

Abstract
Sodium-dependent transporters clear extracellular glutamate in the mammalian CNS. Activation of protein kinase C (PKC) rapidly increases the activity of the neuronal glutamate transporter EAAC1 (excitatory amino acid carrier-1). This effect is associated with redistribution of EAAC1 to the cell membrane and appears to be dependent on a particular PKC subtype, PKCα. In the present study, we sought to determine whether this specificity for regulation of EAAC1 is associated with the formation of EAAC1–PKCα complexes. In C6 glioma cells, activation of PKC with phorbol 12-myristate 13-acetate (PMA) induced formation of EAAC1–PKCα complexes but did not induce formation of complexes with PKCδ, a PKC not thought to regulate EAAC1. Formation of these complexes was blocked by inhibitors of PKC. Confocal microscopy revealed that PMA caused EAAC1 and PKCα to colocalize in clusters at or near the cell surface. The EAAC1–PKCα complexes were also observed in rat brain synaptosomes, demonstrating that this interaction is not restricted to C6 cells. These data demonstrate that EAAC1 and PKCα interact in a PKC-dependent manner that is associated with EAAC1 redistribution. Although PKC activation has been implicated in the regulation of many different neurotransmitter transporters, this study provides the first example of an interaction between a neurotransmitter transporter and PKC. PKCα also forms complexes with GluR2 (glutamate receptor subunit 2) and causes a reduction in the levels of GluR2-containing AMPA receptors at the plasma membrane. Together, these data suggest that PKCα may simultaneously trigger the redistribution of EAAC1 and glutamate receptors.
Keywords: glutamate transporter, EAAC1, trafficking, PKCα, phorbol ester, C6 glioma
Introduction
Glutamate is the predominant excitatory neurotransmitter in the mammalian CNS. An extracellular accumulation of glutamate and the consequent excessive activation of glutamate receptors (GluRs) appears to contribute to the damage observed in acute and chronic neurodegenerative disorders (Choi, 1992). Unlike some other neurotransmitters, there is no evidence for extracellular metabolism of glutamate; instead, extracellular levels are maintained by a family of sodium-dependent transporters, including the following: GLAST [for glutamate/aspartate transporter (EAAT1)], GLT-1 [for glutamate transporter-1 (EAAT2)], EAAC1 [for excitatory amino acid carrier-1 (EAAT3)], EAAT4 (for excitatory amino acid transporter-4), and EAAT5 (Sims and Robinson, 1999; Danbolt, 2001).
Glutamate transporters shape receptor responses by two different mechanisms, binding and active uptake. Binding of glutamate to transporters contributes to the fast component of glutamate removal from the synaptic space. This mechanism requires a high density of transporter molecules surrounding the synaptic cleft to effectively buffer free glutamate available for receptor activation. Active transport is generally considered a slow process compared with the rapid kinetics of some ion channels activated by glutamate, with a turnover time of ∼100 msec, but it appears to be fast enough to shape the activation of other glutamate receptors, particularly those that do not rapidly desensitize or those that couple to G-proteins (Wadiche et al., 1995; Conti and Weinberg, 1999; Diamond, 2001). These observations demonstrate that glutamate transporters are involved in the regulation of synaptic transmission and raise the possibility that regulation of transporters may modulate synaptic transmission.
The activity of most of the glutamate transporters can be regulated by mechanisms that are independent of de novo transporter synthesis (Sims and Robinson, 1999; Danbolt, 2001). In some cases, the changes in activity are associated with insertion or removal of transporter molecules at the plasma membrane. In C6 glioma, activation of protein kinase C (PKC) rapidly (within minutes) increases EAAC1-mediated transport activity. This effect is associated with a redistribution of EAAC1 from subcellular compartments to the plasma membrane (Davis et al., 1998). Using pharmacological approaches combined with downregulation of specific PKC subtypes, we recently developed evidence to suggest that PKCα regulates EAAC1 redistribution and that PKCϵ regulates EAAC1 catalytic efficiency (González et al., 2002). As a first step in the characterization of the involvement of PKCα in the regulation of EAAC1 trafficking, we determined whether EAAC1 and PKCα interact. In the present study, we provide evidence for an interaction between PKCα and EAAC1 that is dependent on PKC activation.
Materials and Methods
Cell culture. C6 glioma cells, a cell line that endogenously expresses EAAC1 and none of the other transporter subtypes, were grown as described previously (Davis et al., 1998).
Preparation of crude synaptosomes. Synaptosomes were prepared from adult rats and resuspended in 5 vol (v/w) of sucrose as described previously (Robinson, 1998). Crude synaptosomes (100 μl containing ∼500 μg of protein) were resuspended in 900 μl of sodium containing buffer (González et al., 2002). PKC inhibitors or vehicle were added, and the synaptosomal suspension was prewarmed to 37°C for 5 min. Phorbol 12-myristate 13-acetate (PMA) (100 nm) was added, and the synaptosomal suspension was kept at 37°C for an additional 30 min. Synaptosomal membranes were recovered by centrifugation at 20,000 × g for 20 min.
Immunoprecipitation and Western blot. C6 cells or synaptosomal pellets were resuspended in 1 ml of lysis buffer (González et al., 2002) and solubilized for 1 hr at 4°C. Lysates were centrifuged at 12,500 rpm to remove cell debris. Supernatants were precleared with 40 μl of protein A-agarose beads (Invitrogen, Grand Island, NY) and gently shaken for 1 hr at 4°C. After centrifugation, an aliquot was saved, and the precleared lysates were incubated overnight with 2 μg (C6 cells) or 3 μg (synaptosomes) of affinity-purified polyclonal rabbit anti-EAAC1 antibody [Alpha Diagnostics International (ADI), San Antonio, TX] at 4°C. Immune complexes were collected after incubation for 2 hr with 30 μl of protein A-agarose slurry. After four washes with lysis buffer, immune complexes were released in 25 μl of 2× sample buffer by boiling at 90–95°C. Immunoprecipitated proteins were resolved by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA). After blocking, membranes were probed with antibodies for PKC (monoclonal mouse anti-PKCα at 1:500, Transduction Laboratories, San Diego, CA; rabbit polyclonal anti-PKCδ at 1:2000, Santa Cruz Biotechnology, Santa Cruz, CA) or EAAC1 (1:5000) and visualized with chemiluminescence.
Immunocytochemistry and confocal microscopy. C6 glioma (plated on glass coverslips) were treated with vehicle or PMA (100 nm) for 30 min, washed with PBS, and fixed with 2% paraformaldehyde for 10 min. After incubation with blocking solution (minimum essential medium containing 15 mm HEPES buffer, 10% fetal bovine serum, and 0.05% sodium azide) for 10 min, anti-EAAC1 (1.25 μg/ml) and mouse anti-PKCα (5 μg/ml) were added for 30 min. Cultures were then incubated with biotinylated donkey anti-rabbit Ig (species specific, 1:100) and rhodamine conjugated donkey anti-mouse Ig (species specific, 1:100) for 30 min, followed by fluorescein-conjugated streptavidin (1:100; all reagents from Jackson ImmunoResearch, West Grove, PA) for 20 min. Coverslips were washed between steps with PBS, postfixed with cold methanol for 8 min, and counterstained with Hoechst H 33258 in PBS (2 μg/ml) for 3 min. Stained cells were mounted in Vectorshield (Vector Laboratories, Burlingame, CA). Immunolabeled cultures were optically sectioned at 0.5 μm intervals with a Leica (Nussloch, Germany) Inverted DM IRE2 HC fluo TCS 1-B-UV microscope coupled to a Leica TCS SP2 spectral confocal system/UV. Controls were run to confirm that the staining was dependent on primary antibodies.
Results
EAAC1 associates with PKCα in C6 cells treated with PMA
Activation of PKC increases the activity and cell surface expression of the glutamate transporter EAAC1 (Davis et al., 1998). Two lines of evidence suggested that PKCα is required for EAAC1 redistribution (González et al., 2002). In the present study, we tested for possible interactions between EAAC1 and PKCα. Using an EAAC1-specific antibody, essentially no PKCα immunoreactivity was detected in immunoprecipitates from control cells; however, treatment of C6 glioma with PMA (100 nm) for 30 min consistently resulted in the recovery of PKCα in EAAC1 immunoprecipitates (Fig. 1A). The increase in PKCα immunoreactivity after PMA treatment was not associated with any difference in the amount of EAAC1 recovered in the immunoprecipitates (Fig. 1A, bottom). The specificity of the EAAC1–PKCα interaction is supported by the absence of PKCα immunoreactivity when an irrelevant antibody (anti-GLT-1; ADI) was used or when the EAAC1 antibody was omitted from the immunoprecipitation procedure (Fig. 1A). In addition, preabsorption of the anti-EAAC1 antibody with the corresponding antigenic peptide (ADI) abolished PKCα immunoreactivity (Fig. 1B). Finally, PKCα was coimmunoprecipitated using an anti-EAAC1 antibody obtained from a different source (Fig. 1C). To determine whether EAAC1 specifically associates with PKCα, immunoprecipitates were probed with antibodies to PKCδ, and, under these conditions, no specific PKCδ immunoreactivity was observed (Fig. 1D), suggesting that that PKC activation with PMA induces the formation of a specific EAAC1–PKCα interaction. We also attempted to determine whether PKCϵ is present in EAAC1 immunoprecipitates, but none was detected (data not shown; n = 3). Because the levels of PKCϵ immunoreactivity are relatively low in C6 glioma (González et al., 2002), we cannot conclusively rule out the possibility that there is a complex but that it is not detectable under the current conditions.
Figure 1.

EAAC1 associates with PKCα in C6 glioma cells. A, Coimmunoprecipitation of PKCα with EAAC1 in C6 glioma cells stimulated with PMA. C6 cells were treated with vehicle or PMA (100 nm) for 30 min. Cell lysates were immunoprecipitated (IP) with an anti-EAAC1 antibody, anti-GLT-1 antibody, or no antibody. Immune complexes were recovered and probed for PKCα immunoreactivity (WB: PKCα). B, Effect of EAAC1 antibody preabsorption with the antigenic peptide. EAAC1 antibody (2 μg) was preabsorbed with 25μg of the corresponding peptide for 2 hr at room temperature. Preabsorbed or non-preabsorbed antibody (IP) was incubated overnight with C6 cells lysate. After recovery, immune complexes were assayed for PKCα immunoreactivity. C, Coimmunoprecipitation of PKCα with EAAC1 using a different anti-EAAC1 antibody. Vehicle- and PMA (100 nm)-treated C6 cells lysates were immunoprecipitated (IP) with an EAAC1 antibody from a different source (Dr. Rothstein, Johns Hopkins University, Baltimore, MD), and the immunoprecipitates were assayed for PKCα immunoreactivity. D, PMA treatment does not induce the interaction of EAAC1 with PKCδ. C6 cell lysates were immunoprecipitated with an anti-EAAC1 antibody, anti-GLT-1 antibody, or no antibody (IP). The immune complexes were probed for PKCδ immunoreactivity. All blots were stripped and reprobed for EAAC1 immunoreactivity (WB: EAAC1) and are representative of three independent experiments.
PKC inhibitors prevent the formation of EAAC1–PKCα complexes
The increase in EAAC1 cell surface expression induced by PMA is abolished by either bisindolylmaleimide II (BIS II), a general PKC inhibitor, or by Gö6976, a selective inhibitor of the classical subtypes of PKC at low micromolar concentrations (Davis et al., 1998; González et al., 2002). Therefore, the effects of PKC inhibitors on formation of EAAC1–PKCα complexes were examined. Either BIS II (10 μm) or Gö6976 (10 μm) prevented the PMA-dependent formation of EAAC1–PKCα complexes (Fig. 2A). As a control, we confirmed that these treatments did not affect the levels of EAAC1 in the immunoprecipitates or the levels of PKCα in the cell lysates (Fig. 2A, middle, bottom). Together, these results strongly suggest that PKC activation is required for formation of EAAC1–PKCα complexes. Because PKCα is the only classical (Gö6976-sensitive) PKC detectable in C6 cells (González et al., 2002), these data suggest that PKCα activation is required.
Figure 2.

Effect of PKC antagonists and PDGF on the EAAC1–PKCα interaction. A, EAAC1–PKCα association is blocked by PKC antagonists. C6 cells were preincubated with BIS II (10 μm) or Gö6976 (10 μm) for 10 min before the addition of PMA (100 nm) for 30 min. EAAC1 immunoprecipitates (IP) were analyzed for PKCα immunoreactivity (WB: PKCα). B, EAAC1–PKCα association is not induced by PDGF. C6 cells were incubated for 30 min with PDGF (20 ng/ml) or PMA (100 nm). Cell lysates were immunoprecipitated with an anti-EAAC1 antibody, and the immune complexes were analyzed for PKCα immunoreactivity. Membranes were stripped and probed for EAAC1 immunoreactivity (WB: EAAC1). In the bottom panels, total cell lysates (25 μg of protein) were probed for PKCα immunoreactivity. The blots are representative of three independent experiments.
Previously, we showed that activation of the platelet-derived growth factor (PDGF) receptor increases the activity and cell surface expression of EAAC1 (Sims et al., 2000). These effects are not blocked by the PKC antagonist BIS II and appear to be independent of PKC activation. Treatment of C6 cells with PDGF did not induce formation of EAAC1–PKCα complexes (Fig. 2B), suggesting that formation of the complex is specific and is not simply related to redistribution of EAAC1.
EAAC1 and PKCα colocalize in C6 cells after PKC activation
Based on these studies, it is not clear whether this complex exists within the cell or at the cell surface. In several different experiments, we attempted to combine biotinylation of cell surface proteins with immunoprecipitation, but we were unable to free the biotinylated transporters from avidin beads with excess biotin. Therefore, we used confocal microscopy to determine whether EAAC1 and PKCα colocalize after activation of PKC. In optical cross-sections of the midsection of the cell, the bulk of EAAC1 immunoreactivity (green) was evenly distributed throughout the interior of the cell, and PKCα immunostaining (red) was predominantly detected in a perinuclear compartment under control conditions (Fig. 3). After PMA treatment, there was a decrease in the intracellular labeling for EAAC1 and PKCα, and both proteins were located at or near the plasma membrane, in which they appear to form clusters and colocalize (yellow). As observed previously, PMA treatment also changed the morphology and increased the “ruffling” of the cells (Davis et al., 1998). It is known that activation of PKC with PMA is associated with the translocation of PKC to the cell membrane. Therefore, it is not surprising that these two plasma membrane-trafficked proteins colocalize at the cell membrane after PMA treatment. The fact that we do not observe significant intracellular colocalization, along with the data that support the formation of complexes between these two proteins, is consistent with the notion that EAAC1 and PKCα interact at or near the cell membrane.
Figure 3.
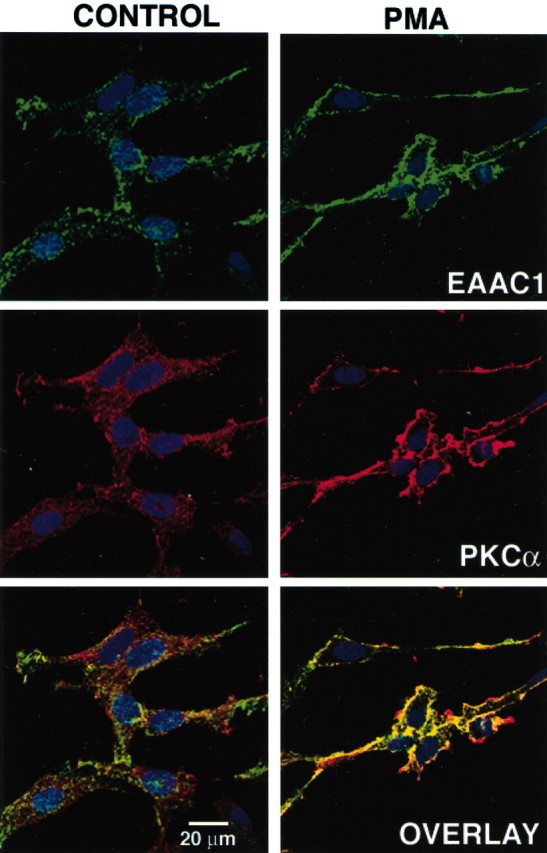
Effect of PMA on EAAC1 and PKCα distribution in C6 cells. After treatment with vehicle or PMA (100 nm) for 30 min, cells were fixed and immunolabeled with rabbit antibodies to EAAC1 (green) and mouse monoclonal antibodies to PKCα (red). Nuclei were stained with Hoechst dye (blue). Serial sections were obtained by confocal microscopy, and optical sections through the middle of the cell, defined by the largest cross-sectional area of the nucleus, are shown. Both control and PMA-treated cells were scanned with the same voltage/offset/pinhole settings and laser power. The images are representative of two independent experiments. In vehicle-treated cultures, the bulk of EAAC1 immunoreactivity is distributed throughout the cytoplasm and PKCα immunoreactivity is predominantly perinuclear. After PMA treatment, the intracellular labeling decreased, and both EAAC1 and PKCα were translocated to the periphery of the cell, where they appear to form clusters and colocalize in the cell surface (yellow). Scale bar, 20μm.
EAAC1 associates with PKCα in brain tissue
To determine whether the association of EAAC1 and PKCα occurs in vivo, EAAC1 was immunoprecipitated from rat cortical synaptosomes. In contrast to our in vitro observations with C6 cells (in which EAAC1–PKCα complexes were not detectable in the absence of PMA), PKCα immunoreactivity was detected in EAAC1 immunoprecipitates in the absence of exogenous PKC activation (Fig. 4A, B). The PKC antagonist BIS II (10 μm) dramatically reduced the amount of PKCα detected, suggesting that endogenous PKC activation causes formation of the complex. The observation that much lower levels of PKCα are detected when synaptosomes are not incubated at 37°C and that this signal increases with length of incubation at 37°C (Fig. 4C) suggests that formation of the complex was triggered during the incubation period. In addition, exogenous activation of PKC with PMA increased the signal for PKCα immunoreactivity, and the PKC antagonist BIS II blocked this effect (Fig. 4A, B). The changes in PKCα immunoreactivity are not produced by a change in the efficiency of the immunoprecipitation, because the amount of EAAC1 immunoreactivity recovered under all conditions was similar (data not shown). To determine whether the formation of EAAC1–PKCα complexes is restricted to cortex, EAAC1 was immunoprecipitated from synaptosomes prepared from several brain regions. PKCα was detected in EAAC1 immunoprecipitates from cerebellum, hippocampus, and midbrain (Fig. 4D). In these regions, application of BIS II (10 μm) reduced the basal complex formation and also prevented the formation of complex induced by exogenous PKC activation. In brainstem, in which the levels of expression for EAAC1 and PKCα were relatively low, PKCα was not detected (data not shown). These results show that the formation of the EAAC1–PKCα complex is detectable in synaptosomes and that endogenous stimuli can also result in formation of this complex.
Figure 4.

Coimmunoprecipitation of EAAC1 with PKCα from rat brain synaptosomes. A, Cortical synaptosomes were preincubated for 5 min at 37°C with BIS II (10 μm) or vehicle, PMA (100 nm) was added, and the incubation was continued for 30 min. Precleared lysate was incubated overnight with 3μg of anti-EAAC1 antibody (IP), and immune complexes were probed for PKCα immunoreactivity (WB: PKCα). B, Densitometric analysis of PKCα immunoreactivity from seven independent experiments performed as described in A. Data are presented as mean ± SEM. *p < 0.05 and ***p < 0.001 compared with PMA treated synaptosomes; **p < 0.001 compared with control synaptosomes; ANOVA. C, Cortical synaptosomes were kept on ice (0 min) or transferred to a 37°C water bath and immediately treated with vehicle or PMA for 15 or 30 min. D, EAAC1 and PKCα interact in different brain regions. Synaptosomes prepared from different brain regions were treated as described in A, and immune complexes were probed for PKCα immunoreactivity. The blots presented are representative of three independent experiments.
Discussion
Previously, we showed that inhibition of PKCα with Gö6976 reduces the PMA-induced increase in EAAC1-mediated activity. Gö6976 abolishes the increase in EAAC1 cell surface expression in C6 glioma and in primary neuronal cultures. In addition, selective downregulation of the PKC subtypes expressed in C6 cells suggests a specific role for PKCα in the regulation of EAAC1 trafficking (González et al., 2002). The specific events involved in EAAC1 redistribution are unknown, but if the involvement of PKCα in EAAC1 redistribution is dependent on phosphorylation of EAAC1 or of an adapter-protein involved in EAAC1 trafficking, the association of EAAC1 and PKCα in a multiprotein complex may be necessary. In C6 cells, treatment with PMA promoted an interaction between EAAC1 and PKCα. This effect was blocked by PKC antagonists, suggesting that the formation of the complex requires PKC activation. In rat brain synaptosomes, the EAAC1–PKCα association was detected in the absence of PMA, and PMA induced an additional increase in the recovery of PKCα immunoreactivity. Both effects were blocked by a PKC antagonist, suggesting that the association may be triggered by endogenous stimulation of PKC activity under physiological and/or pathological conditions.
Several members of the neurotransmitter transporter family are regulated by kinases, phosphatases, and interacting proteins; however, the mechanisms involved in this regulation are unknown. All of the neurotransmitter transporters contain intracellular consensus sequences for phosphorylation, and there is evidence that many members of this family are phosphorylated. In addition, there is some evidence that direct phosphorylation of transporters may be required for the regulation of transporter activity and/or cell surface expression (Blakely and Bauman, 2000; Danbolt, 2001; Foster et al., 2002). Because EAAC1 and PKCα interact, this raises the possibility that, after interaction, PKCα may directly phosphorylate EAAC1 and promote its redistribution. In preliminary studies using C6 cells metabolically labeled with [32P]orthophosphate, we found that PKC activation with PMA promotes the phosphorylation of immunoprecipitable EAAC1, and this effect is blocked by BIS II (data not shown; n = 3), suggesting that PKC phosphorylates EAAC1. However, these data do not prove that direct phosphorylation is required for the regulation of transporter activity or trafficking. An alternative is that PKC phosphorylates an accessory protein that in turn regulates the activity and/or cell surface expression of EAAC1. Finally, it is also possible that the interaction is independent of the phosphorylation of EAAC1 or an adapter protein and that activated PKCα simply has a higher affinity for EAAC1 (or an adapter protein); however, we have not determined whether the PKCα in the complex is catalytically active.
Recently, some proteins that interact with neurotransmitter transporters have been described, and these interactions may either influence PKC signaling or are influenced by PKC signaling. For example, PKC activation modulates an interaction between syntaxin 1A and the γ-aminobutyric acid transporter GAT-1. This interaction appears to regulate both the cellular distribution and catalytic efficiency of GAT-1 (Deken et al., 2000; Horton and Quick, 2001). The PDZ (for postsynaptic density-95/Discs large/zona occludens-1) domain-containing protein PICK1 (for protein interacting with C kinase 1) and the LIM (for Lin-11, Isl-1, and Mec-3) domain-containing protein Hic-5 interact with the dopamine transporter, and these scaffolding proteins may recruit activated PKC and/or focal adhesion kinase to regulate transporter activity or cell surface expression (Torres et al., 2001; Carneiro et al., 2002). Finally, the catalytic subunit of the protein phosphatase 2A has been detected in complexes with the serotonin, norepinephrine, and dopamine transporters. PKC activation results in phosphorylation and sequestration of the serotonin transporter and disrupts the interaction of protein phosphatase 2A with this transporter (Bauman et al., 2000). Despite these efforts, there is currently no evidence that PKC is part of these complexes.
Some proteins that interact with glutamate transporters have also been described recently. These proteins include a member of the LIM family, Ajuba, that associates with GLT-1 (Marie et al., 2002). GTRAP3–18 (glutamate transporter associated protein 3–18) (Lin et al., 2001) and GTRAP48 (Jackson et al., 2001) interact with and regulate the activities of EAAC1 and EAAT4, respectively. In the present study, we describe a novel interaction between the glutamate transporter EAAC1 and PKCα. Although we have evidence that PKCα activation may be required for EAAC1 redistribution (González et al., 2002), at present it is not clear how this interaction might regulate EAAC1 trafficking. Because most cell types express multiple PKC subtypes, it is possible that the interaction simply permits specific regulation of EAAC1 by PKCα. It is also possible that the interaction may alter the rate of recycling and stabilize EAAC1 at the cell membrane. Finally, more complicated models are suggested by the observation that PKCα activation alters both the redistribution of β1-integrin to the cell surface and its endocytosis, with both effects apparently dependent on PKCα–β1-integrin association (Ng et al., 1999).
Activation of PKCα has also been implicated in the regulation of cell surface levels of AMPA receptors containing the GluR2 subunit. After PMA application, activated PKCα is directed to neuronal spines in which GluR2-containing AMPA receptors are located. Here, PKCα and GluR2 interact and AMPA receptors are internalized after phosphorylation. A reduction in the number of postsynaptic AMPA receptors has been implicated as one of the crucial factors for the induction of long-term depression (Barry and Ziff, 2002; Malinow and Malenka, 2002). In hippocampus, GluR2 and EAAC1 are present in the dendritic membranes. Although EAAC1 is not intermixed with GluR2, it is present perisynaptically, immediately outside the synaptic specialization ideally located to restrict the action of glutamate within the precise site of synaptic communication (He et al., 2000). In fact, these transporters are thought to limit the “spillover” between synapses in area CA1 of the hippocampus (Diamond, 2001), and regulation of glutamate transporters has been implicated in long-term potentiation and long-term depression (Brasnjo and Otis, 2001; Levenson et al., 2001). These observations raise the possibility that concerted regulation of receptors and transporters may be a necessary step for the induction of synaptic plasticity. Because activation of the same signaling molecule promotes the redistribution of both GluR2-containing AMPA receptors and the neuronal transporter EAAC1, it is possible that coordinate regulation of transporters and receptors may be required to trigger synaptic plasticity.
Footnotes
This work was supported by National Institutes of Health Grants NS29868 and NS39011. We thank Dr. Jeff Rothstein for generously providing anti-EAAC1 antibody.
Correspondence should be addressed to Dr. Michael B. Robinson, 502N Abramson Pediatric Research Building, 3615 Civic Center Boulevard, Philadelphia, PA 19104-4318. E-mail: robinson@pharm.med.upenn.edu.
Parts of this paper have been published previously in abstract form (González and Robinson, 2002).
Copyright © 2003 Society for Neuroscience 0270-6474/03/235589-05$15.00/0
References
- Barry MF, Ziff EB ( 2002) Receptor trafficking and the plasticity of excitatory synapses. Curr Opin Neurobiol 12: 279–286. [DOI] [PubMed] [Google Scholar]
- Bauman AL, Apparsundaram S, Ramamoorthy S, Wadzinski BE, Vaughan RA, Blakely RD ( 2000) Cocaine and antidepressant-sensitive biogenic amine transporters exist in regulated complexes with protein phosphatase 2A. J Neurosci 20: 7571–7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakely RD, Bauman AL ( 2000) Biogenic amine transporters: regulation in flux. Curr Opin Neurobiol 10: 328–336. [DOI] [PubMed] [Google Scholar]
- Brasnjo G, Otis TS ( 2001) Neuronal glutamate transporters control activation of postsynaptic metabotropic glutamate receptors and influence cerebellar long-term depression. Neuron 31: 607–616. [DOI] [PubMed] [Google Scholar]
- Carneiro AM, Ingram SL, Beaulieu J-M, Sweeney A, Amara SG, Thomas SM, Caron MG, Torres GE ( 2002) The multiple LIM domain-containing adaptor protein Hic-5 synaptically colocalizes and interacts with the dopamine transporter. J Neurosci 22: 7045–7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW ( 1992) Excitotoxic cell death. J Neurobiol 23: 1261–1276. [DOI] [PubMed] [Google Scholar]
- Conti F, Weinberg RJ ( 1999) Shaping excitation at glutamatergic synapses. Trends Neurosci 22: 451–458. [DOI] [PubMed] [Google Scholar]
- Danbolt NC ( 2001) Glutamate uptake. Prog Neurobiol 65: 1–105. [DOI] [PubMed] [Google Scholar]
- Davis KE, Straff DJ, Weinstein EA, Bannerman PG, Correale DM, Rothstein JD, Robinson MB ( 1998) Multiple signaling pathways regulate cell surface expression and activity of the excitatory amino acid carrier 1 subtype of Glu transporter in C6 glioma. J Neurosci 18: 2475–2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deken SL, Beckman ML, Boos L, Quick MW ( 2000) Transport rates of GABA transporters: regulation by the N-terminal domain and syntaxin 1A. Nat Neurosci 3: 998–1003. [DOI] [PubMed] [Google Scholar]
- Diamond JS ( 2001) Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA-1 pyramidal cells. J Neurosci 21: 8328–8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JD, Pananusorn B, Vaughan RA ( 2002) Dopamine transporters are phosphorylated on N-terminal serines in rat striatum. J Biol Chem 277: 25178–25186. [DOI] [PubMed] [Google Scholar]
- González MI, Robinson MB ( 2002) Interaction of protein kinase Cα with the glutamate transporter EAAC1. Soc Neurosci Abstr 28: 441.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González MI, Kazanietz MG, Robinson MB ( 2002) Regulation of the neuronal glutamate transporter excitatory amino acid carrier-1 (EAAC1) by different protein kinase C subtypes. Mol Pharmacol 62: 901–910. [DOI] [PubMed] [Google Scholar]
- He Y, Janssen WGM, Rothstein JD, Morrison JH ( 2000) Differential synaptic localization of the glutamate transporter EAAC1 and glutamate receptor subunit GluR2 in the rat hippocampus. J Comp Neurol 418: 255–269. [PubMed] [Google Scholar]
- Horton N, Quick MW ( 2001) Syntaxin 1A up-regulates GABA transporter expression by subcellular redistribution. Mol Membr Biol 18: 39–44. [PubMed] [Google Scholar]
- Jackson M, Song W, Liu M-Y, Jin L, Dykes-Hoberg M, Lin C-LG, Bowers WJ, Federoff HJ, Sternwels PC, Rothstein JD ( 2001) Modulation of the neuronal glutamate transporter EAAT4 by two interacting proteins. Nature 410: 89–93. [DOI] [PubMed] [Google Scholar]
- Levenson J, Weeber E, Selcher JC, Kategaya LS, Sweatt JD, Eskin A ( 2001) Long-term potentiation and contextual fear conditioning increase neuronal glutamate uptake. Nat Neurosci 5: 155–161. [DOI] [PubMed] [Google Scholar]
- Lin C-LG, Orlov I, Ruggiero AM, Dykes-Hoberg M, Lee A, Jackson M, Rothstein JD ( 2001) Modulation of the neuronal glutamate transporter EAAC1 by the interacting protein GTRAP3–18. Nature 410: 84–88. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC ( 2002) AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci 25: 103–126. [DOI] [PubMed] [Google Scholar]
- Marie H, Billups D, Bedford FK, Dumoulin A, Goyal RK, Longmore GD, Moss SJ, Attwell D ( 2002) The amino terminus of the glial glutamate transporter GLT-1 interacts with the LIM protein Ajuba. Mol Cell Neurosci 19: 152–164. [DOI] [PubMed] [Google Scholar]
- Ng T, Shima D, Squire A, Bastiaens PIH, Gschmeissner S, Humphries MJ, Parker PJ ( 1999) PKCα regulates β1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J 18: 3909–3923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MB ( 1998) Examination of glutamate transporter heterogeneity using synaptosomal preprations. Methods Enzymol 296: 189–202. [DOI] [PubMed] [Google Scholar]
- Sims KD, Robinson MB ( 1999) Expression patterns and regulation of glutamate transporters in the developing and adult nervous system. Crit Rev Neurobiol 13: 169–197. [DOI] [PubMed] [Google Scholar]
- Sims KD, Straff DJ, Robinson MB ( 2000) Platelet-derived growth factor rapidly increases activity and cell surface expression of the EAAC1 subtype of glutamate transporters through activation of phosphatidylinositol 3-kinase. J Biol Chem 274: 5228–5327. [DOI] [PubMed] [Google Scholar]
- Torres GE, Yao W-D, Mohn AR, Quan H, Kim K-M, Levey AI, Staudinger J, Caron MG ( 2001) Functional interaction between monoamine plasma membrane transporters and the synaptic PDZ domain-containing protein PICK1. Neuron 30: 121–134. [DOI] [PubMed] [Google Scholar]
- Wadiche JI, Arriza JL, Amara SG, Kavanaugh MP ( 1995) Kinetics of a human glutamate transporter. Neuron 14: 1019–1027. [DOI] [PubMed] [Google Scholar]