

Abstract
The objective of this study was to evaluate the role of β-adrenergic receptors in modulating associative long-term potentiation (LTP) induced at CA1 synapses. Two independent Schaffer collateral pathways were stimulated in hippocampal slices. The field EPSP (fEPSP) response evoked in one pathway (the weak pathway) was small, whereas a large response, usually 80–90% of the maximum, was evoked in the strong pathway. After recording of the baseline fEPSP evoked at 0.033 Hz, LTP of the weak pathway could be associatively induced by paired stimulation of the weak and strong pathways 100 times at 6 sec intervals, with stimulation of the weak pathway preceded 3–10 msec. However, pairing protocols with an interval between stimulation of the two pathways >10 msec resulted in no LTP. The induced LTP was NMDA receptor dependent, because 50 μm d,l-APV blocked its induction. Bath application of 1 μm isoproterenol enhanced LTP by increasing the window of the stimulation interval up to 15 msec but did not affect the magnitude of the LTP induced by pairing protocols with intervals <10 msec. Similar results were obtained when the experiments were repeated using whole-cell recording. These results suggest that activation of β-adrenergic receptors can enhance associative LTP by increasing the width of the time window rather than the magnitude of the LTP. Enhancement of LTP by β-adrenergic receptors was blocked in slices by pretreatment with inhibitors of protein kinase A or mitogen-activated protein kinase, suggesting that these signaling cascades are involved in this process.
Keywords: associative LTD, associative LTP, β-adrenergic receptors, hippocampus, isoproterenol, synaptic plasticity
Introduction
Long-term potentiation (LTP) is a use-dependent change in synaptic efficacy that has long been considered as the cellular mechanism underlying memory storage in the brain (Bliss and Collingridge, 1993; Malenka and Nicoll, 1999). LTP can be induced using two distinct stimulation paradigms. Homosynaptic LTP can be induced using brief high-frequency stimulation (Bliss and Lomo, 1973), whereas associative LTP can be induced by simultaneously pairing presynaptic stimulation at low frequency with postsynaptic activity, including depolarization of the postsynaptic membrane (Gustafsson et al., 1987), the backpropagating action potential of postsynaptic neurons (Magee and Johnston, 1997; Markram et al., 1997; Bi and Poo, 1998; Feldman, 2000), and activity produced by other synaptic inputs (Levy and Steward, 1983; Kanter and Haberly, 1993). Associatively induced synaptic plasticity is of particular interest, because it is consistent with the simple learning rule originally suggested by Hebb (1949) that synaptic strength increases when there is conjunctive presynaptic and postsynaptic activity. Furthermore, it is also consistent with the concept that the precise timing between presynaptic and postsynaptic activities may be used to encode information in neural networks (Hopfield, 1995; Rieke et al., 1997).
At hippocampal CA1 synapses, induction of both homosynaptic and associative LTPs requires activation of the NMDA subtype glutamate receptor, which, in turn, activates many downstream molecules required for LTP induction (for review, see Roberson et al., 1996). Although LTP induction is dependent on the activation of NMDA receptors, several modulating transmitters, such as adrenergic, cholinergic, and dopaminergic transmitters, in the CNS can modulate its induction. This may be attributable to the fact that signaling cascades activated by modulating transmitter systems often regulate the activity and function of downstream molecules after NMDA receptor activation during LTP induction. For example, it has been reported that the homosynaptic LTP induced at CA1 synapses can be enhanced by stimulation of β-adrenergic receptors (Katsuki et al., 1997; Winder et al., 1999; Yang et al., 2002a), muscarinic receptors (Blitzer et al., 1990; Huerta and Lisman, 1993), and D1/D5 receptors (Otmakhova and Lisman, 1996). However, the effect of these modulating systems on the associative LTP has received less attention. The goal of the present study was to address this issue.
Parts of this paper have been published previously in abstract form (Yang et al., 2002b).
Materials and Methods
The use of animals in this study was in accordance with the guidelines of the local ethical committee for animal research. Male Sprague Dawley rats aged 25–35 d were anesthetized with halothane and decapitated. The brains were quickly removed and placed in ice-cold artificial CSF (ACSF) containing the following (mm): 119 NaCl, 2.5 KCl, 26.2 NaHCO3, 1 NaH2PO4, 1.3 MgSO4, 2.5 CaCl2, and 11 glucose (the pH was adjusted to 7.4 by gassing with 5% CO2–95% O2). Transverse hippocampal slices (450 μm thick) were cut with a vibrating tissue slicer (Campden Instruments, Loughborough, UK) and transferred to an interfacetype holding chamber at room temperature (27°C). The slices were allowed to recover for at least 90 min and then were transferred to an immersion-type recording chamber, perfused at 2 ml/min with ACSF containing 0.1 mm picrotoxin at 30–31°C. To prevent epileptiform discharge of pyramidal neurons, a cut was made at the border between the CA1 and CA3 areas.
For extracellular field potential recording, a glass pipette filled with 3 mNaCl was positioned in the stratum radiatum of the CAl area, and the field EPSP (fEPSP) was recorded. Two bipolar stainless steel electrodes (Frederick Haer Company, Bowdoinham, ME) were placed in the stratum radiatum on opposite sides of the recording pipette to stimulate two separate Schaffer collateral branches. In one pathway, fEPSP was elicited by adjusting the intensity of stimulation to ∼30% of that at which population spikes after fEPSP began to appear; this pathway will be referred to as the “weak” pathway. In the other pathway (the “strong” pathway), the stimulating intensity was adjusted so that 80–90% of the maximal response was elicited. Stable baseline fEPSP activity of the weak pathway was recorded by applying a short-duration voltage pulse (<1 msec) at the determined intensity every 30 sec for at least 10 min. Associative LTP of the weak pathway was then induced by paired stimulation of the weak and strong pathway every 6 sec for 10 min. During the pairing protocol, the delivery of weak stimulation preceded that of strong stimulation by several milliseconds. fEPSP activity of the weak pathway was then elicited by stimulation every 30 sec for an additional 30 min.
For whole-cell recording, patch pipettes were filled with the following (in mm): 116 potassium gluconate, 6 KCl, 2 NaCl, 30 HEPES, 0.5 EGTA, 4 Mg-ATP, and 0.3 Na-GTP (the pH was adjusted to 7.2 with KOH and the osmolarity to 295 mOsm). Recordings were made from pyramidal cells using the blind patch technique (Blanton et al., 1989). The amplifier (Axopatch-1D; Axon Instruments, Foster City, CA) was switched to current-clamp mode once whole-cell recording was obtained, and the Vm was held at approximately −65 mV during recording. A bipolar stainless steel electrode was placed in the stratum radiatum to elicit an EPSP at 0.1 Hz. After a period of stable baseline recording, LTP was induced by pairing an EPSP with a single postsynaptic spike evoked by injection of a 1 msec current pulse every 6 sec for 10 min. During the pairing protocol, the evoking of the EPSP preceded the postsynaptic spike by several milliseconds. EPSP activity was then elicited at 0.1 Hz for an additional 30 min. The serial resistance and input resistance were monitored throughout the recording period by applying a 100 msec negative current pulse of 20–50 pA at 0.1 Hz. Recording was stopped, and the data were discarded if the serial resistance or input resistance varied >30% from the original values during the experiment.
All signals were filtered at 2 kHz using the low-pass Bessel filter provided with the amplifier (Axopatch-1D) and digitized at 5 kHz using a CED micro 1401 interface running Signal software (Cambridge Electronic Design, Cambridge, UK). All drugs were bath applied and were purchased from Sigma (St. Louis, MO), except for APV, timolol, PD98059, and U0126, which were from Tocris-Cookson (Bristol, UK). The initial slopes of the fEPSP and the peak amplitude of the EPSP were measured for data analysis. Synaptic responses were normalized to the average of the values measured over the baseline period. The average size of the slope of the fEPSPs or the peak amplitude of the EPSP recorded between 25 and 30 min after the end of the pairing protocol was used for statistical comparisons. All data are presented as the mean ± SEM and were compared using the paired t test or one-way ANOVA test. The criterion for significance was p < 0.05.
Results
The associative LTP can be induced at CA1 synapses in the hippocampus
After a stable period of baseline recording (10 min), LTP of the weak pathway could be induced by pairing weak pathway stimulation with strong pathway stimulation, the former preceding the latter by 3 msec (LTP, 181 ± 21% of baseline; n = 8; p < 0.01; paired t test) (Fig. 1A). This configuration of stimulation of the weak pathway preceding stimulation of the strong pathway was used in all the pairing protocols described hereafter. LTP of the weak pathway could also be induced by a pairing protocol in which the interval between the weak and strong pathways was 10 msec (206 ± 9% of baseline; n = 8; p < 0.01; paired t test) (Fig. 1B). When the interval was increased to 15 or 30 msec, no LTP was seen (15 msec, 108 ± 9% of baseline, n = 8, p = 0.13, paired t test; 30 msec, 107 ± 16% of baseline, n = 8, p = 0.7, paired t test) (Fig. 1C,D). Figure 2 summarizes the results obtained from the experiments in this series. As can be seen, for an associative LTP to be successfully induced at the weak pathway, the interval between stimulation of the weak and strong pathways had to be between 3 and 10 msec.
Figure 1.

Associative LTP induced at CA1 synapses. A, LTP induced at the weak pathway using the pairing protocol. The interval between stimulation of the weak and strong pathways was 3 msec, stimulation of the weak pathway preceding stimulation of the strong pathway. B, LTP could also be induced using the same protocol with a 10 msec interval. C, D, No LTP was induced when the interval was increased to 15 (C) or 30 (D) msec. Insets are superimpositions of raw data traces averaged over 10 sweeps recorded immediately before and 25 min after the end of the pairing protocol.
Figure 2.
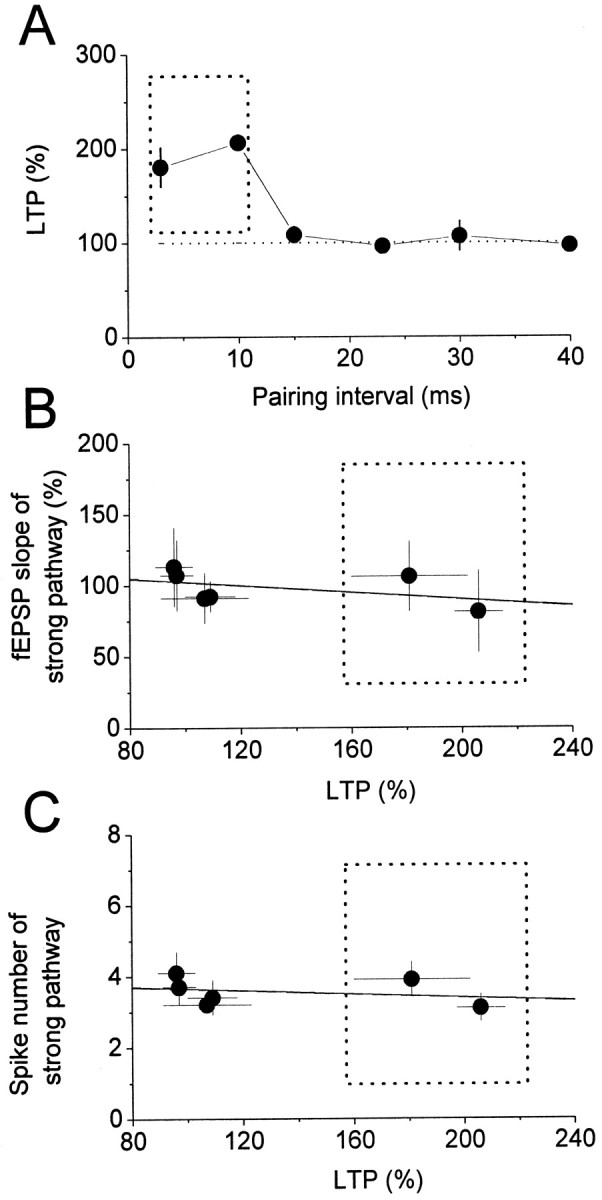
A, Summarized results for the LTP induced by the pairing protocol with different time intervals between stimulation of the weak and strong pathways, with weak leading strong in every case. For LTP to be successfully induced, the interval between the weak and strong pathways had to be within the window marked by the dotted square. The LTPs induced by the pairing protocol using intervals of 23 and 40 msec were, respectively, 96 ± 7% (n = 6) and 97 ± 6% (n = 6) of baselines. B, Relationship between the LTP induced using different intervals and the corresponding normalized mean slope of the fEPSP for the strong pathway; no significant correlation was found (solid line; r = −0.48; p = 0.33). C, Relationship between the LTP induced using different intervals and the corresponding mean number of postsynaptic spikes of the strong pathway; no significant correlation was found (solid line; r = −0.3; p = 0.556). The data in the dotted squares are those using intervals of 3 and 10 msec.
The LTP induced by these pairing protocols is associative and NMDA receptor dependent
Because the intensity for the strong pathway stimulation was adjusted to elicit 80–90% of the maximal synaptic response, it is very likely that the fibers of the Schaffer collateral branch recruited may overlap some fibers that were also recruited by weak stimulation, and the LTP induced in Figure 1, A and B, could be homosynaptic rather than associative. This possibility could be ruled out on the basis of the following observations. First, no LTP was induced when stimulation to the strong pathway (94± 4% of baseline; n = 6; p = 0.34; paired t test) (Fig. 3A) or to the weak pathway (101 ± 6% of baseline; n = 6; p = 0.9; paired t test) (Fig. 3B) was turned off during the pairing protocol. Second, when paired pulses with an interval of 10 msec were delivered to the weak pathway 100 times at 6 sec intervals, no LTP was induced; instead, a small, but significant, long-term depression (LTD) was seen (62 ± 7% of baseline; n = 6; p = 0.003; paired t test) (Fig. 3C). Third, the fEPSP elicited with weak stimulation was too small to achieve synaptic cooperation for the induction of homosynaptic LTP, because three epochs of theta burst stimulation (each epoch consisting of 10 trains of bursts at 5 Hz and each burst containing four pulses at 100 Hz) given every 30 sec to the weak pathway also failed to produce LTP (95 ± 4% of the baseline; n = 6; p = 0.34; paired t test) (Fig. 3D). In addition, we did not find any correlation between the magnitude of the LTP and the slope of the fEPSP elicited by strong stimulation (r =−0.33; p = 0.33) (Fig. 2B). In most cases, there were spikes riding on the fEPSP elicited by strong stimulation, but, again, we did not find any correlation between the magnitude of the LTP and the number of spikes elicited by strong stimulation (r =−0.48; p = 0.56) (Fig. 2C). The results of the above control experiments confirm that the LTP induced by the present pairing protocols is associative.
Figure 3.

LTP induced by the present pairing protocol is associative.A, B, No LTP was induced when only the weak (A) or the strong (B) pathway was stimulated. C, No LTP was induced when paired pulses with an interval of 10 msec were delivered to the weak pathway 100 times at 6 sec intervals. D, No LTP was induced when three trains of theta burst stimulation (TBS) at 100 Hz were delivered to the weak pathway. The insets are superimpositions of raw data traces averaged over 10 sweeps recorded immediately before and 25 min after the end of the pairing protocol or theta burst stimulation.
When 50 μm d, l-APV was included in the perfusion solution, the LTP induced by the pairing protocols using a 3 or 10 msec interval fell, respectively, to 106 ± 6% (n = 4; p = 0.41; paired t test) or 104 ± 7% (n = 4; p = 0.59; paired t test) of baseline. Because the results obtained using the 3 or 10 msec intervals were similar, the pooled results are shown in Figure 4. These results show that the associative LTP induced in the present study was NMDA receptor dependent.
Figure 4.

LTP induced by the present pairing protocol is NMDA receptor dependent. The LTP was blocked by adding 50 μm AP-5 to the ACSF to block NMDA receptors. The results are summarized from experiments using the pairing protocol and intervals of 3 msec (n = 4) or 10 msec (n = 4). The insets are superimpositions of raw data traces averaged over 10 sweeps recorded immediately before and 25 min after the end of the pairing protocol.
Activation of β-adrenergic receptors increases the window of the interval for LTP induction but does not effect the maximal magnitude of the LTP
The LTP induced by the pairing protocol with a 10 msec delay interval was not affected by addition of 1 μm isoproterenol (Iso) to the perfusion solution throughout the experiment (LTP in the presence of Iso, 177 ± 16% of baseline; n = 6; p = 0.11; paired t test) (Fig. 5A). However, the magnitude of the LTP induced with a 15 msec interval was markedly increased in the presence of Iso (LTP, 201 ± 33% of baseline; n = 6; p = 0.01; paired t test) (Fig. 5B). Using a 23 msec interval, Iso had no effect (LTP, 102 ± 18% of baseline; n = 6; p = 0.61; paired t test) (Fig. 5C). Figure 5D summarizes the results for this series of experiments. As can be seen, application of 1 μm Iso throughout recording enhanced the associative LTP by increasing the width of the window for the interval between weak and strong stimulation by ∼50%, i.e., from ∼3–10 msec in the control to ∼3–15 msec in the presence of Iso. Nevertheless, application of 1 μm Iso did not affect the maximal magnitude of the LTP, i.e., it did not affect the LTP induced by the pairing protocol with a 10 msec interval (p = 0.11; ANOVA test) (Fig. 5D), and, although it increased the magnitude of the LTP induced with a 15 msec interval, the final magnitude was no greater than that seen using a 10 msec interval in the absence of Iso.
Figure 5.
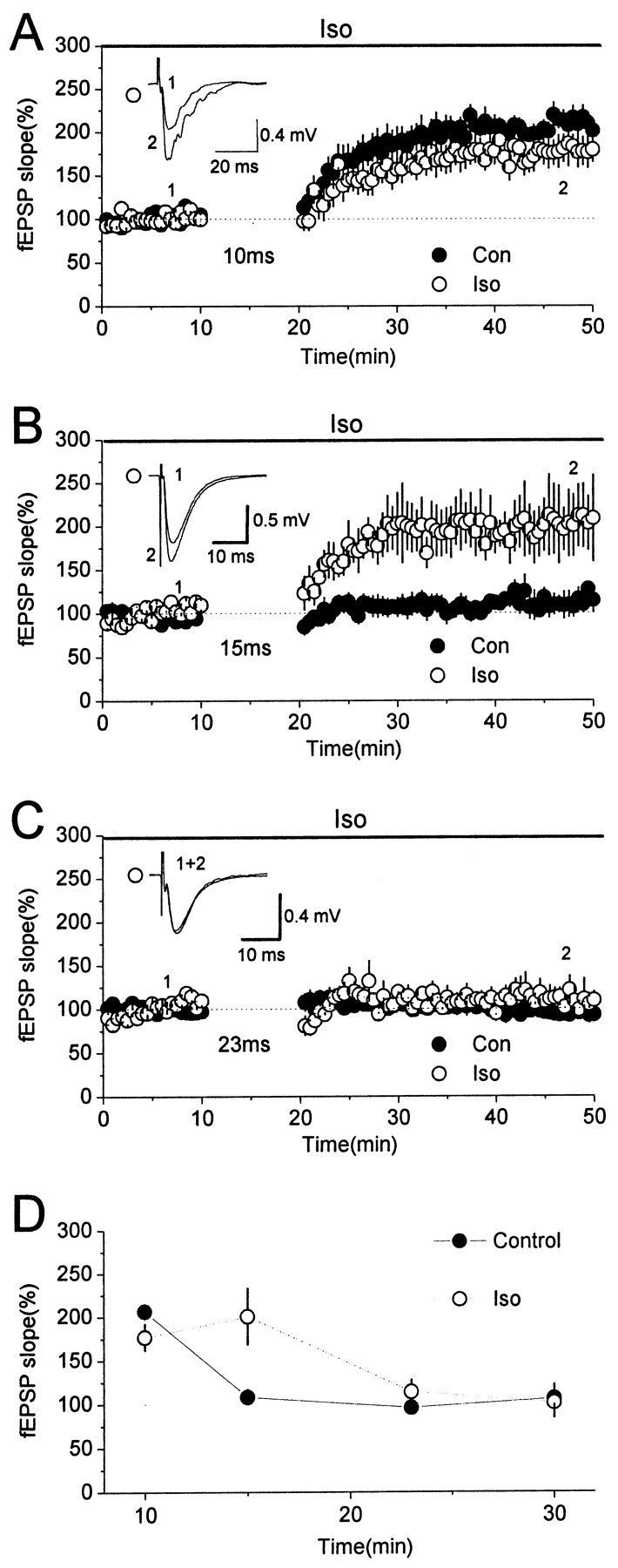
Enhancement of the associative LTP by stimulation of β-adrenergic receptors: field potential recording. A, Application of 1 μm Iso had no significant effect on the LTP induced using the pairing protocol with a 10 msec interval. B, Application of Iso resulted in an LTP induced using the pairing protocol with a 15 msec interval, which did not induce LTP under control conditions.C, Application of Iso had no significant effect when the interval was increased to 23 msec. D, Summarized results for A–C and those using a 30 msec interval (LTP, 101 ± 7% of baseline; n=6). In all of the plots, the open circles are results in the presence of 1 μm Iso, and the filled circles indicate data obtained from control experiments (i.e., results shown in Figs. 1 and 2). There was no significant difference in the LTP induced in control and Iso experiments, except for that using the 15 msec interval (one-way ANOVA test). Insets in A–C are superimpositions of data traces of Iso experiments. Traces were averaged over 10 sweeps recorded immediately before and 25 min after the end of the pairing protocol. Con, Control.
We repeated the experiments using whole-cell recording to confirm these observations. As shown in Figure 6A, the synaptic strength was enhanced when the EPSP was paired with a postsynaptic spike when the EPSP preceded by 7 msec (Fig. 6Aa), but not when the interval was increased to 35 msec (Fig. 6Ab) or 55 msec (Fig. 6Ac). The summarized results from experiments with different EPSP spike intervals (Δt) during pairing are shown in Figure 6C–E. The LTP induced with a Δt ≤ 20 msec was 174 ± 14% of baseline (n = 17; p < 0.001; paired t test) (Fig. 6C, filled circles), whereas no LTP was induced when 20 <Δt ≤ 40 msec (LTP, 104 ± 7% of baseline; n = 12; p = 0.62; paired t test) (Fig. 6D, filled circles) and when Δt > 40 msec (LTP, 104 ± 3% of baseline; n = 10; p = 0.28; paired t test) (Fig. 6E, filled circles). Figure 6F shows the relationship between a given Δt and the resultant change in synaptic strength for all experiments (filled circles). The data were fitted (Fig. 6F, solid curve) using the following equation modified from Song et al. (2000): F(Δt) = 100% + A · e−Δt/τ, where F(Δt) is the synaptic potentiation induced by the pairing protocol with an EPSP-spike interval Δt, A is the scaling factor, and τ is the time constant. τ determines the range of the EPSP-spike interval over which synaptic potentiation occurs, and A determines the maximal amount of synaptic potentiation (Song et al., 2000). The estimated values of A and τ were 164% of baseline and 13.3 msec, respectively. To examine the role of β-adrenergic receptors, recording configuration was kept in cell-attach mode when a seal was obtained and 1 μm Iso was bath applied for 5 min to achieve a stable concentration in the slice, then the seal was interrupted for whole-cell recording, and the experiment was performed as in control conditions. As shown in Figure 6B, the synaptic strength was enhanced when the EPSP was paired with a postsynaptic spike, with the EPSP leading by 10 and 35 msec (Fig. 6Ba,Bb) but not when the interval was increased to 55 msec (Fig. 6Bc). The summarized results from experiments with different values of Δt during pairing are shown in Figure 6C–E. In the presence of Iso, the LTP induced by the pairing protocol with Δt ≤ 20 msec and when 20 <Δt ≤ 40 msec was, respectively, 182 ± 26% (n = 9; p = 0.012; paired t test) (Fig. 6C, open circles) and 171 ± 26% (n = 10; p = 0.019; paired t test) (Fig. 6D, open circles) of baseline, whereas no LTP was induced with Δt > 40 msec (LTP, 108 ± 9%; n = 9; p = 0.35; paired t test) (Fig. 6E, open circles). The relationship between Δt and the resultant change in synaptic strength for all experiments was plotted (Fig. 6F, open circles), and the values of A and τ were estimated as 167% of baseline and 23.7 msec, respectively (Fig. 6F, dotted curve). These results were consistent with the finding using field potential recording; thus, in the following experiments, the field potential recording technique was adopted.
Figure 6.
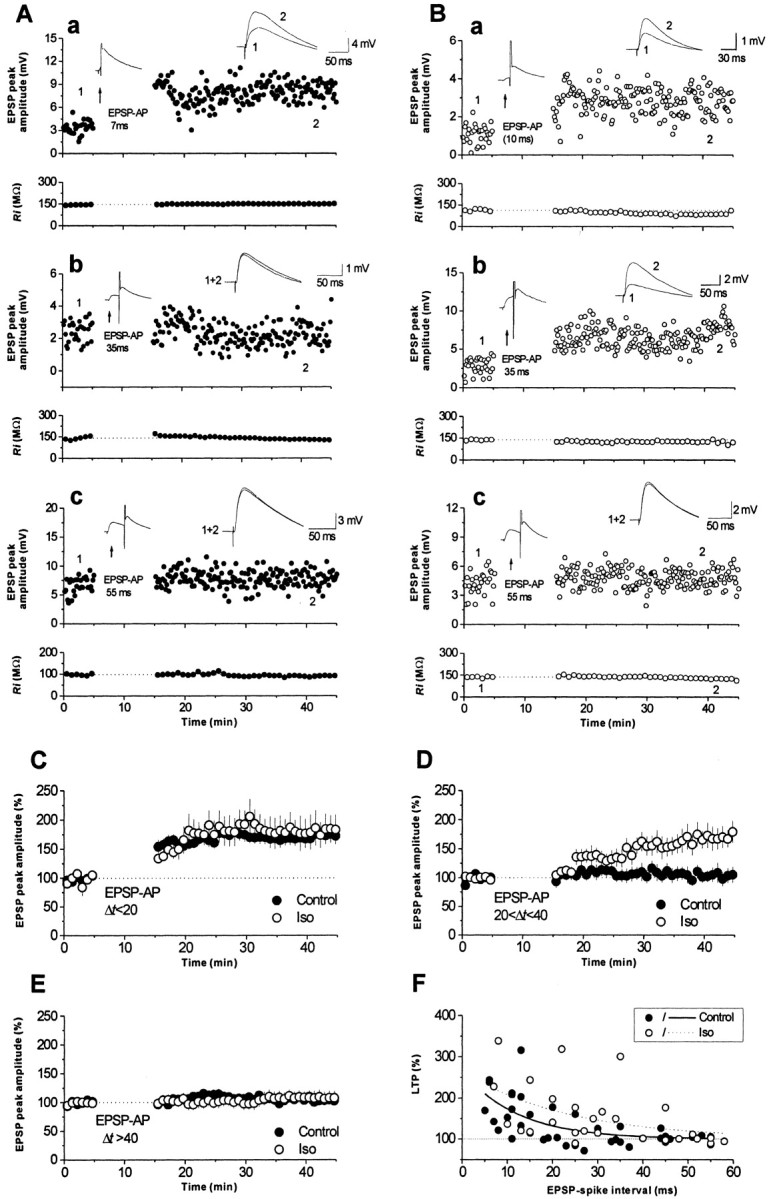
Enhancement of associative LTP by stimulation of β-adrenergic receptors: whole cell recording. A, Three individual experiments performed in normal ACSF, showing the LTP induced using pairing protocols with EPSP-spike intervals of 7 (a), 35 (b), and 55 (c) msec. B, Three individual experiments performed in Iso-containing ACSF, showing the LTP induced with EPSP-spike intervals of 10 (a), 35 (b), and 55 (c) msec. The top panels show EPSP recordings, and the bottom panels show input resistance recordings. The insets in the top panels show traces of the EPSP spike activity during pairing (left) and traces of EPSP activity averaged over 20 sweeps recorded immediately before and 25 min after the end of the pairing protocol. C–E, Summarized results for experiments of which the EPSP spike interval was <20 msec (C), longer than 20 msec and less than 40 msec (D), or longer than 40 msec (E). Filled circles, Normal ACSF; open circles, Iso-containing ACSF. F, Relationship between LTP induced by a given EPSP-spike interval. Each point represents an individual experiment. The solid and dotted lines show the fitting of the data for control (filled circles) and Iso (open circles) experiments with a single exponential decay. AP, Action potential.
Isoproterenol enhanced LTP by activation of β-adrenergic receptors, because the effect was completely blocked by application of 50 μm timolol, a β-adrenergic receptor antagonist (LTP, 103 ± 9% of baseline; n = 9; p = 0.72; paired t test) (Fig. 7A). We also tested whether endogenous norepinephrine was involved in the induction of the associative LTP by testing the effect of 50 μmtimolol on the LTP induced by the pairing protocol using a 10 msec interval and found that it had no effect (Fig. 7B) (p = 0.3; ANOVA test). We also tested LTP induction in slices from animals in which the adrenergic innervation was eliminated by subcutaneous injection of 6-hydroxy-dopamine (6-OHDA) (10 mg/kg bodyweight) once per day for 4 d, starting at birth, with testing being performed at postnatal days 25–35 (Yang et al., 2002a). Figure 7C, which summarizes the results for four slices, shows that the LTP induced by the pairing protocol with a 10 msec interval (193 ± 38% of baseline; n = 4) was no different from that induced by the same pairing protocol in normal slices (p = 0.62; ANOVA test).
Figure 7.

Effect of the endogenous adrenergic system on the associative LTP. A, Application of timolol inhibited LTP induction by the pairing protocol with a 15msec interval in the presence of Iso. B, An LTP was induced by the pairing protocol with a 10 msec interval in the presence of timolol. C, An LTP was induced by the pairing protocol with a 10 msec interval in slices from 6-OHDA-treated animals. The insets are superimpositions of raw data traces averaged over 10 sweeps recorded immediately before and 25 min after the end of the pairing protocol.
Protein kinase A and mitogen activated protein kinase are involved in the enhancement of the associative LTP by β-adrenergic receptor activation
Activation of β-adrenergic receptors raises the concentration of cAMP in the cytoplasm, and c-AMP then activates protein kinase A (PKA). We therefore tested the effect of pretreatment of slices with H-89, a membrane-permeating PKA blocker, on the enhancement of LTP caused by β-adrenergic receptor activation. Because the most dramatic effect of Iso on the LTP was induced using the pairing protocol with a 15 msec interval using field potential recording, this protocol was used. Figure 8A shows the summarized results for seven experiments. As can be seen, in slices preincubated for 2 hr at 35°C with 1 μm H-89, no LTP could be induced using the pairing protocol with a 15 msec interval and 1 μm Iso (LTP, 112 ± 12% of baseline; p = 0.34; paired t test). Because several studies have shown that mitogen-activated protein kinase (MAP kinase), a protein kinase family acting downstream of PKA, is involved in the induction and maintenance of tetanus-induced LTP at synapses in many CNS regions (Liu et al., 1999; Huang et al., 2000; Watabe et al., 2000; Giovannini et al., 2001), we tested the effect of membrane-permeating MAP kinase blockers. The broad-spectrum MAP kinase blocker PD98059 and the extracellular signal-regulated kinase (ERK) -specific blocker U0126 were used. When slices were preincubated for 2 hr at 35°C with PD98059 (50 μm) or U0126 (50 μm), no LTP was induced using the pairing protocol with a 15 msec interval and application of 1 μm Iso [PD98059: LTP, 100 ± 8% of baseline, n = 7, p = 0.9, paired t test (Fig. 8B); U0126: LTP, 98 ± 3% of baseline, n = 6, p = 0.9, paired t test (Fig. 8C)]. The above results suggest that the associative LTP was enhanced by activation of β-adrenergic receptors through activation of PKA, which, in turn, activated ERK kinase. We next asked whether this signaling pathway was also involved in the associative LTP induced by the pairing protocol alone. Figure 8D summarizes the results from seven experiments; as can be seen, no LTP was induced in H-89-pretreated slices by the pairing protocol with a 10 msec interval (LTP, 96 ± 4% of baseline; n = 6; p = 0.45; paired t test). Similar results were obtained in slices pretreated with PD98059 (LTP, 100 ± 5% baseline; n = 6; p = 0.9; paired t test) (Fig. 8E) or with U0126 (LTP, 97 ± 4% baseline; n = 5; p = 0.31; paired t test) (Fig. 8F). These results suggest that induction of the NMDA receptor-dependent associative LTP involves the PKA–ERK kinase signaling pathway.
Figure 8.

PKA and ERK–MAP kinases are involved in the enhancement of the LTP. A–C, The LTP induced by the pairing protocol with a 15 msec interval and application of Iso was blocked if slices were pretreated with H-89 (A), PD98059 (B), or U0126 (C); for comparison, the results of similar experiments performed using untreated slices (i.e., the data shown in Fig. 5B) are superimposed and shown as a solid line (mean) ±dotted lines (SE).D–F, The LTP induced bythe pairing protocol with a 10 msec delay interval was also blocked in slices pretreated with H-89(D), PD98059(E), or U0126 (F); for comparison, the results of similar experiments performed in untreated slices(i.e., the data shown in Fig. 1 B)are super imposed and shown as a solid line (mean) ± dotted lines (SE). The insets in A–F are super impositions of raw data traces averaged over 10 sweeps recorded immediately before and 25 min after the end of the pairing protocol.
Discussion
In this study, we induced an NMDA receptor-dependent associative LTP at a weak Schaffer collateral input onto hippocampus CA1 pyramidal neurons by pairing it with a separate strong Schaffer collateral input. For an LTP to be induced, the interval between the weak and strong inputs had to be within a time window of 10 msec. Activation of β-adrenergic receptors enhanced LTP by increasing the width of the time window to ∼15 msec but did not affect the maximal magnitude of the LTP. PKA and ERK/MAP kinase were involved in the signaling pathway downstream of β-adrenergic receptor activation and were also involved in the induction of LTP under normal conditions.
Associative LTP at CA1 synapses
Associative LTP induced by conjunction of presynaptic and postsynaptic activity has been reported in many CNS synapses. For example, temporal order stimulation applied to the testing and conditioning pathways induces associative LTP in the hippocampus in vivo (Levy and Steward, 1983) and in piriform cortex slices (Kanter and Haberly, 1993). Backpropagating action potentials initiated in the postsynaptic neuron by current injection at somata, together with presynaptic stimulation, result in longterm synaptic modification at CA1 synapses (Magee and Johnston, 1997; Markram et al., 1997), at synapses in slices of the barrel cortex (Feldman, 2000), and in hippocampal cultures (Bi and Poo, 1998; Debanne et al., 1998). Furthermore, the timing requirement for induction of associative LTP–LTD has been fully described for synapses in hippocampal cultures (Bi and Poo, 1998; Debanne et al., 1998) and slices of the rat barrel cortex (Feldman, 2000) and visual cortex (Froemke and Dan, 2002). Bi and Poo (1998) reported that, at synapses in hippocampal cultures, LTP is induced by a pairing protocol with intervals up to 20 msec if the EPSP leads the postsynaptic spike, and LTD is induced in the same interval time window if the spike leads the EPSP. For other synapses, a similar, short (∼20 msec) window has been reported for LTP induction, but a markedly larger window (up to ∼100 msec) is suggested for LTD induction (Debanne et al., 1998; Feldman, 2000) (but see Froemke and Dan, 2002). The present results suggest that a window of ∼10 msec is required for LTP induction at CA1 synapses in hippocampal slices using field potential recording, whereas a window of ∼20 msec is required using whole-cell recording. The results of whole-cell experiments are therefore in agreement with previous reports, but the time window estimated using field potential recording is considerably narrower. This is possibly attributable to the fact that LTP of the weak pathway is induced by conjunction of the weak fEPSP and the postsynaptic spikes evoked by strong pathway stimulation. This is supported by the fact that the mean latency between the stimulation artifact and the first spike is 7.3 ± 0.4 msec. It may also simply be attributable to the use of different recording techniques and pairing strategy.
Role of β-adrenergic receptors
Activation of β-adrenergic receptors has a significant effect on synaptic plasticity at hippocampal CA1 synapses (Katsuki et al., 1997; Yang et al., 2002a), mossy fiber synapses (Huang and Kandel, 1996), perforant synapses of the dentate gyrus (Stanton and Sarvey, 1985), and visual cortex synapses (Kirkwood et al., 1999). As regards homosynaptic plasticity, Katsuki et al. (1997) reported that activation of β-receptors has no effect on the LTP induced by theta burst stimulation at 100 Hz; however, it is reported to enhance the effect of conditioning stimulation at 10 Hz to induce LTP (Yang et al., 2002a). In other words, activation of β-adrenergic receptors enhances synaptic plasticity induced by conditioning stimulation of moderate strength (e.g., 10 Hz stimulation; Katsuki et al., 1997) which produces a total postsynaptic response near the modulation threshold (Θm; Bienenstock et al., 1982; Bear, 1996; Bear and Kirkwood, 1996), but does not affect the LTP induced by high-frequency stimulation, which produces a total postsynaptic response exceeding the Θm. The Θm is defined as the value at which synaptic efficacy is increased if the total postsynaptic response during conditioning stimulation is greater than the Θm, but it is depressed if it is greater than zero but less than the Θm. This concept was originally proposed to explain homosynaptic plasticity, but recent evidence suggests that it can also be applied to associative synaptic plasticity (Abraham et al., 2001). The present results suggest a similar role for β-adrenergic receptors in modulating associative LTP. A pairing protocol with a delay interval just a little longer than the time window might produce a total postsynaptic response that is close to, but not enough to reach, the threshold for LTP induction. However, with the simultaneous activation ofβ-adrenergic receptors, the signaling cascades activated byβ-adrenergic receptors might be able to cooperate to raise the levels of cytosolic molecules required for LTP induction to a point at which LTP can be induced. On the other hand, LTP induced by a pairing protocol with an interval less than the time window would itself be sufficient to raise the levels of cytosolic molecules to those required for LTP induction; therefore, the effect of β-adrenergic receptor activation would be limited.
The results of this study suggest that the endogenous adrenergic system does not have a significant effect on associative LTP in slice preparations because application of timolol did not affect the LTP induced by the pairing protocol with a 10 msec interval (field potential recording), and this pairing protocol could successfully induce LTP in slices taken from 6-OHDA-treated animals (Yang et al., 2002a). Using the same animal model, we reported that endogenous catacholamines may play an important role in modulating homosynaptic LTP but not LTD (Yang et al., 2002a). We proposed that large amounts of norepinephrine or dopamine were released after high-frequency tetanus stimulation and participated in the regulation of LTP induction via β-adrenergic or D1/D5 receptors. LTP induction was therefore impaired in slices lacking adrenergic or dopaminergic innervation. However, only a limited number of adrenergic or dopaminergic fibers are recruited during prolonged low-frequency stimulation for LTD induction, and the LTD is therefore less affected by elimination of the catecholamine system. On the basis of these arguments, it is not surprising that the endogenous effect of the adrenergic system on associative LTP was not significant, because stimuli were delivered at low frequency during the pairing protocol. The hippocampus receives adrenergic innervation mainly from the locus ceruleus in the midbrain (Aston-Jones et al., 1993). Although our results rule out a role of the endogenous adrenergic system in modulating associative LTP, the present results suggest that the chances of success in inducing LTP would be increased if locus ceruleus neurons were simultaneously activated.
Molecular mechanisms
The upstream molecular mechanisms underlying the induction of LTP at CA1 synapses are well known (for review, see Roberson et al., 1996). Evidence from recent studies suggests that, in addition to NMDA receptor activation, an increase in cAMP levels and a subsequent increase in PKA activity in the cytoplasm of postsynaptic neurons are required for LTP induction (Chetaovich and Sweatt, 1993; Makhinson et al., 1999; Otmakhova et al., 2000). Activation of NMDA receptors could result in an increase in the cAMP concentration and PKA activity via activation of adenylyl cyclase types I and VIII (Cooper et al., 1995). These types of adenylyl cyclase can be activated either directly or indirectly via calcium/calmodulin-dependent kinase (Wong et al., 1999) by calcium influx through NMDA receptors and are present at high levels in the hippocampus (Cooper et al., 1995). Recently, it has become clear that MAP kinase is directly involved in the regulation of synaptic plasticity in the CNS (for review, see Impey et al., 1999), including LTP (Liu et al., 1999; Huang et al., 2000; Watabe et al., 2000; Giovannini et al., 2001). In most of these studies, LTP was induced by tetanus stimulation, but the present results provide evidence that the induction of an associative LTP may have similar molecular mechanisms, because the induction of LTP using the pairing protocol with a 10 msec interval was NMDA receptor dependent and was blocked in slices in which PKA or MAP kinase activity was inhibited.
Bi and Poo (1998) proposed that activation of downstream effector molecules could potentially change the time window of synaptic modification. Consistent with this suggestion, our results demonstrated that activation of β-adrenergic receptors facilitated the activation of downstream effector molecules (e.g., PKA and MAP kinase) required for LTP induction and therefore increased the time window for synaptic modification. On the other hand, it has been established that β-adrenergic receptor agonists promote LTP by increasing excitability (Thomas et al., 1996; Watabe et al., 2000), and Iso might therefore have promoted associative LTP simply by increasing the excitability. However, increased excitability alone does not completely fit the observations of the present study. First, the field potential recording results were confirmed by those obtained using whole-cell recording. This technique allows the experimenter to control the membrane potential at a desired range and evoke a single spike during pairing in different experimental conditions, thus limiting the effect of membrane excitability. Second, in the experiments shown in Figure 5, the slices was allowed to equilibrate in Iso-containing ACSF before commencement of recording, and attention was paid to eliciting synaptic responses similar to those elicited under control conditions. The mean slope of the field response evoked at the strong pathway in Figure 5 was 107 ± 12% of that in Figure 1 (p = 0.68), and the mean number of spikes was 3.7 ± 0.3 versus 3.9 ± 0.3 (Fig. 1 vs Fig. 5) (p = 0.54), whereas the mean slope of the field response evoked at the weak pathway was 101 ± 5% of that in Figure 1 (p = 0.63). In addition, a recent study has shown that it is the timing of the first spike, and not the number of spikes, that plays a crucial role in determining the sign and magnitude of synaptic modification when an EPSP is paired with a burst of action potentials (Froemke and Dan, 2002). All of these facts suggest that an increase in excitability alone could not completely account for the enhancing effect of Iso on the associative LTP.
In conclusion, the present results show that the induction of an associative LTP requires the PKA–ERK/MAP kinase signaling cascade, as does induction of an homosynaptic LTP, and that simultaneous activation of β-adrenergic receptors during LTP induction can enhance LTP by increasing the time window of synaptic modification but not the maximal magnitude of the LTP.
Footnotes
This work was supported by grants from the National Science Council of Taiwan (M.-Y.M. and H.-W.Y.) and the Chinese Medical College (M.-Y.M.). Some of the experiments were performed with C.-T. Yen.
Correspondence should be addressed to Dr. Hsiu-Wen Yang, Department of Life Sciences, Chung Shan Medical University, Taichung, 402, Taiwan. E-mail: hwy@csmu.edu.tw.
Copyright © 2003 Society for Neuroscience 0270-6474/03/234173-09$15.00/0
References
- Abraham WC, Mason-Parker SE, Bear MF, Webb S, Tate WT ( 2001) Heterosynaptic metaplasticity in the hippocampus: a BMC-like modifiable threshold for LTP. Proc Natl Acad Sci USA 98: 10924–10929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Jones G, Shipley MT, Grzanna P ( 1993) The locus coeruleus, A5 and A7 noradrenergic cell groups. In: The rat nervous system (Paxinos G, ed), pp 183–213. San Diego: Academic.
- Bear MF ( 1996) A synaptic basis for memory storage in the cerebral cortex. Proc Natl Acad Sci USA 93: 13453–13459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear MF, Kirkwood A ( 1996) Bidirectional plasticity of cortical synapses. In: Cortical plasticity: LTP and LTD (Fazeli MS, Collingridge GL, eds), pp 191–205. Oxford: BIOS Scientific.
- Bienenstock L, Cooper NL, Munro PW ( 1982) Theory for the development of neuron selectivity: orientation specificity and binocular interaction in visual cortex. J Neurosci 2: 32–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi G, Poo M-M ( 1998) Synaptic modification in cultured hippocampal neuron: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci 18: 10464–10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanton MG, Lo Turco JJ, Kriegstein AR ( 1989) Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods 30: 203–210. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL ( 1993) Synaptic model of memory: long term potantiation in the hippocampus. Nature 361: 31–39. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Lomo T ( 1973) Long lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of perforant path. J Physiol (Lond) 232: 331–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzer RD, Gil O, Landau EM ( 1990) Cholinergic stimulation enhances longterm potentiation in the CA1 region of rat hippocampus. Neurosci Lett 118: 207–210. [DOI] [PubMed] [Google Scholar]
- Chetaovich DM, Sweatt JD ( 1993) NMDA receptor activation increases cyclic AMP in area CA1 of the hippocampus via calcium/calmodulin stimulation of adenylyl cyclase. J Neurochem 61: 1933–1942. [DOI] [PubMed] [Google Scholar]
- Cooper DMF, Mons N, Karpen JW ( 1995) Adenylyl cyclase and the interaction between calcium and cAMP signaling. Nature 374: 421–424. [DOI] [PubMed] [Google Scholar]
- Debanne D, Gahwiler BH, Thomson SM ( 1998) Longerm synaptic plasticity between pairs of individual CA3 pyramidal cells in rat hippocampal slice cultures. J Physiol (Lond) 507: 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE ( 2000) Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron 27: 45–56. [DOI] [PubMed] [Google Scholar]
- Froemke RC, Dan Y ( 2002) Spike-timing-dependent synaptic modification induced by natural spike trains. Nature 416: 433–438. [DOI] [PubMed] [Google Scholar]
- Giovannini MG, Blitzer RD, Wong T, Asoma K, Tsokas P, Morrison JH, Iyengar R, Landau EM ( 2001) Mitogen-activated protein kinase regulates early phosphorylation and delayed expression of Ca2+/calmodulin-dependent protein kinase II in longterm potentiation. J Neurosci 21: 7053–7062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson B, Wigstrom H, Abraham WC, Huang Y-Y ( 1987) Longterm potentiation in the hippocampus using depolarizing current pulses as the conditioning stimulus to single volley synaptic potentials. J Neurosci 7: 774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebb DO ( 1949) The organization of behavior. New York: Wiley.
- Hopfield JJ ( 1995) Pattern recognition computation using action potential timing for stimulus representation. Nature 376: 33–36. [DOI] [PubMed] [Google Scholar]
- Huang YY, Kandel ER ( 1996) Modulation of both the early and the late phase of mossy fiber LTP by activation of β-adrenergic receptors. Neuron 16: 611–617. [DOI] [PubMed] [Google Scholar]
- Huang Y-Y, Martin KC, Kandel ER ( 2000) Both protein kinase A and mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of longterm potentiation. J Neurosci 20: 6317–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta PT, Lisman JE ( 1993) Heightened synaptic plasticity of hippocampal CA1 neurons during a cholinergically induced rhythmic state. Nature 364: 723–725. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Storm DR ( 1999) Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron 23: 11–14. [DOI] [PubMed] [Google Scholar]
- Kanter D, Haberly LB ( 1993) Associative longterm potentiation in piriform cortex slices requires GABAa blockade. J Neurosci 13: 2477–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuki H, Izumi Y, Zorumski CF ( 1997) Noradrenergic regulation of synaptic plasticity in the hippocampal CA1 region. J Neurophysiol 77: 3013–3020. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Rozas C, Kirkwood J, Perez F, Bear MF ( 1999) Modulation of long term synaptic depression in visual cortex by acetylcholine and norepinephrine. J Neurosci 19: 1599–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy WB, Steward O ( 1983) Temporal contiguity requirements for longterm associative potentiation/depression in the hippocampus. Neuroscience 8: 791–797. [DOI] [PubMed] [Google Scholar]
- Liu J, Fukunaga K, Yamamoto H, Nishi K, Miyamoto E ( 1999) Differential roles of Ca2+/calmodulin-dependent protein kinase II and mitogen-activated protein kinase activation in hippocampal longterm potentiation. J Neurosci 19: 8292–8299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D ( 1997) A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neuron. Science 275: 209–213. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA ( 1999) Long term potentiation—a decade of progress? Science 285: 1870–1875. [DOI] [PubMed] [Google Scholar]
- Makhinson M, Chotiner JK, Watson JB, O'Dell TJ ( 1999) Adenylyl cyclase activation modulates activity-dependent changes in synaptic strength and Ca2+/calmodulin-dependent kinase II autophosphorylation. J Neurosci 19: 2500–2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Sakmann B ( 1997) Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science 275: 213–215. [DOI] [PubMed] [Google Scholar]
- Otmakhova NA, Lisman JE ( 1996) D1/D5 dopamine receptors activation increases the magnitude of early longterm potentiation at CA1 hippocampal synapses. J Neurosci 23: 7478–7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otmakhova NA, Otmakhov N, Mortenson LH, Lisman JE ( 2000) Inhibition of cAMP pathway decreases early longterm potentiation at CA1 hippocampal synapses. J Neurosci 20: 4446–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieke F, Warland D, de Ruyter van Steveninck R, Bialek W ( 1997) Spikes: exploring the neural code. Cambridge, MA: MIT.
- Roberson ED, English JD, Sweatt JD ( 1996) Second messengers in LTP and LTD. In: Cortical plasticity: LTP and LTD (Fazeli MS, Collingridge GL, eds), pp 35–60. Oxford: BIOS Scientific.
- Song S, Miller KD, Abbott LF ( 2000) Competitive Hebbian learning through spike-timing-dependent plasticity. Nat Neurosci 3: 919–926. [DOI] [PubMed] [Google Scholar]
- Stanton PK, Sarvey JM ( 1985) Depletion of norepinephrine, but not serotonin, reduces long term potentiation in the dentate gyrus of rat hippocampal slices. J Neurosci 5: 2169–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Moody T, Makhinson M, O'Dell TJ ( 1996) Activity-dependent β-adrenergic modulation of low-frequency stimulation-induced LTP in the hippocampal CA1 region. Neuron 17: 475–482. [DOI] [PubMed] [Google Scholar]
- Watabe AM, Zaki PA, O'Dell TJ ( 2000) Coactivation of β-adrenergic and cholinergic receptors enhances the induction of longterm potentiation and synergistically activates mitogen-activated protein kinase in the hippocampal CA1 region. J Neurosci 20: 5924–5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER ( 1999) ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron 24: 715–726. [DOI] [PubMed] [Google Scholar]
- Wong ST, Athos J, Figueroa XA, Pineda VV, Schaefer ML, Chavkin CC, Muglia LJ, Storm DR ( 1999) Calcium-stimulated adenylyl cyclase activity is critical for hippocampus-dependent long term memory and late phase LTP. Neuron 23: 787–798. [DOI] [PubMed] [Google Scholar]
- Yang H-W, Lin Y-W, Yen C-D, Min M-Y ( 2002a) Change in bidirectional plasticity at CA1 synapses in hippocampal slices taken from 6-hydroxydopamine treated rats: the role of endogenous norepinephrine. Eur J Neurosci 16: 1117–1128. [DOI] [PubMed] [Google Scholar]
- Yang H-W, Lin Y-W, Yen C-D, Min M-Y ( 2002b) Activation of β-adrenergic recepors enhances timing-based longterm potentiation (LTP) at hippocampal CA1 synapses. J Physiol (Lond) 543P: 30P. [Google Scholar]