

Abstract
The knock-out analyses of neuregulin and its receptors have indicated that they play essential roles in Schwann cell development. However, the role they play in oligodendrocyte development in vivo has remained unclear, because such knock-out animals die before CNS myelination begins. We examined the role of neuregulin signaling in the CNS by generating transgenic mice that express a dominant-negative mutant of the ErbB2 receptor among oligodendrocytes, using an MBP promoter. The transgenic mice exhibited widespread hypomyelination, resulting from a reduction in oligodendrocyte differentiation. The number of progenitors was conversely increased in the transgenic mice. We report that a reduction in oligodendrocyte differentiation is attributed in part to apoptosis of oligodendrocyte progenitors as they exit the cell cycle. A significant reduction in the number of p27+ oligodendrocyte precursors in the transgenic mice supports this conclusion. Taken together, these data suggest that for oligodendrocyte progenitors, ErbB2 signaling plays a role in governing a properly timed exit from the cell cycle during development into myelinating oligodendrocytes.
Keywords: ErbB2, neuregulin, p27, cell-cycle exit, differentiation, oligodendrocytes
Introduction
The development of oligodendrocytes is regulated by both cell intrinsic factors as well as cell extrinsic factors. Cell intrinsic factors include oligodendrogenic genes, olig-1, olig-2, and Sox10 (Lu et al., 2002; Stolt et al., 2002; Zhou et al., 2001), which control cell fate specification in the oligodendrocyte lineage. Extrinsic factors include PDGF, basic FGF, neurotrophin-3, insulin-like growth factor, jagged-1, and neuregulin (Noble et al., 1988; Richardson et al., 1988; Gard and Pfeiffer, 1993; Barres et al., 1994, 1993; Canoll et al., 1996; Wang et al., 1998; Vartanian et al., 1999; Park et al., 2001). These extrinsic factors influence different aspects of the maturation of oligodendrocytes, as they proliferate, exit the cell cycle, differentiate, and begin myelination. Progress through these different developmental stages can be followed by various surface markers. Dividing oligodendrocyte progenitors are positive for A2B5 immunoreactivity in vitro (Raff et al., 1983), and PDGF receptor α and NG2 (chondroitin sulfate proteoglycan) in vivo (Levine and Stallcup, 1987; Hall et al., 1996; Nishiyama et al., 1996). As they develop into proliferative pro-oligodendrocytes, they begin to express O4 antigen on the cell surface, and as they exit the cell cycle, they express galactosylcerebroside (GalC) that is detected by O1 antibody (Gard and Pfeiffer, 1990). These premyelinating oligodendrocytes express myelin basic protein (MBP), and they continue to express MBP as they myelinate axons.
It has been shown that axons or axon-derived factors play essential roles for different aspects of oligodendrocyte development, such as proliferation, differentiation, and survival, in addition to myelination (Barres and Raff, 1999). Of the extrinsic factors released by the axons, neuregulin (NRG) is of particular importance for many aspects of oligodendrocyte development. NRG is expressed by neurons during development (Chen et al., 1994; Meyer et al., 1997), and when its action was blocked in vivo, the axons no longer supported the survival of oligodendrocytes in the optic nerve (Fernandez et al., 2000), suggesting that NRG is indeed an axon-derived factor that is required for oligodendrocyte survival. In culture, NRG appears to play diverse, sometimes contrary roles. When added to cultured oligodendrocyte progenitors, NRG promoted the proliferation of O4+ cells and inhibited their differentiation into MBP+ populations (Canoll et al., 1996, 1999). However, when ErbB2, the major signaling receptor for NRG, was deleted, O4+ cells were unaffected in spinal cord explant cultures (Park et al., 2001). Instead, there was a dramatic loss of O1+ and MBP+ cells, suggesting that NRG promotes differentiation (Park et al., 2001). The reason for the disparate results needs additional analysis, but the difference between tissue explant versus dissociated culture conditions may be partly responsible for the discrepancy, because the signaling input from the extracellular matrix components can affect NRG action in culture (Colognato et al., 2002).
In this report, we sought to examine the role of NRG/ErbB2 in vivo, by perturbing the signaling ability of ErbB2 among only oligodendrocytes, through the expression of a dominant-negative ErbB2 receptor under MBP promoter in transgenic mice. Although knock-out mice have been generated to analyze the function of NRG signaling, clarification of its role in vivo has remained untenable, because knock-out mice of NRG-1 and their receptors, ErbB2, ErbB3, and ErbB4, die before myelination begins in the CNS, mainly of heart defects (Gassmann et al., 1995; Lee et al., 1995; Meyer and Birchmeier, 1995; Riethmacher et al., 1997; Morris et al., 1999). Here, we demonstrate that when ErbB2 signaling was attenuated, the mice developed a widespread hypomyelination in the CNS. Hypomyelination was the result of a reduction in oligodendrocyte differentiation that arose, at least in part, from the progenitors failing to exit the cell cycle at appropriate times. Taken together, these data suggest that ErbB2 signaling is critical for oligodendrocyte differentiation in vivo.
Materials and Methods
Generation of transgenic constructs and identification of transgenic mice: MBP-p75–ErbB1KD. The cDNA for the p75–ErbB1 KD construct has been described previously (Harrington et al., 2002). The p75–ErbB1 KD was placed under a 9 kb MBP by (1) placing the p75–ErbB1 KD within the exon 3 of the β-globin gene, and (2) ligating the p75–ErbB1 KD fragment and the flanking sequences containing the exon 2–intron 2 of the β-globin gene with the 9 kb MBP promoter. For the first step, the p75–ErbB1 KD in pCR II vector was digested with SpeI and SalI, blunted, and placed into EcoRI-digested pMG2 vector. The pMG2 vector contains 1.8 kb of MBP promoter that was linked to exon 2, intron 2, and exon 3 of the β-globin gene (Gow et al., 1992). This ligation places the p75–ErbB1 KD cDNA into the third exon (see Fig. 2A). For the second step, the fragment containing the 52 bp MBP promoter, exon 2, intron 2, and the p75–ErbB1 KD within the exon 3 of the β-globin gene was subsequently isolated by XmaI and NotI digestion, and placed into pBS–MBP9 vector that contained 9 kb MBP promoter with no exons or introns. The resulting construct is depicted in Figure 2A. To prepare a fragment for microinjection, the construct was digested with SalI and NotI, which yields a fragment that contains the 9 kb MBP promoter, exon 2, intron 2, and the p75–ErbB1 KD within the exon 3 of the β-globin gene. In this construct, translation begins at the start of the transgene. The transgenic mice were produced at the Keck Genetics Research Facility at the Neurobiotechnology Center of the Ohio State University, using methods described previously (Hogan et al., 1994). Transgenic founders were identified by PCR.
Figure 2.

The expression of the p75–ErbB1 KD follows the pattern of endogenous MBP expression during development. A, A diagram of the transgenic p75–ErbB1 KD construct under the MBP promoter. The location of PCR primers that were used for identification of transgene (TG) is indicated by arrows. The angled arrow indicates the start of transcription. Translation starts at the beginning of the transgene. B, The expression of the p75–ErbB1 KD during development. MBP staining was first observed at P5 in the white-matter tracts of the cerebellum; it increased as development proceeded. The p75–ErbB1 KD expression was followed by HA staining in transgenic littermates. Note the reduction in MBP staining in TG compared with NTG at each developmental time point analyzed. Scale bar, 20 μm. C, The p75–ErbB1 KD is targeted to the cell surface in culture. Mouse oligodendrocytes were stained live with anti-HA to detect the p75–ErbB1 KD on the cell surface. The NTG cultures do not show HA staining. Mouse oligodendrocyte cultures were as described previously (Harrington et al., 2002). Scale bars, 12.5 μm.
EM analyses. For transmission electron microscopy (TEM) of the tissues, mice were perfused with 2% paraformaldehyde and 2% glutaraldehyde in 0.1 m Na cacodylate buffer, pH 7.2. The tissues were dissected, rinsed in 0.1 m Na cacodylate buffer, and placed in 1% osmium and 0.1 m Na cacodylate for 1–1.5 hr at room temperature. The tissues were stained en bloc for 1 hr in 2% uranyl acetate and embedded in Spurr resin after the dehydration procedures. Sections were cut at 80 nm using an Ultracut E ultramicrotome (Leica, Deerfield, IL), and collected on 300-mesh grids. Sections were stained in 2% uranyl acetate and Reynolds lead citrate before observation in a Philips (Wilmington, DE) CM 12 TEM at 60 kV.
Immunohistochemistry. Mice were perfused with 3% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.2. The brain was dissected, postfixed for several hours at room temperature, and placed in 20% sucrose and 0.1 m PB at 4°C. The brain was cut at 20 μm thicknesses on a cryostat in the sagittal plane. For staining with antibodies, the sections were incubated in blocking solution containing 1% BSA, 0.3% Triton X-100, and 10% horse serum for mouse monoclonal antibody or 10% goat serum for rabbit polyclonal antibody in 0.1 m PB for 2 hr at room temperature. The primary antibodies were incubated in 5% horse or goat serum, 0.1% BSA in 0.1 m PB overnight at room temperature in the presence of 0.02% sodium azide. The positive staining was detected using Alexa-488- or Cy3-conjugated secondary antibodies, or biotinylated secondary antibodies that were later visualized using streptavidin-Cy3 or Alexa-488. The sections were mounted using Vectashield containing 4′,6′-diamidino-2-phenylindole to stain the nuclei (Vector Laboratories, Burlingame, CA). For staining with anti-bromodeoxyuridine (BrdU), mice were injected with BrdU at 25 μg/g three times at 2 hr intervals. Two hours after the last injection, mice were perfused. BrdU staining was detected essentially as described previously (Seri et al., 2001). For double staining with BrdU and ErbB1, the sections were first incubated overnight with anti-ErbB1, and processed for BrdU as described previously (Seri et al., 2001), and incubated overnight with anti-BrdU. The signal from ErbB1 and BrdU was visualized as described above. The antibodies used for this study are ErbB1 and p27 (Santa Cruz Biotechnology, Santa Cruz, CA), MBP, NG2, and neurofilament (Chemicon, Temecula, CA), adenomatous polyposis (CC1) (Oncogene Sciences, Uniondale, NY), BrdU (BD Biosciences, Bedford, MA), 2′,3′-cyclin nucleotide 3′-phosphodiesterase (CNPase) (Sternberger Monoclonals, Lutherville, MD), proliferating cell nuclear antigen (PCNA) (Dako, Carpinteria, CA), HA and p75 (Babco, Richmond, CA), O1 (a gift from Dr. Patrick Wood, University of Miami, Miami, FL), 192 (an anti-p75; a gift from Dr. Phil Barker, McGill University, Montreal, Canada), and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) (Roche Molecular Biochemicals, Indianapolis, IN).
Immunoprecipitation/Western blot analyses. Mouse brains were homogenized in a lysis buffer containing 1% Nonidet P-40, 20 mm Tris, pH 8.0, 137 mm NaCl, 0.5 mm EDTA, 10% glycerol, 10 mm Na2P2O7, 10mm NaF, 1 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mm vanadate, and 1 mm phenylmethylsulfonyl fluoride. The procedures for immunoprecipitation and Western blotting were essentially as described previously (Yoon et al., 1998). The antibodies used for this study are ErbB1, ErbB2, ErbB3, and p27 (Santa Cruz Biotechnology), phosphotyrosine antibodies (4G10, Upstate Biotechnology, Lake Placid, NY; PY20, Transduction Laboratories Transduction Laboratories, Lexington, KY; and PY99, Santa Cruz Biotechnology). NRG was purchased from R & D Systems (Minneapolis, MN).
Receptor autokinase assays. The ErbB2 receptors in the mouse brain lysates were immunoprecipitated using antibodies to ErbB2 (Santa Cruz Biotechnology), and the ErbB2 receptor immune complexes were washed in HNTG buffer containing 20 mm HEPES, pH 7.4, 150 mm NaCl, 0.1% Triton X-100, 10% glycerol, 10 mm NaF, 1 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mm vanadate, and 1 mm phenylmethylsulfonyl fluoride. The receptor kinase reaction was performed in HNTG buffer plus 5 mm MnCl2, 20 μm ATP, and 20 μCi of [γ-32P]ATP at 4°C for 15 min as described previously (Pinkas-Kramarski et al., 1996). The reaction products were separated on SDS-PAGE, and subjected to autora-diography. For quantification, the bands corresponding to appropriate receptors were cut out from the dried gel and counted in a scintillation counter.
Quantitation of CC1+/TUNEL+, CC1+, and BrdU+cells. Every fourth sagittal section (20 μm in thickness) was processed for immunohistochemistry; positively stained cells were counted from the cerebellar white matter.
Mouse oligodendrocyte cultures. The procedures were performed as described previously (Harrington et al., 2002). After 4–6 d in culture, cells were stained live for the presence of the transgene on the cell surface using anti-HA antibody.
Results
The p75–ErbB1KD construct is described in Figure 1A. The construct comprises two domains: one containing the cytoplasmic domain of ErbB1 with its ATP binding site inactivated, and the other containing the extracellular and transmembrane domains of a neurotrophin receptor, p75. The rationale behind choosing the p75–ErbB1KD as a dominant-negative mutant for ErbB2 is that ErbB1 is a preferred heterodimeric partner for ErbB2 with high affinity, in response to the widest range of ligands in the NRG family (Jones et al., 1999), and the cytoplasmic domain of ErbB1 was shown to efficiently heterodimerize with ErbB2 (Chantry, 1995; Qian et al., 1999; Brennan et al., 2002). In addition, neither p75 nor ErbB1 is expressed among oligodendrocytes lineage during development in vivo (see Fig. 3), so that one could selectively inhibit the signaling ability of endogenous ErbB2 in oligodendrocytes, without interfering with ErbB1 signaling. To prevent the p75–ErbB1KD from binding to the NRG family of ligands, the extracellular and transmembrane domains of ErbB1 were replaced by the corresponding domains of p75. The p75–ErbB1KD can bind neurotrophins, and it has been reported recently that neurotrophin 3 (NT3) inhibits Schwann cell myelination via TrkC in vivo (Cosgaya et al., 2002). Although the role of neurotrophins in oligodendrocytes development in vivo is unclear, NT3 has been shown to have some effect on the proliferation of oligodendrocytes precursors in the optic nerve (Barres et al., 1994) or to promote oligodendrocyte myelination in vitro (Yan and Wood, 2000). Therefore, we tested whether the p75–ErbB1KD can interfere with Trk signaling. For this, 293T cells were transfected with TrkA, and subsequently infected with either the control GFP or the p75–ErbB1KD adenoviruses. The adenovirus carrying the cDNA for the p75–ErbB1KD has been described previously (Harrington et al., 2002). After NGF treatment, the activation status of the immunoprecipitated TrkA was assessed in Western blot analyses using phosphotyrosine antibodies (Fig. 1B, top). The extent of TrkA activation in the presence of the p75–ErbB1KD was similar or identical to the control cells. Similar data were obtained in PC12 cells that express TrkA and p75 (data not shown). Therefore, these data suggest that the p75–ErbB1KD does not interfere with Trk signaling.
Figure 1.

The p75–ErbB1 KD associates with ErbB2 and inhibits NRG-dependent activation of ErbB2 in 293 cells. A, Experimental strategy of the p75–ErbB1 KD construct. The p75–ErbB1 KD would heterodimerize with ErbB receptors and inhibit tyrosine phosphorylation of the associated ErbB receptors. The p75–ErbB1 KD itself does not bind EGF, because the ligand-binding domain is replaced with the corresponding domain of p75. B, The p75–ErbB1 KD does not affect TrkA signaling in the presence of NGF. 293T cells were transfected with TrkA, and subsequently infected with the control GFP or p75–ErbB1 KD adenovirus. After 5 min of treatment with NGF, the lysates were immunoprecipitated with pan-Trk antibody, and the activation status of TrkA was detected using 4G10 antibody. The same blot was reprobed for the presence of the transfected TrkA using pan-Trk antibody in IP/W. Note that tyrosine phosphorylation of TrkA is not affected in the presence of the p75–ErbB1 KD. The presence of the p75–ErbB1 KD is shown as an HA blot. C, The p75–ErbB1 KD associates only with ErbB2, but not with ErbB3 or ErbB4 in 293T cells. 293T cells were transfected with ErbB2, 3, or 4, and subsequently infected with the control GFP or p75–ErbB1 KD adenovirus. After immunoprecipitation with antibodies against ErbB2, 3, or 4, or HA tag included in the p75–ErbB1 KD, the associated p75–ErbB1 KD was detected with anti-HA antibody. A portion of the lysates was subjected in parallel to IP/W, using ErbB2, 3, or 4 antibodies as controls. D, The p75–ErbB1 KD inhibits NRG-dependent activation of ErbB2. 293T cells were transfected with ErbB4 to facilitate activation of the endogenous ErbB2 with NRG, and subsequently infected with the control GFP or p75–ErbB1 KD adenovirus. After 5 min of treatment of soluble NRG, the lysates were immunoprecipitated with ErbB2 antibody and the activation status of ErbB2 was detected using PY99 antibody. The presence of the p75–ErbB1 KD was detected with HA antibody in IP/W. Note that tyrosine phosphorylation of ErbB4 is also reduced in the presence of the p75–ErbB1 KD. E, ErbB4 is present in the p75–ErbB1 KD–ErbB2 complex. 293T cells were transfected with Flag–ErbB4 to facilitate activation of the endogenous ErbB2 with NRG, and subsequently infected with the control GFP or p75–ErbB1 KD adenovirus. After 5 min of treatment of soluble NRG, the lysates were immunoprecipitated with ErbB2 antibody, and the presence of ErbB4 and the p75–ErbB1 KD was probed with anti-Flag or HA antibodies. W:PY, Phosphotyrosine.
Figure 3.
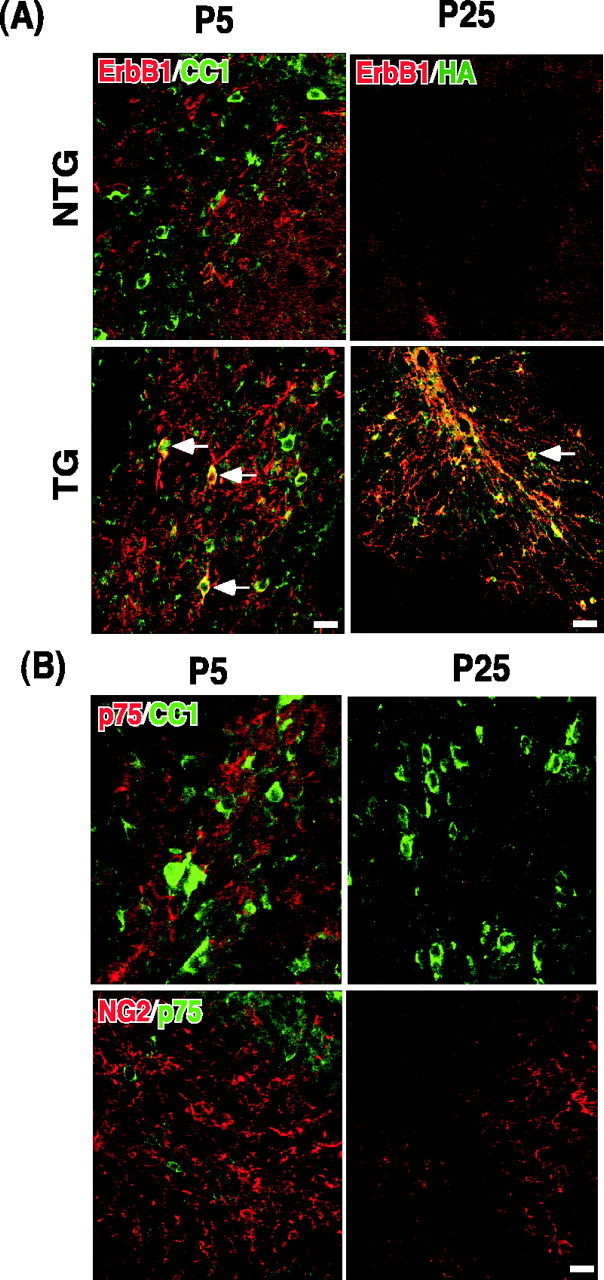
ErbB1 and p75 are not expressed among oligodendrocyte lineage. A, ErbB1 is not expressed in oligodendrocyte lineage. Sagittal brain sections from P5 and P25 mice were analyzed for ErbB1 expression among CC1 + cells. Staining with TG sections is shown as a positive control, because ErbB1 antibody detects the cytoplasmic domain of the human ErbB1 that is present in the p75–ErbB1 KD. At P5, there is some positive staining for ErbB1 in the cerebellar white matter, but it did not colocalize with CC1. These stainings represent migrating cerebellar granule neurons. In TG, subpopulations of CC1 + cells begin to stain for ErbB1 at P5 (arrows). At P25, there is no ErbB1 staining in the cerebellar white matter, nor is there HA staining. In TG at P25, however, almost every cell body positive for ErbB1 is also stained for HA (arrow). B, p75 is not expressed in oligodendrocyte lineage. Sagittal brain sections from P5 and P25 rats were analyzed for p75 expression among oligodendrocytes using CC1 and NG2 as oligodendrocyte markers. At P5 there were many p75 +, CC1 +, and NG2 + cells in the cerebellar white matter, but p75 staining did not colocalize with CC1 or NG2. At P25 there was no p75 staining detected in the cerebellar white matter. Scale bars, 12.5 μm.
The ability of the p75–ErbB1KD to function as a dominant-negative mutant to ErbB2 was characterized in 293T cells. To test its association with ErbB receptors, 293T cells were transfected with ErbB2, 3, or 4 and subsequently infected with either the GFP control or the p75–ErbB1KD adenovirus. After immunoprecipitation with antibodies against ErbB2, 3, or 4, the p75–ErbB1KD was found to be in a complex with ErbB2, but not ErbB3 or 4 (Fig. 1C). We next tested whether the association with the p75–ErbB1KD results in modulation of ErbB2 signaling in the presence of NRG. For this, 293T cells were transfected with ErbB4 to activate endogenous ErbB2 in 293T cells, because ErbB2 does not bind NRG directly. Twenty-four hours after infection with the p75–ErbB1KD or GFP adenoviruses, cells were treated for 10 min with 25 ng/ml of soluble NRG. The extent of ErbB2 activation was measured in immunoprecipitation/Western blot analyses (IP/W), using ErbB2 antibody for the immunoprecipitation and phosphotyrosine antibodies for the Western blot. In control cells, NRG induced tyrosine phosphorylation of ErbB2 and ErbB4 (Fig. 1D). However, in the presence of the p75–ErbB1KD there was significant attenuation of the NRG-mediated tyrosine phosphorylation of ErbB2. These results indicate that the p75–ErbB1KD can function as a dominant-negative mutant for the endogenous ErbB2, a major signaling receptor in NRG signaling (Graus-Porta et al., 1997). It should be noted that the extent of tyrosine phosphorylation in ErbB4 was also attenuated in the presence of the p75–ErbB1KD, although ErbB4 does not interact with the p75–ErbB1KD directly (Fig. 1C,D). This result suggests that ErbB4 was present in the p75–ErbB1KD–ErbB2 complex. As predicted, ErbB4 was detected in the immune complex with the p75–ErbB1KD when we immunoprecipitated ErbB2 (Fig. 1E). These results suggest that the p75–ErbB1KD can block overall NRG signaling in oligodendrocytes, which express mainly ErbB2 and ErbB4 (Canoll et al., 1996, 1999).
To assess the role of NRG–ErbB2 signaling in oligodendrocyte development in vivo, we introduced the p75–ErbB1KD under MBP promoter (Fig. 2A). The MBP promoter contains 9 kb of 5′ sequence as described previously (Forghani et al., 2001). From a pronuclear injection, four independent founder lines that expressed the p75–ErbB1KD were obtained, one of which died as a result of severe seizures soon after weaning. The three remaining founders exhibited posterior tremor, shivering, mild gait difficulty, and/or seizures. These behavioral phenotypes began near postnatal day 10 (P10), persisted for 2–3 months, and then stopped. For this study, two independent lines were analyzed. Both lines exhibited the same phenotype, although the extent of the phenotype differed because of different levels of the p75–ErbB1KD expression.
We first examined whether the expression of the p75–ErbB1KD followed the expression pattern of endogenous MBP. Sagittal brain sections from the transgenic mice (TG) were stained for HA to detect the presence of the p75–ErbB1KD (Fig. 2B), whereas the brains from both TG and nontransgenic control (NTG) littermates were analyzed for endogenous MBP expression at P5, P10, P15, P20, and P25 (Fig. 2B). For the analyses, we focused on the white-matter tract in the cerebellum. The level of the endogenous MBP was weak at P5, and its expression increased gradually, developing into an adult pattern of white-matter MBP expression at P25. The p75–ErbB1KD expression pattern was also detected weakly at P5 based on HA staining, with a gradual increase into adulthood. This result suggests that the p75–ErbB1KD expression followed the endogenous MBP expression pattern in a correct temporal manner. Using a 3.2 kb MBP promoter linked to LacZ, Foran and Peterson (1992) have similarly reported that P6 was the earliest time of LacZ expression in the cerebellum. In TG, the endogenous MBP expression was also first detected at P5, suggesting that the temporal course of MBP expression was not affected by the presence of the transgene. Although the level of MBP increased in TG as development proceeded, its level at each developmental time point was reduced compared with its littermate controls. These results suggest potential hypomyelination in TG, which will be discussed further below. We next tested whether the p75–ErbB1KD is targeted to the plasma membrane by culturing oligodendrocytes and staining them live for the surface-bound chimeric receptor. Oligodendrocytes from TG were stained for HA, whereas those in NTG were not (Fig. 2C), confirming that the p75–ErbB1KD is correctly targeted to the plasma membrane.
To ensure that there is no cross talk between the p75–ErbB1KD and ErbB1 or p75 in vivo, we examined whether oligodendrocytes express p75 or ErbB1 during development in the CNS. For endogenous ErbB1 expression, we stained sagittal brain sections from P5 and P25 with ErbB1 antibody and CC1, a marker for oligodendrocytes. In our study, CC1 staining is almost identical to GalC staining, a postmitotic marker for oligodendrocytes (Richardson et al., 1988; Gard and Pfeiffer, 1990). In NTG cerebellum at P5, there were numerous CC1+ cells, but their staining did not colocalize with ErbB1, which was mainly expressed among migrating cerebellar granule cells at that time (Fig. 3A). However, in TG, subpopulations of CC1+ cells were stained for ErbB1 (Fig. 3A, arrows in TG), detecting the cytoplasmic domain of the p75–ErbB1KD. This is better shown in TG cerebellar staining at P25, where ErbB1 staining overlapped completely with HA staining. At P25 in NTG, however, there was no expression of ErbB1 in the white-matter tract of the cerebellum. Thus, these data demonstrate that ErbB1 is not expressed in oligodendrocyte lineage during development.
The expression of p75 during oligodendrocyte development was examined using rat brain sections, because of antibody compatibility. In the white-matter tract of the cerebellum at P5, there were many p75+ cells, but its staining did not colocalize with either CC1 or NG2 (Fig. 3B). The cells that are p75+ at that time represent migrating cerebellar granule cells. At P25 there was no p75 staining detected in the cerebellar white matter, although many cells were positive for CC1. Therefore, these data indicate that p75 is not expressed in oligodendrocyte lineage during normal development, although injury can induce p75 expression among oligodendrocytes in the CNS or culturing itself can induce p75 (Beattie et al., 2002).
The behavioral phenotype and the MBP expression level in our transgenic mice indicated a defect in myelination. Therefore, we processed the adult spinal cord of the two transgenic lines for the electron microscopy (EM) analyses. In both transgenic lines, there was a reduction in myelinated fibers in the ventral funiculi (Fig. 4A). The extent of hypomyelination was first quantified by counting the number of axon fibers that were myelinated regardless of thickness using the TG-2 line. In NTG, 70% of fibers within all funiculi were myelinated and 30% were unmyelinated. In TG this was reversed, with 35% of fibers myelinated and 65% unmyelinated (Fig. 4B). Although thickness was not considered when making the counts, it should be noted that even among the myelinated fibers, the myelin sheath is much thinner in TG than in NTG. Therefore, we evaluated the ratio of myelin thickness to axon diameter. In NTG, the myelin thickness increased as the axon diameter increased, but in TG-2, the increase in the axon diameter was not accompanied in a similar proportion (Fig. 4C). Based on MBP staining of sagittal brain sections, the hypomyelination phenotype was widespread in the CNS. An example is shown in Figure 4D, with a focus on the corticospinal tract and the cerebellum (see also Fig. 2B). It should be noted in Figure 4D that reduced neurofilament staining accompanied the reduction in MBP staining, confirming a tight relationship between axons and oligodendrocyte development as proposed previously (Barres and Raff, 1999). Taken together, these results indicate that the p75–ErbB1KD mice exhibited a hypomyelination defect in the CNS.
Figure 4.
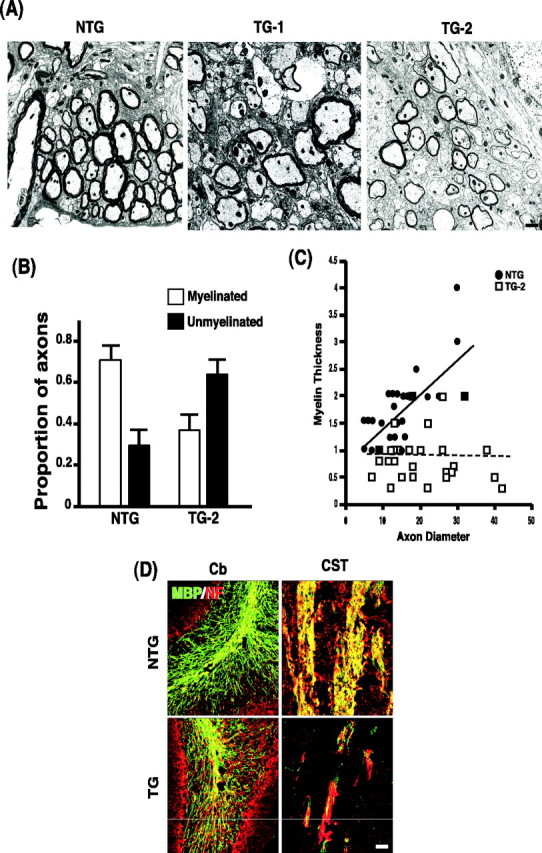
Widespread hypomyelination in the p75–ErbB1 KD mice. A, Representative EM pictures from two transgenic lines and a littermate control. Scale bar, 400 nm. B, Quantification of hypomyelination phenotype in the spinal cord of TG-2 line. The ratio of myelinated to unmyelinated axon fibers is reversed from 7 to 3 in NTG to 3.5 to 6.5 in TG-2. For quantification of myelinated axons from the ventral funiculus of the lumbar spinal cord, two rows of four grids (eight grids total) were prepared from tissues adjacent to the ventral medial fissure for the generation of electron photomicrographs. The data are from three sets of NTG and TG mice at P15. Note that the myelin sheath is also thinner among the myelinated fibers in TG. C, Relationship between the myelin thickness and axon diameter of the myelinated axons. The data are from 30 randomly selected myelinated axons from each group. D, The extent of hypomyelination in the cerebellum (Cb) and in the corticospinal tract (CST). Sagittal sections from P25 mice were processed for double immunohistochemistry with MBP and neurofilament (NF) antibodies. Scale bar, 12.5 μm.
We next examined whether the hypomyelination phenotype was attributable to perturbation of ErbB2 signaling in vivo. The p75–ErbB1KD was present in immune complexes of ErbB2, but not of ErbB3 or ErbB4, when the brain and spinal cord lysates were analyzed (Fig. 5A, top). This result suggests that the p75–ErbB1KD interacts with only ErbB2 in vivo, as in 293T cells. The total levels of these three receptors were very similar when comparing TG with NTG (Fig. 5A, bottom), suggesting that the expression of the p75–ErbB1KD did not affect the overall level of these receptors. The consequence of this physical association between the p75–ErbB1KD and the endogenous ErbB2 was examined. For this, ErbB2 was immunoprecipitated, and the ability of ErbB2 to phosphorylate its oligomeric partners was assessed by adding 32P-γATP to the immune complexes. In TG, there was an ∼45% reduction in the amount of radiolabeled ErbB2 compared with its littermate control (Fig. 5B). A similar reduction in tyrosine phosphorylation of ErbB2 was also observed in TG compared with controls (data not shown). These data indicate that ErbB2 signaling was significantly attenuated in vivo in the presence of the p75–ErbB1KD.
Figure 5.

Attenuation of ErbB2 signaling by the p75–ErbB1KDin vivo. A, The p75–ErbB1KD associates with the endogenous ErbB2, but not with ErbB3 or ErbB4 in vivo. The brain and the spinal cord lysates from adult mice were subjected to IP using ErbB2, 3, or 4 or HA antibodies followed by Western blotting with anti-HA to detect associated p75–ErbB1KD. Note that the level of three ErbB receptors did not change in the presence of the p75–ErbB1KD. B, The p75–ErbB1KD inhibits the signaling ability of the endogenous ErbB2 by 45%. The brain lysates were subjected to receptor autokinase assays using 32P-γATP, after IP with ErbB2. The quantification data are from 12 independent assays, using 10–12 different sets of adult littermates. A representative picture of the assay is shown. A parallel IP/Western blot with ErbB2 antibody was performed as a control. C, The p75–ErbB1KD was not phosphorylated by the endogenous ErbB2 in vivo. The brain lysates were subjected to IP with ErbB2, followed by Western with anti-phospho ErbB1 (top). There was no band at the position of the p75–ErbB1KD (arrow). The asterisk represents a nonspecific band that is present in all the lysates. The p75–ErbB1KD was also not phosphorylated by the endogenous ErbB2 in autokinase assays. The arrow points to the position of the p75–ErbB1KD. Error bars indicate SEM.
When the p75–ErbB1KD associates with ErbB2, ErbB2 can potentially phosphorylate the p75–ErbB1KD at tyrosine residues. This could lead to the recruitment of ErbB1 adaptor molecules and subsequent signaling events. To determine whether the cytoplasmic ErbB1KD domain in the p75–ErbB1KD is phosphorylated at tyrosine residues on association with ErbB2, we immunoprecipitated ErbB2 and blotted with anti-phosphoErB1. There was no tyrosine phosphorylation in the ErbB1KD domain that associated with ErbB2, in two independent TG lines (Fig. 5C, top). Because the phosphoErbB1 antibody detects only the phosphorylation status of tyrosine 845, it is possible that other tyrosine residues may become phosphorylated by association. For this, we subjected the ErbB2 immune complexes to an autokinase assay to detect the overall phosphorylation status of the p75–ErbB1KD, as was done in Figure 5B. Even with increasing amounts of lysates, there was no phosphorylation of the p75–ErbB1KD (Fig. 5C, bottom, arrow). These data suggest that the cytoplasmic ErbB1KD domain in the p75–ErbB1KD is not phosphorylated by the endogenous ErbB2. This lack of cross-phosphorylation may suggest that ligand binding is necessary for the proper conformational change and subsequent autophosphorylation between receptor tyrosine kinases.
We next examined the outcome of attenuated ErbB2 signaling in oligodendrocyte development. Brain sections taken from different developmental time points were stained with differentiation markers, such as O1, and CNPase. MBP staining is shown in Figure 2B. The data with O1 staining in the white-matter tract of the cerebellum is shown in Figure 6A, but similar data were obtained with CNPase as well as MBP staining throughout the CNS (corpus callosum, corticospinal tract, brainstem, spinal cord). O1 staining increased as development proceeded both in TG and NTG, but at each time point, there was a significant reduction in O1 staining in TG compared with NTG. By adulthood, the number of mature oligodendrocytes was reduced by 47% in TG compared with NTG (Fig. 6B). This reduction in oligodendrocyte differentiation could result from a decrease in the number of progenitors. Therefore, we assessed the extent of proliferation using BrdU staining. In the white-matter tract of the cerebellum in both NTG and TG, the number of BrdU+ cells decreased as development proceeded (Fig. 6C,D). However, at each developmental stage analyzed, the number of BrdU+ cells in TG was higher than that in NTG. Similarly, the number of cells that were positive for both NG2, a marker for oligodendrocyte progenitors (Levine and Stallcup, 1987; Nishiyama et al., 1999), and PCNA, a marker for proliferating cells, also increased in TG compared with NTG (Fig. 6E). These results suggest that the increase in BrdU+ cells in TG represent, at least in part, the increase in NG2+PCNA+ oligodendrocyte progenitors. That there is a reduction in differentiated oligodendrocytes, although the number of progenitors increased, suggests that these cells must undergo premature cell death at some point, ultimately halting the course of their development. To assess the extent of apoptosis, we performed TUNEL assays at P5–P25. There were no changes in TUNEL+ cells between NTG and TG, except at P5 and P10. In the white-matter tracts of the cerebellum at P5, there was an increase in TUNEL+ cells in TG compared with NTG (Fig. 7A). The average increase in TUNEL+ cells was 2.1-fold. To assess apoptosis in the oligodendrocyte lineage cells only, we performed double staining of TUNEL and CC1. Although staining identical populations, CC1 antibody differs from GalC antibody in that it mainly stains the cell bodies without the extensive staining of the myelin processes, rendering cell counting feasible in tissue sections. In TG, the proportion of TUNEL+ cells among CC1+ cells increased 3.6-fold compared with NTG (Fig. 7B, left). Representative pictures are also shown (Fig. 7B, right). These results suggest that the cells committed to the oligodendrocyte lineage undergo apoptosis in TG, in which ErbB2 signaling is significantly attenuated. Taken together, these data indicate that ErbB2 signaling is critical in early differentiation in oligodendrocyte development in vivo.
Figure 6.

A subpopulation of postmitotic oligodendrocytes undergoes apoptosis. A, Reduction in oligodendrocyte differentiation in the p75–ErbB1 KD mice. Sagittal brain sections were stained for O1 at different developmental stages. O1 staining was reduced in the white-matter tracts of the cerebellum in the transgenic mice compared with NTG at every developmental stage analyzed. A similar pattern was also observed with CNPase staining (data not shown). Scale bar, 20 μm. B, Reduction in the number of mature oligodendrocytes in the p75–ErbB1 KD mice. Quantification of CC1 + cells was done from the white-matter tract of the cerebellum using P25 mice (n = 3). Error bars indicate SEM. C, An increase in BrdU incorporation in the p75–ErbB1 KD. Sagittal brain sections were stained with anti-BrdU. BrdU staining was increased in the white-matter tracts of the cerebellum in the p75–ErbB1 KD mice compared with NTG at every developmental stage analyzed. Scale bar, 20 μm. D, Quantification of BrdU + cells in the cerebellar white-matter tract during development. Error bars indicate SEM. E, Increased proliferation of NG2 + cells in the transgenic mice. The P5 brain sections were stained for NG2 and PCNA. Note the increase in the number of NG2 +/PCNA + cells in the white-matter tracts in the cerebellum (arrows). Scale bar, 12.5 μm.
Figure 7.

An increase in apoptosis of oligodendrocytes in the p75–ErbB1 KD mice. A, Quantification of TUNEL + cells at P5 (n = 3). Representative pictures from the cerebellum white matter (right). Error bars indicate SEM. B, Quantification of TUNEL + cells among CC1 + cells at P5(n=3). For quantification, see Materials and Methods. Representative pictures from the cerebellar white matter (right). An arrow points to TUNEL +CC1 + cells that were counted. Scale bar, 12.5 μm.
The level of p27, a cell cycle inhibitor, has been shown to increase as oligodendrocytes differentiate in culture (Durand et al., 1997). The critical role of p27 in oligodendrocyte development has been revealed by the analyses of p27–/– mice. In the absence of p27, proliferation of oligodendrocyte progenitors was prolonged with a concomitant decrease in the differentiated phenotype, suggesting that p27 is critical for oligodendrocyte progenitors to exit the cell cycle during their development (Casaccia-Bonnefil et al., 1997). In addition, the precocious expression of p27 into oligodendrocyte progenitors caused premature growth arrest in the presence of the mitogens, suggesting that p27 is sufficient for the cell cycle exit (Tikoo et al., 1998). Because the attenuated ErbB2 signaling resulted in an increase in proliferation, with a less differentiated phenotype, we examined whether the p27 level has been altered in the transgenic mice. In NTG, p27 was first detected at P5, with a gradual increase in its level as development proceeded (Fig. 8A). However, in TG p27 was not detected at P5, but was detected beginning at P10 at a much lower level than that found in NTG (Fig. 8A). After P15, there was not much difference in the p27 level between NTG and TG, suggesting that the biggest difference in the p27 level was found at P5 and P10, the times when oligodendrocytes begin to differentiate. A representative picture at P10 is shown (Fig. 8B). In TG, not only was the number of CC1+/p27+ or CNPase+/p27+ cells much lower than in NTG, but also the intensity of p27 staining in individual cells was significantly lower than the level found in NTG. This result is in agreement with what was reported in cultured optic nerve oligodendrocytes, in which a higher p27 level in a given precursor correlated with its increased commitment to differentiate (Durand et al., 1997). Therefore, our data suggest that oligodendrocytes that were undergoing apoptosis at P5–P10 may represent a population that failed to exit the cell cycle at the appropriate time, under attenuated ErbB2 signaling. In this scenario, the surviving population would divide two or more times when the p27 level is reduced, then differentiate into myelinating oligodendrocytes. If this is the case, the p75–ErbB1KD should be expressed among proliferating oligodendrocyte progenitors. Indeed, some of the cells that express p75–ErbB1KD were positive for BrdU at P5-P10 (Fig. 8C).
Figure 8.
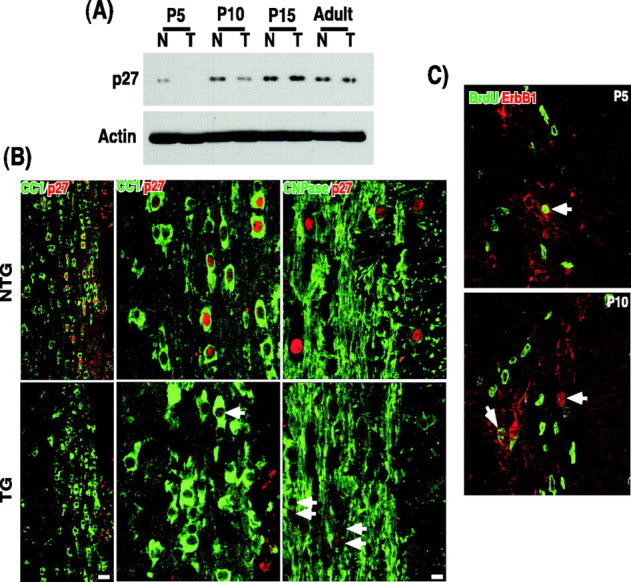
P27 level is significantly decreased in the p75–ErbB1 KD mice. A, Quantification of p27 in Western blot analyses. Actin blot was used as a control. B, The spinal cord sections taken from P10 mice were stained for CC1 and p27 or CNPase and p27. Left, Low-magnification pictures; scale bar, 20μm. Middle and right, High-magnification pictures; scale bar, 8.3μm. Note that some cells in the p75–ErbB1 KD mice have p27 in the nucleus, but its level is much lower than that found in NTG (arrows). C, The p75–ErbB1 KD is expressed among some dividing oligodendrocyte progenitors. The p75–ErbB1 KD was detected by ErbB1 staining in the cerebellar white matter at P5 and P10. Note that the cells that are positive for ErbB1 contain BrdU immunoreactivity in their nucleus.
Discussion
In this report, we provide data showing that NRG signaling plays critical, early roles in oligodendrocyte differentiation during development in vivo. With a 45% reduction in ErbB2 signaling, there was a 47% decrease in the number of mature oligodendrocytes in the adult. This reduction in myelinating oligodendrocytes did not stem from a reduction in oligodendrocyte progenitors, but from the apoptotic death of cells that appeared to have failed to exit the cell cycle at appropriate times. This conclusion is based on our data showing that there was an increase in the proliferation of NG2+ oligodendrocyte progenitors and a significant reduction in p27 level in the transgenic mice with attenuated ErbB2 signaling. Taken together, these data suggest that NRG signaling via ErbB2 plays a role in regulating cell cycle exit timing during oligodendrocyte development.
Vartanian et al. (1994) first reported that NRG signaling increased the number of O1+ oligodendrocytes in culture. Later, using spinal cord explant cultures, Park et al. (2001) demonstrated that ErbB2 signaling is required for oligodendrocyte differentiation. In culture, ErbB2–/– spinal cord explants failed to generate any O1+ differentiated oligodendrocytes, whereas O4+ progenitors were unaffected (Park et al., 2001). O1+ cells represent postmitotic populations (Richardson et al., 1988; Gard and Pfeiffer, 1990) that are derived from O4+ cells in oligodendrocyte lineage (Sommer and Schachner, 1982). Therefore, these data are in agreement with our findings that oligodendrocyte cell cycle completion requires ErbB2. However, the analyses of NRG–/– mice in a similar experimental paradigm yielded strikingly different results. In the absence of NRG, the establishment of the oligodendrocyte lineage never began in culture, which was made evident by a complete absence of O4+ cells, an earlier marker for oligodendrocyte lineage (Vartanian et al., 1999). These data suggest that NRG is essential for the commencement of oligodendrocyte lineage development, at least in culture. However, when ErbB2 was absent in slice culture (Park et al., 2001), the initial development of the lineage was unaffected, but its differentiation potential was compromised. It is quite plausible that a member of the ErbB receptor family other than ErbB2 plays a role in the generation of the oligodendrocyte lineage. ErbB4 is a likely candidate for this, because its exclusive activation was reported with NRG treatment among O2A progenitors in culture (Vartanian et al., 1997).
It should be noted that in our transgenic mice, ErbB2 signaling is perturbed only after oligodendrocytes begin to express MBP. Our approach is similar to the Krox-20-cre-mediated conditional deletion of ErbB2 among committed, promyelinating, and myelinating Schwann cells (Garratt et al., 2000). In that study, perturbation of ErbB2 signaling resulted in hypomyelination of sciatic nerves beginning at P15, suggesting that ErbB2 signaling is also required for Schwann cell myelination, as it is for oligodendrocyte myelination. The effect of ablating ErbB2 among Schwann cell precursors is also similar to what we have found in oligodendrocytes. At P3.5, the number of BrdU+ cells increased by ∼30%, with a fourfold to fivefold increase in TUNEL+ cells (Garratt et al., 2000). Because Krox-20 is expressed as Schwann cells make a transition from being precursors to being committed Schwann cells, it is possible that a loss of ErbB2 might have also affected the cell cycle exit of Schwann cells as well in these mice. However, changes in cell-cycle parameters were not examined in ErbB2-deleted Schwann cells (Garratt et al., 2000).
Because the perturbation of ErbB2 signaling coincided with oligodendrocyte differentiation in our transgenic mice, we cannot completely rule out the possibility that ErbB2 signaling also plays a role in the proliferation of oligodendrocyte progenitors in vivo. This case appears especially plausible, considering that in Schwann cells, NRG/ErbB2 provides both a mitogenic signal as well as cues critical for myelination (Morris et al., 1999; Woldeyesus et al., 1999; Garratt et al., 2000). It should be noted that NRG acted as a mitogen for dissociated oligodendrocyte progenitors in culture (Canoll et al., 1996; Shi et al., 1998). These data differ from the analyses of spinal cord explants from the ErbB2 null mice in culture, which suggested that ErbB2 plays a role in oligodendrocyte differentiation (Park et al., 2001). This discrepancy may be explained by a recent report that coactivation of integrin and NRG signaling in culture causes a reversal in NRG action from blocking to promoting oligodendrocyte differentiation (Colognato et al., 2002). In spinal cord explant cultures, integrin signaling was probably maintained intact, whereas in dissociated culture conditions, its extracellular matrix network was completely lost, disabling integrin signaling. In line with these data, the number of MBP+ cells was reduced by ∼50% in α6 integrin receptor null mice (Colognato et al., 2002). This reduction in oligodendrocyte differentiation appears to be attributable to the apoptotic death of GalC+ cells (Colognato et al., 2002). These results from α6-integrin receptor null mice are quite similar to what we presented in our study; with 45% reduction in ErbB2 signaling in vivo, there was an increase in apoptotic death of premyelinating CC1+ cells, which contributed to the reduction in fully differentiated MBP+ oligodendrocytes. Although the effect on ErbB2 signaling was not directly assessed in α6-integrin receptor null mice, these data imply that indirect inhibition of NRG signaling by removing integrin signaling resulted in the inhibition of oligodendrocyte differentiation in vivo. Clarification of the question as to whether NRG signaling also plays a role in the proliferation of oligodendrocyte progenitors in vivo will require selective deletion of NRG/ErbB2 among oligodendrocyte progenitors.
The p75–ErbB1KD construct can bind neurotrophins via the extracellular and transmembrane domains of p75 (Harrington et al., 2002). Although our 293T data indicate that the p75–ErbB1KD does not affect overall TrkA signaling, we cannot completely rule out that the hypomyelination effect with the p75–ErbB1KD construct is in part influenced by potentially blocking neurotrophin signaling in vivo. When tested in culture, NT3 was mitogenic to optic-nerve-derived oligodendrocyte progenitors (Barres et al., 1994), whereas it enhanced oligodendrocyte myelination in vitro (Yan and Wood, 2000). BDNF, in contrast, did not have much effect in culture, whereas there was a significant reduction in the number of myelinated axons in the optic nerve in BDNF–/– mice (Cellerino et al., 1997). This hypomyelination defect in BDNF–/– mice was attributed to an indirect effect, because BDNF affects the electrical firing of retinal ganglion cells (Cellerino et al., 1997). BDNF was shown recently to promote Schwann cell myelination in vivo by acting on p75 expressed on Schwann cells, whereas NT3 exerted the opposite effect by activating TrkC (Cosgaya et al., 2002). In oligodendrocyte development, it is not known whether the observed effect of BDNF and NT3 requires p75 or Trk receptors. Oligodendrocytes express p75 in culture (Harrington et al., 2002), but they do not express p75 during development in vivo, as we have shown here. Therefore, any effect of neurotrophins in vivo would be mediated by Trk receptors. At least in culture, the p75–ErbB1KD construct does not affect TrkA signaling, and we failed to observe any significant difference in the extent of Trk activation based on tyrosine phosphorylation or Trk autokinase assays between NTG and TG (data not shown). Alternatively, neurotrophins could augment ErbB2 signaling indirectly. It was shown in the neuromuscular junction that neurotrophins modulate the NRG level, thus affecting ErbB2 signaling indirectly (Loeb and Fischbach, 1997; Loeb et al., 2002). However, we did not observe any significant difference in the NRG RNA or protein level between NTG and TG (data not shown). Clarification of the role that neurotrophins and the receptors play in CNS myelination would require future in-depth analyses.
In conclusion, we report that ErbB2 signaling plays a critical role in oligodendrocyte differentiation in vivo, in part by regulating the onset of terminal differentiation in oligodendrocyte development. Perturbation of such ErbB2 signaling leads to a widespread hypomyelination in the CNS.
Footnotes
This work was supported by grants from the American Cancer Society and the Whitehall Foundation, and by National Institutes of Health Grant RO1 NS39472-01 (S.O.Y.). We thank Dr. Alexander Gow for the pMG2 construct; Dr. Alan Peterson for the 9 kb MBP promoter constructs; Dr. Nancy Hynes for ErbB2, ErbB3, and ErbB4 constructs; Dr. Lin Mei for Flag-ErbB4 construct; Kathy Wolken for the EM analyses; Dr. Jan Parker-Thornberg for generating transgenic mice; and Drs. Doug L. Falls, Kuo-Fen Lee, Pilar Perez, and Bruce Carter for critical comments of this manuscript.
Correspondence should be addressed to Dr. Sung Ok Yoon, The Neurobiotechnology Center and Department of Molecular and Cellular Biochemistry, 184 Rightmire Hall, 1060 Carmack Road, Ohio State University, Columbus, OH 43210. E-mail: yoon.84@osu.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235561-11$15.00/0
References
- Barres BA, Raff MC ( 1999) Axonal control of oligodendrocyte development. J Cell Biol 147: 1123–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, Schmid R, Sendnter M, Raff MC ( 1993) Multiple extracellular signals are required for long-term oligodendrocyte survival. Development 118: 283–295. [DOI] [PubMed] [Google Scholar]
- Barres BA, Raff MC, Gaese F, Bartke I, Dechant G, Barde YA ( 1994) A crucial role for neurotrophin-3 in oligodendrocyte development. Nature 367: 371–375. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM, Bresnahan JC, Hempstead BL, Yoon SO ( 2002) ProNGF Induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron 36: 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan PJ, Kumagai T, Berezov A, Murali R, Greene MI ( 2002) HER2/Neu: mechanisms of dimerization/oligomerization. Oncogene 21: 328. [DOI] [PubMed] [Google Scholar]
- Canoll PD, Musacchio JM, Hardy R, Reynolds R, Marchionni MA, Salzer JL ( 1996) GGF/neuregulin is a neuronal signal that promotes the proliferation and survival and inhibits the differentiation of oligodendrocyte progenitors. Neuron 17: 229–243. [DOI] [PubMed] [Google Scholar]
- Canoll PD, Kraemer R, Teng KK, Marchionni MA, Salzer JL ( 1999) GGF/neuregulin induces a phenotypic reversion of oligodendrocytes. Mol Cell Neurosci 13: 79–94. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Tikoo R, Kiyokawa H, Friedrich Jr V, Chao MV, Koff A ( 1997) Oligodendrocyte precursor differentiation is perturbed in the absence of the cyclin-dependent kinase inhibitor p27Kip1. Genes Dev 11: 2335–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellerino A, Carroll P, Thoenen H, Barde YA ( 1997) Reduced size of retinal ganglion cell axons and hypomyelination in mice lacking brain-derived neurotrophic factor. Mol Cell Neurosci 9: 397–408. [DOI] [PubMed] [Google Scholar]
- Chantry A ( 1995) The kinase domain and membrane localization determine intracellular interactions between epidermal growth factor receptors. J Biol Chem 270: 3068–3073. [PubMed] [Google Scholar]
- Chen MS, Bermingham-McDonogh O, Danehy Jr FT, Nolan C, Scherer SS, Lucas J, Gwynne D, Marchionni MA ( 1994) Expression of multiple neuregulin transcripts in postnatal rat brains. J Comp Neurol 349: 389–400. [DOI] [PubMed] [Google Scholar]
- Colognato H, Baron W, Avellana-Adalid V, Relvas JB, Baron-Van Evercooren A, Georges-Labouesse E, ffrench-Constant, C ( 2002) CNS integrins switch growth factor signalling to promote target-dependent survival. Nat Cell Biol 4: 833–841. [DOI] [PubMed] [Google Scholar]
- Cosgaya JM, Chan JR, Shooter EM ( 2002) The neurotrophin receptor p75NTR as a positive modulator of myelination. Science 298: 1245–1248. [DOI] [PubMed] [Google Scholar]
- Durand B, Gao FB, Raff M ( 1997) Accumulation of the cyclin-dependent kinase inhibitor p27/Kip1 and the timing of oligodendrocyte differentiation. EMBO J 16: 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez PA, Tang DG, Cheng L, Prochiantz A, Mudge AW, Raff MC ( 2000) Evidence that axon-derived neuregulin promotes oligodendrocyte survival in the developing rat optic nerve. Neuron 28: 81–90. [DOI] [PubMed] [Google Scholar]
- Foran DR, Peterson AC ( 1992) Myelin acquisition in the CNS of the mouse revealed by an MBP-Lac Z transgene. J Neurosci 12: 4890–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forghani R, Garofalo L, Foran DR, Farhadi HF, Lepage P, Hudson TJ, Tretjakoff I, Valera P, Peterson A ( 2001) A distal upstream enhancer from the myelin basic protein gene regulates expression in myelin-forming Schwann cells. J Neurosci 21: 3780–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gard AL, Pfeiffer SE ( 1990) Two proliferative stages of the oligodendrocyte lineage (A2B5+O4– and O4+GalC–) under different mitogenic control. Neuron 5: 615–625. [DOI] [PubMed] [Google Scholar]
- Gard AL, Pfeiffer SE ( 1993) Glial cell mitogens bFGF and PDGF differentially regulate development of O4+GalC– oligodendrocyte progenitors. Dev Biol 159: 618–630. [DOI] [PubMed] [Google Scholar]
- Garratt AN, Voiculescu O, Topilko P, Charnay P, Birchmeier C ( 2000) A dual role of ErbB2 in myelination and in expansion of the Schwann cell precursor pool. J Cell Biol 148: 1035–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassmann M, Casagranda F, Orioli D, Simon H, Lai C, Klein R, Lemke G ( 1995) Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 378: 390–394. [DOI] [PubMed] [Google Scholar]
- Gow A, Friedrich VL, Lazzarini RA ( 1992) Myelin basic protein gene contains separate enhancers for oligodendrocyte and Schwann cell expression. J Cell Biol 119: 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graus-Porta D, Beerli RR, Daly JM, Hynes NE ( 1997) ErbB2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J 16: 1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A, Giese NA, Richardson WD ( 1996) Spinal cord oligodendrocytes develop from ventrally derived progenitor cells that express PDGF alpha-receptors. Development 122: 4085–4094. [DOI] [PubMed] [Google Scholar]
- Harrington AW, Kim JY, Yoon SO ( 2002) Activation of Rac GTPase by p75 is necessary for c-jun N-terminal kinase-mediated apoptosis. J Neurosci 22: 156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Costantini F, Lacy E ( 1994) Manipulating the mouse embryo: a laboratory manual, Ed 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Jones JT, Akita RW, Sliwkowski MX ( 1999) Binding specificities and affinities of EGF domains for ErbB receptors. FEBS Lett 447: 227–231. [DOI] [PubMed] [Google Scholar]
- Lee KF, Simon H, Chen H, Bates B, Hung MC, Hauser C ( 1995) Requirement for neuregulin receptor erbB2 in neural and cardiac development. Nature 378: 394–398. [DOI] [PubMed] [Google Scholar]
- Levine JM, Stallcup WB ( 1987) Plasticity of developing cerebellar cells in vitro studied with antibodies against the NG2 antigen. J Neurosci 7: 2721–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JA, Fischbach GD ( 1997) Neurotrophic factors increase neuregulin expression in embryonic ventral spinal cord neurons. J Neurosci 17: 1416–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb JA, Hmadcha A, Fischbach GD, Land SJ, Zakarian VL ( 2002) Neuregulin expression at neuromuscular synapses is modulated by synaptic activity and neurotrophic factors. J Neurosci 22: 2206–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH ( 2002) Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 109: 75–86. [DOI] [PubMed] [Google Scholar]
- Meyer D, Birchmeier C ( 1995) Multiple essential functions of neuregulin in development. Nature 378: 386–390. [DOI] [PubMed] [Google Scholar]
- Meyer D, Yamaai T, Garratt A, Riethmacher-Sonnenberg E, Kane D, Theill LE, Birchmeier C ( 1997) Isoform-specific expression and function of neuregulin. Development 124: 3575–3586. [DOI] [PubMed] [Google Scholar]
- Morris JK, Lin W, Hauser C, Marchuk Y, Getman D, Lee KF ( 1999) Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron 23: 273–283. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Chang A, Trapp BD ( 1999) NG2+ glial cells: a novel glial cell population in the adult brain. J Neuropathol Exp Neurol 58: 1113–1124. [DOI] [PubMed] [Google Scholar]
- Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB ( 1996) Interaction between NG2 proteoglycan and PDGF α-receptor on O2A progenitor cells is required for optimal response to PDGF. J Neurosci Res 43: 315–330. [DOI] [PubMed] [Google Scholar]
- Noble M, Murray K, Stroobant P, Waterfield MD, Riddle P ( 1988) Platelet-derived growth factor promotes division and motility and inhibits premature differentiation of the oligodendrocyte/type-2 astrocyte progenitor cell. Nature 333: 560–562. [DOI] [PubMed] [Google Scholar]
- Park SK, Miller R, Krane I, Vartanian T ( 2001) The erbB2 gene is required for the development of terminally differentiated spinal cord oligodendrocytes. J Cell Biol 154: 1245–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkas-Kramarski R, Soussan L, Waterman H, Levkowitz G, Alroy I, Klapper L, Lavi S, Seger R, Ratzkin BJ, Sela M, Yarden Y ( 1996) Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J 15: 2452–2467. [PMC free article] [PubMed] [Google Scholar]
- Qian X, O'Rourke DM, Fei Z, Zhang HT, Kao CC, Greene MI ( 1999) Domain-specific interactions between the p185(neu) and epidermal growth factor receptor kinases determine differential signaling outcomes. J Biol Chem 274: 574–583. [DOI] [PubMed] [Google Scholar]
- Raff MC, Miller RH, Noble M ( 1983) A glial progenitor cell that develops in vitro into an astrocyte or an oligodendrocyte depending on culture medium. Nature 303: 390–396. [DOI] [PubMed] [Google Scholar]
- Richardson WD, Pringle N, Mosley MJ, Westermark B, Dubois-Dalcq M ( 1988) A role for platelet-derived growth factor in normal gliogenesis in the central nervous system. Cell 53: 309–319. [DOI] [PubMed] [Google Scholar]
- Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C ( 1997) Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature 389: 725–730. [DOI] [PubMed] [Google Scholar]
- Seri B, Garcia-Verdugo JM, McEwen BS, Alvarez-Buylla A ( 2001) Astrocytes give rise to new neurons in the adult mammalian hippocampus. J Neurosci 21: 7153–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Marinovich A, Barres BA ( 1998) Purification and characterization of adult oligodendrocyte precursor cells from the rat optic nerve. J Neurosci 18: 4627–4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer I, Schachner M ( 1982) Cell that are O4 antigen-positive and O1 antigen-negative differentiate into O1 antigen-positive oligodendrocytes. Neurosci Lett 29: 183–188. [DOI] [PubMed] [Google Scholar]
- Stolt CC, Rehberg S, Ader M, Lommes P, Riethmacher D, Schachner M, Bartsch U, Wegner M ( 2002) Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev 16: 165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikoo R, Osterhout DJ, Casaccia-Bonnefil P, Seth P, Koff A, Chao MV ( 1998) Ectopic expression of p27Kip1 in oligodendrocyte progenitor cells results in cell-cycle growth arrest. J Neurobiol 36: 431–440. [PubMed] [Google Scholar]
- Vartanian T, Corfas G, Li Y, Fischbach GD, Stefansson K ( 1994) A role for the acetylcholine receptor-inducing protein ARIA in oligodendrocyte development. Proc Natl Acad Sci USA 91: 11626–11630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian T, Fischbach G, Miller R ( 1999) Failure of spinal cord oligodendrocyte development in mice lacking neuregulin. Proc Natl Acad Sci USA 96: 731–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian T, Goodearl A, Viehover A, Fischbach G ( 1997) Axonal neuregulin signals cells of the oligodendrocyte lineage through activation of HER4 and Schwann cells through HER2 and HER3. J Cell Biol 137: 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Sdrulla AD, diSibio G, Bush G, Nofziger D, Hicks C, Weinmaster G, Barres BA ( 1998) Notch receptor activation inhibits oligodendrocyte differentiation. Neuron 21: 63–75. [DOI] [PubMed] [Google Scholar]
- Woldeyesus MT, Britsch S, Riethmacher D, Xu L, Sonnenberg-Riethmacher E, Abou-Rebyeh F, Harvey R, Caroni P, Birchmeier C ( 1999) Peripheral nervous system defects in erbB2 mutants following genetic rescue of heart development. Genes Dev 13: 2538–2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Wood PM ( 2000) NT-3 weakly stimulates proliferation of adult rat O1 –O4 + oligodendrocyte-lineage cells and increases oligodendrocyte myelination in vitro. J Neurosci Res 62: 329–335. [DOI] [PubMed] [Google Scholar]
- Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV ( 1998) Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci 18: 3273–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Choi G, Anderson DJ ( 2001) The bHLH transcription factor Olig2 promotes oligodendrocyte differentiation in collaboration with Nkx2.2. Neuron 31: 791–807. [DOI] [PubMed] [Google Scholar]