

Abstract
Caspase-11 is a key regulator of caspase-1 and caspase-3 activation under pathological conditions. We show here that the expression of caspase-11 is upregulated in the spinal cord of superoxide dismutase 1 (SOD1) G93A transgenic mice, a mouse model of amyotrophic lateral sclerosis (ALS), before the onset of motor dysfunction and remains at the high levels throughout the course of disease. The caspase-1- and caspase-3-like activities, as well as the level of interleukin-1β, were significantly reduced in the spinal cord of symptomatic caspase-11–/–;SOD1 G93A mice compared with that of caspase-11+/–; SOD1 G93A mice. However, neurodegeneration, inflammatory responses, and the disease onset and progression in SOD1 G93A transgenic mice were not altered by the ablation of caspase-11 gene. Thus, although caspases may contribute to certain aspects of pathology in this mouse model of ALS, their inhibition is not sufficient to prevent neurodegeneration. Our study urges caution when considering the inhibition of caspases as a direct therapeutic method for the treatment of chronic neurodegenerative diseases.
Keywords: ALS, motor neuron degeneration, neurodegeneration, SOD1, caspase, apoptosis
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurological disorder characterized by selective degeneration of upper and lower motor neurons, leading to paralysis and death at ∼1–5 years after the onset (Cleveland and Rothstein, 2001). The disease has both sporadic and familial forms, with the latter accounting for ∼5% of the cases. In 1993, Rosen et al. demonstrated that several mutations in the Cu2+/Zn2+ superoxide dismutase (SOD1) are causally responsible for a subset of familial ALS (FALS). Consistent with a causal role of SOD1 mutations in FALS, transgenic mice expressing human SOD1 mutants develop age-dependent progressive motor neuron degeneration with cellular pathological features similar to that of human ALS.
Recent studies suggested caspase-mediated apoptosis as a possible mechanism for motor neuron degeneration in ALS (Friedlander et al., 1997; Kostic et al., 1997; Li et al., 2000). Caspases are actively regulated cysteine proteases responsible for the signal transduction and execution of apoptosis (Cryns and Yuan, 1998). Activation of caspase-1 and caspase-3 in the spinal cords of mutant SOD1 mice and ALS patients has been reported previously (Martin, 1999; Li et al., 2000; Pasinelli et al., 2000; Vukosavic et al., 2000). More recently, activation of caspase-7, -8, and -9 in a mouse model at end stage of the disease has been also reported (Guégan et al., 2001, 2002). Moreover, inhibition of caspase(s) with peptide inhibitors or by introducing a dominant-negative mutant of caspase-1 delayed the onset and progression of the disease symptoms in G93A SOD1 (G93A) transgenic mice (Friedlander et al., 1997; Li et al., 2000), suggesting the importance of caspase-mediated apoptosis in the ALS pathology.
The possible involvement of both caspase-1 and caspase-3 in the pathogenesis of ALS strongly suggested the involvement of caspase-11 in this process because caspase-11 is a key upstream regulator of both caspase-1 and -3 under pathological conditions (Wang et al., 1998; Kang et al., 2000, 2002). Caspase-11–/– mice are defective in interleukin-1β (IL-1β) maturation and resistant to lipopolysaccharide (LPS)-induced septic shock (Wang et al., 1998). Apoptosis and caspase-3 activation by LPS shock or ischemic brain injury are also defective in caspase-11–/– mice (Kang et al., 2000, 2002). Thus, caspase-11 serves as an initiator caspase in apoptosis under these pathological conditions.
Although there have been reports of caspase activation and of beneficial effect of general caspase inhibition in mouse models of ALS, a critical role of individual caspases in mutant SOD1-mediated neurodegeneration has not been examined in different caspase mutant mice. Therefore, it remains to be resolved whether the activation of caspases and the resulting apoptosis play an indispensable role in mediating neurodegeneration in ALS. Furthermore, the contribution of caspase-mediated toxicity in relation to the early events of pathogenesis in mutant SOD1 transgenic mice, such as mitochondrial dilation, neurofilament abnormality, and glutamate transporter downregulation in astrocytes, remains unclear.
In the present study, we provide evidence that caspase-11 plays a critical role in caspase-1 and -3 activation during pathogenesis of G93A mice, a mouse model of FALS (Gurney et al., 1994). However, deletion of caspase-11 was found to have no effect on the disease onset, progression, or inflammatory response in this model of FALS. Our results bring into the question the proposed causal role of these caspases in the pathogenesis–neurodegeneration in FALS.
Materials and Methods
Transgenic mice. Mouse lines expressing human SOD1 mutant G93A [C57BL/6J-TgN(SOD1-G93A)1Gurdl] were obtained from The Jackson Laboratory (Bar Harbor, ME). Caspase-11-deficient mice have been described previously (Wang et al., 1998). To minimize the variations in genetic background, one SOD1 G93A transgenic male mouse was mated with one female caspase-11–/– mouse to generate F1. An F1 male mouse (caspase-11+/–;SOD1 G93A) was mated with two caspase-11–/– mice and one caspase-11+/– mouse to generate all of the genotypes for comparison. A total of five litters were used for clinical assessment. Mice were maintained in a pathogen-free environment, and experiments on mice were conducted according to the protocols approved by the Harvard Medical School Animal Care Committee.
Clinical assessment. The phenotypes of the G93A mice were assessed daily for resting tremor and every other day for upper and lower limb weakness based on their ability to hold on to the cage bar for 20 sec as described previously (Gurney et al., 1994).
Immunoblots. For immunoblotting, tissue samples were pulverized in liquid N2, and the resulting tissue powder was solubilized in 0.7 ml of radioimmunoprecipitation assay lysis buffer (150 mm NaCl, 1% Triton X-100, 0.1% SDS, and 1% sodium deoxycholate in 50 mm Tris-HCl, pH 7.4) with protease inhibitor mixtures (Boehringer Mannheim, Mannheim, Germany). Tissue lysates were processed for immunoblot as described previously (Kang et al., 2000). Antibodies against human- and mouse-specific SOD1 were obtained from Chemicon (Temecula, CA).
Caspase activity assay. Freshly isolated spinal cords were pulverized in the liquid nitrogen-chilled mortar. Caspase activity was determined using methods described by Kang et al. (2000).
Immunohistochemistry, terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling, and Nissl staining. Immunostaining of anti-MacI (macrophage antigen I) (Caltag, Burlingame, CA), anti-GFAP (glial fibrillary acidic protein), or anti-NeuN (neuronal-specific nuclear protein) (Chemicon, Temecula, CA) antibody and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay were done using methods described by Kang et al. (2000). Nissl staining was performed using cresyl violet following the standard method.
Results
Caspase-11 regulates caspase-1 and -3 activation in the spinal cords of G93A mice
Activation of caspase-1 and caspase-3 in the spinal cord of presymptomatic G93A mice has been reported previously (Li et al., 2000). Because caspase-11 is an upstream regulator of caspase-1 and caspase-3 (Wang et al., 1998; Kang et al., 2000), we examined the possibility that caspase-11 is also involved in the pathogenesis of ALS by mediating the activation of caspase-1 and -3. To test this possibility, we first examined the induction of caspase-11 in the spinal cords of the G93A mice by immunoblot assay (Fig. 1A). We found that the expression of both 38 and 43 kDa caspase-11 products was induced as early as 60 d of age in the spinal cords of G93A mice, preceding the onset of first detectable motor deficits, and the levels of caspase-11 protein continued to increase with the peak level at approximately day 120. We also examined the expression of caspase-1 using an antibody originally generated in our laboratory and demonstrated to recognize the full-length and activated caspase-1 (Wang et al., 1998). This antibody was later shown by Li et al. (2000) to be an anti-active caspase-1-specific antibody in the spinal cord of G93A mice. In contrast, we have not found this antibody to be specific for active caspase-1 in the spinal cord or other tissues of wild-type or G93A mice. This antibody recognizes 45 kDa pro-caspase-1 in the spleens of wild-type mice but not in caspase-1–/– mice (Kang et al., 2000), indicating the authenticity of this band. It also recognizes a 35 kDa protein, most likely a nonspecific cross-reactive protein because it is also present in caspase-1–/– spleens. In addition, the antibody recognizes a caspase-1 20 kDa protein fragment in the spinal cord of G93A mice at low levels starting at ∼60 d of age, consistent with reports of active caspase-1 being present in the spinal cords of G93A mice (Li et al., 2000; Pasinelli et al., 2000). Furthermore, our analysis demonstrated a clear induction of full-length caspase-1 expression starting from 60 d of age and continuing to increase as the disease progresses (Fig. 1A). Low levels of active caspase-3 were first detected at day 60 in G93A mice, at approximately the time of caspase-11 induction. However, no signs of caspase-1 and -3 activation were detected in the caspase-11–/–; G93A spinal cords (data not shown). These results imply that caspase-11, in addition to caspase-1 and -3, may be involved in regulating inflammatory reaction and/or apoptosis during the disease onset and progression of this mouse model of ALS.
Figure 1.
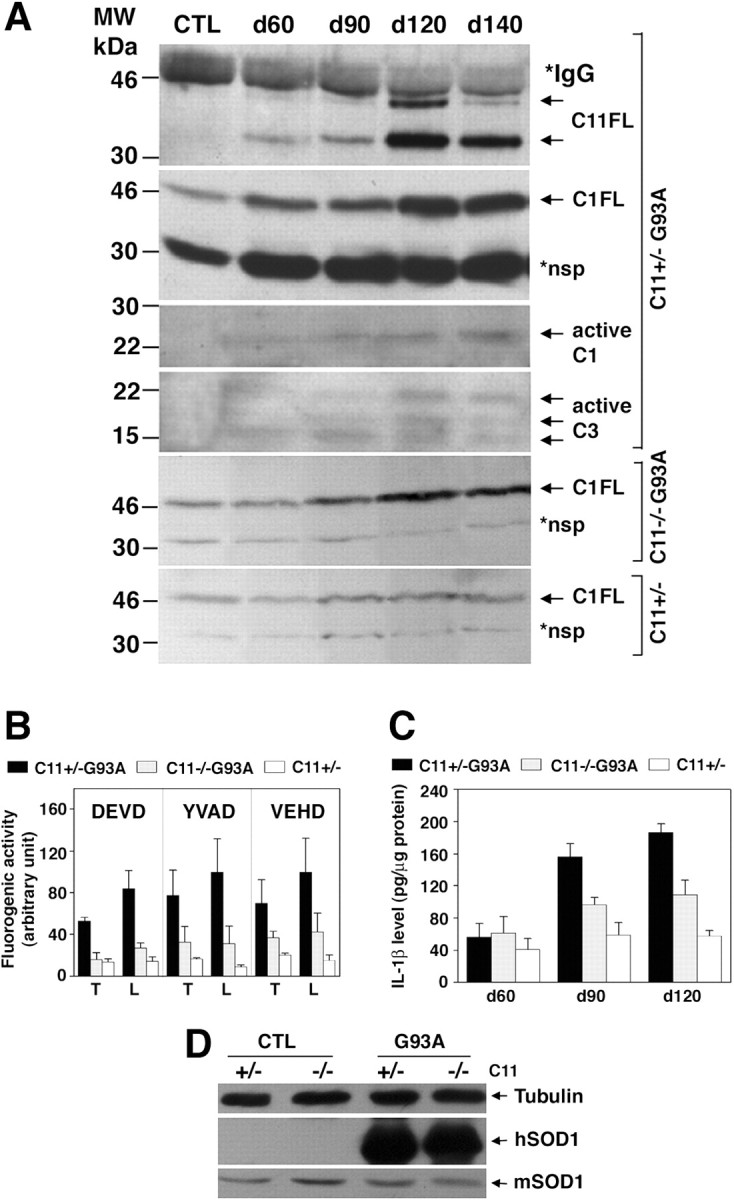
Upregulation of caspase-11 at the disease onset in G93A mice and reduction of caspase-1 and caspase-3 activation in caspase-11–/–;G93A double-mutant mice.A, Upregulation of caspase-11 and appearance of active caspase-1 and caspase-3 fragments in the G93A spinal cords. Two hundred micrograms of the spinal cord lysates of G93A mice at indicated days of age were immunoprecipitated using monoclonal anti-caspase-11 antibody and blotted with the same antibody for detection of the two caspase-11 full-length products of 43 and 38 kDa (C11FL). Control (CTL) lysates were prepared from a 140-d-old wild-type mouse. Eighty micrograms of the samples were also immunoblotted with monoclonal anti-caspase-1 antibody for detection of full-length caspase-1 (C1FL) and the active form (active C1) and with anti-active caspase-3 antibody (active C3). *IgGH, IgG heavy chain; *nsp, nonspecific. Nonspecific bands serve as a loading control.B, Reduction of acDEVDase, acYVADase, and acVEHDase activity in the absence of caspase-11 in the caspase-11–/–; G93A spinal cords. At 120 d of age, thoracic (T) and lumbar (L) spinal cord lysates were prepared from caspase-11+/–; G93A (C11+/–G93A), caspase-11–/–; G93A (C11–/–G93A), and caspase-11+/– (C11+/–) mice. The caspase-3-, caspase-1-, and caspase-11-like enzyme activities were measured using fluorogenic substrates acDEVD-amc (DEVD), acYVAD-amc (YVAD), and acVEHD-amc (VEHD), respectively, in the lysates (n = 4; mean ± SD). C, Reduction of IL-1β level in the absence of caspase-11 in the caspase-11–/–; G93A spinal cords. Spinal cords from caspase-11; G93A mice were taken at indicated days of age, and the level of mature IL-1β was measured by ELISA (n = 4; mean±SD).D, The expression of the human mutant SOD1 protein is not affected by the absence of caspase-11 in the double-mutant mice. Sixty microgram samples of the spinal cord lysates from the 90-d-old caspase-11; G93A mice were immunoblotted with human- and mouse-specific anti-SOD1 antibodies for the detection of introduced and endogenous SOD1 proteins, respectively.
To examine whether caspase-11 indeed regulates caspase-1 and/or -3 in the spinal cord of G93A mice, we crossed caspase-11–/– and G93A mice and compared the profiles of caspase activities in the symptomatic (day 120) spinal cord lysates of littermate caspase-11–/–; G93A and caspase-11+/–; G93A mice (Fig. 1B). Consistent with the activation of caspase-1 and caspase-3 and the caspase-11 induction profile by immunoblot (Fig. 1A), acYVAD-amc (caspase-1-like), acDEVD-amc (caspase-3-like), and acVEHD-amc (caspase-11-like) cleavage activities (Thornberry et al., 1997; Kang et al., 2000) were elevated in the lumbar and thoracic spinal cords of G93A mice (Fig. 1B). In caspase-11–/–; G93A mice, the absence of caspase-11 resulted in a significant reduction of caspase-3- and caspase-1-like activities, as well as caspase-11-like activity (Fig. 1B). The reduced cleavage of the caspase-11 substrate, acVEHD-amc, in the caspase-11–/–; G93A mice compared with that of G93A mice demonstrates that caspase-11 is indeed activated in the G93A spinal cord.
The secretion and maturation of IL-1β is another downstream indicator of caspase-11– caspase-1 pathway (Kuida et al., 1995; Wang et al., 1998). We determined the levels of IL-1β in wild-type control, caspase-11+/–; G93A, and caspase-11–/–; G93A mice by ELISA (Fig. 1C). The increase in IL-1β levels was first detected at approximately day 90 and remained at higher levels at later time points. The levels of IL-1β in the spinal cords of caspase-11–/–; G93A mice were consistently lower than that of caspase-11+/–; G93A mice. This result indicates that caspase-11 also plays a key role in regulating IL-1β levels in the G93A mice.
To examine whether the ablation of caspase-11 gene affected the expression level of the human mutant SOD1 protein in the double-mutant mice, we compared the levels of the mutant protein in the spinal cord lysates of caspase-11+/– and –/–; G93A mice. As shown in Figure 1D, the loss of caspase-11 did not alter the expression of the mutant SOD1 protein in the double-mutant mice.
Together, these results suggest that caspase-11 is activated and regulates the activities of its downstream caspases-1 and -3 and also IL-1β levels during the pathogenesis of this model of FALS.
Neurodegeneration and inflammatory response proceed in G93A mice in the absence of caspase-11
The evidence of activation of caspase-1 and -3 by caspase-11 in G93A mice prompted us to compare the inflammatory response and cell death in G93A spinal cords in the presence or absence of caspase-11. The spinal cords of G93A mice have been found to exhibit signs of inflammatory responses, as evidenced by the significant increases in the number of microglia and astrocytes after the initiation of pathology (Hall et al., 1998). Because the level of IL-1β was reduced in the absence of caspase-11 in G93A mice, we reasoned that the absence of caspase-11 might reduce the inflammatory response in G93A mice. We determined and compared the numbers of microglia and astrocytes using MacI and GFAP as their respective markers in the spinal cords of caspase-11+/–; G93A and caspase-11–/–; G93A mice. As shown in Figure 2A, the spinal cords of G93A mice showed dramatic increases in the number of MacI-positive microglia and GFAP-positive astrocytes, with no significant differences in the absence or presence of caspase-11. This result suggests that, despite the significant reduction of caspase activation and IL-1β levels in caspase-11–/–; G93A mice, the inflammatory response induced by mutant SOD1 was not significantly altered.
Figure 2.
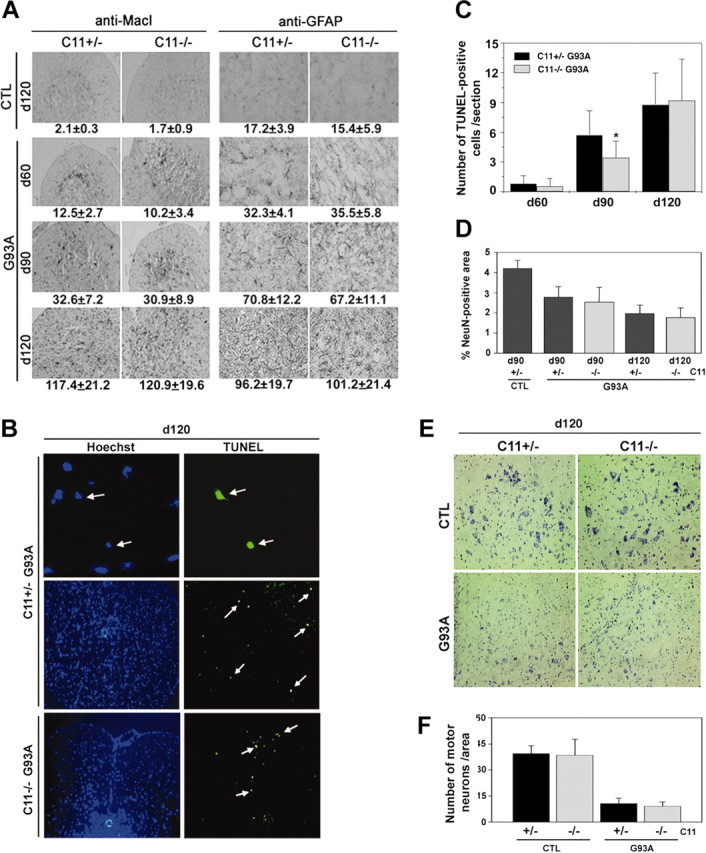
The lack of caspase-11 failed to block neurodegeneration in the G93A mice. A, Glial activation is not affected by the absence of caspase-11. The lumbar spinal cords from caspase-11 single mutant (CTL) and caspase-11; G93A double-mutant mice were taken at indicated days of age and processed for immunohistochemistry using anti-MacI antibody to detect microglial cells and anti-GFAP antibody to detect astrocytes. The cells positive for anti-MacI or anti-GFAP were counted in the white matter of the spinal cord sections, and the numbers are shown at the bottom of each panel (n = 5 for single mutants; n = 12 for double mutants; mean ± SD). Original magnification for the anti-MacI staining, 20×; anti-GFAP staining, 40×. B, TUNEL-positive apoptotic cell death in G93A mice is not significantly affected by the absence of caspase-11. Spinal cords of indicated genotypes and days of age were processed for TUNEL. Arrows indicate the TUNEL-positive cells (top panels, 63× magnification; bottom panels, 20× magnification). Note the condensed nuclei of TUNEL-positive cells in the top panels. C, The numbers of TUNEL-positive cells were determined from the lumbar sections by direct counting (n = 30). *p < 0.05 indicates significantly different (Student's t test). D, Neuronal damage–loss is not protected by the absence of caspase-11. Total neuronal area was measured by staining the spinal cord sections with a neuronal marker anti-NeuN and scanning the positive area by Northern Exposure (n = 10; mean ± SD) and was confirmed by visual inspection. E, F, Motor neuron loss is not protected by caspase-11 deficiency in G93A mice. Number of motor neurons was determined by Nissl-staining the spinal cord sections (E) and counting the large Nissl-positive neurons (F). Three 120-d-old mice were used for each genotype, and 10 tissue sections of lumbar spinal cord were counted for each mouse. Data represent the mean of pooled counting with SD.
To further examine a possible effect of caspase-11 deficiency on the neurodegeneration in G93A mice, we looked for evidence of apoptosis by TUNEL staining. A small increase in the number of TUNEL-positive cells was detected in the lumbar spinal cord of G93A mice as the disease progressed (Fig. 2B,C), whereas considerably fewer cells were positive for TUNEL in the nontransgenic littermate control spinal cords (data not shown). TUNEL-labeled cells were not concentrated in the ventral horns in which degenerating motor neurons are localized but scattered mostly in the margin of the white matter. TUNEL-positive cells had indeed shrunken nucleus, as shown in Figure 2B (arrows), a hallmark of apoptotic cells. Interestingly, there was a slight but consistent reduction in the number of TUNEL-positive cells in the absence of caspase-11 at day 90. However, at day 120 when the disease is in the mid to late stage, the difference in the number of apoptotic cells disappeared between the two groups (Fig. 2C). Thus, the effect of caspase-11 deficiency on apoptosis during the disease progression in G93A mice may be overcome by alternative mechanisms.
To examine the effect of caspase-11 deficiency on neurodegeneration in G93A mice, we estimated the numbers of remaining neurons by immunostaining for NeuN, a marker for neurons (Mullen et al., 1992), in both dorsal and ventral horns of lumbar spinal cords of caspase-11–/–; G93A mice, caspase-11+/–; G93A mice, and nontransgenic control mice. As shown in Figure 2D, total neuronal damage–loss was evident, but there was no significant difference in the number of neurons between caspase-11–/–; G93A and caspase-11+/–; G93A mice. Because there is a possibility that the neurons damaged during the development of pathology may downregulate the expression of NeuN, we examined the loss of motor neurons in the ventral horns of lumbar spinal cords of end stage mice by Nissl staining. As shown in Figure 2, E and F, the number of large Nissl-stained motor neurons was strikingly reduced in the G93A mutant mice regardless of the presence or absence of caspase-11. These results suggest that caspase-11-independent mechanisms account for the neuronal loss in this disease model. Alternatively, in the absence of caspase-11, neurons may die through an alternative pathway.
Consistent with the rate of neuronal loss and biochemical analysis, clinical assessment of disease onset, progression, and the survival rate of G93A mice were not changed in the absence of caspase-11 (Fig. 3), clearly indicating a dissociation between the caspase-11 pathway and neurodegeneration.
Figure 3.

Neurological outcome is not improved by the absence of caspase-11 in the G93A mice. A, The age of disease onset as assessed by the rotarod test in the indicated genotypes. B, The life spans of G93A mice were not affected by the absence or presence of caspase-11. C, A summary of average ages at the onset of motor deficit and life span with mice of indicated genotypes.
Discussion
In this study, we provide evidence showing that, although caspase-11 expression is induced before the onset of motor deficits in G93A mice, caspase-11 activation is not critically involved in the neurodegeneration. Because the lack of caspase-11 resulted in the significant inhibition of caspase-1 and caspase-3 activation, our results suggest that caspase-1 and caspase-3 activation is not crucial for neurodegeneration in this mouse model of ALS. Furthermore, because caspase-11 plays a key role in regulating IL-1β maturation in LPS-stimulated mice (Wang et al., 1998) and in G93A mice (this study), our results further suggest that IL-1β is not a key determinant in mediating neurodegeneration (or the inflammatory response) in this model. This is consistent with the report demonstrating that the neural degeneration and accelerated death of G37R SOD1 mice are not altered in the background of IL-1β–/– mice (Nguyen et al., 2001). Thus, our study provides a cautionary note when considering caspases as targets for the treatment of chronic neurodegenerative diseases such as ALS.
The expression of caspase-11 has been found to be induced under multiple pathological conditions, including LPS-induced septic shock (Wang et al., 1996), the experimental autoimmune encephalomyelitis (EAE) as a mouse model of multiple sclerosis (Hisahara et al., 2001), brain ischemia induced by middle cerebral artery occlusion as a mouse model of stroke (Kang et al., 2000), and G93A transgenic mice (this study). In contrast to the findings described in this paper, caspase-11–/– mice were highly resistant to LPS-induced apoptosis and lethality (Wang et al., 1998; Kang et al., 2002), EAE (Hisahara et al., 2001), and apoptosis induced by ischemic brain injury (Kang et al., 2000). These results suggest that the expression of G93A SOD1 may induce neurodegeneration through multiple pathways that may involve multiple caspases and/or caspase-independent pathways.
Systemic and/or localized inflammation is a prominent feature of LPS-induced septic shock (Raetz and Whitfield, 2002), EAE (Hisahara et al., 2001), and G93A mice (Hall et al., 1998). The levels of IL-1β were significantly reduced in caspase-11–/– mice compared with wild type when induced with LPS injection or EAE (Wang et al., 1998; Hisahara et al., 2001). The infiltration of CD3ϵ+ T cells in the spinal cord was significantly lower in EAE-induced caspase-11–/– mice (Hisahara et al., 2001). In contrast, although the levels of IL-1β were also reduced in the absence of caspase-11 in the G93A mice, the infiltration of microglia and astrocytes into the spinal cord of G93A mice was not affected by the caspase-11 deficiency, indicating that the overall inflammatory response proceeded in the absence of caspase-11. Because the complete elimination of IL-1β in IL-1β–/– mice did not alter the disease course of SOD1 G37R mice (Nguyen et al., 2001), the most likely interpretation is that G93A induces inflammatory responses through multiple or redundant pathways.
Our study cannot rule out, however, that the residual caspase activity in caspase-11–/– background sufficient for neurodegeneration and caspases other than caspase-1 and caspase-3 may be involved in mediating neuronal degeneration in G93A mice. Motor neurons expressing mutant SOD1 have been shown to have an enhanced sensitivity toward Fas-induced cell death (Raoul et al., 2002), although the critical role of Fas pathway in mediating motor neuron degeneration has not been evaluated. In this regard, it is worth noting that the activation of caspase-8 and -9 was found only at the end stage of mutant SOD-expressing transgenic mice (Guégan et al., 2001, 2002).
The lack of obvious TUNEL positivity in degenerating motor neurons in the ventral horns of G93A mice, however, suggests that the motor neuron degeneration in these mice may be mediated through caspase-independent mechanisms. It has been well documented that ALS motor neurons undergo vacuolarization of the rough endoplasmic reticulum and the mitochondria, which is atypical for apoptosis. This may reflect a mitochondrial dysfunction at a very early stage even before the onset of motor deficits in these mouse models (Gurney et al., 1994; Wong et al., 1995; Kong and Xu, 1998), possibly through a Bax-mediated process (Guégan et al., 2001). Bax has been shown to induce caspase-independent cell death (Xiang et al., 1996). However, the weak protection by Bcl-2 overexpression in a mouse model of ALS (Kostic et al., 1997) suggests an involvement of alternative cell death mechanisms as well.
Although degenerating motor neurons are not TUNEL positive, a few cells in the white matter of G93A transgenic mice are TUNEL positive. This result may suggest that cell types other than motor neurons may undergo apoptosis as a secondary response to motor neuron degeneration, which is partially regulated by caspase-11. Other caspases or caspase-independent mechanisms may be involved in the death of these cells as well. We showed previously that introduction of dominant-negative caspase-1 (C281G) mutant in G93A mice delayed the disease onset and progression (Friedlander et al., 1997). Because caspase-1 interacts with caspase-11 (Wang et al., 1998), this dominant-negative mutant of caspase-1 may act by inhibiting caspase-11. However, because the deletion of caspase-11 did not prevent neurodegeneration in the G93A mice, it is possible that caspase-1C281G may act by inhibiting caspases other than caspase-1, -11, or -3 or by another unknown gain-of-function process.
The exact parallels between SOD1 mutant transgenic mice and human ALS are not clear because of the difference in the temporal courses of disease development. Although the activation of caspases and apoptosis in this mouse model of ALS may reflect a secondary degenerative response, it remains to be examined whether inhibition of selective caspases may be beneficial for treatment of ALS in human. In particular, considering that onset and progression of ALS in humans occur over a much longer period of time, blocking apoptotic cell death might still prove beneficial because it may expand a therapeutic window for treatments. Nevertheless, our study provides a cautionary note when considering caspases as a direct therapeutic target for treatment of ALS.
Footnotes
This work was supported by grants from the National Institute on Aging and the Amyotrophic Lateral Sclerosis Association (J.Y.). We thank Christian Mahlke for technical assistance in mouse genotyping, Hong Zhu for generating monoclonal antibodies, and the Yuan laboratory members for helpful advice.
Correspondence should be addressed to Junying Yuan, Department of Cell Biology, Harvard Medical School, 240 Longwood Avenue, Boston, MA 02115. E-mail: jyuan@hms.harvard.edu
I. Sanchez's present address: Department of Anatomy and Neurobiology, Boston University School of Medicine, 715 Albany Street, Boston, MA 02118.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235455-06$15.00/0
References
- Cleveland DW, Rothstein JD ( 2001) From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2: 806–819. [DOI] [PubMed] [Google Scholar]
- Cryns V, Yuan J ( 1998) Proteases to die for. Gen Dev 12: 1551–1570. [DOI] [PubMed] [Google Scholar]
- Friedlander RM, Brown RH, Gagliardini V, Wang J, Yuan J ( 1997) Inhibition of ICE slows ALS in mice. Nature 388: 31. [DOI] [PubMed] [Google Scholar]
- Guégan C, Vila M, Rosoklija G, Hays AP, Przedborski S ( 2001) Recruitment of the mitochondrial-dependent apoptotic pathway in amyotrophic lateral sclerosis. J Neurosci 21: 6569–6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guégan C, Vila M, Teissman P, Chen C, Onteniente B, Li M, Friedlander RM, Przedborski S ( 2002) Instrumental activation of bid by caspase-1 in a transgenic mouse model of ALS. Mol Cell Neurosci 20: 553–562. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, Chen W, Zhai P, Sufit R, Siddique T ( 1994) Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 264: 1772–1775. [DOI] [PubMed] [Google Scholar]
- Hall ED, Oostveen JA, Gurney ME ( 1998) Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia 23: 249–256. [DOI] [PubMed] [Google Scholar]
- Hisahara S, Yuan J, Momoi T, Okano H, Miura M ( 2001) Caspase-11 mediates oligodendrocyte cell death and pathogenesis of autoimmune-mediated demyelination. J Exp Med 193: 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SJ, Wang S, Hara H, Peterson EP, Namura S, Amin-Hanjani S, Huang Z, Srinivasan A, Tomaselli KJ, Thornberry NA, Moskowitz MA, Yuan J ( 2000) Dual role of caspase-11 in mediating activation of caspase-1 and caspase-3 under pathological conditions. J Cell Biol 149: 613–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SJ, Wang S, Kuida K, Yuan J ( 2002) Distinct downstream pathways of caspase-11 in regulating apoptosis and cytokine maturation during septic shock response. Cell Death Differ 9: 1115–1125. [DOI] [PubMed] [Google Scholar]
- Kong J, Xu Z ( 1998) Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci 18: 3241–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic V, Jackson-Lewis V, de Bilbao F, Dubois-Dauphin M, Przedborski S ( 1997) Bcl-2: prolonging life in a transgenic mouse model of familial amyotrophic lateral sclerosis. Science 277: 559–562. [DOI] [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA ( 1995) Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science 267: 2000–2003. [DOI] [PubMed] [Google Scholar]
- Li M, Ona VO, Guégan C, Chen M, Jackson-Lewis V, Andrews LJ, Olszewski AJ, Stieg PE, Lee JP, Przedborski S, Friedlander RM ( 2000) Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science 288: 335–339. [DOI] [PubMed] [Google Scholar]
- Martin LJ ( 1999) Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J Neuropathol Exp Neurol 58: 459–471. [DOI] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM ( 1992) NeuN, a neuronal specific nuclear protein in vertebrates. Development 116: 201–211. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Julien JP, Rivest S ( 2001) Induction of proinflammatory molecules in mice with amyotrophic lateral sclerosis: no requirement for proapoptotic interleukin-1beta in neurodegeneration. Ann Neurol 50: 630–639. [DOI] [PubMed] [Google Scholar]
- Pasinelli P, Houseweart MK, Brown Jr RH, Cleveland DW ( 2000) Caspase-1 and -3 are sequentially activated in motor neuron death in Cu, Zn superoxide dismutase-mediated familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 97: 13901–13906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetz CR, Whitfield C ( 2002) Lipopolysaccharide endotoxins. Annu Rev Biochem 71: 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul C, Estevez AG, Nishimune H, Cleveland DW, deLapeyriere O, Henderson CE, Haase G, Pettmann B ( 2002) Motor neuron death triggered by a specific pathway downstream of Fas potentiation by ALS-linked SOD1 mutations. Neuron 35: 1067–1083. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek, Cayabyab A, Gaston S, Tanzi R, Halperin JJ, Herzfeldt B, Van den Berg R, Hung WY, et al. ( 1993) Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362: 59–62. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Rano TA, Peterson EP, Rasper DM, Timkey T, Garcia-Calvo M, Houtzager VM, Nordstrom PA, Roy S, Vaillancourt JP, Chapman KT, Nicholson DW ( 1997) A combinatorial approach defines specificities of members of the caspase family and granzyme B. Functional relationships established for key mediators of apoptosis. J Biol Chem 272: 17907–17911. [DOI] [PubMed] [Google Scholar]
- Vukosavic S, Stefanis L, Jackson-Lewis V, Guégan C, Romero N, Chen C, Dubois-Dauphin M, Przedborski S ( 2000) Delaying caspase activation by Bcl-2: a clue to disease retardation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci 20: 9119–9125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Miura M, Jung Y, Zhu H, Gagliardini V, Shi L, Greenberg AH, Yuan J ( 1996) Identification and characterization of Ich-3, a member of the interleukin-1beta converting enzyme (ICE)/Ced-3 family and an upstream regulator of ICE. J Biol Chem 271: 20580–20587. [DOI] [PubMed] [Google Scholar]
- Wang S, Miura M, Jung YK, Zhu H, Li E, Yuan J ( 1998) Murine caspase-11, an ICE-interacting protease, is essential for the activation of ICE. Cell 92: 501–509. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL ( 1995) An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14: 1105–1116. [DOI] [PubMed] [Google Scholar]
- Xiang J, Chao DT, Korsmeyer SJ ( 1996) BAX-induced cell death may not require interleukin 1 beta-converting enzyme-like proteases. Proc Natl Acad Sci USA 93: 14559–14563. [DOI] [PMC free article] [PubMed] [Google Scholar]