

Abstract
The human immunodeficiency virus type 1 (HIV-1) envelope protein gp120 has been implicated in the pathogenesis of HIV-1 dementia. Thus, inhibition of gp120 activity could reduce HIV toxicity in the brain. We have used primary cultures of rat cerebellar granule cells to examine mechanisms whereby gp120 causes cell death and to characterize neuroprotective agents. gp120 induced a time- and concentration-dependent apoptotic cell death, which was caspase-3-mediated but caspase-1 independent, and was totally blocked by the irreversible caspase-3-like protease inhibitor N-acetyl-Asp-Glu-Val-Asp-chloromethylketone. Caspase-3 activation was observed only in neurons that internalize gp120, indicating that internalization is key to gp120 toxicity. Because brain-derived neurotrophic factor (BDNF) prevents caspase-3-mediated neuronal cell death, we examined whether BDNF could prevent gp120-mediated apoptosis. Preincubation of neurons with BDNF before the addition of gp120 reduced caspase-3 activation, and consequently rescued 80% of neurons from apoptosis. Most importantly, BDNF reduced the levels of CXC chemokine receptor-4 (CXCR4), a receptor that mediates HIV-1 gp120-induced apoptosis. This effect correlated with the ability of BDNF to reduce gp120 internalization and apoptosis. Moreover, BDNF blocked the neurotoxic effect of stromal-derived factor-1α, a natural ligand for CXCR4, further establishing a correlation between neuroprotection and downregulation of CXCR4. We propose that BDNF may be a valid therapy to slow down the progression of HIV/gp120-mediated neurotoxicity.
Keywords: apoptosis, BDNF, caspase-3, CXCR4, CCR5, SDF-1α
Introduction
A large percentage of patients suffering from acquired immunodeficiency syndrome (AIDS) develop human immunodeficiency virus type 1 (HIV-1)-related cognitive-motor complex, a neurological disease characterized by dementia, memory deficits, and motor impairment (Brew et al., 1995; Price, 1996). These behavioral symptoms appear to be the direct consequence of neuronal cell loss, dendritic simplification, and decreased synaptic density (Masliah et al., 1997). Several studies have indicated that the viral envelope glycoprotein, gp120, which is shed from HIV-infected macrophages during viral replication, may be the etiological agent of neuronal loss observed in postmortem brains of HIV-positive patients. Indeed, gp120 causes cell death in various neuronal populations in vitro, including midbrain dopaminergic neurons, hippocampal, cortical, and cerebellar granule neurons (Lipton et al., 1991; Dawson et al., 1993; Savio and Levi, 1993; Bennett et al., 1995; Meucci and Miller, 1996; Kaul and Lipton, 1999) as well as in vivo in rodent brains (Bagetta et al., 1996; Bansal et al., 2000).
Little is known about the mechanisms whereby gp120 induces neuronal cell death and related treatments to block this effect. Recent evidence has shown that gp120-mediated cytotoxicity may occur via apoptosis through interaction with the seven-transmembrane domain chemokine receptor CXC chemokine receptor-4 (CXCR4) (Davis et al., 1997; Herbein et al., 1998; Hesselgesser et al., 1998; Klein et al., 1999; Kozak et al., 1999). Several lines of independent investigations have shown that this apoptotic cell death is triggered by activation of the proapoptotic protease caspase-3. In fact, gp120 activates caspase-3 in human embryonic kidney (Biard-Piechaczyk et al., 2000), endothelial cells (Ullrich et al., 2000), and rat cerebellar granule cells (Bachis et al., 2000). Moreover, in cerebrocortical cultures, inhibition of caspase-3 prevents gp120-induced apoptosis (Garden et al., 2002). These findings are important, considering that inhibitors of caspase-3 activity can rescue neurons from the apoptotic cycle. Neurotrophic factors are a class of such inhibitors. In particular, brain-derived neurotrophic factor (BDNF), one member of the neurotrophin family of multifunctional neurotrophic factors capable of regulating neuronal development and survival (Reichardt, 2001), has been shown to prevent ischemia and glutamate-mediated neuronal apoptosis by inhibiting caspase-3 activation (Han et al., 2000; Bachis et al., 2002). Thus, it seems conceivable that this trophic factor may prevent gp120-mediated apoptosis and therefore rescue neurons from programmed cell death.
The aim of the present study was to investigate whether gp120 evokes caspase-3-dependent apoptotic cell death of postnatal CNS neurons such as cerebellar granule cells, and if so, whether BDNF rescues these neurons. We report a new neuroprotective property of BDNF and the potential molecular and cellular mechanisms of this effect.
Materials and Methods
Cell culture. Cerebellar granule cells were prepared from 8-d-old Sprague Dawley rat pups (Taconic Farms, Germantown, NY) as described previously (Brandoli et al., 1998; Bachis et al., 2001). Briefly, neurons were plated onto poly-l-lysine (1%)-precoated 100 mm plastic dishes at a density of 2.5 × 10 6 cells/ml and grown in basal medium Eagle (Invitrogen, Grand Island, NY) containing 2 mm glutamine, 10% fetal calf serum, 25 mm KCl, 100 μg/ml gentamicin, and 10,000 U/ml penicillin–streptomycin. Cells were maintained at 37°C in 5% CO2 and 95% O2. Cytosine arabinoside (10 μm) was added 24 hr after cell plating to inhibit glial proliferation. At the time of the experiments, these cultures were composed of ∼96% neurons and ∼4% non-neuronal cells, such as astrocytes, oligodendrocytes, and endothelial cells. Compounds were added to the cultures at 8 d in vitro. After the addition of gp120 or other compounds, cultures were kept in the same medium until analysis of cell viability. Sister cultures that received medium containing heat-inactivated gp120 were used as a control.
Compounds and chemokines. Human recombinant BDNF, neurotrophin-3 (NT-3), and nerve growth factor (NGF) were all purchased from Promega Biotechnologies (Madison, WI). gp120 (SF2 and IIIB strains) and AMD3100 (AMD) were obtained from the National Institutes of Health AIDS Research and Reference Reagent Program. Stromal-derived factor (SDF)-1α, was obtained from Sigma (St. Louis, MO) and N-acetyl-Asp-Glu-Val-Asp-chloromethylketone (DEVDK) was purchased from Calbiochem (La Jolla, CA).
Cell survival. The percentage of surviving neurons in the presence of trophic factors and/or gp120 was estimated using the biochemical and histological assays listed below.
MTT. The activity of mitochondrial dehydrogenases [3(4,5-dimethylthiazol-2-yl)-2.5-diphenyltetrazolium bromide] (MTT) assay was used to determine cell death/survival. This assay was carried out according to the manufacturer's specifications (MTT Kit I, Boehringer Mannheim, Indianapolis, IN) as described previously (Bachis et al., 2001, 2002).
Hoechst 33342/propidium iodide. Cultures were incubated simultaneously with 5 μm Hoechst 33342 (staining healthy cells) and 5 μg/ml propidium iodide (staining apoptotic and necrotic cells) (both from Sigma) (Hoechst/PI), for 15 min in a water bath at 37°C. Reaction was visualized with the Nikon (Tokyo, Japan) inverted fluorescent microscope ECLIPSE TE300.
TUNEL. Apoptotic neurons were examined by in situ terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL), and activated caspase-3 as described previously (Bachis at al., 2001, 2002). In brief, neurons were plated onto 12 mm round, 1 mm thick precoated glass coverslips. Cells were fixed with 4% paraformaldehyde, postfixed in 2:1 ethanol:acetic acid, washed, and equilibrated according to the instructions of the manufacturer (ApopTag; Serological Corporation, Norcross, GA). Neurons were then incubated with terminal deoxynucleotidyl transferase enzyme in the presence of digoxigenin-labeled deoxyNTP, followed by anti-digoxigenin (fluorescein conjugate) antibody and mounted using Vectashield Mounting Medium with 4′,6′-diamidino-2-phenylindole (DAPI) or propidium iodide (Vector Laboratories, Burlingame, CA) as counterstaining. Reaction was visualized with the ECLIPSE TE300 inverted fluorescent microscope.
Analysis of gp120 internalization by confocal microscopy. Cerebellar granule cells were exposed to biotin-conjugated gp120 (bcgp120, IIIB strain; Immunodiagnostics, Woburn, MA) for different times, washed, and then fixed with 4% paraformaldehyde for 15 min. To determine which cell type internalizes gp120, cultures were immunostained overnight with cell-type-specific antibodies recognizing glial fibrillary acidic protein (GFAP, 1:250 dilution) for astrocytes, myelin/oligodendrocyte specific protein (1:100) for oligodendrocytes (all from Chemicon, Temecula, CA), combined with NeuroTrace red fluorescent Nissl stain (1: 150; Molecular Probes, Eugene, OR) for 20 min at room temperature. Cells were then incubated with 7-amino-4-methylcoumarin-3-acetic acid secondary antibody (1:500 dilution; Vector Laboratories) to detect GFAP or other markers of non-neuronal cells for 4 hr at 4°C. Bcgp120 was visualized by fluorescein–streptavidin (1:1000 dilution; Vector Laboratories). In addition, cells were stained with an antibody against microtubule-associated protein-2 (MAP-2; 1:500; Chemicon) to reveal neuronal cell bodies and processes. Cells were then incubated with a Texas-red-conjugated secondary antibody (Vector Laboratories), and bcgp120 was visualized by fluorescein–streptavidin. Coverslips were then mounted using Prolong antifade kit (Molecular Probes) or Vectashield mounting medium with DAPI. Cells were imaged by confocal attofluor ratio vision spinning disk confocal microscopy (ATTO Bioscience, Rockville, MD) mounted on Olympus IX17 inverted microscope (Olympus Optical, Tokyo, Japan), using a blue DAPI filter (excitation, 360 nm; emission, 470 nm), a green FITC filter (excitation, 488 nm; emission, 530 nm), and a Texas red filter (excitation, 570 nm; emission, 645 nm). Images were captured and analyzed using the MetaMorph Imaging System (Universal Imaging, Downingtown, PA).
Caspase-3 immunocytochemistry. For neurofilament and caspase-3 staining, cultures were fixed with 4% paraformaldehyde and incubated overnight with an antibody against mouse 160 kDa neurofilament (1: 350; Sigma) and a cleaved-caspase-3 antibody (1:150 dilution; Cell Signaling, Beverly, MA) for 48 hr at 4°C. Cells were washed and then incubated with fluorescein and Texas red secondary antibodies. Coverslips were then mounted using Vectashield mounting medium with DAPI. For caspase-3 and gp120 internalization, neurons exposed to bcgp120 were incubated with a cleaved caspase-3 antibody followed by fluorescein–streptavidin and Texas red secondary antibody to visualize gp120 and caspase-3, respectively. Immunofluorescence was analyzed with a Nikon (Tokyo, Japan) ECLIPSE TE300 inverted fluorescent microscope. Optronics Magnafire software (Optronics, Goleta, CA) was used to analyze immunoreactive cells.
Caspase-3- and caspase-1-like activity. Neurons were plated onto 100 mm dishes. Caspase-3- and caspase-1-like activities were measured in cell lysates using the caspase-3 and caspase-1 colorimetric assay protease kits (Chemicon) following the manufacturer's instructions. In brief, neurons were lysed for 10 min in ice-cold lysis buffer, containing the following: 150 mm NaCl, 20 mm Tris HCl, pH 7.2, 1% Triton X-100, and 1 mm dithiothreitol. After removal of cellular debris by centrifugation, protein levels in the lysates (cytosolic extract) were measured by the Bradford Coomassie blue colorimetric assay (Bio-Rad, Hercules, CA) and equalized accordingly to obtain 150 μg of cytosolic extract per sample. For caspase-3, samples were incubated with 200 μm caspase-3 substrate N-acetyl-Asp-Glu-Val-Asp-p-nitroanilide at 37°C for 2 hr. For caspase-1, samples were incubated with 200 μm of caspase-1 substrate Tyr-Val-Ala-Asp-p-nitroanilide at 37°C for 2 hr. Samples were analyzed at 400 nm in a microtiter plate reader.
Western blot. Cerebellar granule cells were washed twice in phosphate buffer, harvested in 50 mm Tris HCl, pH 7.0, and pelleted at 3000 × g. Pellets were homogenized in the same buffer with a polytron, centrifuged at 100,000 × g for 20 min, resuspended in 1 ml of Tris HCl, and stored at –20°C. After removal of cellular debris by centrifugation, protein levels in the lysates were measured by the Bradford colorimetric assay. Equal amounts of proteins were loaded onto a 15% SDS-polyacrylamide gel. Proteins were transferred on a nitrocellulose membrane and blocked with Tris-buffered saline (TBS-T; 25 mm Tris and 1% Tween) containing 5% milk powder. Blots were incubated overnight with rabbit affinity-purified polyclonal antibodies anti-CXCR4 or anti-CC-chemokine receptor-5 (CCR5) (1:1000 dilution; Chemicon) in TBS-T plus 5% milk. After several washes with TBS-T, blots were then incubated with the secondary antibody, peroxidase-conjugated anti-rabbit IgG (dilution 1:10,000; Santa Cruz Biotechnology, Santa Cruz, CA). Immunoreactivity was detected by enhanced chemiluminescence (Amersham Biosciences, Arlington Heights, IL). The blot was stripped and reprobed with an antibody against the 200 kDa form of neurofilament (1:1000; Chemicon) to demonstrate that an equal amount of proteins were present in each lane.
Data analysis. Hoechst/PI-, TUNEL-, and caspase-3-positive cells were counted in randomly selected fields using a 40× objective and Optronics Magnafire software. Approximately 1500 nuclei were counted per condition in each experiment, and experiments were repeated at least three times. Because the number of TUNEL- or caspase-3-positive cells in cultures exposed to medium alone (70 ± 10) or heat-inactivated gp120 (72 ± 9) was not significantly different, control cells were defined as those exposed to heat-inactivated gp120. Data are expressed as a percentage of control (means ± SEM). Statistical analysis was performed using ANOVA and the Scheffé test for multiple comparisons. Semiquantitative analysis of CXCR4, CCR5, and neurofilament immunoreactivity was performed by Western blot analysis. Immunoreactive bands on the autoradiograph were scanned with a laser densitometer as described previously (Brandoli et al., 1998). The value of CXCR4/CCR5 immunoreactivity was divided by that of neurofilament and expressed as a percentage of control.
Results
gp120 causes neuronal cell death in the absence of glia
In the CNS, gp120 toxicity appears to depend predominantly on the activation of microglia chemokine receptors rather than on a direct effect on neurons (Kaul and Lipton, 1999; Bezzi et al., 2001). To test this hypothesis we carried out time- and dose-dependent studies of gp120 toxicity using primary cultures of cerebellar granule cells from 8-d-old rat pups. This neuronal preparation contains essentially neurons because it is grown in the presence of cytosine arabinoside and glial cells are present only as a contaminant (Brandoli et al., 1998; Bachis et al., 2001). Neurons were exposed to control medium containing heat-inactivated gp120 or to medium containing various concentrations of gp120; then cell survival was measured by MTT assay, Hoechst/PI staining and in situ TUNEL 24 hr later. gp120 evoked a dose-dependent increase in MTT activity starting from a concentration of 200 pm (Fig. 1A). The increase in MTT activity correlated with the increased number of TUNEL- and Hoechst/PI-positive cells (Fig. 1A). To establish the temporal profile of this effect, neurons were exposed to gp120 (5 nm) for various times. gp120 induced a time-dependent decrease in cell viability measured by all three assays starting at 12 hr and culminating at 48 hr (Fig. 1B). Thus, gp120 causes cell death even when cultures contain few glial cells.
Figure 1.
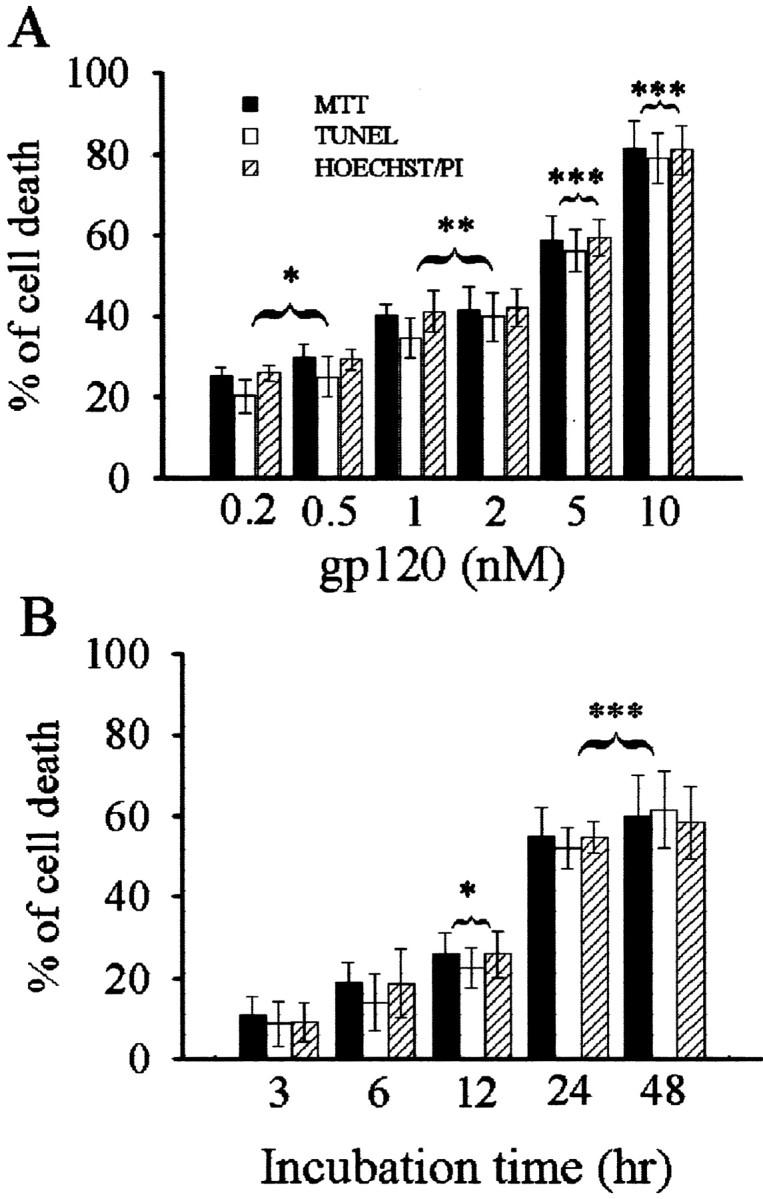
gp120 induces neuronal cell death. Cerebellar granule cells were exposed for 24 hr to gp120 at the indicated doses (A) or to 5 nm gp120 for the indicated times (B). Control cells were exposed to heat-inactivated gp120. Cell survival was analyzed by MTT and TUNEL assays, and the number of dead cells was determined by Hoechst/PI staining. Neurons exposed to medium alone or to inactivated gp120 showed a similar number of cell deaths (5%). *p < 0.05, **p < 0.01, and ***p < 0.001 versus control (n = 12 each point).
gp120 activates caspase-3
The concordance of cell death measured by both TUNEL and MTT assays is remarkable, suggesting that the main form of cell death evoked by gp120 is apoptosis. However, TUNEL alone may not distinguish between necrosis and apoptosis. Thus, to confirm apoptosis, we examined whether gp120 activates caspases. Exposure of cerebellar granule cells to gp120 (5 nm) evoked a significant increase in caspase-3-like activity within 3 hr, an effect that peaked between 6 and 12 hr (Fig. 2A). Caspase-1 activity was not affected at any time examined (Fig. 2A). Inhibition of caspase activity with a relatively specific, irreversible caspase-3-like protease inhibitor, DEVDK (100 μm), prevented gp120-mediated neuronal cell death (Fig. 2B). These data confirm that gp120-mediated neurotoxicity involves a caspase-3-dependent pathway.
Figure 2.
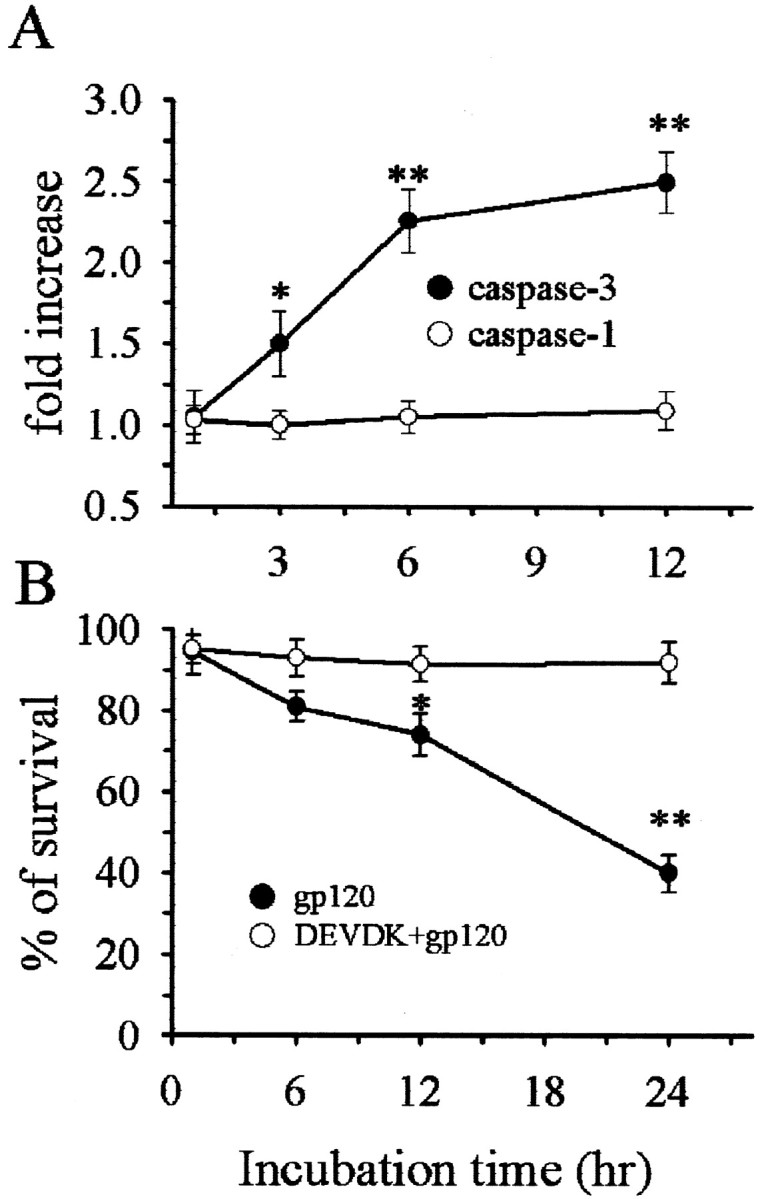
Caspase-3 but not caspase-1 is induced by gp120. A, Cerebellar granule cells were exposed to gp120 (5 nm) and then caspase-3 and caspase-1 activity were measured at the indicated times. B, Neurons were exposed to DEVDK 5 min before gp120 and cell survival measured by counting Hoechst/PI-positive cells at the indicated times. *p < 0.05 and **p < 0.001 versus control (n = 9 each point).
gp120 is internalized primarily by neurons
In non-neuronal cells, internalization of gp120 has been shown to be a prerequisite for its cytotoxic activity (Cefai et al., 1992; Misse et al., 1999). However, primary cultures of cerebellar granule cells contain few non-neuronal cells. To examine whether these non-neuronal components may mediate the neurotoxic effect of gp120, we first determined which cell type internalizes gp120 by confocal microscopy. To better visualize gp120 internalization, cultures were exposed to biotin-conjugated gp120 (bcgp120, 5 nm) for 6 or 12 hr, fixed, and then incubated with fluorescein–streptavidin to detect gp120 in combination with selected markers for neuronal and non-neuronal cells. Analysis of confocal images revealed that bcgp120 immunofluorescence was associated with Nissl-positive neurons only (Fig. 3A). Indeed, GFAP-positive cells, as well as other non-neuronal cells (data not shown), were gp120-negative (Fig. 3A). To test whether neurons internalize gp120, cell body and neuronal processes were visualized using a MAP-2 antibody followed by fluorescein–streptavidin to detect gp120. Cell bodies (Fig. 3B) of MAP-2 positive cells were also gp120-positive. Three-dimensional reconstruction of confocal images revealed that gp120 positivity was inside of MAP-2 positive cells, suggesting that this viral protein, in our experimental conditions, is internalized by neurons.
Figure 3.

gp120 is internalized by neurons. Confocal microscopic analysis of cerebellar granule cells exposed to bcgp 120.A, Cultures were exposed to bcgp 120 for 12 hr and then stained for GPAF (blue) and Nissl (red). gp120 was visualized using fluorescein streptavidin (green). gp120 is only in Nissl-positive cells (20× magnification). B, Cerebellar granule cells were exposed to bcgp120 for 3 hr. Cells were stained for MAP-2 (red). Coverslips were the mounted using DAPI (blue). gp120 was visualized using fluorescein streptavidin (green). gp120 (yellow, overlay red and green) is found primarily in the cytoplasm around the nucleus (60× magnification).
Caspase-3 activation occurs in neurons internalizing gp120
To determine whether the neurotoxic effect of gp120 occurs in cells that internalize this protein, we examined whether neurons internalizing gp120 were also positive for activated caspase-3. Cultures were exposed to inactivated bcgp120 (Fig. 4A) or bcgp120 (Fig. 4B) for 6 hr, fixed, and analyzed for gp120 and activated caspase-3. At least 80% of neurons exposed to gp120 were both gp120- and caspase-3-positive. To examine whether internalization of gp120 is the cause or the effect of cell death, cultures were exposed to bcgp120 for various times and then double-labeled for gp120 and activated caspase-3. Neuronal internalization of gp120 started as early as 30 min and was statistically significant by 1 hr when no neurons were caspase-3-positive (Fig. 4C). By 3 hr, internalization was observed in a greater number of neurons (∼50%). At this time, at least 20% of neurons were also caspase-3-positive (Fig. 4C). Thus, internalization of gp120 occurs at least 1 hr before caspase-3 activation. Together, the apoptotic evidence and internalization studies suggest that granule cell neurons are the major target of gp120.
Figure 4.
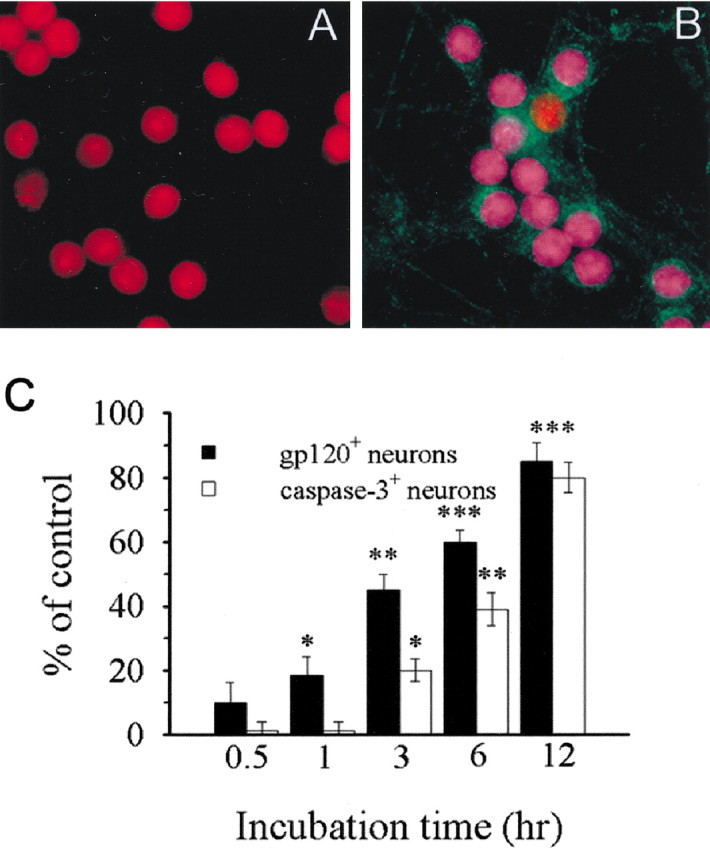
gp120 internalization precedes caspase-3 activation. Cerebellar granule cells were exposed to inactivated gp120 (A) or gp120 (B) for 6 hr and then stained for gp120 (green) and cleaved caspase-3 (blue). After that, Nissl staining was performed (red). Purple (overlay blue and red) indicates caspase-3-positive neurons (20× magnification). C, Graphic representation of the number of caspase-3- and gp120-positive cells. Cultures were exposed to gp120 for the indicated times and then analyzed for cleaved-caspase-3 and gp120 immunoreactivity. The images were captured and the number of gp120- and caspase-3 positive cells counted. The percentage of caspase-3-positive neurons increases with time after exposure to gp120. *p < 0.05, **p < 0.01, and ***p < 0.001 versus control (n = 12 each time point).
BDNF prevents gp120 neurotoxicity
BDNF is a neurotrophic factor that prevents caspase-3-mediated apoptosis in cerebellar granule cells in vitro (Bachis et al., 2002) and in the brain in vivo (Han et al., 2000). Therefore, we determined whether BDNF could prevent gp120-mediated apoptosis. Cultures were exposed to BDNF (50 ng/ml) for 12 hr before the addition of gp120 (5 nm) and then fixed 12 hr later for immuno-histochemical detection of neurofilament and activated caspase-3. Activated caspase-3 was undetectable in control cells (Fig. 5A) but was easily detected in at least 80% of neurons exposed to gp120 (Fig. 5B). In cultures exposed to BDNF before gp120, few neurons were positive for activated caspase-3 (Fig. 5C). To establish the temporal profile of the neuroprotective effect of BDNF, neurons were exposed to BDNF concomitantly or several hours before gp120 and cell death was measured by caspase-3 activation 6 hr later or by Hoechst/PI staining 24 hr later. BDNF evoked a time-dependent reduction of gp120-mediated caspase-3-like activity (Fig. 5D) as well as cell death (Fig. 6). Maximal neuroprotection was obtained when BDNF was added several hours before gp120. Indeed, pretreatment of neurons with BDNF for 24 hr before gp120 abolished gp120-mediated neurotoxicity (Figs. 5D, 6). Instead, when BDNF was added concomitantly with gp120 or 3 hr before gp120, no neuroprotection was observed (Figs. 5D, 6). The effect of BDNF was specific because neither NT-3 nor NGF prevented gp120-mediated cell death (data not shown).
Figure 5.

BDNF prevents gp120 activation of caspase-3. Cultures were exposed to inactivated gp120 for 6 hr (A), gp120 for 6 hr (B), or BDNF (50 ng/ml) 12 hr (C) before gp120. Cells were then fixed and stained for cleaved caspase-3 (red) and neurofilament (green). Cells were counterstained using DAPI (blue). Purple (overlay blue and red) indicates caspase-3 positive neurons. Scale bar, 35 μm. D, Cultures were exposed to BDNF concomitantly (0) or before gp120 for the indicated times. Caspase-3 activity was measured in cell lysates 6 hr after gp120. *p < 0.05 and **p < 0.001 versus gp120 (n = 9 each time point).
Figure 6.

BDNF inhibits gp120 neurotoxicity. Cultures were exposed to BDNF (50 ng/ml) concomitantly (0) or before gp120 for the indicated times. Cell survival was measured by Hoechst/PI staining 24 hr after gp120. *p < 0.05 and **p < 0.001 versus gp120 (n = 9 each time point).
BDNF decreases gp120 internalization by decreasing CXCR4 receptor levels
BDNF may inhibit gp120 neurotoxicity by interacting with gp120 intracellular signal(s) or with the primary event of gp120 cellular entry. Thus, we examined whether BDNF could reduce internalization of gp120. Cerebellar granule cells were exposed to BDNF for different times before the addition of bcgp120; internalization and caspase-3 immunoreactivity were then determined 6 hr later. In cells pretreated with BDNF, internalization of gp120 and caspase-3 activity were drastically reduced (Fig. 7B) compared with gp120-treated cells (Fig. 7A), supporting the hypothesis that inhibition of gp120 internalization reduces apoptosis.
Figure 7.
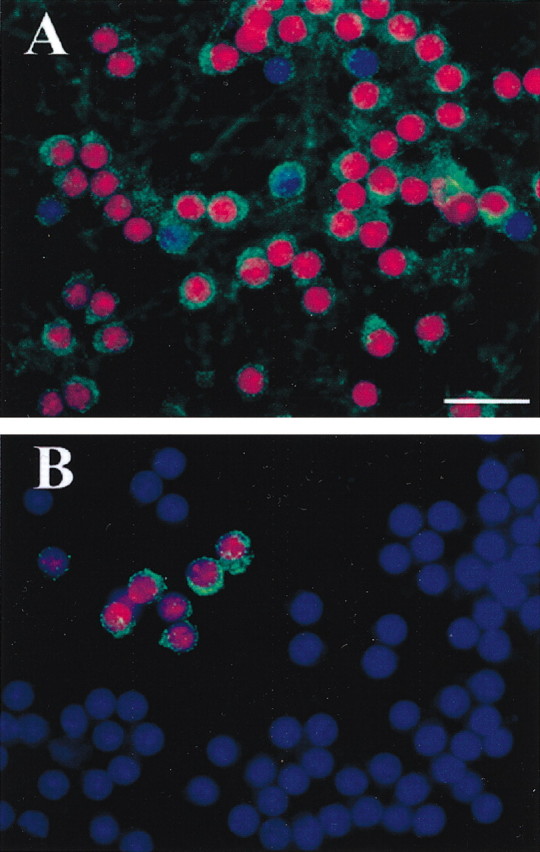
BDNF prevents gp120 internalization. Neurons were exposed to bcgp120 for 6 hr (A) or BDNF for 12 hr (B) before the addition of bcgp120. gp120 internalization (green) and cleaved caspase-3 immunoreactivity (red) were determined 6 hr later. Cells were counterstained using DAPI (blue). In BDNF-treated cultures the number of neurons positive for caspase-3 (purple, overlay blue and red) and gp120 is drastically reduced. Scale bar, 25 μm.
HIV-1, through gp120, binds to CD4 and to members of the chemokine receptor family, such as CXCR4 and CCR5, which act as co-receptors and allow viral entry and, consequently, the lethal infection of the cells (Tarasova et al., 1998; Wang et al., 1998; van der Meer et al., 2000). Thus, we hypothesized that BDNF could reduce the synthesis of CXCR4, the receptor involved in gp120-mediated apoptosis in various non-neuronal cells (Herbein et al., 1998; Hesselgesser et al., 1998; Klein et al., 1999; Kozak et al., 1999; Biard-Piechaczyk et al., 2000). We first examined the effect of BDNF on CXCR4 because the gp120 SF2 and IIIB strains of HIV-1 used in this study interact preferentially with CXCR4 receptors (Weissman et al., 1997). Western blot analyses revealed that BDNF evoked a time-dependent reduction of CXCR4 immunoreactivity beginning at 4 hr and continuing at least up to 12 hr (Fig. 8). Stripping and reprobing blots with an antibody against the 200 kDa form of neurofilament (NF) revealed no changes in the levels of this protein (Fig. 8A), suggesting that the effect of BDNF on CXCR4 is not attributable to a generalized decrease of protein levels. Moreover, to further examine the specificity of BDNF, we determined the levels of CCR5 (Fig. 8). No changes were observed in the levels of this receptor at any time examined. Thus, BDNF appears to affect only CXCR4 expression.
Figure 8.

BDNF decreases CXCR4 levels. Neurons were exposed to BDNF for the indicated times before the addition of gp120 and lysates prepared 6 hr later. A, Example of Western blot analysis of CXCR4 and CCR5 immunoreactivity in control and BDNF-treated (12 hr) neurons. B, Time course analysis of BDNF effect on CXCR4 and CCR5 levels. *p < 0.05, ** p < 0.01, and ***p < 0.001 (n = 6 per time point).
CXCR4 antagonist AMD3100 inhibits gp120 neurotoxicity
The ability of BDNF to decrease CXCR4 levels and therefore rescue neurons from gp120 toxicity suggests that agonists and antagonists of this receptor should mimic and decrease gp120-mediated cell death, respectively. This hypothesis was tested by exposing neurons to SDF-1α, the natural ligand of CXCR4 (Rossi and Zlotnik, 2000) alone or in combination with AMD, a specific antagonist of CXCR4 (Donzella et al., 1998; Labrosse et al., 1998; Lazarini et al., 2000). Cell death was measured 24 hr later using Hoechst/PI staining. SDF-1α (50 nm) induced neuronal cell death that was quantitatively similar to gp120 (Fig. 9). AMD (5 nm), added 30 min before the addition of gp120 or SDF-1α, prevented both gp120- and SDF-1α-mediated neuronal apoptosis (Fig. 9). No effect on cell viability was observed with AMD alone (Fig. 9). Therefore, CXCR4 activation plays a crucial role in the neurotoxic effect of gp120.
Figure 9.

CXCR4 receptor mediates gp120-induced cell death. Cultures were exposed to AMD 30 min before gp120 or SDF-1α. BDNF was added 12 hr before SDF-1α. Cell death was measured 24 hr later by Hoechst/PI. #p < 0.001 versus control; *p < 0.001 versus gp120 or SDF-1α'3b n = 9 each point.
To further establish a correlation between CXCR4 and BDNF neuroprotective effect, cultures were also incubated with BDNF 12 hr before SDF-1α. BDNF prevented SDF-1α-mediated neurotoxicity (Fig. 9). Thus, by establishing a correlation between ligand-induced cell death and antagonist-mediated cell survival, our data also indicate that BDNF neuroprotective property involves CXCR4 receptor downregulation.
Discussion
BDNF is a neurotrophic factor known to prevent and limit neurotoxin- or lesion-induced neuropathological damage in selected neuronal populations. For instance, BDNF enhances the survival of cholinergic neurons of the basal forebrain after a fimbria fornix lesion (Koliatsos et al., 1994) and protects neurons of the nigrostriatal pathway against the toxic properties of N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine or 6-hydroxydopamine (Hyman et al., 1991; Altar et al., 1992; Spina et al., 1992). However, BDNF also prevents the cell death of cerebellar granule cells against glutamate toxicity (Lindholm et al., 1993; Courtney et al., 1997; Marini et al., 1998; Bachis et al., 2002). Here we report that BDNF neurotrophic activity can be extended to neuroprotection against the HIV-1 envelope glycoprotein gp120, indicating that this neurotrophin has a survival action on a broad population of neurons and against a variety of potential neurological insults.
Activation of caspase-mediated apoptosis is an event that plays an important pathophysiological role in gp120-mediated cell death in macrophages, T-cells, and human embryonic kidney cells (Herbein et al., 1998; Biard-Piechaczyk et al., 2000). More recently, apoptosis has been described for neurons exposed to gp120 (Meucci et al., 2000; Bachis et al., 2000; Garden et al., 2002). Initiator caspases, such as caspase-8 and caspase-9, but not pro-inflammatory caspases, such as caspase-1 are induced by gp120 (Biard-Piechaczyk et al., 2000; Garden et al., 2002). In this study, we confirm that gp120 activates caspase-3 but not caspase-1, also in cerebellar granule neurons. Moreover, BDNF and DEVDK, two compounds with anti-caspase-3-like properties, prevented gp120-mediated neuronal cell death, further suggesting the involvement of effector caspases such as caspase-3 in gp120 neurotoxicity. However, the temporal profile of BDNF neuroprotection is different from that of the caspase-3 inhibitor DEVDK. Indeed, BDNF prevented gp120-mediated caspase-3 activation and cell death with a time delay of at least 6 hr, suggesting a mechanism different from a direct inhibition of caspase activity. Based on data showing a correlation between the ability of BDNF to reduce both the internalization of gp120 into neurons and the number of apoptotic cells, we propose that BDNF may prevent gp120-mediated activation of caspase-3 by reducing gp120 internalization.
The number of receptors present on the plasma membrane regulates ligand responses. We have reported previously that BDNF limits glutamate toxicity by downregulating the expression of NMDA-sensitive receptors (Brandoli et al., 1998) and suggested that the neuroprotective properties of BDNF against neurotoxicity are attributable in part to its ability to modulate NMDA receptor function. Therefore, to gain insight into the neuroprotective effect of BDNF against gp120, we tested the hypothesis that this neurotrophin prevents gp120-mediated cell death by downregulating the chemokine receptor CXCR4, a principal co-receptor used by HIV to gain entry into target cells. Moreover, CXCR4 has been shown to play a key role in mediating gp120 cytotoxicity in immune cell types (Herbein et al., 1998) and in neurons (Meucci et al., 2000), even in the absence of cell-surface CD4 (Hesselgesser et al., 1998), a glycoprotein necessary for HIV-1 entry into T-cells. We show that BDNF reduces the levels of CXCR4 protein with a temporal profile that matches its ability to prevent gp120-mediated cell death. This effect of BDNF was specific because the levels of NF and CCR5 did not change even after long-term exposure to the neurotrophin, suggesting that the BDNF-mediated decrease in CXCR4 levels is not attributable to a generalized inhibition of protein synthesis. Therefore, we propose that the neuroprotective activity of BDNF against gp120 may rely on its ability to decrease the availability of CXCR4 receptor and, therefore, gp120 cellular entry and, consequently, to reduce sensitivity of selected neuronal populations to this viral glycoprotein. This mechanism may be the main molecular event whereby BDNF reduces gp120 toxicity. However, additional studies are needed to prove this hypothesis.
SDF-1α is the natural ligand for CXCR4. In non-neuronal cells SDF-1α is as potent as gp120 in inducing CXCR4 responses, including internalization (Tarasova et al., 1998). In cortical neurons in culture, SDF-1α can induce cell death (Kaul and Lipton, 1999). Therefore, we used this ligand, alone or in combination with AMD, a selective antagonist of CXCR4, to examine whether CXCR4 underlies the toxic effect of gp120, and therefore, to further strengthen the hypothesis that downregulation of this receptor underlies the neuroprotective mechanism of BDNF. SDF-1α-mediated cell death was quantitatively similar to that evoked by gp120. Thus, activation of CXCR4 can lead to the cell death of various neuronal populations. On the contrary, blockade of CXCR4 prevents neuronal cell death. In fact, the toxic effect of both SDF-1α and gp120 was prevented by AMD. It is important to note that AMD does not interact with gp120 (Donzella et al., 1998); therefore, this biological activity relies solely on its ability to block gp120 binding to CXCR4. A similar “indirect antagonistic” effect was obtained with BDNF. In fact, BDNF blocked the neurotoxic effect of SDF-1α, although BDNF required a longer temporal delay in comparison to AMD. Thus, potential therapeutic targets for HIV-1 neuropathogenesis could involve reducing CXCR4 activation by specific antagonists or decreasing CXCR4 synthesis by neurotrophic factors.
Glutamate, through NMDA receptor activation, has been suggested to play a major role in acute neurodegenerative processes after gp120 (Lipton et al., 1991). Previously, we have established a link between the neuroprotective activity of BDNF against glutamate and its ability to reduce NMDA-receptor function in cerebellar granule cells (Brandoli et al., 1998). This correlation raises the issue of whether the alteration in NMDA receptor seen after BDNF also underlies the neuroprotective property of this neurotrophin against gp120. The data presented here dispute this assumption. In fact, in these cultures, the caspase-3-dependent cell death by both NMDA and glutamate (Bachis et al., 2001, 2002) is temporally distinct from that of gp120. Indeed, gp120-mediated toxicity requires a time frame considerably longer than that of glutamate. Moreover, the NMDA-receptor antagonist MK-801, or the cytokine interleukin-10, which we showed previously to inhibit glutamate toxicity in these experimental conditions (Bachis et al., 2001) failed to prevent gp120-mediated cell death (in preparation). All these considerations support our hypothesis that CXCR4, and not NMDA receptor, may be the primary target of gp120 in mediating neuronal cell death.
In this study, we demonstrated that gp120-mediated apoptosis is caspase-3 dependent. This finding is therapeutically relevant because increased caspase-3 immunoreactivity has been described in the postmortem brain tissues of HIV-positive patients (James et al., 1999) as well as in human fetal CNS cultures exposed to gp120 (Zheng et al., 1999). Moreover, gp120 activation of the caspase cascade contributes to neuronal cell death in cerebrocortical cultures (Garden et al., 2002). Therefore, the intrinsic ability of BDNF to reduce caspase-3 activation (Han et al., 2000; Bachis et al., 2002) raises a legitimate hope that this molecule can be used as a drug to limit HIV-1-mediated neurodegeneration. Moreover, the ability of BDNF to affect ongoing modifications in the terminal fields of BDNF-sensitive neurons, including synapses, renders this neurotrophin particularly attractive as a therapy for AIDS because axonal and synaptic pathology is commonly seen in AIDS patients (Masliah et al., 1997). It will be important to determine whether BDNF also prevents neuronal apoptosis in vivo.
Footnotes
This work was supported by National Institutes of Health Grant NS40670. We thank Dr. Baggi Somasundaram (ATTO Bioscience) for help with the confocal attofluor ratio vision image and Randi Goodnight for assistance in computer programs.
Correspondence should be addressed to Dr. Italo Mocchetti, Georgetown University Medical Center, Research Building, Room EP04, Box 571464, Washington, DC 20057. E-mail:moccheti@georgetown.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235715-08$15.00/0
References
- Altar AC, Boylan CB, Jackson C, Hershenson S, Miller J, Wiegand SJ, Lindsay RM, Hyman C ( 1992) Brain-derived neurotrophic factor augments rotational behavior and nigrostriatal dopamine turnover in vivo. Proc Natl Acad Sci USA 89: 11347–11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachis A, DeBernardi MA, Mocchetti I ( 2000) Brain derived neurotrophic factor protects cerebellar granule cells against gp120-mediated cell death. Soc Neurosci Abstr 26: 89.8. [Google Scholar]
- Bachis A, Colangelo AM, Vicini S, Doe PP, De Bernardi MA, Brooker G, Mocchetti I ( 2001) Interleukin-10 prevents glutamate-mediated cerebellar granule cell death by blocking caspase-3-like activity. J Neurosci 21: 3104–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachis A, Rabin SJ, Del Fiacco M, Mocchetti I ( 2002) Gangliosides prevent excitotoxicity through activation of TrkB receptor. Neurotox Res 3: 225–234. [DOI] [PubMed] [Google Scholar]
- Bagetta G, Corasaniti MT, Aloe L, Berliocchi L, Costa N, Finazzi-Agro A, Nistico G ( 1996) Intracerebral injection of human immunodeficiency virus type 1 coat protein gp120 differentially affects the expression of nerve growth factor and nitric oxide synthase in the hippocampus of rat. Proc Natl Acad Sci USA 93: 928–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, Booze RM ( 2000) Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res 879: 42–49. [DOI] [PubMed] [Google Scholar]
- Bennett BA, Rusyniak DE, Hollingsworth CK ( 1995) HIV-1 gp120-induced neurotoxicity to midbrain dopamine cultures. Brain Res 705: 168–176. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Domercq M, Brambilla L, Galli R, Schols D, De Clercq E, Vescovi A, Bagetta G, Kollias G, Meldolesi J, Volterra A ( 2001) CXCR4-activated astrocyte glutamate release via TNFα: amplification by microglia triggers neurotoxicity. Nat Neurosci 4: 702–710. [DOI] [PubMed] [Google Scholar]
- Biard-Piechaczyk M, Robert-Hebmann V, Richard V, Roland J, Hipskind RA, Devaux C ( 2000) Caspase-dependent apoptosis of cells expressing the chemokine receptor CXCR4 is induced by cell membrane-associated human immunodeficiency virus type 1 envelope glycoprotein (gp120). Virology 268: 329–344. [DOI] [PubMed] [Google Scholar]
- Brandoli C, Sanna A, De Bernardi MA, Follesa P, Brooker G, Mocchetti I ( 1998) Brain derived neurotrophic factor and basic fibroblast growth factor down-regulate NMDA receptor function in cerebellar granule cells. J Neurosci 18: 7953–7961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew BJ, Rosenblum M, Cronin K, Price RW ( 1995) AIDS dementia complex and HIV-1 brain infection: clinical-virological correlations. Ann Neurol 38: 563–570. [DOI] [PubMed] [Google Scholar]
- Cefai D, Ferrer M, Serpente N, Idziorek T, Dautry-Varsat A, Debre P, Bismuth G ( 1992) Internalization of HIV glycoprotein gp120 is associated with down-modulation of membrane CD4 and p56lck together with impairment of T cell activation. J Immunol 149: 285–294. [PubMed] [Google Scholar]
- Courtney MJ, Akeman KEO, Coffey ET ( 1997) Neurotrophins protect cultured cerebellar granule neurons against the early phase of cell death by a two-component mechanism. J Neurosci 17: 4201–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CB, Dikic I, Unutmaz D, Hill CM, Arthos J, Siani MA, Thompson DA, Schlessinger J, Littman DR ( 1997) Signal transduction due to HIV-1 envelope interactions with chemokine receptors CXCR4 or CCR5. J Exp Med 186: 1793–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Dawson Tm, Uhl GR, Snyder SH ( 1993) Human immunodeficiency virus type 1 coat protein neurotoxicity mediated by nitric oxide in primary cortical cultures. Proc Natl Acad Sci USA 90: 3256–3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzella GA, Schols D, Lin SW, Esté JA, Nagashima KA, Maddon PJ, Allaway GP, Sakmar TP, Henson G, De Clercq E, Moore JP ( 1998) AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat Med 4: 72–77. [DOI] [PubMed] [Google Scholar]
- Garden GA, Budd SL, Tsai E, Hanson L, Kaul M, D'Emilia DM, Friedlander RM, Yuan J, Masliah E, Lipton SA ( 2002) Caspase cascades in human immunodeficiency virus-associated neurodegeneration. J Neurosci 22: 4015–4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han BH, D'Costa A, Back SA, Parsadanian M, Patel S, Shah AR, Gidday JM, Srinivasan A, Deshmukh M, Holtzman DM ( 2000) BDNF blocks caspase-3 activation in neonatal hypoxia-ischemia. Neurobiol Dis 7: 38–53. [DOI] [PubMed] [Google Scholar]
- Herbein G, Mahlkmecht U, Batliwalla F ( 1998) Apoptosis of CD8+ T cells is mediated by macrophages through interaction of HIV gp120 with chemokine receptor CXCR4. Nature 395: 189–194. [DOI] [PubMed] [Google Scholar]
- Hesselgesser J, Taub D, Baskar P, Greenberg M, Hoxie J, Kolson DL, Horuk R ( 1998) Neuronal apoptosis induced by HIV-1 gp120 and the chemokine SDF-1α is mediated by the chemokine receptor CXCR4. Curr Biol 8: 595–598. [DOI] [PubMed] [Google Scholar]
- Hyman C, Hofer M, Barde Y-A ( 1991) BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 350: 369–377. [DOI] [PubMed] [Google Scholar]
- James HJ, Sharer LR, Zhang Q, Wang HG, Epstein LG, Reed JC, Gelbard HA ( 1999) Expression of caspase-3 in brains from paediatric patients with HIV-1 encephalitis. Neuropathol Appl Neurobiol 25: 380–386. [DOI] [PubMed] [Google Scholar]
- Kaul M, Lipton SA ( 1999) Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci USA 96: 8212–8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RS, Williams KC, Alvarez-Hernandez X, Westmoreland S, Force T, Lackner AA, Luster AD ( 1999) Chemokine receptor expression and signaling in macaque and human fetal neurons and astrocytes: implications for the neuropathogenesis of AIDS. J Immunol 163: 1636–1646. [PubMed] [Google Scholar]
- Koliatsos V, Price DL, Gouras GK, Cayouette M, Burton LE, Winslow JW ( 1994) Highly selective effects of nerve growth factor, brain derived neurotrophic factor and neurotrophin-3 on intact and injured basal forebrain magnocellular neurons. J Comp Neurol 343: 247–262. [DOI] [PubMed] [Google Scholar]
- Kozak SL, Kuhmann SE, Platt EJ, Kabat D ( 1999) Roles of CD4 and coreceptors in binding, endocytosis, and proteolysis of gp120 envelope glycoproteins derived from human immunodeficiency virus type 1. J Biol Chem 274: 23499–23507. [DOI] [PubMed] [Google Scholar]
- Labrosse B, Berlot A, Heveker N, Sol N, Schols D, De Clercq E, Alizon M ( 1998) Determinants for sensitivity of human immunodeficiency virus coreceptor CXCR4 to the bicyclam AMD3100. J Virol 72: 6381–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarini F, Casanova P, Tham TN, De Clercq E, Arenzana-Seisdedos F, Baleux F, Dubois-Dalcq M ( 2000) Differential signalling of the chemokine receptor CXCR4 by stromal cell-derived factor 1 and the HIV glycoprotein in rat neurons and astrocytes. Eur J Neurosci 12: 117–125. [DOI] [PubMed] [Google Scholar]
- Lindholm D, Dechant G, Heisenberg CP, Thoenen H ( 1993) BDNF is a survival factor for cultured rat cerebellar granule neurons and protects against glutamate-induced neurotoxicity. Eur J Neurosci 5: 1455–1464. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Sucher NJ, Kaiser PK, Dreyer EB ( 1991) Synergistic effects of HIV coat protein and NMDA receptor-mediated neurotoxicity. Neuron 7: 111–118. [DOI] [PubMed] [Google Scholar]
- Marini AM, Rabin SJ, Lipski R, Mocchetti I ( 1998) Activity-dependent release of BDNF underlies the neuroprotective effect of NMDA. J Biol Chem 273: 29394–29399. [DOI] [PubMed] [Google Scholar]
- Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, Mallory M, Achim CL, McCutchan JA, Nelson JA, Atkinson JH, Grant I ( 1997) Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders: HNRC Group: the HIV Neurobehavioral Research Center. Ann Neurol 42: 963–972. [DOI] [PubMed] [Google Scholar]
- Meucci O, Miller RJ ( 1996) gp120-induced neurotoxicity in hippocampal pyramidal neuron cultures: protective action of TGFβ1 J Neurosci 16: 4080–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O, Fatatis A, Simen AA, Miller RJ ( 2000) Expression of CX3CR1 chemokine receptors on neurons and their role in neuronal survival. Proc Natl Acad Sci USA 97: 8075–8080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misse D, Cerutti M, Noraz N, Jourdan P, Favero J, Devauchelle G, Yssel H, Taylor N, Veas F ( 1999) A CD4-independent interaction of human immunodeficiency virus-1 gp120 with CXCR4 induces their cointernalization, cell signaling, and T-cell chemotaxis. Blood 93: 2454–2462. [PubMed] [Google Scholar]
- Price RW ( 1996) Neurological complications of HIV infection. Lancet 348: 445–452. [DOI] [PubMed] [Google Scholar]
- Reichardt LF ( 2001) Neurotrophic factors: a window into the nervous system. In: Neurobiology of the neurotrophins (Mocchetti I, ed), pp 3–28. Johnson City, TN: Graham.
- Rossi D, Zlotnik A ( 2000) The biology of chemokines and their receptors. Annu Rev Immunol 18: 217–242. [DOI] [PubMed] [Google Scholar]
- Savio T, Levi G ( 1993) Neurotoxicity of HIV coat protein gp120, NMDA receptors, and protein kinase C: a study with rat cerebellar granule cell culture. J Neurosci Res 34: 265–272. [DOI] [PubMed] [Google Scholar]
- Spina MB, Squinto SP, Miller J, Lindsay RM, Hyman C ( 1992) Brain-derived neurotrophic factor protects dopamine neurons against 6-hydroxidopamine and N-methyl-4-phirylpiridinium ion toxicity. Involvement of glutathione system. J Neurochem 59: 99–106. [DOI] [PubMed] [Google Scholar]
- Tarasova NI, Stauber RH, Michejda CJ ( 1998) Spontaneous and ligand-induced trafficking of CXC-chemokine receptor 4. J Biol Chem 273: 15883–15696. [DOI] [PubMed] [Google Scholar]
- Ullrich CK, Groopman JE, Ganju RK ( 2000) HIV-1 gp120- and gp160-induced apoptosis in cultured endothelial cells is mediated by caspases. Blood 96: 15883–15886. [PubMed] [Google Scholar]
- van der Meer P, Ulrich AM, Gonzalez-Scarano F, Levi E ( 2000) Immuno-histochemical analysis of CCR2, CCR3, CCR5, and CXCR4 in the human brain: potential mechanisms for HIV dementia. Exp Mol Pathol 69: 192–201. [DOI] [PubMed] [Google Scholar]
- Wang WK, Dudek T, Zhao YJ, Brumblay HG, Essex M, Lee TH ( 1998) CCR5 coreceptor utilization involves a highly conserved arginine residue of HIV type 1 gp120. Proc Natl Acad Sci USA 95: 5740–5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman D, Rabin RL, Arthos J, Rubbert A, Dybul M, Swofford R, Venkatesan S, Farber JM, Fauci AS ( 1997) Macrophage-tropic HIV and SIV envelope proteins induce a signal through the CCR5 chemokine receptor. Nature 389: 981–985. [DOI] [PubMed] [Google Scholar]
- Zheng J, Ghorpade A, Niemann D, Cotter RL, Thylin MR, Epstein L, Swartz JM, Shepard RB, Liu X, Nukuna A, Gendelman HE ( 1999) Lymphotropic virions affect chemokine receptor-mediated neural signaling and apoptosis: implications for human immunodeficiency virus type 1-associated dementia. J Virol 73: 8256–8267. [DOI] [PMC free article] [PubMed] [Google Scholar]