development , 1067–1073. [DOI] [PubMed] [Google Scholar]
- Yap CC, Winckler B. (2015). Adapting for endocytosis: roles for endocytic sorting adaptors in directing neural development. Front Cell Neurosci , 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu A, Rual JF, Tamai K, Harada Y, Vidal M, He X, Kirchhausen T. (2007). Association of Dishevelled with the clathrin AP-2 adaptor is required for Frizzled endocytosis and planar cell polarity signaling. Dev Cell , 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Jones SM, Yamoah EN, Lundberg YW. (2008). Otoconin-90 deletion leads to imbalance but normal hearing: a comparison with other otoconia mutants. Neuroscience , 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Keen JH. (2008). Gyrating clathrin: highly dynamic clathrin structures involved in rapid receptor recycling. Traffic , 2253–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen L, Jiang S, Feng L, Bright NA, Peden AA, Seymour AB, Novak EK, Elliott R, Gorin MB, Robinson MS, et al. (1999). Abnormal expression and subcellular distribution of subunit proteins of the AP-3 adaptor complex lead to platelet storage pool deficiency in the pearl mouse. Blood , 146–155. [PubMed] [Google Scholar]
- Zlatic SA, Tornieri K, L’Hernault SW, Faundez V. (2011). Clathrin-dependent mechanisms modulate the subcellular distribution of class C Vps/HOPS tether subunits in polarized and nonpolarized cells. Mol Biol Cell , 1699–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.