

Abstract
Traumatized axons possess an extremely limited ability to regenerate within the adult mammalian CNS. The myelin-derived axon outgrowth inhibitors Nogo, oligodendrocyte-myelin glycoprotein, and myelin-associated glycoprotein, all bind to an axonal Nogo-66 receptor (NgR) and at least partially account for this lack of CNS repair. Although the intrathecal application of an NgR competitive antagonist at the time of spinal cord hemisection induces significant regeneration of corticospinal axons, such immediate local therapy may not be as clinically feasible for cases of spinal cord injury. Here, we consider whether this approach can be adapted to systemic therapy in a postinjury therapeutic time window. Subcutaneous treatment with the NgR antagonist peptide NEP1–40 (Nogo extracellular peptide, residues 1–40) results in extensive growth of corticospinal axons, sprouting of serotonergic fibers, upregulation of axonal growth protein SPRR1A (small proline-rich repeat protein 1A), and synapse re-formation. Locomotor recovery after thoracic spinal cord injury is enhanced. Furthermore, delaying the initiation of systemic NEP1–40 administration for up to 1 week after cord lesions does not limit the degree of axon sprouting and functional recovery. This indicates that the regenerative capacity of transected corticospinal tract axons persists for weeks after injury. Systemic Nogo-66 receptor antagonists have therapeutic potential for subacute CNS axonal injuries such as spinal cord trauma.
Keywords: Nogo, Nogo-66 receptor antagonist, axon regeneration, spinal cord injury, trauma, serotonergic fiber, SPRR1A, NEP1–40 peptide
Introduction
Adult mammalian CNS trauma interrupts axonal continuity and results in persistent functional deficits. The success or failure of axonal growth and reconnection with synaptic targets is a principal determinant of recovery in conditions such as spinal cord injury (SCI). CNS axonal regeneration is determined by both cell-autonomous and cell-extrinsic factors. Adult retinal axons can extend if provided with a favorable environment, but they do so with less alacrity than embryonic or peripheral neurons (Goldberg et al., 2002). Rapid peripheral axon regeneration is supported by the induction of regeneration-associated genes such as GAP-43 (Bomze et al., 2001) and SPRR1A (small proline-rich repeat protein 1A) (Bonilla et al., 2002), which are not upregulated after CNS axotomy. Changes in growth state may be mediated by increased levels of cAMP (Neumann et al., 2002; Qiu et al., 2002) and by the signal transducer and activator of transcription signaling (Liu and Snider, 2001).
There is also clear evidence that the environment surrounding CNS axons limits their growth. Although this may include a paucity of growth-promoting factors, such as neurotrophins (Schnell et al., 1994; Coumans et al., 2001), the presence of CNS inhibitors of axonal regeneration is now well documented. The glial scar produced by astrocytes is laden with inhibitory chondroitin sulfate proteoglycans (CSPG) (Snow et al., 1990; Dou and Levine, 1994; Davies et al., 1999). The digestion of CSPGs can provide a level of axon growth and recovery (Bradbury et al., 2002).
Several lines of evidence have identified CNS myelin components as prominent inhibitors of adult axon regeneration. CNS axons extend robustly into peripheral nerve grafts lacking oligodendrocytes (Richardson et al., 1980) or into the myelin-free CNS (Savio and Schwab, 1990). Three molecules of CNS myelin have been identified as axon-regeneration inhibitors: Nogo (Chen et al., 2000; GrandPre et al., 2000; Prinjha et al., 2000), myelin-associated glycoprotein (MAG) (Savio and Schwab, 1990; McKerracher et al., 1994; Mukhopadhyay et al., 1994), and oligodendrocyte myelin glycoprotein (Wang et al., 2002a). All three purified proteins inhibit axon outgrowth from mature neurons in vitro. Remarkably, a single neuronal protein, the Nogo-66 receptor (NgR) binds all three proteins and appears to be the primary mediator of their action (Fournier et al., 2001; Domeniconi et al., 2002; Liu et al., 2002; Wang et al., 2002a). The p75 lowaffinity neurotrophic receptor protein has been implicated in transducing a myelin/NgR signal to the axonal interior (Wang et al., 2002b; Wong et al., 2002).
The presence of myelin-derived inhibitors suggests that blocking their action might allow the intrinsic growth potential of CNS axons to be unmasked, with resultant functional recovery after adult CNS axotomy. Indeed, neutralizing Nogo A with IN-1 antibody can induce CNS axon regeneration and improve recovery after various lesions (Bregman et al., 1995; Thallmair et al., 1998). Genetic studies of MAG depletion have met with mixed results (Bartsch et al., 1995; Schafer et al., 1996). Targeting the axonal NgR with a competitive antagonist compound has the potential to block the action of the three known myelin inhibitors. Indeed, intrathecal application of an NgR competitive antagonist, NEP1–40 (Nogo extracellular peptide, residues 1–40), produces significant axonal regrowth after spinal cord hemisection injury (GrandPre et al., 2002). However, local drug delivery initiated at the time of SCI may be logistically difficult for the treatment of many SCI patients. In the present study, we also demonstrate the therapeutic potential of an NgR antagonist peptide for SCI by systemic peptide administration at a postinjury time window.
Materials and Methods
Spinal cord dorsal hemisection and corticospinal fiber tracing. All surgical procedures and postoperative care were performed in accordance with guidelines of the Yale Animal Care and Use Committee. Adult female C57BL/6 mice (8–10 weeks of age, Charles River, Wilmington, MA) were deeply anesthetized with intramuscular ketamine (100 mg/kg) and intraperitoneal xylazine (15 mg/kg). A complete laminectomy was performed, and the dorsal part of spinal cord was fully exposed at levels T6 and T7 (GrandPre et al., 2002). A dorsal overhemisection was performed at T6 with a 30 gauge needle and a pair of microscissors to completely sever the dorsal and dorsolateral corticospinal tracts (CSTs). The depth of the lesion (1.0 mm) was ensured by passing the marked needle several times across the dorsal part of the spinal cord. The muscle layers over the laminectomies were sutured, and the skin on the back was closed with surgical staples. To trace the CSTs, a burr hole overlying the cerebral cortex on the right side was made into the skull. Biotin dextran amine [BDA; molecular weight (MW), 10,000; 10% in PBS] (Molecular Probes, Eugene, OR) was applied at four injection sites at a depth of 0.7 mm from the cortical surface. For the animals receiving treatment 7 d after SCI, the BDA injection was conducted on day 28 after spinal cord lesion.
NgR antagonist peptide administration. The NgR antagonist NEP1–40 peptide (acetyl-RI YKGVIQAIQKSDEGHPFRAYLESEVAISEELVQKYSNS-amide) was synthesized (GrandPre et al., 2002). We tested eight groups of animals (10–12 animals per group): four groups with subcutaneous injections starting at the time of initial injury, two with intraperitoneal injections given 3–4 hr after injury, and two with more delayed subcutaneous injections starting 7 d after SCI. In the animals treated immediately after initial injury, an Alzet osmotic minipump (model 2002, Alza, Mountain View, CA) was implanted after the hemisection of dorsal spinal cord and positioned to deliver reagents to the subcutaneous space. The pump was filled with vehicle (83% PBS plus 17% DMSO) or NEP1–40 in the vehicle. The vehicle or NEP1–40 was delivered continuously at a rate of 0.6 μl/hr for 14 d. NEP1–40 was administered at doses of 1.2, 3.9, and 11.6 mg · kg−1 · d−1. For the animals receiving 3 hr delayed treatment, the first injection of vehicle or vehicle plus peptide (11.6 mg · kg−1 · d−1) was administered intraperitoneally 3–4 hr after spinal cord lesion, and the same dose of peptide was given once daily for another 14 d. For the mice receiving posttraumatic treatment that was further delayed, the minipump (Alzet model 2002) was implanted subcutaneously 7 d after hemisection to deliver vehicle or vehicle plus NEP1–40 (11.6 mg · kg−1 · d−1) continuously for 2 weeks. In this study, 91 mice underwent the procedure: 34 received vehicle and 57 received NEP1–40.The mortality rate was 19.8% during the first postoperative week and did not differ significantly between groups.
Histology and behavioral analysis. Animals starting treatment at the time of SCI or 3 hr after SCI were perfused transcardially 20 d after injury with PBS, followed by 4% paraformaldehyde. For the animals receiving treatment 7 d after SCI, the perfusion was performed on day 42 after the hemisection injury. The spinal cord was postfixed overnight. The spinal cord 4 mm rostral and 4 mm caudal to the lesion site (8 mm long) was embedded in a glutaraldehyde-polymerized albumin matrix and cut parasagittally on a vibratome (30μm thick). Transverse sections (50μm) were collected from the spinal cord 5–7 mm rostral and 5–7 mm caudal to the injury site. The freefloating sections were preincubated with 0.5% BSA/TBS for 1 hr and then processed with avidin-HRP (Vector Laboratories, Burlingame, CA), followed by a nickel-enhanced diaminobenzidine reaction (GrandPre et al., 2002). To visualize the lesion area, some sections were double-stained with antibodies directed against myelin basic protein (MBP; Sternberger Monoclonals, Lutherville, MD). The sections were mounted, dehydrated, and covered with mounting medium. Some transverse sections were immunostained with antibodies against SPRR1A (Bonilla et al., 2002), and 5-HT (Diasorin, Stillwater, MN). For other transverse sections 8–9 mm caudal to the lesion, tracer BDA was visualized with a streptavidin-Alexa Fluor 594 conjugate (Molecular Probes), and these sections were double-stained with primary antibodies against synaptophysin (Sigma, St. Louis, MO) or MBP. Double-stained sections were analyzed by confocal microscopy. For axon quantification, sections from the first five to seven animals processed were analyzed for each treatment group.
The Basso–Beattie–Bresnahan (BBB) locomotor rating scale (Basso et al., 1996), footprint analysis, and inclined grid walking were tested by two individuals. For footprint analysis, the walking patterns of mouse hindpaws were recorded with ink during a continuous locomotion across a 50 cm runway, and stride length on each side and strike width were calculated (Metz et al., 2000). For inclined grid walking, the mice were trained to climb a wire grid (35 cm long with 2.54 cm squares) at a slope of 45°. The number of instances in which the hindpaw dropped below the grid plane was scored for each excursion from bottom to top. Throughout the surgery, behavioral testing, and histologic analysis, researchers were unaware of the identity of the compound in the minipump or syringe.
Results
Systemic NEP1–40 results in extensive CST sprouting rostral to SCI
Previous studies have indicated that the blood–brain barrier is absent for weeks to months in an area of spinal cord trauma (Schnell et al., 1999). This suggested to us that systemic administration of the NEP1–40 peptide (MW, 4625) might be as efficacious as intrathecal treatment. We delivered NEP1–40 or vehicle subcutaneously to mice with midthoracic dorsal hemisection via an osmotic minipump implanted a few minutes after cord injury. Doses of 0, 1.2, 3.9, or 11.6 mg · kg−1 · d−1 were administered for the first 2 weeks of a 3 week survival period. These doses produced no evidence of overt toxicity. The integrity of descending CST was assessed by injecting BDA into the motor cortex on the right side. In the animals receiving vehicle injection, the prominent dorsal CST (dCST) is tightly bundled rostral to the lesion (Fig. 1A), and a few dorsolateral fibers are visible at the ipsilateral site. A few BDA-labeled collateral sprouts project into the gray matter, particularly in the ventral cord (Fig. 1B), but the sprouting is confined to the side of the cord contralateral to the tracer injection. In contrast, sections from injured animals treated with the NgR antagonist NEP1–40 exhibit an entirely different BDA labeling pattern. Rostral to the lesion site, a high density of fibers outside of the BDA-labeled dorsal CST (Fig. 1C,D) is present in all of the NEP1–40-treated samples. Ectopic fibers extend throughout the gray matter area, and some fibers reach into the lateral and dorsolateral white matter (Fig. 1C,D). A smaller number of fibers (5–10 sprouts per transverse section) are seen on the opposite side of the spinal cord in the gray matter (ipsilateral to the tracer injection site). The number of collateral sprouts is increased 10-fold in the NgR antagonist-treated animals at a dose of 11.6 mg · kg−1 · d−1 (Fig. 1G). The extent of axon growth is dose-dependent, with significant sprouting detectable at a dose of 1.2 mg · kg−1 · d−1. The degree of sprouting rostral to the lesion in mice treated subcutaneously compares favorably with that observed in rats treated intrathecally with NEP1–40 (GrandPre et al., 2002).
Figure 1.
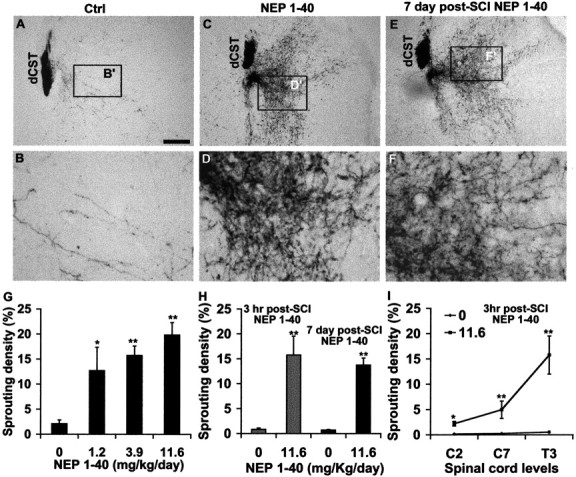
Systemic NEP1–40 promotes dCST sprouting in transverse sections rostral to the dorsal hemisection of the spinal cord. A, B, The BDA-labeled dorsal CST is visualized in a transverse section 5–7 mm rostral to the hemisection as a discrete bundle in a vehicle-treated animal. Several BDA-labeled fibers project into the gray matter area. Dorsal is up in this and all transverse sections.C,D,A transverse section 5–7 mm rostral to the hemisection from a NEP1–40-treated animal(subcutaneous administration via minipump) exhibits a high density of sprouts extending laterally into the gray matter, some of them into the white matter area.E,F,A transverse section 5–7 mm rostral to the hemisection from an animal receiving NEP1–40 treatment 7 d after hemisection (subcutaneous administration via minipump) exhibits an identical dCST sprouting pattern as for immediate peptide treatment. B′, D′, and F′ are shown at higher magnification in B, D, and F, respectively. G, The sprouting density of BDA-labeled fibers outside of the dorsal CST is illustrated for several doses of immediate subcutaneous NEP1–40 treatment mice. H, Bar graph indicates the sprouting density of BDA-labeled fibers outside of the dorsal CST from 3 hr intraperitoneal delayed treatment in mice (gray bars) and 7 d subcutaneous delayed administration in mice (black bars). I, The number of ectopic CST sprouts at the C2, C7, and T3 spinal levels from 3 hr intraperitoneal posttreatment animals. Means ± SEM from seven mice in each group are reported. Scale bar:(inA)A,C,E,100μm;B,D,F,25μm. The NEP1–40 values are statistically different from control. *p< 0.05; **p < 0.01; Student's t test.
Delayed NEP1–40 induces extensive axonal sprouting rostral to SCI
When subcutaneous infusions begin at the time of lesion, it is probable that hours to days elapse before peak intrathecal levels are achieved. Thus, it seems plausible that initiation of treatment during the first few hours is not critical. Moreover, the mechanism of NEP1–40 is thought to be dependent on the chronic blockade of axon growth inhibitors, and not on acute neuroprotection. For these reasons, we considered whether the peptide might promote axonal growth when administered in a therapeutic rather than a prophylactic time frame. Systemic NEP1–40 administration was delayed for 3–4 hr after the spinal cord hemisection. To avoid a second period of anesthesia and surgery, once-daily intraperitoneal injections of NEP1–40 were used rather than subcutaneous minipump infusion. This delayed systemic treatment produced the same degree and pattern of CST sprouting as did the immediate subcutaneous and intrathecal infusions (Fig. 1H, gray bars).
To consider the extent of this window of NEP1–40 responsiveness, we began vehicle or NgR antagonist peptide systemic administration 7 d after trauma. Treatment with a full week delay yielded a CST sprouting pattern at 42 d after injury that is remarkably similar to the NEP1–40 treatment immediately after injury at 20 d (Fig. 1E,F). Quantification of these sprouts revealed essentially equal degrees of BDA-labeled CST sprouting (Fig. 1H, black bar).
Examination of parasagittal longitudinal sections from 1 to 4 mm rostral to the lesion reveals that dCST fibers exhibit a large number of branching sprouts in the ventral gray matter area of the NEP1–40-treated animals (Fig. 2C–E) in contrast to the vehicle-treated group (Fig. 2A,B). Counts of such fibers show a >10-fold increase in sprouting density after peptide treatment (Fig. 2F). Sections from animals receiving 7 d delayed systemic NEP1–40 display a similar degree and pattern of CST sprouting (Fig. 2G–I). These findings suggest that systemic NEP1–40 treatment several hours or several days after injury induces almost the same sprouting response as does treatment immediately after injury.
Figure 2.
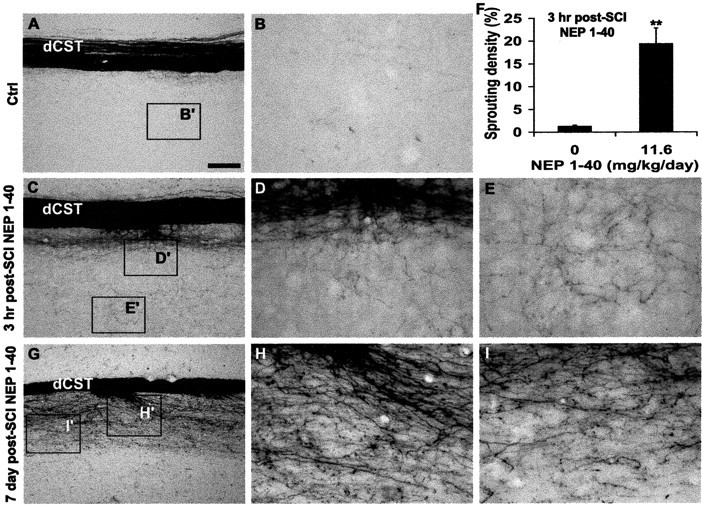
Delayed systemic NEP1–40 stimulates CST sprouting in rostral parasagittal sections. Sections 1–4 mm rostral to the dorsal hemisection of the spinal cord from the control group (A, B) reveal the longitudinal dCST (A) and few BDA-labeled fibers in the gray matter (B). Many collateral sprouts with a branching pattern are seen in the sections receiving intraperitoneal NEP1–40 treatment 3 hr after injury (C–E). Quantification of sprouting from longitudinal sections 1–4 mm rostral to spinal cord lesion indicates that the sprouting density of dorsal CST in the animals receiving NEP1–40 3 hr after treatment is more than 10 times greater than that in the time-matched controls (F). Longitudinal sections (G–I) from mice receiving NEP1–40 treatment 7 d after hemisection illustrate a similar spouting pattern as for 3 hr delayed drug delivery. In all the sagittal sections rostral is to the left and dorsalisup.B′,D′,E′,I′, and H′ are shown at higher magnification in B,D,E,I, and H, respectively. Scale bar: (inA) A,C,G,100μm; B, D, E, H, I,25μm. Means ± SEM from seven mice in each group are reported. The NEP1–40 values are statistically different from control. **p < 0.01; Student's t test.
Systemic NEP1–40 is expected to access the CNS and promote sprouting because of the increased permeability in and around the SCI site. To assess the distribution of NEP1–40 effects, we examined sections ranging from the level of the injury rostral to the cervicomedullary junction. The degree of ectopic sprouting is graded along the neuraxis, with the greatest sprouting closer to the lesion site (Fig. 1I). It may be that this distribution is attributable to increased NEP1–40 access near the injury and/or the proximity of the site of sprouting to the axotomy itself. Nonetheless, it is clear that the effects of the systemic NEP1–40 are significant >20 mm rostral to the injury itself.
CST axonal sprouting induced by systemic NEP1–40 crosses the lesion site into the distal spinal cord
Delayed systemic injection of the NgR antagonist peptide induces the rostral CST sprouting. But do these fibers cross the lesion site into the caudal spinal cord to provide functional recovery? Parasagittal sections from vehicle-treated mice show no CST fibers extending beyond the injury site (Fig. 3A,B). Similar sections from delayed systemic NEP1–40-treated mice contain numerous CST fibers that cross the lesion and project into the distal gray and white matter areas in a highly branched pattern (Fig. 3C–F). Immunohistochemical localization of MBP demonstrates that the interruption of myelinated tracts extends from the dorsal surface past the central canal (Fig. 3C, inset). In the vicinity of the lesion, the course of these fibers was typically tortuous (Fig. 3D–F, see Fig. 5B) and quite distinct from the normal straight fibers in the rostral CST. Collaterals and arborized fibers are most frequently seen in the gray matter area. Reconstructions of consecutive sections as camera lucida drawings display the overall distribution pattern of the regenerating CST fibers several millimeters from the lesion (Fig. 4). In control animals, no CST axons extend past the lesion (Fig. 4 A). In the animals treated with systemic NEP1–40 after a 3 hr delay (Fig. 4B–D), some of the regenerating axons bypass the lesion site through remaining tissue bridges of the ventral and ventrolateral gray and white matter, whereas other CST fibers cross the transection area via the lesioned dorsal and dorsolateral part of the spinal cord into distal regions. Notably, in the mice treated with systemic NEP1–40 7 d after SCI, a similar regeneration pattern is seen as in animals treated 3 hr after injury (Fig. 4 E).
Figure 3.
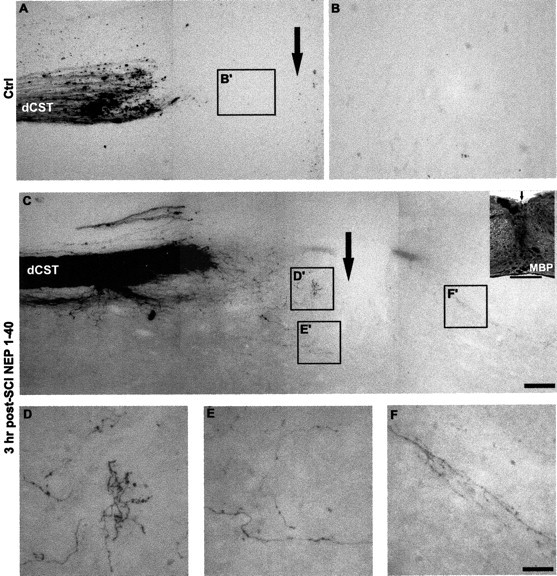
Systemic NEP1–40 promotes dCST growth through the SCI site. A,B,A parasagittal section containing the transection site (arrow) from a vehicle-treated animal demonstrates the transection of BDA-labeled dCST fibers at the injury site. No fibers extend into the area of the trauma or beyond it. C, A section from an animal receiving intraperitoneal NEP1–40 demonstrates a similar degree of dorsal hemisection injury, but numerous branched, sprouting fibers are observed in the areas around the transection site. The higher magnifications of these areas in D′–F′ illustrate the meandering course of the regenerating CST fibers in the NEP1–40-treated animals (D–F). The inset in C immunostained for MBP illustrates the disruption of staining in the hemisection site. Scale bars: (in C) A, C, 100 μm (A, C); (in F) B, D–F, 25 μm; inset, 500 μm.
Figure 5.
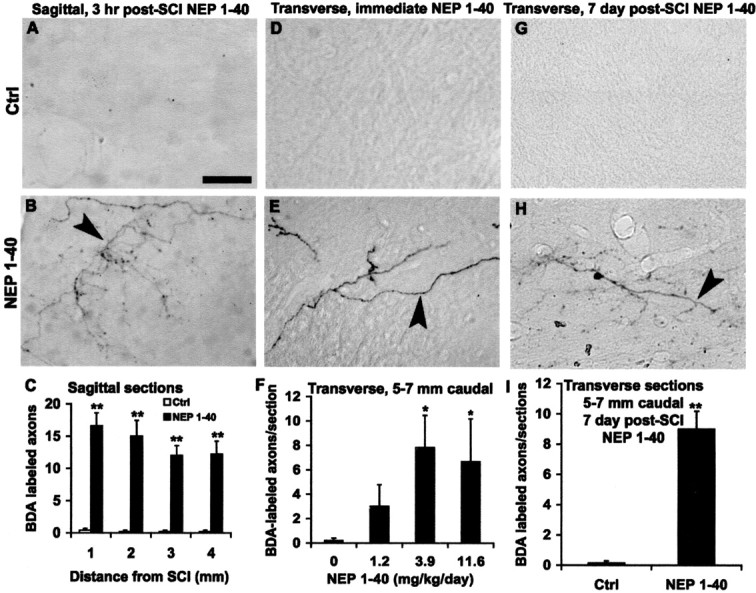
Systemic NgR antagonist peptide induces numerous distal regenerating CST fibers. A, B, Parasagittal sections 1–4 mm distal to the hemisection display the numerous branching fibers (arrowhead) in the peptide-treated mice (B), in contrast to no fibers in the control sections (A). C, Quantification of CST fibers is illustrated at various distances caudal to the injury site for the group given treatment 3 hr after trauma. D, E, Transverse sections at a level 5–7 mm caudal to the lesion illustrate no fibers in the gray matter of the dorsal spinal cord from the vehicle-treated animals (D), but a large number of fibers with branching patterns in the NEP1–40 group are seen (arrowhead) ventral to dorsal CST (E). F, CST fiber counts at a level of 5–7 mm caudal to the lesion from transverse sections indicate a greater number in NEP1–40-treated groups (subcutaneous application) than in the control group. G, H, Transverse sections 5–7 mm caudal to the lesion show a number of BDA-labeled fibers (H, arrowhead) from mice treated with NEP1–40 7 d after SCI delivery, but no fibers in the sections from the time-matched vehicle-treated animals (G). I, Graph shows the number of BDA-labeled CST fibers 5–7 mm distal to hemisection from animals receiving NEP1-40 treatment 7 d after injury. Scale bar, 25 μm. In all the graphs, means ± SEM from seven mice in each group are reported. The NEP1–40 values are statistically different from the control. *p < 0.05; **p < 0.01; Student's t test.
Figure 4.
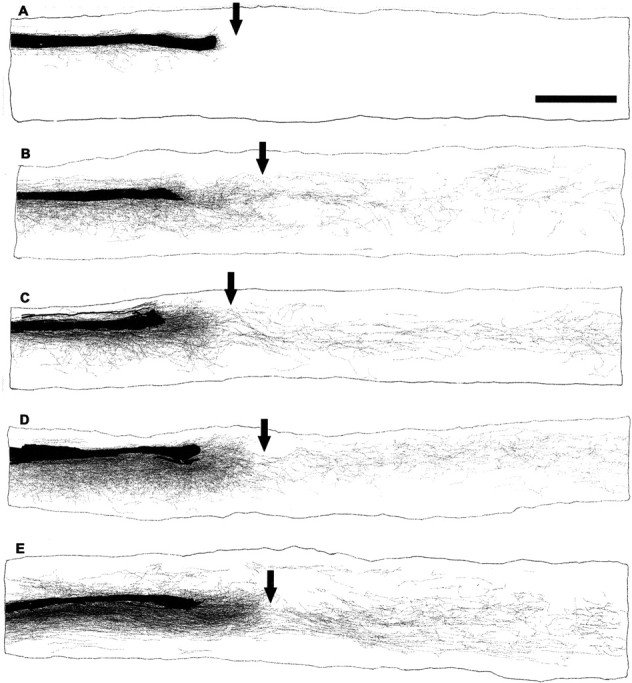
Camera lucida reconstructions of consecutive parasagittal sections around the lesion site. The injury site is indicated by an arrow. Control animals show no regenerative growth (A). In contrast, NEP1–40 treatment 3 hr (B–D) or 7 d (E) after trauma induces a high density of sprouting of the lesioned dorsal CST fibers. Some of these regenerated fibers grow around and through the lesion, and project into the distal spinal cord, particularly into the gray matter areas. Scale bar, 1 mm.
The reconstructions demonstrate 9–24 BDA-labeled regenerated fibers coursing in the rostrocaudal axis at any one level between 1 and 4 mm caudal to the lesion in each NEP1–40-treated mouse (Fig. 5C). These sections demonstrate that axons sprout for millimeters beyond the lesion site in the presence of NEP1–40. For transverse sections 5–7 mm below the injury site, NEP1–40 dose-dependently increases the number of CST fibers (Fig. 5D–F). The fiber counts for the systemic NEP1–40-treated animals are approximately half of those at more proximal levels in the sagittal sections. In the more delayed NEP1–40-treated mice (NEP1-40 delivery 7 d after injury), a similar pattern and degree of CST regeneration is observed in sections 5–7 mm distal to hemisection (Fig. 5G–I).
For regenerating CST fibers to contribute to functional recovery, they must form synapses in the caudal spinal cord. Anatomically, synapse formation by CST fibers can be assessed by examining the colocalization of BDA-positive varicosities with synaptophysin. Although ventral fibers are not severed in this model and might theoretically produce such labeling, no double-labeling was observed in over 100 such lumbar sections from untreated SCI mice. This is because of the extremely rare occurrence of ventral spared CST fibers in this mouse model, as opposed to a rat model (Weidner et al., 2001). It is most likely that the easily identified BDA/synaptophysin double-labeling in the NEP1–40-treated samples (Fig. 6A) are associated with fibers regenerating under the influence of the peptide treatment and are consistent with synapse formation. Not surprisingly, these caudal sprouting CST fibers are unmyelinated at this point, 3 weeks after injury (Fig. 6B).
Figure 6.

Synaptophysin and MBP distribution relative to caudal CST fibers in NEP1–40-treated mice. A, Confocal imaging displays the double-staining for BDA-labeled regenerating CST fibers (red) and synaptophysin (green) in the ventral horn of transverse spinal cord at a level of 8–9 mm caudal to the lesion site from NEP1–40-treated mice. Note the localization of synaptophysin to varicosities (arrowheads) of BDA-positive fibers in the lumbar level of spinal cord.B, Transverse section of ventral gray matter 8–9 mm caudal to the hemisection stained for BDA (red) and MBP (green) illustrates that NEP1–40-induced CST fibers in the distal spinal cord are not myelinated 20 d after SCI. Scale bars, 25 μm.
NEP1–40 induces SPRR1A expression and serotonergic fiber sprouting after dorsal spinal cord hemisection
Evidence for axonal regeneration by systemic NEP1–40 treatment can be detected by molecular as well as anatomical methods. One molecular correlate of axonal regeneration is the induction of “growth-associated proteins,” such as SPRR1A (Bonilla et al., 2002). Although peripheral nerve injury induces SPRR1A dramatically, central injury such as SCI does not do so (Bonilla et al., 2002). The combination of systemic NEP1–40 treatment and central axotomy leads to the induction of SPRR1A expression in spinal interneurons and motoneurons both rostral and caudal to the injury (Fig. 7A,B). Numerous SPRR1A-positive axons are also detected in the NEP1–40-treated injured spinal cords (data not shown). In addition to providing additional evidence for NEP1–40-induced axon regeneration, these data suggest that NgR activation may limit axon growth both by acute inhibitory effects on axon growth and by chronic suppression of gene expression patterns associated with successful regeneration. Because SPRR1A expression was not apparent in the motor cortex or in CST fibers (data not shown), this mechanism does not appear to be essential for NEP1–40-induced sprouting after axotomy.
Figure 7.
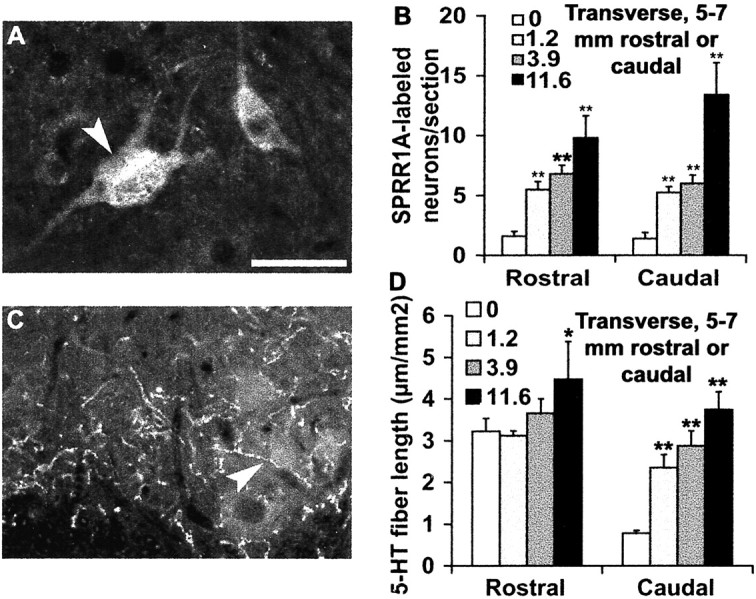
Systemic administration of NEP1–40 induces an upregulation of SPRR1A and distal serotonin fiber sprouting. A, A ventral horn transverse section from 5–7 mm caudal to the lesion site displays the positive neurons (arrowhead) stained with regeneration-related protein SPRR1A from NEP1–40-treated animals. B, SPRR1A-positive neuronal counts at a level of 5–7 mm rostral or 5–7 mm caudal to the lesion demonstrate a significant increase in the number of immunoreactive cells in the NEP1–40-treated groups. C, A transverse section of ventral horn 5–7 mm distal to the hemisection illustrates the serotonin fibers stained with antibody against 5-HT (arrowhead). D, Immunoreactive serotonin fiber length in the ventralhorn at a level of 5–7 mm rostral or 5–7 mm caudal to injury site was measured. The means ± SEM from seven mice in each group are reported. The NEP1–40 values are statistically different from the control. *p < 0.05; **p < 0.01; Student's t test. Scale bar: (in A) A, C, 25 μm.
Both descending raphespinal and CST fibers contribute to locomotor function. Dorsal hemisection injures a majority of the serotonergic fibers, decreasing the density of these fibers by >75% in the ventral horn in this mouse SCI model (Fig. 7D). Systemic NEP1–40 dose-dependently restores serotonin fiber density in the ventral horn of the caudal spinal cord to the levels present before injury (Fig. 7C). Thus, the growth-promoting effects of NEP1–40 treatment are not limited to one axonal pathway.
Systemic treatment with NgR antagonist peptide improves locomotor function
Do mice treated with systemic NEP1–40 walk better than control animals after injury? A standardized rat locomotor scoring system was used to assess outcome (Basso et al., 1996). A dose-dependent benefit of systemic NEP1–40 treatment was detectable by 17 d after injury (Fig. 8A). The intermediate dose was nearly as effective as the maximal dose of 11.6 mg · kg−1 · d−1. Although less than half of the vehicle-treated mice (4 of 9) were capable of weight-bearing on the hindlimbs at 17 d after injury, all (14 of 14) mice treated with 3.9 or 11.6 mg · kg−1 · d−1 were weight-bearing at this stage. In contrast to rats treated intrathecally with NEP1–40, no benefit of NEP1–40 was detectable at the earliest stages after injury (GrandPre et al., 2002). This may reflect a more delayed time for systemic NEP1–40 to reach therapeutic levels in the spinal cord.
Figure 8.

Systemic NgR antagonist peptide improves functional recovery after dorsal hemisection. A, The locomotor BBB score is reported as a function of time after dorsal hemisection in the vehicle-or NEP1–40-treated animals initiated at the time of SCI. B, The BBB score is plotted as a function of time after SCI in control or NEP1–40 animals receiving treatment 7 d after trauma. C, Two examples of the representative footprints from control (top) or NEP1–40 (bottom) animals treated 7 d after SCI are shown. The double-headed arrows indicate strike length for one step. D, Footprint analysis reveals a shorter stride length in control mice than uninjured or injured + NEP1–40 mice(treatment 7 d after injury), where as no differences are found in the stride width among the three groups. E, Hindlimb errors during inclined grid climbing are reported as a function of post-SCI time in the control or NEP1–40-treated groups (treatment after 7 d). In all the graphs, means± SEM from seven mice in each group are reported. The NEP1–40 values are statistically different from the control in A, B, and E. The control values are statistically different from no-SCI or SCI + NEP1–40 mice in D. *p < 0.05; **p < 0.01; Student's t test.
Our CST tracing experiments indicate that the delayed blockade of NgR with systemic NEP1–40 1 week after axotomy stimulates extensive anatomical regeneration of injured spinal cord descending axons. To determine whether this therapeutic strategy benefits late functional recovery, we examined a battery of behavioral tests. The locomotor BBB score in control animals reaches a stable level of 13 at 3 weeks after the hemisection (Fig. 8B). The slight differences of BBB recovery between these controls and the controls receiving vehicle treatment at the time of injury (Fig. 8A) may be attributable to the toxic effects of systemic DMSO (Cavaletti et al., 2000). At 6 weeks after SCI, all the control mice have frequent, consistent weight-supported plantar steps, but most of them (5 of 7) have only occasional forelimb–hindlimb coordination, with rotation of the predominant paw position when making initial contact with a surface. In contrast, the locomotor score of the delayed systemic NEP1–40-treated mice continues to increase between 2 and 6 weeks after trauma. At 6 weeks after injury, most of the delayed NEP1–40-treated animals (8 of 9) have consistent forelimb–hindlimb coordination and parallel paw position at initial contact with the testing surface.
Two other behavioral tests were used to additionally characterize the performance of the delayed systemic NEP1–40-treated mice. Analysis of hindpaw footprints in vehicle-treated mice demonstrates that stride length is significantly decreased at 6 weeks after SCI, but the stance width is close to normal. For the NEP1–40 mice, the stride length at 6 weeks after injury is close to that of uninjured mice (Fig. 8C,D). In a second test, mice climbed a grid placed at a 45° angle to vertical, and excursions of the hindlimbs below the plane of the grid were counted (Metz et al., 2000). Uninjured mice make no errors on this test (data not shown). In control mice, there are numerous errors, with only minimal improvement between 2 and 6 weeks after injury. In contrast, mice treated systemically with NgR antagonist peptide exhibit a progressive improvement in grid climbing during this period, with the majority of the improvement occurring between 2 and 4 weeks after injury (Fig. 8E).
Discussion
Encouraging progress toward promoting axonal regeneration in the damaged spinal cord has been achieved recently by a variety of methods. Suppression of the action of myelin-derived growth inhibitors (Bregman et al., 1995; Thallmair et al., 1998; GrandPre et al., 2002), reduction of the scar-issue barrier (Bradbury et al., 2002), enhancement of the growth potential of neurons (Bomze et al., 2001; Dergham et al., 2002; Neumann et al., 2002; Qiu et al., 2002), replacement of cells (Li et al., 1997; Ramon-Cueto et al., 2000; Takami et al., 2002), and supplementation with neurotrophic factors (Schnell et al., 1994; Coumans et al., 2001) have been shown to stimulate axonal regrowth and/or functional recovery of injured spinal cord. Nevertheless, these therapies have been administered locally and initiated at the time of spinal cord lesions in most instances. From a clinical point of view, such immediate local therapy may pose logistical problems for the treatment of most human SCIs. In the present study, we show that subcutaneous or intraperitoneal treatment with NgR antagonist peptide induces extensive axonal sprouting and significantly improves locomotive functional recovery. It is likely that NEP1–40 enters the spinal cord through a focally disrupted blood–brain barrier at the SCI site. To our knowledge, this is the first report to describe a systemic therapy to promote axonal regeneration after CNS trauma.
We also show that delayed NEP1–40 treatment stimulates axonal sprouting and promotes locomotor recovery. The therapeutic window for NgR antagonist treatment of subacute SCI is at least 1 week in these studies. At this time point, there is no decrease in the degree of axonal sprouting compared with immediate therapy with NEP1–40. Whether responsiveness declines after several weeks or months in chronic SCI remains to be examined. It is clear that the inherent regenerative capacity of lesioned cortical motor neurons persists at least into the subacute stage of axotomy. In this regard, our results are consistent with the previous reports showing axonal regrowth in chronic injury treated with combinations of neurotrophic factors, fetal spinal cord transplantation, and IN1 antibody (Ye and Houle, 1997; von Meyenburg et al., 1998; Coumans et al., 2001). Our findings demonstrate an extended therapeutic window for the systemic treatment of SCI with Nogo receptor antagonists.
NEP1–40 was identified as an NgR competitive antagonist (GrandPre et al., 2002), and this antagonism is the most probable in vivo mode of action. Several factors argue against an alternative mechanism based on a beneficial immune response to NEP1–40 administration resembling that described for anti-Nogo or antimyelin antibodies (Bregman et al., 1995; Thallmair et al., 1998; Huang et al., 1999). First, the peptide was administered as a lowmolecular-weight soluble compound without adjuvant; therefore, it is highly unlikely to incite an antibody response. Second, immunization studies with a glutathione S-transferase–Nogo-66 protein containing the NEP1–40 sequence in the presence of adjuvant yield no detectable antibody titer to Nogo and no enhanced axonal regenerative response after SCI (S. M. Strittmatter, unpublished observation). Third, a much lower dose of intrathecal NEP1–40 produces an outcome essentially identical to that of systemic NEP1–40 (GrandPre et al., 2002).
SCI is a relatively specific model for studying CNS axon regeneration because the majority of the SCI-related anatomical and functional deficit is attributable to axonal interruption. Several SCI models, including contusions, compressions, and transections (incomplete or complete), have been used for examining experimental treatments. Contusion or compression injuries are widely accepted because they resemble the complex lesions of many clinical SCI cases. Because such models usually produce central gray matter necrosis with variable white matter damage, the discreteness of the anatomical result and its correlation with physiological performance is limited. In contrast, incomplete spinal cord transection, such as dorsal hemitransection, interrupts a group of defined pathways, making it easier to investigate regeneration-promoting therapy (Huang et al., 1999; Brosamle et al., 2000; Coumans et al., 2001; Qiu et al., 2002). Moreover, this model has been used frequently for studying the regeneration-related behavioral alterations (Huang et al., 1999; Bradbury et al., 2002; Dergham et al., 2002; Schucht et al., 2002). Such spinal injuries produce less scar tissue and tissue cavitation in mice than in larger mammals (Steward et al., 1999). Because the primary target of NEP1–40 treatment is myelin-derived inhibition rather than scar-derived inhibition of axon regeneration, the benefits may be more pronounced in mice than in other species. From the current study, it is clear that systemic NEP1–40 treatment induces dramatic improvements in mouse locomotor scores. When administered during the first 2 weeks, NgR antagonist treatment induces a return to weight-bearing on the hindlimbs (BBB score, >9) that is twice as rapid as in control mice. When the NgR antagonist is administered during the 1–3 week period after SCI, significant and persistent locomotor improvements in the 2–6 week period are observed in several measures. Scores of hindlimb coordination and placement are improved compared with vehicle-treated animals (BBB score), stride length returns to that of uninjured mice, and the ability to climb a grid is close to that of uninjured animals.
After SCI, the extent of motor function improvement depends on both the reorganization of segmental circuitry and the restoration of supraspinal input. Our morphologic studies are consistent with previous studies (Bregman et al., 1993) in demonstrating that segmental mechanisms play a major role in the locomotor recovery of spinally injured rodents under control conditions, because no CST regrowth is detected. In principle, enhanced plasticity at the segmental level might contribute to functional improvement in the NgR antagonist-treated mice. However, we have no anatomical data to support this hypothesis. There are several lines of evidence indicating that the restoration of supraspinal input to the lumbar cord participates in the functional improvement. First, BDA traces regenerated fibers across the lesion site and into the caudal part of the spinal cord after NgR antagonist treatment. For example, at 1–4 mm caudal to the transection site, 9–24 regenerated fibers were seen in each NEP1–40-treated mice. Near the lesion site, some regenerating dCST fibers circumvent the lesion through the ventral part of gray or white matter of the cord, whereas others course through the lesioned dorsal or lateral spinal cord. Even at 5–7 mm distal to SCI, 5–18 BDA-labeled fibers are detectable in transverse sections of NEP1–40-treated mice. The total number of regenerated axons may be much greater than that observed here, because this anterograde BDA tracing method labels only a fraction of total CST fibers (Brosamle and Schwab, 1997). Second, the synaptic marker synaptophysin is localized to the varicosities of BDA-labeled axons at the lumbar levels of spinal cord, demonstrating anatomical reconnection between sprouting CST fibers and distal spinal cord neurons. On a numerical basis, the camera lucida reconstructions suggest that a majority of such distal CST fibers in systemic NEP1–40-treated mice derive from sprouted dCST fibers. However, it remains plausible that distal sprouts from the rare ventral CST (vCST) fibers are also involved in the synaptic reorganization after NEP1–40 application. Spontaneous structural plasticity in vCST is known to correlate with rat functional recovery after SCI (Weidner et al., 2001). Third, the serotonergic axon growth induced by peptide NEP1–40 demonstrates that NgR antagonism can promote regeneration in multiple descending pathways that may play a role in locomotor recovery. The widespread neuronal distribution of the NgR (Wang et al., 2002c) makes it likely that additional fiber systems are responsive to NEP1–40 and contribute to the improved performance of spinal cord-injured mice after NEP1–40 treatment. In particular, the sprouting of injured rubrospinal fiber may also contribute to functional improvements. Similarly, the reorganization of spared pathways, such as descending ventrolateral fibers (Brustein and Rossignol, 1998), may also contribute to functional recovery (Raineteau et al., 2001). Together, the CST tracing, the 5-HT staining and the BDA/synaptophysin colocalization indicate the restoration of supraspinal inputs plays a significant role in the pronounced functional recovery after systemic NgR antagonist treatment.
The present study reveals that systemic NgR antagonist peptide stimulates anatomical sprouting and caudal growth of both CST and raphespinal fibers, and induces upregulation of growth-associated proteins in the spinal cord. Importantly, systemic administration of NgR antagonist, even applied in the subacute/chronic stage, yields a concomitant improvement in locomotor function. The ability of systemic NEP1–40 treatment in mice to match intrathecal effects in rats supports the notion that the blood–brain barrier breakdown after trauma provides a therapeutic window for the administration of regeneration-promoting reagents that might not otherwise gain access to the CNS. It is clear that subacute traumatic CNS axonal injuries can be successfully treated in mice by the delayed systemic administration of an NgR antagonist. The NEP1–40 peptide selectively blocks Nogo-66 action (GrandPre et al., 2002) but not myelin-associated glycoprotein action (Liu et al., 2002) at the NgR. Thus, more general NgR antagonists might be even more effective systemic therapeutics for promoting recovery after adult CNS axotomies such as SCI.
Footnotes
This work was supported by grants from the Christopher Reeve Paralysis Foundation (S.L.) and from the National Institutes of Health, the McKnight Foundation for Neuroscience, and Biogen, Inc.(S.M.S.).S.M.S.is an Investigator of the Patrick and Catherine Weldon Donaghue Medical Research Foundation. We thank Yiguang Fu for expert technical assistance.
Correspondence should be addressed to Dr. Stephen M. Strittmatter, Department of Neurology and Section of Neurobiology, Yale University School of Medicine, P.O. Box 208018, New Haven, CT 06520. E-mail: stephen.strittmatter@yale.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/234219-09$15.00/0
References
- Bartsch U, Bandtlow CE, Schnell L, Bartsch S, Spillmann AA, Rubin BP, Hillenbrand R, Montag D, Schwab ME, Schachner M ( 1995) Lack of evidence that myelin-associated glycoprotein is a major inhibitor of axonal regeneration in the CNS. Neuron 15: 1375–1381. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC, Anderson DK, Faden AI, Gruner JA, Holford TR, Hsu CY, Noble LJ, Nockels R, Perot PL, Salzman SK, Young W ( 1996) MASCIS evaluation of open field locomotor scores: effects of experience and teamwork on reliability. Multicenter Animal Spinal Cord Injury Study. J Neurotrauma 13: 343–359. [DOI] [PubMed] [Google Scholar]
- Bomze HM, Bulsara KR, Iskandar BJ, Caroni P, Skene JH ( 2001) Spinal axon regeneration evoked by replacing two growth cone proteins in adult neurons. Nat Neurosci 4: 38–43. [DOI] [PubMed] [Google Scholar]
- Bonilla IE, Tanabe K, Strittmatter SM ( 2002) Small proline-rich repeat protein 1A is expressed by axotomized neurons and promotes axonal outgrowth. J Neurosci 22: 1303–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB ( 2002) Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature 416: 636–640. [DOI] [PubMed] [Google Scholar]
- Bregman BS, Kunkel-Bagden E, Reier PJ, Dai HN, McAtee M, Gao D ( 1993) Recovery of function after spinal cord injury: mechanisms underlying transplant-mediated recovery of function differ after spinal cord injury in newborn and adult rats. Exp Neurol 123: 3–16. [DOI] [PubMed] [Google Scholar]
- Bregman BS, Kunkel-Bagden E, Schnell L, Dai HN, Gao D, Schwab ME ( 1995) Recovery from spinal cord injury mediated by antibodies to neurite growth inhibitors. Nature 378: 498–501. [DOI] [PubMed] [Google Scholar]
- Brosamle C, Schwab ME ( 1997) Cells of origin, course, and termination patterns of the ventral, uncrossed component of the mature rat corticospinal tract. J Comp Neurol 386: 293–303. [DOI] [PubMed] [Google Scholar]
- Brosamle C, Huber AB, Fiedler M, Skerra A, Schwab ME ( 2000) Regeneration of lesioned corticospinal tract fibers in the adult rat induced by a recombinant, humanized IN-1 antibody fragment. J Neurosci 20: 8061–8068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustein E, Rossignol S ( 1998) Recovery of locomotion after ventral and ventrolateral spinal lesions in the cat: I. Deficits and adaptive mechanisms. J Neurophysiol 80: 1245–1267. [DOI] [PubMed] [Google Scholar]
- Cavaletti G, Oggioni N, Sala F, Pezzoni G, Cavalletti E, Marmiroli P, Petruccioli MG, Frattola L, Tredici G ( 2000) Effect on the peripheral nervous system of systemically administered dimethylsulfoxide in the rat: a neurophysiological and pathological study. Toxicol Lett 118: 103–107. [DOI] [PubMed] [Google Scholar]
- Chen MS, Huber AB, van der Haar ME, Frank M, Schnell L, Spillmann AA, Christ F, Schwab ME ( 2000) Nogo-A is a myelin-associated neurite outgrowth inhibitor and an antigen for monoclonal antibody IN-1. Nature 403: 434–439. [DOI] [PubMed] [Google Scholar]
- Coumans JV, Lin TT, Dai HN, MacArthur L, McAtee M, Nash C, Bregman BS ( 2001) Axonal regeneration and functional recovery after complete spinal cord transection in rats by delayed treatment with transplants and neurotrophins. J Neurosci 21: 9334–9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SJ, Goucher DR, Doller C, Silver J ( 1999) Robust regeneration of adult sensory axons in degenerating white matter of the adult rat spinal cord. J Neurosci 19: 5810–5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L ( 2002) Rho signaling pathway targeted to promote spinal cord repair. J Neurosci 22: 6570–6577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domeniconi M, Cao Z, Spencer T, Sivasankaran R, Wang K, Nikulina E, Kimura N, Cai H, Deng K, Gao Y, He Z, Filbin M ( 2002) Myelin-associated glycoprotein interacts with the nogo66 receptor to inhibit neurite outgrowth. Neuron 35: 283. [DOI] [PubMed] [Google Scholar]
- Dou CL, Levine JM ( 1994) Inhibition of neurite growth by the NG2 chondroitin sulfate proteoglycan. J Neurosci 14: 7616–7628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier AE, GrandPre T, Strittmatter SM ( 2001) Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature 409: 341–346. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Klassen MP, Hua Y, Barres BA ( 2002) Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science 296: 1860–1864. [DOI] [PubMed] [Google Scholar]
- GrandPre T, Nakamura F, Vartanian T, Strittmatter SM ( 2000) Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature 403: 439–444. [DOI] [PubMed] [Google Scholar]
- GrandPre T, Li S, Strittmatter SM ( 2002) Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature 417: 547–551. [DOI] [PubMed] [Google Scholar]
- Huang DW, McKerracher L, Braun PE, David S ( 1999) A therapeutic vaccine approach to stimulate axon regeneration in the adult mammalian spinal cord. Neuron 24: 639–647. [DOI] [PubMed] [Google Scholar]
- Li Y, Field PM, Raisman G ( 1997) Repair of adult rat corticospinal tract by transplants of olfactory ensheathing cells. Science 277: 2000–2002. [DOI] [PubMed] [Google Scholar]
- Liu BP, Fournier A, GrandPre T, Strittmatter SM ( 2002) Myelin-associated glycoprotein as a functional ligand for the nogo-66 receptor. Science 297: 1190–1193. [DOI] [PubMed] [Google Scholar]
- Liu RY, Snider WD ( 2001) Different signaling pathways mediate regenerative versus developmental sensory axon growth. J Neurosci 21: RC164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerracher L, David S, Jackson DL, Kottis V, Dunn RJ, Braun PE ( 1994) Identification of myelin-associated glycoprotein as a major myelin-derived inhibitor of neurite growth. Neuron 13: 805–811. [DOI] [PubMed] [Google Scholar]
- Metz GA, Merkler D, Dietz V, Schwab ME, Fouad K ( 2000) Efficient testing of motor function in spinal cord injured rats. Brain Res 883: 165–177. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT ( 1994) A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 13: 757–767. [DOI] [PubMed] [Google Scholar]
- Neumann S, Bradke F, Tessier-Lavigne M, Basbaum AI ( 2002) Regeneration of sensory axons within the injured spinal cord induced by intraganglionic cAMP elevation. Neuron 34: 885–893. [DOI] [PubMed] [Google Scholar]
- Prinjha R, Moore SE, Vinson M, Blake S, Morrow R, Christie G, Michalovich D, Simmons DL, Walsh FS ( 2000) Inhibitor of neurite outgrowth in humans. Nature 403: 383–384. [DOI] [PubMed] [Google Scholar]
- Qiu J, Cai D, Dai H, McAtee M, Hoffman PN, Bregman BS, Filbin MT ( 2002) Spinal axon regeneration induced by elevation of cyclic AMP. Neuron 34: 895–903. [DOI] [PubMed] [Google Scholar]
- Raineteau O, Fouad K, Noth P, Thallmair M, Schwab ME ( 2001) Functional switch between motor tracts in the presence of the mAb IN-1 in the adult rat. Proc Natl Acad Sci USA 98: 6929–6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramon-Cueto A, Cordero MI, Santos-Benito FF, Avila J ( 2000) Functional recovery of paraplegic rats and motor axon regeneration in their spinal cords by olfactory ensheathing glia. Neuron 25: 425–435. [DOI] [PubMed] [Google Scholar]
- Richardson PM, McGuinness UM, Aguayo AJ ( 1980) Axons from CNS neurons regenerate into PNS grafts. Nature 284: 264–265. [DOI] [PubMed] [Google Scholar]
- Savio T, Schwab ME ( 1990) Lesioned corticospinal tract axons regenerate in myelin-free rat spinal cord. Proc Natl Acad Sci USA 87: 4130–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer M, Fruttiger M, Montag D, Schachner M, Martini R ( 1996) Disruption of the gene for the myelin-associated glycoprotein improves axonal regrowth along myelin in C57BL/Wlds mice. Neuron 16: 1107–1113. [DOI] [PubMed] [Google Scholar]
- Schnell L, Schneider R, Kolbeck R, Barde YA, Schwab ME ( 1994) Neurotrophin-3 enhances sprouting of corticospinal tract during development and after adult spinal cord lesion. Nature 367: 170–173. [DOI] [PubMed] [Google Scholar]
- Schnell L, Fearn S, Klassen H, Schwab ME, Perry VH ( 1999) Acute inflammatory responses to mechanical lesions in the CNS: differences between brain and spinal cord. Eur J Neurosci 11: 3648–3658. [DOI] [PubMed] [Google Scholar]
- Schucht P, Raineteau O, Schwab ME, Fouad K ( 2002) Anatomical correlates of locomotor recovery following dorsal and ventral lesions of the rat spinal cord. Exp Neurol 176: 143–153. [DOI] [PubMed] [Google Scholar]
- Snow DM, Lemmon V, Carrino DA, Caplan AI, Silver J ( 1990) Sulfated proteoglycans in astroglial barriers inhibit neurite outgrowth in vitro. Exp Neurol 109: 111–130. [DOI] [PubMed] [Google Scholar]
- Steward O, Schauwecker PE, Guth L, Zhang Z, Fujiki M, Inman D, Wrathall J, Kempermann G, Gage FH, Saatman KE, Raghupathi R, McIntosh T ( 1999) Genetic approaches to neurotrauma research: opportunities and potential pitfalls of murine models. Exp Neurol 157: 19–42. [DOI] [PubMed] [Google Scholar]
- Takami T, Oudega M, Bates ML, Wood PM, Kleitman N, Bunge MB ( 2002) Schwann cell but not olfactory ensheathing glia transplants improve hindlimb locomotor performance in the moderately contused adult rat thoracic spinal cord. J Neurosci 22: 6670–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thallmair M, Metz GA, Z'Graggen WJ, Raineteau O, Kartje GL, Schwab ME ( 1998) Neurite growth inhibitors restrict plasticity and functional recovery following corticospinal tract lesions. Nat Neurosci 1: 124–131. [DOI] [PubMed] [Google Scholar]
- von Meyenburg J, Brosamle C, Metz GA, Schwab ME ( 1998) Regeneration and sprouting of chronically injured corticospinal tract fibers in adult rats promoted by NT-3 and the mAb IN-1, which neutralizes myelin-associated neurite growth inhibitors. Exp Neurol 154: 583–594. [DOI] [PubMed] [Google Scholar]
- Wang KC, Koprivica V, Kim JA, Sivasankaran R, Guo Y, Neve RL, He Z ( 2002a) Oligodendrocyte-myelin glycoprotein is a Nogo receptor ligand that inhibits neurite outgrowth. Nature 417: 941–944. [DOI] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z ( 2002b) P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420: 74–78. [DOI] [PubMed] [Google Scholar]
- Wang X, Chun SJ, Treloar H, Vartanian T, Greer CA, Strittmatter SM ( 2002) Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J Neurosci 22: 5505–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner N, Ner A, Salimi N, Tuszynski MH ( 2001) Spontaneous corticospinal axonal plasticity and functional recovery after adult central nervous system injury. Proc Natl Acad Sci USA 98: 3513–3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM ( 2002) A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci 5: 1302–1308. [DOI] [PubMed] [Google Scholar]
- Ye JH, Houle JD ( 1997) Treatment of the chronically injured spinal cord with neurotrophic factors can promote axonal regeneration from supraspinal neurons. Exp Neurol 143: 70–81. [DOI] [PubMed] [Google Scholar]