

Abstract
Voltage-gated chloride channels have recently been implicated as being important for cell proliferation and invasive cell migration of primary brain tumors cells. In the present study we provide several lines of evidence that glioma Cl– currents are primarily mediated by ClC-2 and ClC-3, two genes that belong to the ClC superfamily. Transcripts for ClC-2 thru ClC-7 were detected in a human glioma cell line by PCR, whereas only ClC-2, ClC-3, and ClC-5 protein could be identified by Western blot. Prominent ClC-2, -3, and -5 channel expression was also detected in acute patient biopsies from low- and high-grade malignant gliomas. Immunogold electron microscopic studies as well as digital confocal imaging localized a portion of these ClC channels to the plasma membrane. Whole-cell patch-clamp recordings show the presence of two pharmacologically and biophysically distinct Cl– currents that could be specifically reduced by 48 hr exposure of cells to channel-specific antisense oligonucleotides. ClC-3 antisense selectively and significantly reduced the expression of outwardly rectifying current with pronounced voltage-dependent inactivation. Such currents were sensitive to DIDS (200–500 μm) and 5-nitro-2-(3-phenylpropylamino) benzoic acid (165 μm). ClC-2 antisense significantly reduced expression of inwardly rectifying currents, which were potentiated by hyperpolarizing prepulses and inhibited by Cd2+ (200–500 μm). Currents that were mediated by ClC-5 could not be demonstrated. We suggest that ClC-2 and ClC-3 channels are specifically upregulated in glioma membranes and endow glioma cells with an enhanced ability to transport Cl–. This may in turn facilitate rapid changes in cell size and shape as cells divide or invade through tortuous extracellular brain spaces.
Keywords: ClC channel, brain tumor, patch clamp, antisense knockdown, cell migration, cell proliferation
Introduction
Most primary brain tumors are derived from glial cells and are collectively referred to as gliomas. This heterogeneous group of tumors includes astrocytomas, glioblastomas, and oligodendrogliomas among others. Their precise lineage relationship to glial cells and the mechanisms underlying their malignant transformation are poorly understood (Linskey, 1997). In addition to their uncontrolled proliferation, glioma cells show an unusual propensity to disperse from the tumor site and invade the healthy brain tissue (Merzak et al., 1994; Merzak and Pilkington, 1997). These characteristics make gliomas elusive targets for surgical management (Kaba and Kyritsis, 1997).
In many aspects, migrating glioma cells mirror the migration of progenitor cells during embryonic brain development (Levison et al., 1993; Amberger et al., 1997; Simpson and Armstrong, 1999), suggesting that they may recapitulate some features of gliogenesis or neurogenesis (Amberger et al., 1997; Noble and Mayer-Pröschel, 1997). Therefore, glioma cells may serve as a model system for studying the mechanisms of cell migration. Migration and invasion within the spatial constraints of the mature brain require special adaptations for these invading cells. For example, glioma cells appear to undergo shape changes as they squeeze through narrow extracellular brain spaces (Soroceanu et al., 1999; Ransom et al., 2001). Glioma cell shrinkage can be inhibited by Cl– channel blockers that render cells unable to invade, suggesting that this process requires Cl– channel-mediated fluid secretion (Soroceanu et al., 1999; Ransom et al., 2001). Hence, Cl– channels may be instrumental in regulating cell volume in the context of glioma cell invasion, a possibly unappreciated aspect of glioma biology. Cl– channels have also been implicated in the growth control of a number of cell types including Schwann cells (Wilson and Chiu, 1993; Pappas and Ritchie, 1998), C6 glioma cells (Rouzaire-Dubois et al., 2000), rat aortic smooth muscle cells (Wang et al., 2002), and mouse liver cells (Wondergem et al., 2001).
Several studies have reported on the expression of Cl– channels in glioma cells, some requiring volume changes for activation (Jackson and Strange, 1993, 1995; Bakhramov et al., 1995; Ullrich and Sontheimer, 1996; Bordey and Sontheimer, 1998; Ullrich et al., 1998; Rouzaire-Dubois et al., 1999; Soroceanu et al., 1999; Ransom et al., 2001); however, the molecular identity of these channels has yet to be elucidated. The most diverse and well studied Cl– channel family currently includes 10 members (ClC-0... ClC-7, ClC-Ka, and ClC-Kb) that share between 30 and 80% sequence identity. Five of these channels, ClC-2, ClC-3, ClC-5, ClC-6, and ClC-7 have been unequivocally identified in brain (Kawasaki et al., 1994; Brandt and Jentsch, 1995), and two, ClC-2 and ClC-3, have been suggested to be involved in cell volume regulation (Coca-Prados et al., 1996; Bond et al., 1998). These channels may therefore be candidates in the search for Cl– channels that facilitate glioma cell invasion.
In the present study we set out to examine the expression and functional activity of endogenous voltage-gated Cl– channels in glioma cells. We demonstrate the expression of ClC-2, ClC-3, and ClC-5 at the mRNA and protein levels. Additionally, whole-cell patch-clamp recordings show two distinct Cl– currents that can be attributed to ClC-2 and ClC-3, respectively, using antisense knock-down strategies.
Materials and Methods
Cell culture. All experiments were performed on the glioma cell lines D54-MG [glioblastoma multiforme (GBM), World Health Organization (WHO) grade IV], a gift from Dr. D. Bigner (Duke University), U251-MG (GBM; a gift from Dr. Y. Gillespie (University of Alabama at Birmingham). U-138 (GBM), U118 (GBM), U87 (GBM), and STTG-1 (anaplastic astrocytoma, WHO grade III) were obtained from the American Type Tissue Collection (Rockville, MD). Cells were cultured in either DMEM/F12 (Invitrogen, Grand Island, NY) supplemented with 7% fetal calf serum (FCS) (Hyclone, Logan, UT) or DMEM supplemented with 10% FCS. No difference was observed between cells cultured in either media.
Electrophysiology. Whole-cell voltage-clamp recordings were obtained via standard methods (Hamill et al., 1981). Patch pipettes were made from thin-walled (outer diameter 1.5 mm, inner diameter 1.12 mm) borosilicate glass (TW150F-4; WPI, Sarasota, Fl) and had resistances of 3–5 MΩ. Recordings were made on the stage of an inverted Nikon Diaphot microscope equipped with Hoffman Modulation Contrast Optics. Current recordings were obtained with an Axopatch 200A amplifier (Axon Instruments, Foster City, CA). Current signals were low-pass filtered at 2 kHz and were digitized on-line at 10–20 kHz, using a Digidata 1200 digitizing board (Axon Instruments) interfaced with an IBM-compatible computer (Dell XPS R400). Data acquisition and storage were conducted with the use of pClamp 8.2 (Axon Instruments). Cell capacitances and series resistances were measured directly from the amplifier, with the upper limit for series resistance being 10 MΩ, and series resistance compensation was adjusted to 80% to reduce voltage errors. Liquid junction potentials produced by test solutions were minimized by grounding the recording chamber via an agar salt bridge (4% agar, 500 mm KCl). Cells were plated on glass coverslips in a 24-well plate, and recordings were made 24–100 hr after plating. For antisense experiments cells were transfected 48 hr after plating, and recordings were made 48 hr after transfection. Outward currents were elicited by a voltage protocol that stepped the membrane from a holding potential of –40 mV for 40 msec and then to voltages ranging from –60 mV to 120 mV for 180 msec. Inward currents were activated from a protocol that stepped from the holding potential of –40 mV for 40 msec then stepped from –140 mV to 20 mV for 800 msec. Inward currents were activated by holding the cell at –120 mV for a minimum of 10 sec before the inward current protocol.
Solutions. Unless stated otherwise KCl pipette solution was used with 2 mm TEA in the extracellular bathing solution to block outward K + currents. The standard KCl pipette solution contained (in mm): 145 KCl, 1 MgCl2, 10 EGTA, 10 HEPES sodium salt, pH adjusted to 7.3 with Tris-base. CaCl2 (0.2 mm) was added to the pipette solution just before recording, resulting in a free-calcium concentration of 1.9 nm. Cells were perfused continuously at room temperature with a saline solution containing (in mm): 125 NaCl, 5.0 KCl, MgSO4, 1.0 CaCl2, 1.6 Na2HPO4, 0.4 Na2H2PO4, 10.5 glucose, 32.5 HEPES acid, and 2 TEA, pH adjusted to 7.4 with NaOH. The osmolarity of this solution was ∼300 mOsm. Drugs were added directly to these solutions, and unless stated otherwise all drugs were purchased from Sigma (St. Louis, MO). When CdCl2 was added to the bath solution, phosphates and sulfates were omitted to prevent precipitation of CdPO4 and CdSO4. Similar results were observed when KCl pipette solution and 2 mm TEA in the bathing solution were replaced with a CsCl pipette solution (KCl in pipette replaced with 145 mm CsCl). For ion replacement studies, Cl – ions in the bathing solution were replaced with an equal amount of the substituting ion.
PCR. Total RNA was extracted from D54-MG cells using Trizol (Invitrogen) using the manufacturer's protocol, treated with DNase (Promega) using the manufacturer's protocol, alcohol extracted with phenol/chloroform/isoamyl, precipitated, and resuspended in 1 mm sodium citrate, pH 6.4 (Ambion). Starting with 2 μg total D54 RNA as template, cDNA was synthesized using 500 ng random hexamers at 70°C for 10 min before placing the reaction on ice. Tris-HCl (14.7 mm), pH 8.3, 22 mm KCl, 0.9 mm MgCl2, 12 mm dithiothreitol, 0.6 mm each dNTP and 12 U Superscript Reverse Transcriptase II (Invitrogen) were added in a final volume of 17 μl, and the reaction was incubated at 25°C for 15 min, 42°C for 120 min, and 92°C for 2 min. For (–) reverse transcriptase (RT) reactions, water was substituted for the Superscript RT II. After the RT reaction was complete, the cDNA was precipitated using 0.1 vol of 5 m ammonium acetate and 2.5 vol of 100% EtOH at –20°C for at least 2 hr. Precipitated reactions were pelleted by centrifugation and resuspended in 10 μl of water. Half of the cDNA was used as template for each PCR reaction. DNA was amplified by adding 100 ng each gene specific primers and Platinum PCR Supermix (Invitrogen: 22 mm Tris-HCl, pH 8.4, 55 mm KCl, 1.65 mm MgCl2, 220 μm each dNTP and 22 U/ml TaqDNA polymerase with Platinum Taq antibody) for a final reaction volume of 50 μl. PCR cycling conditions were as follows: an initial denaturation step of 94°C for 5 min, 94°C for 1 min, annealing at 57°C for 1 min for ClC-1, and elongation at 72°C for 1 min. A final elongation step of 10 min at 72°C occurred on the last cycle. All PCR reactions were cycled 30 times except for ClC-4, which required 35 cycles. For ClC-2 and ClC-4, the annealing temperature used was 55°C; for ClC-3, -5, -6, and -7 the temperature was 50°C. The PCR primers for ClC-1, ClC-2, ClC-4, and ClC-7 were created with molecular biology software (Vector NT and Gene-Tool); ClC-3 and ClC-5 primers have been published previously (Enz et al., 1999) (rat and human are homologous for these primers), and ClC-6 has also been published previously (Eggermont et al., 1997). Primer sets for ClC-1, ClC-2, and ClC-6 span introns. Primers for all but ClC-1 were produced by Invitrogen; ClC-1 primers were produced by IDT Technologies.
Western blot analysis. Cells were lysed using RIPA buffer [50 mm TrisCl, pH 7.5, 150 mm NaCl, 1% Nondet P-40 (NP-40), 0.5% sodium deoxycholate, 1% SDS] for 30 min supplemented with protease inhibitor mixture obtained from Sigma. Homogenates were centrifuged for 5 min at 12,000 × g at 4°C. Protein quantification was performed on the supernatant using a DC protein assay kit from Bio-Rad (Hercules, CA). Protein was boiled for 5 min in Laemmli-SDS sample buffer containing 600 mm β-mercaptoethanol. Equal amounts of protein were loaded into each lane of a 7.5 or 4–20% gradient precast acrylamide SDS-PAGE gel (Bio-Rad). Proteins were separated at 120 V constant. Gels were transferred onto polyvinylidene difluoride paper (Millipore, Bedford, MA) at 200 mA constant for 2 hr at room temperature, and membranes were blocked in blocking buffer (5% nonfat dried milk, 2% bovine serum albumin, and 2% normal goat serum in TBS plus 0.1% Tween 20). Blots were incubated in primary antibody according to manufacturer's instructions. The membranes were then rinsed three times for 10 min and then incubated with HRP-conjugated secondary antibodies for 90 min. Blots were once again washed three times for 10 min and developed with enhanced chemiluminesence (Amersham Biosciences, Arlington Heights, IL) on Hyperfilm (Amersham Biosciences). For negative controls blots were stripped and reprobed with the appropriate control peptide incubated with antibody according to manufacturer's instructions. Recent controversy has focused on the specificity of voltage-gated chloride channel antibodies. For that reason we chose to use two sets of antibodies to confirm Western blot and immunocytochemistry results. One set of ClC-2, ClC-3, and ClC-5 polyclonal antibodies was obtained from Alpha Diagnostics (San Antonio, TX). Alternative ClC-2 and ClC-3 antibodies were obtained from Alomone Labs (Jerusalem, Israel), and ClC-5 was a generous gift from Thomas Jentsch (University of Hamburg, Hamburg, Germany). Actin and secondary HRP-conjugated antibodies were purchased from Sigma.
Immunocytochemistry. Cells plated on coverslips (12 mm round; Macalaster Bicknell, New Haven, CT) were washed two times with PBS and fixed with 4% paraformaldehyde for 15 min. Cells were then washed two more times with PBS and then permeabilized in PBS, 0.3% Triton X-100, and 3% goat serum [permeabilization buffer (PB)] for 30 min. Primary antibody was diluted in PB and added according to manufacturer's suggestion overnight at 4°C. The cells were washed three times in PBS before adding an FITC-conjugated goat anti-rabbit secondary antibody (Molecular Probes, Eugene, OR) diluted at 1:500 in PB for 1 hr at room temperature. Cells were then washed two times with PBS, washed once with DAPI (10 –4 mg/ml; Sigma), and diluted in PBS for 5 min. DAPI was rinsed off with PBS, and then cells were mounted onto clean coverslips with Gel/Mount (Biomedia Corporation, Forest City, CA). Fluorescent images (400 and 1000×) were acquired on a Leica DMRB fluorescent microscope (Leica, Heerbrugg, Germany). Digital confocal images (400 nm sections) were acquired with a Zeiss Axiovert 200M (München, Germany).
Immunogold electron microscopy. D54-MG cells were fixed in 4% paraformaldehyde for 30 min and in 0.25% glutaraldehyde for 30 min at room temperature and then permeabilized with 0.1% Triton X-100 in PIPES for 45 sec at room temperature. After rinsing and blocking, the cells were incubated with anti-ClC antibodies (Alomone) ClC-2, ClC-3, and Jentsch ClC-5 (1:100) for 4 hr at 4°C, washed, and incubated with 6 nm gold-labeled goat anti-rabbit IgG (1:10; Electron Microscopy Sciences) overnight at 4°C. Cells were rinsed, incubated with 1% OsO4 for 60 min at room temperature, dehydrated, and embedded in SPURR's resin (Electron Microscopy Sciences). Ultrathin sections (<90 nm) obtained on a Reichert Ultracut S (Leica, Heerbrugg, Germany) were contrasted with uranyl acetate and lead citrate and then examined on a JOEL 100 CX electron microscope (Joel, Peabody, MA).
Immunohistochemistry. Human glioma tissues with pathology reports were obtained from three separate sources: The Cooperative Human Network (Eastern and Southern Divisions), the Brain Tumor Tissue Bank (London, UK, and Ontario, Canada), and the University of Alabama at Birmingham Brain Bank (Birmingham, AL). Frozen tissue samples were cryosectioned into 6–8 μm. The basic procedure for fixing and staining fresh-frozen tissue slices has been described previously (Lyons et al., 2002). Consecutive slices of human tissue were stained with primary antibodies, ClC-2, ClC-3, and ClC-5 (Alomone ClC-2 and ClC-3; Jentsch ClC-5) overnight. After rinsing the next day, an immunoperoxidase staining system, EnVision+ Kit (Dako Corporation, Carpenteria, CA) was applied to the tissues for 1 hr at room temperature. Here, an HRP enzyme is conjugated to a secondary rabbit antibody. After rinsing, the binding was detected colorimetrically through reaction with 3′,3′-diaminobenzadine tetrahydrochloride (peroxidase substrate DAB kit, Vector Labs, Burlingame, CA). The slices were rinsed and counterstained with Methyl green (Vector Labs) for 7 min at 55°C. The process was completed with three EtOH rinses and three xylene rinses before slices were permanently mounted under coverslips for microscopic evaluation. Each tissue section was evaluated by comparing ClC staining with the ClC – controls for a minimum of three times each. Representative fields were chosen for documentation. Staining patterns were compared between consecutive slices as well as with other patient samples from the same tumor type. The staining from each slide was compared with controls and scored by at least two independent evaluators for each patient sample.
Antisense and nonsense oligonucleotide knockdown. Phosphorothiate-modified, 5′ end fluorescein-tagged antisense oligonucleotide primers were purchased from Invitrogen Custom Primers (Rockville, MD). The antisense oligonucleotide primer sequences used were as follows: ClC2: 5′-CGCCGCGGCCGCCAT-3′; ClC3: 5′-TCCATTTGTCATTGT-3′. ClC-3 antisense will eliminate both the short and long form of ClC-3 (Shimada et al., 2000). Both sequences used were specific for each protein and not found to be conserved in any other protein. A nonsense primer sequence was constructed from 15 randomized bases (5′-CCGTATGACCGCGCC-3′) and served as an experimental control. For electrophysiological antisense experiments, tumor cells were plated on glass coverslips in a 24-well plate at a density of ∼15,000 cells per well and allowed to grow for 2 d before transfection. Oligonucleotide primers (0.5–2 μg/ml final concentration) were used in combination with lipofectin. Cells were incubated with DNA/lipofectin for 6 hr with Opti-MEM serum-free media (Cancer Center, University of Alabama at Birmingham) according to the manufacturer's instructions and then replaced with normal serum containing media. Antisense-treated cells were selected by their fluorescence. The recording set up was equipped with epifluorescence (Nikon, Garden City, NY). For Western blotting, cells were transfected 24 hr after plating (25–100 μg/ml). Twenty-four hours after transfection serum-containing media was added to the dishes. Cells were harvested and protein lysates were collected 48 hr after transfection.
Statistical analysis. Current–responses to varied voltage steps and ramps were analyzed and measured in Clampfit (Axon Instruments); the resulting raw data were graphed and plotted in Origin 6.0 (MicroCal, Northampton, MA). Unless stated otherwise, all values are reported ± SE, with n being the number of cells sampled. Two-tailed t tests were performed to evaluate statistical significance, and p values are given in Results (Origin). The constant field potential equation PX/PCl = [Cl]o * e –(ΔErev(zF/RT)/[X]o (Hille, 1992) and shifts in reversal potential were used to calculate relative permeability ratios of iodide, bromide, and glutamate to chloride (PI/PCl, PBr/PCl, PGluc/PCl). Here, X is the substituting anion, ΔErev is the shift in reversal potential when switching from extracellular chloride [Cl]o to the substituting anion [X]o.
Results
Glioma cells express two biophysically and pharmacologically distinct Cl– currents
To examine the expression of Cl– channels in glioma cells, we first examined whole-cell currents elicited from cultured D54-MG glioma cells by patch clamp. These recordings were obtained under iso-osmotic conditions, as were all subsequent recordings. To avoid activation of swelling activated currents, we maintained the pipette osmolarity 10% below that of bath solution. To ensure that we were indeed recording Cl– currents, we initially replaced intracellular K+ with Cs+. However, prolonged recordings with CsCl-containing pipette solutions lead to extensive membrane blebbing, a phenomenon that we have not observed in other cells using identical solutions. In turn, blebbing often resulted in the spontaneous activation or enhancement of outward and inward currents. These currents were reminiscent of swelling-activated Cl– currents described previously in these cells (Ransom et al., 2001). To avoid any contamination of our recordings with these spontaneously occurring, swelling activated currents, we subsequently obtained recordings using a KCl pipette solution while inhibiting K+ currents with extracellular TEA (2 mm), which inhibited outward K+ currents as shown previously (Ransom and Sontheimer, 2001). A representative recording (Fig. 1A) demonstrates that outwardly rectifying currents exhibit time-dependent inactivation and particularly that the inactivating current was sensitive to gluconate. Both gluconate and glutamate show decreased permeation and were used to pharmacologically inhibit outward Cl– currents (Ransom et al., 2001). Representative whole-cell traces show that these outward Cl– currents were also sensitive to DIDS (200–500 μm) and 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB) (165 μm) (Fig. 1B,D). Of the mean, normalized peak gluconate-sensitive current, 72% was inhibited by DIDS (500 μm), whereas 70% was inhibited by NPPB (165 μm) (Fig. 1C,E). Moreover, the gluconate-sensitive current showed a significant degree of outward rectification and reversed at the chloride equilibrium potential (Fig. 1C,E). The small residual current that remained after gluconate application was sensitive to neither K+ nor Cl– channel blockers, and we presume it to be a nonspecific leak conductance.
Figure 1.

Voltage-dependent outward Cl– currents in human glioma cells. A, Representative examples of whole-cell Cl– currents that were evoked with voltage steps from –60 to +100 mV from a holding potential of –40 mV (in the presence of 2 mm TEA to block outward potassium currents). Traces demonstrate currents before and after gluconate, and subtraction of the two traces yielded the gluconate-sensitive current. B, Whole-cell currents using the same voltage step protocol before and after DIDS (500 μm). C, I–V plot of peak gluconate-sensitive current before and after DIDS (500 μm). D, Whole-cell Cl– currents before and after NPPB (165 μm). E, I–V plot of peak gluconate-sensitive currents before and after application of NPPB (165 μm). B, D, E, CsCl pipette solution.
When we altered the stimulus protocol and applied hyperpolarizing voltage steps ranging from –80 to –140 mV, we observed small inward currents (Fig. 2). It has been demonstrated that inward Cl– currents can be enhanced if voltage steps are preceded by a 30 sec prepulse to –120 mV (Bond et al., 1998). Indeed, these small inward currents showed increased activation after a hyperpolarizing prepulse. These currents were inwardly rectifying and demonstrated time-dependent activation at voltage steps more negative than –80 mV (Fig. 2A,B). As would be expected for inward Cl– currents (outward movement of Cl–), replacement of extracellular Cl– with gluconate did not inhibit these currents, and removal of the extracellular 5 mm KCl had no effect (data not shown). Pooled data demonstrate sensitivity to Cd2+ (200 μm) and marked inward rectification (Fig. 2B).
Figure 2.

Voltage-dependent inward Cl– currents in human glioma cells. A, Inward currents were evoked with voltage steps from –140 to +20 mV from a holding potential of –40 mV. Cells were hyperpolarized to –120 mV for a minimum of 20 sec to increase activation of inward current. Representative traces of inward Cl– current before (top) and after Cd2+ (200 μm) (middle) and the subtracted Cd2+ sensitive current (bottom) are shown. B, I–V plot of Cd2+ (200 μm)-sensitive Cl– currents evoked from the same voltage step protocol. Currents returned after washout of Cd2+, and removal of the 5 mm [K]o or addition of 200 μm Ba2+ had no effect (data not shown).
Although specific Cl– channel blockers are few, differential sensitivity of ClC channels to DIDS, 9-AC, NPPB, niflumic acid, tamoxifen, and Cd2+ has been useful for the pharmacological characterization of ClC Cl– channels. For example, inward Cl– currents mediated by ClC-2 are typically sensitive to Cd2+ or Zn2+ (Clark et al., 1998; Enz et al., 1999; Nehrke et al., 2002), whereas outward Cl– currents thought to be mediated by ClC-3 are typically inhibited by DIDS or NPPB (Duan et al., 1997; von Weikersthal et al., 1999). Glioma cell outwardly rectifying Cl– currents were consistently found to be sensitive to gluconate, DIDS, and NPPB (Fig. 1B–E). The gluconate-sensitive current (after subtraction) was characterized by pronounced time-dependent inactivation and voltage dependence. By contrast, inward currents were inhibited by Cd2+ (Fig. 2B,C). These Cd2+-sensitive currents showed significant inward rectification and were activated at steps more negative than –80 mV.
Another defining feature of Cl– channels is their permeability to a number of halide and non-halide anions. Indeed, the relative permeability to I–, Br–, and F– has been used as a distinguishing feature of ClC channels and ICLswell. We therefore examined whether replacement of extracellular Cl– with other anions could sustain these Cl– currents. Indeed, both I– and Br– produced slightly larger outward currents while reducing the inward current; additionally, outward currents were reduced in gluconate (Fig. 3). Relative permeability of ions through channels are typically derived from shifts in tail current reversal potentials; however, ClC channel gating is coupled to the permeating anion, disallowing this approach (Pusch et al., 1995). If one infers the relative permeability from changes in reversal potential, our reversal potential shifts are suggestive of a permeability sequence of I–>Br–>Cl–>glutamate with PI–/PCl– = 1.6, PBr–/PCl– = 1.3, and Pglut–/PCl– = 0.24 (Fig. 3, Table 1).
Figure 3.

Effects of halide ion replacement on D54 glioma cells. Representative recording of Cl– current from a linear voltage ramp protocol (–160 to +160 mV, holding at –40 mV) is shown. Extracellular Cl– (thick black line) in the bath solution was replaced with 130 mm NaI (gray line), NaBr (black dashed line), or Na-gluconate (light gray line). Inset magnifies the region around the reversal potential.
Table 1.
Chloride ion replacement shifts the reversal potential in D54 glioma cells
|
Ion |
Number of cells (n) |
Shift in reversal potential (mV)a ± SD |
PX-/PCl- |
|---|---|---|---|
| Iodide | 9 | -12.5 ± 5.3 | 1.64 |
| Bromide | 8 | -6.6 ± 4.7 | 1.30 |
| Glutamate
|
4
|
36.6 ± 3.9
|
0.24
|
Relative to chloride.
Glioma cells and acute patient biopsies show expression of ClC-2, -3, and -5
We were ultimately interested in determining whether the above described currents could be mediated by known Cl– channels of the ClC family. In an initial effort to examine this question, we used RT-PCR using specific primers for ClC-1 through ClC-7 (Table 2) and mRNA from D54-MG glioma cells. RT-PCR performed with the specific primers yielded fragments of the predicted molecular weight (Table 2) for ClC-2, -3, -4, -5, -6, and -7, but lacked transcripts for ClC-1 (Fig. 4). The latter has been shown to be specific for muscle (Jentsch et al., 1995). It has been demonstrated previously that there are multiple splice variants of ClC-6. The primer pair that we used had been characterized previously, and RT-PCR analysis with this primer pair yielded fragments at 520 and 350 base pairs. The two bands correspond to the presence or absence of a particular exon (Eggermont et al., 1997).
Table 2.
Primer sets used to RT-PCR ClC channels from D54 glioma cells
|
Gene |
Sequence (5′-3′) |
Product length |
Accession number |
|---|---|---|---|
| ClC-1 | se GCATCTGTGCTGCTGTCCTC | 410 bp | NM_000083 |
| as GACACCGAGCATGACTTGGC | |||
| ClC-2 | se GGGGGCCCAGTGTCACCAGGAAC | 556 bp | NM_004366 |
| as CGGGGGAGGCCATGACGGGAGTG | |||
| ClC-3 | se CCTCTTTCCAAAGTATAGCAC | 552 bp | AF172729 |
| as TTACTGGCATTCATGTCATTTC | |||
| ClC-4 | se GCGGGCAGGATGGTGGGAATTG | 650 bp | AF170492 |
| as GCGCCGCAGCTTCAGGGGATGT | |||
| ClC-5 | se GGAACATCCTGTGCCACTG | 543 bp | X91906 |
| as AATCACAGAGCTTGGAGGAG | |||
| ClC-6 | se GTTTAACTTCCCCTATTTCC | 519 bp | NM_001286 |
| as GCATTCTCCTAACACCATCG | 353 bp | ||
| ClC-7 | se GGGCGTGGTGGGCGGTGTG | 472 bp | Z67743 |
|
|
as CGCCCCCGTGAGGTAGGACAGG
|
|
|
Figure 4.

RT-PCR of ClC-1 through ClC-7 in D54 glioma cells. Lane 1 is a 100 bp marker (Invitrogen). Lanes denoted with + are RT-PCR reactions with primers for the designated ClC channel. Lanes denoted with – are identical reactions with water substituted for reverse transcriptase. Using D54-MG total RNA as a template, only the muscle-specific ClC-1 primers did not yield a product. ClC-2 through ClC-7 mRNA was present in D54-MG cells as judged by amplification of the expected size PCR products using gene-specific primers.
To examine channel expression at the protein level, we performed Western blots on glioma cell lysates (D54, U251, U158, U87, and STTG1) using antibodies to ClC-2, -3, -4, and -5 (Fig. 5). We did not observe any immunoreactivity with ClC-4 (data not shown); however, we consistently saw bands corresponding to ClC-2, -3, and -5. Because there remains significant controversy concerning the specificity of these antibodies (Stobrawa et al., 2001), we went to great efforts to confirm their specificity and hence the validity of our results. Specifically, we used at least two different sources for each antibody (Alomone: ClC-2, and -3; T. Jentsch: ClC-5; Alpha Diagnostics: ClC-2, -3, -5). Representative blots with six tumor cell lines that were probed with antibodies obtained from Alpha Diagnostics are shown in Figure 5A. For comparison, two glioma cells lines, D54 and STTG-1, were probed with Alomone's ClC-2 and ClC-3 and Jentsch ClC-5 antibodies (Fig. 5B). Note that similar bands were observed in both sets of blots at the appropriate molecular weights. To confirm the specificity of each antibody and as a negative control, the blots were probed with the antibodies preabsorbed with the matching peptide. Under these conditions all bands seen were essentially eliminated (data not shown).
Figure 5.
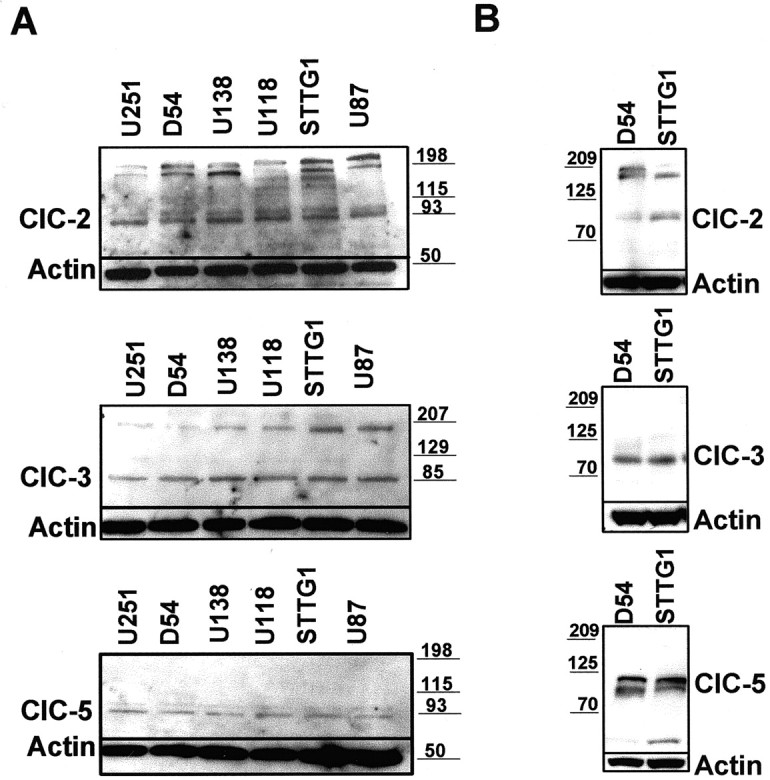
Western blot analysis demonstrating expression of ClC-2, ClC-3, and ClC-5 in the human glioma cell lines U251, D54, U138, U118, STTG1, and U87. A, Top, Alpha Diagnostics ClC-2 antibody recognizes a doublet at ∼90 kDa and several bands at a higher molecular weight, possibly multimers of ClC-2. A, Middle, Alpha Diagnostics ClC-3 antibody recognizes a prominent band at ∼85 kDa and a much lighter band ∼15 kDa higher. A, Bottom, Alpha Diagnostics ClC-5 antibody recognizes a prominent band at ∼90 kd and a lighter band at a slightly higher molecular weight. B demonstrates similar results with Alomone's ClC-2 and ClC-3 antibodies and Jentsch ClC-5 antibody. All blots either were probed with actin (Sigma) as a loading control with the designated antibody or they were stripped and reprobed.
To illustrate the distribution of ClC-2, ClC-3, and ClC-5 in human glioma cells, we used immunocytochemistry with FITC-conjugated secondary antibodies. Distinct localization was observed for each, with differential staining of the cytoplasm versus cell surface (Fig. 6). Figure 6, A and B, demonstrates 400× and 1000× images, respectively. The images in Figure 6C are digital confocal images. All images shown were probed with Alpha Diagnostics antibodies; however, similar results were obtained with the alternate set of antibodies. Interestingly, all three ClC channels (ClC-2, -3, and -5) appear to associate prominently with lamellipodia at the leading edges of the cells, and overall they appear in clusters on the membrane. In addition, both Alpha Diagnostics and Jentsch ClC-5 antibody recognized large vesicular type structures in the cell cytoplasm.
Figure 6.
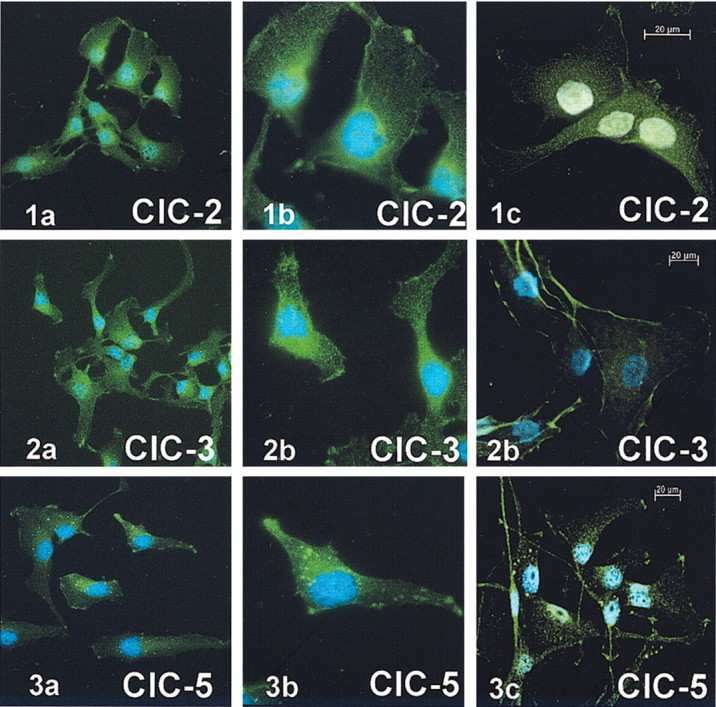
Immunoreactivity for ClC-2, ClC-3, and ClC-5 demonstrates intracellular and plasma membrane labeling of D54 glioma cells (Alpha Diagnostics). Left panels are 400 × images; middle panels are 1000 × images. Right panels demonstrate 400 nm sections of digital confocal images. Similar results were observed with Alomone ClC-2 and ClC-3 and Jentsch ClC-5 antibodies.
To further confirm the surface expression of these channels, we obtained immunogold electron microscopy (EM) images from D54 glioma cells in which ClC-2, -3, and -5 were each conjugated to 6 nm gold particles. As is demonstrated in Figure 7A–C (white arrows), immunoreactivity for each channel was found in surface clusters.
Figure 7.

Immunogold EM with 6 nm gold particles localizes ClC-2, ClC-3, and ClC-5 to the plasma membrane of glioma cells. A–C show localization of a portion of ClC-2, ClC-3, and ClC-5 with the plasma membrane of human glioma cells (Alomone ClC-2 and ClC-3 and Jentsch ClC-5).
Because the above studies were performed on cultured cells, we sought to confirm these findings by examining ClC expression in acute biopsies from patients with glioblastoma multiforme and pilocytic astrocytoma. Several such biopsies were examined, and a representative example of each tumor type is illustrated in Figure 8. Paraffin sections of these biopsies were stained with ClC antibodies, followed by secondary antibodies detected with DAB (a brown reaction product). These studies show prominent expression of ClC-2, -3, and -5 and by and large confirmed our findings in cultured cells. Taken together, our biochemical and immmunohistochemical studies suggest that ClC-2, -3, and -5 are expressed in glioma cells in vivo and that, at least in isolated glioma cells, a significant percentage of these channels is localized in the plasma membrane.
Figure 8.

Human biopsy samples stain positive for ClC-2, ClC-3, and ClC-5. Consecutive cryostat sections of frozen patient samples of a glioblastoma tumor (A) and a pilocytic astrocytoma tumor (B) were immunohistochemically stained with antibodies to ClC-2 (a1, b1), ClC-3 (a2, b2), and ClC-5 (a3, b3) and detected with a DAB reaction (a brown reaction product). The slices were counterstained with methyl green to detect cell nuclei. The control stainings (a4, b4) were performed under identical conditions omitting only the primary antibodies.
Antisense studies suggest that glioma Cl– currents are mediated in part by ClC-2 and ClC-3
We next sought to determine whether any of the currents observed in glioma cells (Figs. 1, 2, 3) could be attributed to defined ClC channels. Because of the current profiles, we hypothesized that the inactivating, outwardly rectifying currents were attributable to ClC-3, whereas the activating, inwardly rectifying currents were attributable to ClC-2. The lack of specific Cl– channel blockers led us to use antisense knockdown techniques to investigate our hypothesis. We used specific antisense primers for ClC-2 and ClC-3 (sequences given in Materials and Methods). D54-MG cells were recorded 48 hr after transfection with fluorescently tagged antisense and nonsense oligonucleotides. Current densities of successfully transfected cells (identified by their fluorescence) were analyzed. Representative traces for ClC-3 antisense knockdown demonstrated a significant reduction in whole-cell currents (Fig. 9A). Mean current densities of nonsense- and antisense-treated cells exhibited significant reductions in whole-cell currents at potentials that typically show the greatest activation (50% at the peak current, p < 0.01) (Fig. 9B). The specificity of the ClC-3 antisense oligonucleotides is demonstrated in Figure 9C. When an equal amount of protein is loaded (as evidenced by actin loading control), ClC-3 protein was dramatically reduced when D54 cells were treated with the ClC-3 antisense oligonucleotides.
Figure 9.

ClC-3 antisense oligonucleotides inhibit whole-cell outward Cl– currents in D54 glioma cells. A, Whole-cell outward currents from a representative control (nonsense- or ClC-2 antisense-treated cell) and ClC-3 antisense-treated cell before and after Na-gluconate bath solution and the subtracted gluconate-sensitive current. Currents elicited by voltage step protocol are shown in the inset. B, Antisense treatment significantly reduced whole-cell outward Cl– currents at peak voltages: +80, 40%; +100 mV, 48%; +120 mV, 50%. Interestingly, the current that remained appeared to be the leak current that was not sensitive to gluconate (n=number of cells examined). C, Western blot analysis demonstrates the specificity of the ClC-3 antisense oligonucleotides (lane 1, nonsense-treated cells; lane 2, ClC-2 antisense-treated cells; lane 3, ClC-3 antisense-treated cells; lane 4, ClC-3 antisense-treated cells with a threefold higher concentration of DNA). Of note, for electrophysiology experiments ClC-3 was used at a three- to fourfold higher concentration than ClC-2 (see Materials and Methods).
Cells treated with antisense to ClC-2 demonstrated a marked reduction in inward current (Fig. 10A). Once again, peak current densities at hyperpolarized potentials were significantly reduced by 60% (p < 0.03) (Fig. 10B). Interestingly, treatment with ClC-2 antisense oligonucleotides also increased the input resistance of these cells (1239 ± 270 MΩ), with average values for nonsense-treated cells of 485 ± 99 MΩ and ClC-3 antisense-treated cells = 598 ± 101 MΩ (n = 14 control, n = 10 ClC-2; p < 0.01, relative to nonsense). Consistent with this change, the leak current in these cells appeared to be smaller, suggesting that ClC-2 contributed to the resting conductance. Alternatively, secondary effects caused by antisense treatment may have affected other membrane properties such as trafficking of other channels to the membrane that are typically open at rest. We do not believe this is the result of nonspecific antisense toxicity, because this would cause a decrease in the mean input resistance, making the cell appear leaky, which is the opposite of what we observed. Antisense specificity for ClC-2 is demonstrated by Western blot in Figure 10D. Only cells treated with antisense oligonucleotides for ClC-2 demonstrate a decrease in the amount of ClC-2 protein.
Figure 10.

ClC-2 antisense oligonucleotides inhibit inward Cl– currents and increase input resistance in D54 glioma cells. A, Whole-cell outward Cl– currents from a representative nonsense- and ClC-2 antisense-treated cell before and after application of Cd2+ (200 μm)and the Cd2+ -sensitive subtracted currents. Currents were elicited by the voltage-step protocol shown in the inset. B, Antisense treatment significantly reduced inward Cl– currents at peak voltages: –140 mV before hyperpolarization, 60%; 140 mV after hyperpolarization, 58%; after hyperpolarization at –120 mV, 55%; after hyperpolarization at –100 mV, 60%. C, Treatment of D54 glioma cells with ClC-2 antisense oligonucleotides significantly increased the input resistance of these cells relative to nonsense or ClC-3 antisense-treated cells (p < 0.01 relative to nonsense) (n = number of cells examined). D, Once again Western blotting demonstrated the specificity of the ClC-2 antisense oligonucleotides. Only cells treated with ClC-2 antisense oligonucleotides show a decrease in ClC-2 protein (lane 1, nonsense-treated cells; lane 2, ClC-2 antisense-treated cells; lane 3, ClC-3 antisense-treated cells).
The reduction in whole-cell currents of cells treated with antisense suggested that both ClC-2 and ClC-3 contribute to distinctly different Cl– currents in D54 glioma cells.
Of note, antisense knockdown specifically interrupts the synthesis of new protein but has no affect on existing protein. Hence the effective depletion of functional channels in the membrane depends primarily on the turnover of these proteins. We currently do not know the turnover time for ClC channels. However, for voltage-gated Na+ channels, a half-life of 26 hr in neuroblastoma cells (Waechter et al., 1983) and2dinrat neurons has been reported (Schmidt and Catterall, 1986). Therefore, complete elimination of channel synthesis would only reduce currents by 50% every 24–48 hr, a value that compares favorably with the effect that we observed in our knockdown experiments.
Discussion
In the present study we demonstrate the presence of a subset of ClC genes and their proteins in cultured glioma cells. Specifically, we provide evidence for expression of ClC-2, -3, and -5 protein in glioma cell membranes, often associated with lamellipodia. Importantly, the same complement of channels was observed in acute biopsies from patients who had these tumors removed surgically. We overcame the absence of specific pharmacological drugs for ClC channels subtypes through the use of an antisense knockdown strategy. These studies suggest that Cd2+-sensitive inward Cl– currents can be reduced with ClC-2 antisense, whereas outwardly rectifying, DIDS-sensitive currents are selectively reduced after ClC-3 knockdown.
Cl– channels have been referred to as “the problem children of ion channels” (Clapham, 2001), and the field harbors considerable controversies. This is primarily attributable to the absence of specific pharmacological inhibitors for Cl– channels and great deal of concerns surrounding the specificity of antibodies for their detection of channel proteins. Single-channel properties that might clarify some of theses issues are scarce because of the small conductances of these channels. Also, essentially all of our current knowledge on ClC channels is derived from their recombinant expression and characterization in Xenopus oocytes. To add to the controversy, it has been suggested that overexpression of ClC channels causes upregulation of endogenous Xenopus channels (for review, see Schmieder et al., 2002). In light of these issues, we went to great lengths to establish the specificity of our reagents and approach. For example, the use of multiple commercial and noncommercial antibodies for Western blots and immunocytochemistry yielded convergent results. In addition, Western blotting demonstrates the specificity of our knockdown strategy by only reducing either ClC-2 or ClC-3 protein levels.
Biophysical and pharmacological properties of ClC currents in glioma cells
The signature features reported for recombinant ClC-2 currents are (1) inward rectification, (2) time-dependent activation, (3) potentiation by negative holding potentials, and (4) sensitivity to Cd2+ and Zn2+ (Clark et al., 1998; Enz et al., 1999; Nehrke et al., 2002). Our pharmacological and biophysical characterization of inwardly rectifying Cl– currents in glioma cells is consistent with these reported features for ClC-2. Our knock-down studies selectively reduced these inward currents by 60%, demonstrating that these currents were mediated at least partially by ClC-2. Interestingly, ClC-2 knockdown was associated with a marked increase in the input resistance of the cells. This suggests that ClC-2 may contribute to the high Cl– permeability in these cells. Indeed, we and others have shown that such a high resting Cl– conductance is common for proliferating glial cells that have undergone neoplastic transformation or in response to injury (gliosis) (for review, Walz, 2002). We did not see a comparable change in the resting Cl– conductance after the knockdown of ClC-3.
Currents thought to be mediated by ClC-3 have been described as outwardly rectifying and DIDS and NPPB sensitive, and they often show pronounced voltage-dependent inactivation (Duan et al., 1997; von Weikersthal et al., 1999). Consistent with these features, glioma cells expressed outwardly rectifying DIDS- and NPPB-sensitive currents. After antisense treatment, outward peak currents were reduced by 50%, with the voltage-dependent inactivating current component nearly eliminated. The fact that these currents were selectively reduced after antisense treatment with ClC-3-specific oligonucleotides strongly suggests that they were mediated at least in part by ClC-3. The strategy pursued in our studies was very similar to that used in several recent publications demonstrating the selective loss of outwardly rectifying currents after ClC-3 antisense treatment in HeLa, Xenopus oocytes, and bovine epithelial cells (Wang et al., 2000; Hermoso et al., 2002).
Although we were able to detect ClC-5 at both the mRNA and protein levels, we lacked conclusive biophysical evidence for functional channels in glioma cells. As reported previously, ClC-5 gives rise to outwardly rectifying currents that are unaffected by all known Cl– channel inhibitors (Mo et al., 1999). When we recorded in the simultaneous presence of DIDS or NPPB and TEA, we occasionally recorded a small, time- and voltage-dependent outward current that may be attributed to ClC-5 (data not shown). The low probability of seeing these currents in glioma cells (<10%) made it impossible to study them by antisense knockdown approaches. ClC-5 has been reported to be found in endocytotic vesicles (Gunther et al., 1998), yet when expressed in oocytes, ClC-5 mediated plasma membrane currents (Friedrich et al., 1999). Our studies show some localization on the cell surface of glioma cells by immunocytochemistry and immunogold EM. The functional significance, if any, of such membrane-associated ClC-5 in glioma cells is unclear and warrants further study.
We routinely used ion replacements to verify that the recorded currents were indeed carried by Cl–. We always observed a potentiation of outward currents by Br– and I– as was reported for recombinant ClC-3 (Duan et al., 1997), and currents were always greatly diminished in gluconate or glutamate. A significant, albeit controversial body of literature exists on the relative permeability of ClC channels to other anions (for review, see Fahlke, 2001). However, because ion permeation and gating are believed to be coupled processes in these channels, it is impossible to accurately determine the relative permeability (Pusch et al., 1995). Moreover, we were recording endogenous currents, in which more than one channel population contributed to the overall Cl– current. Hence, we are not comfortable drawing any conclusions from our ion replacement studies; instead, we are using them as a means to isolate true Cl– currents.
Localization of ClC channels
ClC-2 appears to be a ubiquitous Cl– channel that has been identified previously on the plasma membrane of many cell types. In the nervous system, ClC-2 channels are found on the end feet of astrocytes and on the cell body, axons, and dendrites of hippocampal neurons, where they have been implicated in chloride homeostasis and Cl– movements associated with GABAergic synaptic transmission (Sik et al., 2000). Although ClC-3 and ClC-5 have been identified in brain (Steinmeyer et al., 1995; von Weikersthal et al., 1999; Stobrawa et al., 2001), recent papers suggest that both ClC-3 and ClC-5 are almost exclusively channels associated with intracellular vesicles (Gunther et al., 1998; Stobrawa et al., 2001). We therefore felt compelled to study the precise localization of ClC channels in glioma cells in greater detail. Our immunocytochemical and confocal studies localize these channels in lamellipodia, and our immunogold EM studies unequivocally identify clusters of ClC-2, ClC-3, and ClC-5 immunoreactivity in the cell membrane. It is possible that membrane expression of ClC channels is found primarily in dividing, highly motile cells and hence absent from normal brain cells. As discussed further below, Cl– channel function has been implicated in both cell proliferation and cell migration.
Functional implications
Chloride channels have been implicated in a multitude of cellular functions that include osmoregulation, salt secretion, cell migration, and cell proliferation (for review, see Jentsch et al., 2002). Of these functions, the role of Cl– channels in the regulation of cell volume in response to a changing osmotic environment is the most well studied. However, unequivocal molecular identification of the underlying Cl– channels has been a tedious task. A significant number of reports attribute cell volume regulation to a ubiquitous but elusive Cl– channel named IClswell (Nilius et al., 1998). Nevertheless, some studies provide compelling evidence for a partial role of ClC-2 and ClC-3 in volume regulation. For example, knockdown of endogenous ClC-3 in HeLa cells (Hermoso et al., 2002), bovine epithelial cells (Wang et al., 2000), or oocytes impairs regulatory volume decrease. Inhibition of ClC-2 by functional blocking antibodies impairs volume regulation in rat hepatoma cells (Roman et al., 2001), knockdown of ClC-3 by antisense oligonucleotides eliminates swelling-activated currents (von Weikersthal et al., 1999), and mutation of a single amino acid (serine 51) alters volume activation of ClC-3 (Duan et al., 1999). However, transgenic knock-out mice for either ClC-2 or ClC-3 have not been able to detect any defects in cell volume regulation (Stobrawa et al., 2001; Arreola et al., 2002; Nehrke et al., 2002). This apparent discrepancy may be attributable to a compensatory mechanism and must be reconciled by future studies.
For other aspects of biology, Cl– channel function has been less well studied, yet a few studies have implicated Cl– channels in cell shape changes that may occur in conjunction with cell division or cell migration. For example, it has been demonstrated that cell division is associated with a transient increase in cell volume (Premack and Gardner, 1991; Garber and Cahalan, 1997) and can be inhibited by Cl– channel blockers (Voets et al., 1995; Phipps et al., 1996; Schlichter et al., 1996). Increased Cl– channel activity has been shown to coincide with entry into the cell cycle in human cervical cancer cells (Shen et al., 2000), and Cl– channel blockers have been shown to modulate Schwann cell proliferation (Wilson and Chiu, 1993; Pappas and Ritchie, 1998). Similarly, the proliferation of C6 glioma cells, rat aortic smooth muscle cells, and mouse liver cells is inhibited after ClC-3 knockdown (Rouzaire-Dubois et al., 2000; Wondergem et al., 2001; Wang et al., 2002). These studies suggest that the inability to regulate cell volume may be the underlying mechanism that leads to impaired cell proliferation. Indeed, in astrocytes it has been demonstrated that cell swelling activates mitogen-activated kinases that in turn modulate astrocytic Cl– channels (Crépel et al., 1998). The most direct link to date of a molecularly identified Cl– channel and cell proliferation–maturation comes from two recent studies in Caenorhabditis elegans. Here the activity of CLH-3, an ortholog of ClC-2, is required to induce oocyte maturation (Rutledge et al., 2001). The activation of CLH-3 channels occurs by serine–threonine dephosphorylation via a type 1 protein phosphatase (Rutledge et al., 2002), a feature that was also been demonstrated for rat ClC-2. These examples demonstrate intriguing functional interactions of Cl– channels with the cell cycle machinery.
Other evidence suggests that Cl– channels may also serve important functions in the context of cell migration. In rat astrocytes, changes in cell morphology are sufficient to induce Cl– currents (Lascola and Kraig, 1996), with cytoskeletal actin being responsible for gating of Cl– channels (Lascola et al., 1998). Calcium levels, which increase in migratory cells (Pastan et al., 1992; Komuro and Rakic, 1998), have been shown to affect the localization of voltage-gated Cl– channels (Lascola et al., 1998; Huang et al., 2001). Moreover, the translocation of cell processes has been suggested to require local volume increases in those parts of the cell that are in the active process of locomotion (Ehrengruber et al., 1996; Voura et al., 1998). More specifically, it is believed that Cl– and K+ enter at the leading edge of a lamellipodia, leading to local swelling that is obligatory for the translocation of this cellular process (Schneider et al., 2000). A similar requirement for Cl– channel-mediated Cl– efflux has been shown for migrating glioma cells. These cells have a significant resting Cl– conductance that is obligatory for migration (Ransom et al., 2001), because pharmacological inhibition of Cl– channels by either Cd2+ or NPPB impaired glioma migration (Soroceanu et al., 1999; Ransom et al., 2001). Although indirect, these findings are consistent with a contribution of Cd2+-sensitive ClC-2 channels and possibly NPPB-sensitive ClC-3 channels to glioma cell migration. It appears that these Cl– channels allow the secretion of Cl– along with obligated water to accomplish cell shrinkage, which in turn facilitates glioma cell invasion into narrow extracellular spaces. Other immature neuronal or glial precursor cells or stem cells in the mature brain may similarly invoke Cl– channel function during cell migration. Clearly, further studies are necessary to elucidate the role of Cl– channels in cell migration–invasion in general, and the specific contribution of ClC channels in this context warrants further study.
Footnotes
This work was supported by National Institutes of Health Grant RO1-NS36692 and Human Development Grant P30HD-38985. We thank Dr. Thomas Jentsch for providing the ClC-5 channel-specific antibodies. We also thank Jessy Deshane, Patricia Ritch, Tara Spears, and Ed Phillips for technical assistance.
Correspondence should be addressed to Dr. Harald Sontheimer, 1719 Sixth Avenue South, Civitan International Research Center 545, Birmingham, AL 35294. E-mail: sontheimer@uab.edu.
S. Schade's present address: Transmolecular Inc., 3800 Colonnade Parkway, Suite 240, Birmingham, AL 35243.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235572-11$15.00/0
References
- Amberger VR, Avellana-Adalid V, Hensel T, Baron-Van Evercooren A, Schwab ME ( 1997) Oligodendrocyte-type 2 astrocyte progenitors use a metalloendoprotease to spread and migrate on CNS myelin. Eur J Neurosci 9: 151–162. [DOI] [PubMed] [Google Scholar]
- Arreola J, Begenisich T, Nehrke K, Nguyen HV, Park K, Richardson L, Yang B, Schutte BC, Lamb FS, Melvin JE ( 2002) Secretion and cell volume regulation by salivary acinar cells from mice lacking expression of the Clcn3 Cl(–) channel gene. J Physiol (Lond) 545: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhramov A, Fenech C, Bolton TB ( 1995) Chloride current activated by hypotonicity in cultured human astrocytoma cells. Exp Physiol 80: 373–389. [DOI] [PubMed] [Google Scholar]
- Bond TD, Ambikapathy S, Mohammad S, Valverde MA ( 1998) Osmosensitive Cl– currents and their relevance to regulatory volume decrease in human intestinal t84 cells: outwardly vs. inwardly rectifying currents. J Physiol (Lond) 511: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H ( 1998) Electrophysiological properties of human astrocytic tumor cells in situ: enigma of spiking glial cells. J Neurophysiol 79: 2782–2793. [DOI] [PubMed] [Google Scholar]
- Brandt S, Jentsch TJ ( 1995) Clc-6 and Clc-7 are two novel broadly expressed members of the Clc chloride channel family. FEBS Lett 377: 15–20. [DOI] [PubMed] [Google Scholar]
- Clapham D ( 2001) How to lose your hippocampus by working on chloride channels. Neuron 29: 1–3. [DOI] [PubMed] [Google Scholar]
- Clark S, Jordt SE, Jentsch TJ, Mathie A ( 1998) Characterization of the hyperpolarization-activated chloride current in dissociated rat sympathetic neurons. J Physiol (Lond) 506: 665–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coca-Prados M, Sanchez-Torres J, Peterson-Yantorno K, Civan MM ( 1996) Association of Clc-3 channel with Cl– transport by human nonpigmented ciliary epithelial cells. J Membr Biol 150: 197–208. [DOI] [PubMed] [Google Scholar]
- Crépel V, Panenka W, Kelly MEM, MacVicar BA ( 1998) Mitogen-activated protein and tyrosine kinases in the activation of astrocyte volume-activated chloride current. J Neurosci 18: 1196–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Winter C, Cowley S, Hume JR, Horowitz B ( 1997) Molecular identification of a volume-regulated chloride channel. Nature 390: 417–421. [DOI] [PubMed] [Google Scholar]
- Duan D, Cowley S, Horowitz B, Hume JR ( 1999) A serine residue in Clc-3 links phosphorylation-dephosphorylation to chloride channel regulation by cell volume. J Gen Physiol 113: 57–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermont J, Buyse G, Voets T, Tytgat J, De Smedt H, Droogmans G, Nilius B ( 1997) Alternative splicing of ClC-6 (a member of the CIC chloride-channel family) transcripts generates three truncated isoforms one of which, ClC-6c, is kidney-specific. Biochem J 325: 269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrengruber MU, Deranleau DA, Coates TD ( 1996) Shape oscillations of human neutrophil leukocytes: characterization and relationship to cell motility. J Exp Biol 199: 741–747. [DOI] [PubMed] [Google Scholar]
- Enz R, Ross BJ, Cutting GR ( 1999) Expression of the voltage-gated chloride channel ClC-2 in rod bipolar cells of the rat retina. J Neurosci 19: 9841–9847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahlke C ( 2001) Ion permeation and selectivity in ClC-type chloride channels. Am J Physiol Renal Physiol 280: F748–F757. [DOI] [PubMed] [Google Scholar]
- Friedrich T, Breiderhoff T, Jentsch TJ ( 1999) Mutational analysis demonstrates that ClC-4 and ClC-5 directly mediate plasma membrane currents. J Biol Chem 274: 896–902. [DOI] [PubMed] [Google Scholar]
- Garber SS, Cahalan MD ( 1997) Volume-regulated anion channels and the control of a simple cell behavior. Cell Physiol Biochem 7: 229–241. [Google Scholar]
- Gunther W, Luchow A, Cluzeaud F, Vandewalle A, Jentsch TJ ( 1998) Clc-5, the chloride channel mutated in Dent's disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc Natl Acad Sci USA 95: 8075–8080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ ( 1981) Improved patch-clamp techniques for high resolution current recording from cells and cell free membrane patches. Pflügers Arch 391: 85–100. [DOI] [PubMed] [Google Scholar]
- Hermoso M, Satterwhite CM, Andrade YN, Hidalgo J, Wilson SM, Horowitz B, Hume JR ( 2002) ClC-3 is a fundamental molecular component of volume-sensitive outwardly rectifying Cl– channels and volume regulation in HeLa cells and Xenopus laevis oocytes. J Biol Chem 277: 40066–40074. [DOI] [PubMed] [Google Scholar]
- Hille B ( 1992) Ionic channels in excitable membranes. Sunderland, MA: Sinauer.
- Huang P, Liu J, Di A, Robinson NC, Musch MW, Kaetzel MA, Nelson DJ ( 2001) Regulation of human ClC-3 channels by multifunctional Ca2+/calmodulin-dependent protein kinase. J Biol Chem 276: 20093–20100. [DOI] [PubMed] [Google Scholar]
- Jackson PS, Strange K ( 1993) Volume-sensitive anion channels mediate swelling-activated inositol and taurine efflux. Am J Physiol 265: 489–500. [DOI] [PubMed] [Google Scholar]
- Jackson PS, Strange K ( 1995) Characterization of the voltage-dependent properties of a volume-sensitive anion conductance. J Gen Physiol 105: 661–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ, Günther W, Pusch M, Schwappach B ( 1995) Properties of voltage-gated chloride channels of the ClC gene family. J Physiol (Lond) [Suppl] 482: 19S–25S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ, Stein V, Weinreich F, Zdebik AA ( 2002) Molecular structure and physiological function of chloride channels. Physiol Rev 82: 503–568. [DOI] [PubMed] [Google Scholar]
- Kaba SE, Kyritsis AP ( 1997) Recognition and management of gliomas. Drugs 53: 235–244. [DOI] [PubMed] [Google Scholar]
- Kawasaki M, Uchida S, Monkawa T, Miyawaki A, Mikoshiba K, Marumo F, Sasaki S ( 1994) Cloning and expression of a protein kinase C-regulated chloride channel abundantly expressed in rat brain neuronal cells. Neuron 12: 597–604. [DOI] [PubMed] [Google Scholar]
- Komuro H, Rakic P ( 1998) Orchestration of neuronal migration by activity of ion channels, neurotransmitter receptors, and intracellular Ca 2+ fluctuations. J Neurobiol 37: 110–130. [PubMed] [Google Scholar]
- Lascola CD, Kraig RP ( 1996) Whole-cell chloride currents in rat astrocytes accompany changes in cell morphology. J Neurosci 16: 2532–2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lascola CD, Nelson DJ, Kraig RP ( 1998) Cytoskeletal actin gates a Cl – channel in neocortical astrocytes. J Neurosci 18: 1679–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levison SW, Chuang C, Abramson BJ, Goldman JE ( 1993) The migrational patterns and developmental fates of glial precursors in the rat subventricular zone are temporally regulated. Development 119: 611–622. [DOI] [PubMed] [Google Scholar]
- Linskey ME ( 1997) Glial ontogeny and glial neoplasia: the search for closure. J Neurooncol 34: 5–22. [DOI] [PubMed] [Google Scholar]
- Lyons SA, O'Neal J, Sontheimer H ( 2002) Chlorotoxin, a scorpion-derived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia 39: 162–173. [DOI] [PubMed] [Google Scholar]
- Merzak A, Pilkington GJ ( 1997) Molecular and cellular pathology of intrinsic brain tumours. Cancer Metastasis Rev 16: 155–177. [DOI] [PubMed] [Google Scholar]
- Merzak A, McCrea S, Koocheckpour S, Pilkington GJ ( 1994) Control of human glioma cell growth, migration and invasion in vitro by transforming growth factor β1 Br J Cancer 70: 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo L, Hellmich HL, Fong P, Wood T, Embesi J, Wills NK ( 1999) Comparison of amphibian and human ClC-5: similarity of functional properties and inhibition by external pH. J Membr Biol 168: 253–264. [DOI] [PubMed] [Google Scholar]
- Nehrke K, Arreola J, Nguyen HV, Pilato J, Richardson L, Okunade G, Baggs R, Shull GE, Melvin JE ( 2002) Loss of hyperpolarization-activated Cl(–) current in salivary acinar cells from Clcn2 knockout mice. J Biol Chem 277: 23604–23611. [DOI] [PubMed] [Google Scholar]
- Nilius B, Prenen J, Voets T, Eggermont J, Droogmans G ( 1998) Activation of volume-regulated chloride currents by reduction of intracellular ionic strength in bovine endothelial cells. J Physiol (Lond) 506: 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble M, Mayer-Pröschel M ( 1997) Growth factors, glia and gliomas. J Neurooncol 35: 193–209. [DOI] [PubMed] [Google Scholar]
- Pappas CA, Ritchie JM ( 1998) Effect of specific ion channel blockers on cultured Schwann cell proliferation. Glia 22: 113–120. [PubMed] [Google Scholar]
- Pastan I, Chaudhary V, Fitzgerald DJ ( 1992) Recombinant toxins as novel therapeutic agents. Annu Rev Biochem 61: 331–354. [DOI] [PubMed] [Google Scholar]
- Phipps DJ, Branch DR, Schlichter LC ( 1996) Chloride-channel block inhibits T lymphocyte activation and signaling. Cell Signal 8: 141–149. [DOI] [PubMed] [Google Scholar]
- Premack BA, Gardner P ( 1991) Role of ion channels in lymphocytes. J Clin Immunol 11: 225–238. [DOI] [PubMed] [Google Scholar]
- Pusch M, Ludewig U, Rehfeldt A, Jentsch TJ ( 1995) Gating of the voltage-dependent chloride channel CIC-O by the permeant anion. Nature 373: 527–531. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Sontheimer H ( 2001) BK channels in human glioma cells. J Neurophysiol 85: 790–803. [DOI] [PubMed] [Google Scholar]
- Ransom CB, O'Neal JT, Sontheimer H ( 2001) Volume-activated chloride currents contribute to the resting conductance and invasive migration of human glioma cells. J Neurosci 21: 7674–7683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman RM, Smith RL, Feranchak AP, Clayton GH, Doctor RB, Fitz JG ( 2001) ClC-2 chloride channels contribute to HTC cell volume homeostasis. Am J Physiol Gastrointest Liver Physiol 280: G344–G353. [DOI] [PubMed] [Google Scholar]
- Rouzaire-Dubois B, Bostel S, Dubois JM ( 1999) Evidence for several mechanisms of volume regulation in neuroblastoma x glioma hybrid NG108–15 cells. Neuroscience 88: 307–317. [DOI] [PubMed] [Google Scholar]
- Rouzaire-Dubois B, Milandri JB, Bostel S, Dubois JM ( 2000) Control of cell proliferation by cell volume alterations in rat C6 glioma cells. Pflügers Arch 440: 881–888. [DOI] [PubMed] [Google Scholar]
- Rutledge E, Bianchi L, Christensen M, Boehmer C, Morrison R, Broslat A, Beld AM, George AL, Greenstein D, Strange K ( 2001) CLH-3, a ClC-2 anion channel ortholog activated during meiotic maturation in C. elegans oocytes. Curr Biol 11: 161–170. [DOI] [PubMed] [Google Scholar]
- Rutledge E, Denton J, Strange K ( 2002) Cell cycle- and swelling-induced activation of a Caenorhabditis elegans ClC channel is mediated by CeGLC-7alpha/beta phosphatases. J Cell Biol 158: 435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlichter LC, Sakellaropoulos G, Ballyk B, Pennefather PS, Phipps DJ ( 1996) Properties of K + and Cl – channels and their involvement in proliferation of rat microglial cells. Glia 17: 225–236. [DOI] [PubMed] [Google Scholar]
- Schmidt JW, Catterall WH ( 1986) Biosynthesis and processing of the a-subunit of the voltage-sensitive sodium channel in rat brain neurons. Cell 46: 437–445. [DOI] [PubMed] [Google Scholar]
- Schmieder S, Lindenthal S, Ehrenfeld J ( 2002) Cloning and characterisation of amphibian ClC-3 and ClC-5 chloride channels. Biochim Biophys Acta 1566: 55–66. [DOI] [PubMed] [Google Scholar]
- Schneider SW, Pagel P, Rotsch C, Danker T, Oberleithner H, Radmacher M, Schwab A ( 2000) Volume dynamics in migrating epithelial cells measured with atomic force microscopy. Pflügers Arch 439: 297–303. [DOI] [PubMed] [Google Scholar]
- Shen MR, Droogmans G, Eggermont J, Voets T, Ellory JC, Nilius B ( 2000) Differential expression of volume-regulated anion channels during cell cycle progression of human cervical cancer cells. J Physiol (Lond) 529: 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Li X, Xu G, Nowak DE, Showalter LA, Weinman SA ( 2000) Expression and canalicular localization of two isoforms of the Clc-3 chloride channel from rat hepatocytes. Am J Physiol Gastrointest Liver Physiol 279: G268–276. [DOI] [PubMed] [Google Scholar]
- Sik A, Smith RL, Freund TF ( 2000) Distribution of chloride channel-2-immunoreactive neuronal and astrocytic processes in the hippocampus. Neuroscience 101: 51–65. [DOI] [PubMed] [Google Scholar]
- Simpson PB, Armstrong RC ( 1999) Intracellular signals and cytoskeletal elements involved in oligodendrocyte progenitor migration. Glia 26: 22–35. [PubMed] [Google Scholar]
- Soroceanu L, Manning TJ Jr, Sontheimer H ( 1999) Modulation of glioma cell migration and invasion using Cl – and K + ion channel blockers. J Neurosci 19: 5942–5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmeyer K, Schwappach B, Bens M, Vandewalle A, Jentsch TJ ( 1995) Cloning and functional expression of rat Clc-5, a chloride channel related to kidney disease. J Biol Chem 270: 31172–31177. [DOI] [PubMed] [Google Scholar]
- Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, Bosl MR, Ruether K, Jahn H, Draguhn A, Jahn R, Jentsch TJ ( 2001) Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 29: 185–196. [DOI] [PubMed] [Google Scholar]
- Ullrich N, Sontheimer H ( 1996) Biophysical and pharmacological characterization of chloride currents in human astrocytoma cells. Am J Physiol 270: C1511–C1521. [DOI] [PubMed] [Google Scholar]
- Ullrich N, Bordey A, Gillespie GY, Sontheimer H ( 1998) Expression of voltage-activated chloride currents in acute slices of human gliomas. Neuroscience 83: 1161–1173. [DOI] [PubMed] [Google Scholar]
- Voets T, Szucs G, Droogmans G, Nilius B ( 1995) Blockers of volume-activated Cl– currents inhibit endothelial cell proliferation. Pflügers Arch 431: 132–134. [DOI] [PubMed] [Google Scholar]
- von Weikersthal SF, Barrand MA, Hladky SB ( 1999) Functional and molecular characterization of a volume-sensitive chloride current in rat brain endothelial cells. J Physiol (Lond) 516: 75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voura EB, Sandig M, Kalnins VI, Siu C ( 1998) Cell shape changes and cytoskeleton reorganization during transendothelial migration of human melanoma cells. Cell Tissue Res 293: 375–387. [DOI] [PubMed] [Google Scholar]
- Waechter CJ, Schmidt JW, Catterall WA ( 1983) Glycosylation is required for maintenance of functional sodium channels in neuroblastoma cells. J Biol Chem 258: 5117–5123. [PubMed] [Google Scholar]
- Walz W ( 2002) Chloride/anion channels in glial cell membranes. Glia 40: 1–10. [DOI] [PubMed] [Google Scholar]
- Wang GL, Wang XR, Lin MJ, He H, Lan XJ, Guan YY ( 2002) Deficiency in ClC-3 chloride channels prevents rat aortic smooth muscle cell proliferation. Circ Res 91: E28–E32. [DOI] [PubMed] [Google Scholar]
- Wang L, Chen L, Jacob TJ ( 2000) The role of Clc-3 in volume-activated chloride currents and volume regulation in bovine epithelial cells demonstrated by antisense inhibition. J Physiol (Lond) 524: 63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GF, Chiu SY ( 1993) Mitogenic factors regulate ion channels in Schwann cells cultured from newborn rat sciatic nerve. J Physiol (Lond) 470: 501–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wondergem R, Gong W, Monen SH, Dooley SN, Gonce JL, Conner TD, Houser M, Ecay TW, Ferslew KE ( 2001) Blocking swelling-activated chloride current inhibits mouse liver cell proliferation. J Physiol (Lond) 532: 3–72. [DOI] [PMC free article] [PubMed] [Google Scholar]