

Abstract
To determine the importance of the NMDA receptor (NMDAR) in pain hypersensitivity after injury, the NMDAR1 (NR1) subunit was selectively deleted in the lumbar spinal cord of adult mice by the localized injection of an adenoassociated virus expressing Cre recombinase into floxed NR1 mice. NR1 subunit mRNA and dendritic protein are reduced by 80% in the area of the virus injection, and NMDA currents, but not AMPA currents, are reduced 86–88% in lamina II neurons. The spatial NR1 knock-out does not alter heat or cold paw-withdrawal latencies, mechanical threshold, or motor function. However, injury-induced pain produced by intraplantar formalin is reduced by 70%. Our results demonstrate conclusively that the postsynaptic NR1 receptor subunit in the lumbar dorsal horn of the spinal cord is required for central sensitization, the central facilitation of pain transmission produced by peripheral injury.
Keywords: NMDA receptor, conditional knock-out, Cre–loxP, formalin pain, spinal cord dorsal horn, synaptic transmission, central sensitization
Introduction
Glutamate receptors are the major transducer of excitatory neurotransmission, the means by which information on injury in the periphery is conveyed, via primary sensory neurons, to the CNS, where the sensation of pain is realized (Dubner and Ruda, 1992; Woolf and Costigan, 1999). The spinal cord dorsal horn (SCDH) is the initial site of integration for ascending and descending information on pain (Woolf and Salter, 2000). Intense or sustained noxious stimuli that are associated with tissue injury result in a temporal summation of postsynaptic depolarizations, which have been argued to remove the voltage-dependent magnesium block of NMDA ionotropic glutamate receptors, allowing calcium influx into the postsynaptic cell (Woolf and Salter, 2000). This influx in turn will activate calcium-sensitive intracellular signal cascades (Woolf and Costigan, 1999) that lead to the phosphorylation of the NMDA and other receptor–ion channels, initiating prolonged increases in the excitability of spinal cord neurons, a process described as central sensitization (Woolf and Costigan, 1999; Woolf and Salter, 2000). Central sensitization results in increased synaptic efficacy, and the recruitment of normally subliminal inputs leads to the spread of pain sensitivity beyond the site of injury (secondary hyperalgesia) and the generation of pain in response to low-threshold inputs (allodynia) (Woolf and Salter, 2000). The mechanisms underlying this pain hypersensitivity have features that resemble the synaptic plasticity in the hippocampus that underlies learning and memory, for which NMDA receptors (NMDARs) also have been shown to be crucial (Hollmann and Heinemann, 1994; Tsien et al., 1996; Dingledine et al., 1999). Pharmacological antagonism of NMDA receptors (Woolf and Thompson, 1991; Yamamoto and Yaksh, 1992) does not usually interfere with the nociceptive response to normal non-tissue-damaging noxious stimuli (basal pain sensitivity) but reduces the initiation and maintenance of central sensitization (Woolf and Salter, 2000). However, the evidence has relied on drugs (competitive and noncompetitive NMDA receptor antagonists) for which the specificity, site, duration, and extent of action on local targets are either not fully known or cannot be controlled. In the hippocampus, disruption of NMDA receptor function by a deletion of the NMDAR1 (NR1) subunit of the multimeric NMDA receptor, conditionally restricted to the CA1 region of adult mice, has provided evidence for the essential role of the NMDA receptor in the acquisition of spatial memories (Tsien et al., 1996). The Cre–loxP recombination system provides an approach for spatial and temporal control of gene deletion in the CNS (Tsien et al., 1996; Kaspar et al., 2002b). An intramolecular recombination catalyzed by Cre results in the deletion of an essential DNA sequence between the two loxP sites, which have been introduced into the gene of interest at a location that does not interfere with transcription (Tsien et al., 1996). We modified the approach of Tsien et al. (1996) by using a recombinant adenoassociated virus (rAAV) (Kaspar et al., 2002b) to deliver Cre recombinase to the lumbar SCDH of an adult floxed mouse, allowing the specific role of SCDH NMDA receptors in injury-induced pain to be determined.
Materials and Methods
The floxed NMDA receptor 1 subunit mutant mouse
Mice, which were homozygous for the floxed NR1 subunit (fNR1) gene, were as described previously (Tsien et al., 1996). These mice have a loxP site placed in the intron that lies between exons 10 and 11 and a second loxP site downstream after exon 22, the last exon. The two loxP sequences flank a region of the NR1 gene that encodes the four membrane domains [three transmembrane (TM) domains and the M2 recurrent loop] and the entire C-terminal sequence of the polypeptide chain (Tsien et al., 1996). Adult (30–40 gm) fNR1 mice of both sexes were used in these studies. The animals used for the breeding of the fNR1 line were tested for homozygosity of the loxP sites using the Southern blot procedure, and the MAX-BAX (Charles River Laboratories, Wilmington, MA) background strain characterization procedure was used to identify breeders that were at least 92–95% C57BL/6 background.
The Cre-expressing rAAV vector
The vector construction, viral production, and purification were as described by Kaspar et al. (2002b). The rAAV is a single-stranded DNA parvovirus (∼4.7 kb) that is engineered without viral coding sequences. The inserted transgene included a cytomegalovirus promoter and the coding sequences for a fusion protein of green fluorescent protein (GFP)-Cre. Control animals received the same dosage of a rAAV-GFP vector that lacked the coding sequence for Cre recombinase.
Intraparenchymal administration of rAAV
Initial experiments demonstrated that when the viral vector is administered into the CSF via an intrathecal injection, very little if any uptake of the virus into spinal cord occurred (data not shown). Therefore, the virus was microinjected [intraparenchymal injection (IPI)] directly into the SCDH. The mouse was anesthetized with ketamine–xylazine and placed in a spinal frame that supported the abdomen and pelvis, and a laminectomy was used to remove spinous process VL2 and part of VL3. Three unilateral injections of 1 μl (1 × 10 6 viral particles/μl) were administered 0.5 mm apart, at a depth of 0.3 mm (SCDH), using a glass pipette with a 40-μm-diameter tip attached to a 5 μl Hamilton syringe. The syringe was mounted on a microinjector (model 5000; David Kopf Instruments, Tujunga, CA) attached to a stereotaxic unit (model 960; David Kopf Instruments). Mice were evaluated as described below after at least 14 d had elapsed after the IPI administration of the vehicle (PBS), rAAV-GFP-Cre, or rAAV-GFP, as described in Results.
Light-microscopic immunocytochemistry
Animals were perfused transcardially with 4% paraformaldehyde. The spinal cord, with attached dorsal root ganglia (DRGs), was dissected and placed into 4% paraformaldehyde for 1 hr before being cryoprotected in 30% sucrose for a minimum of 72 hr. The spinal cord was measured and cut into 3 mm sections, frozen into a mold with optimal cutting temperature compound, and then cut on a cryostat into 20 or 12 μm sections for immunocytochemistry and in situ hybridization, respectively. Spinal cord sections were incubated with antibodies (Abs) to the C terminus of NR1 (1:500; Ab 1516), NR2A (1:1000; Ab 1555P), and NR2B (1:1000; Ab 1557P) (each from Chemicon International, Temecula, CA); with antibodies to the C terminus of GFP (1:3000; 8363-2) [Clontech (Palo Alto, CA) or Molecular Probes Inc. (Eugene, OR)] (1:1000); or with antibodies to the C terminus of neuronal-specific nuclear protein [NeuN; 1:400; monoclonal antibody (mAb) 377; Chemicon International]. The sections were incubated in appropriate biotinylated secondary antibodies (BA-1000 and BA-9200; Vector Laboratories, Burlingame, CA), and immunoreactivity was detected with the avidin–biotin–peroxidase complex (ABC)–3,3-diaminobenzidinetetrahydrochloride technique (Hsu et al., 1981). To test the specificity of the immunolabeling, control slides were exposed to diluted normal goat serum instead of the primary antibody.
NR1 in situ hybridization
Slide-mounted spinal cord and DRG sections (12 μm) were hybridized with a 2.2 kb [33P]UTP-labeled (Perkin-Elmer Life Sciences, Boston, MA) antisense or sense probe directed to the region of the NR1 subunit that includes the entire sequence that is flanked by the loxP sites. Hybridization was performed according to methods described previously (Simmons et al., 1989). After the posthybridization washes, slides were dried by dehydration, dipped in NTB-2 emulsion from Eastman Kodak (Rochester, NY), and incubated at 4°C for 16–18 d. Slides were then developed and counterstained with hematoxylin and eosin. The reduction in SCDH NR1 labeling was determined by comparing ipsilateral with contralateral NR1 mRNA labeling in sections obtained from the injection site (a distance spanning ∼1.0 mm), in sections rostral and caudal to the injection site, where the GFP labeling was substantially reduced (the edge), and at the midpoint between the injection site and the edge. Labeling of mRNA and immunolabeling was estimated using the Metamorph software program (Universal Imaging, Downingtown, PA).
Electron microscopy
Three untreated and three fNR1 mice treated with IPI of rAAV-GFP-Cre (∼2 weeks previously) were deeply anesthetized with sodium pentobarbital (150 mg/kg, i.p.) and perfused through the ascending aorta with 5 ml of heparin-saline, followed by 40 ml of 3.75% acrolein (Polysciences, Warrington, PA) and 2% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. The spinal cord was removed and cut into 3-mm-thick blocks, which were stored in acrolein-paraformaldehyde for 20 min. Transverse sections (40 μm thick) were cut through the lumbar spinal cord, incubated in 1% sodium borohydride, freeze-thawed, and incubated in 0.5% bovine serum albumin (BSA), as described by Milner and Drake (2001).
For immunolabeling, sections were placed in a mouse anti-NR1 mAb (PharMingen, San Diego, CA) diluted 1/50 in 0.1% BSA for 40–48 hr at 4°C. This antibody has been characterized for specificity using the present labeling protocol (Wang et al., 1999a; Milner and Drake, 2001). NR1 was localized using the ABC method (Hsu et al., 1981), as described previously by Milner and Drake (2001). The secondary antibody was biotinylated horse anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA), and 3,3′-diaminobenzidine was used to visualize labeling. Sections for electron microscopy were embedded in EMbed (Electron Microscopy Sciences, Fort Washington, PA), and ultrathin sections were prepared as described by Drake and Milner (1999). Final preparations were analyzed on a Philips CM10 electron microscope. Figures were generated from digital images or scanned negatives using a Macintosh 8500/120, Adobe Photoshop 6.0 (Adobe Systems, San Jose, CA), and Quark X-Press 3.2.
Sections from untreated mice were examined qualitatively to establish the distribution of NR1 labeling and optimal labeling conditions. From each of three treated mice, one section near the injection site was analyzed quantitatively for NR1-labeled profiles. Lamina I-II was examined ipsilateral and contralateral to the injection site, and semirandom fields in lamina II were chosen on the basis of good morphological preservation and location within 50 μm of the plastic–tissue interface. Ten digital photographs of lamina II fields (totaling 2220 μm2) per dorsal horn were captured. All profiles with NR1-immunoperoxidase labeling were counted and identified using nomenclature and morphological definitions consistent with those of Peters et al. (1991). Profiles that lacked identifying characteristics were classified as “unknown.”
Electrophysiology
Spinal cord slice preparation. Lumbosacral spinal cords were removed under urethane anesthesia (1.5 gm/kg, i.p.) from fNR1 mice that received an IPI of either rAAV-GFP (n = 4) or rAAV-GFP-Cre (n = 5) or from age-matched untreated C57BL/6 mice (n = 6). The isolated spinal cord was placed in preoxygenated ice-cold Krebs' solution (in mm: 117 NaCl, 3.6 KCl, 2.5 CaCl2, 1.2 MgCl2, 1.2 NaH2PO4, 25 NaHCO3, and 11 glucose), and a 600-μm-thick transverse slice with attached dorsal root (right side in the area of rAAV injection) was cut using a vibrating microslicer (DTK1500; Dosaka, Kyoto, Japan) (Yoshimura and Jessell, 1989; Kohno et al., 1999). The spinal cord slice was placed on nylon mesh in the recording chamber, perfused with Krebs' solution (10 ml/min), and saturated with 95% O2 and 5% CO2 at 36 ± 1°C.
Patch-clamp recording from lamina II neurons. Whole-cell patch-clamp recordings were made from neurons located in lamina II [substantia gelatinosa (SG)] of the SCDH (Yoshimura and Nishi, 1993; Moore et al., 2002). In adult spinal cord slices, the SG is readily identifiable as a translucent region. For whole-cell recording, the resistance of patch pipettes was 5–10 MΩ when filled with the internal solution (in mm: 110 Cs2SO4, 0.5 CaCl2, 2 MgCl2, 5 EGTA, 5 HEPES, 5 TEA, and 5 ATP-Mg salt). Membrane currents were amplified with an Axopatch 200A amplifier (Axon Instruments, Foster City, CA) in voltage-clamp mode. Signals were filtered at 2 kHz and digitized at 5 kHz. Data were collected and analyzed with pClamp8 software (Axon Instruments). In all neurons, the frequency and amplitude of spontaneous EPSCs were recorded.
Dorsal root stimulation. Evoked EPSCs were recorded in lamina II neurons by dorsal root stimulation voltage clamped to -70 (AMPA) or + 40 mV (NMDA), respectively (Yoshimura and Nishi, 1993; Kohno et al., 1999). Dorsal root stimulation demonstrates a purely synaptic response that has a fast AMPA receptor-mediated and a slow NMDA receptor-mediated response. In spinal cord slices from untreated, rAAVGFP, and rAAV-GFP-Cre mice, when A fibers were fully activated (100 μA, 0.05 msec), AMPA receptor-mediated primary afferent-evoked EPSCs were collected (-70 mV). To test the NMDA synaptic response, NMDA receptor-mediated synaptic EPSCs were observed in untreated and rAAV-GFP-Cre mice in the presence of a glycine receptor antagonist (strychnine; 1 μm), a GABAA receptor antagonist (bicuculline; 20 μm), and an AMPA/kainate receptor antagonist (CNQX; 20 μm).
Behavioral tests
Motor coordination. Each mouse was placed on a rotarod (Life Sciences IITC, Hialeah, FL) set at 40 rpm (speed level 10). The length of time (seconds) that the mouse remained on the rod was measured. A cutoff of 60 sec was observed (Crawley and Paylor, 1997).
Mechanical stimulus threshold. The threshold to a non-noxious mechanical stimulus was assessed using a set of von Frey hairs. The animal was placed in a Plexiglas cage with mesh flooring suspended above the researcher and left to equilibrate for 15 min. von Frey filaments were applied perpendicularly against the midplantar surface of the foot. The predetermined mean von Frey hair of 2 gm was presented to each foot, and the subsequent presentation of the following filament was determined using the up–down method of Dixon, as described by Chaplan et al. (1994).
Thermal (heat) paw-withdrawal threshold. Thermal paw withdrawal was assessed using a thermal nociceptive stimulus (Hargreaves et al., 1988). Animals were placed in a Plexiglas cage, placed on a preheated glass plate maintained at 30°C, and left to equilibrate for 30 min. A radiant thermal stimulus was focused on the midplantar surface of the hindpaw and the latency (0.1 sec) for the withdrawal of the paw from the heat source was determined automatically. Each paw was evaluated separately, and a maximum cutoff of 15 sec was used. Two thermal stimulus intensities (low and high) were used.
Cold (cooling stimulus) threshold. Cold threshold sensitivity was assessed using the acetone drop application method (Bridges et al., 2001). Animals were placed in Plexiglas cages with mesh flooring at a level above the researcher and left to equilibrate for 15 min. A drop of acetone was placed against the midplantar surface of each hindpaw, and a positive response (score of 1) was recorded if the animal withdrew (flinched) the paw after application, whereas no response was given a value of 0. The score was the mean number of positive responses observed in five consecutive trials.
Thermal tail withdrawal threshold. A hot water bath was heated to a constant temperature of either 48, 52.5, or 55°C. Mice were held loosely in a towel and the lower half of each mouse's tail was dipped into the bath (Bilsky et al., 1996). A timer was stopped immediately when the mouse flicked its tail. A maximum tail withdrawal latency of 20, 15, and 10 sec, respectively, was used as a cutoff to prevent damage to the tail.
Formalin-induced nociceptive hypersensitivity. Formalin (5%) was administered in a volume of 20 μl with a 25 μl glass syringe (Hamilton Company, Reno, NV) into the right hindpaw of a lightly restrained mouse using a sterile 30 gauge, 0.5 inch needle. Immediately after the formalin injection, the mouse was placed in a test chamber and observed continuously for the next 60 min. The total time spent licking (seconds) was recorded in 5 min intervals and divided into 0–10 min (phase 1) and 10–60 min (phase 2) nociceptive responses, respectively (Elliott et al., 1995).
Statistics
The behavioral and electrophysiology data were analyzed by an ANOVA followed by the Student–Newman–Keuls test (multiple groups) or the t test (two groups). The electron microscopic data (number of NR1-labeled profiles per dorsal horn on the injected vs contralateral side) were compared using a Mann–Whitney rank–sum test. The level of significance was p ≤ 0.05.
Results
Viral transduction and recombination in the SCDH
Two weeks after IPI of rAAV-GFP-Cre into the adult mouse lumbar spinal cord, a highly localized pattern of expression of GFP was observed, one that was restricted to the ipsilateral dorsal horn and part of the ipsilateral ventral horn (Fig. 1A) and that persisted for as long as 10 months after administration of the viral vector. Region-specific Cre-mediated recombination was evident where reduction in NR1 mRNA labeling was measured in an adjacent section (Fig. 1B) by in situ hybridization using an anti-sense riboprobe, the sequence of which spans the loxP sites that will be deleted by the Cre-mediated recombination. The loxP sequences flank a region of the NR1 gene that encodes the four membrane domains (three TM domains and the M2 recurrent loop) and the entire C-terminal sequence of the polypeptide chain (Tsien et al., 1996). Three injections of rAAV-GFP-Cre at 0.5 mm intervals result in a segment-specific expression of GFP and an 80% depletion of the NR1 mRNA that extends for 3.75 ± 0.25 mm (SEM) (n = 13) rostrocaudally to incorporate the L4, L5, and L6 spinal segments. These are the target areas that comprise the sciatic sensory distribution from the paw to the dorsal horn (Woolf and Swett, 1984). The extent of the GFP label correlated almost perfectly with the area of reduced NR1 mRNA (Fig. 1A,B). Using this injection protocol, the entire ipsilateral dorsal horn was effectively depleted of NR1 mRNA, leaving the contralateral dorsal horn and nonlumbar spinal cord completely intact. The ratio of ipsilateral to contralateral labeling of NeuN (Fig. 1C) at 94.3 ± 7.5% (SEM) is not significantly different, indicating no loss of neurons. SCDH sections from animals that received the control vector (rAAV-GFP) (n = 5) showed no difference in the ratio percentage of ipsilateral to contralateral labeling of NR1 mRNA (96.3 ± 7.8% SEM) or NeuN (109.2 ± 8.7% SEM).
Figure 1.
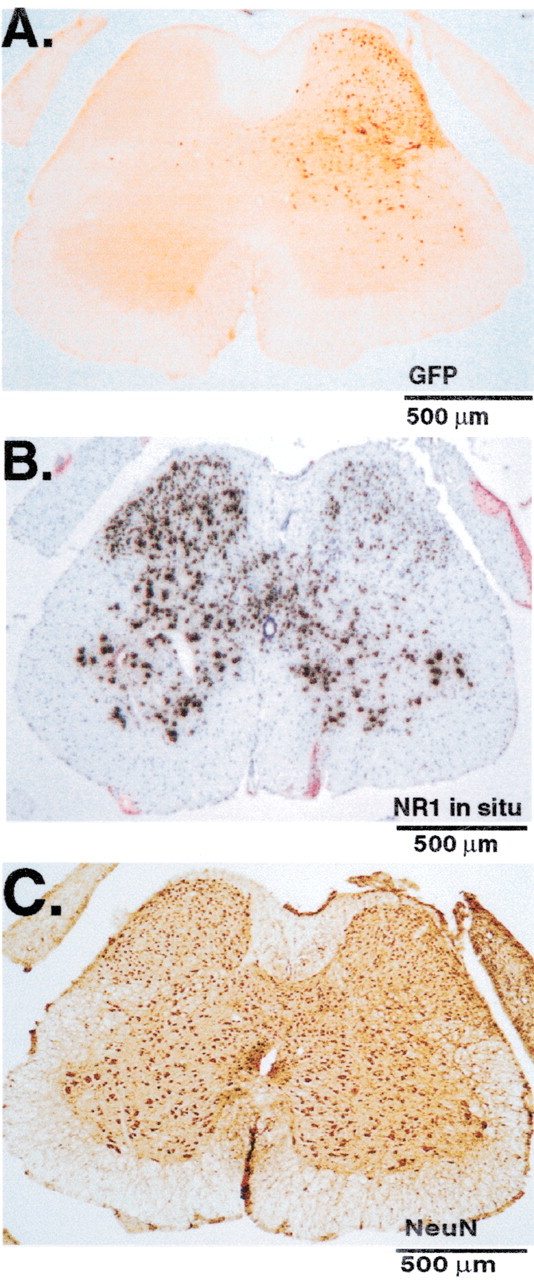
IPI of rAAV-GFP-Cre into the SCDH of a floxed NR1 mouse results in viral transduction, Cre-mediated recombination, and a spatiotemporal knock-out of the NR1 gene. A, B, On the side ipsilateral to the injection of rAAV-GFP-Cre, viral transduction results in the expression of GFP immunolabeling and the disappearance of NR1 mRNA as measured by in situ hybridization. C, In the same tissue, the expression of the neuronal marker NeuN is unaffected when ipsilateral and contralateral sides are compared.
NR1 labeling of individual neurons in lamina II was evident on the contralateral side (Fig. 2A, arrows) and absent in lamina II of the rAAV-GFP-Cre-injected side. Overall, much of the labeling was diffuse, and the morphological types of cells that contained labeling could not be determined. Therefore, an ultra-structural analysis was conducted to quantitatively determine changes in NR1 protein. At the ultrastructural level, NR1 labeling was abundant in laminas I and II of untreated fNR1 mice (data not shown) and on the contralateral side of rAAV-GFP-Cretreated mice (Fig. 2B). Of the 876 neuronal NR1-labeled profiles counted in the 2220 μm2 fields on the contralateral side, most were dendrites (81%), followed by axons (9%), terminals (6%), and somata (4%) (Aicher et al., 1997, 2002). Labeled dendrites were of all sizes, indicating diffuse NR1 distribution along the dendritic tree. Labeling in the larger dendrites and somata was consistent with NR1 expression in lamina II neurons, whereas the smaller dendrites may have originated from superficial and/or deeper neurons. Most NR1-labeled axons were small and unmyelinated. Labeled terminals were heterogeneous, ranging in size from small to large and forming symmetric synapses, asymmetric synapses, or no synapses. On the rAAV-GFP-Cre-injected side, the mean numbers of NR1-labeled dendritic profiles were reduced by 80% (Fig. 2C) (p < 0.05). A similar reduction was found for NR1-labeled somata and axons. Profiles on the rAAVGFP-Cre-injected side that lacked NR1 labeling were not degenerating, indicating that the absence of NR1 labeling was not attributable to death or damage to NR1-expressing cells.
Figure 2.
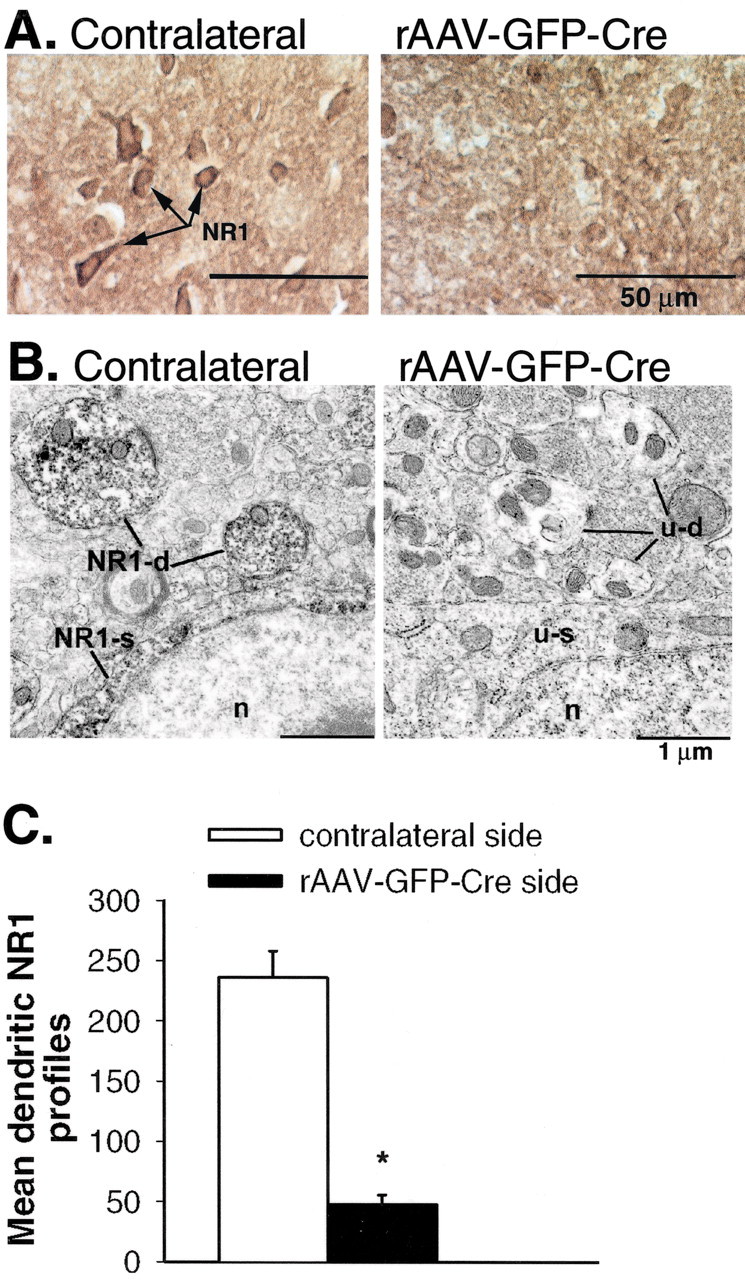
IPI of rAAV-GFP-Cre into the SCDH of a floxed NR1 mouse results in a decrease in NR1 protein that is confined to the ipsilateral (rAAV-GFP-Cre-injected) side of the SCDH. A, The NR1 protein immunolabeling (light microscopy) that is seen on the contralateral (noninjected) side (arrows) is absent on the ipsilateral side. B, Ultrastructural analysis (electron microscopy) of the SCDH of a rAAV-GFP-Cre-injected mouse demonstrates a loss of NR1 protein labeling in dendrites (u-d) and somata (u-s) compared with the contralateral side (NR1-d and NR1-s). N, Nucleus. C, Measurement of the number of mean dendritic NR1 profiles indicates an 80% reduction in NR1 protein labeling on the rAAV-GFP-Cre-injected side. *p ≤ 0.05.
In the SCDH of the adult mouse, mRNAs for only two of the four NR2 subunits, the ϵ1 (NR2A) and ϵ2 (NR2B), have been observed (Watanabe et al., 1994). When we compared the ratio of the immunolabeling of the ipsilateral (rAAV-GFP-Cre injected) and contralateral sides of the dorsal horn in six mice, no difference was observed for either the ϵ1 (96 ± 6.6% SEM) or ϵ2 (96.6 ± 10.0% SEM) subunits (data not shown). Thus, the knock-out of the NR1 subunit does not result in a compensatory change in the NR2 subunits.
NR1 labeling has also been observed in primary sensory neurons of the rat DRG (Shigemoto et al., 1992; Petralia et al., 1994; Wang et al., 1999b). To determine whether IPI administration of rAAV-GFP-Cre was associated with retrograde transport of the vectors, we examined the ipsilateral and contralateral DRG for GFP immunolabeling and NR1 mRNA (in situ) in sections that correspond to the lumbar spinal cord segments, where recombination had occurred after rAAV-GFP-Cre (n = 14) or after rAAV-GFP (n = 9). No labeling for GFP was observed in the DRG (Fig. 3A), nor was any evidence obtained of a decrease in NR1 mRNA on the ipsilateral side compared with the contralateral side (Fig. 3B). We also examined sections from the thoracic and cervical spinal cord and the brain and found no evidence for the expression of GFP. The deletion of the NR1 subunit in fNR1 mice appeared to be confined to SCDH cells, primarily in postsynaptic elements (dendrites and somata), and did not extend to the corresponding DRG cells that constitute the major presynaptic source of NR1.
Figure 3.
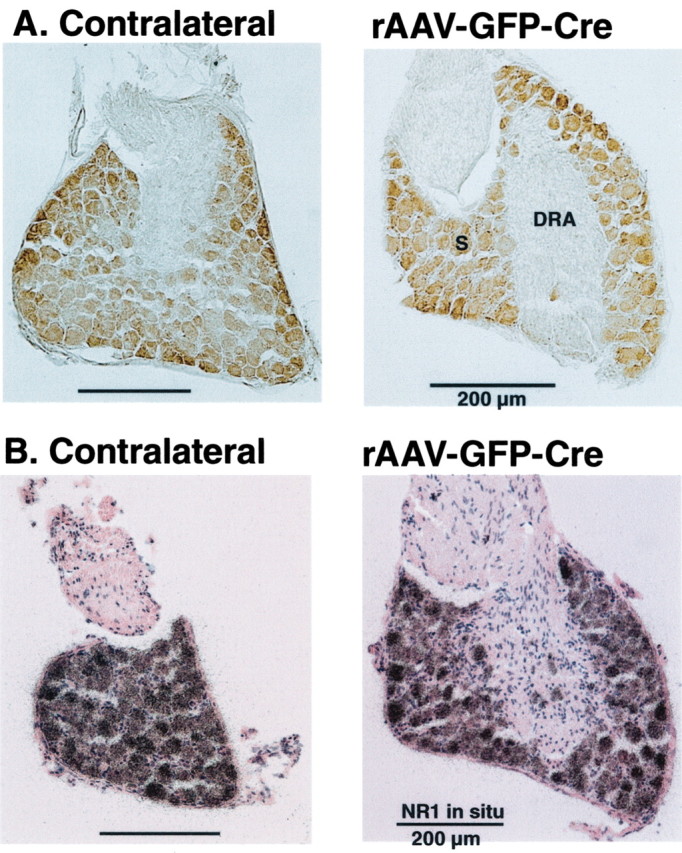
IPI of rAAV-GFP-Cre into the SCDH of a floxed NR1 mouse does not result in retrograde movement of the viral vector to the primary sensory neurons of the corresponding lumbar DRGs. These DRGs represent the presynaptic afferent innervation of the SCDH. A, Immunolabeling for GFP in the primary sensory neurons of the DRGs produced only a light background labeling over the somata (S) and no labeling of the dorsal root afferents (DRA). B, In situ hybridization of the neurons of the DRG produced the same intensity and extent of NR1 mRNA labeling of sensory neuron somata on the ipsilateral and contralateral sides.
Effects of a spatiotemporal knock-out of the NR1 subunit on NMDA and AMPA receptor currents and synaptic current activity in lamina II neurons of the SCDH
We compared the electrophysiological responses of lamina II neurons from C57BL/6 mice (the background strain of our fNR1 mice), which served as an untreated control, with those of fNR1 mice that had received rAAV-GFP or rAAV-GFP-Cre by IPI administration ∼2 weeks before in thick transverse spinal cord slices using the whole-cell patch-clamp recording technique (Kohno et al., 1999; Moore et al., 2002).
Spontaneous EPSC activity, membrane resting potential, and AMPA receptor-mediated current responses were unaffected in rAAV-GFP or rAAV-GFP-Cre mice
The frequency and amplitude of spontaneous activity (EPSCs recorded at -70 mV) observed in lamina II neurons were the same in untreated C57BL/6 mice or fNR1 mice given either viral vector (Fig. 4A). Furthermore, the resting membrane potential (untreated, -64 ± 1 mV, n = 12; rAAV-GFP, -63 ± 2 mV, n = 12, p = 0.66; rAAV-GFP-Cre, -67 ± 1 mV, n = 15, p = 0.12) in viral vector-treated mice was also unaffected (data not shown). To determine the effects of the viral vectors on AMPA receptor function, AMPA (10 μm) was bath-applied to spinal cord slices from untreated, rAAV-GFP, and rAAV-GFP-Cre mice, voltage-clamped at a holding potential of -70 mV in the presence of tetrodotoxin (TTX, 1 μm). Bath application of AMPA evoked exactly the same response in all three groups (Fig. 4B), indicating that this glutamatergic ionotropic receptor was unaffected by the viral injection.
Figure 4.
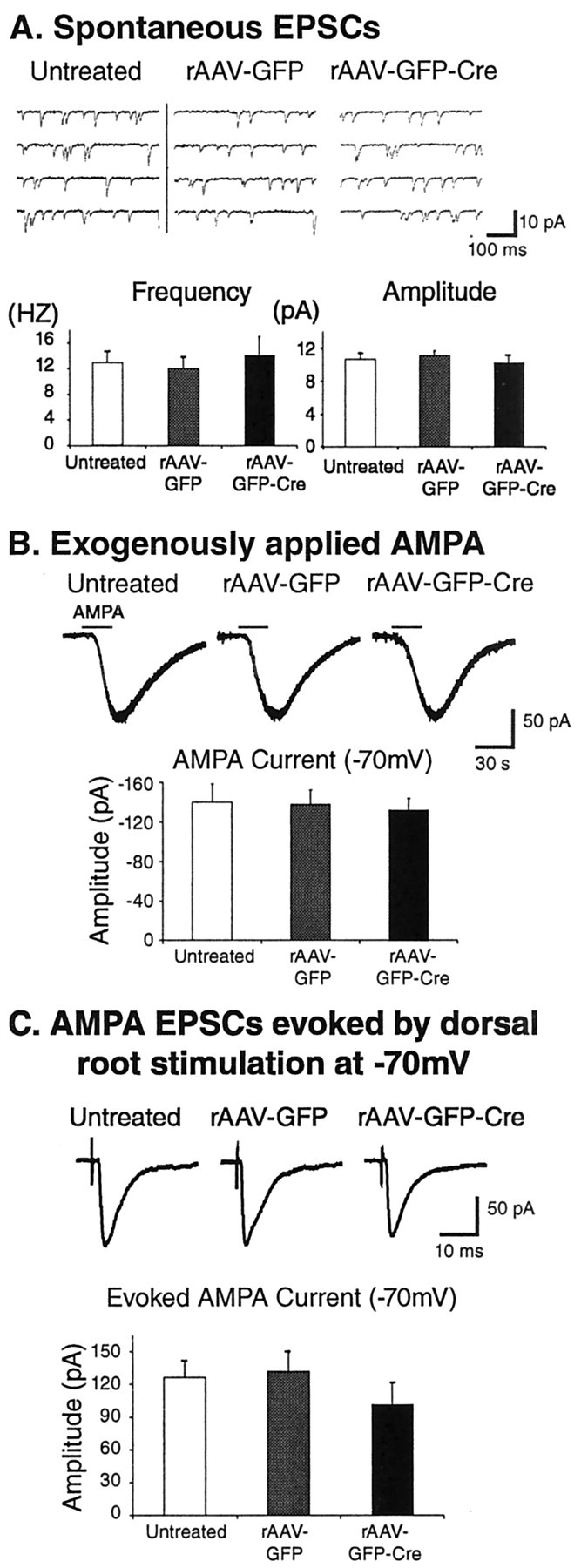
Spontaneous currents, AMPA receptor-mediated currents, and AMPA-mediated synaptic current activity are unaffected in lamina II neurons of the SCDH from rAAV-GFP or rAAV-GFP-Cre mice. A–C, Whole-cell patch-clamp recordings were made in spinal cord slices from C57BL/6 (untreated controls) or floxed NR1 mice that received IPIs of rAAV-GFP or rAAV-GFP-Cre. A, The frequency and amplitude of spontaneous activity (EPSCs recorded at -70 mV) are the same in untreated C57BL/6 mice or floxed NR1 mice given either viral vector. B, AMPA (10 μm) was bath-applied to spinal cord slices from untreated, rAAV-GFP, and rAAV-GFP-Cre mice. The neurons were voltage-clamped at a holding potential of -70 mV in the presence of TTX (1 μm). Bath application of AMPA evoked exactly the same response in all three groups. C, When A fibers were fully activated by dorsal root stimulation, the monosynaptic AMPA receptor-mediated primary afferent-evoked EPSC amplitudes (-70 mV) were similar in all three groups.
Current responses mediated by NMDA receptors were diminished in rAAV-GFP-Cre mice
To investigate whether the viral vectors produced changes in NMDA receptor function, NMDA (50 μm) was also bath-applied to these same spinal cord slices from untreated, rAAV-GFP, and rAAV-GFP-Cre mice, with the voltage of the lamina II neurons clamped at four different holding potentials (-70, -40, 0, and +40 mV) in the presence of TTX (1 μm) to inhibit synaptic effects resulting from the application of NMDA. Exogenously applied NMDA elicits an inward current at -70 or -40 mV and induces an outward current at +40 mV (Fig. 5A). Most neurons from untreated and rAAV-GFP mice had NMDA receptor components at -70 mV (untreated, -45 ± 8 pA, n = 11; rAAV-GFP, -34 ± 8 pA, n = 7, p = 0.40), -40 mV (untreated, -59 ± 8 pA, n = 10; rAAV-GFP, -49 ±6 pA, n = 7, p = 0.33), or +40 mV (untreated, 102 ± 18 pA, n = 12; rAAV-GFP, 77 ± 15 pA, n = 6, p = 0.37) (Fig. 5A). In contrast, the neurons from rAAV-GFPCre-injected mice exhibited minimal or absent NMDA receptor responses at -70 mV (-6 ± 2 pA; n = 10; p < 0.01) (Fig. 5A), -40 mV (-8 ± 2 pA; n = 8; p < 0.01) (Fig. 5A), or +40 mV (17 ± 5 pA; n = 10; p < 0.01) (Fig. 5A). The reduction in NMDA receptor current responses after applied NMDA averaged 86% (Fig. 5A). Therefore, the characteristic current–voltage relationship was abolished in animals that received rAAV-GFP-Cre (Fig. 5B).
Figure 5.
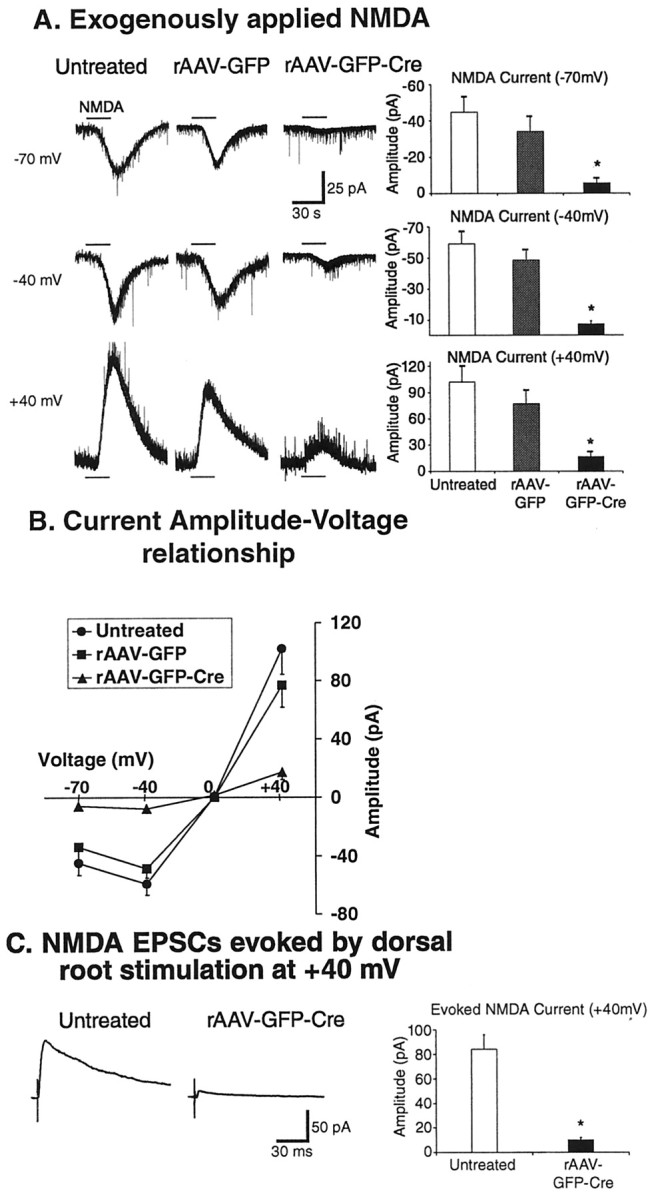
Current and synaptic responses mediated by NMDA receptors were diminished in rAAV-GFP-Cre mice. A–C, Whole-cell patch-clamp recordings were made in spinal cord slices from C57BL/6 (untreated controls) or floxed NR1 mice that received IPIs of rAAV-GFP or rAAV-GFP-Cre. These recordings were made from the same cells as those examined for AMPA responses (Fig. 4). A, NMDA (50 μm) was bath-applied to spinal cord slices from untreated, rAAV-GFP, and rAAV-GFP-Cre mice, with the voltage of the lamina II neurons clamped at four different holding potentials (-70, -40, 0, and +40 mV) in the presence of TTX (1 μm) to inhibit synaptic effects resulting from the application of NMDA. Exogenously applied NMDA elicits an inward current at -70 or -40 mV and induces outward current at +40 mV. Most neurons from untreated and rAAV-GFP mice had NMDA receptor components at-70,-40,or+40mV.Incontrast,the neurons from rAAV-GFP-Cre-injected mice exhibited minimal or absent NMDA receptor responses at each of these potentials(*p < 0.01).B,TheI–Vthat is characteristic of the NMDA ion channel is present in untreated and rAAV-GFP mice but absent in rAAV-GFP-Cremice. C,NMDA receptor-mediated primary afferent-evoked EPSC amplitudes(at+40mV) are significantly decreased in rAAV-GFP-Cre mice compared with untreated mice. The responses were measured in the presence of a glycine receptor antagonist (strychnine;1 μm), aGABAA receptor antagonist (bicuculline; 20 μm), and an AMPA/kainate receptor antagonist (CNQX; 20 μm). (*p < 0.01).
Synaptic responses mediated by the NMDA receptor, but not the AMPA receptor, were diminished in rAAV-GFP-Cre mice
In spinal cord slices from untreated, rAAV-GFP, and rAAV-GFPCre mice, when A fibers were fully activated by dorsal root stimulation, the monosynaptic AMPA receptor-mediated primary afferent-evoked EPSC amplitudes (-70 mV) were similar in all three populations (untreated, 126 ± 16 pA, n = 9; rAAV-GFP, 132 ± 19 pA, n = 5, p = 0.83; rAAV-GFP-Cre, 102 ± 20 pA, n = 7, p = 0.35) (Fig. 4C). Next, we investigated NMDA receptor-mediated synaptic responses from untreated and rAAV-GFP-Cre mice in the presence of a glycine receptor antagonist (strychnine; 1 μm), a GABAA receptor antagonist (bicuculline; 20 μm), and an AMPA/kainate receptor antagonist (CNQX; 20 μm). Under these conditions, NMDA receptor-mediated primary afferent-evoked EPSC amplitudes (at +40 mV) were significantly decreased by 88% in rAAV-GFP-Cre mice (untreated, 84 ± 12 pA, n = 11; rAAV-GFP-Cre, 7 ± 2 pA, n = 7, p < 0.01) (Fig. 5C).
Effects of a spatiotemporal knock-out of the NR1 subunit on motor coordination and acute mechanical and thermal stimuli
Because fNR1 mice have been reported to grow and mate normally and to exhibit behavior responses, including spatial memory, that are indistinguishable from those of wild-type mice (Tsien et al., 1996), we focused our evaluation on any consequences that might result from the IPI procedure. To do this, we compared groups of fNR1 mice before and at 2–4 weeks after IPI of either the control vector (rAAV-GFP) or the Cre-expressing vector (rAAV-GFP-Cre). The rotarod test measures the ability of a mouse to maintain balance and demonstrate motor coordination (Crawley and Paylor, 1997). Motor coordination was not affected by IPI of either viral vector (Fig. 6A). Next, we determined whether IPI affected non-injury-inducing noxious stimuli, including that resulting from placing the tail in water maintained at three different elevated temperatures, the latency to respond to an acutely painful low-intensity or high-intensity thermal stimulus delivered to the paw, the paw threshold response to a nonpainful mechanical stimulus (von Frey filaments), or the paw threshold response to a cold stimulus (evaporating acetone). Compared with the responses before IPI, the administration of either viral vector did not alter the latency for thermal tail withdrawal at 48, 52.5, or 55°C (Fig. 6B); the latency for thermal paw withdrawal at low (Fig. 6C) or high intensity (Fig. 6D); the mechanical stimulus threshold (Fig. 6B); or the cold stimulus threshold (Fig. 6F). When the paw ipsilateral to the IPI was compared with the contralateral paw, no difference was observed in the responses to acute mechanical or thermal stimuli (Fig. 6C–F). Neither the IPI procedure for delivering the virus nor the spatiotemporal knock-out of NR1 affected non-injury-inducing stimuli.
Figure 6.
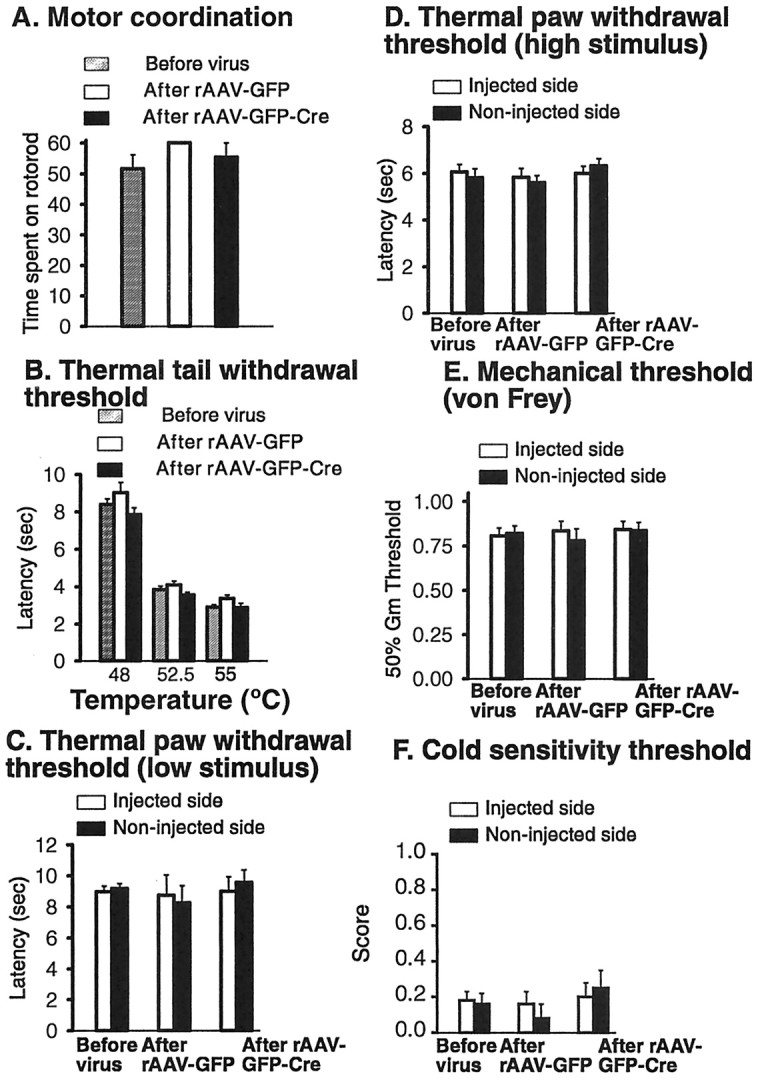
A spatiotemporal knock-out of NR1 in the SCDH of floxed NR1 mice does not affect motor coordination and non-injury-inducing stimuli. A, Motor coordination was measured using the rotarod test. B, A thermal stimulus was applied to the tail (thermal tail withdrawal threshold) at 48, 52.5, and 55°C. C, D, A brief thermal stimulus of low (C) and higher (D) intensity was applied to the paw. E, Mechanical (tactile) stimuli were applied using von Frey hairs. F, Cold sensitivity was measured by applying a drop of acetone to the paw.
Effects of a spatiotemporal knock-out of the NR1 subunit on injury-induced pain
Formalin was injected into the paws of fNR1 mice that had received either the vehicle for the vector (PBS) 2–4 weeks previously (n = 11), rAAV-GFP (n = 6), or rAAV-GFP-Cre (n = 9) by IPI. Intraplantar formalin evokes two phases of spontaneous pain-related behavior: an immediate, short-lasting phase attributable to the direct irritant effects of the chemical, and a slower-onset, longer-lasting phase that reflects a combination of ongoing sensory input and central sensitization (Coderre et al., 1990; Coderre and Melzack, 1992; Puig and Sorkin, 1995; McCall et al., 1996). The formalin-induced pain (licking and biting the affected paw) that occurred during phase 2 (10–60 min after the formalin injection) was decreased by 73% in the rAAV-GFP-Cre group but not in the vehicle-treated and rAAV-GFP-treated animals (Fig. 7A). Because no difference was observed between vehicle and rAAV-GFP treatments, these two control groups were combined so that we could better compare the responses at 5 min intervals in the control mice (n = 17) and rAAV-GFP-Cre-treated mice (n = 9). Figure 7B shows that during the peak of the phase 2 response (15–35 min after formalin), the rAAV-GFP-Cre-treated group showed significantly less injury-induced pain.
Figure 7.
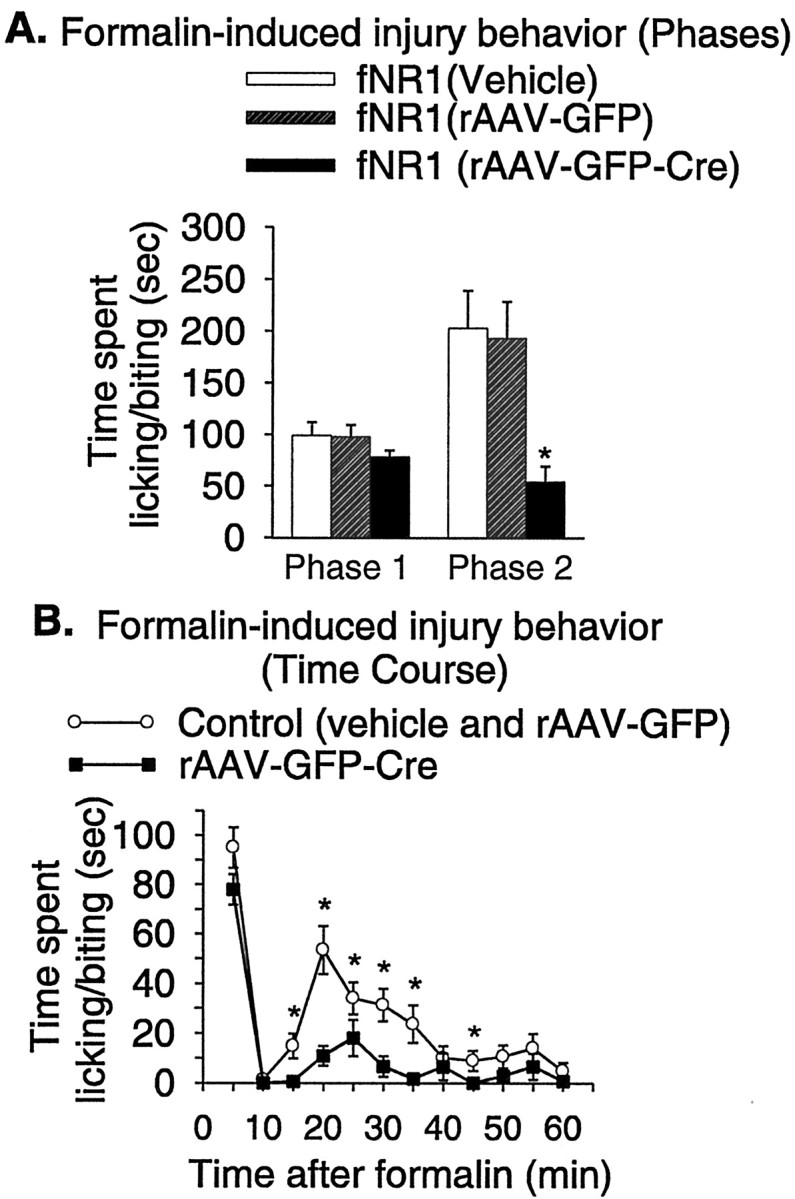
A spatiotemporal knock-out of NR1 in the SCDH of fNR1 mice significantly decreases the painful response to an injury-inducing stimulus measured during phase 2 of the formalin test. A, B, Intraplantar injection of formalin (5%) results in licking activity that can be divided into two phases, phase 1, which encompasses the first 0–10 min, and a second phase, which begins at 10 min and continues to 60 min (phase 2). A, During phase 1, the area under the response to formalin over the time curve is the same in fNR1 mice that received vehicle, rAAV-GFP, or rAAV-GFP-Cre treatment. During phase 2, the response of rAAV-GFP-Cre mice is reduced 70% compared with vehicle and rAAV-GFP mice (*p ≤ 0.05). B, When the data are presented as the time course of the response at 5 min intervals, rAAV-GFP-Cre mice show a diminished pain response at 5–35 (and at 45) min after formalin (*p ≤ 0.05).
Discussion
A conditional knock-out can delete a gene both spatially and temporally (Tsien et al., 1996), avoiding the complications resulting from deletion of the gene in systems other than those that affect the response, gene redundancy during development, or even embryonic lethality (Tsien et al., 1996). The original approach used (Tsien et al., 1996), although successful, requires the mating of a mutant mouse containing the loxP sites in the gene of interest (a floxed mouse) with a transgenic mouse that expresses the Cre recombination enzyme under the control of a region-specific promoter. However, region-specific promoters for the SCDH are not yet available, and expression of the Cre transgene integrated at different genomic locations often varies because of position variegation effects, necessitating the generation and analysis of multiple transgenic founder lines (Kaspar et al., 2002b). Thus, we used rAAV to deliver Cre recombinase and GFP to the target cells in the SCDH.
Viral vectors administered into CSF are taken up by meningeal cells, so that viral transduction of spinal cord cells after intrathecal administration is minimal (Mannes et al., 1998). However, microinjection of viral vectors directly into the brain or spinal cord (Kaplitt and Makimura, 1997; Peel et al., 1997; Mannes et al., 1998; Kaspar et al., 2002b) results in significant neuronal transduction. For rAAV-GFP-Cre, maximum recombination in the brain requires 7–14 d after injection (Kaspar et al., 2002b), and we found no difference in GFP expression and NR1 mRNA reduction in SCDH between 14 d and 6 weeks. Using multiple injections, we were able to transduce most cells in the target area, resulting in a maximum reduction of 80% in NR1 mRNA and protein. This estimate of the percentage of reduction probably represents an underestimate of the actual reduction in postsynaptic NR1, because presynaptic NR1 is not affected and therefore contributes residual NR1 protein and mRNA. As in the brain (Kaspar et al., 2002b), viral transduction was neurotropic, with GFP expression confined to NeuN-positive cells and absent in glia (GFAP-positive cells). Kaspar et al. (2002b) and this study found no evidence of neuronal toxicity after rAAV-GFP-Cre at the light-microscopic level, and we also observed none at the ultrastructural level. Although retrograde transport of rAAV vectors and transgene expression in specific projection neurons in the entorhinal cortex and substantia nigra has been reported previously (Kaspar et al., 2002a), we did not observe transport of either rAAV vector to the sensory neurons of the DRG that innervate the lumbar SCDH, to thoracic or cervical spinal cord, or to supraspinal sites. Thus, rAAV-GFP-Cre appears to produce a loss of NR1 that is confined to SCDH somata and dendrites. These SCDH components comprise the postsynaptic targets of primary afferents that originate in the DRG.
The specific functional consequences of the deletion of NR1 demonstrate the distinct advantages of the use of rAAV vectors to produce a spatiotemporal gene knock-out in the adult CNS. This approach can be readily adapted to other receptor–ion channels in the nervous system and will allow the examination of receptor function localized to a particular target area to be examined without the confounds that occur with constitutive knock-outs, thereby providing an important new strategy for defining receptor function without requiring specific antagonists, which is of enormous benefit in the evaluation of the many new targets that mass genomic screens are revealing. In addition, the spatial localization of vector transduction to somata of SCDH allows conclusions as to the primary postsynaptic locus of the functional receptor with a degree of certainty not possible with current pharmacological approaches.
Native NMDA receptors are hetero-oligomers of subunits (Sugihara et al., 1992; Hollmann and Heinemann, 1994) that, in the mouse, include NR1 (ζ1), ϵ1–ϵ4 (NR2A–NR2D), and NR3 (Dingledine et al., 1999). In rat dorsal horn NMDA, single-cell reverse transcriptase-PCR has detected the NR1 subunit and each of the four NR2B subunits, with the NR2B subunit being the predominant NR2 subunit (Karlsson et al., 2002). Studies of expressed NMDA receptors (Dingledine et al., 1999) and neurons of a CA1 hippocampal NR1 knock-out mouse (Tsien et al., 1996) show that the NR1 subunit is required to generate characteristic NMDA currents, and our results are compatible with this, showing a loss of response both to bath-applied NMDA and to NMDA receptor-mediated synaptic currents after NR1 deletion.
Our results demonstrate that NMDA receptors mediate critical aspects of the intensity coding that differentiates the responses to injury-inducing and nondamaging noxious stimuli (Woolf and Costigan, 1999). The application of acute, high-intensity, nondamaging thermal stimuli to the skin (e.g., paw or tail) activates high-threshold primary afferents, C and Aδ fibers, that result within seconds in transient, well localized pain. We found that these responses, along with those to normal mechanical (tactile) stimuli, do not require the presence of functional NMDA receptors in the spinal cord. Synaptic transmission after these stimuli is mediated by AMPA receptors, which generate fast EPSCs. In the NR1 knock-out mice, AMPA receptor-mediated currents are not altered in the cells in which NMDA-mediated currents are abolished. Thus, the fast, early warning protective pain system is preserved in the absence of NMDA receptors. These results are consistent with the observation that NMDA receptor antagonists given systemically or intrathecally do not modify baseline responses to acutely painful stimuli (Yaksh, 1999).
Noxious tissue-damaging stimuli generate a complex cascade of transmitters and modulators in the dorsal horn that initiate activity-dependent neuroplastic changes that result in pain hypersensitivity (Woolf and Costigan, 1999). NMDA receptors open more slowly and remain open longer than AMPA receptors, and this slow time course allows for a summation of membrane excitability by successive C fiber-induced depolarizations that remove the voltage-dependent Mg2+ block of the NMDA receptor, amplifying the response to each subsequent input, a phenomenon described as windup (Woolf and Costigan, 1999). Open NMDA channels allow a large Na+ and Ca2+ influx through the postsynaptic membrane, resulting in the slow-rising, long-duration EPSC and an increase in intracellular Ca2+. The increase in intracellular Ca2+ produces a cascade of molecular events leading to central sensitization. Post-translational changes in dorsal horn neurons include the activation of protein kinases that phosphorylate membrane-bound NMDA receptors and alter their functional properties, increasing membrane excitability. The phosphorylation of the NMDA receptor results in changes in NMDA receptor channel kinetics and a reduction in its voltage-dependent Mg2+ block (Chen and Huang, 1992; Wang and Salter, 1994; Yu et al., 1997). Both of these changes augment subsequent responsiveness to synaptically released glutamate, increasing synaptic strength and enabling previously subthreshold inputs to drive the output of the cell (Woolf and King, 1990). This effective increase in gain alters receptive field properties (Cook et al., 1987; Simone et al., 1989; Woolf and King, 1990) and pain sensitivity, causing hypersensitivity far beyond the site and duration of the C fiber activating stimulus that initiated the central sensitization (Torebjork et al., 1992). NMDA receptor antagonists can block windup, central sensitization, and the consequent pain hypersensitivity (Woolf and Thompson, 1991; Woolf and Costigan, 1999). However, determining whether the effect is pre-synaptic or postsynaptic and limited to the lumbar dorsal horn, as well as the extent of its contribution, has not been possible to establish using pharmacological techniques.
The formalin test is the most widely used method for assessing the antihypersensitivity efficacy of NMDA receptor antagonists (Coderre and Melzack, 1992; Yaksh, 1999). The immediate response to intraplantar formalin results from the stimulation of primary afferent nociceptors (Puig and Sorkin, 1995; McCall et al., 1996), whereas the latter response, phase 2, is generated by a reduced but continuing stimulation of peripheral nociceptors (Puig and Sorkin, 1995; McCall et al., 1996), activation of inflammatory mediators (Taylor et al., 2000), and central sensitization triggered by the phase 1 input from the periphery and maintained by the input associated with phase 2 (Coderre et al., 1990; Coderre and Melzack, 1992; Henry et al., 1999; Woolf and Costigan, 1999). Pharmacological studies using centrally applied receptor antagonists indicate a contribution of NMDA as well as neurokinin (NK1) receptors in the behavioral response seen during phase 2 of formalin (Coderre and Melzack, 1992; Yaksh, 1999). However, these studies cannot discriminate between a presynaptic versus postsynaptic action of the drug, ensure that all of the available receptors have been blocked, or ensure that those that are blocked are limited to the dorsal horn. Our results point to a critical role for postsynaptic NMDA receptors in the lumbar dorsal horn in injury-induced pain and unambiguously identify the in vivo anatomical locus of the responsible NMDA receptors. Furthermore, our results relate directly to the continuing controversy over the relative contribution to phase 2 pain of continued peripheral input (Puig and Sorkin, 1995; Henry et al., 1999), NK1 receptors, and NMDA receptors (Elliott et al., 1994; Shimoyama et al., 1997). Our results demonstrate that the NMDA receptor is the key player in the pain hypersensitivity expressed during phase 2 of the formalin response.
Localized deletion of NR1 preserves the responses to acute nondamaging stimuli while attenuating injury-induced responses. This finding suggests that spinally administered compounds that remain localized, including NMDA receptor antagonists, NR1 antisense (Rydh-Rinder et al., 2001; Yukhananov et al., 2002), or RNA interference targeted to NR1 may yield safe and useful therapeutic procedures for pain that have not been achieved with available NMDA receptor antagonists (Chizh et al., 2001).
Footnotes
This work was supported in part by National Institute on Drug Abuse (NIDA) Grants DA01457 and DA00198 to C.E.I., NIDA Training Grant DA07274 to J.B., National Institute of Neurological Disorders and Stroke Grants NS41073 to M.O., NS Training Grant T3207348 to D.H., and NS38253 and NS39518 to C.J.W. We thank S. Franklin, D. Vetter, K. Manova, S. Tonegawa, I. Perez-Otano, G. Royle, H. Zhu, J. Wagner, and L. Levin for advice and/or assistance.
Correspondence should be addressed to Charles E. Inturrisi, Department of Pharmacology, Weill Medical College of Cornell University, 1300 York Avenue, New York, NY 10021. E-mail: ceintur@med.cornell.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235031-10$15.00/0
References
- Aicher SA, Sharma S, Cheng PY, Pickel VM ( 1997) The N-methyl-d-aspartate (NMDA) receptor is postsynaptic to substance P-containing axon terminals in the rat superficial dorsal horn. Brain Res 772: 71–81. [DOI] [PubMed] [Google Scholar]
- Aicher SA, Goldberg A, Sharma S ( 2002) Co-localization of mu opioid receptor and N-methyl-d-aspartate receptor in the trigeminal dorsal horn. J Pain 3: 203–210. [DOI] [PubMed] [Google Scholar]
- Bilsky EJ, Inturrisi CE, Sadee W, Hruby VJ, Porreca F ( 1996) Competitive and non-competitive NMDA antagonists block the development of antinociceptive tolerance to morphine, but not to selective mu or delta opioid agonists in mice. Pain 68: 229–237. [DOI] [PubMed] [Google Scholar]
- Bridges D, Ahmad K, Rice AS ( 2001) The synthetic cannabinoid WIN55,212-2 attenuates hyperalgesia and allodynia in a rat model of neuropathic pain. Br J Pharmacol 133: 586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL ( 1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Method 53: 55–63. [DOI] [PubMed] [Google Scholar]
- Chen L, Huang LY ( 1992) Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature 356: 521–523. [DOI] [PubMed] [Google Scholar]
- Chizh BA, Headley PM, Tzschentke TM ( 2001) NMDA receptor antagonists as analgesics: focus on the NR2B subtype. Trends Pharmacol Sci 22: 636–642. [DOI] [PubMed] [Google Scholar]
- Coderre TJ, Melzack R ( 1992) The contribution of excitatory amino acids to central sensitization and persistent nociception after formalin-induced tissue injury. J Neurosci 12: 3665–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coderre TJ, Vaccarino AL, Melzack R ( 1990) Central nervous system plasticity in the tonic pain response to subcutaneous formalin injection. Brain Res 535: 155–158. [DOI] [PubMed] [Google Scholar]
- Cook AJ, Woolf CJ, Wall PD, McMahon SB ( 1987) Dynamic receptive field plasticity in rat spinal cord dorsal horn following C-primary afferent input. Nature 325: 151–153. [DOI] [PubMed] [Google Scholar]
- Crawley JN, Paylor R ( 1997) A proposed test battery and constellations of specific behavioral paradigms to investigate the behavioral phenotypes of transgenic and knockout mice. Horm Behav 31: 197–211. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF ( 1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7–61. [PubMed] [Google Scholar]
- Drake CT, Milner TA ( 1999) Mu opioid receptors are in somatodendritic and axonal compartments of GABAergic neurons in rat hippocampal formation. Brain Res 849: 203–215. [DOI] [PubMed] [Google Scholar]
- Dubner R, Ruda MA ( 1992) Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci 15: 96–103. [DOI] [PubMed] [Google Scholar]
- Elliott K, Hynansky A, Inturrisi CE ( 1994) Dextromethorphan attenuates and reverses analgesic tolerance to morphine. Pain 59: 361–368. [DOI] [PubMed] [Google Scholar]
- Elliott KJ, Brodsky M, Hynansky AD, Foley KM, Inturrisi CE ( 1995) Dextromethorphan suppresses both formalin-induced nociceptive behavior and the formalin-induced increase in spinal cord c-fos mRNA. Pain 61: 401–409. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J ( 1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32: 77–88. [DOI] [PubMed] [Google Scholar]
- Henry JL, Yashpal K, Pitcher GM, Chabot J, Coderre TJ ( 1999) Evidence for tonic activation of NK-1 receptors during the second phase of the formalin test in the rat. J Neurosci 19: 6588–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S ( 1994) Cloned glutamate receptors. Annu Rev Neurosci 17: 31–108. [DOI] [PubMed] [Google Scholar]
- Hsu SM, Raine L, Fanger H ( 1981) Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem 29: 577–580. [DOI] [PubMed] [Google Scholar]
- Kaplitt MG, Makimura H ( 1997) Defective viral vectors as agents for gene transfer in the nervous system. J Neurosci Methods 71: 125–132. [DOI] [PubMed] [Google Scholar]
- Karlsson U, Sjodin J, Angeby Moller K, Johansson S, Wikstrom L, Nasstrom J ( 2002) Glutamate-induced currents reveal three functionally distinct NMDA receptor populations in rat dorsal horn: effects of peripheral nerve lesion and inflammation. Neuroscience 112: 861–868. [DOI] [PubMed] [Google Scholar]
- Kaspar BK, Erickson D, Schaffer D, Hinh L, Gage FH, Peterson DA ( 2002a) Targeted retrograde gene delivery for neuronal protection. Mol Ther 5: 50–56. [DOI] [PubMed] [Google Scholar]
- Kaspar BK, Vissel B, Bengoechea T, Crone S, Randolph-Moore L, Muller R, Brandon EP, Schaffer D, Verma IM, Lee KF, Heinemann SF, Gage FH ( 2002b) Adeno-associated virus effectively mediates conditional gene modification in the brain. Proc Natl Acad Sci USA 99: 2320–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Kumamoto E, Higashi H, Shimoji K, Yoshimura M ( 1999) Actions of opioids on excitatory and inhibitory transmission in substantia gelatinosa of adult rat spinal cord. J Physiol (Lond) 518: 803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannes AJ, Caudle RM, O'Connell BC, Iadarola MJ ( 1998) Adenoviral gene transfer to spinal-cord neurons: intrathecal vs. intraparenchymal administration. Brain Res 793: 1–6. [DOI] [PubMed] [Google Scholar]
- McCall WD, Tanner KD, Levine JD ( 1996) Formalin induces biphasic activity in C-fibers in the rat. Neurosci Lett 208: 45–48. [DOI] [PubMed] [Google Scholar]
- Milner TA, Drake CT ( 2001) Ultrastructural evidence for presynaptic mu opioid receptor modulation of synaptic plasticity in NMDA-receptor-containing dendrites in the dentate gyrus. Brain Res Bull 54: 131–140. [DOI] [PubMed] [Google Scholar]
- Moore KA, Kohno T, Karchewski LA, Scholz J, Baba H, Woolf CJ ( 2002) Partial peripheral nerve injury promotes a selective loss of GABAergic inhibition in the superficial dorsal horn of the spinal cord. J Neurosci 22: 6724–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peel AL, Zolotukhin S, Schrimsher GW, Muzyczka N, Reier PJ ( 1997) Efficient transduction of green fluorescent protein in spinal cord neurons using adeno-associated virus vectors containing cell type-specific promoters. Gene Ther 4: 16–24. [DOI] [PubMed] [Google Scholar]
- Peters A, Palay SL, Webster HD ( 1991) The fine structure of the nervous system. New York: Oxford UP.
- Petralia RS, Yokotani N, Wenthold RJ ( 1994) Light and electron microscope distribution of the NMDA receptor subunit NMDAR1 in the rat nervous system using a selective anti-peptide antibody. J Neurosci 14: 667–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig S, Sorkin LS ( 1995) Formalin-evoked activity in identified primary afferent fibers: systemic lidocaine suppresses phase-2 activity. Pain 64: 345–355. [DOI] [PubMed] [Google Scholar]
- Rydh-Rinder M, Berge OG, Hokfelt T ( 2001) Antinociceptive effects after intrathecal administration of phosphodiester-, 2′-O-allyl-, and C-5-propyne-modified antisense oligodeoxynucleotides targeting the NMDAR1 subunit in mouse. Brain Res Mol Brain Res 86: 23–33. [DOI] [PubMed] [Google Scholar]
- Shigemoto R, Ohishi H, Nakanishi S, Mizuno N ( 1992) Expression of the mRNA for the rat NMDA receptor (NMDAR1) in the sensory and autonomic ganglion neurons. Neurosci Lett 144: 229–232. [DOI] [PubMed] [Google Scholar]
- Shimoyama N, Shimoyama M, Elliott KJ, Inturrisi CE ( 1997) d-Methadone is antinociceptive in the rat formalin test. J Pharmacol Exp Ther 283: 648–652. [PubMed] [Google Scholar]
- Simmons DM, Arriza JL, Swanson LW ( 1989) A complete protocol for in situ hybridization of messenger RNAs in brain and other tissues with radiolabeled single-stranded RNA probes. J Histotechnol 12: 169–181. [Google Scholar]
- Simone DA, Baumann TK, Collins JG, LaMotte RH ( 1989) Sensitization of cat dorsal horn neurons to innocuous mechanical stimulation after intradermal injection of capsaicin. Brain Res 486: 185–189. [DOI] [PubMed] [Google Scholar]
- Sugihara H, Moriyoshi K, Ishii T, Masu M, Nakanishi S ( 1992) Structures and properties of seven isoforms of the NMDA receptor generated by alternative splicing. Biochem Biophys Res Commun 185: 826–832. [DOI] [PubMed] [Google Scholar]
- Taylor BK, Peterson MA, Roderick RE, Tate J, Green PG, Levine JO, Basbaum AI ( 2000) Opioid inhibition of formalin-induced changes in plasma extravasation and local blood flow in rats. Pain 84: 263–270. [DOI] [PubMed] [Google Scholar]
- Torebjork HE, Lundberg LE, LaMotte RH ( 1992) Central changes in processing of mechanoreceptive input in capsaicin-induced secondary hyperalgesia in humans. J Physiol (Lond) 448: 765–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien JZ, Huerta PT, Tonegawa S ( 1996) The essential role of hippocampal CA1 NMDA receptor-dependent synaptic plasticity in spatial memory. Cell 87: 1327–1338. [DOI] [PubMed] [Google Scholar]
- Wang H, Gracy KN, Pickel VM ( 1999a) Mu-opioid and NMDA-type glutamate receptors are often colocalized in spiny neurons within patches of the caudate-putamen nucleus. J Comp Neurol 412: 132–146. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhang RX, Wang R, Qiao JT ( 1999b) Decreased expression of N-methyl-d-aspartate (NMDA) receptors in rat dorsal root ganglion following complete Freund's adjuvant-induced inflammation: an immunocytochemical study for NMDA NR1 subunit. Neurosci Lett 265: 195–198. [DOI] [PubMed] [Google Scholar]
- Wang YT, Salter MW ( 1994) Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature 369: 233–235. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Mishina M, Inoue Y ( 1994) Distinct spatiotemporal distributions of the N-methyl-d-aspartate receptor channel subunit mRNAs in the mouse cervical cord. J Comp Neurol 345: 314–319. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Costigan M ( 1999) Transcriptional and posttranslational plasticity and the generation of inflammatory pain. Proc Natl Acad Sci USA 96: 7723–7730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, King AE ( 1990) Dynamic alterations in the cutaneous mechano-receptive fields of dorsal horn neurons in the rat spinal cord. J Neurosci 10: 2717–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW ( 2000) Neuronal plasticity: increasing the gain in pain. Science 288: 1765–1769. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Swett JE ( 1984) The cutaneous contribution to the hamstring flexor reflex in the rat: an electrophysiological and anatomical study. Brain Res 303: 299–312. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Thompson SW ( 1991) The induction and maintenance of central sensitization is dependent on N-methyl-d-aspartic acid receptor activation: implications for the treatment of postinjury pain hypersensitivity states. Pain 44: 293–299. [DOI] [PubMed] [Google Scholar]
- Yaksh TL ( 1999) Spinal systems and pain processing: development of novel analgesic drugs with mechanistically defined models. Trends Pharmacol Sci 20: 329–337. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Yaksh TL ( 1992) Comparison of the antinociceptive effects of pre- and posttreatment with intrathecal morphine and mk801, an NMDA antagonist, on the formalin test in the rat. Anesthesiology 77: 757–763. [DOI] [PubMed] [Google Scholar]
- Yoshimura M, Jessell TM ( 1989) Primary afferent-evoked synaptic responses and slow potential generation in rat substantia gelatinosa neurons in vitro J Neurophysiol 62: 96–108. [DOI] [PubMed] [Google Scholar]
- Yoshimura M, Nishi S ( 1993) Blind patch-clamp recordings from substantia gelatinosa neurons in adult rat spinal cord slices: pharmacological properties of synaptic currents. Neuroscience 53: 519–526. [DOI] [PubMed] [Google Scholar]
- Yu XM, Askalan R, Keil II GJ, Salter MW ( 1997) NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science 275: 674–678. [DOI] [PubMed] [Google Scholar]
- Yukhananov R, Guan J, Crosby G ( 2002) Antisense oligonucleotides to N-methyl-d-aspartate receptor subunits attenuate formalin-induced nociception in the rat. Brain Res 930: 163–169. [DOI] [PubMed] [Google Scholar]