

Abstract
Oxidative stress is believed to be the cause of cell death in multiple disorders of the brain, including perinatal hypoxia/ischemia. Glutamate, cystine deprivation, homocysteic acid, and the glutathione synthesis inhibitor buthionine sulfoximine all cause oxidative injury to immature neurons and oligodendrocytes by depleting intracellular glutathione. Although vitamin K is not a classical antioxidant, we report here the novel finding that vitamin K1 and K2 (menaquinone-4) potently inhibit glutathione depletion-mediated oxidative cell death in primary cultures of oligodendrocyte precursors and immature fetal cortical neurons with EC50 values of 30 nm and 2 nm, respectively. The mechanism by which vitamin K blocks oxidative injury is independent of its only known biological function as a cofactor for γ-glutamylcarboxylase, an enzyme responsible for posttranslational modification of specific proteins. Neither oligodendrocytes nor neurons possess significant vitamin K-dependent carboxylase or epoxidase activity. Furthermore, the vitamin K antagonists warfarin and dicoumarol and the direct carboxylase inhibitor 2-chloro-vitamin K1 have no effect on the protective function of vitamin K against oxidative injury. Vitamin K does not prevent the depletion of intracellular glutathione caused by cystine deprivation but completely blocks free radical accumulation and cell death. The protective and potent efficacy of this naturally occurring vitamin, with no established clinical side effects, suggests a potential therapeutic application in preventing oxidative damage to undifferentiated oligodendrocytes in perinatal hypoxic/ischemic brain injury.
Keywords: glutathione depletion, oxidative stress, cell death, vitamin K, neuron, oligodendrocyte, white matter, cystine deprivation, menaquinone-4, cerebral palsy
Introduction
Oxidative mechanisms of injury are important in many neurological disorders, including cerebral damage caused by ischemia/reperfusion. Hypoxic/ischemic injury to the developing brain is a major neurological disorder in the perinatal period. Neurological deficits in survivors include mental retardation, seizures, and cerebral palsy. The neuropathology of perinatal hypoxic/ischemic brain injury is complex and differentially involves gray and white matter regions, depending on the type and extent of the injury, the age of the infant, and the developmental stage of cerebral vascularity (Kinney and Armstrong, 1997). In the premature infant, the cerebral white matter is particularly susceptible to ischemic injury, resulting in periventricular leukomalacia (PVL) and cerebral palsy, i.e., preferential damage to the cerebral white matter that is mainly populated at that age with premyelinating oligodendrocytes (OLs) (Volpe 1997, 2001; Back et al., 2001). We demonstrated previously that these premyelinating OLs are more sensitive to oxidative damage than are mature, myelinating cells (Back et al., 1998). There is now emerging evidence suggesting that oxidative stress plays a key role in the pathogenesis of PVL (Haynes et al., 2003).
Glutathione (GSH) depletion is widely used as a cellular model of oxidative stress (Murphy et al., 1989, 1990; Kane et al., 1993; Ratan et al., 1994). Reduced GSH is the major intracellular antioxidant and plays a pivotal role in maintaining cellular redox homeostasis. Depletion of GSH results in a time-dependent accumulation of endogenous reactive oxygen species (ROS) and subsequent oxidative stress. GSH depletion is the underlying mechanism by which glutamate induces receptor-independent cell death in neurons and OLs (Murphy et al., 1989, 1990; Oka et al., 1993). Glutamate promotes cystine efflux or competitively blocks cystine uptake via an exchanger/antiporter, resulting in loss of intracellular cystine and GSH. Similarly, cystine-free culture medium, or the GSH synthesis inhibitor buthionine sulfoximine (BSO), results in loss of GSH and oxidative cell death (Yonezawa et al., 1996; Back et al., 1998). A decrease in tissue GSH concentration has been demonstrated after in vivo ischemia/hypoxia (Orwar et al., 1994; Shivakumar et al., 1995; Wallin et al., 2000) and excitotoxicity (Floreani et al., 1997). Furthermore, a GSH prodrug, YM737, provides protection against cerebral ischemia in rats (Yamamoto et al., 1993).
Our finding of the protective effect of vitamin K against oxidative stress induced by GSH depletion was made serendipitously in the course of our investigation into the mechanism of cell death. Vitamin K is a family of fat-soluble vitamins composed of a naphthoquinone with isoprenylacyl side chains of varying lengths and is required for proper synthesis of vitamin K-dependent proteins involved in blood coagulation and bone metabolism (Suttie, 1991; Furie et al., 1999). It is a cofactor for a single known enzyme, γ-glutamylcarboxylase, that catalyzes the posttranslational conversion of glutamic acid to γ-carboxyglutamic acid (Gla) in vitamin K-dependent proteins. The enzyme is also an epoxidase converting vitamin K to vitamin K epoxide during formation of Gla. The carboxylase is expressed in the CNS during embryogenesis, even before its expression in the liver, the site for production of many vitamin K-dependent proteins (Romero et al., 1998). The physiological roles of vitamin K and the carboxylase in the CNS are not characterized. It is well known that vitamin K is developmentally deficient in neonates, especially in preterm infants whose risk for PVL is especially high. Vitamin K status is important in the maintenance of normal biosynthesis of lipid sulfatide, an important component of CNS myelin, in young animals (Sundaram et al., 1996). Moreover, exposure to the vitamin K antagonist warfarin in utero causes CNS malformations and mental retardation (Hall et al., 1980). These observations suggest a possible unidentified role of vitamin K in the developing brain. In this study, we report the discovery of a novel and potent protective action of vitamin K in preventing oxidative injury to primary OL precursors and to immature cortical neurons that appears to be independent of the current known function of vitamin K.
Materials and Methods
Materials. DMEM, HBSS, Earle's balanced salt solution (EBSS), FBS, penicillin, and streptomycin were purchased from Life Technologies, Inc. Antibody against polyADP-ribose was obtained from Chemicon International Inc. (Temecula, CA). 2-Chloro-vitamin K1 was a generous gift from Dr. John W. Suttie (University of Wisconsin, Madison, WI). Primary rat hepatocytes were obtained from Dr. Timothy R. Billiar (University of Pittsburgh, Pittsburgh, PA). Unless otherwise specified, all other chemicals were from Sigma (St. Louis, MO).
OL cultures. Primary rat OLs were prepared from the cerebral hemispheres of Sprague Dawley rats at postnatal days 1–2 using a shaking method as described with modifications (McCarthy and de Vellis, 1980; Oka et al., 1993). Briefly, forebrains free of meninges were chopped into 1 mm 3 blocks and placed into HBSS containing 0.01% trypsin and 10 μg/ml DNase. After digestion for 15 min at 37°C, the tissue was collected by centrifugation and triturated with the plating medium DMEM20S containing DMEM, 20% FBS, and 1% penicillin-streptomycin, and passed through a 70 μm sieve. Cells were plated onto poly-d-lysine-coated 75 cm 2 flasks at a density of 1 pup brain per flask. Cultures were fed with fresh DMEM20S medium every other day for 10–11 d at 37°C in a humid atmosphere of 5% CO2 and 95% air.
To isolate OLs, the co-culture flasks were shaken for 1 hr at 200 rpm at 37°C to remove adherent microglia/macrophages, and the cultures were washed with the same medium and subjected to shaking at 200 rpm overnight (18–22 hr) to separate OLs from the astrocyte layer. The suspension was plated onto uncoated Petri dishes and incubated for 1 hr at 37°C to further remove residual microglia and astrocytes that adhere to the dishes. The OLs were then collected by passing through a 15 μm sieve and centrifuged. Isolated OLs were plated onto poly-ornithine (50 μg/ml)-coated culture plates, i.e., 96-well plates (at a density of 3.3 × 10 3 cell/well, for cell survival assay), 24-well plates with glass coverslips (1.74 × 10 4 cell/well, for imaging), and 60 mm plates (2.75 × 10 5 cell/plate, for enzyme assay). Purified OLs were cultured for 7–9 d in a serum-free basal-defined medium (BDM; DMEM containing 0.1% BSA, 50 μg/ml apo-transferrin, 50 μg/ml insulin, 30 nm sodium selenite, 10 nm d-biotin, 10 nm hydrocortisone, 200 μm l-cystine, 10 ng/ml PDGF, and 10 ng/ml basic FGF). At 7–9 d, the OL cultures, primarily composed of progenitors and pre-OLs [A2B5+,O4+, myelin basic protein - (MBP -)] but not mature OLs (MBP +), were used in this study. The purity of OL cultures was consistently >95% OLs with <1% astrocyte contamination.
Oxidative stress in cell cultures. To induce oxidative stress, unless specified otherwise, BDM medium without cystine (Cys -) was used. Vitamin K1 and menaqunone-4 were made fresh in anhydrous DMSO as a 1000× working solution. The final concentration of DMSO in culture medium was 0.1%, and had no effect on cell viability, proliferation, or morphology. The final concentration of vitamin K was as indicated. When glutamate toxicity was examined, the concentration of cystine in the culture medium was 20 μm instead of the normal 200 μm, because previous results showed that glutamate toxicity inversely correlates with the concentration of cystine in the culture medium (Murphy et al., 1989; Oka et al., 1993). BDM containing 20 μm cystine was not toxic to OL precursors.
Neuronal cultures. Primary embryonic cortical neurons from E14 rat cerebral hemispheres were isolated and cultured according to a method described previously (Wang et al., 1998). The neurons were plated in poly-d-lysine-coated 96-well culture plates at a density of 2.4 × 10 4 cells/well and used after 24–48 hr of plating.
Cell survival assay. Cell survival was determined after treatment for 22–24 hr using Alamar Blue (Southern Biotech, Birmingham, AL), a tetrazolium dye that is reduced by living cells to a colored product. This assay is similar in principle to the MTT cell viability assay and has been previously validated as an accurate measure of survival of OLs in our culture system (Back et al., 1999). All results of cell death assays were also confirmed by visual inspection under a light microscope. Briefly, culture medium in the 96-well plate was aspirated, and cells were incubated with 200 μl of assay solution prepared by diluting 100× stock solution of Alamar Blue into EBSS for 2 hr at 37°C. The fluorescence of the assay solution, reflecting cell viability, was measured with a fluorescence plate reader (FluoroCount; Packard Instrument Co., Meridian, CT) using an excitation wavelength of 560 nm and an emission wavelength of 590 nm. All survival assays were performed in triplicate, and cell viability was expressed as mean ± SD. Intracellular GSH levels were determined by a colorimetric method as described previously (Back et al., 1998).
Intracellular free radical accumulation measurement. Intracellular free radical generation was evaluated with dichlorohydrofluorescin diacetate (DCFH-DA) and dihydrorhodamine 123 (Rho123; Molecular Probes, Eugene, OR) (LeBel et al., 1992; Wang and Joseph, 1999). DCFH-DA and Rho123 were prepared as 100 mm and 10 mm stocks, respectively, in dimethyl sulfoxide and stored in the dark at -20°C. After cells were treated for 10–15 hr, DCFH-DA (100 μm) or dihydrorhodamine (10 μm) was added directly to the cells and incubated with the cells for 20 min at 37°C. The extracellular DCFH-DA or dihydrorhodamine was then removed by washing the cells twice with EBSS. The fluorescence of the cells loaded with DCFH-DA was measured using a multiwell fluorescence plate reader (excitation λ = 485 nm; emission λ = 530 nm). For fluorescent imaging of oxidized Rho123, the oxidized form of dihydrorhodamine, cells were immediately visualized using a digital fluorescent microscope equipped with a 40× oil immersion objective. Cells were visualized by excitation at 490 nm and emission at 515 nm. For all images, the microscope settings, such as brightness, contrast, and exposure time were held constant to compare the relative intensity of oxidized Rho123 across all treatment conditions.
Antioxidant activity assay. Antioxidant activity of various reagents was assayed with two different methods. In the first method, we monitored at 517 nm the disappearance of the optical absorbance of stable-free radical 1,1-diphenyl-2-picrylhydrazyl (DPPH) on reaction with test compounds (Blois, 1958). The rate of the reaction represents the antioxidant potency of test agents. The known free radical scavenger Trolox was used as a positive control. Briefly, 10 μl of test reagents at indicated final concentrations were added to 300 μl of 100 μm DPPH. Optical absorbance of DPPH at 517 nm was immediately monitored for 5 min. In the second method, we determined Trolox equivalent activity concentration of the test agents according to the method of Rice-Evans and Miller (1994). The inhibition of the absorbance of the radical cation formation of 2,2′-azinobis (3-ethylbenzothiazoline 6-sulfonate) (ABTS +·) was monitored continuously for 30 min at 660 nm in PBS containing 150 μm ABTS, 2.5 μm metmyoglobin, 75 μm H2O2, and the indicated concentration of test reagent.
Immunocytochemistry and immunofluorescence microscopy. Cells were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature, washed three times with PBS, and blocked with TBST (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 0.1% Triton X-100) containing 5% goat serum for 1 hr at room temperature. The coverslips were incubated with mouse monoclonal antibodies A2B5 (1:100 dilution; American Type Culture Collection, Manassas, VA), O4, O1 (1:100 dilution; gifts from Dr. Steven E. Pfeiffer, University of Connecticut Health Center, Farmington, CT), MBP (1:500 dilution; Boehringer Mannheim, Indianapolis, IN), CD11 (1:100), or glial fibrillary acidic protein GFAP (1:4000) for 1 hr. After three to four washes at 5 min each, the appropriate secondary antibody conjugated with either fluorescein or Oregon Green (Molecular Probes Inc.) was added to the coverslips and incubated for 1 hr. After extensive washes with TBST, nuclei were stained by adding Hoechst 33258 at a final concentration of 2 μg/ml for 1 min. After three more washes, the coverslips were mounted onto glass slides with FluoroMount and kept in the dark at 4°C. Cell images were captured with a fluorescence microscope (Nikon Eclipse E800) equipped with a Spot RT digital camera (Diagnostic Instruments, Inc.).
Cell homogenization and solubilization of microsomal proteins. Three to 5 × 10 7 OL precursors or primary rat hepatocytes (Li et al., 1999) were first rinsed with cold PBS and scraped into buffer A (PBS, 20% glycerol, and 1× protease inhibitor mixture) (Furie et al., 1997). Microsomes were isolated by homogenization and differential ultracentrifugation, and microsomal proteins were extracted with buffer A supplemented with 0.5% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) and 0.2% phosphatidylcholine, as described previously (Furie et al., 1997). Protein concentration was measured using the lipid- and detergent-compatible Dc protein assay (Bio-Rad, Hercules, CA) with BSA as standard.
γ-Glutamylcarboxylase and vitamin K epoxidase assay. Vitamin K-dependent carboxylase activity of the microsomal protein preparations was evaluated by measuring vitamin K-dependent incorporation of 14CO2 into the synthetic peptide substrate FLEEL over 30 min at 25°C (Ulrich et al., 1988). Purified recombinant Flag-tagged bovine carboxylase was used as a standard, as described previously (Furie et al., 1997). The assay mixture (125 μl) contained 25 mm 3-(N-morpholino)propanesulfonic acid, pH 7.0, 0.5 mm NaCl, 0.16% CHAPS, 0.16% phosphatidylcholine, 8 mm dithiothreitol, 222 μm chemically reduced vitamin K1 (Abbott Laboratories, North Chicago, IL), and 1.4 mm NaH 14CO3 (10 μCi; Amersham). The reaction was initiated by adding the reaction mixture to microsomal preparation or recombinant carboxylase. 14CO2 incorporation in the absence of vitamin K was considered background and was subtracted. Inhibition of recombinant carboxylase by 2-Cl-vitmain K1 was determined using increasing the molar ratio of 2-Cl-vitamin K1 over the cofactor vitamin K1 in the assay mixture.
Vitamin K epoxidase activity was determined as described previously (Sugiura et al., 1997). For assay of vitamin K epoxide, NaH 14CO3 was replaced by NaHCO3. On completion of the carboxylase reaction, the reaction mixture was extracted with 250 μl of ethanol and then with 750 μl of hexane. The organic and aqueous phases were separated by centrifugation at 1000 × g for 10 min. The organic phase was removed, and the solvent was evaporated to dryness. The residue was redissolved in 200 μl of methanol. Half of this solution was injected onto a reverse-phase C18 HPLC column (Hypersil ODS; 5 μm; 4.6 × 250 mm; Custom LC, Houston, TX) using a Beckman model 126 HPLC equipped with a Beckman model 168 diode array detector. The column was developed with a mobile phase of 10% dichloromethane/90% methanol that had been saturated with nitrogen. Vitamin K derivatives were detected at 226 nm, and vitamin K epoxide was quantitated using a purified synthetic standard of 2,3-vitamin K1 epoxide. The identity of vitamin K epoxide in the HPLC fraction was further confirmed by mass spectroscopy.
Results
Characterization of primary OLs in culture
OLs differentiate along a specific lineage, progressing through four stages recognized by specific antibodies: progenitors (A2B5+ or NG2+), pre-OLs (O4+), immature OLs (O1+), and mature OLs (MBP+) (Pfeiffer et al., 1993). Each developmental stage possesses a characteristic morphology. Primary OL cultures used in this study were grown for 7–9 d in vitro. The majority of cells were OL progenitors (A2B5+) and pre-OLs (A2B5+, O4+). Immunocytochemical characterization confirmed that the majority of OLs in our cultures were positive for the surface markers recognized by the monoclonal antibodies A2B5 and O4 and were negative for O1 (Fig. 1). There were only a few mature, differentiated OLs expressing MBP. These OLs (A2B5+, O4+, MBP -) are referred to as OL precursors in this study. Contamination by astrocytes (GFAP +) and microglia (CD11 +) was <2% (Fig. 1).
Figure 1.
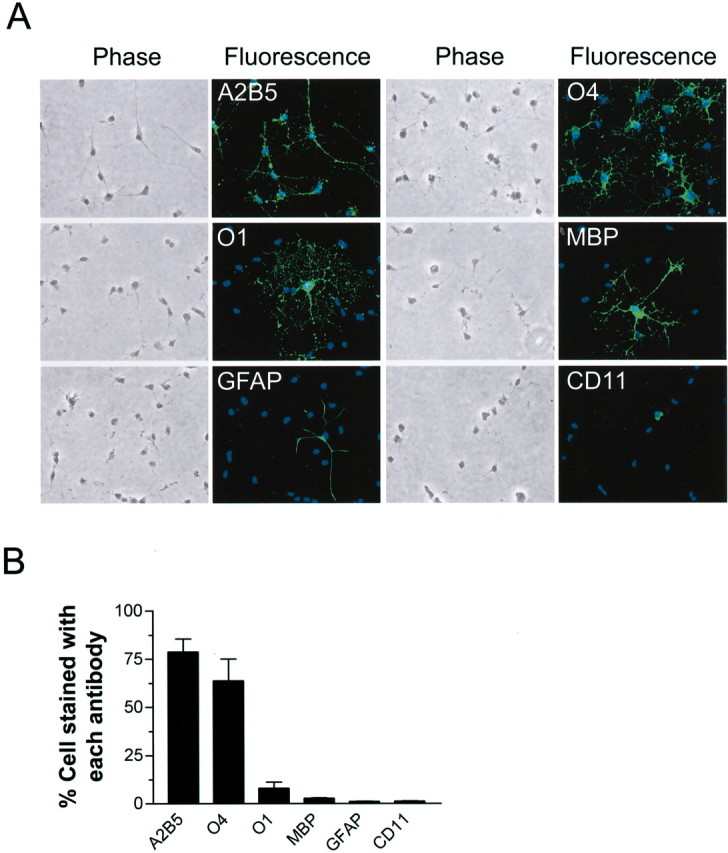
Characterization of primary OL cultures.A,Representative microphotographs of morphology and immunocytochemistry of cells (7–9 d in vitro) labeled with indicated antibodies. B, Composition of developing OL cultures as determined by immunostaining for indicated markers. Total cell number was determined by counting all cells labeled with the nuclei dye Hoechst 33258. Values represent mean ± SEM from three separate experiments.
Vitamin K1 and menaquinone (MK)-4 protect against cystine depletion-induced oxidative death to OL precursors
Our discovery of a protective effect of vitamin K against oxidative stress-induced cell death was made serendipitously as follows. Previously, we showed that OL precursors undergo oxidative stress-induced cell death after GSH depletion caused by removal of cystine from the culture medium (Yonezawa et al., 1996; Back et al., 1998). In pursuing the mechanism of the cell death in this model, we initially hypothesized that activation of poly(ADP-ribose) polymerase (PARP) is involved in the toxicity, because it is known that free radicals can cause extensive DNA-strand breakage and activation of the DNA repair enzyme PARP. Activation of this enzyme, however, also results in ATP depletion, energy failure, and cell death (Szabo and Dawson, 1998). We found, however, that the known PARP inhibitors, 3-methoxybenzamide and 3-aminobenzamide, did not protect against OL death induced by cystine depletion (data not shown), indicating that PARP activation is not involved in this model. Consistent with this result, polyADP-riboslyation of multiple proteins was not observed by Western blotting analysis with antibody against polyADP-ribose (data not shown), nor was cellular ATP depletion observed before cell lysis (data not shown). In addition to PARP inhibitors, we tested in parallel the effect of inhibitors of mono-ADP-ribosyltransferase (MART), activation of which has not been linked to energy failure. Unexpectedly, we found complete protection with the MART inhibitors (Banasik et al., 1992), vitamin K1 (phylloquinone), and vitamin K2 (MK-4). Cystine deprivation for 18–20 hr caused complete cell death in cultures (Fig. 2B). In the presence of 0.1 μm vitamin K1 or MK-4, however, the cell death was completely prevented (Fig. 1A, B). Interestingly, other known MART inhibitors, such as novobiocin, stearic acid, palmitic acid, coumermycin, and m-iodobenzylguanidine, had no or limited protective effect on cystine depletion-induced cell death and were associated with cytotoxicity at high concentrations (Fig. 2A).
Figure 2.

Vitamin K protected against cystine deprivation-induced OL death independent of MART. A, Effects of various MART inhibitors on cystine depletion-induced OL death. OLs were subjected to either normal medium (Cys +) or cystine-free medium (Cys -) in the presence of indicated concentrations of various MART inhibitors. Cell viability was evaluated after 24 hr. Results are representative of four independent experiments. B, Phase contrast photomicrographs of OLs exposed to normal or cystine-depleted medium with or without 0.1μm vitamin K1 or MK-4 for 18 hr. Magnification, 400×. Data are representative of at least 10 separate experiments. C, Vitamin K prevented OL death in a concentration-dependent manner. Cell viability was assayed 24 hr after cystine deprivation in the presence of increasing concentrations of K1 and MK-4. Values are mean ± SEM of six separate experiments.
We next examined the concentration dependence of the protective effect of vitamin K1 and MK-4. Surprisingly, vitamin K completely abolished cystine depletion-mediated OL death at remarkably low concentrations with EC50 ≈30 nm and 2 nm for K1 and MK-4, respectively (Fig. 2C). These EC50 values for protecting OLs were much lower than their IC50 for inhibiting MART (i.e., 1.9 μm and 13 μm for K1 and MK-4, respectively) in vitro (Banasik et al., 1992). Interestingly, the effective concentrations obtained in this cell death model are close to the normal adult human serum vitamin K levels (2.9 ± 1.4 nm) (Mummah-Schendel and Suttie, 1986). The order of potency of K1 and MK-4 to protect OLs against cystine deprivation was opposite to their order of potency to inhibit MART. Therefore, these results suggest that vitamin K exerts its protective effect by a mechanism other than MART inhibition in these cultures.
Vitamin K1 and MK-4 prevent GSH depletion-induced oxidative death to both primary OLs and immature neurons
Methods other than cystine depletion are often used to induce intracellular GSH depletion, and it was of interest to test whether vitamin K1 and MK-4 could prevent oxidative toxicity in these models. Glutamate has been shown to cause oxidative toxicity in immature cortical neurons and OLs by decreasing intracellular cystine and, thus, GSH (Murphy et al., 1989, 1990; Oka et al., 1993). Consistent with our previous observations (Oka et al., 1993), OLs exposed to glutamate underwent cell death (Fig. 3A). This toxicity of glutamate was completely prevented by 0.1 μm vitamin K1 or 0.1 μm MK-4 (Fig. 3A). Similarly, blocking GSH biosynthesis with 1 mm BSO, an inhibitor for the rate-limiting enzyme γ-glutamylcysteine synthetase, induced oxidative cell death that also was completely blocked by vitamin K1 or MK-4 (Fig. 3A). The protective effect of vitamin K1 and MK-4 on oxidative cell death was not specific to OLs, because their presence also completely inhibited glutamate-induced neuronal death (Fig. 3B). In addition, we found that vitamin K1 and MK-4 also blocked homocysteic acid- and BSO-induced neuron degeneration (Fig. 3B).
Figure 3.
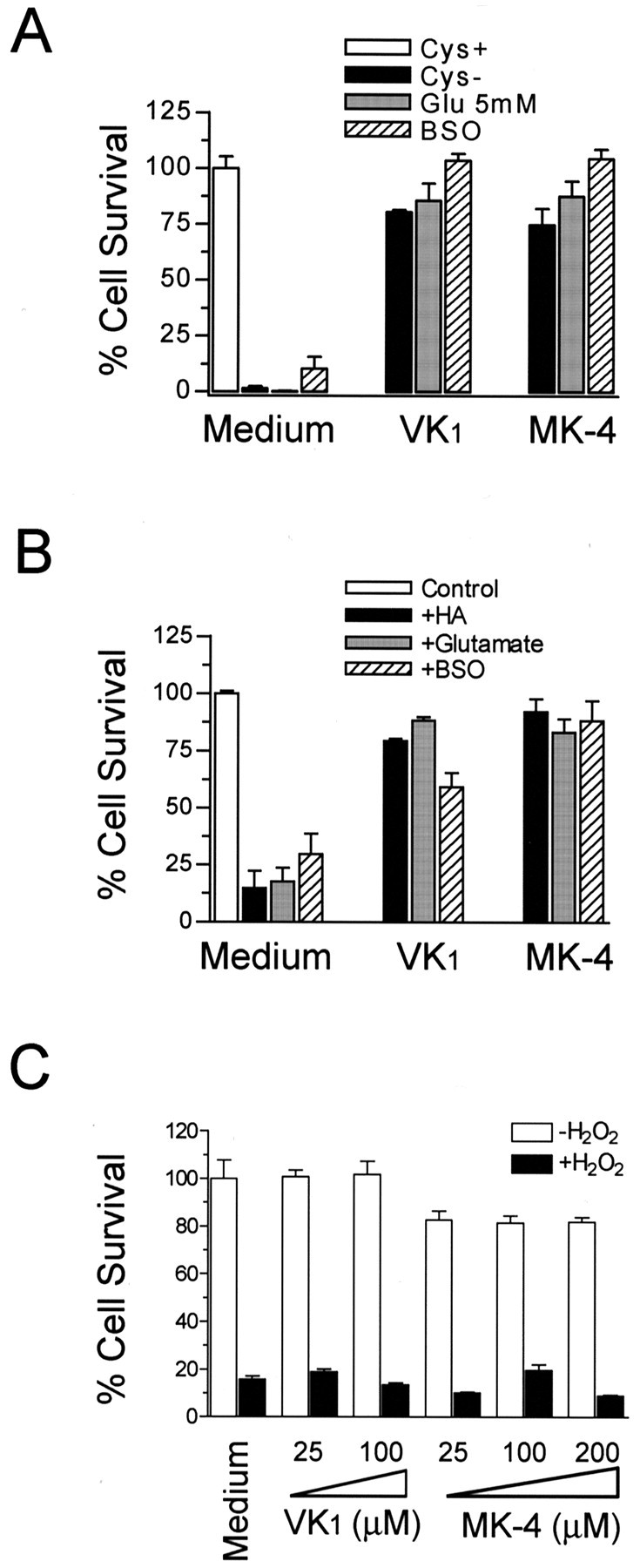
Vitamin K1 and MK-4 protected both OL precursors and neurons from cell death induced by various GSH depletion methods. A, Vitamin K prevented OL cell death induced by cystine depletion, by glutamate (5 mm), and by BSO (1 mm). B, Vitamin K1 and MK-4 prevented immaturecorticalneuronaldeathinducedbyhomocysteicacid(HA,2.5mm),glutamate(5mm), or BSO (1 mm). Primary cells were incubated with indicated agents for 24 hr in the presence or absence of K1 (0.1μm) or MK-4 (0.1μm), and cell viability was analyzed after 24 hr. Results are mean ± SEM of three separate experiments. C, Effect of vitamin K1 and MK-4 on H2O2-induced toxicity. OL precursors were incubated with or without 800μm H2O2 in the presence of indicated concentrations of vitamin K for 15 hr, and cell viability was evaluated. Data are representative of at least four independent experiments with similar results.
Taken together, these data demonstrate that both vitamin K1 and MK-4 are potent inhibitors of oxidative cell death induced by GSH depletion in primary OL precursor cultures and in neuronal cultures. In addition to GSH depletion-mediated cell death, we also examined the effect of vitamin K on exogenous oxidant H2O2-induced cell death. At concentrations ranging from 0.01 μm to 200 μm, vitamin K1 and MK-4 did not prevent H2O2-induced OL death (Fig. 3C). They also did not block kainate-, menadione-, or nitric oxide-induced cytotoxicity to the OL precursors (data not shown).
Both the naphthoquinone ring and the 3′-side chain are required for vitamin K protection
There are two naturally occurring forms of vitamin K, phylloquinone (vitamin K1) and MKs (vitamin K2) (Fig. 4A). Both K1 and K2 are characterized by a naphthoquinone ring and an aliphatic side chain. Vitamin K2 is a series of naphthoquinones with side chains of varying numbers of isoprenoid units (n = 2–13; when n = 4, K2 is also called MK-4). Although vitamin K1 is the predominant form of vitamin K in the liver, heart, and pancreas, MK-4 is the predominant form of vitamin K in the brain (Thijssen et al., 1996) and can be synthesized from vitamin K1, probably by a cellular enzymatic conversion mechanism that has yet to be identified (Thijssen et al., 1996; Davidson et al., 1998). To examine the structural requirements for vitamin K protection in culture, we tested several commercially available compounds that share some similarity with K1 and MK-4 (Fig. 4A). Menadione, also known as vitamin K3, contains the same naphthoquinone structure as vitamin K1 and K2 but without the 3′-side chain. This compound was ineffective in the protection of OL precursors against cystine deprivation (Fig. 4B) and was, in fact, quite toxic to OLs at a higher concentration (>2 μm; data not shown). Similar results were obtained with immature neurons (data not shown). Another lipophilic vitamin, retinoic acid, that has an aliphatic side chain but lacks the naphthoquinone structure also did not show any protection of OL precursors against cystine deprivation (data not shown). Taken together, these results demonstrate that both the nathphoquinone ring structure and the isoprenoid chain are essential for inhibition of GSH depletion-mediated cell death.
Figure 4.
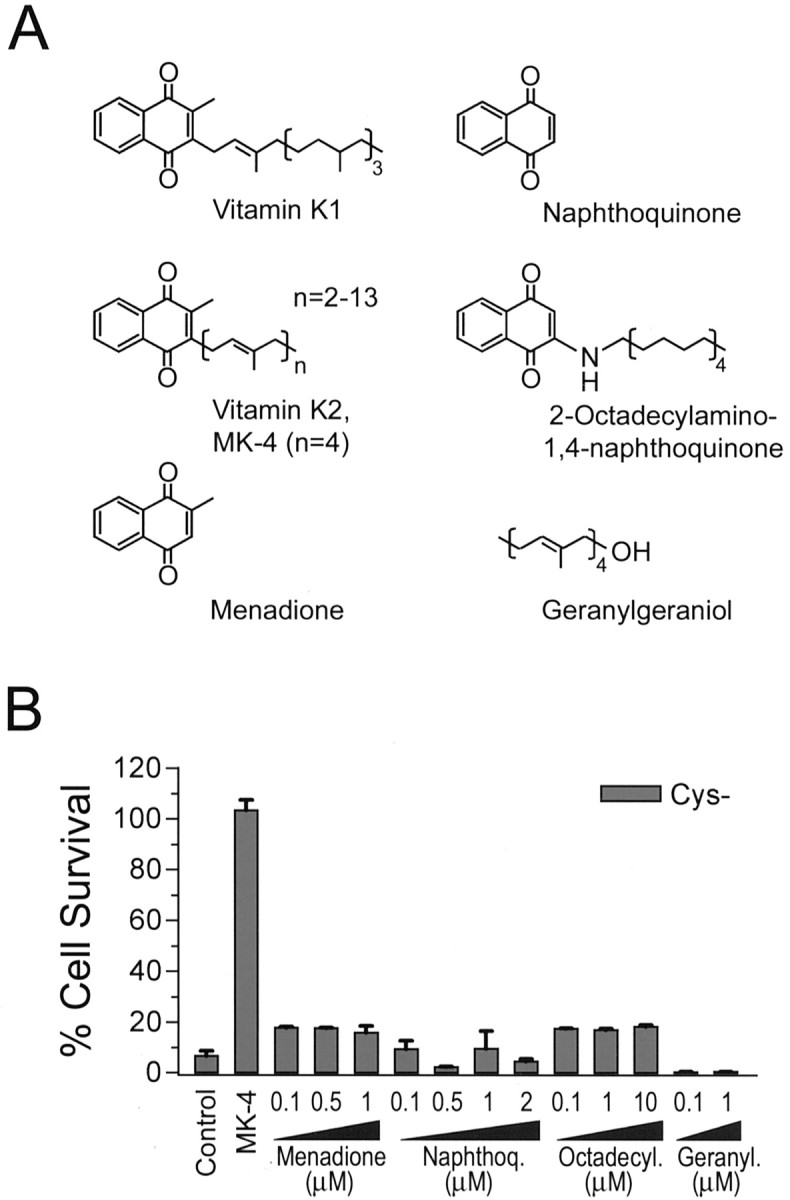
Structural requirement for vitamin K-dependent protection against cystine depletion-induced cell death. A, Structures of vitamin K1, K2, and compounds tested in B for their effects on cystine depletion-induced OL death. B, Effect of various compounds on OL cell death induced by cystine depletion. Representative results of two to three independent experiments are shown. No cytotoxicity to OLs in normal culture medium was seen with the test compounds at the concentration used.
Vitamin K protection against oxidative cell death is independent of vitamin K-dependent γ-glutamylcarboxylase
To explore further the mechanism by which vitamin K1 and MK-4 protect against oxidative cell injury, we investigated whether this protection is mediated by the known mechanism for the biological actions of vitamin K. As mentioned above, vitamin K is an essential cofactor for γ-glutamylcarboxylase, an enzyme that catalyzes the incorporation of CO2 into certain glutamic acid residues of about a dozen proteins that are involved in blood coagulation, bone metabolism, and cell growth and survival (Suttie, 1985; Furie et al., 1999). This posttranslational formation of Gla plays an important biological role in protein function. For example, the highly negatively charged Gla residues are required for Ca2+-dependent phospholipid binding of all coagulation factors, a requirement for their biological activity. The carboxylase requires the reduced, hydroquinone form (KH2) of vitamin K as the actual cofactor. In the course of carboxylation, KH2 is stoichiometrically oxidized by the carboxylase to vitamin K-2, 3-epoxide, which is then reduced by a NADPH-dependent, warfarin-sensitive vitamin K reductase back to the quinone (K) and hydroquinone (KH2) forms (the so-called vitamin K cycle). During this cycle, KH2 is continuously regenerated. Blockade of vitamin K epoxide cycling back to vitamin K by warfarin and dicoumarol analogs prevents efficient carboxylation of vitamin K-dependent proteins and, thus, forms the basis of their action as anticoagulants.
To test for a role of vitamin K-dependent γ-glutamylcarboxylase in protection from oxidative cell death, the effect of warfarin on the protective function of vitamin K was investigated. Treatment of OLs with increasing concentrations of warfarin or dicoumarol did not block the protective effect of vitamin K, suggesting that regeneration of KH2 is not required for the observed protection and that carboxylation may not be involved (Fig. 5A). It is possible, however, that even in the absence of efficient recycling, enough vitamin K is present to generate carboxylation of a protein(s) that confers protection. Because there is evidence suggesting the existence of a warfarin-resistant vitamin K reductase (Suttie, 1985), we next tested the effect of 2-chloro-vitamin K1 (Cl-K1), an analog of K1 and a direct inhibitor of the carboxylase in vitro and in vivo (Lowenthal et al., 1960; Suttie, 1985). Even at over 100-fold excess of K1, Cl-K1 did not prevent vitamin K1 protection against cystine depletion-induced cell death (Fig. 5B). At higher concentrations (140 μm), Cl-K1 became cytotoxic. To prove that Cl-K1 indeed directly inhibits carboxylase enzyme activity at the concentrations used in our cultures, we incubated purified recombinant carboxylase with increasing molar ratios of Cl-K1 to vitamin K1 and determined the carboxylase activity. We found that Cl-K1 is indeed a potent inhibitor of the vitamin K-dependent carboxylase (Fig. 5C). Consistent with the above data, we found no measurable vitamin K-dependent carboxylase activity or epoxidase activity using a purified microsome preparation from cultured OLs. Primary adult rat hepatocytes, the major cell type expressing the carboxylase, were used as a positive control in these experiments (Fig. 5D, E). Therefore, we conclude that vitamin K blocks GSH depletion-induced oxidative cell death by a mechanism independent of carboxylation.
Figure 5.

Vitamin K protected OLs via a mechanism independent of vitamin K-dependentγ-glutamylcarboxylase.A,Inhibitors of vitamin K cycling, warfarin and dicoumarol, had no effect on vitamin K protection. OL precursors were induced to undergo cystine depletion-induced cell death in the presence or absence of indicated treatment for 24 hr, and cell viability was assayed. Concentrations used were: vitamin K1, 0.1μm; MK-4, 0.1μm; warfarin, 500μm; dicoumarol, 200μm. Results are representative of at least two separate experiments. B, Carboxylase inhibitor Cl-K1 did not reverse vitamin K protection. Cells were treated as indicated with or without Cl-K1 (0–140 μm) and vitamin K1 (0.2 μm) for 24 hr, and cell viability was analyzed. C, Cl-K1 directly inhibited enzymatic activity of purified γ-glutamylcarboxylase in vitro. Purified γ-glutamylcarboxylase was incubated in the presence of increasing concentrations of Cl-K1 versus K1 and assayed in duplicates for its enzyme activity. Results are mean ± SEM of three independent experiments. D, E, OL precursors express little, if any, γ-glutamylcarboxylase and vitamin K epoxidase activity. Microsomal proteins were extracted from cultured primary OLs, and vitamin K-dependent carboxylase and epoxidase activity of the microsomal preparation were analyzed. Primary rat hepatocyte microsomal preparation was performed in parallel as positive control for the enzyme assays. Results are representative of two independent experiments.
Persistent presence of MK-4 in culture is not required for its potent protection
We next asked whether pretreatment of cells with MK-4 would render them resistant to subsequent oxidative insult, because this would have potential therapeutic implications in vivo. Both vitamin K1 and MK-4 were readily uptaken by OLs as determined by HPLC analysis (data not shown). In experiments in which cells were treated with 0.1 μm MK-4, washed extensively, and then subjected to cystine deprivation, it was observed that pretreatment with 0.1 μm MK-4 for 1 hr was sufficient to provide complete protection against subsequent cystine depletion-induced cell death (Fig. 6A). Interestingly, similar pretreatment with vitamin K1 was not effective in preventing subsequent cystine depletion-induced cell death.
Figure 6.

Persistent presence of MK-4 was not required for protection. A, Effect of pretreatment with MK-4 on subsequent cystine depletion-induced cell death. OL precursors were preincubated with or without MK-4 (0.1 μm) for 1 hr in cystine-free medium, followed by wash three times with cystine-free medium, when indicated. Cell viability was determined 24 hr later, and the percentage of cell survival was based on the control in which cells were maintained in normal medium. Results are one representative of three independent experiments with similar results. B, Effect of vitamin K treatment at various time intervals after initiation of cystine depletion. Cells were treated with increasing amounts of K1 or MK-4 (μm) at the time of cystine depletion or the indicated time after cystine depletion. The total cystine deprivation time is 24 hr, and the cell viability was evaluated. l-Cystine (200 μm) was added back to the cystine-free medium at 8 hr after cystine depletion and was used as a control. Results are representative of three experiments.
To examine the time dependence of the protective action of vitamin K against the cystine depletion-induced cell death pathway, increasing concentrations of vitamin K1 and MK-4 were added to OLs at indicated intervals after the initiation of cystine deprivation, and cell viability was determined after 24 hr of cystine depletion (Fig. 6B). Concentrations as low as 100 nm for K1 or 10 nm for MK-4 were completely protective when added at the onset of cystine deprivation (i.e., added at T = 0 hr). Vitamin K1, however, lost its protective potency when added after 6 hr. MK-4 at10 nm was no longer protective if added after 6 hr, but MK-4 at 100 nm was still fully protective. MK-4 at 100 nm became only marginally effective when added after 8 hr. A higher concentration of MK-4 (1 μm) was required to inhibit cell death when added 8 hr after cystine depletion. Because it has been found that ROS became detectable only 10–15 hr after the onset of cystine deprivation (data not shown) (Back et al., 1998), these data suggest that for maximal effectiveness vitamin K must be present before the generation of ROS.
Vitamin K and MK-4 do not reverse decreases in intracellular GSH level but completely prevent ROS generation
Accumulation of intracellular oxygen free radicals occurs with intracellular GSH depletion initiated by exposure to glutamate, BSO, homocysteic acid, or cystine depletion of the medium (Murphy et al., 1989, 1990; Back et al., 1998). Thus, we tested whether vitamin K prevents the ROS accumulation caused by GSH depletion. Accumulation of intracellular ROS was assessed by oxidation of Rho123 or DCFH-DA. Rho123 reacts with oxidants to form the red fluorescent product rhodamine 123 (Henderson and Chappell, 1993). DCFH-DA, a cell-permeable, nonfluorescent dye, is deesterified, retained inside cells, and becomes fluorescent on oxidation (Royall and Ischiropoulos, 1993). Vitamin K1 and MK-4 completely inhibited the intracellular accumulation of ROS resulting from cystine deprivation (Fig. 7A, B), without sparing cells from depletion of intracellular GSH (Fig. 7D). Consistent with the hypothesis that vitamin K prevents death by decreasing the cellular oxidant burden, MK-4 (1 μm) rescued the cells when introduced to cells 8 hr after the onset of cystine deprivation (Fig. 6B); this rescue was associated with a significant decrease in ROS accumulation (Fig. 7C).
Figure 7.

Vitamin K blocked cystine deprivation-induced free radical accumulation. OLs were subjected to Cys + or Cys - medium as indicated, with or without vitamin K1 or MK-4 for 10–15 hr, and cells were then loaded with DCFH-DA (A) or Rho123 (B), and the free radical production was evaluated as described. Both DCFH-DA and Rho123 are nonfluorescent but become fluorescent on oxidation. DCF fluorescence was measured using a microplate reader, and oxidized Rho123 was visualized under a fluorescence microscope with fixed exposure settings to compare the relative production of ROS among cells with different treatments. C, Effect of vitamin K on ROS accumulation when added 8 hr after initiation of cystine depletion. Cells were first subjected to cystine deprivation for 8 hr, followed by the addition of MK-4. ROS accumulation in cells was evaluated 3 hr later, as inA.D,Effect of vitam in K on the loss of intracellular GSH induced by cystine depletion. Cells were treated as indicated for 15 hr, and the GSH level was evaluated as described. E, F, Vitamin K did not react with free radicals. Free radicals are generated by DDPH in solution and have absorption at 517 nm. The scavenging of the radicals was monitored spectrophotometrically after the addition of vitamin K or Trolox. Trolox was used as a positive control. Trolox rapidly scavenged free radicals, whereas vitamin K was unable to react directly with the radicals, and the absorption of free radical remained constant over time (E). Trolox equivalent antioxidant activity was determined by monitoring inhibition by specified reagents of ABTS cation formation with H2O2 as oxidant trigger as described (F). Data are representative of at least three independent experiments.
We next examined the possibility of a direct interaction between vitamin K and ROS. Unlike α-tocopherol and its soluble analog Trolox, vitamin K itself is not known to be an antioxidant (Vervoort et al., 1997). To evaluate the antioxidant potential of vitamin K1 and MK-4, we used two widely applied radical scavenging assays (Blois, 1958; Rice-Evans and Miller, 1994). In the first assay, we tested the ability of vitamin K1 and MK-4 to quench the stable lipoxyl radical DPPH. In the presence of vitamin K1 and MK-4 at 5 and 25 μm, the optical absorbance of a solution of DPPH remained constant. In contrast, Trolox, a known antioxidant, rapidly and efficiently scavenged DPPH, as indicated by the loss of DPPH absorbance (Fig. 7E). The second assay, the Trolox equivalent activity assay, is based on the inhibition by antioxidants of the formation of the radical cation ABTS+·. The ABTS radical cation is formed by the interaction of ABTS with ferrylmyoglobin radical species (·X - [FeIV = O]), generated by the activation of metmyoglobin with H2O2. Antioxidant compounds suppress the absorbance of the ABTS radical cation to an extent and on a time scale depending on their antioxidant capacities. Vitamin K1 and MK-4 at concentrations as high as 100 μm had no antioxidant activity, whereas tocopherol and Trolox efficiently prevented ABTS+· formation (Fig. 7F).
Discussion
We report here the discovery of a novel role of vitamin K in preventing GSH depletion-induced oxidative injury to primary OL precursors and to immature cortical neurons. To our knowledge, this is the first study demonstrating a protective function of vitamin K against oxidative stress in any cell.
We show that at low nanomolar concentrations, vitamin K1 and MK-4 block cell death induced by various methods in a concentration-dependent manner: cystine depletion of culture medium, or exposure to excess glutamate, homocysteic acid, or BSO (Fig. 3). All of these treatments result in depletion of intracellular GSH and accumulation of ROS. In this model, oxidative stress occurs because of the gradual and progressive accumulation of ROS within the cells. We also tested the protective efficacy of vitamin K in another model of cell death involving oxidative stress. Vitamin K1 and MK-4 had no effect on cell death induced by exogenous H2O2. The oxidative stress and the death mechanisms in these two models are likely to be different because exogenous H2O2 presents a rapid and intense insult.
Vitamin K is required for the vitamin K-dependent γ-glutamylcarboxylase that catalyzes a functionally important posttranslational modification of a relatively small number of vitamin K-dependent proteins, including transmembranous proteins with unknown functions, and Gas6 (growth arrest-specific gene product 6) with a survival-promoting function (Tsaioun, 1999). Although the involvement of carboxylase was not explored, low concentrations of vitamin K were found to have a trophic effect on primary cortical neurons cultured in the absence of serum (Nakajima et al., 1993). On the basis of these observations and current understanding of vitamin K metabolism, it was intriguing to hypothesize that the protective action of vitamin K1 and MK-4 against oxidative injury was because of vitamin K-dependent carboxylation of a protein that promotes cell survival. Several lines of evidence presented here, however, demonstrated that this hypothesis was incorrect. First, even at high concentrations, the vitamin K antagonists warfarin and dicoumarol, which inhibit vitamin K reductase, vitamin K cycling, and consequent carboxylation by depleting KH2, had no effect on the protective function of vitamin K (Fig. 5A). Second, 2-Cl-vitamin K1, an irreversible inhibitor of vitamin K-dependent γ-glutamylcarboxylase, did not block the protective action of vitamin K (Fig. 5B). The inhibitory effectiveness of 2-Cl-vitamin K1 was confirmed in an enzyme assay using purified γ-glutamylcarboxylase (Fig. 5C). Third, neither OLs nor immature cortical neurons express vitamin K-dependent γ-glutamylcarboxylase activity (Fig. 5D and data not shown). Moreover, the vitamin K epoxidase activity, required for the carboxylation reaction, also was not present in these cells (Fig. 5E and data not shown). Finally, the reported Km value of γ-glutamylcarboxylase for reduced K is in the range of 20–60 μm (Stanley et al., 1997), 1000-fold higher than the concentration required for protection by vitamin K. Therefore, we conclude that vitamin K prevents oxidative cell death via a mechanism independent of γ-glutamylcarboxylase.
Depletion of GSH is a necessary step for cell death induced by glutamate exposure or cystine deprivation (Murphy et al., 1989; Back et al., 1998). The cystine deprivation-induced loss of intracellular GSH is not prevented by treatment with vitamin K. The generation of ROS, however, is completely inhibited (Fig. 7). The origin of the ROS and the molecular events that lead to ROS generation and cell death in our model are not known and require further investigation. Other protective agents, such as the antioxidants idebenone and α-tocopherol, protect against glutamate depletion-induced or cystine depletion-induced oxidative cell death via preventing ROS accumulation without blocking the loss of GSH (Murphy et al., 1989, 1990; Oka et al., 1993; Yonezawa et al., 1996). Studies from several groups showed only weak activity (IC50 >> 100 μm) of vitamin K species (including menadione) as inhibitors of lipid peroxidation in various test systems (Wills, 1972; Canfield et al., 1985; Talcott et al., 1985; Ohyashiki et al., 1991). Consistent with these observations, vitamin K1 and MK-4 do not possess any antioxidant activity in the two antioxidant assays used here (Fig. 7E, F). Others, however, have also shown that the reduced form of vitamin K, hydroquinone KH2, has potent anti-lipid peroxidation activity in solution (Mukai et al., 1993) and in microsomes (Vervoort et al., 1997). Whether KH2 can act as an antioxidant in cells has not been studied previously. Nevertheless, it is possible that the prevention of ROS generation in OLs by vitamin K is mediated through a mechanism involving redox cycling of this vitamin. Direct detection of KH2 in cells has not been technically possible because KH2 is readily oxidized during extraction. Whether vitamin K is actually converted to KH2 in the OLs is currently unknown, but it appears unlikely. Warfarin, an inhibitor for vitamin K epoxidase/reductase, and dicoumarol, a potent inhibitor of quinone reductase responsible for the two-electron reduction of quinone to hydroquinone, had no effect on the vitamin K-dependent protection, even at concentrations 100-fold higher than those required to block the reductase (Fig. 5). Furthermore, staining for quinone reductase activity (Murphy et al., 1998) revealed only minor positivity in these OL precursors but profound activity in primary astrocytes, and this activity was completely blocked by the inhibitor dicoumarol (J. Li, A. Greene, J. J. Volpe, and P. A. Rosenberg, unpublished observations). Because vitamin K has also been shown to be metabolized into unidentified derivatives (Canfield et al., 1987; Ross et al., 1991), it remains possible that the protective activity of vitamin K observed in our study may because of one of its unknown metabolites, rather than vitamin K per se.
Another possibility for the action of vitamin K in preventing GSH depletion-induced cell death is that vitamin K at low concentrations may interact with early signaling events leading to ROS production. Several lines of evidence suggest that GSH depletion-induced oxidative death is mediated through a specific cell death signaling pathway. First, the transcriptional and translational inhibitors actinomycin D and cycloheximide block cystine deprivation-induced OL death (J. Li, J. J. Volpe, and P. A. Rosenberg, unpublished observations). Second, the tyrosine kinase inhibitor geldanamycin (Xiao et al., 1999) prevents GSH depletion-induced neuronal death. Third, inhibitors of the MEK signaling pathway protect against oxidative death in neurons (Stanciu et al., 2000). Although the detailed molecular cell death pathway in these oxidative injury models awaits thorough investigation, it has been suggested that vitamin K modulates cellular signaling transduction pathways in other systems. Using a chick embryogenesis model, Saxena et al. (1997) demonstrated the existence of a vitamin K1-dependent protein–tyrosine phosphorylation cascade that is sensitive to the alteration in levels or metabolism of vitamin K1. Ni et al. (1998) also showed increased tyrosine phosphorylation of specific proteins in cells treated with vitamin K analogs. Furthermore, the vitamin K-dependent protein, Gas6, binds to its tyrosine kinase receptor and activates a series of intracellular signaling pathways that are responsible for its survival-promoting effect in Schwann cells and fibroblasts (Goruppi et al., 1996, 1999; Li et al., 1996; Nakano et al., 1996). Neither tyrosine kinase inhibitors (such as genistein) nor phosphatidylinositol 3′-kinase (PI3K) inhibitors (LY9804902 and wortmanin), however, prevented the protective effect of vitamin K in OL cultures (J. Li, J. J. Volpe, and P. A. Rosenberg, unpublished observations), suggesting that the Gas6-PI3K pathway is not involved in our system. Again, this finding is consistent with our conclusion that the function of vitamin K is independent of carboxylation.
Discovery of new approaches to prevent oxidative stress-mediated injury to OLs is important for designing clinical strategies against PVL (Volpe, 2001). Using a primary cell culture system in which the OL lineage closely corresponds to that present in human cerebral white matter at a developmental stage most vulnerable to PVL (Back et al., 2001), we and others have demonstrated that these cells are exquisitely sensitive to GSH depletion-mediated oxidative injury (Back et al., 1998), AMPA–kainate-receptor activation (Rosenberg et al., 2003), and hypoxia/hypoglycemia-mediated cell death (Fern and Moller, 2000). The finding of a potent protective action of vitamin K1 and MK-4 against oxidative injury has potentially important clinical implications. OL precursors are the targets of white matter injury in neonatal hypoxia/ischemia rodent models (Back et al., 2002). Because neonates, and especially preterm infants, are developmentally vitamin K deficient and are at a substantial risk for hemorrhagic diseases, including intracranial hemorrhage, it is recommended by the American Academy of Pediatrics that vitamin K be given to newborns at birth to prevent hemorrhage. The ability of vitamin K to cross the blood brain barrier, its safety, and the current use in newborns make it an attractive potential therapeutic molecule for the prevention and treatment of PVL, for which no current treatment is available. The efficacy of vitamin K in preventing hypoxic/ischemic white matter injury in a neonatal animal model is currently under investigation.
In summary, we demonstrate for the first time that oxidative cell death induced by GSH depletion in primary OL precursors and in primary cortical neurons can be prevented by nanomolar concentrations of vitamin K1 and MK-4. The cytoprotective effect of K vitamins in this model is independent of their known biological role in carboxylation. They do not prevent the loss of intracellular GSH caused by cystine depletion but markedly inhibit ROS accumulation and, thus, cell death. These results suggest a new approach to developing potential preventative and therapeutic strategies for neurological diseases in which GSH depletion-induced oxidative stress plays a role.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants HD18655 and NS38475 (to J.J.V.), a Hearst Foundation Award (to J.L.), United Cerebral Palsy Foundation Research Grant R-737 (to J.L.), a fellowship from the Howard Hughes Medical Institute (to J.C.L.), and NIH Grant HL42443 (to B.C.F.). We thank Dr. Hannah C. Kinney for discussions and comments on this manuscript. We are grateful to Dr. John W. Suttie for reagents and helpful discussions and to Amanda Greene for technical assistance.
Correspondence should be addressed to Dr. Paul A. Rosenberg, Department of Neurology, Enders 3, Children's Hospital, 300 Longwood Avenue, Boston, MA 02115. E-mail: paul.rosenberg@tch.harvard.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235816-11$15.00/0
References
- Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ ( 1998) Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci 18: 6241-6253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Khan R, Gan X, Rosenberg PA, Volpe JJ ( 1999) A new Alamar Blue viability assay to rapidly quantify oligodendrocyte death. J Neurosci Methods 91: 47-54. [DOI] [PubMed] [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC ( 2001) Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci 21: 1302-1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, Chricton CA, Xanthoudakis S, Tam J, Arvin KL, Holtzman DM ( 2002) Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci 22: 455-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banasik M, Komura H, Shimoyama M, Ueda K ( 1992) Specific inhibitors of poly(ADP-ribose) synthetase and mono(ADP-ribosyl)transferase. J Biol Chem 267: 1569-1575. [PubMed] [Google Scholar]
- Blois MS ( 1958) Antioxidant determinations by the use of a stable free radical. Nature 181: 1199-1200. [Google Scholar]
- Canfield LM, Davy LA, Thomas GL ( 1985) Anti-oxidant/pro-oxidant reactions of vitamin K. Biochem Biophys Res Commun 128: 211-219. [DOI] [PubMed] [Google Scholar]
- Canfield LM, Johnson TM, Martin GS, Gunn JM ( 1987) Absorption and metabolism of vitamin K in Swiss 3T3 mouse fibroblasts–a model system for study of vitamin K absorption and metabolism. Biochem Biophys Res Commun 147: 731-739. [DOI] [PubMed] [Google Scholar]
- Davidson RT, Foley AL, Engelke JA, Suttie JW ( 1998) Conversion of dietary phylloquinone to tissue menaquinone-4 in rats is not dependent on gut bacteria. J Nutr 128: 220-223. [DOI] [PubMed] [Google Scholar]
- Fern R, Moller T ( 2000) Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci 20: 34-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floreani M, Skaper SD, Facci L, Lipartiti M, Giusti P ( 1997) Melatonin maintains glutathione homeostasis in kainic acid-exposed rat brain tissues. FASEB J 11: 1309-1315. [DOI] [PubMed] [Google Scholar]
- Furie BC, Kuliopulos A, Roth DA, Sugiura I, Walsh CT, Furie B ( 1997) Purification of native bovine carboxylase and expression and purification of recombinant bovine carboxylase. Methods Enzymol 282: 333-346. [DOI] [PubMed] [Google Scholar]
- Furie B, Bouchard BA, Furie BC ( 1999) Vitamin K-dependent biosynthesis of gamma-carboxyglutamic acid. Blood 93: 1798-1808. [PubMed] [Google Scholar]
- Goruppi S, Ruaro E, Schneider C ( 1996) Gas6, the ligand of Axl tyrosine kinase receptor, has mitogenic and survival activities for serum starved NIH3T3 fibroblasts. Oncogene 12: 471-480. [PubMed] [Google Scholar]
- Goruppi S, Ruaro E, Varnum B, Schneider C ( 1999) Gas6-mediated survival in NIH3T3 cells activates stress signalling cascade and is independent of Ras. Oncogene 18: 4224-4236. [DOI] [PubMed] [Google Scholar]
- Hall JG, Pauli RM, Wilson KM ( 1980) Maternal and fetal sequelae of anticoagulation during pregnancy. Am J Med 68: 122-140. [DOI] [PubMed] [Google Scholar]
- Haynes RL, Folkerth RD, Keefe RJ, Sung I, Swzeda L, Rosenberg PA, Volpe JJ, Kinney HC ( 2003) Nitrosative and oxidative injury to premyelinating oligodendrocytes in periventricular leukomalacia. J Neuropathol Exp Neurol 62: 441-450. [DOI] [PubMed] [Google Scholar]
- Henderson LM, Chappell JB ( 1993) Dihydrorhodamine 123: a fluorescent probe for superoxide generation? Eur J Biochem 217: 973-980. [DOI] [PubMed] [Google Scholar]
- Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Ord T, Bredesen DE ( 1993) Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science 262: 1274-1277. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Armstrong DD ( 1997) Perinatal neurophathology. In: Greenfields's neurophathology (Graham DI, Lantos PL, eds), pp 537-600. London: Arnold.
- LeBel CP, Ischiropoulos H, Bondy SC ( 1992) Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol 5: 227-231. [DOI] [PubMed] [Google Scholar]
- Li J, Bombeck CA, Yang S, Kim YM, Billiar TR ( 1999) Nitric oxide suppresses apoptosis via interrupting caspase activation and mitochondrial dysfunction in cultured hepatocytes. J Biol Chem 274: 17325-17333. [DOI] [PubMed] [Google Scholar]
- Li R, Chen J, Hammonds G, Phillips H, Armanini M, Wood P, Bunge R, Godowski PJ, Sliwkowski MX, Mather JP ( 1996) Identification of Gas6 as a growth factor for human Schwann cells. J Neurosci 16: 2012-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenthal J, MacFarlane JA, McDonald KM ( 1960) The inhibition of the antidotal activity of vitamin K1 against coumarin anticoagulant drugs by its chloro analogue. Experientia 16: 428-429. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J ( 1980) Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol 85: 890-902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai K, Morimoto H, Kikuchi S, Nagaoka S ( 1993) Kinetic study of free-radical-scavenging action of biological hydroquinones (reduced forms of ubiquinone, vitamin K and tocopherol quinone) in solution. Biochim Biophys Acta 1157: 313-331. [DOI] [PubMed] [Google Scholar]
- Mummah-Schendel LL, Suttie JW ( 1986) Serum phylloquinone concentrations in a normal adult population. Am J Clin Nutr 44: 686-689. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT ( 1989) Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2: 1547-1558. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Schnaar RL, Coyle JT ( 1990) Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J 4: 1624-1633. [PubMed] [Google Scholar]
- Murphy TH, So AP, Vincent SR ( 1998) Histochemical detection of quinone reductase activity in situ using LY83583 reduction and oxidation. J Neurochem 70: 2156-2164. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Furukawa S, Hayashi K, Yamada A, Kawashima T, Hayashi Y ( 1993) Age-dependent survival-promoting activity of vitamin K on cultured CNS neurons. Brain Res Dev Brain Res 73: 17-23. [DOI] [PubMed] [Google Scholar]
- Nakano T, Kawamoto K, Higashino K, Arita H ( 1996) Prevention of growth arrest-induced cell death of vascular smooth muscle cells by a product of growth arrest-specific gene, gas6. FEBS Lett 387: 78-80. [DOI] [PubMed] [Google Scholar]
- Ni R, Nishikawa Y, Carr BI ( 1998) Cell growth inhibition by a novel vitamin K is associated with induction of protein tyrosine phosphorylation. J Biol Chem 273: 9906-9911. [DOI] [PubMed] [Google Scholar]
- Ohyashiki T, Yabunaka Y, Matsui K ( 1991) Antioxidant effects of vitamin K homologues on ascorbic acid/Fe 2+-induced lipid peroxidation of lecithin liposomes. Chem Pharm Bull 39: 976-979. [DOI] [PubMed] [Google Scholar]
- Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ ( 1993) Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J Neurosci 13: 1441-1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orwar O, Li X, Andine P, Bergstrom CM, Hagberg H, Folestad S, Sandberg M ( 1994) Increased intra- and extracellular concentrations of γ-glutamylglutamate and related dipeptides in the ischemic rat striatum: involvement of glutamyl transpeptidase. J Neurochem 63: 1371-1376. [DOI] [PubMed] [Google Scholar]
- Pfeiffer SE, Warrington AE, Bansal R ( 1993) The oligodendrocyte and its many cellular processes. Trends Cell Biol 3: 191-197. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Murphy TH, Baraban JM ( 1994) Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem 62: 376-379. [DOI] [PubMed] [Google Scholar]
- Rice-Evans C, Miller NJ ( 1994) Total antioxidant status in plasma and body fluids. Methods Enzymol 234: 279-293. [DOI] [PubMed] [Google Scholar]
- Romero EE, Velazquez-Estades LJ, Deo R, Schapiro B, Roth DA ( 1998) Cloning of rat vitamin K-dependent γ-glutamyl carboxylase and developmentally regulated gene expression in postimplantation embryos. Exp Cell Res 243: 334-346. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Dai W, Gan XD, Ali S, Fu J, Back BA, Sanchez RM, Segal MM, Follett PL, Jensen FE, Volpe JJ ( 2003) Mature myelin basic protein-expressing oligodendrocytes are insensitive to kainate toxicity. J Neurosci Res 71: 237-245. [DOI] [PubMed] [Google Scholar]
- Ross PJ, Shearer MJ, Diplock AT, Schey SA ( 1991) A fibroblast cell culture model to study vitamin K metabolism and the inhibition of vitamin K epoxide reductase by known and suspected antagonists. Br J Haematol 77: 195-200. [DOI] [PubMed] [Google Scholar]
- Royall JA, Ischiropoulos H ( 1993) Evaluation of 2′,7′-dichlorofluorescin and dihydrorhodamine 123 as fluorescent probes for intracellular H2O2 in cultured endothelial cells. Arch Biochem Biophys 302: 348-355. [DOI] [PubMed] [Google Scholar]
- Saxena SP, Fan T, Li M, Israels ED, Israels LG ( 1997) A novel role for vitamin K1 in a tyrosine phosphorylation cascade during chick embryogenesis. J Clin Invest 99: 602-607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivakumar BR, Kolluri SV, Ravindranath V ( 1995) Glutathione and protein thiol homeostasis in brain during reperfusion after cerebral ischemia. J Pharmacol Exp Ther 274: 1167-1173. [PubMed] [Google Scholar]
- Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW, DeFranco DB ( 2000) Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem 275: 12200-12206. [DOI] [PubMed] [Google Scholar]
- Stanley TB, Stafford DW, Olivera BM, Bandyopadhyay PK ( 1997) Identification of a vitamin K-dependent carboxylase in the venom duct of a Conus snail. FEBS Lett 407: 85-88. [DOI] [PubMed] [Google Scholar]
- Sugiura I, Furie B, Walsh CT, Furie BC ( 1997) Propeptide and glutamate-containing substrates bound to the vitamin K-dependent carboxylase convert its vitamin K epoxidase function from an inactive to an active state. Proc Natl Acad Sci USA 94: 9069-9074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaram KS, Fan JH, Engelke JA, Foley AL, Suttie JW, Lev M ( 1996) Vitamin K status influences brain sulfatide metabolism in young mice and rats. J Nutr 126: 2746-2751. [DOI] [PubMed] [Google Scholar]
- Suttie JW ( 1985) Vitamin K-dependent carboxylase. Annu Rev Biochem 54: 459-477. [DOI] [PubMed] [Google Scholar]
- Suttie JW ( 1991) Vitamin K. In: Handbook of vitamins, Ed 2 (Machlin LJ, ed), pp 145-194. New York and Basel: Marcel Dekker.
- Szabo C, Dawson VL ( 1998) Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci 19: 287-298. [DOI] [PubMed] [Google Scholar]
- Talcott RE, Smith MT, Gianni DD ( 1985) Inhibition of microsomal lipid peroxidation by naphtoquinones: structure-activity relationships and possible mechanisms of action. Arch Biochem Biophys 241: 88-94. [DOI] [PubMed] [Google Scholar]
- Thijssen HH, Drittij-Reijnders MJ, Fischer MA ( 1996) Phylloquinone and menaquinone-4 distribution in rats: synthesis rather than uptake determines menaquinone-4 organ concentrations. J Nutr 126: 537-543. [DOI] [PubMed] [Google Scholar]
- Tsaioun KI ( 1999) Vitamin K-dependent proteins in the developing and aging nervous system. Nutr Rev 57: 231-240. [DOI] [PubMed] [Google Scholar]
- Ulrich MM, Furie B, Jacobs MR, Vermeer C, Furie BC ( 1988) Vitamin K-dependent carboxylation. A synthetic peptide based upon the γ-carboxylation recognition site sequence of the prothrombin propeptide is an active substrate for the carboxylase in vitro J Biol Chem 263: 9697-9702. [PubMed] [Google Scholar]
- Vervoort LM, Ronden JE, Thijssen HH ( 1997) The potent antioxidant activity of the vitamin K cycle in microsomal lipid peroxidation. Biochem Pharmacol 54: 871-876. [DOI] [PubMed] [Google Scholar]
- Volpe JJ ( 1997) Brain injury in the premature infant–from pathogenesis to prevention. Brain Dev 19: 519-534. [DOI] [PubMed] [Google Scholar]
- Volpe JJ ( 2001) Neurology of the newborn, Ed 4. Philadelphia: Saunders.
- Wallin C, Puka-Sundvall M, Hagberg H, Weber SG, Sandberg M ( 2000) Alterations in glutathione and amino acid concentrations after hypoxiaischemia in the immature rat brain. Brain Res Dev Brain Res 125: 51-60. [DOI] [PubMed] [Google Scholar]
- Wang H, Joseph JA ( 1999) Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic Biol Med 27: 612-616. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Chung HJ, Schnuer J, Pratt K, Zable AC, Kavanaugh MP, Rosenberg PA ( 1998) High affinity glutamate transport in rat cortical neurons in culture. Mol Pharmacol 53: 88-96. [DOI] [PubMed] [Google Scholar]
- Wills ED ( 1972) Effects of vitamin K and naphtoquinones on lipid peroxide formation and oxidative demethylation by liver microsomes. Biochem Pharmacol 21: 1879-1883. [DOI] [PubMed] [Google Scholar]
- Xiao N, Callaway CW, Lipinski CA, Hicks SD, DeFranco DB ( 1999) Geldanamycin provides posttreatment protection against glutamate-induced oxidative toxicity in a mouse hippocampal cell line. J Neurochem 72: 95-101. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sakamoto N, Iwai A, Yatsugi S, Hidaka K, Noguchi K, Yuasa T ( 1993) Protective actions of YM737, a new glutathione analog, against cerebral ischemia in rats. Res Commun Chem Pathol Pharmacol 81: 221-232. [PubMed] [Google Scholar]
- Yonezawa M, Back SA, Gan X, Rosenberg PA, Volpe JJ ( 1996) Cystine deprivation induced oligodendroglial death: rescue by free radical scavengers and by a diffusible glial factor. J Neurochem 67: 566-573. [DOI] [PubMed] [Google Scholar]