

Abstract
Cultured embryonic cortical progenitor cells will mimic the temporal differentiation pattern observed in vivo, producing neurons first and then glia. Here, we investigated the role of two endogenously produced growth factors, the neurotrophins brain-derived neurotrophic factor and neurotrophin-3 (NT-3), in the early progenitor-to-neuron transition. Cultured cortical progenitors express BDNF and NT-3, as well as their receptors TrkB (tyrosine kinase receptor B) and TrkC. Inhibition of these endogenously expressed neurotrophins using function-blocking antibodies resulted in a marked decrease in the survival of cortical progenitors, accompanied by decreased proliferation and inhibition of neurogenesis. Inhibition of neurotrophin function also suppressed the downstream Trk receptor signaling pathways, PI3-kinase (phosphatidyl inositol-3-kinase) and MEK—ERK (MAP kinase kinase—extracellular signal-regulated kinase), indicating the presence of autocrine—paracrine neurotrophin:Trk receptor signaling in these cells. Moreover, specific inhibition of these two Trk signaling pathways led to distinct biological effects; inhibition of PI3-kinase decreased progenitor cell survival, whereas inhibition of MEK selectively blocked the generation of neurons, with no effects on survival or proliferation. Thus, neurotrophins made by cortical progenitor cells themselves signal through the TrkB and TrkC receptors to mediate cortical progenitor cell survival and neurogenesis via two distinct downstream signaling pathways.
Keywords: MAPK, PI3-kinase, Akt, BDNF, NT-3, FGF2, neurogenesis, gliogenesis, neural apoptosis, proliferation
Introduction
The generation of differentiated neurons and glial cells from proliferating mammalian neural stem or progenitor cells is a complex process involving an interplay between intrinsic cellular programs and extrinsic cues such as growth factors. This complex process has perhaps been best studied in the developing cortex. In rodents, stem and progenitor cells proliferate within the cortical ventricular zone and then differentiate into neurons during midgestation and into glial cells during late gestation and early postnatal life. Remarkably, cortical progenitor cells isolated at the onset of neurogenesis and plated in serum-free conditions will reproduce this temporal in vivo differentiation pattern, generating neurons first and glia second (Qian et al., 2000).
Study of this system has led to identification of a number of key intracellular proteins that are essential for proliferation and differentiation of cortical progenitor cells, including the pRb family (Slack et al., 1998; Toma et al., 2000; Ferguson et al., 2002), neurogenic and gliogenic basic helix-loop-helices (HLHs) (Lu et al., 2000, 2001; Nieto et al., 2001; Sun et al., 2001), the inhibitory HLH Id2 (Lasorella et al., 2000; Toma et al., 2000), and the C/EBP (CAAT enhancer-binding protein) family of transcription factors (Ménard et al., 2002). In addition, some of the signaling pathways that allow extrinsic cues to regulate these intracellular proteins have been identified. For example, ciliary neurotrophic factor (CNTF) and leukemia inhibitory factor signal via the JAK—STAT (Janus-activated kinase—signal transducer and activator of transcription) pathway to promote the differentiation of glial cells (Sauvageot and Stiles, 2002), whereas exogenous plate PDGF (Park et al., 1999) promotes the differentiation of neurons via activation of an MEK—RSK—C/EBP (MAP kinase kinase—ribosomal S6 kinase—CAAT enhancer-binding protein) pathway (Ménard et al., 2002). Moreover, although the intracellular signaling pathways have not yet been elucidated, fibroblast growth factor 2 (FGF2) is known to be an essential survival and proliferation factor for cortical progenitors both in vivo (Vaccarino et al., 1999; Raballo et al., 2000) and in vitro (Ghosh and Greenberg, 1995; Lukaszewicz et al., 2002).
One class of growth factors that might play a role in regulating cortical progenitor cell biology are the neurotrophins. At least two members of the neurotrophin family, BDNF and neurotrophin-3 (NT-3), along with their preferred tyrosine kinase receptors (TrkB and TrkC), are expressed in the cortical ventricular/subventricular zones at the onset of cortical neurogenesis (Maisonpierre et al., 1990; Fukumitsu et al., 1998). Moreover, culture work has suggested that NT-3 might selectively regulate cell cycle exit and neuronal differentiation in cortical progenitors (Ghosh and Greenberg, 1995; Lukaszewicz et al., 2002). However, although animals lacking either single neurotrophins or their Trk receptors do display some CNS phenotypes, including cortical abnormalities (Minichiello and Klein, 1996; Alcantara et al., 1997; Martinez et al., 1998; Ringstedt et al., 1998; Kahn et al., 1999; Xu et al., 2000; Lotto et al., 2001), the precise role of neurotrophins produced by cortical precursors is still unclear, as are the receptor—signaling mechanisms that underlie these biological effects.
Here we investigated these questions using cortical progenitor cells isolated at the onset of neurogenesis in vivo. We demonstrated previously that these cells are all dividing, nestin-positive precursors at the time of isolation and that a significant number of them differentiate into postmitotic neurons over the first week in vitro. Our studies here indicate that, as seen in vivo, cultured cortical precursors express the neurotrophins BDNF and NT-3, as well as their preferred TrkB and TrkC receptors, and that this autocrine—paracrine neurotrophin loop is essential for progenitor cell survival. Moreover, these endogenously produced neurotrophins signal via Trk receptors to activate the PI3-kinase—Akt and MEK—ERK pathways, and these pathways subserve distinct functions, with PI3-kinase being essential for progenitor survival and MEK for the differentiation of neurons but not glial cells. Thus, cortical progenitors rely on endogenously produced neurotrophins and distinct Trk-mediated signaling pathways for multiple aspects of their biology, including survival and neurogenesis.
Materials and Methods
Primary cultures of cortical progenitors and neurons. The preparation of cortical progenitors from mouse embryos has been described in detail previously (Slack et al., 1998; Gloster et al., 1999; Toma et al., 2000; Ménard et al., 2002). Briefly, cortical tissue, obtained from embryonic day 12.5 (E12.5) to E13.5 CD1 mice, was dissected in ice-cold HBSS (Invitrogen, Gaithersburg, MD) and then transferred into 37°C Neurobasal medium (Invitrogen) containing 500 μm glutamine, 2% B27 supplement, and 1% penicillin—streptomycin (Invitrogen); this medium was supplemented with 40 ng/ml FGF2 (Collaborative Research, Bedford, MA), except when mentioned. The tissue was mechanically triturated with a plastic pipette into small clusters of cells that were plated in six-well tissue culture dishes (Nunc, Naperville, IL) or chamber slides precoated with laminin and poly-d-lysine (Collaborative Research). Cell density was 1,500,000 and 100,000 cells per well, respectively. Two to 3 hr after plating, progenitor cells were treated as follows: with either PD98059 or LY294002 (both from Biomol, Plymouth Meeting, PA) in 1% DMSO (final concentration); acutely stimulated with 150 ng/ml NGF (Cedarlane Laboratories, Hornby, Ontario, Canada), BDNF, NT-3, NT-4 (all from Peprotech, Rocky Hill, NJ), or FGF2 for 15 min; cultured in the presence of 50 ng/ml CNTF (Peprotech); or treated with 20 μg/ml neurotrophin function-blocking antibodies (Promega, Madison, WI) or 40 μg/ml control chicken IgY (Promega). The same amount of antibody or control IgY were readded 24 hr later. Cultures were maintained at 37°C in a 5% CO2 incubator.
Mature postmitotic cortical neurons were obtained as described previously (Pozniak et al., 2002; Wartiovaara et al., 2002). Briefly, E16—E17 mouse cortices were mechanically dissociated into a single-cell suspension in the same medium as for the progenitor cells without FGF2. The cells were plated for3dinthe same conditions as for cortical progenitors, and then one-half of the medium was removed and replaced with fresh medium supplemented with a final concentration of 7 μm cytosine arabinoside (Sigma, St. Louis, MO). Two days later, the cells were treated and/or harvested.
Immunocytochemistry, transferase-mediated biotinylated UTP nick end labeling, and quantitation. Immunocytochemistry and 5-bromo-2-deoxyuridine (BrdU) incorporation protocols have been described previously (Toma et al., 2000; Wartiovaara et al., 2002). Primary antibodies were mouse anti-nestin (1:400; Chemicon, Temecula, CA), mouse anti-Ki67 (1:200; PharMingen, San Diego), mouse anti-MAP2 (microtubule-associated protein 2) (1:400; Sigma), rabbit anti-βIII-tubulin (1:1000; Research Diagnostics, Flanders, NJ), mouse anti-HuD (1:300; Molecular Probes, Eugene, OR), and rabbit anti-TrkB (1:500; Santa Cruz Biotechnology, Santa Cruz, CA). Cells [2 d in vitro (DIV), or less when mentioned] were washed with HEPES-buffered saline (HBS), pH 7.4, fixed for 20 min with 4% paraformaldehyde (Sigma), permeabilized for 5 min in 0.2% NP-40 (Roche Products, Hertforshire, UK) in HBS, and then blocked for 1—2 hr at room temperature with buffer containing 6% normal goat serum (NGS) and 0.5% bovine serum albumin (BSA) (Jackson ImmunoResearch, West Grove, PA). Cells were then incubated at 4°C overnight with primary antibodies in HBS containing 3% NGS and 0.25% BSA. After washing with HBS, cells were incubated at room temperature for 1 hr with either indocarbocyanine (Cy3)-conjugated goat anti-mouse or anti-rabbit IgG (1:600), or FITC-conjugated anti-mouse or anti-rabbit IgG (1:200; Jackson ImmunoResearch) secondary antibodies (as necessary) prepared in HBS containing 3% goat serum and 0.25% BSA. Samples were then washed with HBS, counterstained for 2 min with Hoechst 33258 (1:2000; Sigma), and mounted with GelTol (Fisher Scientific, Houston, TX).
For the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) experiments, cells were washed, fixed, and permeabilized as mentioned above. TUNEL reaction was performed for1hrat37°C. Each 100 μl of TUNEL reaction mixture contained 20 μl of 5× terminal deoxynucleotidyl transferase (TdT) buffer, 0.9 μlofTdT enzyme (both from Promega), and 1 μl of biotin-16-deoxyuracil triphosphate (Boehringer Mannheim, Mannheim, Germany). After the TUNEL reaction, preparations were washed with HBS and incubated for 1 hr at room temperature with dichlorotriazinyl aminofluorescein or Cy3-conjugated streptavidin (Jackson ImmunoResearch) diluted 1:2000 in HBS. In those experiments in which cells were double labeled, cells were then blocked with 3% NGS and 0.5% BSA for 1 hr, and immunocytochemical analysis was performed as above.
For BrdU incorporation analysis, 10 μm BrdU (Boehringer Mannheim) was added overnight directly to the media of cultured progenitor cells. After washing with HBS, the cells were fixed for 30 min with 70% ethanol, air dried for 1 min, treated for 10 min with 2N HCl, and then treated for 10 min with 0.1 m borate buffer (Na2B4O7-H2O, pH 8.5). Preparations were washed three times with 0.5% Tween 20 and 1% BSA in HBS, before incubation overnight at 4°C with mouse anti-BrdU (1: 150; Chemicon) in HBS containing 3% NGS and 0.5% BSA. After washing, the slides were incubated for 1 hr at room temperature with Cy3-conjugated anti-mouse IgG, counterstained with Hoechst, and mounted.
For quantitation, four to six random fields of each treatment (per experiment) were captured and processed. Digital image acquisition and analysis was performed with the Northern Eclipse software (Empix, Mississauga, Ontario, Canada) using a Sony (Tokyo, Japan) XC-75CE CCD video camera. In all graphs, error bars indicate the SD, and the statistics were performed using one-way ANOVA with the Student—Newman—Keuls method.
Western blot analysis and immunoprecipitations. For biochemical analysis, cortical progenitors (4 hr to 2 DIV) and neurons (6 DIV) were washed with ice-cold HBSS and lyzed directly in the dish with either TBS lysis buffer (137 mm NaCl, 20 mm Tris, pH 8, and 1% NP-40, and 10% glycerol) or radioimmunoprecipitation assay buffer (50 mm Tris, pH 7.2, 150 mm NaCl, 2 mm EDTA, 1% NP-40, 1% Na deoxycholate, and 0.1% v/v SDS) supplemented with protease inhibitor mixture (Boehringer Mannheim) and 1.5 mm sodium vanadate. Lysates were scraped into Eppendorf Scientific (Westbury, NY) tubes, rocked for 10 min at 4°C, and cleared by centrifugation. Protein concentration was determined using the bicinchoninic acid assay (Pierce, Rockford, IL) and BSA as a standard. Equal amounts of protein (25–50 μg) were boiled in sample buffer, separated by 10–15% SDS-PAGE gels, and transferred to 0.2 μm nitrocellulose membrane for 3 hr at 0.75 A. Membranes were blocked in 5% skim milk powder in TBS-T (TBS plus 0.5% Tween 20) or in 3% BSA in TBS-T (for 4G10) for 2 hr at room temperature and then incubated overnight at 4°C with primary antibody. Antibodies used were as follows: anti-phospho(Ser473) Akt (1:1000; Cell Signaling Technology, Beverly, MA); anti-Akt (1:1000; Cell Signaling Technology); anti-phospho-(Thr183/Tyr185) ERK (1:2500; Promega), anti-ERK (1:5000; Santa Cruz Biotechnology), anti-cleaved caspase 3 (1:1000; Cell Signaling Technology), anti-cyclin E (cycE) (2 μg/ml; Upstate Biotechnology, Lake Placid, NY), anti-cyclin-dependent kinase 2 (cdk2) (1:1000; Santa Cruz Biotechnology), anti-neuronal-specific enolase (1:2000; Polysciences, Warrington, PA), anti-neurofilament medium (1:1000; Chemicon), anti-glial fibrillary acidic protein (GFAP) (1:2000; Dako, High Wycombe, UK), anti-phosphotyrosine (4G10; 1:100; Upstate Biotechnology), and anti-TrkB and TrkC (1:500; Santa Cruz Biotechnology). After washing with TBS-T, membranes were incubated with secondary antibodies, HRP-conjugated goat anti-mouse or anti-rabbit (1:10,000; Boehringer Mannheim), in blocking solution for 2 hr at room temperature. Detection was performed using the ECL chemiluminescence reagent (Amersham Biosciences, Arlington Heights, IL) and XAR x-ray films (Eastman Kodak, Rochester, NY). Membranes were subsequently stripped with stripping buffer (70 mm Tris, pH 6.8, 2% v/v SDS, and 0.7% v/v β-mercaptoethanol) for 5–10 min at 55°C, were extensively washed with Milli-Q water (Millipore, Bedford, MA), reblocked, and reprobed as described above.
For immunoprecipitations, FGF2 was washed out for 1 hr from cortical progenitors (2 DIV), and both cortical progenitors and neurons were stimulated for 10 min with 100 ng/ml NGF, BDNF, NT-3, or NT-4. Equal amounts of protein were incubated with 3 μl of the pan-Trk antibody (203b) (Hempstead et al., 1992) for 2 hr at 4°C and then incubated for the same time period with 25 μl of protein A-Sepharose (Sigma). The precipitated proteins were collected by centrifugation, washed three times with TBS lysis buffer, boiled with sample buffer, and loaded on a 7.5% SDS-PAGE gel.
RNA extractions and reverse transcription-PCR analysis. Extraction of RNA from cultured cortical progenitors (1 DIV), neurons (6 DIV), or sympathetic neurons (Wartiovaara et al., 2002) was performed using TRIZOL (Invitrogen) according to the protocol of the manufacturer. Total samples were then treated with DNase (Promega) for 30 min at 37°C. The absence of genomic DNA contamination was assessed by performing PCR for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Farah et al., 2000). RNA (5–10 ng) was reverse transcribed using Moloney murine leukemia virus reverse transcriptase (Fermentas, Hanover, MD) and oligo-dT (Promega) following the protocol of the manufacturer. The primers used and amplification conditions were as described previously (Benoit et al., 2001). PCR products were separated on a 1.5% agarose gel and sized with a 100 bp DNA ladder (Promega).
Results
Cortical progenitor cells express and are responsive to neurotrophins
To address the potential role of neurotrophins in cortical neurogenesis, we examined cortical progenitors that were isolated from E12.5–E13.5 mouse cortex and plated in the presence of FGF2. We showed previously (Slack et al., 1998; Gloster et al., 1999; Toma et al., 2000; Ménard et al., 2002) that, on plating, virtually all of these cells are dividing, nestin-positive progenitors and that, over the ensuing 5 d, many of them exit the cell cycle to become postmitotic neurons. To ask whether endogenously produced growth factors such as the neurotrophins might be involved in these events, we initially measured the effect of cell density on cortical progenitor cell survival. Serial dilutions of progenitor cells plated in 40 ng/ml FGF2 led to a proportional increase in apoptosis (Fig. 1a), suggesting the presence of endogenously produced survival factors.
Figure 1.

Mouse-derived cortical progenitor cells express BDNF, NT-3, TrkB, and TrkC. a, TUNEL analysis of cortical progenitors plated at decreasing cell density. Progenitors were plated at varying dilutions in the presence of FGF2 (40ng/ml) and, 2 d later, were analyzed for apoptosis. *p < 0.05; **p < 0.01; ***p < 0.001 (ANOVA). Error bars indicate SDs. b,c, RT-PCR analysis for the presence of the mRNAs (b) for the neurotrophins BDNF, NT-3, and NGF (c), for the neurotrophin receptors TrkB and TrkC in cortical progenitors (CORTICAL PROGENITOR), postmitotic cortical neurons (PM), mouse E15 brain (E15 Br), mouse adult brain (Ad Br), and cultured sympathetic neurons (SCG). GAPDH was used as a loading control in all cases. Similar results were obtained with three independent cortical progenitor cell RNA preparations (1 or 2 DIV; in the presence of 40 ng/ml FGF2). d, Immunocytochemical analysis for TrkB in cycling cortical progenitors (cultured with 40 ng/ml FGF2) and postmitotic cortical neurons. The top three panels are photographs of the same field and represent immunostaining for TrkB (left, red), the proliferation marker Ki67 (middle, green), and the merge of these two panels (right), along with a Hoechst counterstain (blue) to show all nuclei in the field (left and right). The bottom three panels are photographs of the same field and represent immunostaining for TrkB (left, red), the late neuronal marker MAP2 (middle, green), and the merge of these two panels (right), along with Hoechst (blue, left and right). Scale bar, 50 μm. e, Western blot analysis for TrkB and TrkC. Cortical progenitors (CORTICAL PROGENITOR) cultured for 4hr with 40 ng/ml FGF2 or postmitotic cortical neurons (PM) were harvested and immunoprecipitated with an antibody that recognizes all full-length members of the Trk family (IP:Pan-Trk), and then the immunoprecipitates were analyzed by Western blot analysis for TrkB (left) or TrkC (right). Arrow indicates the immunoreactive Trk receptor bands, and size markers are indicated on the right.
The neurotrophins BDNF and NT-3 and their preferred TrkB and TrkC receptors are expressed within the ventricular zone of embryonic rats (Maisonpierre et al., 1990; Fukumitsu et al., 1998), suggesting that they could participate as autocrine—paracrine survival factors in cortical progenitors. To verify that these neurotrophins were expressed by cultured murine cortical progenitors, we first performed reverse transcription (RT)-PCR analysis on total RNA isolated from cells cultured for either 1 or 2 d (Fig. 1b,c). This analysis indicated that cortical progenitors selectively express the neurotrophins BDNF and NT-3, but not NGF (Fig. 1b), as well as the TrkB and TrkC receptors (Fig. 1c), in agreement with previous in vivo studies. To determine the percentage of cells expressing the TrkB receptor, we then performed double-label immunocytochemical analysis for TrkB and for nestin, a marker for progenitor cells (data not shown), for Ki67, a protein expressed in dividing cells (Fig. 1d) (Scholzen and Gerdes, 2000; Kee et al., 2002), or for MAP2, a marker for postmitotic neurons (Fig. 1d). This analysis demonstrated that the majority of both progenitors and newly born neurons were positive for TrkB. Coincubation of the TrkB antibody with an excess of the control peptides abolished immunoreactivity (data not shown). Western blot analysis confirmed expression of full-length forms of both TrkB and TrkC by cortical progenitors as soon as 4 hr after plating (Fig. 1e).
To verify that the Trk receptors expressed by cortical progenitors were functional, we washed and then stimulated cortical progenitors for 10 min with each of the four members of the neurotrophin family and looked for Trk receptor activation. As a positive control, we used postmitotic cortical neuron cultures. Immunoprecipitation of total Trk proteins followed by Western blot analysis for phosphotyrosine demonstrated that BDNF, NT-3, and NT-4, the ligands for TrkB and TrkC, led to Trk receptor activation in both cortical progenitors and neurons. In contrast, NGF, which binds to TrkA, had no effect (Fig. 2a, left). Trk receptor activation was also observed in both cortical progenitors (2 DIV) and postmitotic cortical neurons maintained in media, without washing and without the addition of exogenous neurotrophins (Fig. 2a, right), consistent with the endogenous expression of BDNF and NT-3. To confirm this result, we examined activation of Akt and the ERKs, signaling proteins known to be activated downstream of tyrosine kinase receptors such as the Trks. Cortical progenitors were cultured in the absence of exogenous FGF2 and then were stimulated for 15 min with NGF, BDNF, NT-3, or FGF2 4 hr after plating. Western blot analysis demonstrated that BDNF, NT-3, and FGF2, but not NGF, caused a small, but consistent increase in the activated, phosphorylated form of Akt, along with a robust increase in phosphorylation of the ERKs (Fig. 2b). Immunocytochemical analysis demonstrated that >95% of the cells in these cultures were nestin positive at the time of stimulation (data not shown), confirming that cortical progenitors respond to BDNF, NT-3, and NT-4 via the TrkB and TrkC receptors.
Figure 2.

Cortical progenitors are responsive to exogenous and endogenous neurotrophins. a, Left, Cortical progenitors cultured for 2 d (CORTICAL PROGENITOR) with 40 ng/ml FGF2, postmitotic cortical neurons (PM) were washed and then stimulated for 10 min with one of the four neurotrophins, NGF, BDNF, NT-3 or NT-4 and immunoprecipitated with an antibody to all Trk receptors (IP: Pan-Trk), and the immunoprecipitates were then analyzed by Western blot analysis with an antibody to phosphotyrosine (Probe: P-Tyr). The arrow indicates the phosphotyrosine-positive band migrating at the size of the Trk receptors. BDNF, NT-3, and NT-4, but not NGF, induce Trk tyrosine phosphorylation. Right, Cortical progenitors (CORTICAL PROGENITOR; 2 DIV) cultured with FGF2 (40 ng/ml) and postmitotic cortical neurons (PM) were analyzed for endogenous Trk receptor activation, as described above, without washing or exogenous neurotrophin stimulation. b, Cortical progenitor cells, cultured for 4 hr (without FGF2), were acutely stimulated with NGF, BDNF, NT-3, or FGF2 and analyzed by Western blot for the activation of Akt and ERKs, using phosphorylation-specific Akt (P-Akt) and ERK (P-ERK) antibodies. The same blots were then reprobed for total Akt and ERK protein as a loading control. BDNF, NT-3, and FGF2 all caused increased Akt and ERK phosphorylation, relative to cells that were either unstimulated (CTL) or stimulated with NGF. c, Western blot analysis for Akt and ERK activation in cortical progenitors cultured for 18 hr in the presence of FGF2 (40 ng/ml) and antibodies specific for BDNF (anti-BDNF; 20 μg/ml), NT-3 (anti-NT-3; 20 μg/ml), or control anti-chicken IgY (Ctl-IgY; 40 μg/ml). Western blots were first probed for the activated, phosphorylated forms of Akt and the ERKs (P-Akt and P-ERKs) and then reprobed for total Akt and ERK protein as a control for equal amounts of protein.
Endogenous neurotrophins are essential for cortical progenitor cell survival, proliferation, and neuronal differentiation
To examine the potential role of these endogenously produced neurotrophins, we used function-blocking antibodies for BDNF and NT-3 (Kohn et al., 1999). Using these antibodies, we first asked whether endogenous BDNF and/or NT-3 made any contribution to activation of downstream signaling pathways when progenitors were cultured in our normal culture conditions, in the presence of exogenous FGF2. Progenitors were cultured and treated for 18 hr with 20 μg/ml anti-BDNF or anti-NT-3 or, as a control, 40 μg/ml control chicken IgY. Western blot analysis of these treated cells for phospho-Akt and phospho-ERK revealed that, even when progenitors were cultured in exogenous FGF2, blocking BDNF or NT-3 led to a significant decrease in activation of both of these signaling proteins (Fig. 2c). Thus, endogenous BDNF and NT-3 contribute significantly to the activation of these two pathways in cortical progenitors.
We next asked about the effects of blocking endogenous neurotrophins on cortical progenitor cell biology. To perform these experiments, progenitors were immediately cultured in FGF2 with or without anti-BDNF and/or anti-NT-3. Initially, we examined cell survival by TUNEL. This analysis revealed that cortical progenitors cultured in the presence of anti-BDNF or NT-3 showed a marked increase in apoptosis (Fig. 3a,c), with ∼40–50% of cells being TUNEL positive. When both anti-BDNF and anti-NT-3 were added together, a larger increase in TUNEL-positive cells was observed (Fig. 3c) (p < 0.05), although this increase was not additive, suggesting that BDNF and NT-3 act to support the survival of overlapping populations of progenitors. A statistically similar decrease in cell survival of ∼40% was obtained when Trk receptor signaling was blocked using 200 nm of the pharmacological inhibitor K252a (two separate experiments; data not shown).
Figure 3.
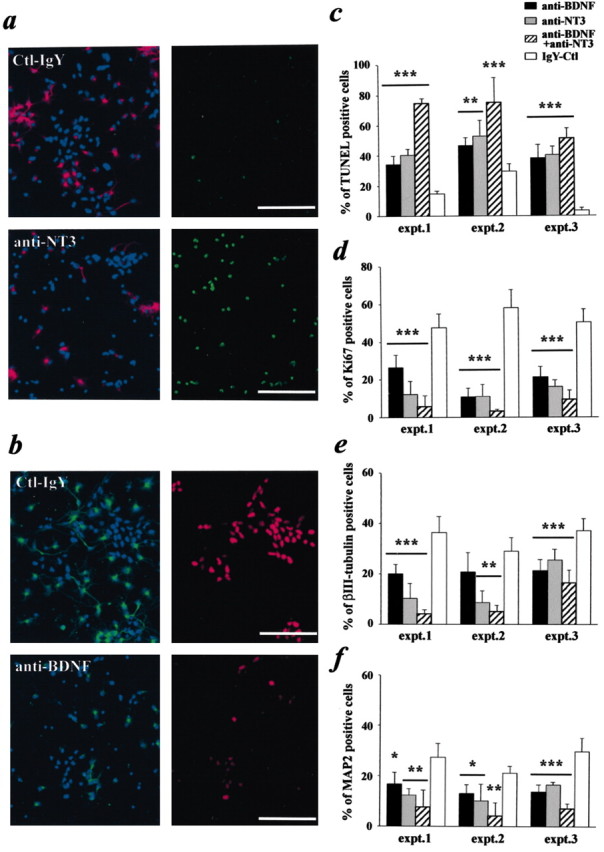
Inhibition of endogenous neurotrophins decreases cortical progenitor cell survival, proliferation, and differentiation into neurons. Cortical progenitor cells cultured in the presence of FGF2 (40 ng/ml) were treated for 2d with control IgY (40 μg/ml), anti-NT-3 (20 μg/ml), anti-BDNF (20 μg/ml), or the combination of anti-NT-3 and anti-BDNF (20 μg/ml each). a, Double-label immunocytochemistry for the neuronal marker MAP2 (left, red; counterstained with Hoechst in blue) and TUNEL (right, green). Cells were treated for 2 d with either anti-NT-3 (bottom panels) or control IgY antibody (top panels). b, Double-label immunocytochemistry for βIII-tubulin (left, green; cells are counterstained with Hoechst in blue) and the Ki67 antigen, a marker for proliferating cells (right, red). Scale bars: a, b, 100 μm. c, Quantitation of three individual experiments (expt.) assessing cellular apoptosis performed as shown in a using TUNEL. d, Quantitation of three individual experiments assessing cellular proliferation performed as that shown in b using Ki67 as a marker for dividing cells. e, f, Quantitation of three individual experiments assessing neurogenesis performed as those shown in a and b using both the early neuronal marker βIII-tubulin and the later neuronal marker MAP2. In c—f, *p < 0.05; **p < 0.01; ***p < 0.001 (ANOVA). Error bars indicate SDs. Ctl, Control.
To confirm that this increase in apoptosis was attributable to the death of cortical progenitors and not just newly born neurons, we performed similar experiments and quantitated the percentage of cells with apoptotic nuclear morphology (determined by staining with Hoechst) that expressed or did not express the early neuron-specific marker βIII-tubulin (neurons and progenitor cells, respectively) (Fig. 3b). This analysis revealed that, in cultures treated with the control IgY antibody, ∼11 and 3% of progenitors and neurons, respectively, had apoptotic nuclei. In contrast, in cultures treated with anti-BDNF, ∼45% of progenitors and 25% of neurons were apoptotic, whereas in those treated with anti-NT-3, ∼55 and 19% of progenitors and neurons were apoptotic, respectively. Thus, the majority of cells dying after treatment with anti-BDNF or anti-NT-3 were progenitors, although neuronal apoptosis was also increased, consistent with previous observations (Ghosh et al., 1994).
This dramatic effect on progenitor cell biology was confirmed when we examined cell proliferation and neuronal differentiation. To assess proliferation, we immunostained progenitors for Ki67, an antigen that is highly expressed by mitotically active cells throughout the cell cycle (Scholzen and Gerdes, 2000; Kee et al., 2002). These experiments demonstrated that the number of Ki67-immunoreactive cells was greatly reduced in the presence of anti-BDNF and/or anti-NT-3 (Fig. 3b,d). Similar results were obtained for neuronal differentiation, as assayed by immunostaining for βIII-tubulin, an early neuronal gene, and MAP2, a late neuronal marker (Fig. 3a,b,e,f); the addition of anti-BDNF and/or anti-NT-3 significantly reduced the number of neurons present in these cultures. Thus, endogenous neurotrophin signaling is essential for progenitor cell survival, and FGF2 alone cannot substitute for these endogenous neurotrophins with regard to survival. Whether endogenously produced neurotrophins play an equally important role in promoting progenitor cell proliferation or differentiation cannot be determined because the observed decreases in these two parameters may be because of decreased survival.
PI3-kinase and MEK signaling pathways subserve distinct functions in cortical progenitor cells
To ask how neurotrophins might signal through the TrkB and TrkC receptors to mediate progenitor cell survival, we initially focused on the PI3-kinase and MEK—ERK pathways, both of which are activated by endogenous neurotrophins, as shown in Figure 2b,c, and both of which have been implicated in survival of other neural cells (Kaplan and Miller, 1997, 2000). To inhibit these pathways, we used two well characterized pharmacological inhibitors, LY294002 (to inhibit PI3-kinase) and PD98059 (to inhibit MEK). We first verified the ability of these two compounds to selectively inhibit the appropriate pathways in progenitors; cells were treated with different concentrations of one of these two compounds for 4 hr and were then assessed for phosphorylation of Akt and the ERKs, which are downstream substrates of PI3-kinase and MEK, respectively. Western blot analysis revealed that 50 or 100 μm LY294002 specifically inhibited Akt but not ERK phosphorylation, whereas 50 μm PD98059 decreased ERK but not Akt phosphorylation, as predicted (Fig. 4a).
Figure 4.
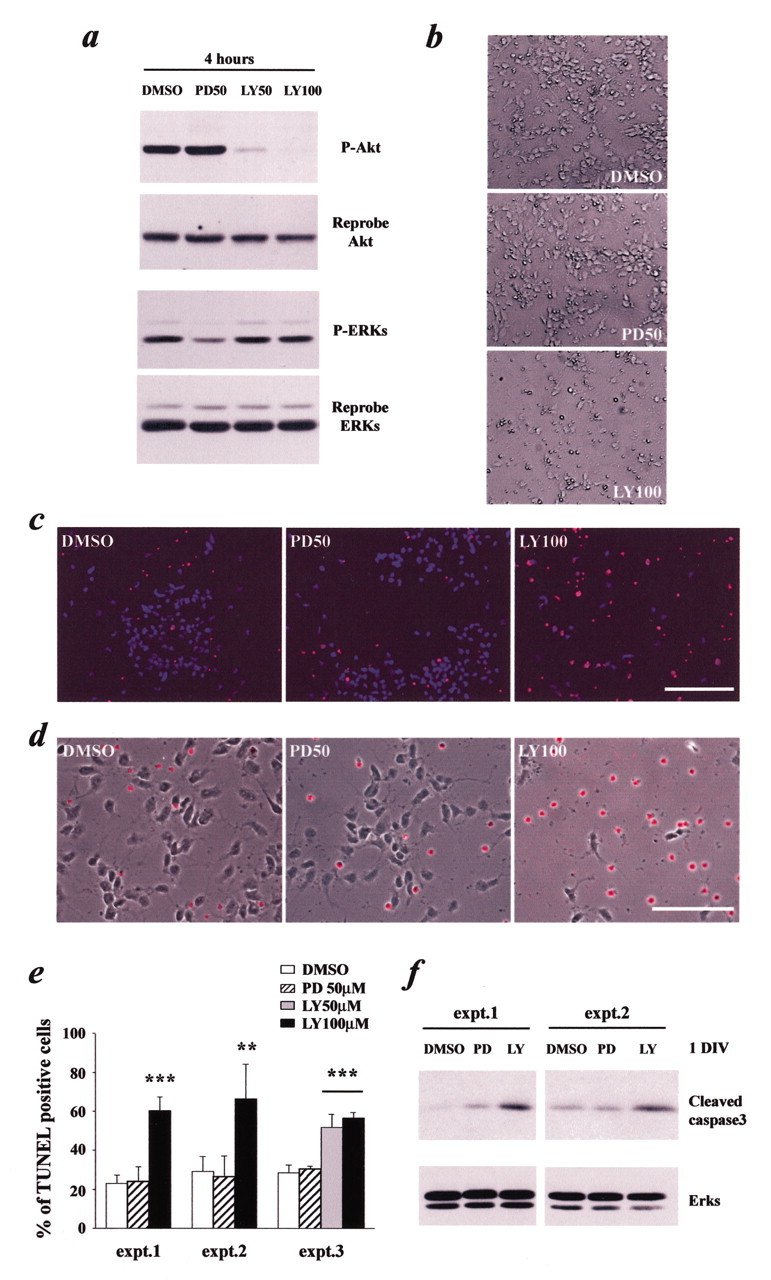
Cortical progenitor cell survival depends on PI3-kinase but not MEK activation. a, Western blot analysis to ascertain the efficacy and specificity of treatment with the pharmacological inhibitors PD98059 and LY294002. Progenitors were cultured in the presence of FGF2 (40 ng/ml) with DMSO (1%), PD98059 (50 μm), or LY294002 (50–100 μm) for 4 hr, and lysates were analyzed by Western blots for the active, phosphorylated form of Akt or the ERKs (P-Akt, P-ERKs). The blots were then reprobed for total Akt and ERK protein as a loading control. b—d, Analysis of apoptosis in cortical progenitors (in the presence of 40 ng/ml FGF2) treated for 2 d with DMSO, PD98059 (PD50), or LY294002 (LY100), as assessed by phase microscopy of living cells (b) or by TUNEL analysis (c, d). Cells were counterstained with Hoechst (c) or combined with the phase picture (d) to show all nuclei or cells in the field, respectively. Scale bars: c, 100 μm; d, 50 μm. e, Quantitation of three individual experiments (expt.) similar to that shown in c.**p < 0.01; ***p < 0.001 (ANOVA). Error bars indicate SDs. f, Western blot analysis for the active, cleaved form of caspase-3 in cortical progenitors cultured for 1 d in the presence of FGF2 (40 ng/ml) with DMSO, PD98059, or LY294002. Two independent experiments are shown. The blots were reprobed for total ERK protein as a loading control.
We then used these inhibitors to ask whether either of these two pathways was important for survival, proliferation, or differentiation of cortical progenitors; cells were cultured for2dinthe presence of FGF2 with or without LY294002 or PD98059. Phase microscopy of living cultures, along with analysis of these cells for TUNEL, revealed that inhibition of PI3-kinase with 50 or 100 μm LY294002 had a profound effect on progenitor cell survival (Fig. 4b—e); 50–70% of cells in these cultures were TUNEL positive. Similar results were obtained with a second pharmacological inhibitor of PI3-kinase, wortmannin (data not shown). In contrast, inhibition of MEK had no effect on cell survival (Fig. 4b—e). To confirm that inhibition of PI3-kinase, but not MEK, selectively increased apoptosis of progenitors, we also examined caspase 3 activation. Western blot analysis revealed that 1 d of treatment with LY294002 caused an increase in levels of the cleaved, active form of caspase-3, whereas PD98059 had no significant effect (Fig. 4f). Thus, PI3-kinase, but not MEK, is essential for survival of cortical progenitors.
We next examined the potential role of these two pathways in proliferation of cortical progenitors by measuring BrdU incorporation and by immunostaining for Ki67. For the BrdU studies, progenitors were cultured in FGF2 with or without LY294002 or PD98059 and were then pulsed with BrdU overnight before analysis by immunostaining. These experiments demonstrated that inhibition of MEK had no effect on either BrdU incorporation (Fig. 5a) or on the percentage of cells expressing Ki67 (Fig. 5b,c) but that inhibition of PI3-kinase greatly reduced both of these parameters (Fig. 5a—c). Additional support for the conclusion that PI3-kinase, but not MEK, was important for survival and proliferation in these cultures derived from the finding that total cell number was unaffected by MEK inhibition, whereas PI3-kinase inhibition reduced total cell number by 40–50% (Fig. 5e).
Figure 5.

Cortical progenitor cell proliferation is reduced when PI3-kinase, but not MEK, is inhibited. Cortical progenitors cultured in the presence of FGF2 (40 ng/ml) were treated for 2 d with DMSO (1%), PD98059 (50 μm; PD50), or LY294002 (100 μm; LY100). a, Immunocytochemical analysis for BrdU (red), after an overnight pulse of BrdU immediately before analysis. Cells were counterstained with Hoechst (blue). b, Immunostaining for the Ki67 antigen for proliferating cells (green). Cells were counterstained with Hoechst (blue). Scale bars: a, b, 100 μm. c, Quantitation of three individual experiments (expt.) similar to that shown in b. d, Western blot analysis for two S-phase markers, cycE and cdk2, in cortical progenitors (CP) and postmitotic cortical neurons (PM). The blot was reprobed for total ERK protein as a loading control. e, Quantitation of total cell number per field, as assessed by Hoechst staining. The number of cells correspond to the average of 15 randomly captured fields per treatment per experiment. ***p < 0.001 (ANOVA). Error bars indicate SDs.
To confirm these findings biochemically, we also performed Western blot analysis for two known S-phase markers, cycE and cdk2 (for review, see Ekholm and Reed, 2000). This analysis revealed that, as predicted, both cycE and cdk2 were highly expressed in cortical progenitors cultured for 2 d, whereas they were virtually absent in cultures of postmitotic cortical neurons (Fig. 5d). Treatment with PD98059 did not significantly alter levels of either of these proteins; however, LY294002 led to a great decrease in both (Fig. 5d), confirming the immunocytochemical results. Thus, MEK is not important for either survival or proliferation of cortical progenitors. Whether PI3-kinase is important for proliferation cannot be determined from these experiments because of its important role in progenitor cell survival.
Finally, we asked whether either of these two pathways was important for differentiation of neurons from cortical progenitors. Progenitors were cultured for 2 d in the presence of FGF2 with or without PD98059 or LY294002 and then were immunostained for three different panneuronal proteins, βIII-tubulin and HuD (Clayton et al., 1998), two early neuronal markers, and MAP2, a late neuronal protein. This analysis revealed that treatment with PD98059 significantly decreased the percentage of cells expressing all three of these neuronal proteins (Fig. 6a—d), a result similar to what we observed previously when MEK was inhibited in cortical progenitors using a dominant-negative MEK construct (Ménard et al., 2002). Confirmation of this result was obtained by Western blot analysis of similar cultures for two additional neuron-specific proteins, neuron-specific enolase and the medium neurofilament protein (Fig. 6e). Both immunocytochemistry (Fig. 6a—d) and Western blot analysis (Fig. 6e) indicated that a similar inhibition was observed for treatment with LY294002. However, this latter effect may be attributable to the decrease in cell survival observed when PI3-kinase is inhibited.
Figure 6.

Differentiation of neurons but not astrocytes from cortical progenitors is dependent on MEK activation. a—e, Cortical progenitors cultured in the presence of FGF2 (40 ng/ml) were treated for 2 d with DMSO (1%), PD98059 (50 μm; PD50), or LY294002 (100 μm; LY100). a, Immunocytochemical analysis for the early panneuronal marker HuD (red). Cells were counterstained with Hoechst (blue). b, c, Immunostaining for the late panneuronal marker MAP2 (green). Cells were counterstained with Hoechst (blue) or combined with the phase picture of the field (c). Scale bars: a, b, 100 μm; c, 50 μm. d, Quantitation of four individual experiments (expt.) similar to those shown in a and b, analyzing the proportion of cells expressing HuD, MAP2, or βIII-tubulin. **p < 0.01; ***p < 0.001 (ANOVA). Error bars indicate SDs. e, Western blot analysis for the neuronal markers neuron-specific enolase (NSE) and neurofilament-medium (NFM) in cortical progenitors differentiated for 2 d (CP) and in postmitotic cortical neurons (PM) as a control. The blots were reprobed for total ERK protein as a loading control. f, Western blot analysis for the astrocyte-specific protein GFAP in cortical progenitors cultured in the presence of FGF2 (40 ng/ml) with or without CNTF (50 ng/ml) for 3 d in the presence or absence of 50 μm PD98059. Mixed cortical cultures (grown in the absence of cytosine arabinoside), which contain both neurons and astrocytes (PM-CA), were used as a positive control. The blot was reprobed for total ERK protein as a loading control.
Cortical progenitor cells normally only generate astrocytes when they are stimulated with the gliogenic factor CNTF (Park et al., 1999; Ménard et al., 2002) or after long periods of time in culture (Qian et al., 2000). To ask whether the inhibition of MEK, which blocks progenitors from becoming neurons, also alters the generation of astrocytes, we examined these cultures for expression of GFAP. Immunocytochemical analysis demonstrated that progenitors cultured in either the presence or absence of 50 μm PD98059 for 3 d did not generate GFAP-positive glial cells (two separate experiments; data not shown), a finding confirmed biochemically by Western blot analysis (Fig. 6f). Moreover, even when progenitors were stimulated with CNTF, which induces astrocyte formation, inhibition of MEK had little or no effect on GFAP expression (Fig. 6f). Because inhibition of the MEK pathway blocks the generation of neurons but not astrocytes from cortical progenitors and because it has no effect on progenitor cell survival or proliferation, these results suggest that MEK activation functions specifically to bias cortical progenitors to become neurons in the present culture conditions.
Inhibition of neurotrophin, PI3-kinase, or MEK signaling has the same effect on cortical progenitors in the absence of exogenous FGF2
Studies presented here indicate that inhibition of endogenous neurotrophins have a profound effect on basal activation of the PI3-kinase—Akt and MEK—ERK pathways even when progenitors were cultured in the presence of FGF2 (Fig. 2c). Nonetheless, the effects of MEK and PI3-kinase inhibition observed here could formally be attributable to inhibition of FGF2 signaling alone. To directly address this possibility, we performed a series of experiments in the absence of exogenous FGF2. Initially, as a baseline, cortical progenitors were cultured for 2 d with and without FGF2, and the cells were assayed for survival, proliferation, and neuronal differentiation. Results of these experiments confirmed, as described previously (Ghosh and Greenberg, 1995; Lukaszewicz et al., 2002), that exogenous FGF2 was essential to promote maximal survival and proliferation of cultured cortical progenitors (Fig. 7a). In the absence of FGF2, the percentage of TUNEL-positive cells was somewhat increased, and the percentage of Ki67-positive cells was decreased by >50%. Somewhat surprisingly, the percentage of βIII-tubulin but not MAP2-positive neurons increased (Fig. 7a), reflecting either an inhibitory effect of FGF2 on early neurogenesis or a preferential loss of proliferating progenitor cells in the absence of FGF2.
Figure 7.

Inhibition of endogenous neurotrophins, PI3-kinase, and MEK have similar effects in the absence of exogenous FGF2. a, Cortical progenitors were cultured for 2 d in the presence or absence of exogenous FGF2 (40 ng/ml), before analysis of apoptosis by TUNEL staining, proliferation by immunostaining for Ki67, or differentiation of neurons by immunocytochemistry forβIII-tubulin or MAP2. b, Cortical progenitors were treated with control IgY (40 μg/ml; IgY-Ctl), anti-NT-3 (20 μg/ml), or anti-BDNF (20 μg/ml) in the absence of exogenous FGF2 for 2 d before analysis similar to that shown in a. Graphs are representative results from one of three independent experiments. c, Cortical progenitors were treated with DMSO (1%), PD98059 (50 μm; PD), or LY294002 (50–100 μm; LY) for 2 d in the absence of exogenous FGF2 before analysis as in a. Graphs are representative results from one of three independent experiments. In all three panels, **p < 0.01; ***p < 0.001 (Student's t test in a; ANOVA in b and c). Error bars indicate SDs.
We then assayed these same parameters in cortical progenitors maintained without FGF2 but in the presence of function-blocking neurotrophin antibodies. These studies revealed that the survival of progenitors in the absence of FGF2 was primarily attributable to the autocrine—paracrine production of BDNF and NT-3; ∼80% of progenitors were TUNEL positive when either of these two neurotrophins were inhibited using function-blocking antibodies (Fig. 7b). Coincidentally with this dramatic increase in apoptosis, both the basal rate of proliferation and the proportion of neurons were reduced to <10% (Fig. 7b), effects likely attributable to the massive apoptosis.
Finally, we asked what effect inhibition of either PI3-kinase or MEK had on progenitors in the absence of FGF2. As seen with the inhibition of endogenous neurotrophins, the inhibition of PI3-kinase increased the percentage of TUNEL-positive cells to nearly 80% (Fig. 7c) while at the same time decreasing both the number of proliferating, Ki67-positive cells and the number of newly born neurons (Fig. 7c). In contrast, as would have been predicted, inhibition of MEK with 50 μm PD98059 had no effect on either survival or proliferation but significantly decreased the percentage of βIII-tubulin and MAP2-positive neurons that were generated (Fig. 7c). Although these studies do not definitively show that the neurotrophins are the only autocrine—paracrine factors that activate MEK and PI3-kinase in cortical progenitors, they strongly support the idea that endogenously produced neurotrophins mediate their effects via two distinct signaling proteins, PI3-kinase for survival and MEK for neurogenesis.
Discussion
The results presented here support three major conclusions. First, our data demonstrate the existence of a neurotrophin/Trk receptor autocrine—paracrine loop in cortical progenitor cells, in which the neurotrophins BDNF and NT-3 signal via the TrkB and/or TrkC receptors to activate their downstream intracellular effectors PI3-kinase and MEK. Second, using neurotrophin function-blocking antibodies, we show that endogenous BDNF and NT-3 are essential for cortical progenitor cell survival, even in the presence of exogenous FGF2. Third, our results with specific pharmacological inhibitors demonstrate that the Trk-mediated signaling pathways, PI3-kinase—Akt and MEK—ERK, subserve distinct biological functions in cortical progenitors. PI3-kinase is essential for survival, whereas MEK activation is essential for neurogenesis but not gliogenesis. Thus, cortical progenitors rely on endogenously produced neurotrophins and distinct Trk-mediated signaling pathways for multiple aspects of their biology, including survival and neurogenesis.
During embryogenesis, the proliferating precursor cells in the developing cortical neuroepithelium are exposed to a variety of different cues, which are integrated by these cells so that an appropriate number of cortical neurons are ultimately generated. These cues include growth factors, such as the neurotrophins FGF2 and epidermal growth factor (Ghosh and Greenberg, 1995; Burrows et al., 1997; Vaccarino et al., 1999; Raballo et al., 2000; Lukaszewicz et al., 2002), neurotransmitters, such as GABA, glutamate, acetylcholine, and PACAP (pituitary adenylate cyclase-activating polypeptide) (LoTurco et al., 1995; Antonopoulos et al., 1997; Haydar et al., 2000; Ma et al., 2000; Li et al., 2001; Suh et al., 2001), and a variety of cell contact-mediated signals, such as those involving Notch (Chambers et al., 2001; Shen et al., 2002). Together, these cues interact with the intrinsic cellular machinery to determine precursor cell survival, proliferation, and differentiation into neurons versus glia. Data presented here, together with previous work on FGF2 and PDGF, support the idea that growth factors that signal via receptor tyrosine kinases play a key role in regulating all three of these processes. FGF2 is clearly essential for precursor cell proliferation (Ghosh and Greenberg, 1995; Vaccarino et al., 1999; Raballo et al., 2000; Lukaszewicz et al., 2002), whereas the current work indicates that autocrine and/or paracrine NT-3 and BDNF function within the cortical neuroepithelium to promote survival. Moreover, previous work indicates that receptor tyrosine kinase signaling is essential for the genesis of neurons from cortical progenitor cells (Park et al., 1999; Ménard et al., 2002). The finding that the same class of signaling receptors promotes survival, proliferation, and differentiation raises the issue of biological specificity. In this regard, one of the surprising results reported here is that at least two distinct responses are subserved by different downstream signaling pathways, with PI3-kinase promoting survival and MEK promoting neurogenesis. Such segregation provides a mechanism whereby the integrated signal resulting from coactivation of a number of receptor tyrosine kinases could regulate precursor cell biology differentially.
In addition to the autocrine—paracrine effects reported here for cortical progenitor cells, neurotrophins continue to play an essential role for postmitotic cortical neurons, regulating survival, growth, phenotype, and ultimately, connectivity (Kaplan and Miller, 1997, 2000). In contrast to the peripheral nervous system, in which single neurotrophins play essential roles in regulating the biology of specific neuronal populations, such as NGF with sympathetic neurons (for review, see Klein, 1994; Snider, 1994), multiple neurotrophins appear to play redundant and overlapping roles for central neurons. Data presented here suggest that this is also true for cortical progenitors because these cells express both TrkB and TrkC in culture and in vivo (Maisonpierre et al., 1990; Ghosh and Greenberg, 1995; Fukumitsu et al., 1998), and BDNF and NT-3 apparently play similar roles in regulating cell survival (data presented here). It is likely that this functional overlap between neurotrophins, and potentially between other receptor tyrosine kinase ligands, such as FGF2, explains the relatively modest phenotypes observed in the developing cortex of single neurotrophin knock-out animals (Jones et al., 1994; Ringstedt et al., 1998; Kahn et al., 1999). Support for this idea derives from animals lacking TrkB and/or TrkC, in which numerous CNS abnormalities have been described previously (Minichiello and Klein, 1996; Alcantara et al., 1997; Martinez et al., 1998; Ringstedt et al., 1998; Kahn et al., 1999; Xu et al., 2000; Lotto et al., 2001). It will be interesting to further examine animals deficient in both receptors for deficits in cortical precursor cell survival and proliferation.
Our data indicate that the PI3-kinase pathway is a major survival pathway for cortical progenitor cells, a finding reminiscent of previous findings with neurotrophin-induced survival of postmitotic neurons (Kaplan and Miller, 1997, 2000). Specifically, PI3-kinase is the major survival pathway for cortical neurons under normal culture conditions (Hetman et al., 1999; Pozniak et al., 2002), suggesting that the PI3-kinase—Akt pathway is a conserved survival pathway throughout nervous system development. Additional support for this conclusion derives from a study using a nestin-driven conditional knock-out of the Pten tumor suppressor gene, whose product normally antagonizes the function of PI3-kinase; these animals displayed hyperactivation of Akt accompanied by increased proliferation and decreased apoptosis of telencephalic ventricular zone progenitors, with a resultant twofold increase in brain size and cell number by birth (Groszer et al., 2001). As has been reported for other CNS neurons, the PI3-kinase—Akt pathway could promote survival of cortical progenitors through the inhibition of Forkhead family members, BAD (Bcl-2-associated death protein), or glycogen synthase kinase-3β (Datta et al., 1997; Brunet et al., 1999; Hetman et al., 2000, 2002).
In contrast to the PI3-kinase pathway, the MEK pathway plays no role in cortical progenitor survival or proliferation but, instead, specifically regulates neurogenesis. These data are in agreement with our recent findings using a dominant-negative MEK adenovirus, in which we demonstrated that MEK was necessary for neurogenesis when cortical progenitors were stimulated with PDGF (Ménard et al., 2002), strongly supporting the idea that multiple receptor tyrosine kinases use the MEK pathway as a positive neurogenic signal. Moreover, data presented here showing that MEK inhibition had little or no effect on CNTF-induced astrocyte formation argues that MEK activation is not a generic differentiation pathway but is specific for neurogenesis. What are the downstream proneurogenic targets of MEK? One major target is the C/EBP family of transcription factors, which are essential for cortical progenitor cells to become neurons (Ménard et al., 2002). The lack of survival effect by the inhibition of the MEK pathway is reminiscent of studies using postmitotic cortical neurons. In these neurons, the MEK pathway was neuroprotective only during stress-induced apoptosis and not under the basal culture conditions (Hetman et al., 1999). Additional studies will be required to determine whether MEK signaling contributes to cortical progenitor survival under pathological conditions.
A number of studies have investigated previously the role of neurotrophins in cortical progenitor cell biology. One study reported that exogenous NT-3 promoted generation of MAP2-positive neurons and that a function-blocking NT-3 antibody inhibited neurogenesis (Ghosh and Greenberg, 1995), whereas another reported that exogenous NT-3 promoted cell cycle exit (Lukaszewicz et al., 2002). Although we have not examined the effects of exogenously added neurotrophins here, except to show that they cause an induction of the MEK—ERK and PI3-kinase—Akt pathways, such data are consistent with our finding that neurotrophin signaling via MEK is essential for neurogenesis. It was impossible, however, in the studies reported here to ask whether function-blocking antibodies to BDNF or NT-3 inhibited neurogenesis because these growth factors were essential for progenitor cell survival.
In summary, results presented here strongly support the idea that endogenously produced BDNF and NT-3 signal via TrkB and TrkC on cortical progenitor cells to promote survival and neurogenesis. They do so via two distinct and separable signaling pathways, the PI3-kinase and MEK pathways, which are common downstream substrates of many receptor tyrosine kinases. On the basis of these findings, we propose that neurotrophins play an important autocrine—paracrine role in determining progenitor cell biology in vivo in the embryonic telencephalon. Moreover, we propose that the PI3-kinase and MEK pathways provide points of convergence for multiple ligands that use tyrosine kinase receptors, such as FGF2, PDGF, and the neurotrophins, and, in so doing, provide one way that the complex extracellular environment of the neuroepithelium can be integrated to dynamically regulate survival, proliferation, and ultimately, the generation of neurons during development.
Footnotes
This work was supported by research grants from the Canadian Institute of Health Research (CIHR) to F.D.M.F.D.M. is a CIHR senior scientist, and F.B.H. is supported by CIHR and McGill Tomlinson studentships. We thank all of the members of the Miller laboratory for technical support and helpful discussions.
Correspondence should be addressed to Freda Miller, Black 3403, Hospital for Sick Children Research Institute, 555 University Avenue, Toronto, Ontario, Canada M5G 1X8. E-mail: fredam@sickkids.ca.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235149-12$15.00/0
References
- Alcantara S, Frisen J, del Rio JA, Soriano E, Barbacid M, Silos-Santiago I ( 1997) TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell death. J Neurosci 17: 3623–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonopoulos J, Pappas IS, Parnavelas JG ( 1997) Activation of the GABAA receptor inhibits the proliferative effects of bFGF in cortical progenitor cells. Eur J Neurosci 9: 291–298. [DOI] [PubMed] [Google Scholar]
- Benoit BO, Savarese T, Joly M, Engstrom CM, Pang L, Reilly J, Recht LD, Ross AH, Quesenberry PJ ( 2001) Neurotrophin channeling of neural progenitor cell differentiation. J Neurobiol 46: 265–280. [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME ( 1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868. [DOI] [PubMed] [Google Scholar]
- Burrows RC, Wancio D, Levitt P, Lillien L ( 1997) Response diversity and the timing of progenitor cell maturation are regulated by developmental changes in EGFR expression in the cortex. Neuron 19: 251–267. [DOI] [PubMed] [Google Scholar]
- Chambers CB, Peng Y, Nguyen H, Gaiano N, Fishell G, Nye JS ( 2001) Spatiotemporal selectivity of response to Notch1 signals in mammalian forebrain precursors. Development 128: 689–702. [DOI] [PubMed] [Google Scholar]
- Clayton GH, Perez GM, Smith RL, Owens GC ( 1998) Expression of mRNA for the elav-like neural-specific RNA binding protein, HuD, during nervous system development. Brain Res Dev Brain Res 109: 271–280. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME ( 1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231–241. [DOI] [PubMed] [Google Scholar]
- Ekholm SV, Reed SI ( 2000) Regulation of G(1) cyclin-dependent kinases in the mammalian cell cycle. Curr Opin Cell Biol 12: 676–684. [DOI] [PubMed] [Google Scholar]
- Farah MH, Olson JM, Sucic HB, Hume RI, Tapscott SJ, Turner DL ( 2000) Generation of neurons by transient expression of neural bHLH proteins in mammalian cells. Development 127: 693–702. [DOI] [PubMed] [Google Scholar]
- Ferguson KL, Vanderluit JL, Hebert JM, McIntosh WC, Tibbo E, MacLaurin JG, Park DS, Wallace VA, Vooijs M, McConnell SK, Slack RS ( 2002) Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J 21: 3337–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumitsu H, Furukawa Y, Tsusaka M, Kinukawa H, Nitta A, Nomoto H, Mima T, Furukawa S ( 1998) Simultaneous expression of brain-derived neurotrophic factor and neurotrophin-3 in Cajal-Retzius, subplate and ventricular progenitor cells during early development stages of the rat cerebral cortex. Neuroscience 84: 115–127. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME ( 1995) Distinct roles for bFGF and NT-3 in the regulation of cortical neurogenesis. Neuron 15: 89–103. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME ( 1994) Requirement for BDNF in activity-dependent survival of cortical neurons. Science 263: 1618–1623. [DOI] [PubMed] [Google Scholar]
- Gloster A, El-Bizri H, Bamji SX, Rogers D, Miller FD ( 1999) Early induction of Talpha1 alpha-tubulin transcription in neurons of the developing nervous system. J Comp Neurol 405: 45–60. [DOI] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X, Wu H ( 2001) Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo Science 294: 2186–2189. [DOI] [PubMed] [Google Scholar]
- Haydar TF, Wang F, Schwartz ML, Rakic P ( 2000) Differential modulation of proliferation in the neocortical ventricular and subventricular zones. J Neurosci 20: 5764–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempstead BL, Rabin SJ, Kaplan L, Reid S, Parada LF, Kaplan DR ( 1992) Overexpression of the trk tyrosine kinase rapidly accelerates nerve growth factor-induced differentiation. Neuron 9: 883–896. [DOI] [PubMed] [Google Scholar]
- Hetman M, Kanning K, Cavanaugh JE, Xia Z ( 1999) Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J Biol Chem 274: 22569–22580. [DOI] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z ( 2000) Role of glycogen synthase kinase-3β in neuronal apoptosis induced by trophic withdrawal. J Neurosci 20: 2567–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetman M, Hsuan SL, Habas A, Higgins MJ, Xia Z ( 2002) ERK1/2 antagonizes glycogen synthase kinase-3beta-induced apoptosis in cortical neurons. J Biol Chem 277: 49577–49584. [DOI] [PubMed] [Google Scholar]
- Jones KR, Farinas I, Backus C, Reichardt LF ( 1994) Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell 76: 989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn MA, Kumar S, Liebl D, Chang R, Parada LF, De Vellis J ( 1999) Mice lacking NT-3, and its receptor TrkC, exhibit profound deficiencies in CNS glial cells. Glia 26: 153–165. [PubMed] [Google Scholar]
- Kaplan DR, Miller FD ( 1997) Signal transduction by the neurotrophin receptors. Curr Opin Cell Biol 9: 213–221. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD ( 2000) Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10: 381–391. [DOI] [PubMed] [Google Scholar]
- Kee N, Sivalingam S, Boonstra R, Wojtowicz JM ( 2002) The utility of Ki-67 and BrdU as proliferative markers of adult neurogenesis. J Neurosci Methods 115: 97–105. [DOI] [PubMed] [Google Scholar]
- Klein R ( 1994) Role of neurotrophins in mouse neuronal development. FASEB J 8: 738–744. [DOI] [PubMed] [Google Scholar]
- Kohn J, Aloyz RS, Toma JG, Haak-Frendscho M, Miller FD ( 1999) Functionally antagonistic interactions between the TrkA and p75 neurotrophin receptors regulate sympathetic neuron growth and target innervation. J Neurosci 19: 5393–5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasorella A, Noseda M, Beyna M, Yokota Y, Iavarone A ( 2000) Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature 407: 592–598. [DOI] [PubMed] [Google Scholar]
- Li BS, Ma W, Zhang L, Barker JL, Stenger DA, Pant HC ( 2001) Activation of phosphatidylinositol-3 kinase (PI-3K) and extracellular regulated kinases (Erk1/2) is involved in muscarinic receptor-mediated DNA synthesis in neural progenitor cells. J Neurosci 21: 1569–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotto RB, Asavaritikrai P, Vali L, Price DJ ( 2001) Target-derived neurotrophic factors regulate the death of developing forebrain neurons after a change in their trophic requirements. J Neurosci 21: 3904–3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoTurco JJ, Owens DF, Heath MJ, Davis MB, Kriegstein AR ( 1995) GABA and glutamate depolarize cortical progenitor cells and inhibit DNA synthesis. Neuron 15: 1287–1298. [DOI] [PubMed] [Google Scholar]
- Lu QR, Yuk D, Alberta JA, Zhu Z, Pawlitzky I, Chan J, McMahon AP, Stiles CD, Rowitch DH ( 2000) Sonic hedgehog-regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron 25: 317–329. [DOI] [PubMed] [Google Scholar]
- Lu QR, Cai L, Rowitch D, Cepko CL, Stiles CD ( 2001) Ectopic expression of Olig1 promotes oligodendrocyte formation and reduces neuronal survival in developing mouse cortex. Nat Neurosci 4: 973–974. [DOI] [PubMed] [Google Scholar]
- Lukaszewicz A, Savatier P, Cortay V, Kennedy H, Dehay C ( 2002) Contrasting effects of basic fibroblast growth factor and neurotrophin 3 on cell cycle kinetics of mouse cortical stem cells. J Neurosci 22: 6610–6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Maric D, Li BS, Hu Q, Andreadis JD, Grant GM, Liu QY, Shaffer KM, Chang YH, Zhang L, Pancrazio JJ, Pant HC, Stenger DA, Barker JL ( 2000) Acetylcholine stimulates cortical precursor cell proliferation in vitro via muscarinic receptor activation and MAP kinase phosphorylation. Eur J Neurosci 12: 1227–1240. [DOI] [PubMed] [Google Scholar]
- Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD ( 1990) NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron 5: 501–509. [DOI] [PubMed] [Google Scholar]
- Martinez A, Alcantara S, Borrell V, Del Rio JA, Blasi J, Otal R, Campos N, Boronat A, Barbacid M, Silos-Santiago I, Soriano E ( 1998) TrkB and TrkC signaling are required for maturation and synaptogenesis of hippocampal connections. J Neurosci 18: 7336–7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ménard C, Hein P, Paquin A, Savelson A, Yang XM, Lederfein D, Barnabé-Heider F, Mir AA, Sterneck E, Peterson AC, Johnson PF, Vinson C, Miller FD ( 2002) An essential role for a MEK-C/EBP pathway during growth factor-regulated cortical neurogenesis. Neuron 36: 597–610. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Klein R ( 1996) TrkB and TrkC neurotrophin receptors cooperate in promoting survival of hippocampal and cerebellar granule neurons. Genes Dev 10: 2849–2858. [DOI] [PubMed] [Google Scholar]
- Nieto M, Schuurmans C, Britz O, Guillemot F ( 2001) Neural bHLH genes control the neuronal versus glial fate decision in cortical progenitors. Neuron 29: 401–413. [DOI] [PubMed] [Google Scholar]
- Park JK, Williams BP, Alberta JA, Stiles CD ( 1999) Bipotent cortical progenitor cells process conflicting cues for neurons and glia in a hierarchical manner. J Neurosci 19: 10383–10389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozniak CD, Barnabé-Heider F, Rymar VV, Lee AF, Sadikot AF, Miller FD ( 2002) p73 is required for survival and maintenance of CNS neurons. J Neurosci 22: 9800–9809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Shen Q, Goderie SK, He W, Capela A, Davis AA, Temple S ( 2000) Timing of CNS cell generation: a programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron 28: 69–80. [DOI] [PubMed] [Google Scholar]
- Raballo R, Rhee J, Lyn-Cook R, Leckman JF, Schwartz ML, Vaccarino FM ( 2000) Basic fibroblast growth factor (Fgf2) is necessary for cell proliferation and neurogenesis in the developing cerebral cortex. J Neurosci 20: 5012–5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringstedt T, Linnarsson S, Wagner J, Lendahl U, Kokaia Z, Arenas E, Ernfors P, Ibanez CF ( 1998) BDNF regulates reelin expression and Cajal-Retzius cell development in the cerebral cortex. Neuron 21: 305–315. [DOI] [PubMed] [Google Scholar]
- Sauvageot CM, Stiles CD ( 2002) Molecular mechanisms controlling cortical gliogenesis. Curr Opin Neurobiol 12: 244–249. [DOI] [PubMed] [Google Scholar]
- Scholzen T, Gerdes J ( 2000) The Ki-67 protein: from the known and the unknown. J Cell Physiol 182: 311–322. [DOI] [PubMed] [Google Scholar]
- Shen Q, Zhong W, Jan YN, Temple S ( 2002) Asymmetric numb distribution is critical for asymmetric cell division of mouse cerebral cortical stem cells and neuroblasts. Development 129: 4843–4853. [DOI] [PubMed] [Google Scholar]
- Slack RS, El-Bizri H, Wong J, Belliveau DJ, Miller FD ( 1998) A critical temporal requirement for the retinoblastoma protein family during neuronal determination. J Cell Biol 140: 1497–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider WD ( 1994) Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell 77: 627–638. [DOI] [PubMed] [Google Scholar]
- Suh J, Lu N, Nicot A, Tatsuno I, DiCicco-Bloom E ( 2001) PACAP is an anti-mitogenic signal in developing cerebral cortex. Nat Neurosci 4: 123–124. [DOI] [PubMed] [Google Scholar]
- Sun Y, Nadal-Vicens M, Misono S, Lin MZ, Zubiaga A, Hua X, Fan G, Greenberg ME ( 2001) Neurogenin promotes neurogenesis and inhibits glial differentiation by independent mechanisms. Cell 104: 365–376. [DOI] [PubMed] [Google Scholar]
- Toma JG, El-Bizri H, Barnabé-Heider F, Aloyz R, Miller FD ( 2000) Evidence that helix-loop-helix proteins collaborate with retinoblastoma tumor suppressor protein to regulate cortical neurogenesis. J Neurosci 20: 7648–7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccarino FM, Schwartz ML, Raballo R, Nilsen J, Rhee J, Zhou M, Doetschman T, Coffin JD, Wyland JJ, Hung YT ( 1999) Changes in cerebral cortex size are governed by fibroblast growth factor during embryogenesis. Nat Neurosci 2: 246–253. [DOI] [PubMed] [Google Scholar]
- Wartiovaara K, Barnabé-Heider F, Miller FD, Kaplan DR ( 2002) N-myc promotes survival and induces S-phase entry of postmitotic sympathetic neurons. J Neurosci 22: 815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu B, Zang K, Ruff NL, Zhang YA, McConnell SK, Stryker MP, Reichardt LF ( 2000) Cortical degeneration in the absence of neurotrophin signaling: dendritic retraction and neuronal loss after removal of the receptor TrkB. Neuron 26: 233–245. [DOI] [PubMed] [Google Scholar]