

Abstract
Estrogen influences the development and function of the nervous system through estrogen receptor-dependent changes in gene expression and by rapidly influencing diverse intracellular signaling pathways. We have investigated the influence of estradiol on developing neonatal rat cerebellar neurons in primary culture and found that low concentrations of 17β-estradiol (17β-E2), 17α-E2, 17β-E2-BSA, and ICI182,780 stimulated phosphorylation of the extracellular signal-regulated kinases 1/2 (ERK1/2) mitogen-activated protein kinases (MAPK). Neither testosterone nor progesterone increased ERK1/2 phosphorylation. The effects of the estrogens were specific to the ERK1/2 MAPK pathway and were blocked by U0126, an inhibitor of the ERK1/2 MAPK kinase (MEK1/2). Compared with control cultures, significant MAPK-dependent decreases in viable granule cell numbers were observed in dissociated explant cultures of developing cerebellar neurons 24–96 hr after pulse treatment with 10 pm 17β-E2 or 10 nm ICI182,780. In contrast, continuous exposure to 10 pm 17β-E2 significantly increased granule cell numbers. Analysis of bromodeoxyuridine incorporation revealed that a 15 min pulsed treatment with 10 pm 17β-E2 increased mitogenesis, whereas continuous exposure to the same concentration of 17β-E2 was anti-mitotic. Estradiol did not increase caspase activity; however, significant increases in cellular permeability and lysis were observed. Cell lysis and death were independent of the pan-caspase inhibitor zVAD-fmk but were blocked fully by the irreversible calpain inhibitor PD150606. These results indicate that rapid activation of the ERK1/2 MAPK pathway by low concentrations of 17β-E2 induces oncotic/necrotic, but not apoptotic, programmed cell death in a subpopulation of developing granule cells and increased mitogenesis of the granule cell neuroblasts refractory to estrogen-induced neurotoxicity.
Keywords: cerebellum, development, ERK, estradiol, estrogen, granule cell, MAPK, necrosis, neuron, neurotoxicity, oncosis
Introduction
In the brain 17β-estradiol (17β-E2) is an important regulator of development and function of neurons and glia. Many estrogenic actions are mediated via intracellular estrogen receptors (ERs) that function as ligand-activated transcription factors to regulate the expression of estrogen-responsive genes (Evans, 1988; Paech et al., 1997). Numerous studies also have demonstrated that estradiol and other estrogenic compounds can activate a diverse array of intracellular signaling mechanisms rapidly via membrane-initiated mechanisms. For example, 17β-E2 can activate adenylate cyclase rapidly, increase intracellular [Ca2+], activate phospholipase C to generate inositol 1,4,5-trisphosphate and diacylglycerol, stimulate nitric oxide synthase to generate nitric oxide, and activate the extracellular signal-regulated kinases 1/2 (ERK1/2) mitogen-activated protein kinase (MAPK) pathway (for review, see Falkenstein et al., 2000; Belcher and Zsarnovszky, 2001). The molecular mechanisms of these rapid membrane-initiated actions are not understood fully. In some cell types rapid estradiol effects may be initiated by the binding of 17β-E2 at membrane-associated ERs that are related closely to the “classical” intracellular receptors (Levin, 1999, 2001; Norfleet et al., 1999; Razandi et al., 1999; Watson et al., 1999). However, there is additional evidence indicating that some rapid actions are independent of the intracellular ERs and may involve the activation of G-protein-coupled receptor-dependent mechanisms (Falkenstein et al., 2000; Filardo et al., 2000; Schmidt et al., 2000; Nadal et al., 2001).
Previous studies from our laboratory have demonstrated that estrogen receptors are expressed in the developing and adult rat cortex and cerebellum and that the expression of those receptors is regulated dynamically throughout critical periods of cortical and cerebellar development (Belcher, 1999; Jakab et al., 2001; Zsarnovszky and Belcher, 2001). To investigate whether estradiol impacts developing neurons from the hindbrain, we determined the effects that exposure to estradiol and other related steroids have on the growth factor-regulated MAPK signaling pathways in primary cultures of developing and mature cerebellar granule cells. Specifically, we have characterized the pharmacological effects that various steroid hormones and the estradiol derivatives 17α-E2 and ICI182,780 (a pure ER antagonist) have on rapid MAPK signaling in mature cultures of cerebellar granule cells. In dissociated explant cultures of immature granule cells the effects on proliferation and viability of pulsed or continuous exposure to low concentrations of estradiol and ICI182,720 were determined also. The results of our studies indicate that picomolar to nano molar concentrations of these estrogens rapidly and specifically can induce a transient increase in ERK1/2 phosphorylation. We further found that brief exposures to these low concentrations of 17β-E2 or ICI182,780 result in a MAPK-dependent increase in oncotic/necrotic granule cell death, but estradiol also causes increased mitosis in the surviving granule cell neuroblasts. In contrast, continuous exposure to estradiol was anti-mitotic; however, this treatment resulted in increased granule cell viability, presumably via ER-mediated neuroprotective mechanisms that were independent of ERK1/2. In mature postmitotic granule cell neurons 17β-E2 did not influence viability or cell death.
Materials and Methods
Animals. Timed pregnant Sprague Dawley rats were obtained from the supplier (Charles River, Wilmington, MA) at least 48 hr before they gave birth. The postnatal age of the litter was calculated by using the day on which pups first appeared as postnatal day 0 (P0). On P7–P9 the sex and weight of each age-matched animal were determined and recorded before granule cell preparation. At this age the mean weight of the pups was 18–20 gm; pups weighing significantly less than the average were excluded from study. All animal procedures were performed in accordance with protocols approved by the University of Cincinnati Institutional Animal Care and Use Committee and followed National Institutes of Health guidelines.
Preparation of primary cultures of cerebellar neurons. Primary cerebellar cultures were prepared from P7–P9 male or female Sprague Dawley rat pups without enzymatic treatment and were maintained under serum- and steroid-free conditions as previously described (Wong et al., 2001). Cerebellar cells were diluted serially in an appropriate volume of culture media and seeded at an initial density of 1.5 × 10 5 granule cells/cm 2. For analysis of mature primary cerebellar granule cells a final concentration of 10 μm cytosine β-d-arabinofuranoside (AraC; Sigma, St. Louis, MO) was added 24 hr after seeding to inhibit the proliferation of non-neuronal cells; treatments were performed after 7 d in culture. Cell counting indicated that greater than ∼90–95% of the cells contained in these cultures were granule cell neurons (Wong et al., 2001). Dissociated primary explant cultures were prepared identically to primary cultures of granule cell neurons except that AraC was not added and drug treatments were performed immediately after cell attachment.
Drug treatments. Cerebellar cultures were exposed for various times to 1,3,5(10)-estratrien-3,17β-diol (17β-estradiol), 1,3,5(10)-estratrien-3,17α-diol (17α-estradiol), 4-pregnen-3,20-dione (progesterone, Sigma), 4-androsten-17β-ol-3-one (testosterone, Steraloids, Newport, RI), or 7 α-[9-(4,4,5,5,5-pentafluoro-pentylsulphinyl)nonyl]oestra-1,3,5(10)-triene-3,17β-diol (ICI182,780) (Tocris Cookson, Ellisville, MO) at indicated concentrations (serially diluted in DMSO vehicle). 1,3,5(10)-Estratrien-3,17β-diol 17-hemisuccinate/BSA (E2-BSA; Sigma) and BSA (1 μm, fraction V, BSA; USB, Amersham Biosciences, Cincinnati, OH) were diluted in PBS, pH 7.4, with potentially contaminating free estradiol removed by microfiltration (30 kDa cutoff; Micron YM-30, Millipore, Bedford, MA). For pulse-chase estradiol treatments, after incubation with drug, the treatment medium was removed; then the cultures were washed with phenol red-free HBSS (Invitrogen, Carlsbad, CA) or PBS, and fresh estrogen-free culture medium was added.
Positive controls for ERK1/2 activation included treatment with 10% fetal calf serum (Invitrogen) or brain-derived neurotrophic factor (BDNF; 100 ng/ml; Promega, Madison, WI); for activation of p38 and JNK the cultures were exposed to 30 ng/ml anisomycin for 30 min. The involvement of the MAPK signaling pathway was assessed in cultures that were pretreated for 30 min with the MEK1 inhibitor U0126 (10 μm; Promega). Additional control or antagonist treatments included staurosporine (100 nm; Sigma), zVAD-fmk (12.5 μm; Promega), and PD150606 (50 μm; Calbiochem, La Jolla, CA), with each serially diluted to the desired concentrations in the appropriate vehicle. In experiments that used antagonists the vehicle-treated negative controls were exposed to concentrations of vehicle identical to those present in experimental cultures receiving both antagonist and agonist.
Generation of cerebellar cell lysates and Western blot analysis. After treatment the medium was aspirated, and the attached cells were washed with ice-cold HBSS or PBS. Cells were detached on ice with cold 2 mm EDTA in PBS and collected at 2°C by centrifugation at 600 × g for 10 min. Pelleted cells were resuspended and homogenized on ice in (in mm) 20 Tris-HCl, pH 7.5, 150 NaCl, 1 PMSF, 1 EDTA, 1 EGTA, 2.5 sodium pyrophosphate, 1 β-glycerol phosphate, and 1 Na3VO4 plus 1 mg/ml Pefabloc, 10 μg/ml leupeptin, 10 μg/ml pepstatin, 1 μg/ml aprotinin, 1% Triton X-100, 0.05% sodium deoxycholate, and phosphatase inhibitor mixture 1 (Sigma). Homogenates were sonicated on ice for 5 sec a total of six times. Alternatively, cell lysis was achieved by three freeze/thaw cycles in liquid N2. Lysates were cleared by centrifugation at 14,000 × g for 1 min at 2°C. Total protein present in each lysate was quantified by using a modified Lowry assay (DC protein assay; Bio-Rad, Hercules, CA).
SDS-PAGE, Western blotting, and densitometric analysis were performed by standard protocols (Jakab et al., 2001). For ERK1/2 analysis 5–10 μg/lane of each protein lysate was fractionated on 10% gels by SDS-PAGE and then electrotransferred to nitrocellulose or polyvinylidene difluoride membranes. For analysis of the stress-activated protein kinase/Jun terminal kinase (SAPK/JNK) and p38 MAP kinase activation, 50 μg/lane of control or cerebellar lysate was analyzed. Membranes were blocked with 5% nonfat dry milk for 1 hr in TBS-T and incubated with appropriate phospho-specific antisera (Table 1) overnight at 4°C. Antigens bound by primary antibodies were detected with appropriate HRP-conjugated anti-IgG secondary antibodies (1:30,000 dilution; Kirkegaard and Perry Laboratories, Gaithersburg, MD), and immunoreactive bands were visualized onto preflashed x-ray film by enhanced chemiluminescence, using the SuperSignal West Pico Substrate (Pierce, Rockford, IL). Multiple exposures of each blot were collected, and those in the linear range of the film were used for densitometric analysis. Digital images of appropriate films were captured with the EDAS290 imaging system (Kodak, New Haven, CT), and the optical density of each immunoreactive band was determined with Kodak 1D Image Analysis software. Optical densities were calculated as arbitrary units after local area background subtraction, normalized to the density of the phospho-independent MAPK immunoreactivity, and reported as fold induction relative to control.
Table 1.
Primary antisera
|
Antigen |
Source |
Species |
Designation |
Dilution |
|---|---|---|---|---|
| Phospho-ERK1/2 (Thr202/Tyr204) | Cell Signaling | Rabbit | 9101 | 1:1000; 1:400a |
| ERK1 (K-23) | Santa Cruz | Rabbit | SC-94 | 0.2 μg/ml |
| ERK2 (C-14) | Santa Cruz | Rabbit | SC-154 | 0.2 μg/ml |
| Phospho-p38 (Thr180/Tyr182) | Cell Signaling | Rabbit | 9211 | 1:1000 |
| p38 MAP kinase | Cell Signaling | Rabbit | 9212 | 1:1000 |
| Phospho-SAPK/JNK (Thr183/Tyr185) | Cell Signaling | Rabbit | 9251 | 1:1000 |
| SAPK/JNK | Cell Signaling | Rabbit | 9252 | 1:1000 |
| GFAP
|
Sigma
|
Mouse
|
GA5
|
1:1000a
|
Dilution used for immunocytochemistry.
Immunocytochemical analysis of MAPK activation. Cerebellar cell cultures were prepared from 8-d-old female Sprague Dawley rat pups as described above and seeded into poly-l-lysine-coated eight-well chamber slides (Lab Tek; Nunc, Naperville, IL) at a density of 100,000–140,000 cells/cm 2 in a final volume of 300 μl/well. Cultures were maintained in a humidified incubator in 5% CO2 at 37°Cfor7dinthe presence of a final concentration of 10 μm AraC. On the seventh day in culture one-half of the culture medium was removed from each experimental and vehicle control well. A final concentration of 2 × 10-11m 17β-estradiol or an equal volume of DMSO vehicle was added to the removed medium, mixed, and readded to the wells. Culture media were mixed gently by orbital rotation for 30 sec and incubated in 5% CO2 at 37°C for 13 min. This treatment resulted in a total exposure time of 15 min. A third group of control cultures remained untreated. After incubation the cultures were fixed with -20°C methanol for 20 min and washed with PBS (3 × 10 min). Cultures then were incubated at room temperature for 2 hr with anti-phospho-ERK1/2 and anti-glial fibrillary acidic protein (GFAP) primary antibodies (Table 1). After 3 × 10 min washes in PBS the cultures were incubated with FITC-conjugated goat anti-mouse and Texas Red-conjugated goat anti-rabbit IgG antisera (Jackson ImmunoResearch Laboratories, West Grove, PA), each at a dilution of 1:60 in PBS/0.2% Triton X-100. Cultures were washed thoroughly in PB and then coverslipped with Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA). Microscopic examination of immunostained material was performed with a Nikon TE 2000 inverted microscope. Fluorescence of FITC, Texas Red, and DAPI was examined by epifluorescence microscopy, using a filter configuration for sequential excitation/emission imaging via 488, 568, and 405 nm channels (Chroma Technology, Brattleboro, VT). Digital images were captured with a SpotRT (Media Cybernetics, Silver Spring, MD) CCD camera, using Image-Pro Plus version 4.5 software. Final graphics were generated and labeled with Photoshop version 6.01 (Adobe, San Jose, CA) software, with triple-stained material generated by an overlay of the individual blue, red, and green images.
Analysis of granule cell viability. Dissociated explant cultures were seeded onto 24-well plates and exposed to drug or vehicle for the indicated times. For each sample the culture medium was collected before cell counting, and the amount of lactate dehydrogenase (LDH) released into the medium as a result of cell lysis was determined as previously described (Wong et al., 2001). Cells then were washed and detached by incubation on ice for 2 min with 200 μl of ice-cold 2 mm EDTA in PBS, pelleted by centrifugation at 600 × g, resuspended into an appropriate volume of HBSS, and lightly stained with trypan blue. Viable granule cell numbers were determined by direct cell counting of small (∼5 μm diameter) trypan blue-excluding granule cells with a hematocytometer, using standard methods. For each experiment the mean ± SEM of four independent samples was determined for each treatment group; triplicate cell counts were made for each sample, with each experiment repeated at least three times. Granule cell viability also was assessed by the previously described MTS reduction assay (Wong et al., 2001).
Analysis of propidium iodide permeability. Cell cultures were seeded and maintained as described above in 60 mm cell culture dishes in 3 ml of growth medium. At 3 hr after seeding the granule cell cultures were pulse-treated for 15 min with a final concentration of 10-11m 17β-estradiol or an equal volume of DMSO vehicle. After treatment the medium was removed, and the cultures were washed two times with steroid-free growth medium. A final volume of 3 ml of steroid-free growth medium was added, and the cultures were incubated at 37°C in 5% CO2 until the required time after treatment. At each time point that was analyzed, a final concentration of 1.5 μm propidium iodide (PI; Molecular Probes, Eugene, OR) was added to each culture. After a 4 min incubation period digital red fluorescent images were captured, and PI-permeable cell numbers were determined (Image-Pro software, version 4.5) for each sample. Results for each time point are reported as the mean values from six randomly selected 0.27 mm 2 fields from three different samples.
Analysis of granule cell proliferation: BrdU incorporation. Dissociated explant cultures were seeded into 96-well plates without AraC, allowed to attach for 2 hr, treated with 10-11m 17β-estradiol or DMSO vehicle for 15 min, washed with HBSS, and then incubated with fresh medium containing a final concentration of 10 μm 5-bromo-2′-deoxyuridine (BrdU). Additional cultures were exposed to 10-11m estradiol or vehicle for the entire incubation period. The amount of BrdU incorporated into newly synthesized DNA at each time point was monitored by an ELISA-based approach (BrdU Labeling and Detection Kit III, Roche Molecular Biochemicals, Indianapolis, IN).
Analysis of caspase-3 activity. Dissociated explant cultures were seeded in 60 mm dishes at 1.5 × 10 5/cm 2 and allowed to attach for 2 hr. Cultures were treated with 17β-estradiol for 15 min and washed with HBSS; treatment medium was replaced with fresh steroid-free medium and then incubated for 24 hr. Staurosporine-treated (100 nm) cultures were used as positive controls, and additional control cultures were treated with the pan-caspase inhibitor zVAD-fmk (12.5 μm). After incubation the cell lysates were prepared, and caspase-3 activity was determined for 40 μgof lysate on the basis of the liberation of a colored p-nitroaniline (pNA) from the caspase-3 substrate Ac-DEVD-pNA (Colorimetric CaspACE Assay System, Promega). Specific caspase-3 activity (pmol of pNA/hr per microgram of protein) of each sample was calculated from a standard pNA curve generated from a dilution series of known concentrations of pNA.
Statistical analysis. Unless noted otherwise, all data that have been presented are representative of at least three independent experiments. Statistical analysis was conducted with a Student's t test or by one-way ANOVA with post-test comparison, using Newman–Keuls or Dunnett's multiple comparison tests as appropriate. The level of statistical significance between treatment groups is indicated as follows: *p < 0.05; **p < 0.01; ***p < 0.001. Data were analyzed with Excel (Microsoft, Redmond, WA) and GraphPad Prism version 3.01 (GraphPad Software, San Diego, CA)
Results
Preliminary studies with an accepted physiological concentration of estradiol (10-8 M) indicated that the ERK1/2 MAPK signaling pathway in primary cultures of cerebellar neurons was variably responsive to estradiol treatment (data not shown). Dose–response analysis of rapid 17β-E2-induced ERK1/2 activation was performed to characterize fully the rapid activation of ERK1/2 by estradiol (Fig. 1). In granule cell cultures that were exposed to various concentrations of 17β-E2 for 10 min, analysis of multiple experiments revealed that 17β-E2 stimulated significant ERK1/2 activation in two different concentration ranges. An initial biphasic or hormetic stimulation of ERK phosphorylation was observed at 10-11 to 10-10 M, and a second significant increase in MAPK signaling was induced by high (10-7 to 10-6 M) concentrations of 17β-E2 (Fig. 1A,B). A highly variable and insignificant responsiveness was observed at the 10-8m concentration.
Figure 1.

The dose dependency and kinetics of 17β-estradiol-induced ERK1/2 activation. A, Estradiol dose–response analysis: representative Western blots. Shown are protein lysates (∼5 μg/lane) from cerebellar granule cell cultures treated for 10 min with vehicle (V10′) or increasing concentrations of 17β-E2 (10-14 to 10-7m). Phospho-ERK-IR was detected with antiserum specific for the dually phosphorylated (top panel) active form of ERK1/2 (pERK). For normalization the blots were reanalyzed with phosphorylation state-independent ERK1/2 antiserum. Estradiol concentrations are indicated above each lane, and the positions of ERK1-IR and ERK2-IR are indicated to the left of each panel. B, Estradiol dose–response analysis: densitometric analysis. Levels of 17β-E2-induced ERK1/2 activation, expressed as fold increase above the baseline of vehicle-treated control,were determined by densitometric analysis of pERK1-IR and pERK2-IR. Results are expressed as the means ± SEM (n = 3–9) of the fold induction as compared with control from nine independent experiments. Baseline levels are indicated with a dashed line. C, D, Time course analysis. ERK1/2 phosphorylation was increased significantly 10 min after the addition of 10-11m 17β-E2. With longer 17β-E2 exposures the phosphorylation rapidly returned to baseline or lower levels. For each experiment the results are normalized to total ERK-IR and are the mean fold induction ± SEM of at least four different experiments. *,***See Materials and Methods.
At the lowest concentration of 17β-E2 that induces maximal effects (10-11 M), the time dependence of 17β-E2-stimulated ERK1/2 phosphorylation was investigated (Fig. 1C,D). Increased ERK1/2 phosphorylation was often detectable as early as 2 min after treatment, with increases reaching significance by 10 min. Increases in ERK1/2 phosphorylation were transient and returned to baseline levels or below after 15 min of exposure. Neither the time course nor dose dependency of 17β-E2-induced ERK1/2 activation was influenced by the sex of the animal from which the granule cell cultures were derived.
To determine whether cell-impermeable 17β-E2 elicits a rapid increase in ERK activation, we treated granule cell cultures with 17β-E2 covalently bound to BSA (E2-BSA). As was observed for free 17β-E2, after a 10 min treatment with E2-BSA (17β-E2 concentration of 10-11 M) a rapid increase in ERK1/2 phosphorylation was observed (Fig. 2A), suggesting that 17β-E2-induced ERK1/2 activation is initiated at the extracellular surface of the plasma membrane.
Figure 2.

Analysis of the specificity and nature of 17β-estradiol-induced ERK1/2 activation. A, Cell-impermeable 17β-E2-BSA induces ERK1/2 phosphorylation. Shown are representative Western blots of protein lysates sequentially probed for pERK1/2 and total ERK1/2. Lysates were isolated from granule cell cultures after a 10 min treatment with equal molar concentrations of BSA or BSA-conjugated 17β-E2 (final 17β-E2 concentration of 10-11m). B, Representative Western blots of cell lysates from cultures treated with 10-11m 17β-E2 (E2-10′) or vehicle (V) for 10 min with or without a 30 min preexposure to 10 μm U0126 or vehicle. The presence of U0126 blocks estrogen-induced ERK phosphorylation, indicating that estrogen effects are MEK-dependent and mediated via the MAPK pathway. C, Analysis of estradiol-induced SAPK/JNK MAPK activation. Representative Western blot analyses demonstrate that treatment with 10-11m 17β-E2 does not induce detectable increases rapidly in SAPK/JNK MAPK activation. HEK 293 cell lysates exposed (lane 1) or unexposed (lane 2) to UV light and granule cell cultures treated for 30 min with 30 ng/ml anisomycin (lane 3) served as controls. Granule cell cultures were treated with vehicle for 10 min (lane 4; V) or 10-11m 17β-estradiol for 10 and 30 min (lanes 5, 6, respectively). The top panel was probed with antiserum specific for the phosphorylated forms of JNK1 and JNK2. The bottom panel was probed with antiserum that recognizes JNK2 independently of phosphorylation state. The positions of the 54 and 46 kDa JNK1 and JNK2 immunoreactive bands are indicated. D, Analysis of estradiol-induced p38 MAPK activation. Representative Western blot analyses demonstrate that treatment with 10-11m 17β-E2 does not induce detectable increases in p38 MAPK activation. Activation of the p38 MAP kinase was analyzed with phospho-p38 antiserum (p-p38) as described for JNK1/2 and similarly was found to be unaffected by 17β-E2 treatment. All presented results are representative of at least three independent experiments.
The involvement and specificity of the MAPK signaling pathway also was confirmed. In the presence of 10 μm U0126, a specific inhibitor of the upstream MAPK kinase (MEK), ERK1/2 responsiveness to 17β-E2 was abrogated completely (Fig. 2B). Western blot analysis that used phosphorylation state-specific antisera for the SAPK/JNK and p38 MAP kinases indicated that SAPK/JNK and p38 were not activated by 17β-E2 treatment (Fig. 2C,D), thus demonstrating that the rapid MAPK response to estradiol is specific to the ERK1/2 MAPK pathway.
Because these granule cell cultures contain a small number (<5% of the total cells) of contaminating glia-like cells (Wong et al., 2001), immunofluorescence analysis of 17β-E2-induced ERK activation was performed to identify unambiguously which cell type was responsive to 17β-E2. In cultures treated with 10-11m 17β-E2 for 15 min, increased MAPK phosphorylation was detected in only the small granule cell neurons (Fig. 3A–D). Further, in cultures immunostained for both phosphorylated ERK1/2 and the glia-specific antigen GFAP, increased phosphorylated ERK1/2 was observed only in the GFAP-negative granule cells (Fig. 3E,F), clearly demonstrating that in granule cell neurons, but not cerebellar glia, ERK1/2 phosphorylation was increased rapidly by estradiol treatment.
Figure 3.

Immunofluorescent analysis of the cell-type specificity of estradiol-induced rapid MAPK phosphorylation in cerebellar granule cell cultures. Shown are representative photomicrographs of control (left) and experimental (right) cerebellar granule cell cultures treated with vehicle or 10-11m 17β-E2 for 15 min. Bright-field images of control (A) and 17β-E2-treated (B) cultures and corresponding epifluorescent images were immunostained for phospho-ERK1/2 and visualized with a Texas Red-conjugated goat anti-rabbit secondary antibody (C, control; D, 17β-E2-treated). Estradiol treatment resulted in increased cytoplasmic and nuclear staining of granule cells (white arrows), whereas only scattered cells with markedly lower levels of fluorescence could be seen in untreated or vehicle-treated cultures (white arrowheads). Asterisk indicates a dead granule cell that shows homogeneous autofluorescence. Phospho-ERK1/2 immunoreactivity was not detected in GFAP-immunopositive (green fluorescence) glia present in vehicle-treated (E) or E2-treated (F) cultures, whereas increased phospho-ERK1/2 staining is apparent in the granule cells (F, white arrow). Cell nuclei were stained blue with DAPI. Scale bars, 15 μm.
To characterize further the pharmacological properties of 17β-E2-induced ERK1/2 phosphorylation in cerebellar granule cells, we examined the responsiveness to other steroids. As was observed for 17β-E2, the “nonestrogenic” estradiol isomer 17α-estradiol (17α-E2) rapidly induced ERK1/2 phosphorylation (Fig. 4A). At concentrations that induced maximal effects, 17α-E2 and 17β-E2 were comparably efficacious; however, 17α-E2 was markedly less potent than 17β-E2, with a single peak of maximal activation observed in the concentration range of 10-9 to 10-8 M. The phosphorylation state of ERK1/2 in cerebellar granule cells was unresponsive to progesterone or testosterone across a concentration range of 10-13 to 10-6m (Fig. 4C,D).
Figure 4.

Dose–response analysis of ERK1/2 activation by 17α-estradiol, progesterone, and testosterone. Shown are representative Westernresults of ERK1/2 activation analysis in primary cultures of cerebellar granule cells treated for 10 min with increasing concentrations of the nonestrogenic estrogen isomer 17α-estradiol (A), progesterone (B), or testosterone (C). ERK1/2 phosphorylation was induced rapidly by 17α-E2. Maximal activation was right-shifted by two orders of magnitude as compared with 17β-E2, with significant (p < 0.05; n = 3) stimulation of ERK1/2 phosphorylation at 10-9 and 10-8m 17α-E2 but with insignificant effects at the higher concentrations. Neither progesterone nor testosterone stimulated ERK1/2 phosphorylation. Results are representative of at least three experiments.
When cultures were cotreated or pretreated for 5 min with 1 μm ICI182,780, 17β-E2-induced MAPK phosphorylation was not blocked at any 17β-E2 concentrations that were tested (data not shown). However, when cultures were pretreated with ICI182,780 for 30 min, increases in ERK1/2 activation were not detectable 10 or 30 min after treatment with 10-11m 17β-E2 (Fig. 5A). On the basis of the rapid and transient kinetics of estradiol-induced ERK1/2 activation, we hypothesized that ICI182,780 also was inducing rapid and transient ERK1/2 phosphorylation. Analysis of ICI182,780 dose dependency experiments demonstrated that a 10 min exposure to 10-9 to 10-7m ICI182,780 increased ERK1/2 phosphorylation (Fig. 5B). Further, time course studies indicated that 10-8m ICI182,780 induced a significant rapid and transient increase in ERK1/2 phosphorylation with temporal characteristics similar to those observed for 17β-E2 (Fig. 5C,D). Rather than acting as a true competitive antagonist, these results suggest that, in the presence of higher concentrations of ICI182,780, 10-11m 17β-E2 cannot restimulate ERK1/2 signaling after transient ICI182,780-induced ERK1/2 phosphorylation.
Figure 5.

Analyses of ICI182,780-mediated activation of ERK1/2. A, ICI182, 780 blockade of E2-mediated MAPK stimulation; Western blot analyses of 17β-E2-induced ERK1/2 phosphorylation in cell lysates from cultures pretreated for 30 min with 1 μm ICI182,780. This increased duration of ICI182,789 pretreatment abrogates estradiol-stimulated ERK1/2 phosphorylation. B, ICI182,780 dose–response analysis. Shown are representative Western blot results of the dose dependency of rapid ICI182, 780-induced ERK1/2 phosphorylation. In primary cultures of cerebellar granule cells treated for 10 min with increasing concentrations of ICI182, 780, rapid increases in ERK1/2 phosphorylation are observed with concentrations between 10-9 and 10-7m, indicating that ICI182,780 is a highly potent agonist of rapid ERK1/2 phosphorylation. C, D, Time course analysis of ERK1/2 phosphorylation stimulated by 10-8m ICI182,780 revealed that ERK1/2 phosphorylation was increased significantly at 10 and 15 min after the addition of 10-8m ICI182,780, and with longer exposure ERK1/2 phosphorylation rapidly returned to baseline levels. For each experiment the results are normalized to total ERK-IR and are the mean fold induction ± SEM of six different experiments. *,***See Materials and Methods.
Direct cell counting of viable granule cells at different times after either a 15 min pulse treatment of 10-11m 17β-E2 followed by an estrogen-free chase period or after continuous exposure to 10-11m 17β-E2 indicated that this low concentration of estradiol significantly altered the number of viable cells present. In control cultures and cultures continuously exposed to 17β-E2 significant increases in viable cells were detected 24 hr after treatment. In pulse-treated cultures increases in the number of viable granule cells were not observed until 72 hr after treatment (Fig. 6A). In all cases by 72 hr after treatment granule cell numbers had reached a plateau. In terms of the number of viable granule cells, chronic treatment with 17β-E2 resulted in significant increases in viable granule cells after 48 hr in culture. In contrast, there were significantly fewer viable granule cells in pulse-treated cultures at all time points (Fig. 6A).
Figure 6.
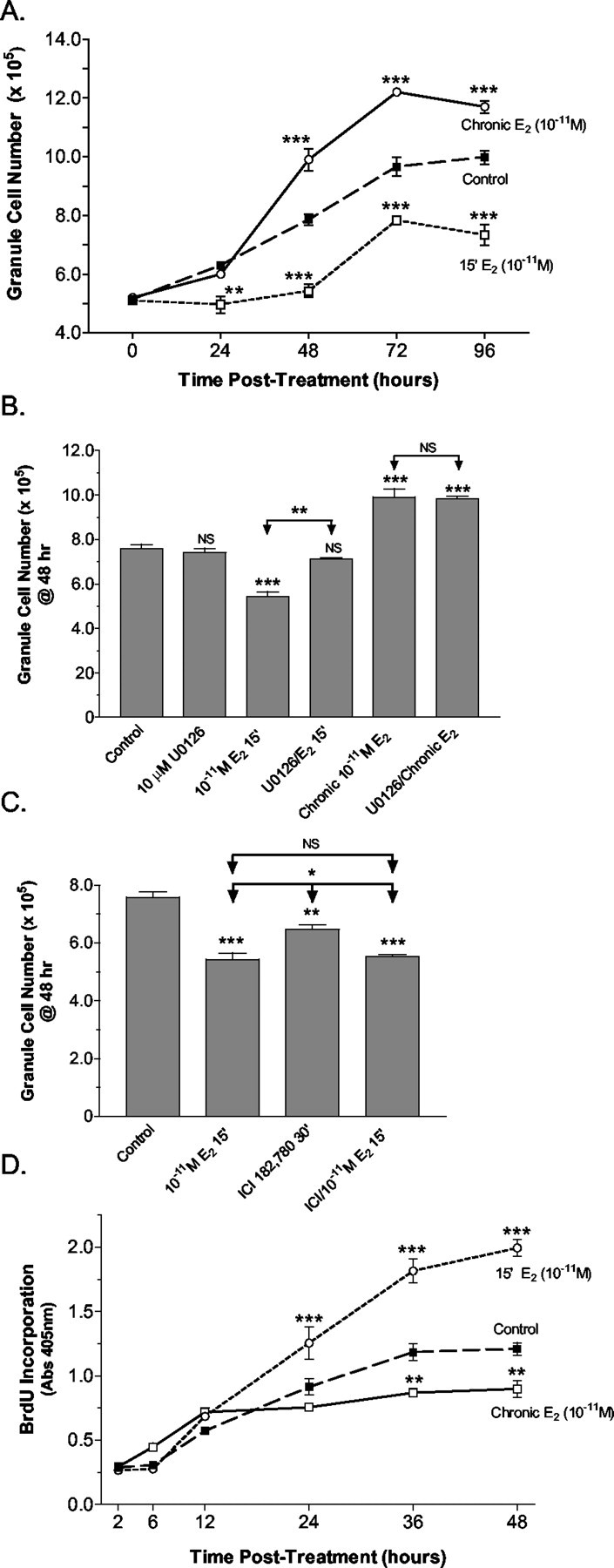
Analysis of 17β-estradiol effects on developing granule cell viability and mitogenesis. A, At different time points after a 15 min pulsed (15′ E2) or continuous (Chronic E2) treatment with 10-11m 17β-E2 or vehicle control, the number of viable cells present in freshly dissociated granule cell explant cultures was determined by direct cell counting of viable granule cells. After 48 hr of treatment a significant increase in granule cell numbers resulted from continuous estrogen exposure. In pulse-treated cultures significant decreases in viable cells were observed, with net increases in cell numbers not observed until 72 hr after treatment. Values are average cell number per culture ± SEM from three independent experiments. No significant difference was observed between cultures that were control pulse-treated or treated continuously with vehicle; as a result, the values for the controls were combined. For each time point the level of significance between the cell numbers determined for the control and each experimental group is indicated. B, The role of 17β-E2-induced MAPK activation was demonstrated by pretreatment of the cultures with the MEK1inhibitor U0126. Direct counting of viable granule cells indicated that at the time of maximal differences in viable granule cell numbers (48 hr) U0126 significantly blocked 17β-E2-induced decreases in viable granule cells. Pretreatment with U0126 did not influence increased granule cell viability resulting from chronic exposure to 10-11m 17β-E2. U0126 alone had no effect on granule cell viability. Values are average cell number per culture ± SEM from three independent experiments. The level of significance between control and experimental treatment groups is indicated directly above the error bar (NS, no significant difference). Levels of significance between the 17β-E2and U0126 + 17β-E2 treatment groups are indicated with arrows. C, A significant reduction in granule cell viability was detected 48 hr after a 30 min treatment with 1 μm ICI182,780, which was augmented further by an additional 15 min exposure to 10-11m 17β-E2. Levels of significance are indicated as above, with values represented as average cell number per culture ± SEM from three independent experiments. D, At different time points after a 15 min pulsed (15′ E2) or continuous (Chronic E2) treatment with 10-11m 17β-E2 or vehicle, the amount of BrdU incorporated into newly synthesized DNA of dividing cerebellar cells was assessed via an ELISA-based method. The results of the analysis of BrdU incorporation after continuous or pulse exposures in dissociated cerebellar explants to 10-11m 17β-E2 reveal that pulsed exposures induced additional rounds of mitosis, whereas chronic exposure was anti-mitogenic when compared with vehicle-treated control cultures. Values are expressed as the means ± SEM from three independent experiments, with the level of significance for the difference between control and each experimental value indicated. *,**,***See Materials and Methods.
The role of ERK1/2 activation in the observed changes in granule cell numbers was investigated by determining whether U0126 could block the 17β-E2-induced changes in granule cell viability (Fig. 6B). At 48 hr after treatment (the time of maximal differences in viable granule cell numbers) significantly fewer viable granule cells were present in cultures pulse-treated with 17β-E2. In the presence of 10 μm U0126, however, 15 min of exposure to 10-11m 17β-E2 had no significant influence on the number of viable granule cells. In cultures chronically treated with 17β-E2 for 48 hr the presence of U0126 during the entire incubation period did not inhibit 17β-E2-induced increases in viable granule cell numbers. These results indicate that pulsed 17β-E2 treatment caused a decrease in granule cell numbers that was mediated by rapid ERK1/2 activation, whereas the effects of chronic 17β-E2 treatment were independent of MEK–ERK signaling.
As with 17β-E2, pulsed treatment with 1 μm ICI182,780 resulted in a significant decrease in viable granule cells 48 hr after treatment (Fig. 6C). When ICI182,780 treatment was followed by an additional 15 min exposure to 10-11m 17β-E2 before the chase period, an additional significant decrease in cell numbers was observed. This result suggests that ICI182,780 does not inhibit 17β-E2-dependent decreases in granule cell viability, but rather the effects of ICI182,780 and estradiol are additive.
To determine whether pulsed or continuous 17β-E2 exposures differentially influence the mitogenesis of immature granule cell precursors, we analyzed DNA synthesis during the first 48 hr in culture by measuring BrdU incorporation. Surprisingly, the results of BrdU incorporation experiments revealed that exposure to 10-11m 17β-E2 for 15 min was mitogenic compared with vehicle-treated control cultures (Fig. 6D). In contrast, chronic exposure to the same concentration of 17β-E2 significantly decreased granule cell mitogenesis.
In vivo and in culture the cerebellar granule cells normally undergo apoptotic and oncotic/necrotic forms of programmed cell death during development (Wood et al., 1993; Villalba et al., 1997; Tanaka and Marunouchi, 1998). Therefore, we initially sought to determine whether increased granule cell death occurred after pulsed estradiol treatment. Initial observations revealed increased cytoplasmic vacuolation of granule cell neurons in estradiol-treated cultures, a feature consistent with oncotic cell death (Kitanaka and Kuchino, 1999). Because the loss of plasma membrane integrity is an additional hallmark of oncotic forms of cell death, the permeability to propidium iodide and the release of LDH into the culture media were used to determine whether increases in oncosis/necrosis were induced by pulsed E2 treatment. Significant increases in PI permeability were detected at 3, 12, and 24 hr after a 15 min pulse treatment with 10-11m E2 (Fig. 7A–C). However, at 48 and 72 hr after treatment, times when rapid and significant increases in viable granule cells and granule cell mitogenesis occur (Figs. 5, 6), PI permeability was decreased significantly as compared with control (Fig. 7C).
Figure 7.
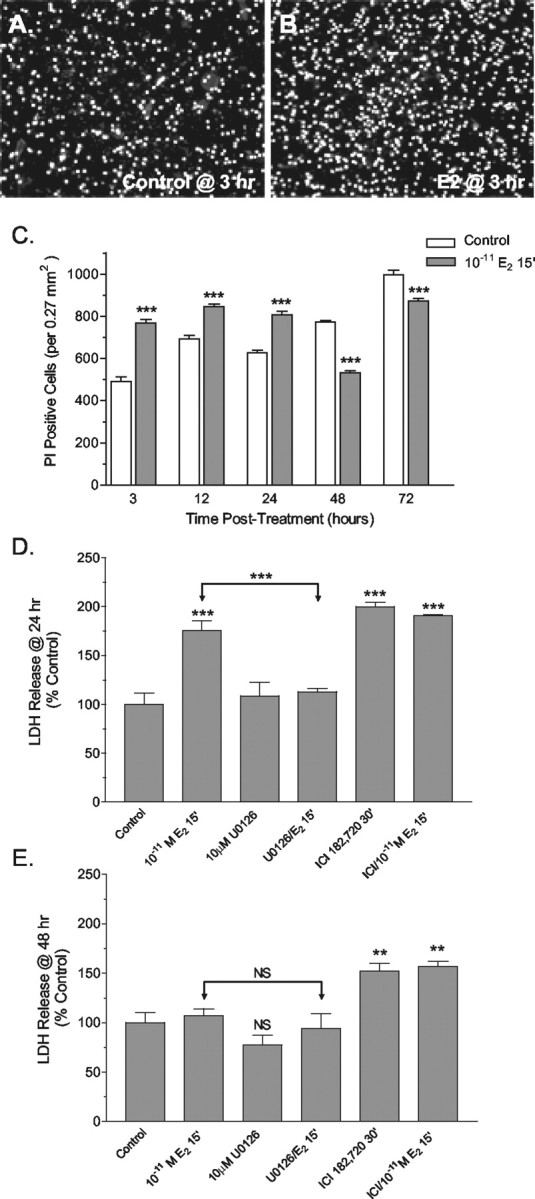
Effects of pulsed 17β-E2 treatment on granule cell permeability. Compared with control cultures (A), propidium iodide (PI) permeability, a measure of oncotic cell death, significantly increased in cultures of granule cells 3 hr after a 15 min exposure to 10-11M 17β-E2 (B). C, Quantitative determination of the number of PI-permeable granule cells revealed that estradiol treatment induced significant changes in permeability. Increased oncotic profiles were observed at the 3, 12, and 24 hr post-treatment time points, whereas at the 48 and 72 hr time points significant decreases in PI permeability were observed. Results are reported as the means ± SEM for six 0.27 mm 2 fields randomly selected from three different samples for each time point. At 24 hr (D) and 48 hr (E) after 15 min of exposure to 10-11m 17β-E2 the amount of LDH released into culture media was determined and used as a measure of necrotic cell death. LDH release was increased significantly in E2-treated cultures at 24 hr, but not at 48 hr. Pretreatment with U0126 completely blocked E2-induced increases in LDH release at 24 hr, indicating that E2 was acting via activation of the MAPK pathway. At both times a robust increase in LHD release was observed after a 30 min exposure to 1 μm ICI182,780, an effect not accentuated by an additional 15 min of exposure to ICI182, 780 and 10-11m E2. Results are expressed as the means ± SEM (n = 8–9), with significance assessed by a one-way ANOVA and Dunnett's or Newman–Keuls post-test. The level of significance between control and experimental treatment groups is indicated directly above the error bar (NS, no significant difference). **,***See Materials and Methods.
Analysis of LDH at 24 hr after a 15 min pulsed exposure to 10-11m 17β-E2 revealed that a significant MAPK-dependent increase in LDH release also occurred (Fig. 7D). Pulsed treatment with 1 μm ICI 182,720 also resulted in significant LDH release; this effect was not increased further by the addition of 17β-E2. Similar to the results obtained for the analysis of PI permeability, in 17β-E2-treated cultures decreased LDH release was detected at the 48 hr time point (Fig. 7E). However, at 48 hr a significant LDH release was detected in cultures treated with ICI182,780. This last observation suggests that later proliferative and protective actions of pulsed 17β-E2 treatment may involve ligand-activated transactivational activity that involves the ERs. In mature cultures of granule cells neither viability nor LDH release was influenced by either pulsed or continuous exposure to any concentration of 17β-E2 between 10-11 and 10-6m (Fig. 8A–D).
Figure 8.

Effects of 17β-estradiol on mature cerebellar granule cells. Changes in granule cell permeability were assessed by LDH release, and granule cell viability was determined by the MTS reduction assay in mature granule cell cultures 24 hr after either a 15 min pulsed treatment or a continuous exposure to different concentrations of 17β-E2 from 10-11 to 10-6m. At all of the estradiol concentrations that were analyzed, no differences in cell lysis or viability were detected in response to either pulsed 15 min exposures (A, B) or a continuous 24 hr exposure (C, D). Results are expressed as the means ± SEM (n = 24), with significance assessed by a one-way ANOVA and Dunnett's post-test.
Results of chromosomal fragmentation analysis and counting of fluorescently labeled condensed chromatin structures confirmed that granule cells in both vehicle and 17β-E2-treated cultures were undergoing apoptotic-like programmed cell death. However, neither chronic nor pulsed 17β-E2 treatments appeared to increase apoptotic cell morphologies (data not shown). To confirm that caspase-dependent apoptosis was not increased by a 15 min pulsed treatment with 10-11m E2, we determined caspase activity at 24 hr after treatment and found it to be unaffected by 17β-E2 treatment (Fig. 9A). Treatment with the nonspecific PKC inhibitor staurosporine resulted in a robust increase in caspase activity. In staurosporine-, vehicle-, and 17β-E2-treated cultures the caspase activity was inhibited completely by the pan-caspase inhibitor zVAD-fmk. Nearly identical levels of caspase activity were detected in vehicle and 17β-E2-treated cultures. Caspase activity similarly was unaffected by a blockade of MAPK signaling before E2 treatment (Fig. 9A).
Figure 9.
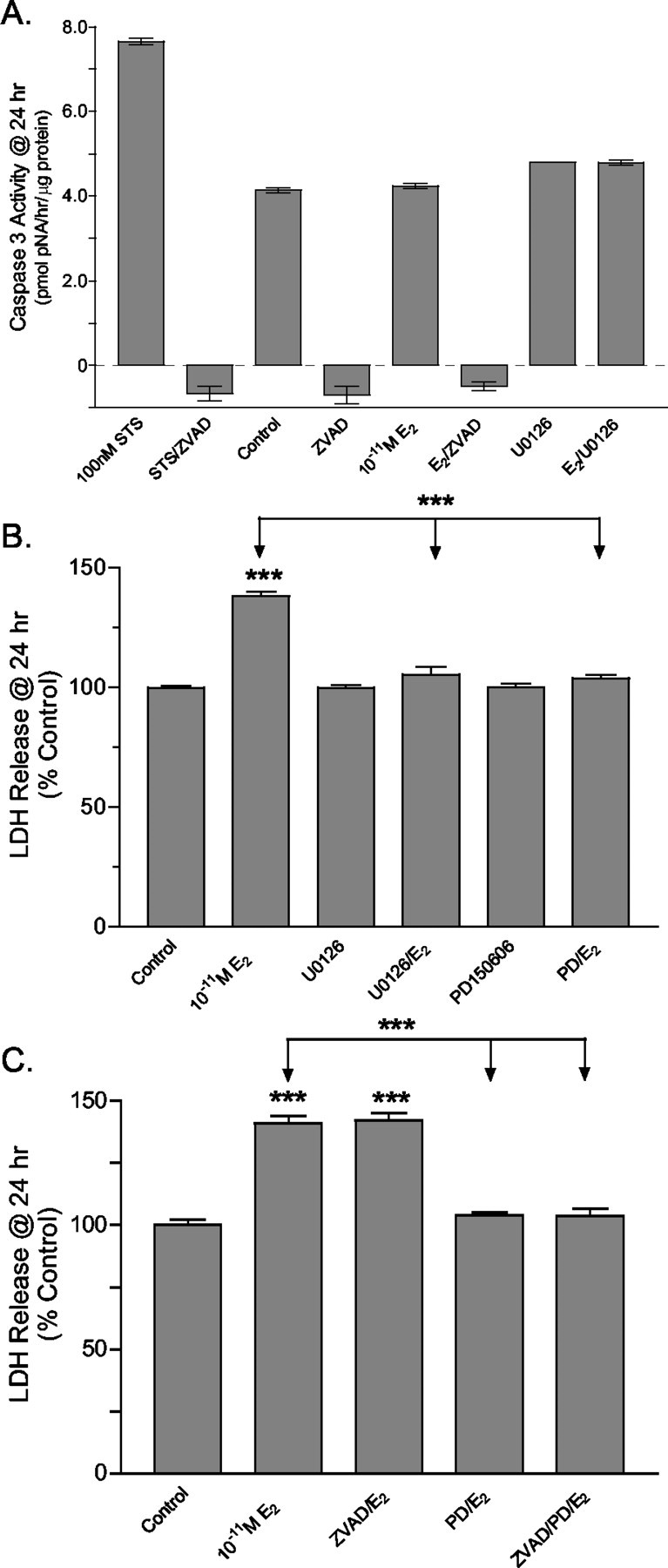
Analysis of the role of caspase or calpain activity in the mechanism of 17β-E2-mediated granule cell neurotoxicity. A, Immature granule cell cultures were pulse-treated with 10-11m 17β-E2, and caspase activity was determined 24 hr after treatment. Compared with control, 17β-E2 treatment did not increase caspase activity. The nonspecific PKC inhibitor staurosporine (STS) was used as a positive control to stimulate apoptosis by activation of caspases and resulted in a robust increase in caspase activity. In staurosporine-, vehicle-, and 17β-E2-treated cultures the caspase activity was inhibited completely by the pan-caspase inhibitor zVAD-fmk (12.5 μm). Caspase activity similarly was unaffected by the blockade of MAPK signaling before 17β-E2 treatment. B, At 24 hr after 15 min of exposure to 10-11m E2 the amount of LDH released into culture media was determined as an indicator of oncotic cell death. LDH release was increased significantly in 17β-E2-treated cultures. Pretreatment with U0126 (10 μm) completely blocked E2-induced increases in LDH release, confirming that 17β-E2 was acting via activation of the MAPK pathway. The irreversible calpain inhibitor PD150606 (50 μm) similarly blocked 17β-E2-induced LDH release. C, 17β-E2-induced LDH release was independent of the pan-caspase inhibitor zVAD-fmk. Compared with control, significant increases in LDH release were observed in 17β-E2-treated cultures with and without the addition of zVAD-fmk. In the presence of PD150606 the LDH release in cultures treated with 17β-E2 was not significantly different from control. ZVAD-fmk did not enhance PD150606 blockade of LDH release. Results indicate that 17β-E2-mediated LDH release is a MAPK-dependent consequence of necrosis-like programmed cell death and not a secondary consequence of apoptosis. In all cases the results are expressed as the means ± SEM (n = 24), with significance assessed by a one-way ANOVA and Dunnett's or Newman–Keuls post-test. The level of significance between control and experimental treatment groups is indicated directly above the error bar. ***See Materials and Methods.
Finally, the role of Ca2+-activated cysteine proteases, calpains, for which the intracellular activation is known to play an important role in oncotic cell death (Yamashima, 2000; Razandi et al., 2002), was investigated 24 hr after a 15 min exposure to 10-11m E2. Compared with control, significant increases in LDH release were observed in 17β-E2-treated cultures with and without the addition of zVAD-fmk (Fig. 9B,C), indicating that estradiol-induced LDH release was independent of caspase activity. Pretreatment with U0126 completely blocked 17β-E2-induced increases in LDH release, further indicating that 17β-E2 was acting via activation of the MAPK pathway. The irreversible calpain inhibitor PD150606 similarly blocked 17β-E2-induced LDH release (Fig. 9B), and PD150606 blockade of LDH release was not enhanced by the inhibition of caspase activity by zVAD-fmk (Fig. 9C). Together, those results show that 17β-E2-mediated LDH release is a MAPK-dependent consequence of oncotic-like programmed cell death and not a secondary consequence of increased apoptosis.
Discussion
The present study provides data indicating that 17β-E2, 17α-E2, and the ER antagonist ICI182,780 activate ERK1/2 MAPK signaling in developing and mature cerebellar neurons. The dose–response characteristics for each estrogen-like compound were hormetic (Calabrese and Baldwin, 2002), as indicated by an inverted U-shaped dose–response curve with low-concentration agonist effects and antagonist-like or no effects at higher concentrations. For 17β-E2, ERK1/2 phosphorylation was increased significantly at low physiological concentrations; this stimulation was lost between 1 and 10 nm and observed again at pharmacological concentrations.
The bimodal nature of the effects observed at physiological and again at supraphysiological estradiol concentrations may be mediated via different mechanisms. At 10-8m 17β-E2, a concentration often considered experimentally physiological, no significant stimulation of ERK1/2 signaling was observed as a result of a high frequency in failure to respond. This increased variability likely resulted from 10-8m E2 inducing effects intermediate to physiological dose inhibition and supraphysiological dose activation. The most efficacious concentrations (10-11 to 10-10 M) correspond closely with the bioavailable concentration of free 17β-E2 in the neonatal rat brain (Montano et al., 1995). Thus in regions of the neonatal rat brain not expressing aromatase (Horvath and Wikler, 1999), 10-11m may reflect the physiological concentration of free estradiol.
Temporal analysis indicated that 17β-E2 activates ERK1/2 in a rapid and transient manner reminiscent of EGF-mediated ERK1/2 signaling in PC12 cells (Gotoh et al., 1990). As in PC12 cells this transient increase in ERK phosphorylation may arise from formation of the short-lived scaffolding protein complexes necessary for MAPK activation (Kao et al., 2001). Alternatively, transient activation may result from PKA-mediated negative feedback regulation of Raf activity (Filardo, 2002). Although further investigation into the mechanism of rapid estrogen-induced ERK signaling is necessary, it is clear that, in studies lacking a well characterized dose–response curve and/or time course analyses, the potential for misinterpretation of false-negative results is high.
The ability of estradiol to increase ERK signaling in cerebellar neurons contrasts with previous studies reporting that 10 nm 17β-E2 could not stimulate ERK1/2 phosphorylation in organotypic cerebellar cultures (Singh et al., 1999, 2000). These differences may have resulted from differences in the steroid and neurotrophic stimulation received during culture. Alternatively, a failure to respond to 10 nm E2 is consistent with our observed variable responsiveness at this concentration. Because of the rapid and transient nature of the response, slow or unequal exposure of cells may be especially problematic in organotypic cultures in which close cell–cell interactions may induce barriers to rapid diffusion.
The estrogen receptor antagonist ICI182,780 is a 7α-alkylamide analog of 17β-E2, consisting of a central estradiol moiety with a long side chain extending from C7 (Wakeling and Bowler, 1992; Parisot et al., 1995; Wakeling, 1995). With binding in the ligand-binding domain, the 7α side chain of ICI182,780 influences the conformation of helix 12 to block AF2 function (Pike et al., 1999, 2001; Egner et al., 2001). Micromolar concentrations of this transactivational antagonist often have been used to discriminate whether or not rapid estrogen actions are mediated via a classical ER. However, if rapid effects are ER-mediated, the conformational changes required to stimulate ERK signaling may differ from those involved with AF2 transactivation. The effects of ICI182,780 binding at a distinct class of ERs that mediate rapid E2 actions are not known. In either case there is no a priori reason for ICI182,780 binding to induce conformational effects that block rapid signaling mechanisms.
The finding that ICI182,780 is a highly efficacious agonist of ERK1/2 phosphorylation in cerebellar neurons is especially significant. Because dose-dependent hormetic inhibition was observed at the high concentrations of ICI182,780 used experimentally, the ability of ICI182,780 to block rapid E2 effects may only mimic true ER antagonism. Further, ICI182,780 blockade often is assessed after a pretreatment period; this preexposure could result in the examination of endpoints at times long after a transient response. Thus hormetic high-dose inhibition and/or desensitization after pretreatment, rather than competitive inhibition, may explain the variable ability of ICI182,780 to block rapid estrogen actions in other experimental systems (Watters et al., 1997; Gu et al., 1999; Kuroki et al., 2000).
Depending on the duration of exposure, 17β-E2 can impose contrasting effects on mitogenesis and viability in granule cell neuroblasts. Chronic exposure to 17β-E2 was anti-mitotic, yet an increase in viable granule cell number was observed. Those results suggest that chronic estradiol exposure can induce neuroprotective mechanisms that compensate for the anti-mitotic effects. In contrast, pulsed 17β-E2 treatments significantly increased mitogenesis but caused a decrease in viable granule cell numbers. These results suggest that pulsed 17β-E2 exposure increases granule cell death but increases mitogenesis in granule cell neuroblasts that are refractory to estradiol toxicity. The neuroprotective or neurotoxic effects resulting from chronic or pulsed estrogen treatment differed in ERK1/2 dependency. Decreased granule cell viability after pulsed 17β-E2 treatments was MAPK-dependent, whereas the neuroprotective effects of chronic E2 exposure were MAPK-independent. Interestingly, in this one model system the same concentration of estrogen can induce either neuroprotection or neurotoxicity, depending only on the duration of exposure. These opposing effects may underlie fundamental differences between the mechanisms that control longer-term ER-mediated transcriptional effects and those that are involved in rapid intracellular signaling. These differences may have important relevance to the normal fluctuations in hormone concentrations that occur in vivo during development and in adults.
The neuroprotective actions of estradiol that have been observed here likely involve ER-mediated transactivation, as suggested by these effects being MAPK-independent and requiring longer exposure periods. The longer times necessary for the implementation of these protective effects suggest that ER-mediated expression of estrogen-responsive genes may be required. In contrast, the neurotoxicity of pulsed 17β-E2 exposures may not rely on ER transcriptional activities. The pulsed treatment constitutes a very short exposure time, a treatment protocol designed to elicit primarily estrogen-induced rapid effects. Consistent with a central role for E2-stimulated ERK1/2 signaling in these effects was the finding that a blockade of MEK–ERK signaling inhibited E2 neurotoxicity. Because ICI182,780 inhibits ER-mediated transactivation, the fact that ICI182,780 treatment mimicked E2 neurotoxicity suggests further that a mechanism independent of ER transactivation is involved. Because viability and cellular permeability of mature granule cells was not impacted by pulsed or continuous exposure to any concentration of estradiol that was tested, it appears that only immature granule cells are sensitive to the mitogenic and neurotoxic actions of estradiol.
Because the neuroprotective actions of estrogen are well documented (Dubal et al., 1998, 1999; Singer et al., 1999; Han and Holtzman, 2000; Dubal and Wise, 2001; Wise et al., 2001), we chose to focus our studies on understanding in more detail the mechanisms of low-dose E2 neurotoxicity. During cerebellar development granule cell death occurs as a fundamental biological process to eliminate excess cells. On the basis of morphological and biochemical characteristics, cell death mechanisms can be divided into two major types: oncosis/necrosis and apoptosis (Schweichel and Merker, 1973; Kitanaka and Kuchino, 1999). The loss of plasma membrane integrity is a hallmark of oncotic cell death; permeability of PI and LDH release are sensitive indicators of compromised plasma membrane integrity. Increased numbers of PI-stained cells were observed 3–24 hr after pulsed 17β-E2 exposure, and LDH release was increased 24 hr after treatment. However, increased granule cell permeability was not detected at 48–72 hr after treatment, a time that coincides with significant increases in granule cell numbers and mitogenesis. Similar to direct cell-counting results, LDH release was dependent on ERK1/2 signaling, but was not blocked by ICI182,780, suggesting that classical estrogen-responsive gene expression was not involved. Brief exposures to ICI182,780 alone increased LDH release, suggesting that ICI182,780-induced activation of MAPK results in estrogen-like neurotoxicity. Because 17β-E2 exposures did not increase caspase-3 activity significantly and inhibition of caspase activity did not block estrogen-induced effects, E2 neurotoxicity in immature granule cells was not classically apoptotic. Those results are consistent with our Western blotting studies, which indicate that estrogen treatment does not influence proapoptotic JNK1/2 or p38 MAPK signaling.
Increased calpain activity and a lack of increased caspase activity during cell death are other distinguishing features of oncotic/necrotic cell death. Calpains are Ca2+-dependent neutral proteases that participate in essential and pathological cellular functions (Sorimachi et al., 1997; Schnellmann and Williams, 1998). The blockade of 17β-E2-induced LDH release by the calpain inhibitor PD150606 suggests that oncotic cell death is induced in a subset of developing cerebellar neurons. Although calpain inhibitors are cytoprotective (Nixon et al., 1994) and calpains act to weaken the cytoskeleton before cell lyses (Sorimachi et al., 1997), the mechanism of E2-mediated ERK activation of calpains is unknown. In other systems EGF receptor activation of ERK1/2 results in increased intracellular calcium concentrations as a prerequisite for calpain activation (Grewal et al., 1999; Glading et al., 2001), whereas ERK1/2 activity also has been proposed to enhance calpain activity via a PKA-dependent mechanism (Shiraha et al., 2002). Although the mechanism through which E2-stimulated ERK signaling induces calpain-dependent oncotic granule cell death is unknown, these mechanisms normally may function to regulate mature cerebellar neuron numbers and may have important implications in understanding the action of xenoestrogens and phytoestrogens in the developing brain.
The results presented here may lend insight to the role of estrogen in the developing cerebellum and potentially other brain regions. Developing neurons may respond to changing estrogen levels via rapid mechanisms to regulate the numbers of neurons by removing those neuroblasts that remain sensitive because of improper synaptic connections or that have not received appropriate biochemical signals. The subpopulations of neuroblasts refractory to the estrogenic insult may undergo further mitogenesis and subsequently continue along their developmental pathways. In more mature cells estrogen exposure then may become neuroprotective and aid cellular survival during the developmental progression. Thus estrogen may play numerous and sometimes opposing roles in developing and mature neurons that could be a direct consequence of the ability of this steroid hormone to elicit ER-mediated changes in gene expression and to alter intracellular signaling rapidly. The pure antagonist of ER transactivation ICI182,780 might prove to be a useful pharmacological tool for experimentally separating rapid effects from the combined rapid and “classic” transcriptional effects of estrogen.
Footnotes
These studies were supported by National Institutes of Health Grants R01 NS37795 and R01 NS42798, funded by the National Institute of Neurological Disorders and Stroke, and by a pilot study grant funded by the National Institute of Environmental Health Sciences through the University of Cincinnati Center for Environmental Genetics (P30-ES06096).
Correspondence should be addressed to Dr. Scott M. Belcher, Department of Pharmacology and Cell Biophysics, University of Cincinnati College of Medicine, 231 Albert Sabin Way, P.O. Box 670575, Cincinnati, OH 45267-0575. E-mail: scott.belcher@uc.edu.
J. K. Wong's present address: Division of Toxicological Sciences, Department of Environmental Sciences, Johns Hopkins School of Hygiene and Public Health, 615 North Wolfe Street, Baltimore, MD 21205.
Copyright © 2003 Society for Neuroscience 0270-6474/03/234984-12$15.00/0
References
- Belcher SM ( 1999) Regulated expression of estrogen receptor α and β mRNA in granule cells during development of the rat cerebellum. Brain Res Dev Brain Res 115: 57–69. [DOI] [PubMed] [Google Scholar]
- Belcher SM, Zsarnovszky A ( 2001) Estrogenic actions in the brain: estrogen, phytoestrogens, and rapid intracellular signaling mechanisms. J Pharmacol Exp Ther 299: 408–414. [PubMed] [Google Scholar]
- Calabrese EJ, Baldwin LA ( 2002) Hormesis: the dose–response revolution. Annu Rev Pharmacol Toxicol 43: 175–197. [DOI] [PubMed] [Google Scholar]
- Dubal DB, Wise PM ( 2001) Neuroprotective effects of estradiol in middle-aged female rats. Endocrinology 142: 43–48. [DOI] [PubMed] [Google Scholar]
- Dubal DB, Kashon ML, Pettigrew LC, Ren JM, Finklestein SP, Rau SW, Wise PM ( 1998) Estradiol protects against ischemic injury. J Cereb Blood Flow Metab 18: 1253–1258. [DOI] [PubMed] [Google Scholar]
- Dubal DB, Shughrue PJ, Wilson ME, Merchenthaler I, Wise PM ( 1999) Estradiol modulates Bcl-2 in cerebral ischemia: a potential role for estrogen receptors. J Neurosci 19: 6385–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egner U, Heinrich N, Ruff M, Gangloff M, Mueller-Fahrnow A, Wurtz JM ( 2001) Different ligands–different receptor conformations: modeling of the hERα LBD in complex with agonists and antagonists. Med Res Rev 21: 523–539. [DOI] [PubMed] [Google Scholar]
- Evans RM ( 1988) The steroid and thyroid hormone receptor superfamily. Science 240: 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falkenstein E, Tillmann HC, Christ M, Feuring M, Wehling M ( 2000) Multiple actions of steroid hormones: a focus on rapid, nongenomic effects. Pharmacol Rev 52: 513–556. [PubMed] [Google Scholar]
- Filardo EJ ( 2002) Epidermal growth factor receptor (EGFR) transactivation by estrogen via the G-protein-coupled receptor, GPR30: a novel signaling pathway with potential significance for breast cancer. J Steroid Biochem Mol Biol 80: 231–238. [DOI] [PubMed] [Google Scholar]
- Filardo EJ, Quinn JA, Bland KI, Frackelton Jr AR ( 2000) Estrogen-induced activation of Erk-1 and Erk-2 requires the G-protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol 14: 1649–1660. [DOI] [PubMed] [Google Scholar]
- Glading A, Uberall F, Keyse SM, Lauffenburger DA, Wells A ( 2001) Membrane proximal ERK signaling is required for M-calpain activation downstream of epidermal growth factor receptor signaling. J Biol Chem 276: 23341–23348. [DOI] [PubMed] [Google Scholar]
- Gotoh Y, Nishida E, Yamashita T, Hoshi M, Kawakami M, Sakai H ( 1990) Microtubule-associated protein (MAP) kinase activated by nerve growth factor and epidermal growth factor in PC12 cells. Identity with the mitogen-activated MAP kinase of fibroblastic cells. Eur J Biochem 193: 661–669. [DOI] [PubMed] [Google Scholar]
- Grewal SS, York RD, Stork PJ ( 1999) Extracellular signal-regulated kinase signaling in neurons. Curr Opin Neurobiol 9: 544–553. [DOI] [PubMed] [Google Scholar]
- Gu Q, Korach KS, Moss RL ( 1999) Rapid action of 17β-estradiol on kainate-induced currents in hippocampal neurons lacking intracellular estrogen receptors. Endocrinology 140: 660–666. [DOI] [PubMed] [Google Scholar]
- Han BH, Holtzman DM ( 2000) BDNF protects the neonatal brain from hypoxic–ischemic injury in vivo via the ERK pathway. J Neurosci 20: 5775–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath TL, Wikler KC ( 1999) Aromatase in developing sensory systems of the rat brain. J Neuroendocrinol 11: 77–84. [DOI] [PubMed] [Google Scholar]
- Jakab RL, Wong JK, Belcher SM ( 2001) Estrogen receptor-β immunoreactivity in differentiating cells of the developing rat cerebellum. J Comp Neurol 430: 396–409. [DOI] [PubMed] [Google Scholar]
- Kao SC, Jaiswal RK, Kolch W, Landreth GE ( 2001) Identification of the mechanisms regulating the differential activation of the MAPK cascade by epidermal growth factor and nerve growth factor in PC12 cells. J Biol Chem 276: 18169–18177. [DOI] [PubMed] [Google Scholar]
- Kitanaka C, Kuchino Y ( 1999) Caspase-independent programmed cell death with necrotic morphology. Cell Death Differ 6: 508–515. [DOI] [PubMed] [Google Scholar]
- Kuroki Y, Fukushima K, Kanda Y, Mizuno K, Watanabe Y ( 2000) Putative membrane-bound estrogen receptors possibly stimulate mitogen-activated protein kinase in the rat hippocampus. Eur J Pharmacol 400: 205–209. [DOI] [PubMed] [Google Scholar]
- Levin ER ( 1999) Cellular functions of the plasma membrane estrogen receptor. Trends Endocrinol Metab 10: 374–377. [DOI] [PubMed] [Google Scholar]
- Levin ER ( 2001) Genome and hormones: gender differences in physiology. Invited review: cell localization, physiology, and nongenomic actions of estrogen receptors. J Appl Physiol 91: 1860–1867. [DOI] [PubMed] [Google Scholar]
- Montano MM, Welshons WV, vom Saal FS ( 1995) Free estradiol in serum and brain uptake of estradiol during fetal and neonatal sexual differentiation in female rats. Biol Reprod 53: 1198–1207. [DOI] [PubMed] [Google Scholar]
- Nadal A, Ropero AB, Fuentes E, Soria B ( 2001) The plasma membrane estrogen receptor: nuclear or unclear? Trends Pharmacol Sci 22: 597–599. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Saito KI, Grynspan F, Griffin WR, Katayama S, Honda T, Mohan PS, Shea TB, Beermann M ( 1994) Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer's disease. Ann NY Acad Sci 747: 77–91. [DOI] [PubMed] [Google Scholar]
- Norfleet AM, Thomas ML, Gametchu B, Watson CS ( 1999) Estrogen receptor-α detected on the plasma membrane of aldehyde-fixed GH3/B6/F10 rat pituitary tumor cells by enzyme-linked immunocytochemistry. Endocrinology 140: 3805–3814. [DOI] [PubMed] [Google Scholar]
- Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS ( 1997) Differential ligand activation of estrogen receptors ERα and ERβ at AP1 sites. Science 277: 1508–1510. [DOI] [PubMed] [Google Scholar]
- Parisot JP, Hu XF, Sutherland RL, Wakeling A, Zalcberg JR, DeLuise M ( 1995) The pure antiestrogen ICI182,780 binds to a high-affinity site distinct from the estrogen receptor. Int J Cancer 62: 480–484. [DOI] [PubMed] [Google Scholar]
- Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, Engstrom O, Ljunggren J, Gustafsson JA, Carlquist M ( 1999) Structure of the ligand-binding domain of oestrogen receptor β in the presence of a partial agonist and a full antagonist. EMBO J 18: 4608–4618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike AC, Brzozowski AM, Walton J, Hubbard RE, Thorsell AG, Li YL, Gustafsson JA, Carlquist M ( 2001) Structural insights into the mode of action of a pure antiestrogen. Structure 9: 145–153. [DOI] [PubMed] [Google Scholar]
- Razandi M, Pedram A, Greene GL, Levin ER ( 1999) Cell membrane and nuclear estrogen receptors (ERs) originate from a single transcript: studies of ERα and ERβ expressed in Chinese hamster ovary cells. Mol Endocrinol 13: 307–319. [DOI] [PubMed] [Google Scholar]
- Razandi M, Oh P, Pedram A, Schnitzer J, Levin ER ( 2002) ERs associate with and regulate the production of caveolin: implications for signaling and cellular actions. Mol Endocrinol 16: 100–115. [DOI] [PubMed] [Google Scholar]
- Schmidt BM, Gerdes D, Feuring M, Falkenstein E, Christ M, Wehling M ( 2000) Rapid, nongenomic steroid actions: a new age? Front Neuroendocrinol 21: 57–94. [DOI] [PubMed] [Google Scholar]
- Schnellmann RG, Williams SW ( 1998) Proteases in renal cell death: calpains mediate cell death produced by diverse toxicants. Ren Fail 20: 679–686. [DOI] [PubMed] [Google Scholar]
- Schweichel JU, Merker HJ ( 1973) The morphology of various types of cell death in prenatal tissues. Teratology 7: 253–266. [DOI] [PubMed] [Google Scholar]
- Shiraha H, Glading A, Chou J, Jia Z, Wells A ( 2002) Activation of M-calpain (calpain II) by epidermal growth factor is limited by protein kinase A phosphorylation of M-calpain. Mol Cell Biol 22: 2716–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer CA, Figueroa-Masot XA, Batchelor RH, Dorsa DM ( 1999) The mitogen-activated protein kinase pathway mediates estrogen neuroprotection after glutamate toxicity in primary cortical neurons. J Neurosci 19: 2455–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Setalo Jr G, Guan XP, Warren M, Toran-Allerand CD ( 1999) Estrogen-induced activation of mitogen-activated protein kinase in cerebral cortical explants: convergence of estrogen and neurotrophin signaling pathways. J Neurosci 19: 1179–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh M, Setalo G, Guan X, Frail DE, Toran-Allerand CD ( 2000) Estrogen-induced activation of the mitogen-activated protein kinase cascade in the cerebral cortex of estrogen receptor-α knock-out mice. J Neurosci 20: 1694–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorimachi H, Ishiura S, Suzuki K ( 1997) Structure and physiological function of calpains. Biochem J 328: 721–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Marunouchi T ( 1998) Immunohistochemical analysis of developmental stage of external granular layer neurons which undergo apoptosis in postnatal rat cerebellum. Neurosci Lett 242: 85–88. [DOI] [PubMed] [Google Scholar]
- Villalba M, Bockaert J, Journot L ( 1997) Concomitant induction of apoptosis and necrosis in cerebellar granule cells following serum and potassium withdrawal. NeuroReport 8: 981–985. [DOI] [PubMed] [Google Scholar]
- Wakeling AE ( 1995) Use of pure antioestrogens to elucidate the mode of action of oestrogens. Biochem Pharmacol 49: 1545–1549. [DOI] [PubMed] [Google Scholar]
- Wakeling AE, Bowler J ( 1992) ICI182,780, a new antioestrogen with clinical potential. J Steroid Biochem Mol Biol 43: 173–177. [DOI] [PubMed] [Google Scholar]
- Watson CS, Norfleet AM, Pappas TC, Gametchu B ( 1999) Rapid actions of estrogens in GH3/B6 pituitary tumor cells via a plasma membrane version of estrogen receptor-α. Steroids 64: 5–13. [DOI] [PubMed] [Google Scholar]
- Watters JJ, Campbell JS, Cunningham MJ, Krebs EG, Dorsa DM ( 1997) Rapid membrane effects of steroids in neuroblastoma cells: effects of estrogen on mitogen-activated protein kinase signaling cascade and c-fos immediate early gene transcription. Endocrinology 138: 4030–4033. [DOI] [PubMed] [Google Scholar]
- Wise PM, Dubal DB, Wilson ME, Rau SW, Liu Y ( 2001) Estrogens: trophic and protective factors in the adult brain. Front Neuroendocrinol 22: 33–66. [DOI] [PubMed] [Google Scholar]
- Wong JK, Kennedy PR, Belcher SM ( 2001) Simplified serum- and steroid-free culture conditions for the high-throughput viability analysis of primary cultures of cerebellar neurons. J Neurosci Methods 110: 45–55. [DOI] [PubMed] [Google Scholar]
- Wood KA, Dipasquale B, Youle RJ ( 1993) In situ labeling of granule cells for apoptosis-associated DNA fragmentation reveals different mechanisms of cell loss in developing cerebellum. Neuron 11: 621–632. [DOI] [PubMed] [Google Scholar]
- Yamashima T ( 2000) Implication of cysteine proteases calpain, cathepsin, and caspase in ischemic neuronal death of primates. Prog Neurobiol 62: 273–295. [DOI] [PubMed] [Google Scholar]
- Zsarnovszky A, Belcher SM ( 2001) Identification of a developmental gradient of estrogen receptor expression and cellular localization in the developing and adult female rat primary somatosensory cortex. Brain Res Dev Brain Res 129: 39–46. [DOI] [PubMed] [Google Scholar]