

Abstract
The 75 kDa neurotrophin receptor (p75NTR) and two neurotrophin receptor homologs (NRH1, NRH2) constitute a subfamily of the nerve growth factor/tumor necrosis factor receptor superfamily. NRH1 coexists with p75NTR in fish, amphibians, and birds but is absent in mammals, whereas NRH2 exists only in mammals. Unlike p75NTR and NRH1, NRH2 lacks a canonical extracellular ligand binding domain. The similarity of NRH2 to the product of metalloproteinase cleavage of p75NTR prompted us to examine the cleavage of p75NTR in greater detail. p75NTR, NRH1, and NRH2 undergo multiple proteolytic cleavages that ultimately release cytoplasmic fragments. For p75NTR, cleavage in the extracellular domain by a PMA-inducible membrane metalloproteinase is followed by cleavage within or near the transmembrane domain, releasing the intracellular domain into the cytoplasm. This processing resembles the α- and γ-secretase-mediated processing of β-amyloid precursor protein and the similar processing of Notch. Although neurotrophins did not regulate p75NTR processing, theα- andγ-secretase-mediated cleavage of p75 is modulated by receptor tyrosine kinases (Trks) TrkA and TrkB but not TrkC. Surprisingly, although NRH1 and NRH2 also undergo proteolytic cytoplasmic release of intracellular domains, a different protease mediates the cleavage. Furthermore, whereas the p75NTR soluble intracellular domain accumulates only in the presence of proteasome inhibitors, the equivalent fragment of NRH2 is stable and localizes in the nucleus. Because soluble intracellular domains of p75NTR and NRH2 were found to activate NF-κB in concert with TNF receptor associated factor 6 (TRAF6), we propose that cleavage of these proteins may serve conserved cytoplasmic and nuclear signaling functions through distinct proteases.
Keywords: proteolysis, p75NTR, neurotrophin, Trk, regulated intramembrane proteolysis, RIP, NF-κB
Introduction
The 75 kDa neurotrophin receptor (p75NTR) (Johnson et al., 1986; Radeke et al., 1987) is a member of the NGF/tumor necrosis factor (TNF) receptor superfamily, which is defined by an extracellular domain (ECD) containing a repeated cysteine-rich motif. Apart from a death domain motif present in several family members including p75NTR (Feinstein et al., 1995; Liepinsh et al., 1997), there is little homology of intracellular domains (ICDs) among these receptors. However, two novel genes encode proteins with substantial sequence homology to p75NTR in cytoplasmic and transmembrane domains, defining a gene subfamily. We refer to these as neurotrophin receptor homolog 1 (NRH1) and NRH2 (Hutson and Bothwell, 2001). Xenopus NRH1 has also been designated “fullback” (GenBank accession AF131890), and rat NRH2 has been called “PLAIDD,” for “p75-like apoptosis-inducing death domain protein” (Frankowski et al., 2002).
p75NTR is activated by all four neurotrophins. p75NTR is frequently coexpressed with receptor tyroisne kinases (Trks) TrkA, TrkB, or TrkC neurotrophin receptors, with which it associates (Wolf et al., 1995; Bibel et al., 1999), and this association alters the signaling properties of both partners by poorly understood mechanisms (Roux and Barker, 2002). p75NTR also associates with NgR, a receptor for Nogo, myelin associated glycoprotein (MAG), and oligodendrocyte myelin glycoprotein (OMgp), and mediates the signaling responses to these proteins (Wang et al., 2002; Wong et al., 2002). Despite these multiple functions, p75NTR signaling mechanisms are poorly understood.
Although proteolytic shedding of the p75NTR ECD by Schwann cells was reported more than a decade ago (DiStefano and Johnson, 1988), the function of shedding remains obscure. DiStefano et al. (1993) did not examine the fate of the predicted membrane-resident C-terminal fragment (m-CTF). However, the possibility that this fragment may be functionally significant is suggested by recent findings that a p75NTR splice variant lacking a neurotrophin-binding domain is functionally important (von Schack et al., 2001) and by the recognition that NRH2 structurally resembles the p75NTR m-CTF.
The unidentified protease mediating p75NTR ECD shedding is typical of a class of membrane metalloproteinases, known as “sheddases,” with diverse and important functions (Hooper et al., 1997; Peschon et al., 1998). Recently, a novel mode of receptor processing has been recognized-regulated intramembrane proteolysis (RIP) (Brown et al., 2000). Exemplified by Notch, RIP involves receptor cleavage near the membrane junction of the ECD, typically by a membrane metalloproteinase of the A disintegrin and metalloproteinase (ADAM) family. This cleavage triggers a second cleavage within the transmembrane domain, typically by a presenilin-containing protease complex, releasing the intracellular domain (ICD), which often traffics to the nucleus as a transcriptional regulator. By analogy to β-amyloid precursor protein, another example of RIP, the initial and secondary cleaving activities will be referred to here as α- and γ-secretase.
These considerations led us to investigate whether p75NTR is subject to RIP and whether NRH1 and NRH2 are processed similarly. We report here that p75NTR, NRH1, and NRH2 undergo proteolytic processing, generating soluble ICD fragments. Processing of p75NTR requires sequential action of α- and γ-secretase, whereas similar processing of NRH1 and NRH2 does not use γ-secretase and does not require previous cleavage by α-secretase. We provide evidence that soluble ICDs of these receptors may have cytoplasmic and nuclear signaling functions.
Materials and Methods
Vector construction. PIND-hp75 was constructed by digesting a 1.7 kb insert of pBAP-hp75 with SalI and BamHI, blunting the SalI site by Klenow, and ligating into the vector pIND (Invitrogen, Carlsbad, CA) via BamHI and a HindIII site that had been blunted by Klenow fill in. Myc-tagged expression constructs were generated by subcloning PCR amplified Xenopus NRH1 and mouse NRH2 into the vector pCS2+MT (Roth et al., 1991) via the ClaI site by adding ClaI linkers using the primers 5′-ATC CCA TCG ATA TGG AAA TGA GGG GCC CAC GTT TAA CC and 5′-TTT AAA TCG ATA CAC CAC AGA GCT GGC ATC ATT TGC GCT for xNRH1 and 5′-ATC CCA TCG ATA TGC TTT ATA ACG TCA GCA AAG GT and 5′-TTT AAA TCG ATA CAC CAC CGA GGA GCT CTC AGC TG for mNRH2, creating a C-terminal fusion to six tandem repeats of the myc epitope. C-terminal green fluorescent protein (GFP) fusions to NRH1 and NRH2 were made by subcloning via the ClaI sites of the MT fusions, with mNRH2 subcloned into the ClaI site of pCS2-GFP CE, and xNRH1 subcloned into the ClaI site of pCS2-GFP-XLT (gifts of R. T. Moon, University of Washington). C-terminal GFP fusion to human p75 NTR was created by adding a 3′ SpeI linker to human p75 using the primer 5′-AAA ACT AGT CAC CGG GGA TGT GGC AGT GGA and T7 for a 5′ primer with pCDNA3-hp75 as a template, followed by digestion of the PCR product with EcoR1 and SpeI and ligating into the EcoR1–XbaI sites of pCS2-GFP XLT. PCMV-FLAG-rp75 ICD was created by digesting the p75 ICD from pGEX-rp75ICD via EcoR1 (5′) and SalI (3′) sites and subcloning into the vector pFLAG-CMV-6c (Sigma) via EcoR1 and SalI sites, creating a FLAG fusion on the N terminus of the intracellular domain. PCMV-FLAG-mNRH2 ICD was generated by subcloning the NRH2 ICD from a GST fusion in pGEX4T1 via the EcoR1 and SalI sites of pCMV-FLAG-6c (Sigma), creating a FLAG fusion on the N terminus of the intracellular domain. The original fusion in pGEX4T1 was made by PCR using the BamH1 linker 5′-ATA GGA TCC AAA TGC TGG CGC TCA CAT and SalI linker 5′-ATA GTC GAC CGG CAC TCA CAC CAC CGA. pCDNA-mNRH2L was constructed by subcloning a 1.2 kb insert of mNRH2 from pBS-mNRH2 via KpnI and NotI and ligating into pCDNA3. PCDNA3-rNRH2 sec was created by subcloning the rNRH2 sec cDNA from clone jlrIc9b4 in pMet7 into pCDNA3.1 via BamH1 and NotI sites. PRK-TNF receptor associated factor 6 (TRAF6) was a gift of David Goeddel (Tularik, Inc., San Francisco, CA); PCDNA3-rat p75 NTR, pGEX-rp75 ICD, pCMV5-rTrkA and pCMV5-rTrkB were gifts of Moses Chao (Skirboll Institute, New York University). PMEX–rTrkC was a gift of Pantelis Tsoulfas (University of Miami, Miami, FL). Xenopus NRH1 used for subcloning was clone 26E7.1, obtained from C. Niehrs (Max Plank Institute, Heidelberg, Germany). Full-length mouse NRH2L cDNA (clone jtmxa18f2) and rat NRH2S (clone jlrIc9b4) were a gift of Millenium Pharmaceuticals (Cambridge, MA).
Generation of Ponasterone A-inducible stable cell lines. Human p75NTR cloned into the pIND vector (Invitrogen) was transfected into EcR293 cells [human embryonic kidney (HEK) 293 cells stably transfected with the ecdysone receptor (Invitrogen)] and put under selection using both 500 μg/ml Zeocin and 500 μg/ml geneticin (G418). Individual clones were isolated and expanded and then assayed for low basal expression and high inducible expression of p75NTR using Ponasterone A (Invitrogen). All experiments reported here were done using p75 clone 5. pIND stable cell line culture media was supplemented with 200 μg/ml geneticin, and expression was routinely induced using 5 μm Ponasterone A.
Cell culture, treatments, and transfection. COS, HEK293, RN22F, and pIND-p75 cells were routinely cultured in pH 7.2 DMEM (Invitrogen) containing 10% fetal bovine serum (Hyclone, Logan, UT) and 1% penicillin/streptomycin (Invitrogen) at 37°C. CHO-DG44 cells expressing human BDNF were cultured in DMEM/F12 supplemented with 10 μg/ml transferrin, 10 μg/ml insulin, and 10 nm selenium. Culture supernatant was fractioned on Hi-Trap SP and Sephacryl S200 (Amersham Biosciences, Piscataway, NJ), yielding homogeneously pure proBDNF. For inducible p75NTR expression, pIND cells at 50–60% confluency were induced with 5 μm Ponasterone A for 24 hr before treatments. In cleavage experiments, cells were pretreated for 90 min with 10 μm GM6001 (Calbiochem, San Diego, CA), 25 μm TAPI (Peptides International), 2–10 μm DAPT (gift of M. Wolfe, Harvard Medical School, Boston, MA), 1 μm Epoxomycin (Calbiochem), 5 μm β-clasto-lactacystin (Calbiochem), 1 μm calpastatin (Calbiochem), 26 μm SB203580 (Calbiochem), 100 μm PD98059 (Calbiochem), 100 nm K252a (Calbiochem), or DMSO vehicle control. Cells were stimulated for 40 min with 100 ng/ml PMA (Sigma, St. Louis, MO), 100 ng/ml human NGF (gift of Genentech, San Francisco, CA), human BDNF and NT-3 (gifts of Regeneron), human proNGF (10 ng/ml) (gift of B. Hempstead, Cornell University, New York, NY), or human proBDNF (60 ng/ml) (purified in our laboratory). Cells were then washed one time with cold PBS on ice, lysed in 20 mm Tris 7.6, 1 mm EDTA, 0.5 mm EGTA, 1% Triton X-100, 250 mm sucrose, and 1× protease inhibitor mixture (Sigma), and centrifuged 10 min at 16,000 × g; supernatants were quantified by Bradford assay for use in SDS-PAGE. For transient transfections, HEK293 cells were plated at 70–80% confluency on 35 mm plates and transfected with Lipofectamine 2000 (Invitrogen) using 4 μg of total DNA per well. In cotransfections of p75 and Trk, each 35 mm plate received 2 μg of pCDNA3-hp75 and 2 μg of the respective Trk expression vector. For inhibiting N-linked glycosylation, cells were cultured 24 hr in the presence of 10 μg/ml tunicamycin (Sigma) and then lysed as described. Primary cultures of Schwann cells were done using Schwann cells derived from a mixed primary culture of postnatal day (P) 1 rat trigeminal ganglia, grown on glass coverslips coated with poly-d-lysine and rat laminin (Sigma), cultured in Ham's F12 Media (Invitrogen) containing 10% heat-inactivated fetal bovine serum (Hyclone), 1% penicillin/streptomycin, supplemented with 50 ng/ml NGF. Schwann cells were transfected in 12-well plates using Lipofectamine 2000 and 1.5 μg of DNA per well.
Antibodies. Rabbit polyclonal antisera 9992 against the intracellular domain of p75NTR was a gift of Moses Chao. For generation of an antibody against the NRH2 intracellular domain, Alpha Diagnostic International (San Antonio, TX) synthesized the peptide CQAEAVETMAC-DQMPAYTLLRNW, which was coupled to KLH and used to immunize rabbits. On the basis of high ELISA score, serum from rabbit 5592 was selected for affinity purification against the immunizing peptide covalently coupled to agarose. The monoclonal anti-myc antibody 9E10 was purified from hybridoma culture supernatant using protein G-Sepharose. Trk expression was monitored with the anti-pan Trk antibody sc-139 (Santa Cruz Biotechnology, Santa Cruz, CA.)
Western blotting. Cell lysate (3–20 μg) was separated by SDS PAGE using 4–20% acrylamide gradient gels (Bio-Rad, Hercules, CA). For detection of p75 with 9992 antisera, protein was transferred electrophoretically using the semidry transfer method to polyvinylidene difluoride membrane, Immobilon P (Millipore, Bedford, MA). Membranes were then blocked in 1% nonfat dry milk in TBST (20 mm Tris, 137 mm NaCl, pH 7.4, 0.05% Tween 20). Incubation in a 1:5000 dilution of 9992 antisera specific for the intracellular domain p75 (compliments of Moses Chao) in blocking buffer was performed overnight at 4°C. Membranes were washed once in high-salt TBS (500 mm NaCl) with 0.05% Tween 20 followed by multiple washes in TBST. Secondary goat anti-rabbit HRP-conjugated antibody (Jackson Laboratories) was diluted 1:20,000 in blocking buffer and incubated with the membranes for 1 hr at room temperature. Membranes were then washed as before and developed using ECL (Amersham Biosciences). For NRH1 and NRH2, immunoblotting followed the same procedure except that proteins were transferred to Protran nitrocellulose (Schleicher and Schuell, Keene, NH), and myc-tagged proteins were detected using 0.5 μg/ml mouse monoclonal anti-myc antibody. Untagged NRH2 was detected using 1 μg/ml rabbit polyclonal antisera 5592.
Cell fractionation. After the indicated treatments, cells were lysed in buffer containing (in mm): 10 HEPES, 10 NaCl, 1 KH2PO4, 5 NaHCO3, 5 EDTA, and 1× protease inhibitor mixture, and homogenized by passage through a 22 ga needle. Nuclei were spun down at 325 × g for 10 min, and the resulting supernatant was then microfuged at 16,000 × g for 30 min at 4°C. The supernatant was removed (cytosol), and buffer containing 1% Triton X-100 was added to the pellet (membrane). Nuclear proteins were extracted in high salt buffer with (in mm): 10 HEPES, 0.5 MgCl2, 420 NaCl, 0.2 EDTA, 25% glycerol, and protease inhibitors.
Affinity precipitation of biotinylated cell-surface proteins. Plates (10 cm) of COS cells were transfected by calcium phosphate with mNRH2-MT for 24 hr, washed three times in ice-cold PBS, pH 8.0, placed on ice, and covered with either 2 ml cold PBS containing 1 mg/ml freshly prepared EZ-Link Sulfo-NHS-LC-Biotin (Pierce, Rockford, IL) or PBS alone. Cells were incubated 30 min on ice and then washed one time with 100 mm glycine, followed by three washes of cold PBS. Cells were then lysed in 1 ml lysis buffer [1% NP-40, 0.5% deoxycholate, 50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1× protease inhibitor mixture (Sigma)]. For affinity precipitation, 500 μl of the lysate was incubated with 30 μl ultralink-neutravidin beads (Pierce) for 1 hr at 4°C, followed by four washes in cold lysis buffer. Pelleted beads were then resuspended in 30 μl SDS loading buffer for analysis by PAGE. Affinity-precipitated proteins were detected by Western blot using the 9E10 monoclonal anti-myc antibody, stripped, and reprobed with goat anti-pyruvate kinase (Rockland, Gilbertsville, PA).
Luciferase assays. For luciferase assays, HEK293 cells in 24-well plates were transfected with Lipofectamine 2000 in complete media with 1050 ng total DNA per well. Each well contained 200 ng of pNfKB-luciferase reporter (Stratagene, La Jolla, CA), 50 ng EF1-LACZ for normalization, 750 ng of pCDNA3 empty vector, full-length rp75NTR, mNRH2, FLAG-rp75 ICD, or FLAG-mNRH2 ICD, and either 50 ng of TRAF6 or pCDNA3 empty vector. Cells were harvested for luciferase assay 24 hr after transfection. Data represent the averages of triplicate wells per condition, normalized for transfection efficiency. Statistical significance was determined using a two-tailed Student's t test. The data are means ± SD of triplicate assays normalized to EF1α promoter-driven LacZ expression.
Immunohistochemistry. Bisected embryonic day (E) 20 rat embryos were immersion fixed in Methacarnoy's fixative (10% chloroform, 60% methanol, 30% acetic acid), embedded in paraffin, and sectioned at 10 μm. Slides were deparaffinized in three changes of Xylene; endogenous peroxidase was blocked with 0.75% H2O2 in MeOH; slides were hydrated to DH2O and 0.1 m PBS, pH 7.4. Slides were incubated briefly in 5% bovine serum albumin in PBS before overnight incubation at 4°C in primary antibody (rabbit 5592 anti-NRH2 or rabbit 9992 anti-p75NTR) diluted to 2 μg/ml. Slides were rinsed thoroughly in PBS after primary antibody incubation. Slides were then incubated with biotinylated goat anti-rabbit (Vector Laboratories, Burlingame, CA) at 1:500 for one hr, rinsed in PBS, incubated with streptavidin-HRP (Zymed Laboratories, San Francisco, CA) at 1:800 for 1 hr, rinsed in PBS, followed by rinsing in 0.05 m Tris buffer, pH 7.6. Finally, slides were incubated in a diaminobenzidine (DAB) solution (0.074% 3,3′DAB in 0.05 m Tris buffer with 0.03% H2O2) for 10–15 min at room temperature. Slides were thoroughly rinsed, lightly stained with methyl green, dehydrated, cleared, and coverslipped with Permount. Bright-field images were collected on a Leitz ortholux 2 microscope using a Kodak DC290 digital camera.
Immunocytochemistry. Cultured cells were fixed in 4% PFA for 30 min at room temperature, rinsed in PBS/10 mm glycine/0.1% Triton X-100 for 20 min, and permeabilized with three additional 10 min washes in PBS/0.25% Triton X-100 (PBST). Blocking was done in 10% donkey serum for 1 hr at room temperature, followed by a 3 hr incubation with primary antibody (2 μg/ml 5592 anti-NRH2) diluted in blocking serum. Cells were rinsed three times for 10 min in PBST, and then incubated with 1:500 dilution of Cy3-conjugated donkey anti-rabbit antisera (Jackson ImmunoResearch, West Grove, PA) for 45 min at room temperature. Cells were incubated 15 min in a PBS wash containing 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI), washed three times for 10 min in PBS, and coverslipped with Vectashield (Vector Laboratories). Cells transfected with GFP-tagged constructs were washed one time in cold PBS and fixed in 4% PFA at room temperature for 20 min, washed in PBST, nuclei stained with 1 μg/ml DAPI, washed three times for 10 min in PBST, and coverslipped using Vectashield. Confocal images were collected on a Leica TCS SP/MP confocal multiphoton system using a PL APO 100.0× oil immersion objective.
Results
Structural relationships between p75NTR and homologs
Expressed sequence tag (EST) and genomic databases from various species contain sequences representing two homologs of p75NTR, which we designate NRH1 and NRH2. Alignment of the sequences of these homologs reveals extensive similarity in the transmembrane and cytoplasmic domains (Fig. 1A). For comparison, whereas NRH subfamily members share roughly 30% sequence identity in their intracellular domains across species, the human p75NTR ICD is only 13% identical to that of human TNFR1. NRH1 and NRH2, like p75NTR, possess death domains and a C-terminal motif (SSXV) that is predicted to bind type I PDZ domains. This suggests that signaling mechanisms used by the three proteins may be similar. However, NRH2 lacks the cysteine-rich repeat domain that constitutes the ligand-binding domain of p75NTR. This does not simply reflect failure to identify full-length cDNA clones of NRH2 transcripts, because no cysteine repeat coding sequences are present in the short interval between the 5′ end of the human genomic sequences for NRH2 at chromosomal position 3p21.32a and an adjacent gene, KIF9, nor is a cysteine-repeat coding sequence present in the mouse gene (on chromosome 9, position 15954.94).
Figure 1.

Relationship among p75NTR homologs. A, Sequence alignment reveals extensive sequence identity among mouse p75NTR, Xenopus NRH1, and mouse NRH2 in transmembrane and cytoplasmic domains. p75NTR and NRH1 possess similar cysteine-rich repeat motifs in the extracellular domain, whereas NRH2 lacks this domain. Blue box, Transmembrane domain; green box, p75NTR PEST domain; red box, death domain; black box, PDZ binding motif. B, BLAST scanning of EST and genomic DNA libraries reveals the presence of p75NTR and homologs in various vertebrate orders. p75NTR is present in all vertebrate orders for which data are available, whereas NRH1 is not found in mammals, and NRH2 is found only in mammals. C, Dendrogram modeling phylogenic relatedness of p75NTR and homologs. Sequence alignments generated by the CLUSTAL algorithm were used to generate dendrograms using VectorNTI software. D, Diagram depicting the structural similarity of p75NTR, metalloprotease-cleaved p75NTR, and the NRH subfamily proteins. NRH2 exists in long (L) and short (S) splice variants that resemble predicted C-terminal fragments of α-secretase-like and γ-secretase-like cleavage of p75NTR, respectively.
NRH1 homologs exist in amphibians (Xenopus laevis and Silurana tropicalis), fish (zebrafish and pufferfish), and birds (chickens) but none is present in mammalian EST databases, and none can be detected in the human or mouse genomic databases. In contrast, NRH2 homologs are present among ESTs from diverse mammals (human, mouse, rat, bovine, porcine), but none are present in ESTs or genomic databases of non-mammalian vertebrates. p75NTR homologs can be identified in all vertebrate classes, including fish, amphibians, birds, and mammals. Consequently, p75NTR coexists with NRH1 in fish, amphibians, and birds but coexists with NRH2 in mammals (Fig. 1B). The simplest (but not the only) scenario for the evolution of this gene family is that a gene duplication gave rise to NRH1 and p75NTR early in vertebrate evolution and that NRH2 arose by a deletion mutation of NRH1 around the time of divergence of avian and mammalian lineages. A dendrogram modeling phylogenic relationships of p75NTR and homologs is consistent with this interpretation (Fig. 1C). EST sequences suggest the existence of multiple splice variants of NRH2, with variant sequences at the N terminus. These putative splice variants can be grouped into long splice variants (NRH2L) that are predicted to span the membrane, and a short splice variant (NRH2S), lacking a transmembrane domain, that is predicted to exist as a soluble cytoplasmic protein [and these predictions have been confirmed recently (Frankowski et al., 2002)]. NRH2L and NRH2S are comparable with the predicted C-terminal products of cleavage of p75NTR by α-secretase-like and γ-secretase-like activities (Fig. 1D).
α-secretase and γ-secretase cleavage of p75NTR
To examine possible cleavage fragments of p75NTR we used an antibody against the cytoplasmic domain of p75NTR for Western blot analysis of a stable HEK293 cell line engineered to inducibly express hp75NTR in response to Ponasterone A (pIND p75 cells). Several immunoreactive protein species were detected, and all were derived from p75NTR as indicated by their induction by Ponasterone A (Fig. 2A). In addition to the intact 70–80 kDa receptor (present as a broad band because of heterogenous glycosylation), a major ∼50 kDa component was observed that probably represents immature nascent protein that is not fully glycosylated (Grob et al., 1985). Because a zinc-dependent metalloprotease is implicated in the ectodomain shedding of the p75NTR (DiStefano et al., 1993) and metalloproteases implicated in α-secretase receptor cleavage are often activated by a protein kinase C (PKC)-dependent mechanism (Peschon et al., 1998), we assessed the effect of the PKC activator PMA on p75NTR cleavage. Exposure of cultures to PMA caused the appearance of a faint ∼30 kDa product, only visible with long blot exposures (data not shown). The size of this fragment is consistent with the size expected for an α-secretase-mediated ectodomain cleavage event. (Although the predicted polypeptide molecular weight is ∼24,000, the presence of an additional increment from O-linked carbohydrate is likely.)
Figure 2.

PMA induces proteolytic processing of p75NTR. A, Using pIND-p75 cells, p75NTR expression was induced with 5 μm Ponasterone A (Pon) for 24 hr before addition of inhibitors. Inhibitors of different proteolytic enzymes, epoxomycin (Epo, 1 μm), β-clasto-lactacystin (Lac, 5 μm), calpastatin (Cal, 1 μm), and DAPT (2 μm) were added for 90 min before PMA addition (100 ng/ml, 40 min). Western blots probed with an antibody specific to the cytoplasmic domain of p75NTR revealed a PMA-stimulated proteolysis that results in a small fragment (<) produced by a DAPT-sensitive, γ-secretase-mediated cleavage. This PMA-stimulated cleavage product is derived from an intermittent α-secretase product (*) that visibly accumulates in the presence of the γ-secretase inhibitor DAPT. B, RN22F rat Schwannoma cells, which endogenously express p75NTR, were treated with inhibitors and stimulated with PMA as in A with similar results. C, Primary mouse embryonic fibroblast cells were transfected with p75NTR and treated with Epo and PMA. Without treatment, these cells have a constitutive α-secretase cleavage, and no observable γ-secretase cleavage of p75NTR. Stimulation with PMA fails to trigger additional processing. D, pIND-p75 cells were pretreated with Epo (all lanes) and the p38 MAP kinase inhibitor SB203580 (26 μm), the MEK inhibitor PD98059 (100 μm), the metalloproteinase inhibitor TAPI (25μm), or a DMSO control, and then stimulated with PMA (all lanes). PMA-induced cleavage of p75NTR was inhibited in the presence of either TAPI or PD98059, but not SB203580.
For several instances of RIP, including Notch, the products of the initial and secondary cleavage events do not accumulate in significant quantities, because the product of α-secretase cleavage is rapidly processed by γ-secretase cleavage, and the latter product is quickly subjected to proteasomal degradation (Oberg et al., 2001). The cytoplasmic domain of p75NTR contains a PEST motif, characteristic of proteins that are subject to proteasomal degradation (Fig. 1A). Therefore, we examined whether exposure to proteasome inhibitors might stabilize cleavage products of the receptor. In the absence of PMA, epoxomycin did not produce significant accumulation of any p75NTR cleavage products in pIND-p75 cells (Fig. 2A). However, when treated with PMA, both β-clasto-lactacystin and epoxomycin caused accumulation of a ∼25 kDa fragment that is consistent with the size predicted for a γ-secretase cleavage event (Fig. 2A). Although proteasome inhibitors dramatically increase the amount of the ∼25 kDa fragment detected, small amounts were observed in the absence of proteasome inhibitors when blots were heavily overexposed (data not shown). To assess whether the low abundance of the 25 kDa fragment results from the rapid processing of this fragment by γ-secretase, we exposed cells to a potent and specific γ-secretase inhibitor, DAPT, which eliminated the 25 kDa fragment and caused accumulation of the 30 kDa fragment (Fig. 2A), indicating that the 25 kDa fragment results from γ-secretase-mediated cleavage of the 30 kDa fragment. This cleavage event was also inhibited by other γ-secretase inhibitors, including WPE-89, MW111, and MG132 (data not shown). In contrast, the calpain inhibitor calpastatin had no effect on PMA-induced processing. We next confirmed that the sequential processing of p75NTR occurs in cells that endogenously express p75NTR using RN22F Schwannoma cells. Although low levels of basal α-secretase activity were revealed by the presence of the ∼30 kDa band in untreated RN22F cells (Fig. 2B, left lane), a ∼25 kDa cleavage product was only detectable when cells were stimulated with PMA in the presence of the proteasome inhibitor epoxomycin. As in the pIND-p75 cells, production of this unstable, PMA-inducible 25 kDa fragment was blocked by DAPT, indicating that the conversion of the 30 kDa fragment to the 25 kDa fragment is mediated by a γ-secretase protease (Fig. 2B, right lane). We have noted significant differences in the extent of constitutive α-cleavage of p75NTR in diverse cell lines, as evidenced by comparison of the Schwannoma cells with the inducible pIND-p75 cells. The dependence of cleavage on cellular context is most striking in primary mouse embryonic fibroblasts transfected with p75NTR, where extensive α-secretase-like cleavage occurs constitutively (Fig. 2C). Interestingly, in these cells where the α-product is easily detected, PMA negligibly affected α-secretase processing, and γ-secretase-like cleavage is not observed (Fig. 2C).
α-secretase-like activity cleaving p75NTR resembles ADAM10 and ADAM17
Stimulation of p75NTR cleavage by PMA is consistent with a role for select metalloproteases of the ADAM family. We therefore examined whether the pharmacological profile of the α-secretase was consistent with that of the PMA-inducible ADAMs, ADAM 10 (kuzbanian) and ADAM 17 (TACE). PMA-stimulated cleavage of p75NTR is prevented by TAPI (Fig. 2D), a potent but broadly specific inhibitor of membrane metalloproteases including ADAM10 and ADAM17. Recent reports suggest that PMA activation of ADAM10/17 is mediated by activation of either p38 MAP kinase (Gechtman et al., 1999) or ERK MAP kinase (Fan and Derynck, 1999). The PMA-stimulated cleavage of p75NTR is blocked by PD-98059, an inhibitor of ERK activation, but not by SB-203580, an inhibitor of p38 activation (Fig. 2D). Although ERK activation is necessary, ERK alone is not sufficient to induce cleavage, because exposure to other agents that activate ERK, such as epidermal growth factor or fetal bovine serum, did not stimulate processing (data not shown). Therefore, the p75NTR α-secretase is a TAPI-sensitive, zinc-dependent metalloprotease (DiStefano et al., 1993) with sensitivity to PMA that requires the activity of the ERK MAP kinase pathway. This profile resembles ADAM 10 and 17 and suggests that these proteases may mediate p75NTR cleavage, although other metalloproteases may contribute. ADAM10 and ADAM17 appear to function redundantly for both Notch and APP, and also may do so for p75NTR. ADAM17 is not essential for p75NTR cleavage, because cleavage occurs in fibroblasts derived from ADAM17 null mice. These fibroblasts, however, abundantly express ADAM10 (data not shown).
Trans-regulation of p75NTR cleavage
The α-secretase-mediated cleavage of Notch is stimulated by its physiologic ligand, Delta (Lieber et al., 2002). Therefore we examined whether α-secretase cleavage of p75NTR is stimulated by neurotrophins. In pIND-p75 cells expressing p75NTR, but not Trk neurotrophin receptors, NGF, BDNF, and NT3 had no significant effect on α-secretase cleavage of p75NTR (Fig. 3, top panel; all lanes pretreated with epoxomycin). Recent studies have revealed that pro-neurotrophins activate p75NTR-mediated proapoptotic signaling more potently than mature fully processed neurotrophins (Lee et al., 2001; Beattie et al., 2002). However, neither proNGF nor proBDNF influenced α-secretase cleavage of p75NTR (Fig. 3). We next examined whether the coexpression of the Trk neurotrophin receptors altered the processing of p75NTR, because it has been proposed that these receptors physically interact (Huber and Chao, 1995; Wolf et al., 1995; Ross et al., 1996; Bibel et al., 1999). HEK293 cells were cotransfected with p75NTR and TrkA, TrkB, or TrkC, and 24 hr later the cells were pretreated with epoxomycin before stimulation with PMA. Surprisingly, in the absence of PMA treatment, the expression of TrkA results in a robust accumulation of a p75NTR fragment of the same mass as the m-CTF product of α-secretase-mediated cleavage (Fig. 3, bottom panel). A similar but weaker effect was observed for TrkB, whereas TrkC had no effect. Accumulation of m-CTF could result either from stimulation of cleavage of p75NTR by α-secretase or inhibition of cleavage of m-CTF by γ-secretase. The latter mechanism appears to predominate, because the presence of TrkA and TrkB diminishes the production of the ICD fragment. Some contribution of the former mechanism is plausible, however, because TrkA and TrkB signaling activate mitogen-activated protein kinases, which are implicated in the regulation of α-secretase. No neurotrophins were added in the experiments shown, but added neurotrophins did not enhance the effect of TrkA or TrkB in other experiments (data not shown). However, constitutive activation of Trk receptors occurs when they are highly expressed. To assess directly whether Trk receptor signaling is required for modulation of p75NTR processing, we examined the effect of addition of K252a, a selective inhibitor of Trk receptor signaling. Interestingly, K252a did not influence the ability of TrkA to promote accumulation of m-CTF, but K252a inhibited the ability of TrkB to promote accumulation of m-CTF (Fig. 3, bottom panel), suggesting that these two Trk receptors may modulate p75NTR processing by different mechanisms. Although the mere physical presence of TrkA is sufficient to promote m-CTF accumulation, possibly because a p75NTR/TrkA complex shields p75NTR from γ-secretase, active signaling by TrkB is apparently required for it to be effective. It should be noted that we used relatively high concentrations of K252a in these experiments (100 nm) to ensure that inhibition of Trk tyrosine kinase activity was complete. At this concentration, K252a is known to nonselectively inhibit other protein kinases, including protein kinase C. Thus, not surprisingly, we observed that stimulation of p75NTR processing by the protein kinase C activator, PMA, was inhibited by K252a.
Figure 3.

Trk modulation of p75NTR processing. Top panel, p75 expression in pIND-p75 cells was induced overnight, and then cells were pretreated with epoxomycin (all lanes) before 40 min stimulation with the neurotrophins NGF (100 ng/ml), proNGF (10 ng/ml), BDNF (200 ng/ml), proBDNF (60 ng/ml), NT-3 (100 ng/ml), or PMA (100 ng/ml). Where PMA activates bothα and γ cleavage events, neurotrophins fail to directly affect p75NTR processing. Bottom panel, HEK293 cells were cotransfected with p75NTR and TrkA, TrkB, TrkC, or empty vector control and then pretreated with epoxomycin (all lanes) before stimulating p75 cleavage with PMA. The expression of TrkA results in significant accumulation of the p75NTRα-secretase product in the absence of PMA. TrkB produces this result as well, but less intensely, whereas TrkC has no effect. In the absence of Trk expression, PMA stimulates formation of α- and γ-secretase products, but the expression of TrkA or TrkB prevents the PMA-induced γ cleavage, because there is a concomitant accumulation of the α product and decrease in the γ product in the presence of TrkA and TrkB, but not TrkC. Pretreatment with the Trk kinase inhibitor K252a (100 nm) does not eliminate the effect of TrkA expression onα-product stability but does inhibit the effect of TrkB, suggesting alternate modes of influence on p75NTR processing with different dependence on Trk receptor activation. Blots were reprobed with a pan-Trk antibody to show equivalent expression levels (bottom).
Processing of NRH1 and NRH2 is distinct from p75NTR
Similarities in the structure of p75NTR, NRH1, and NRH2, including the remarkable homology of the transmembrane sequences of these proteins, suggested that NRH1 and NRH2 might also be processed by α-secretase and γ-secretase. In HEK293 cells transiently transfected with plasmids encoding NRH1 and NRH2 bearing C-terminal myc-epitope tags (NRH1-MT, NRH2-MT), short exposures of Western blots using an antibody against the myc tag revealed that NRH1-MT was expressed predominantly as a 70–80 kDa protein, with smaller amounts of larger species suggesting dimeric and trimeric aggregates. NRH2-MT was expressed predominantly as a 50–60 kDa protein (Fig. 4A). In addition, longer exposures consistently revealed the existence of several smaller weight fragments in both NRH1 and NRH2. In the absence of any treatment, both NRH1 and NRH2 produce fragments of 25–28 kDa (Fig. 4A). Because the six myc-epitope tag contributes ∼11 kDa to the molecular weight, this mass is consistent with a cleavage event within or just intracellular to the membrane. Unlike NRH1 and p75NTR, the NRH2 sequence diagramed in Figure 1 lacks the hydrophobic leader sequence that typically characterizes type I membrane proteins and secreted proteins. This raised the possibility that the NRH2 cleavage fragments seen in Figure 4A were the result of intracellular proteolysis of a protein that does not correctly target to the cell surface. Initially, therefore, we examined whether NRH2 exists as a cytoplasmic or a membrane protein using a cell-surface biotinylation assay. Using COS cells transiently transfected with the full-length isoform of myc-tagged mouse NRH2, we exposed intact cells to a cell-impermeant biotinylating reagent, followed by precipitation of detergent cell extracts with streptavidin beads and Western blot analysis with antibodies against the myc epitope. The ∼50 kDa band was specifically precipitated, confirming that full-length mNRH2-MT is expressed at the cell surface (Fig. 4B). Notably, the 25 kDa fragment does not precipitate in this assay, confirming that it is intracellular, and provides an internal control for membrane integrity during the biotin labeling. In addition, we validated membrane integrity by reprobing the blot for the cytoplasmic protein pyruvate kinase, which did not precipitate (Fig. 4B). The short putative NRH2 extracellular domain contains predicted sites for both N- and O-linked glycosylation, and the heterogeneous smear observed on the Western blot is typical for a glycosylated protein. We verified that NRH2 is glycosylated by culturing cells with tunicamycin, an inhibitor of N-linked glycosylation. This converted the protein to a smaller, less heterogeneous species (Fig. 4C), confirming the presence of N-linked glycosylation. Glycosylation is atypical of cytoplasmic proteins, confirming that this form of NRH2 is an intrinsic membrane protein. This conclusion is consistent with the results of Frankowski et al. (2002).
Figure 4.

NRH1 and NRH2 are constitutively cleaved at the cell surface. A, Western blot of HEK293 cells transiently transfected with mNRH2-MT and xNRH1-MT shows that full-length NRH1 is expressed as a ∼75–80 kDa protein and NRH2 is ∼55–60 kDa, but both proteins produce fragments too small to represent nascent immature proteins. B, COS cells were transfected with mNRH2-MT and either untreated or cell-surface biotinylated. Cells were washed extensively and lysed, and biotinylated proteins were affinity precipitated using ultralink-neutravidin beads. Full-length NRH2-MT specifically precipitated from cells labeled with biotin, but not unlabeled cells, indicating that the precipitate was specific for biotinylated cell-surface proteins. Blots were reprobed for the cytoplasmic protein pyruvate kinase (P.K.) as a control for the physical integrity of the cells during the labeling procedure. C, Overnight treatment with the glycosylation inhibitor tunicamycin (10 μg/ml) reduced the mass of NRH2-MT from a broad 55–60 kDa band to < 50 kDa, indicating that NRH2 is substantially N-glycosylated, indicative of a cell-surface protein. D, HEK293 cells transfected with either NRH1-MT or NRH2-MT were pretreated for 2 hr with the proteasome inhibitor epoxomycin (1 μm) and/or the γ-secretase inhibitor DAPT (10μm) and then treated with PMA (100 ng/ml) for 40 min before lysis. Neither epoxomycin nor DAPT had any effect on cleavage. PMA induced a novel cleavage in NRH2 producing a ∼40 kDa band (<), but this cleavage was also unaffected by epoxomycin and DAPT. D, PMA-induced cleavage of NRH2 is prevented by 2 hr pretreatment with metalloprotease inhibitors GM6001 (10 μm) and TAPI (25 μm), similar to the PMA-induced cleavage of p75NTR.
In contrast to p75NTR, the cleavage fragments of NRH1 and NRH2 were readily detectible in the absence of proteasome inhibitors (Fig. 4A,D). We examined whether treatment with PMA might increase the formation of the small fragments and whether epoxomycin affected their detection. Unlike p75NTR, the stability of the NRH1 and NRH2 intracellular fragments was not sensitive to epoxomycin, nor was their generation stimulated by PMA (Fig. 4D). Surprisingly, NRH cleavage is also insensitive to the γ-secretase inhibitor DAPT (Fig. 4D) and WPE (data not shown), indicating that this cleavage is not mediated by γ-secretase. A PMA-induced α-secretase-like cleavage of NRH2 does occur (Fig. 4D, <) and is inhibited by metalloprotease inhibitors GM6001 and TAPI (Fig. 4E). However, unlike p75NTR, this cleavage is not a necessary prerequisite for proteolytic release of the ICD, because production of the ICD fragment is insensitive to GM6001 and TAPI (Fig. 4E). We were unable to detect PMA-stimulated cleavage of NRH1 by our methods, although it is possible that an α-secretase cleavage product of NRH1 is hidden within the broad band representing the heterogeneously glycosylated full-length NRH1 protein. Thus, NRH1 and NRH2 are processed to produce soluble ICD fragments like p75NTR, but neither of the two proteases required to produce soluble p75NTR ICD are required to produce soluble ICDs of NRH1 or NRH2, and the ICD fragments of NRH1 and NRH2 are proteolytically more stable. These differences between p75NTR and the NRH proteins are unlikely to result from the C-terminal epitope tags used with the NRH proteins, because we do not see a change in p75NTR processing when it is similarly tagged (data not shown).
Nuclear trafficking of ICD fragments of p75NTR and NRH2
The subcellular distributions of the various proteolytic fragments of p75NTR and NRH2L-MT were assessed by centrifugal fractionation of HEK293 cell extracts into cytoplasmic, nuclear, and membrane fractions, followed by Western blot analysis. As shown in Figure 5, the putative m-CTF fragments reflecting cleavage of p75NTR and NRH2 by an α-secretase-like activity were found in the membrane fractions and not in the cytoplasmic fractions, as predicted. The small amount of m-CTF fragments of p75NTR and NRH2 present in the nuclear fractions probably reflects contamination by membrane, because small amounts of full-length p75NTR and NRH2 are also present in the nuclear fractions. The putative ICD fragments of p75NTR and NRH2 are not present in membrane fractions but are present in cytoplasmic fractions, as predicted. Importantly, significant amounts of the ICD fragments of p75NTR and NRH2 are present in the nuclear fractions, suggesting that these fragments can enter the nucleus.
Figure 5.

Subcellular fractionation of p75NTR and NRH2. A, p75NTR expression was induced (+ Pon) in pIND-p75 cells, and then cells were treated with epoxomycin (Epo, 1 μm), DAPT (2 μm), or PMA (100 ng/ml) as indicated. Cells were then fractionated into cytoplasmic, membrane, and nuclear fractions to examine the distribution of p75NTR cleavage products with antibody 9992. The γ-secretase product appeared in the cytoplasmic and nuclear fractions, whereas the α-secretase product is present in the membrane but not the cytoplasm. Specificity of additional bands in the nuclear fraction (*, X) is questionable because of their presence in the uninduced control. Alternatively, these bands may represent p75NTR in membrane contamination from the Golgi. B, Subcellular fractionation of HEK293 cells either untransfected (–) or transfected with myc-tagged mNRH2L (Tx). Western blot probed with anti-myc antibody indicates cytoplasmic and nuclear localization of the smallest fragment (<<). Larger mass NRH2 localize to membrane fractions (**, ***). As in A, nonspecific bands are present in nuclear fractions (***).
Subcellular distribution of these proteins was also assessed by laser scanning confocal microscopy after transfection of cells with plasmids encoding full-length p75NTR and NRH2L bearing C-terminal GFP tags. In HEK293 cells, both p75NTR–GFP (Fig. 6C,D) and NRH2–GFP (Fig. 6A,B) were most abundantly localized to the cell surface. In addition, GFP was also visible in the nucleus of NRH2–GFP-transfected cells but was not detectable in p75NTR–GFP-transfected cells. We noted the same distribution in various transfected cells, including NIH3T3 cells, HeLa cells, and COS cells (data not shown). Because our analysis by Western blotting suggested that in the absence of PMA stimulation there is little p75NTR cleavage but considerable NRH2 cleavage in 293 cells, we examined the effect of PMA stimulation in the presence of epoxomycin on the subcellular distribution of the GFP-tagged proteins, but failed to see significant changes for either protein (data not shown). This may reflect the difficulty in resolving the comparatively small amount of p75NTR γ product from the abundant full-length protein. To assess the distribution in a cell type that naturally expresses p75NTR and NRH2, we transfected primary cultures of Schwann cells with the GFP-tagged proteins. NRH2–GFP transfected Schwann cells often exhibited prominent nuclear localization (Fig. 6E,F), although this enrichment was not seen in all transfected cells (data not shown). In contrast, although p75NTR–GFP could be detected in the nucleus at low levels, no preferential nuclear accumulation was observed (Fig. 6G,H). These experiments were repeated in the presence of a proteasome inhibitor (epoxomycin) and PMA treatment, but such treatments had no discernible effect, nor did application of a nuclear export inhibitor (leptomycin B) (data not shown). Therefore, consistent with previous studies of Notch, it is difficult to follow the fate of the p75NTR γ-secretase product using conventional means. In contrast, NRH2 is readily seen both at the cell surface and in the nucleus.
Figure 6.
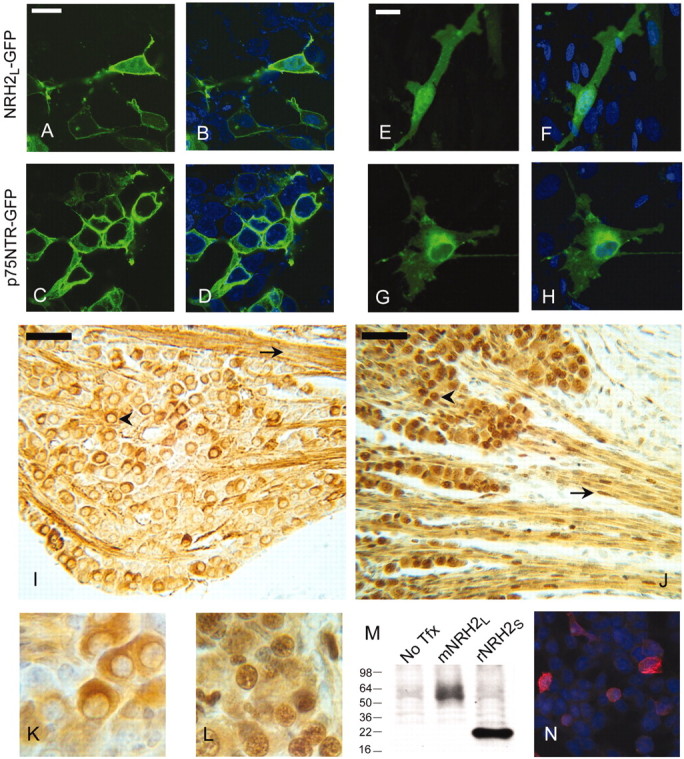
Subcellular distribution of p75NTR and NRH2L. A–D, HEK293 cells were transfected with either NRH2L–GFP (A) or p75NTR–GFP (C) and imaged by confocal microscopy. Comparison with DAPI-stained nuclei (B, D) shows that NRH2–GFP (A, B)is strongest at the cell surface but also has prominent nuclear localization, whereas p75NTR-GFP (C, D) is not detectable in the cell nucleus. E–H, Primary cultures of P0 rat Schwann cells were transfected with NRH2L–GFP (E) or p75NTR–GFP (G) and imaged by confocal microscopy. Comparison with DAPI-stained nuclei (F, H) shows that NRH2-GFP (E, F) shows enriched nuclear localization, and faint nuclear localization is detectable in p75NTR-GFP transfected cells (G, H). I, Bright-field image of DAB-stained immunoreactivity for p75NTR in the trigeminal ganglion of an E20 rat (10 μm section), using an antibody to the intracellular domain of p75NTR (9992). Subsets of trigeminal neurons stain in the soma (arrowhead) but not the nucleus. Immunoreactivity is also prominent among Schwann cells (arrow) but is non-nuclear. J, Immunoreactivity for NRH2 in a section adjacent to that in I, using an antibody specific to the intracellular domain of NRH2 (5592). NRH2 is strongly expressed innearly all sensory neurons of the E20 trigeminal ganglion, with lower levels of expression in Schwann cells. Neuronal labeling was almost always enriched in the nucleus (arrowhead), whereas only subsets of Schwann cells had nuclear labeling (arrow). This labeling was reproduced using several fixatives (Methacarnoys, 10% Formalin, 4% PFA) and both paraffin and cryostat processing. K, Higher-magnification image of representative p75NTR immunostaining of sensory neurons depicted in I. L, Higher-magnification image of representative NRH2 immunostaining of sensory neurons depicted in J. M, Western blot of HEK293 cells transfected with either mouse NRH2L or rat NRH2S using the anti-NRH2 intracellular domain antibody 5592 demonstrates that the antibody recognizes the long and short forms of NRH2, although some background reactivity is seen in untransfected (No Tfx) cell lysates. N, HEK293 cells were transiently transfected with rat NRH2S and immunostained using the 5592 anti-NRH2 antibody and a Cy3-labeled secondary. Untransfected cells were not labeled by the antibody, but transfected cells labeled strongly, confirming that the antibody recognizes the native conformation of NRH2. Scale bars: A, E, 20 μm; I, J, 50 μm.
We next examined the subcellular distribution of p75NTR and NRH2 in vivo. We generated a polyclonal antibody against a region of the NRH2 death domain with poor homology to p75NTR and verified that the antibody specifically recognizes mouse and rat NRH2 (Fig. 6M,N) but not p75NTR (data not shown). Using an anti-p75NTR intracellular domain antibody the specificity of which has been established independently (antibody 9992), we performed immunocytochemistry on tissue sections from developing rat. NRH2 is coexpressed with p75NTR in various cell types of the peripheral nervous system, including spinal and cranial sensory neurons, sympathetic neurons, and Schwann cells, but NRH2 is distributed more widely than p75NTR in the CNS (data not shown). In cells that were found to express both p75NTR and NRH2, such as E18 rat trigeminal ganglion neurons and subpopulations of Schwann cells in the trigeminal nerve, NRH2 immunoreactivity had prominent nuclear localization (Fig. 6J,L), whereas little if any p75NTR immunoreactivity was detected in nuclei (Fig. 6I,K). We observed prominent nuclear staining with the NRH2 antibody in several neural and epithelial cell types, but failed to see prominent nuclear p75NTR immunoreactivity in any cell type in vivo. This is consistent with staining of the p75 9992 antibody of cell lines that naturally express p75NTR, including PC12 cells and RN22F Schwannomas, in which nuclear staining is not evident even in the presence of proteasome inhibitors and PMA (data not shown). In contrast, and in agreement with the results of NRH2–GFP-transfected cells, NRH2 is readily detected in the nucleus. The NRH2 nuclear immunostaining was specific, because previous absorption of the antibody with the immunizing peptide completely eliminated staining (data not shown). These data strongly support a physiologic function for NRH2 nuclear translocation.
Figure 7.

Soluble intracellular domains of p75NTR and NRH2 potentiate NF-κB activation by TRAF6. HEK293 cells were cotransfected with a κB-luciferase reporter and the normalization vector EF1-LacZ, and either TRAF6 (black bars) or an empty vector control (open bars), and full-length rat p75NTR, rat p75NTRICD, full-length mouse NRH2L-MT, mouse NRH2 ICD, or empty vector control. Twenty-four hours after transfection, cells were lysed and assayed for luciferase expression. In the absence of TRAF6 expression, mild activation of NF-κB resulted from the expression of the p75NTR ICD compared with the empty vector control. This degree of activation was dwarfed by the basal activity seen in the presence of TRAF6 expression. When TRAF6 was coexpressed, activation of NF-κB by full-length p75NTR and NRH2 did not significantly differ from the empty vector control. In contrast, soluble p75NTR ICD and NRH2 ICD significantly potentiated NF-κB activation by TRAF6. Two-tailed t test; p75NTR ICD versus vector: *p = 0.012; NRH2 ICD versus vector: *p = 0.007. Error bars represent SD; n = 3.
Soluble ICDs of p75NTR and NRH2 affect endogenous transcriptional signaling pathways
The results above suggest that soluble ICDs of p75NTR and NRH2 localized within the cytoplasm and nucleus might participate in transcriptional regulation, as has been observed for various other membrane proteins that undergo “RIP.” The presence of significant pools of ICDs within the cytoplasm (Fig. 5) raises the possibility that ICDs may also possess cytoplasmic signaling functions. p75NTR is thought to regulate NF-κB-mediated transcriptional regulation (Carter et al., 1996) by interacting with cytoplasmic proteins such as TRAF6, RIP2, and IL-1 receptor associated kinase (IRAK) (Khursigara et al., 1999; Khursigara et al., 2001; Mamidipudi et al., 2002). Consequently, we examined whether soluble ICD forms of p75NTR and NRH2 were competent to function in this context. HEK293 cells expressing a κB luciferase reporter construct were transfected with either full-length or soluble ICD of p75NTR and NRH2 constructs in the presence or absence of cotransfected TRAF6. In the absence of exogenous TRAF6, p75NTR (without added neurotrophin) and NRH2 had little effect on κB-luciferase expression, whereas a soluble ICD of p75NTR modestly stimulated κB-luciferase expression (Fig. 9). Overexpression of TRAF6 in the absence of p75NTR or NRH2 significantly stimulated κB-luciferase expression. In the presence of TRAF6 overexpression, full-length p75NTR and NRH2 did not potentiate NF-κB activation. In contrast, the soluble ICDs of p75NTR and NRH2 significantly stimulated TRAF6 induction of κB-luciferase expression. Thus NRH2, like p75NTR, couples to NF-κB signal transduction, and in the absence of ligand the ICDs of p75NTR and NRH2 couple to NF-κB activation more efficiently than full-length membrane-resident proteins. We were unable to assess whether NF-κB activation by full-length p75NTR and NRH2 requires cleavage by γ-secretase, because inhibitors of γ-secretase directly activated expression of the κB-luciferase reporter (data not shown).
Discussion
α-secretase and γ-secretase
These studies demonstrate that proteolysis releases ICDs from p75NTR, NRH1, and NRH2. For p75NTR this process resembles other examples of RIP. α-secretase action generates m-CTF, which is cleaved by γ-secretase. In contrast, the NRH2 ICD is generated by a protease that is distinct from γ-secretase, in a process that does not require previous cleavage by α-secretase, although α-secretase cleavage of NRH2 does occur. We have not yet identified the enzyme(s) responsible for these α-secretase activities. Cleavage is not eliminated in fibroblasts derived from ADAM17 null mice (data not shown). However, these fibroblasts express ADAM10, which may function redundantly with ADAM17. ADAM10 and ADAM17 do not possess well defined specificity for the local sequence at the site of cleavage but cleave at sites near the outer face of the membrane. The size of the p75NTR m-CTF is consistent with such cleavage. γ-secretase also lacks a rigidly defined sequence specificity, but cleaves within protein transmembrane domains, commonly N-terminal to valine residues, which are abundant in the p75NTR transmembrane domain. Although NRH1 and NRH2 have valine residues at equivalent positions within their transmembrane domains, cleavage of these proteins is not mediated by γ-secretase.
The α-secretase-independent release of NRH2 ICD may occur because the short ECD of NRH2L resembles the α-secretase-truncated ECD of p75NTR. Alternatively, the unidentified protease responsible for NRH2 cleavage may simply lack a requirement for an abbreviated ECD. p75NTR processing is highly dependent on cell type. α-secretase-mediated cleavage in HEK293 cells requires activation by PMA, whereas Schwann cells have substantial constitutive α-secretase cleavage that is further stimulated by PMA, and mouse embryo fibroblasts exhibit strong constitutive α-secretase cleavage, generating m-CTF that is not subject to γ-secretase cleavage. The failure of p75NTR to be cleaved by γ-secretase in these cell lines is surprising, because β-amyloid precursor protein and Notch are efficiently cleaved by γ-secretase in the same cell line. Presenilin functions as a protease in the context of a complex with multiple partners, including nicastrin and Aph-2 (Kopan and Goate, 2002). Perhaps p75NTR association with presenilin requires the presence of another presenilin accessory protein that mouse embryo fibroblasts lack. Alternatively, these cells may express proteins that protect p75NTR from γ-secretase action. Our results indicate that Trks may function in this manner.
p75NTR/Trk interactions
Full-length p75NTR affects the sensitivity (Hempstead et al., 1991; Davies et al., 1993; Barker and Shooter, 1994; Lee et al., 1994; Verdi et al., 1994) and specificity (Mischel et al., 2001) of neurotrophin binding to Trks, whereas p75NTR lacking an ECD is sufficient to enhance the affinity of NGF/TrkA binding (Esposito et al., 2001) and retains the ability to physically interact with TrkB (Bibel et al., 1999). Thus the p75NTR m-CTF fragment may influence Trk receptor function. It is notable that TrkC does not influence the stability of the p75NTR m-CTF fragment, because p75NTR has been shown to interact differently with the three Trks. Although p75NTR enhances TrkA and TrkB autophosphorylation in response to their preferred ligands, an effect on TrkC activation by NT-3 is contested (Hantzopoulos et al., 1994; Vesa et al., 2000). It has been suggested that p75NTR signals cell death in the absence of Trks but potentiates survival in the presence of Trks (for review, see Roux and Barker, 2002). This cross-talk may not reflect an effect of p75NTR on Trks so much as a silencing of p75NTR signaling by Trks (Majdan et al., 2001). In the absence of Trks, truncated and intracellular forms of p75NTR more potently induce neuronal cell death (Coulson et al., 2000; Roux et al., 2001). Therefore, the regulation of p75NTR cleavage by TrkA and TrkB may be one mechanism by which Trks affect p75NTR cell death signaling.
Nuclear trafficking of ICDs
We have presented evidence that the ICDs of p75NTR and NRH2 traffic to the nucleus. Despite similarities, nuclear trafficking of p75NTR and NRH2 ICDs differs in important respects. The p75NTR ICD is unstable in the absence of proteasome inhibitors, whereas NRH2 ICD is stable independently of proteasome inhibition. p75NTR ICD instability is not inconsistent with possible signaling functions because similar instability is characteristic of other examples of functionally relevant RIP-mediated nuclear trafficking (Brown et al., 2000). p75NTR ICD instability may result from the presence of a PEST sequence; NRH2 lacks such a sequence. Even considering the greater stability of the NRH2 ICD, the prominent nuclear localization of NRH2 immunoreactivity in sensory neurons and Schwann cells is notable. It is possible that these cell types preferentially express NRH2 splice variants that consist exclusively of the ICD. The distribution of these splice variants in vivo has not been examined.
Do soluble ICDs signal?
We have demonstrated that artificially generated p75NTR and NRH2 ICDs actively stimulate NF-κB-mediated gene expression. This suggests that physiological release of these ICDs may contribute to NF-κB signaling. We were unable to directly test this possibility by inhibition of γ-secretase, however, because DAPT itself stimulated NF-κB activity. Our finding is not the first evidence for potent signaling capacity of a p75NTR ICD construct. Expression of p75NTR ICD in developing neurons of transgenic mice induced extensive neuronal apoptosis (Majdan et al., 1997), and proapoptotic signaling of p75NTR ICD has been demonstrated in cultured cells (Coulson et al., 2000; Roux et al., 2001). The present study raises the possibility that such ICD-mediated functions may occur physiologically.
The p75NTR death domain has been implicated in NF-κB activation through interaction with RIP2 (Khursigara et al., 2001) or TRAF6 (Khursigara et al., 1999; Wang et al., 2000), Myd88, and IRAK (Mamidipudi and Wooten, 2002). Our finding that TRAF6 cooperates with ICDs of both p75NTR and NRH2 to stimulate NF-κB suggests that the NRH2 death domain recruits similar signaling proteins. Our results suggest, however, that activation may not occur exclusively at the plasma membrane but may also occur within the cytoplasm or even within the nucleus. The p75NTR death domain represents a subtype present in MyD88, p100 NF-κB, p105 NF-κB, and DAP kinase, (Feinstein et al., 1995). p75NTR interacts with MyD88 via an interaction of the death domain of MyD88. The presence within p100 and p105 NF-κB subunits of similar death domains suggests that nuclear trafficking of the p75NTR ICD might directly promote nuclear access of NF-κB proteins.
Nuclear trafficking of p75NTR interacting proteins
Numerous proteins interact with the p75NTR ICD (for review, see Roux and Barker, 2002), and many of these have been suggested to have nuclear functions. These include proteins with multiple zinc finger domains, characteristic of transcription factors [neurotrophin receptor interacting factor (NRIF), TRAF6, TRAF1, SC1], three members of the MAGE gene family (Salehi et al., 2000; Tcherpakov et al., 2002), which have been implicated as transcriptional and cell cycle regulators (Barker and Salehi, 2002), and the apoptosis-inducing protein p75 neurotrophin receptor-associated cell death executor (NADE), which has tightly regulated nuclear import/export (Mukai et al., 2000). Several of these p75NTR-interacting proteins (NADE, NRIF, TRAF6, NRAGE) show enhanced binding to p75NTR in the presence of NGF, and several (NRIF, SC-1) translocate from the cytoplasm to the nucleus in the presence of NGF (Casademunt et al., 1999; Chittka and Chao, 1999). Thus one can envision a model in which neurotrophin binding initially recruits these proteins to p75NTR, after which proteolysis permits nuclear trafficking of these proteins as a complex with the p75NTR ICD. It remains to be determined whether the closely similar ICDs of NRH1 and NRH2 also interact with these proteins.
RIP of p75NTR may also contribute to cytoplasmic signaling processes. Transfection of a soluble p75NTR ICD activates RhoA GTPase and the ICD fragment physically associates with RhoA, whereas such an interaction was not detected with the full-length membrane-resident p75NTR (Yamashita et al., 1999). This suggests the possibility that functional RhoA/p75NTR ICD interactions might be stimulated by proteolytic release of the p75NTR ICD. p75NTR plays an important role in axon guidance, both in axon growth in response to neurotrophins (Yamashita et al., 1999; Bentley and Lee, 2000; Tucker et al., 2001), and in axon repulsion in response to myelin inhibitory factors via association of p75NTR with the Nogo receptor. RhoA activation is implicated in these responses (Wang et al., 2002; Wong et al., 2002; Yamashita et al., 2002). ADAM metalloprotease regulation of p75NTR-mediated axon guidance would be consistent with growing literature suggesting metalloprotease function in axon guidance (McFarlane, 2003).
Although it is our presumption that the functional significance of cleavage of p75NTR by α- and γ-secretases, and by analogy to Notch and similar systems, lies in the active propagation of signals from the cell surface, the alternative possibility that cleavage occurs as a mode of negative regulation of p75NTR signaling cannot be dismissed. However, the observation that NRH2 generates as its primary translation products proteins resembling the products of cleavage of p75NTR by α-secretase and γ-secretase proteases suggests that the p75NTR cleaved products are not simply inactive products of p75NTR degradation. Progress toward understanding the function of p75NTR cleavage and understanding the function of NRH2 may be closely linked. Finally, because several other members of the TNF receptor superfamily also undergo metalloprotease-mediated ECD shedding, it may be productive to examine whether these receptors also undergo subsequent γ-secretase-mediated generation of soluble ICDs.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant R01NS33200 to M.B. K.K. was supported by NIH Institutional Training Grant 5 T32 GM07108. We thank M. Chao (Skirboll Institute, New York University) for providing anti-p75NTR antibody 9992, M. Wolfe (Harvard University) for providing γ-secretase inhibitors, Millenium Pharmaceuticals for providing plasmids encoding rat and mouse NRH2, Genentech for providing NGF, Regeneron for providing BDNF and NT3, B. Hempstead (Cornell Medical College) for providing proNGF, and Immunex Pharmaceuticals for providing ADAM17 null fibroblasts.
Correspondence should be addressed to Mark Bothwell, Department of Physiology and Biophysics, Box 357290, University of Washington, 1959 Pacific Avenue, Seattle, WA 98195. E-mail: mab@u.washington.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235425-12$15.00/0
References
- Barker PA, Salehi A ( 2002) The MAGE proteins: emerging roles in cell cycle progression, apoptosis, and neurogenetic disease. J Neurosci Res 67: 705–712. [DOI] [PubMed] [Google Scholar]
- Barker PA, Shooter EM ( 1994) Disruption of NGF binding to the low affinity neurotrophin receptor p75LNTR reduces NGF binding to TrkA on PC12 cells. Neuron 13: 203–215. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM, Bresnahan JC, Hempstead BL, Yoon SO ( 2002) ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron 36: 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley CA, Lee KF ( 2000) p75 is important for axon growth and Schwann cell migration during development. J Neurosci 20: 7706–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibel M, Hoppe E, Barde YA ( 1999) Biochemical and functional interactions between the neurotrophin receptors Trk and p75NTR. EMBO J 18: 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Ye J, Rawson RB, Goldstein JL ( 2000) Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100: 391–398. [DOI] [PubMed] [Google Scholar]
- Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhauser N, Bohm-Matthaei R, Baeuerle PA, Barde YA ( 1996) Selective activation of NF-kappa B by nerve growth factor through the neurotrophin receptor p75. Science 272: 542–545. [DOI] [PubMed] [Google Scholar]
- Casademunt E, Carter BD, Benzel I, Frade JM, Dechant G, Barde YA ( 1999) The zinc finger protein NRIF interacts with the neurotrophin receptor p75(NTR) and participates in programmed cell death. EMBO J 18: 6050–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittka A, Chao MV ( 1999) Identification of a zinc finger protein whose subcellular distribution is regulated by serum and nerve growth factor. Proc Natl Acad Sci USA 96: 10705–10710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson EJ, Reid K, Baca M, Shipham KA, Hulett SM, Kilpatrick TJ, Bartlett PF ( 2000) Chopper, a new death domain of the p75 neurotrophin receptor that mediates rapid neuronal cell death. J Biol Chem 275: 30537–30545. [DOI] [PubMed] [Google Scholar]
- Davies AM, Lee KF, Jaenisch R ( 1993) p75-deficient trigeminal sensory neurons have an altered response to NGF but not to other neurotrophins. Neuron 11: 565–574. [DOI] [PubMed] [Google Scholar]
- DiStefano PS, Johnson Jr EM ( 1988) Identification of a truncated form of the nerve growth factor receptor. Proc Natl Acad Sci USA 85: 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiStefano PS, Chelsea DM, Schick CM, McKelvy JF ( 1993) Involvement of a metalloprotease in low-affinity nerve growth factor receptor truncation: inhibition of truncation in vitro and in vivo J Neurosci 13: 2405–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito D, Patel P, Stephens RM, Perez P, Chao MV, Kaplan DR, Hempstead BL ( 2001) The cytoplasmic and transmembrane domains of the p75 and Trk A receptors regulate high affinity binding to nerve growth factor. J Biol Chem 276: 32687–32695. [DOI] [PubMed] [Google Scholar]
- Fan H, Derynck R ( 1999) Ectodomain shedding of TGF-alpha and other transmembrane proteins is induced by receptor tyrosine kinase activation and MAP kinase signaling cascades. EMBO J 18: 6962–6972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein E, Kimchi A, Wallach D, Boldin M, Varfolomeev E ( 1995) The death domain: a module shared by proteins with diverse cellular functions. Trends Biochem Sci 20: 342–344. [DOI] [PubMed] [Google Scholar]
- Frankowski H, Castro-Obregon S, del Rio G, Rao RV, Bredesen DE ( 2002) PLAIDD, a type II death domain protein that interacts with p75 neurotrophin receptor. Neuromol Med 1: 153–170. [DOI] [PubMed] [Google Scholar]
- Gechtman Z, Alonso JL, Raab G, Ingber DE, Klagsbrun M ( 1999) The shedding of membrane-anchored heparin-binding epidermal-like growth factor is regulated by the Raf/mitogen-activated protein kinase cascade and by cell adhesion and spreading. J Biol Chem 274: 28828–28835. [DOI] [PubMed] [Google Scholar]
- Grob PM, Ross AH, Koprowski H, Bothwell M ( 1985) Characterization of the human melanoma nerve growth factor receptor. J Biol Chem 260: 8044–8049. [PubMed] [Google Scholar]
- Hantzopoulos PA, Suri C, Glass DJ, Goldfarb MP, Yancopoulos GD ( 1994) The low affinity NGF receptor, p75, can collaborate with each of the Trks to potentiate functional responses to the neurotrophins. Neuron 13: 187–201. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV ( 1991) High-affinity NGF binding requires coexpression of the Trk protooncogene and the low-affinity NGF receptor. Nature 350: 678–683. [DOI] [PubMed] [Google Scholar]
- Hooper NM, Karran EH, Turner AJ ( 1997) Membrane protein secretases. Biochem J 321: 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber LJ, Chao MV ( 1995) A potential interaction of p75 and TrkA NGF receptors revealed by affinity crosslinking and immunoprecipitation. J Neurosci Res 40: 557–563. [DOI] [PubMed] [Google Scholar]
- Hutson LD, Bothwell M ( 2001) Expression and function of Xenopus laevis p75(NTR) suggest evolution of developmental regulatory mechanisms. J Neurobiol 49: 79–98. [DOI] [PubMed] [Google Scholar]
- Johnson D, Lanahan A, Buck CR, Sehgal A, Morgan C, Mercer E, Bothwell M, Chao M ( 1986) Expression and structure of the human NGF receptor. Cell 47: 545–554. [DOI] [PubMed] [Google Scholar]
- Khursigara G, Orlinick JR, Chao MV ( 1999) Association of the p75 neurotrophin receptor with TRAF6. J Biol Chem 274: 2597–2600. [DOI] [PubMed] [Google Scholar]
- Khursigara G, Bertin J, Yano H, Moffett H, DiStefano PS, Chao MV ( 2001) A prosurvival function for the p75 receptor death domain mediated via the caspase recruitment domain receptor-interacting protein 2. J Neurosci 21: 5854–5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Goate A ( 2002) Aph-2/Nicastrin: an essential component of gamma-secretase and regulator of Notch signaling and Presenilin localization. Neuron 33: 321–324. [DOI] [PubMed] [Google Scholar]
- Lee KF, Davies AM, Jaenisch R ( 1994) p75-deficient embryonic dorsal root sensory and neonatal sympathetic neurons display a decreased sensitivity to NGF. Development 120: 1027–1033. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL ( 2001) Regulation of cell survival by secreted proneurotrophins. Science 294: 1945–1948. [DOI] [PubMed] [Google Scholar]
- Lieber T, Kidd S, Young MW ( 2002) Kuzbanian-mediated cleavage of Drosophila Notch. Genes Dev 16: 209–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liepinsh E, Ilag L, Otting G, Ibanez CF ( 1997) NMR structure of the death domain of the p75 neurotrophin receptor. EMBO J 16: 4999–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdan M, Lachance C, Gloster A, Aloyz R, Zeindler C, Bamji S, Bhakar A, Belliveau D, Fawcett J, Miller FD, Barker PA ( 1997) Transgenic mice expressing the intracellular domain of the p75 neurotrophin receptor undergo neuronal apoptosis. J Neurosci 17: 6988–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdan M, Walsh GS, Aloyz R, Miller FD ( 2001) TrkA mediates developmental sympathetic neuron survival in vivo by silencing an ongoing p75NTR-mediated death signal. J Cell Biol 155: 1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamidipudi V, Li X, Wooten MW ( 2002) Identification of interleukin 1 receptor-associated kinase as a conserved component in the p75-neurotrophin receptor activation of nuclear factor-kappa B. J Biol Chem 277: 28010–28018. [DOI] [PubMed] [Google Scholar]
- McFarlane S ( 2003) Metalloproteases: carving out a role in axon guidance. Neuron 37: 559–562. [DOI] [PubMed] [Google Scholar]
- Mischel PS, Smith SG, Vining ER, Valletta JS, Mobley WC, Reichardt LF ( 2001) The extracellular domain of p75NTR is necessary to inhibit neurotrophin-3 signaling through TrkA. J Biol Chem 276: 11294–11301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai J, Hachiya T, Shoji-Hoshino S, Kimura MT, Nadano D, Suvanto P, Hanaoka T, Li Y, Irie S, Greene LA, Sato TA ( 2000) NADE, a p75NTR-associated cell death executor, is involved in signal transduction mediated by the common neurotrophin receptor p75NTR. J Biol Chem 275: 17566–17570. [DOI] [PubMed] [Google Scholar]
- Oberg C, Li J, Pauley A, Wolf E, Gurney M, Lendahl U ( 2001) The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. J Biol Chem 276: 35847–35853. [DOI] [PubMed] [Google Scholar]
- Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA ( 1998) An essential role for ectodomain shedding in mammalian development. Science 282: 1281–1284. [DOI] [PubMed] [Google Scholar]
- Radeke MJ, Misko TP, Hsu C, Herzenberg LA, Shooter EM ( 1987) Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature 325: 593–597. [DOI] [PubMed] [Google Scholar]
- Ross AH, Daou MC, McKinnon CA, Condon PJ, Lachyankar MB, Stephens RM, Kaplan DR, Wolf DE ( 1996) The neurotrophin receptor, gp75, forms a complex with the receptor tyrosine kinase TrkA. J Cell Biol 132: 945–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth MB, Zahler AM, Stolk JA ( 1991) A conserved family of nuclear phosphoproteins localized to sites of polymerase II transcription. J Cell Biol 115: 587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux PP, Barker PA ( 2002) Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol 67: 203–233. [DOI] [PubMed] [Google Scholar]
- Roux PP, Bhakar AL, Kennedy TE, Barker PA ( 2001) The p75 neurotrophin receptor activates Akt (protein kinase B) through a phosphatidylinositol 3-kinase-dependent pathway. J Biol Chem 276: 23097–23104. [DOI] [PubMed] [Google Scholar]
- Salehi AH, Roux PP, Kubu CJ, Zeindler C, Bhakar A, Tannis LL, Verdi JM, Barker PA ( 2000) NRAGE, a novel MAGE protein, interacts with the p75 neurotrophin receptor and facilitates nerve growth factor-dependent apoptosis. Neuron 27: 279–288. [DOI] [PubMed] [Google Scholar]
- Tcherpakov M, Bronfman FC, Conticello SG, Vaskovsky A, Levy Z, Niinobe M, Yoshikawa K, Arenas E, Fainzilber M ( 2002) The p75 neurotrophin receptor interacts with multiple MAGE proteins. J Biol Chem 277: 49101–49104. [DOI] [PubMed] [Google Scholar]
- Tucker KL, Meyer M, Barde YA ( 2001) Neurotrophins are required for nerve growth during development. Nat Neurosci 4: 29–37. [DOI] [PubMed] [Google Scholar]
- Verdi JM, Birren SJ, Ibanez CF, Persson H, Kaplan DR, Benedetti M, Chao MV, Anderson DJ ( 1994) p75LNGFR regulates Trk signal transduction and NGF-induced neuronal differentiation in MAH cells. Neuron 12: 733–745. [DOI] [PubMed] [Google Scholar]
- Vesa J, Kruttgen A, Shooter EM ( 2000) p75 reduces TrkB tyrosine autophosphorylation in response to brain-derived neurotrophic factor and neurotrophin 4/5. J Biol Chem 275: 24414–24420. [DOI] [PubMed] [Google Scholar]
- von Schack D, Casademunt E, Schweigreiter R, Meyer M, Bibel M, Dechant G ( 2001) Complete ablation of the neurotrophin receptor p75NTR causes defects both in the nervous and the vascular systems. Nat Neurosci 4: 1–2. [DOI] [PubMed] [Google Scholar]
- Wang JJ, Rabizadeh S, Tasinato A, Sperandio S, Ye X, Green M, Assa-Munt N, Spencer D, Bredesen DE ( 2000) Dimerization-dependent block of the proapoptotic effect of p75(NTR). J Neurosci Res 60: 587–593. [DOI] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z ( 2002) p75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature 420: 74–78. [DOI] [PubMed] [Google Scholar]
- Wolf DE, McKinnon CA, Daou MC, Stephens RM, Kaplan DR, Ross AH ( 1995) Interaction with TrkA immobilizes gp75 in the high affinity nerve growth factor receptor complex. J Biol Chem 270: 2133–2138. [DOI] [PubMed] [Google Scholar]
- Wong ST, Henley JR, Kanning KC, Huang KH, Bothwell M, Poo MM ( 2002) A p75(NTR) and Nogo receptor complex mediates repulsive signaling by myelin-associated glycoprotein. Nat Neurosci 5: 1302–1308. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Tucker KL, Barde YA ( 1999) Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron 24: 585–593. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Higuchi H, Tohyama M ( 2002) The p75 receptor transduces the signal from myelin-associated glycoprotein to Rho. J Cell Biol 157: 565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]