

Abstract
Aspirin (acetylsalicylic acid) has become one of the most commonly used drugs, given its role as an analgesic, antipyretic and agent for cardiovascular prophylaxis. Several decades of research have provided considerable evidence demonstrating its potential for the prevention of cancer, particularly colorectal cancer. Broader clinical recommendations for aspirin-based chemoprevention strategies have recently been established; however, given the known hazards of long-term aspirin use, larger-scale adoption of an aspirin chemoprevention strategy is likely to require improved identification of individuals for whom the protective benefits outweigh the harms. Such a precision medicine approach may emerge through further clarification of aspirin’s mechanism of action.
Despite greater adoption of population screening and considerable advances in understanding the molecular basis of colorectal neoplasia, colorectal cancer (CRC) remains the second leading cause of cancer deaths in the United States, with an estimated 129,700 new cases expected for 2015 (REF. 1). Aspirin (acetylsalicylic acid) has emerged as perhaps the most promising agent for the chemoprevention of CRC2,3. This is due in large part to remarkably consistent data that have emerged from numerous basic, clinical and epidemiological studies over the past several decades (FIG. 1). The United States Preventive Services Task Force (USPSTF) originally recommended against the use of aspirin for the prevention of CRC in 2007 (REF. 4). However, in 2015, in their updated draft recommendations5 for low-dose aspirin in the primary prevention of cardiovascular disease (CVD), the USPSTF acknowledged that supporting evidence had become so compelling that CRC prevention warranted inclusion in their rationale for routine aspirin use among those aged between 50 and 69 with specific cardiovascular risk profiles6,7 (BOX 1). This decision distinguishes aspirin as the first pharmacological agent to be endorsed for cancer chemoprevention in a population not characterized as having a high risk of developing cancer. Nevertheless, the USPSTF also cautioned against the potential harms associated with regular aspirin use and highlighted the need to clarify the mechanisms by which aspirin prevents the development of colorectal neoplasia.
Figure 1 |. Timeline of notable human studies in aspirin chemoprevention.
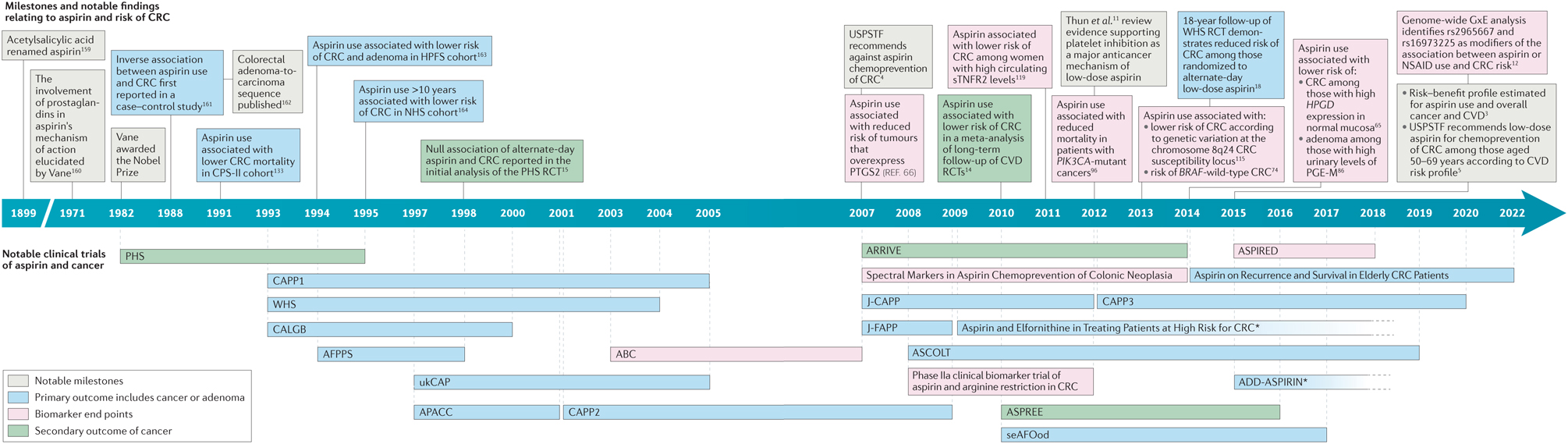
Decades of research have substantially clarified the anticancer effect associated with the use of aspirin. Recent compelling human data have also influenced the United States Preventive Services Task Force (USPSTF) to support the use of aspirin for the primary prevention of colorectal cancer (CRC) in certain subgroups of the US population. Asterisks indicate trials that are ongoing and have an unknown end date. ABC, Aspirin and the Biology of the Colon; AFPPS, Aspirin/Folate Polyp Prevention Study; APACC, Association pour la Prévention par l’Aspirine du Cancer Colorectal; ARRIVE, Aspirin to Reduce Risk of Initial Vascular Events; ASCOLT, Aspirin for Dukes C and High Risk Dukes B Colorectal Cancers; ASPREE, ASPirin in Reducing Events in the Elderly; ASPIRED, ASPirin Intervention for the REDuction of colorectal cancer risk; CALGB, Colorectal Adenoma prevention study originated in the cooperative trials group cancer and Leukaemia Group B; CAPP, Colorectal Adenoma/carcinoma Prevention Programme; CPS-II, Cancer Prevention Study II; CVD, cardiovascular disease; GxE, gene by environment; HPFS, Health Professionals Follow-up Study; HPGD, 15-hydroxyprostaglandin dehydrogenase; J-CAPP, Japan Colorectal Aspirin Polyps Prevention; J-FAPP, Japan Familial Adenomatous Polyposis Prevention; NHS, Nurses’ Health Study; NSAID, non-steroidal anti-inflammatory drug; PGE-M, major metabolite of prostaglandin E2; PHS, Physicians’ Health Study; PIK3CA, PI3K catalytic subunit-α; PTGS2, prostaglandin-endoperoxide synthase 2; RCT, randomized clinical trial; seAFOod, Systematic Evaluation of Aspirin and Fish Oil; sTNFR2, soluble TNF receptor 2; ukCAP, United Kingdom Colorectal Adenoma Prevention; WHS, Women’s Health Study.
Box 1 |. Summary of the United States Preventive Services Task Force draft recommendation.
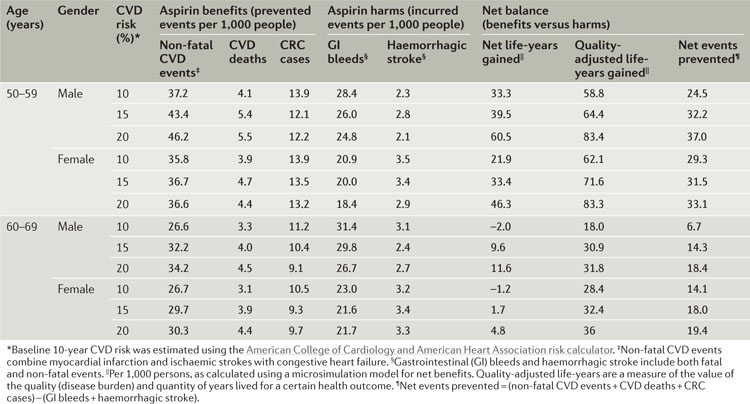
In October 2015, the United States Preventive Services Task Force (USPSTF) released a draft statement5 recommending the use of aspirin for the primary prevention of colorectal cancer (CRC) and cardiovascular disease (CVD). This statement was based on the findings of an evidence review commissioned by the Agency for Healthcare Research and Quality for the USPSTF7 (see the table). The recommendation has received a B grade for adults aged 50–59 years with a 10% or greater 10-year CVD risk, who are not at increased risk for bleeding, have a life expectancy of at least 10 years and are willing to take aspirin for at least 10 years5. For adults aged 60–69 years with similar risk profiles, the recommendation was assigned a C grade5. A B grade indicates that there is “high or moderate certainty that the net benefit is moderate to substantial” and it is suggested that practitioners offer or provide this service5. A C grade indicates that there is “at least moderate certainty that the net benefit is small” and it is suggested that practitioners offer or provide the service to certain patients depending on individual circumstances5. Table adapted from REF. 7 with permission from the Agency for Healthcare Research and Quality. Updated information is available on the USPSTF website.
In this Opinion article, we review the weight of the evidence supporting the chemopreventive potential of aspirin and highlight advances in our understanding of its mechanisms of action. We challenge the notion that aspirin prevents cancer through a single, dominant pathway and propose an integrative multi-pathway model for its mode of action. Furthermore, we highlight how these pathways can be leveraged to develop mechanistic biomarkers for personalized risk stratification. Such biomarkers may then be translated clinically in a precision medicine approach that is crucial to the future of aspirin chemoprevention.
Supporting evidence
The epidemiological evidence supporting the efficacy of aspirin for the prevention of cancer, especially CRC, is substantial2,3,8–11. A recent pooled analysis of 10 cohort and case–control studies demonstrated that aspirin use was associated with a 29% reduction in the incidence of CRC (odds ratio (OR) = 0.71; 95% confidence interval (CI) 0.66–0.77)12. More recently, a study from northern Denmark reported a 27% reduction in CRC risk (OR = 0.73; 95% CI 0.54–0.99) in those who consecutively filled low-dose aspirin prescriptions for more than 5 years13. Beyond cohort and case–control studies, additional data have also emerged through secondary analyses of randomized controlled trials (RCTs) originally conducted to examine the role of aspirin in the prevention of CVD that have subsequently been linked to long-term cancer outcomes (TABLE 1). A meta-analysis of 3 such trials found that aspirin treatment for 5 or more years at doses of 75–300 mg per day reduced the long-term risk of CRC by 24% (hazard ratio (HR) = 0.75; 95% CI 0.56–0.97)14. Because aspirin treatment was randomized, these findings are less likely to be due to confounding effects associated with the reason for use, which may complicate the analysis of data from cohort or case–control studies.
Table 1 |.
Placebo-controlled clinical trials of aspirin in the prevention of colorectal cancer
| Trial name | Primary end point | Inclusion criteria | Aspirin dose | Participants (n) | Median duration of treatment (years) | RR (95% CI) | |
|---|---|---|---|---|---|---|---|
| CRC incidence | CRC mortality | ||||||
| TPT14 | CVD | Men aged 45–69 years without a current or recent history of possible peptic ulceration, a history of possible or definite MI or stroke or other medication incompatible with trial treatment | 75 mg per day | 5,085 | 6.9 | 0.75 (0.56–0.97), pooled with UK-TIA and SALT* | 0.61 (0.43–0.87), pooled with UK-TIA and SALT |
| UK-TIA14* | CVD | Those aged ≥40 years whose last cerebrovascular event had occurred >3 months earlier; who have had a disabling major stroke; patients with potential cardiac sources of embolism who were not taking anticoagulants | 300 or 1,200 mg per day | 2,449 | 4.4 | See pooled results above* | See pooled results above |
| SALT14 | CVD | Those aged 50–79 years who had had a transient ischaemic attack (including amaurosis fugax), minor ischaemic stroke or retinal artery occlusion in the previous 3 months | 75 mg per day | 1,360 | 2.7 | See pooled results above* | See pooled results above |
| BDT‡ | CVD | Male physicians without peptic ulcer, stroke or definite MI | 500 mg per day | 4,959 | 6.0 | 0.70 (0.51–0.97)9 | 0.73 (0.49–1.10)14 |
| PHS15 | CVD and cancer | Male physicians aged 40–84 years who did not regularly use aspirin or other NSAID and did not have a history of MI, stroke, cancer, liver or renal disease, gout, peptic ulcer or contraindications to aspirin | 325 mg every other day§ | 22,071 | 5.0 | 1.03 (0.83–1.28) | Not reported |
| WHS18 | CVD and cancer | Women, aged 45 years or older, with no history of cancer (except for non-melanoma skin cancer), CVD or other major chronic illness | 100 mg every other day | 39,876 | 9.0 | 0.80 (0.67–0.97) | Not reported |
| CAPP2 (REF. 19) | CRC | Carriers of Lynch syndrome | 600 mg per day§ | 861 | 2.1 | 0.63 (0.35–1.13) | Not reported |
BDT, British Doctors Aspirin Trial; CAPP2, Colorectal Adenoma/carcinoma Prevention Programme 2; CI, confidence interval; CRC, colorectal cancer; CVD, cardiovascular disease; MI, myocardial infarction; NSAID, non-steroidal anti-inflammatory drug; PHS, Physicians’ Health Study; RR, relative risk; SALT, Swedish Aspirin Low Dose Trial; TPT, Thrombosis Prevention Trial; UK-TIA, UK-Transient Ischaemic Attack; WHS, Women’s Health Study.
For aspirin doses of 75–300 mg, from Rothwell et al.14.
BDT was not placebo controlled. Comparisons with a ‘no aspirin’ control group.
Trials had a 2 × 2 factorial design. PHS: aspirin with or without β-carotene (50 mg per day). CAPP2: aspirin with or without resistant starch (30 g per day).
Aspirin RCTs with cancer outcomes.
Until recently, RCTs of aspirin that included cancer outcomes as predefined end points were limited in number and generated mostly discouraging findings. The Physicians’ Health Study (PHS), a placebo-controlled RCT of alternate-day 325 mg aspirin among 22,071 male physicians, found no association between aspirin use and CRC incidence over 12 years of follow-up15. The Women’s Health Study (WHS), the largest placebo-controlled RCT of aspirin for primary prevention, compared alternate-day 100 mg aspirin with a median duration of treatment of 9 years16. The results at completion of the originally planned follow-up of 10 years provided no evidence of an effect of aspirin on CRC incidence16. In addition, the Colorectal Adenoma/carcinoma Prevention Programme 2 (CAPP2) trial observed no reduction in the risk of colorectal neoplasia among carriers of Lynch syndrome, a hereditary CRC syndrome, after 2.5 years of planned follow-up in those assigned to 600 mg of aspirin per day17.
However, extended follow-up of RCTs has provided more promising evidence. For example, in the WHS, an inverse association between those randomized to aspirin and CRC incidence emerged10 years after randomization (HR = 0.80; 95% CI 0.67–0.97)18. In the CAPP2 trial, a secondary preplanned analysis, conducted after the first participant reached 10 years of follow-up, did show a significantly reduced risk of CRC in the per-protocol analysis (HR = 0.41; 95% CI 0.19–0.86) and after accounting for the multiple primary events commonly seen among patients with Lynch syndrome (incidence rate ratio (IRR) = 0.56; 95% CI 0.32–0.99)19. Nonetheless, these findings may not be generalizable to sporadic, non-hereditary CRC, as Lynch syndrome tumours arise through a distinct pathway of defective DNA mismatch repair20,21 and account for fewer than 3% of all cases of CRC.
Thus, there has not yet been a definitive RCT investigating the effect of long-term daily aspirin treatment at a range of doses with sporadic CRC as a pre-specified end point over at least 10 years of defined follow-up. Such a trial has been challenging to pursue owing to the large number of subjects and prolonged follow-up required for incident cancers to emerge. Moreover, the conduct of such an RCT would be complicated by problems of long-term compliance with an aspirin regimen, as well as the rising prevalence of aspirin use for CVD prevention, which leads to significant treatment-group crossover over time. The resulting decrease in assigned aspirin use in the treatment group and increase in non-trial aspirin use in the placebo group will dilute a potential randomized treatment effect.
Aspirin RCTs with precancerous outcomes.
In lieu of such studies, RCTs designed to examine the impact of aspirin on colorectal adenomas, the precursor to the majority of CRCs22–25, have provided compelling evidence of causality and the efficacy of aspirin chemoprevention (TABLE 2). Adenoma prevention trials are more feasible, given the relatively short-term follow-up needed to examine the recurrence of adenomas in individuals at higher risk for colorectal neoplasia. Four trials (namely, the Aspirin/Folate Polyp Prevention Study (AFPPS), Association pour la Prévention par l’Aspirine du Cancer Colorectal (APACC), Cancer and Leukaemia Group B (CALGB) and the United Kingdom Colorectal Adenoma Prevention (ukCAP) trial) report a range of reductions in the risk of recurrent adenoma (4–39%) among individuals with a high risk of sporadic CRC26–29. Recently, the Japan Colorectal Aspirin Polyps Prevention (J-CAPP) trial showed that aspirin reduced the recurrence of any adenoma or CRC (relative risk (RR) = 0.60; 95% CI 0.36–0.98)30. Interestingly, in a secondary analysis, prevention of adenoma recurrence seemed to be restricted to non-smokers, providing evidence of a possible interaction with cigarette smoking. This interaction was recently corroborated by a large cross-sectional study of colorectal polyps31 and an RCT of a combined aspirin, calcitriol and calcium pill32. The RCT found no overall association between treatment and adenoma recurrence, but provided evidence of an interaction between treatment effects and smoking status32. Although the study encountered several challenges — including early trial termination, a non-traditional combination drug, high dropout rate and low compliance — the authors also attributed the null association to the relatively large proportion of smokers in the study population. As such, further investigation into lifestyle modifiers of aspirin chemoprevention, such as cigarette smoking, is warranted.
Table 2 |.
Placebo-controlled clinical trials of aspirin in the prevention of colorectal adenoma
| Trial name | Primary end point | Inclusion criteria | Participants (n) | Median duration of follow-up (years) | Aspirin dose | RR (95% CI) | Refs | |
|---|---|---|---|---|---|---|---|---|
| Adenoma | Advanced adenoma | |||||||
| AFPPS | Recurrent adenoma | Recent resection of colorectal adenomas | 1,084 | 3.0 | 81 mg per day | 0.81 (0.69–0.96) | 0.62 (0.39–0.97) | 165 |
| 325 mg per day | 0.96 (0.82–1.12) | 0.86 (0.58–1.30) | ||||||
| Any aspirin | 0.88 (0.77–1.02) | 0.74 (0.52–1.06) | ||||||
| APACC | Recurrent adenoma | Recent resection of colorectal adenomas | 184 | 4.0 | 160 mg per day | 0.88 (0.65–1.19) | 1.19 (0.65–2.21) | 165 |
| 300 mg per day | 1.03 (0.77–1.37) | 0.58 (0.25–1.37) | ||||||
| Any aspirin | 0.95 (0.75–1.21) | 0.91 (0.51–1.60) | ||||||
| CALGB | Adenoma | Recent resection of Dukes’ stage A or B1 CRC or B2 CRC and 5-year disease-free survival | 517 | 1.1 | 325 mg per day | 0.61 (0.44–0.86) | 0.77 (0.29–2.05) | 165 |
| ukCAP | Recurrent adenoma | Recent resection of colorectal adenomas | 853 | 3.0 | 300 mg per day* | 0.79 (0.63–0.99) | 0.63 (0.43–0.91) | 165 |
| J-CAPP | Recurrent adenoma and CRC | Recent resection of colorectal adenomas and CRCs | 311 | 2.0 | 100 mg per day | 0.60 (0.36–0.98), pooled with advanced melanoma | 30 | |
AFPPS, Aspirin/Folate Polyp Prevention Study; APACC, Association pour la Prévention par l’Aspirine du Cancer Colorectal; CALGB, Colorectal Adenoma prevention study originated in the cooperative trials group cancer and Leukaemia Group B; CI, confidence interval; CRC, colorectal cancer; J-CAPP, Japan Colorectal Aspirin Polyps Prevention; RR, relative risk; ukCAP, United Kingdom Colorectal Adenoma Prevention.
Trial had a 2 × 2 factorial design. ukCAP: aspirin with or without folate supplement (0.5 mg per day).
Ongoing aspirin RCTs.
Over the coming years, additional ongoing RCTs (Aspirin to Reduce Risk of Initial Vascular Events (ARRIVE)33, Aspirin for Dukes C and High Risk Dukes B Colorectal Cancers (ASCOLT)34, ASPirin in Reducing Events in the Elderly (ASPREE)35, ASPirin Intervention for the REDuction of colorectal cancer risk (ASPIRED)36, CAPP3 (REF. 37) and Systematic Evaluation of Aspirin and Fish Oil (seAFOod) polyp prevention trial38), combined with continued collection of long-term outcome data from completed trials, are expected to add to the growing evidence in support of aspirin chemoprevention of CRC. All things considered, the ‘perfect’ trial of aspirin and CRC prevention may not be feasible. However, the consistency of both epidemiological and RCT evidence thus far provides a compelling basis from which to more broadly consider the use of aspirin for chemoprevention, especially for CRC, a consensus opinion that is shared by many authorities in the field2,3,8,9,11. Indeed, the current level of ‘imperfect’ evidence was considered sufficiently persuasive to lead the USPSTF to incorporate the effect of aspirin on CRC in its broader recommendation for adults in the United States between the ages of 50 and 69 with a greater than 10% 10-year risk of CVD to consider chronic disease prophylaxis with low-dose aspirin39.
Assessing risk–benefit for chronic aspirin use.
BOX 2 presents a summary of the potential side effects associated with regular aspirin use. Several studies have estimated the magnitude of such risks, particularly gastrointestinal bleeding, through assessment of the severe adverse events reported during RCTs. A meta-analysis of 35 RCTs using 75–325 mg daily doses of aspirin alone estimated an HR for major gastrointestinal bleeding of 1.31 (95% CI 1.21–1.42)40. For average-risk individuals (without a history of bleeding and not taking other anticoagulant or antiplatelet medications), this translates into one or two gastrointestinal bleeding events per 1,000 person-years. Most41, but not all42,43, studies find that such toxicities are largely dose related, with the hazards for bleeding being generally higher in patients treated with 300–325 mg than in those treated with 75–162.5 mg44–47. However, in 2012, a meta-analysis of RCTs by Rothwell and colleagues10 found that major extracranial bleeding (mainly gastrointestinal) associated with aspirin occurred primarily in the short term (<3 years) following initiation of aspirin use, with likelihood increasing with age. They concluded that in the long term (≥3 years), low-dose (<300 mg) aspirin use was not significantly associated with the risk of such events10. Nonetheless, the consideration of such risks highlights the importance of developing a precision medicine strategy to identify those individuals who are most likely to benefit from a prophylactic aspirin regimen.
Box 2 |. Adverse events associated with aspirin use.
In the United States, aspirin is widely available without a prescription. The most commonly reported adverse effects of aspirin use include nonspecific gastrointestinal (GI) symptoms, such as abdominal pain, dyspepsia, and nausea and vomiting157. The most common serious adverse event is GI bleeding39, with aspirin use associated with an odds ratio (OR) for GI bleeding of 1.59 (95% confidence interval (CI) 1.32–1.91) and an increase of 0.29 events per 1,000 person-years of aspirin exposure. Aspirin is also associated with intracranial bleeding (OR = 1.27; 95% CI 0.98–1.66), although the absolute risk increase is lower (0.1 events per 1,000 person-years of aspirin exposure) than the risk of GI bleeding. Bleeding events seem to be correlated with dose and increasing age. The estimated adjusted incidence rate ratio for major bleeding events with each increasing year of age is 1.05 (95% CI 1.05–1.05). For GI symptoms and prophylaxis for bleeding, clinical management may include co-administration of a proton pump inhibitor or H2 blocker158.
People with the following conditions are generally advised against using aspirin39: aspirin allergy or intolerance; active peptic ulcer; bleeding disorders; recent history of GI or intracranial bleeding; renal failure; severe liver disease; or low platelet count (thrombocytopenia). Individuals concurrently using anticoagulants or non-steroidal anti-inflammatory drugs (NSAIDs) may also be cautioned against using aspirin as these may increase the incidence or severity of bleeding events.
Molecular risk stratification
The first efforts to develop precision medicine approaches to improve the therapeutic index for aspirin chemoprevention focused on pharmacogenomics48, and particularly on variants of genes encoding the highly polymorphic CYP2C9 (cytochrome p450 family 2 subfamily C polypeptide 9) and UGT (UDP-glucuronosyltransferase) enzymes, which are responsible for the metabolism of aspirin and non-steroidal anti-inflammatory drugs (NSAIDs). However, data have not been consistent: most49–55, but not all56,57, recent studies have demonstrated no significant modification of aspirin or NSAID responsiveness according to UGT or CYP genotype. An additional precision medicine approach to aspirin chemoprevention may emerge through advances in our understanding of the associated anticancer mechanisms. These biological pathways may be exploited for molecular biomarkers to stratify risk–benefit (TABLE 3). In this section, we review the progress made thus far in elucidating aspirin’s mode of action, which has been facilitated by advances in biobanking, genomics, metabolomics and integrative molecular epidemiology over the past decade.
Table 3 |.
Summary of promising molecular biomarkers associated with aspirin chemoprevention of adenoma or colorectal cancer
| Biomarker | Study population | Study design | Outcome | Cases (n) | Biomarker description | RR (95% CI)* of outcome according to regular aspirin or NSAID use stratified by biomarker | Refs | |
|---|---|---|---|---|---|---|---|---|
| Germline genetic | ||||||||
| rs6983267-T | NHS and HPFS | Nested case–control | CRC | 840 | T allele of SNP |
|
GG: 0.99 (0.70–1.40) | 115 |
| rs2965667-TT | 10 cohorts‡ | GWAS§ | CRC | 8,634 | TT genotype of SNP | TT: 0.63 (0.59–0.68) | TA or AA: 1.76 (1.16–2.66) | 12 |
| rs16973225-AA | AA genotype of SNP | AA: 0.63 (0.59–0.68) | AC or CC: 0.93 (0.75–1.17) | |||||
| rs2920421-GA | CCFR | Case–unaffected-sibling-control study | CRC | 1,621 | GA genotype of SNP | GA: 0.60 (0.45–0.80) |
|
94 |
| rs2430420-GG | AFPPS | Nested cohort within an RCT | Adenoma | 370 | GG genotype of SNP | GG: 0.68 (0.50–0.94)∥ | GA or AA: 0.95 (0.75–1.20)∥ | 154 |
| rs28362380-TT | TT genotype of SNP | TT: 0.75 (0.61–0.92)∥ | TC or CC: 1.32 (0.85–2.06)∥ | |||||
| Colonic mucosa | ||||||||
| HPGD | NHS and HPFS | Prospective cohort | CRC | 270 | HPGD mRNA expression in normal mucosa | High HPGD: 0.49 (0.34–0.71) | Low HPGD: 0.90 (0.63–1.27) | 65 |
| Colorectal tumour | ||||||||
| PIK3CA mutation | NHS and HPFS | Prospective cohort | CRC | 1,226 | PIK3CA (exons 9 and 20) mt tumours | PIK3CA mt: 0.66 (0.48–0.89) | PIK3CA wt: 0.79 (0.69–0.90) | 74,96 |
| CRC-specific survival | 964 | PIK3CA mt: 0.18 (0.06–0.61) | PIK3CA wt: 0.96 (0.69–1.32) | |||||
| BRAF mutation | NHS and HPFS | Prospective cohort | CRC | 1,226 | BRAFV600E mutation status of tumours | BRAF wt: 0.73 (0.64–0.83) | BRAF mt: 1.03 (0.76–1.38) | 74 |
| PTGS2 overexpression | NHS and HPFS | Prospective cohort | CRC | 632 | Positive PTGS2 expression in tumour by IHC | Positive PTGS2: 0.64 (0.52–0.78) | Negative PTGS2: 0.96 (0.73–1.26) | 66 |
| Urine | ||||||||
| PGE-M | NHS | Nested case–control | Adenoma | 420 | Quartiles (Q) of urinary PGE-M | High PGE-M (Q2–Q4): 0.61 (0.43–0.87) | Low PGE-M (Q1): 1.05 (0.50–2.19) | 86 |
| AFPPS | RCT | Adenoma | 328 | Tertiles (T) of urinary PGE-M | High PGE-M (T2 or T3):
|
Low PGE-M (T1):
|
87 | |
| Plasma | ||||||||
| MIC1 | NHS and HPFS | Nested case–control | CRC | 618 | Cohort-specific quintiles of plasma MIC1 | High MIC1 and PTGS2 positive: 0.60 (0.41–0.88) | High MIC1 and PTGS2 negative: 1.21 (0.71–2.07) | 175 |
| sTNFR2 | NHS | Nested case–control | CRC | 280 | ≥Median plasma sTNFR2 | sTNFR2 ≥ median: 0.39 (0.18–0.86) | sTNFR2 < median: 0.86 (0.41–1.79) | 119 |
AFPPS, Aspirin/Folate Polyp Prevention Study; CI, confidence interval; CRC, colorectal cancer; GWAS, genome-wide association study; HPGD, 15-hydroxyprostaglandin dehydrogenase; HR, hazard ratio; IHC, immunohistochemistry; MIC1, macrophage inhibitory cytokine 1; mt, mutant; NSAID, non-steroidal anti-inflammatory drug; PGE-M, major metabolite of prostaglandin E2; PIK3CA, PI3K catalytic subunit-α; PTGS2, prostaglandin-endoperoxide synthase 2; RCT, randomized clinical trial; RR, relative risk; SNP, single nucleotide polymorphism; sTNFR2, soluble tumour necrosis factor receptor 2; wt, wild-type.
All estimates are based on multivariable adjusted models.
Ten cohorts: CCFR, Colon Cancer Family Registry; DACHS, Darmkrebs: Chancen der Verhütung durch Screening Study; DALS, Diet, Activity and Lifestyle Study; HPFS, Health Professionals Follow-up Study; NHS, Nurses’ Health Study; OFCCR, Ontario Familial Colorectal Cancer Registry; PMH-CCFR, Postmenopausal Hormone Study–CCFR; PLCO, Prostate, Lung, Colorectal and Ovarian Cancer Screening Trial; VITAL, Vitamins and Lifestyle; WHI, Women’s Health Initiative.
Case-only interaction analysis;
RR represents 81 mg aspirin versus placebo. No significant effect was measured for 325 mg aspirin.
Prostaglandin synthesis and catabolism.
The capacity of aspirin to irreversibly bind and acetylate, and therefore inhibit, prostaglandin-endoperoxide synthase 1 (PTGS1; also known as COX1) and PTGS2 (also known as COX2) has been widely considered to be central to its chemopreventive mechanisms58–66 (FIG. 2). These enzymes perpetuate pro-inflammatory signals, which promote cellular proliferation, angiogenesis and apoptotic resistance. PTGS enzymes are responsible for the conversion of arachidonic acid into downstream effectors that are further metabolized into prostaglandins (PGs) and related eicosanoids (such as PGE2, PGD2, PGF2, thromboxane A2 (TXA2) and prostacyclin; collectively known as prostanoids). PGs can then bind to a number of downstream targets, including plasma membrane and nuclear receptors.
Figure 2 |. The hypothesized inter-related mechanisms of aspirin chemoprevention.

Aspirin exerts its anticancer effects through several interconnected mechanisms: prostaglandin (PG) synthesis and catabolism in epithelial cells (lower left panel); inhibition of WNT–β-catenin signalling (lower right panel); and inactivation of platelets (upper right panel) and the host immune response (upper left panel). Through direct inhibition of prostaglandin-endoperoxide synthase 2 (PTGS2) at higher doses, aspirin blocks conversion of arachidonic acid to PGE2. Aspirin’s effects may be enhanced by the expression of 15-hydroxyprostaglandin dehydrogenase (HPGD), a metabolic antagonist of PTGS2, through catabolism of PGE2 to PGE-M (the major metabolite of PGE2). PGE2 can activate WNT–β-catenin signalling through paracrine activation of EP2, its plasma membrane receptor. PGE2 can also activate cAMP and protein kinase A (PKA) signalling, further stabilizing cytosolic β-catenin. Aspirin can further inhibit β-catenin through inactivation of protein phosphatase 2A (PP2A), the phosphatase responsible for removing post-translational modifications that target β-catenin for ubiquitylation and subsequent destruction. In colorectal tumorigenesis, PTGS2 and β-catenin are upregulated, leading to increased cellular proliferation, growth and survival. Once in the nucleus, β-catenin forms a transcriptional activation complex with transcription factor 7 like-2 (TCF7L2) and activates effector genes with roles in tumorigenesis, such as MYC and PPARD (which encodes peroxisome proliferator-activated receptor-δ). Genetic variation at the single nucleotide polymorphism (SNP) rs6983267 may impair binding of the β-catenin–TCF7L2 complex to these transcriptional targets. The antiplatelet effects associated with low-dose aspirin are mediated through inhibition of PTGS1. PTGS1 converts arachidonic acid to thromboxane A2 (TXA2), the major metabolite promoting recruitment and activation of platelets. Platelets and inflammatory cells — especially infiltrating neutrophils and fibroblasts — recruited to the colonic epithelium in response to chronic inflammation or mucosal injury, may act as paracrine activators of PTGS2 in the colonic epithelium. The combined antiplatelet and anti-inflammatory effects of aspirin may specifically prevent inflammation-associated tumorigenesis, and aspirin may be particularly effective in individuals with increased circulating levels of inflammatory cytokines. Targets that are discussed as potential biomarkers that may have utility in risk stratification for precision chemoprevention are shown in red. CXCL1, C-X-C motif chemokine 1; IFNγ, interferon-γ; IL, interleukin; MIC1, macrophage inhibitory cytokine 1; PGT, prostaglandin transporter; sTNFR2, soluble TNF receptor 2; TNF, tumour necrosis factor.
Increased synthesis of PGE2, which is directly attributable to PTGS2 activity, has been consistently observed in patients with colorectal neoplasia and has been shown to promote colorectal carcinogenesis67,68. Genetic deletion of Ptgs2 or any of several PGE2 receptor genes results in decreased intestinal tumour incidence and burden in mouse models of CRC69–73. The relevance of this chemopreventive mechanism in humans is further supported by molecular epidemiology studies. Within two large cohorts, regular aspirin use conferred a significant reduction in the risk of cancers that overexpress PTGS2 (RR = 0.64; 95% CI 0.52–0.78) as measured by immunohistochemical staining of the tumour66. However, aspirin use was not associated with the risk of cancers with low or negative PTGS2 expression (RR = 0.96; 95% CI 0.73–1.26; Pheterogeneity = 0.02). More recently, these findings were extended by examining the effect of aspirin on other molecular subtypes, including CRCs with mutations in BRAF, a proto-oncogene in the MAPK signalling cascade74. Aspirin use was associated with lower risk of BRAF-wild-type CRC (HR = 0.73; 95% CI 0.64–0.83) but not BRAF-mutant CRC (HR = 1.03; 95% CI 0.76–1.38); Pheterogeneity = 0.005). In particular, the benefit of aspirin seemed to be most evident among cancers that overexpress PTGS2 and were BRAF wild-type. Because BRAF mutation is strongly associated with cancers that seem to emerge through epigenetic dysregulation and thus the serrated carcinogenesis pathway75–77, these results suggest that those cancers with upregulated MAPK signalling, and perhaps serrated cancers, may be less dependent on PTGS2 or PG-mediated pathways for their growth.
15-hydroxyprostaglandin dehydrogenase (HPGD; also known as 15-PGDH), the primary enzyme that catabolizes PGs, functions as a metabolic antagonist, or ‘brake’, for PTGS2. In vivo experiments have demonstrated that Hpgd-null mice are more susceptible to colon tumorigenesis78 and are insensitive to NSAID-based chemoprevention79. The Adenoma Prevention with Celecoxib RCT provided human evidence that supported the role of HPGD as a crucial mediator of NSAID-based chemoprevention79. Celecoxib, a selective PTGS2 inhibitor, prevented adenoma recurrence only in patients with above-average expression of HPGD transcripts in normal colonic mucosa, suggesting a cooperative role for HPGD and PTGS2 inhibition in reducing the risk of neoplasia. These provocative findings were extended to aspirin in a study of HPGD expression in the adjacent normal mucosa of CRC tumours65. Aspirin use was associated with a reduced risk of CRC in those individuals with high expression of HPGD in normal colon tissue (HR = 0.49; 95% CI 0.34–0.71). Conversely, in individuals with low colonic expression of HPGD there was no protection associated with aspirin use (Pheterogeneity = 0.02). These results highlight the potential importance of HPGD in conferring sensitivity to aspirin and suggest that normal tissue markers such as HPGD mRNA may serve as predictive biomarkers for chemoprevention. If validated in other populations, a clinical strategy could include concurrent biopsies of normal mucosa among individuals who undergo resection of an adenoma during endoscopy. Assessment of the level of expression of HPGD in these biopsies could then be used to predict the likelihood of a response to aspirin administered for prevention of recurrent neoplasia.
Although germline mutations in HPGD have not been consistently associated with carcinogenesis in humans, a recent study demonstrated that alterations in SLCO2A1, the gene that encodes the PG transporter responsible for bringing substrate to HPGD, was associated with marked early-onset colonic neoplasia80. Carriers of this mutation mimic the Hpgd-null mouse phenotype, demonstrating elevated systemic PGE2 (as measured by higher levels of the major urinary metabolite of PGE2, known as PGE-M (11α-hydroxy-9,15-dioxo-2,3,4,5-tetranor-prostane-1,20-dioicacid)81) and resistance to NSAID-based stabilization of PG synthesis80. These results further support the role of disordered PG catabolism in carcinogenesis and suggest that SLCO2A1 may be a potential genetic biomarker for chemoprevention.
Additional evidence of the relevance of PG balance in colorectal neoplasia has been obtained through the application of methods to quantify PGE-M. Urinary PGE-M is widely accepted as the most accurate quantification of in vivo systemic PGE2 levels82. Multiple studies have demonstrated an increased risk of CRC and adenoma in individuals with higher pre-diagnostic urinary PGE-M levels83–85. In addition to supporting the importance of PGs in carcinogenesis, urinary PGE-M levels may also have potential for molecular risk stratification. In the Nurses’ Health Study (NHS), regular use of aspirin, and/or other NSAIDs, was associated with a 39% reduction in adenoma risk among women with high baseline PGE-M levels but not among those with low PGE-M levels86. However, these results were not confirmed by a similar study performed within the AFPPS RCT, which concluded that aspirin chemoprevention of adenoma recurrence was independent of prostanoid levels87. Thus, further studies are needed to determine the potential use of urinary PGE-M as a biomarker for chemoprevention.
Additional PG pathway-based approaches for precision chemoprevention have been discussed in previous reviews48,88. For example, several gene-by-environment (GxE) interaction analyses have been pursued that focus on genetic variants associated with PG or aspirin function54,89–93; however, few of these studies have demonstrated a significant interaction between aspirin use and genetic variants in the context of CRC or adenoma risk. One cohort study did observe that the GA genotype of the single nucleotide polymorphism (SNP) rs2920421, which lies in an intronic region of ALOX12 (which encodes arachidonate 12-lipoxygenase), was associated with a lower risk of CRC only among current NSAID users and not among never users or former users94. This finding requires corroboration in other populations as candidate-gene-based approaches to GxE interaction studies have a high potential for false-positive results.
GxE interaction analyses in the context of genome-wide association studies (GWASs) have provided an important and complementary approach to candidate gene studies, facilitating the discovery of novel genome-wide significant interactions between germline variants and regular aspirin use with risk of CRC. A recent study of 8,634 CRC cases and 8,553 controls from 10 studies identified that the SNP rs2965667 (minor allele frequency (MAF) = 1.7%) seems to predict a differential response to aspirin use with putative, albeit somewhat indirect, relevance to PG synthesis12. For rs2965667, the use of aspirin and NSAIDs was associated with a lower risk of CRC among individuals with the TT genotype (OR = 0.66; 95% CI 0.61–0.70; P = 7.7 × 10−33). By contrast, aspirin use among those with either of the rare TA and AA genotypes (present in only 4% of individuals studied) was associated with an increased risk of CRC (OR = 1.89; 95% CI 1.27–2.81; P = 0.002). This SNP lies proximal to several candidate genes that encode proteins associated with the putative mechanisms of action of aspirin, which may be relevant to the association of aspirin with CRC survival95,96. These genes include: microsomal glutathione S-transferase 1 (encoded by MGST1), which is a member of the membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG) family, is upregulated in several cancers, including CRC, and has high sequence homology with PGE synthase 1 (encoded by PTGES; also known as MGST1L1)97–99; LIM domain only 3 (LMO3), which is a known oncogene for other non-colorectal cancers100,101; and PI3K catalytic subunit 2γ (encoded by PIK3C2G), which is part of the PI3K family. Although these findings are promising, it is important to note that a limitation of the discovery-based GxE interaction approach using GWAS data is the stringent criteria required to achieve statistical significance at a genome-wide level and the possibility that genetic variation in key candidate pathways (such as the PG synthesis pathway) may not be well represented on current GWAS platforms. Thus, a genome-wide GxE interaction approach is associated with a greater likelihood of false-negative results.
Combined, these studies demonstrate multiple associations implicating modulation of PG synthesis as a major mode of action for aspirin chemoprevention and the potential of tissue, urinary and genomic biomarkers associated with PG synthesis for personalized risk stratification.
WNT–β-catenin signalling.
Given the crucial importance of WNT dysregulation in the development of the majority of CRCs21,23, the possible effects of aspirin on this signalling pathway have become a focus of mechanistic investigation. Briefly, this pathway is activated through extracellular WNT ligand binding to surface receptors, which results in cytosolic stabilization of β-catenin and, ultimately, translocation of β-catenin to the nucleus102 (FIG. 2). In the nucleus, β-catenin binds to transcription factor 7 like-2 (encoded by TCF7L2) to form a transcriptional activation complex that leads to the expression of genes affecting cell proliferation and migration. In the absence of ligand, β-catenin becomes ubiquitylated and targeted for degradation through the formation of the destruction complex, which contains axin, adenomatous polyposis coli (APC), β-catenin and glycogen synthase kinase 3β (GSK3β). Aberrant activation of WNT signalling through APC loss-of-function results in cytosolic and nuclear accumulation of β-catenin and is observed in the majority of CRCs21. Aspirin treatment of human CRC cell lines reduces the cytoplasmic pool of β-catenin through inactivation of protein phosphatase 2A (PP2A), which is responsible for dephosphorylating the β-catenin amino acid residues (T41 and S45) that target β-catenin for ubiquitylation103.
Mechanistic investigation into the potential for interaction of PGE2 and WNT signalling has further highlighted the relevance of WNT modulation to aspirin chemoprevention, which has been previously reviewed in detail104. In brief, PGE2 seems to enhance WNT signalling via multiple downstream effectors. Early evidence from in vitro cancer cell models demonstrated that PGE2 stimulates tumour cell growth by binding to its plasma membrane receptor, EP2, thereby activating WNT–β-catenin signalling, in addition to PI3K and AKT signalling pathways105. EP2 binds to axin, displacing it from its association with GSK3β and thereby liberating β-catenin. The activation of PI3K and AKT contributes to β-catenin stabilization through direct phosphorylation and inactivation of GSK3β. More recent evidence from developmental stem cell biology further supports the mechanistic role of PGE2 in β-catenin stabilization through activation of cAMP and protein kinase A (PKA) signalling, which leads to additional inactivation of the destruction complex106. Furthermore, PGE2 increases transcription of PPARD (which encodes peroxisome proliferator-activated receptor-δ (PPARδ)), which is a direct target gene of WNT signalling via the β-catenin–TCF7L2 transcriptional complex107. PPARδ is upregulated in some CRCs107,108 and has been associated with apoptotic resistance in CRC cell lines109. Moreover, genetic deletion of Ppard in ApcMin/+ mouse models of CRC tumorigenesis abrogates the pro-tumorigenic effects of PGE2 (REF. 110). Importantly, in ApcMin/+ mice with functional PPARδ, aspirin derivatives reduced both intestinal tumorigenesis and tumour expression of PPARδ111.
Beyond experimental models, mechanistically based candidate gene studies have provided proof of principle that aspirin may influence WNT signalling in humans. GWASs of CRC susceptibility have consistently identified a SNP, rs6983267, on chromosome 8q24 as an important risk locus112,113: the T allele of rs6983267 was associated with a ~17% reduction in CRC risk. This SNP seems to have a role in WNT signalling by modulating the binding of TCF7L2, which leads to lower expression of the MYC proto-oncogene114. As experimental studies have demonstrated an effect of aspirin on WNT signalling, it follows that genetic variation at this locus may differentially influence aspirin function. Among a nested case–control study, the association of regular aspirin use with CRC differed significantly according to the genotype of this SNP115, with the benefit of aspirin predominantly evident among individuals with at least one T allele (Pinteraction = 0.01). Compared with non-use, aspirin use was associated with ORs of 0.61 (95% CI 0.47–0.79) among those with the GT genotype and 0.52 (95% CI 0.35–0.78) among those with the TT genotype. By contrast, regular aspirin use was not associated with lower risk among individuals with the GG genotype (OR = 0.99; 95% CI 0.70–1.40). Importantly, the T allele was also associated with decreased MYC expression in CRC tissue (Ptrend = 0.03), and among those individuals with at least one protective T allele, aspirin use was specifically associated with lower risk of CRCs positive for nuclear accumulation of β-catenin (OR = 0.44; 95% CI 0.26–0.75; Pinteraction = 0.04). Opportunely, the protective T allele is common (MAF = 0.49 in Europeans; 0.85 in African Americans; 0.61 in Latin Americans)116 and thus most of the population may benefit from aspirin use. Taken together, these data support a mechanistic connection between aspirin chemoprevention and the WNT–β-catenin signalling axis, the functional relevance of 8q24 genetic variation in carcinogenesis and the potential utility of rs6983267 as a mechanistic biomarker.
Inflammatory and immune responses.
The anti-inflammatory properties of aspirin and the establishment of chronic inflammation as a risk factor for CRC have elicited possible mechanistic connections between aspirin chemoprevention, chronic inflammation and modulation of the host immune response63. PTGS2 can be induced at sites of inflammation in response to cytokines (such as interleukin-1α (IL-1α), IL-1β, interferon-γ and tumour necrosis factor (TNF)) produced by inflammatory cells (FIG. 2). Additionally, PGE2 seems to mediate inflammation-associated tumorigenesis in ApcMin/+ mouse models of CRC117. During inflammation-associated tumorigenesis, infiltrating neutrophils and tumour-associated fibroblasts were found to express EP2, TNF, IL-6, C-X-C motif chemokine 1 (CXCL1), PTGS2 and WNT5A118.
Multiple studies have examined the association of CRC risk and circulating inflammatory or immune markers, including IL-6, C-reactive protein (CRP) and soluble TNF receptor 2 (sTNFR2), in the context of aspirin use in human populations. A nested case–control study of women found that plasma sTNFR2 levels, but not IL-6 or CRP levels, were associated with risk of CRC (RR = 1.67; 95% CI 1.05–2.68)119. Among those with high baseline levels of sTNFR2, the use of aspirin or other NSAIDs was associated with lower risk of CRC (RR = 0.39; 95% CI 0.18–0.86). By contrast, women with low baseline levels of sTNFR2 did not experience a preventive benefit associated with initiation of regular use of aspirin or NSAIDs (RR = 0.86; 95% CI 0.41–1.79). These results, however, were not observed in a separate study of men120. It remains unclear whether a true sex difference exists or whether this inconsistency stems from methodological differences between the studies. Furthermore, as shown in recent meta-analyses, the association of IL-6 and CRP levels with CRC has been inconsistent and no studies have reported a significant differential association of aspirin use with CRC according to levels of these markers121,122.
The circulating inflammatory cytokine macrophage inhibitory cytokine 1 (MIC1; also known as growth differentiation factor 15 (GDF15)) may be an important mediator in systemic inflammatory responses and, as a member of the human transforming growth factor-β (TGFβ) superfamily, may have a specific role in colorectal carcinogenesis123,124. A recent cohort study examined plasma levels of MIC1 in the context of CRC risk125. In a comparison of extreme quintiles, higher MIC1 levels were associated with a 93% increased risk of CRC (HR = 1.93; 95% CI 1.27–2.94; Ptrend = 0.004). Furthermore, in exploratory analyses, among individuals with high plasma MIC1 levels, the use of aspirin and NSAIDs was associated with a lower risk of PTGS2-positive (RR = 0.60; 95% CI 0.41–0.88) but not PTGS2-negative CRC (RR = 1.21; 95% CI 0.71–2.07).
GWASs have further implicated a potential role for aspirin in the modulation of immunity and inflammation. The SNP rs16973225 was identified in a genome-wide case-only GxE interaction analysis12. This SNP is located at chromosome 15q25.2, approximately 625 kb upstream of the gene encoding IL-16, which is a multifunctional cytokine with crucial functions in pro-inflammatory processes that has been implicated in inflammatory bowel disease and CRC. This SNP showed a genome-wide significant interaction with use of aspirin and/or NSAIDs (Pinteraction = 8.2 × 10−9). Regular use was associated with a lower risk of CRC among individuals with the AA genotype (OR = 0.66; 95% CI 0.62–0.71; P = 1.9 × 10−30). However, the reduced risk associated with regular aspirin use was nullified among those with the AC or CC genotype (OR = 0.97; 95% CI 0.78–1.20). Taken together, these data suggest that the anticancer effect of aspirin may be mediated at least partially through modulation of chronic inflammation and the immune system.
Platelet-mediated effects.
Unlike epithelial cells, which express both PTGS1 and PTGS2, platelets express only PTGS1 and, as non-nucleated cells, lack the ability to resynthesize the enzyme11. Thus, low-dose aspirin leads to permanent inhibition of platelet-mediated production of TXA2 (FIG. 2). This action has been hypothesized to explain the vascular benefits of aspirin, as TXA2 is the major metabolite that promotes activation and aggregation of platelets, vasoconstriction and vascular smooth muscle cell proliferation126. Recently, the anticancer effects of aspirin have been proposed to be mediated by platelets rather than through a direct effect on epithelial cells11. Such a hypothesis would explain why aspirin, at least in secondary analyses of RCTs of aspirin for CVD prevention, seems to be effective in preventing cancer even at low doses, which would be rapidly metabolized and would generate systemic concentrations not expected to be high enough to directly inhibit epithelial PTGS2 and subsequent production of systemic PGE2. To reconcile this with the highly consistent evidence implicating the importance of PTGS2 in colorectal carcinogenesis, it has been hypothesized that the recruitment of activated platelets to the mucosa in response to inflammatory events or mucosal injury leads to paracrine upregulation of PTGS2 expression in epithelial cells11,127. Thus, even low doses of aspirin, typically considered only sufficient to inhibit platelets, may eventually lead to substantial downstream inhibition of PTGS2 in epithelial and tumour cells.
Additional mechanisms: insights from survival studies.
Beyond its effect on cancer initiation, aspirin may also have an effect on cancer progression. Several studies have shown that aspirin use, particularly after diagnosis, is associated with a lower risk of CRC-specific mortality among patients with established disease10,14,128–133. These associations may also be explained by the effect of aspirin on pathways such as PG synthesis or WNT signalling. For example, aspirin was associated with improved survival, particularly among individuals with index primary tumours that overexpressed PTGS2 by immunohistochemistry compared with those with tumours without PTGS2 overexpression128. However, there may be additional distinct pathways by which aspirin influences cancer progression or metastasis but not initiation134,135. For example, aspirin use does not seem to be differentially associated with the likelihood of developing cancer with activating mutations in PIK3CA (which encodes PI3K catalytic subunit-α)74, the product of which functions in a pathway that seems to share functionally important crosstalk with PTGS2 (REF. 136). However, after diagnosis, aspirin use was associated with dramatically improved survival among individuals with PIK3CA-mutant CRC in a cohort study (HR = 0.18; 95% CI 0.06–0.61)96 and in an analysis of self-selected aspirin use within a clinical trial of adjuvant rofecoxib (HR = 0.11; 95% CI 0.001–0.83)137, supporting the possibility that aspirin may influence distinct biological pathways in the setting of established cancer. Two additional observational studies did not individually support these findings138,139. However, a recent meta-analysis140, which included one of these null studies138, concluded that the association of aspirin with CRC-specific survival does seem to differ according to PIK3CA mutation status. Additional RCTs (ASCOLT34 and ADD-ASPIRIN141) designed to assess the effect of aspirin in patients who have undergone potentially curative surgery for CRC are currently underway and will provide additional opportunities to determine whether PIK3CA or other molecular biomarkers of cancer-specific pathways may be leveraged to predict responsiveness to therapy.
Additional markers for risk stratification.
The results of three RCTs of aspirin in relation to CRC biomarkers have elucidated other potential markers for molecular risk stratification that are associated with alternative pathways. First, a Phase IIb double-blind, placebo-controlled trial among patients with a history of adenoma or CRC examined the effect of aspirin at 325 mg per day for 3 months on spectral biomarkers in macroscopically normal tissue that measure cellular microarchitectural changes associated with the development of neoplasia53,142,143 (TABLE 4). Following aspirin treatment, both spectral slope (SPEC) and fractal dimension (FRAC) trended towards less neoplastic signatures in normal tissue, although only SPEC approached statistical significance53. In secondary analyses, tissue expression of PGE2 correlated with SPEC and FRAC in aspirin users, but there was no differential association according to UGT1A6 (which encodes UDP glucuronosyltransferase 1 family polypeptide A6) genotype.
Table 4 |.
Placebo-controlled clinical trials of aspirin with biomarker end points
| Trial name | Primary end point | Inclusion criteria | Aspirin dose (mg per day) | Participants (n) | Duration of treatment | Primary end point | Secondary end points |
|---|---|---|---|---|---|---|---|
| ABC | Tissue gene expression | Healthy men and women aged 20–45 years who completed a cross-sectional study of diet and aspirin metabolism and were selected on the basis of UGT1A6 genotype | 325 | 44 | 60 days | Null; no significant differences in gene expression were observed by aspirin use or UGT1A6 genotype54 | PGE2 tissue expression was lower in individuals receiving aspirin than in those receiving placebo (P < 0.001)54. Plasma concentrations of 2-hydroxyglutarate was reduced after aspirin use (P = 0.005)144 |
| Spectral Markers in Aspirin Chemoprevention of Colonic Neoplasia53 | SPEC and FRAC | Subjects aged ≤70 years undergoing surveillance colonoscopy for adenoma or CRC resected in the past 6 years | 325 | 72 | 3 months | SPEC = +48.9% from baseline (less neoplastic),
P = 0.055; FRAC = −48.9% from baseline (less neoplastic). P = 0.200 |
PGE2 suppression correlated with SPEC (R = −0.55; P = 0.01), but not FRAC; no significant differences in SPEC or FRAC according to UGT1A6 genotype |
ABC, Aspirin and the Biology of the Colon; CRC, colorectal cancer; FRAC, fractal dimension; PGE2, prostaglandin E2; R, Spearmann’s correlation coefficient; SPEC, spectral slope; UGT, UDP-glucuronosyltransferase.
Second, the Aspirin and the Biology of the Colon (ABC) placebo-controlled crossover trial examined the impact of aspirin treatment for 60 days on colonic epithelial and stromal gene expression among healthy participants predicted to have impaired aspirin glucuronidation on the basis of UGT1A6 genotype54 (TABLE 4). Overall, aspirin did not induce differences in gene expression but did decrease tissue expression of PGE2. A recent secondary metabolomic analysis of plasma collected from these participants identified a potentially interesting association between aspirin use and a reduction in the levels of the oncometabolite 2-hydroxyglutarate144.
Last, aspirin has been hypothesized to act synergistically with ornithine decarboxylase (ODC) inhibitors145. ODC is the enzyme responsible for the synthesis of colonic polyamines, which have been consistently associated with the development of colorectal neoplasia146,147. An RCT to determine the effect of combined treatment with the ODC inhibitor eflornithine and aspirin on adenoma recurrence is currently underway148 and may build on the success of the earlier eflornithine–sulindac combination trial in preventing adenoma recurrence149. Previously, the A allele of SNP rs2302615 in the ODC promoter has been associated with adenoma risk150–152, but its utility in predicting aspirin sensitivity has been inconsistent110,153,154. Most recently, among a sub-cohort of the AFPPS RCT, a targeted genotyping approach that examined SNPs in the ODC promoter identified two novel SNPs, the GG genotype of rs2430420 and the TT genotype of rs28362380, that seem to be potential markers of efficacy with 81 mg per day of aspirin154 (TABLE 3). However, this genetic interaction was not observed with the group receiving a dose of 325 mg. Clearly, studies in additional large populations are warranted to validate the use of ODC SNPs, SPEC and FRAC, and 2-hydroxygluturate as potential risk stratification biomarkers.
Conclusions
The evidence in support of the effect of aspirin on CRC is compelling, forming the basis for a recent clinical recommendation for its use in chemoprevention. We, and others, have shown that the effect of aspirin on CRC is not likely to be mediated by a single mechanism, but instead by several inter-related mechanisms (FIG. 2). Specifically, aspirin may inhibit PTGS-mediated conversion of arachidonic acid to PGE2 either by a direct effect on epithelial cells or through a paracrine effect via inhibition of PTGS1-mediated synthesis of TXA2 in platelets. Inhibition of PGE2 synthesis by aspirin may be enhanced by background upregulation of HPGD. Aspirin may also inhibit WNT signalling either directly or through downregulation of PGE2.
Several genetic, tissue, plasma and urinary biomarkers relevant to these multiple, yet inter-related, pathways seem to have promise as biomarkers for risk stratification to identify those individuals who are most likely to benefit from a long-term aspirin regimen. Nonetheless, despite their potential, several pragmatic test characteristics — including specificity, sensitivity and convenience — are crucial considerations for their adoption in clinical settings. For example, urine- and blood-based biomarkers may be relatively easy to use in the clinic. However, the markers identified to date (such as MIC1 or PGE-M) may lack specificity for neoplasia as they may be generally elevated in response to other inflammatory states. Tissue biomarkers (such as HPGD) may offer greater specificity than urine- or blood-based biochemical markers, but they require invasive biopsy and may be more challenging to obtain in primary prevention settings. Finally, the sensitivity and specificity of germline genetic markers are likely to depend not only on the strength of interaction with aspirin use but also on the allele frequency of a polymorphism in a given population. Moreover, genetic testing may be less easily implemented in clinical settings, given the additional need for interpretation of genetic information and patient counselling155. Thus, further studies are required to prioritize the biomarkers identified to date for implementation in the clinic and to identify additional novel pathways and biomarkers.
To help achieve this goal, we recently launched the ASPIRED trial36. Participants who have recently had an adenoma removed will be enrolled in a double-blind, placebo-controlled RCT of 81 mg or 325 mg of aspirin daily for 8 weeks. Individuals will provide blood, urine, colorectal biopsy, saliva and stool specimens both before and after treatment. These biological specimens will then be used to examine the effect of aspirin on cancer-related biomarkers, including many of those discussed in this Opinion article. Additional ongoing RCTs, including the seAFOod polyp prevention trial38, will also examine the effect of aspirin and fish oil on bioactive lipid mediators, including urinary levels of PGE-M156. Finally, RCTs involving patients at risk for cardiovascular events (ARRIVE33) or other primary outcomes (ASPREE35) may also provide additional evidence to support the efficacy of aspirin in cancer prevention.
In conclusion, the recent USPSTF draft recommendation that recognizes the weight of evidence in support of aspirin for CRC prevention is a crucial first step in realizing a potential broader population-wide impact of aspirin use for chemoprevention. However, it is likely that the future of aspirin chemoprevention will still benefit from molecularly inspired human research aimed at clarification of aspirin’s inter-related anticancer mechanisms. Such studies will be crucial for fulfilling the promise of precision medicine for cancer prevention.
Glossary
- Case–control studies
A type of observational study design in which two groups differ according to outcome (for example, in which those with the disease or condition (cases) are compared with disease-free individuals (controls)). These studies can be nested within a larger cohort with only a subset of the larger control population being used
- Confidence interval
(CI). A measure of the uncertainty associated with a sample estimate for a given parameter
- Events per 1,000 person-years
Also known as the incidence density rate or person–time incidence rate. This approach normalizes event observations to the amount of observation time and is useful if observation times are not constant across a sample population or the risk of an event varies over time
- Fractal dimension
(FRAC). A spectroscopic measure of light scattering due to nanoscale architectural changes in mass density of cellular components that has been associated with early neoplastic changes in epithelial cells
- Glucuronidation
The addition of glucuronic acid to a substrate
- H2 blocker
A drug that inhibits the production of gastric acid by targeting histamine H2 receptors of gastric parietal cells
- Hazard ratio
(HR). A measure of the ratio of the hazard rates (the rate at which an event occurs) for a given outcome (for example, cancer) described by an explanatory variable (for example, aspirin versus placebo)
- Incidence rate ratio
(IRR). A measure of the ratio between the rates of how often an outcome (for example, cancer) occurs in a population at any given time according to an explanatory variable (for example, aspirin versus placebo)
- Odds ratio
(OR). A measure of association representing the odds (the probability of disease divided by 1 minus the probability) of an outcome according to an explanatory variable (for example, aspirin users versus non-users)
- Pheterogeneity
A statistic that measures the significance of the difference (or heterogeneity) between two effect sizes
- Pinteraction
A statistic that measures the significance of the effect of a given exposure or explanatory variable on a second exposure or explanatory variable, and vice versa
- Proton pump inhibitor
A drug that inhibits the production of gastric acid by targeting the proton pump transport activity of gastric parietal cells. Proton pump inhibitors are generally more effective than H2 blockers
- Ptrend
A statistic that measures the significance of a correlation of effect size, either positive or negative, across a continuous or ordinal variable
- Relative risk
(RR). The probability of an outcome (for example, cancer) occurring in one group (for example, aspirin) versus the probability in a comparison group (for example, placebo)
- Serrated carcinogenesis pathway
A carcinogenesis pathway in which colorectal tumours are characterized by epigenetic dysregulation (promoter hypermethylation), BRAF mutation and activation, and often microsatellite instability, rather than adenomatous polyposis coli (APC) mutations and chromosomal instability
- Single nucleotide polymorphism
(SNP). A common type of genetic variation in which a single nucleotide or base occurs at a specific position in the genome that is different from the expected or reference nucleotide
- Spectral slope
(SPEC). A spectroscopic measure of light scattering due to nanoscale architectural changes in the size distribution of cellular components that has been associated with early neoplastic changes in epithelial cells
Contributor Information
David A. Drew, Massachusetts General Hospital and Harvard Medical School, Clinical and Translational Epidemiology Unit, 55 Fruit Street, Bartlett Ext. 9, Boston, Massachusetts 02114, USA.
Yin Cao, Massachusetts General Hospital and Harvard Medical School, Clinical and Translational Epidemiology Unit, and Harvard T.H. Chan School of Public Health, Department of Nutrition, 55 Fruit Street, Bartlett Ext. 9, Boston, Massachusetts 02114, USA..
Andrew T. Chan, Massachusetts General Hospital and Harvard Medical School, Clinical and Translational Epidemiology Unit, Division of Gastroenterology, GRJ-825C, Boston, Massachusetts 02114, USA.
References
- 1.Siegel RL, Miller KD & Jemal A Cancer statistics, 2015. CA Cancer J. Clin 65, 5–29 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Chan AT et al. Aspirin in the chemoprevention of colorectal neoplasia: an overview. Cancer Prev. Res. (Phila.) 5, 164–178 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thorat MA & Cuzick J Prophylactic use of aspirin: systematic review of harms and approaches to mitigation in the general population. Eur. J. Epidemiol 30, 5–18 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Preventive US Services Task Force. Routine aspirin or nonsteroidal anti-inflammatory drugs for the primary prevention of colorectal cancer: U. S. Preventive Services Task Force recommendation statement. Ann. Intern. Med 146, 361–364 (2007). [PubMed] [Google Scholar]
- 5.U.S. Preventive Services Task Force. Draft Recommendation Statement: Aspirin to prevent cardiovascular disease and cancer U.S. Preventive Services Task Force [online] http://www.uspreventiveservicestaskforce.org/Page/Document/draft-recommendation-statement/aspirin-to-prevent-cardiovascular-disease-and-cancer (2015).
- 6.Chubak J, Kamineni A, Buist DSM,Anderson ML & Whitlock EP Aspirin Use for the Prevention of Colorectal Cancer: An Updated Systematic Evidence Review for the U.S. Preventive Services Task Force (Agency for Healthcare Research and Quality (US), 2015). [PubMed] [Google Scholar]
- 7.Dehmer SP, Maciosek MV & Flottemesch TJ Aspirin Use to Prevent Cardiovascular Disease and Colorectal Cancer: A Decision Analysis: Technical Report (Agency for Healthcare Research and Quality (US; ), 2015). [PubMed] [Google Scholar]
- 8.Cuzick J et al. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 10, 501–507 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Flossmann E et al. Effect of aspirin on long-term risk of colorectal cancer: consistent evidence from randomised and observational studies. Lancet 369, 1603–1613 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Rothwell PM et al. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet 379, 1602–1612 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Thun MJ, Jacobs EJ & Patrono C The role of aspirin in cancer prevention. Nat. Rev. Clin. Oncol 9, 259–267 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Nan H et al. Association of aspirin and NSAID use with risk of colorectal cancer according to genetic variants. JAMA 313, 1133–1142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Friis S, Riis AH, Erichsen R, Baron JA& Sorensen HT Low-dose aspirin or nonsteroidal anti-inflammatory drug use and colorectal cancer risk: a population-based, case-control study. Ann. Intern. Med 163, 347–355 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Rothwell PM et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 376, 1741–1750 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Sturmer T et al. Aspirin use and colorectal cancer: post-trial follow-up data from the Physicians’ Health Study, Ann. Intern. Med 128, 713–720 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Cook NR et al. Low-dose aspirin in the primary prevention of cancer: the Women’s Health Study: a randomized controlled trial. JAMA 294, 47–55 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Burn J et al. Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome.N. Engl. J. Med 359, 2567–2578 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Cook NR, Lee IM, Zhang SM, Moorthy MV, & Buring JE Alternate-day, low-dose aspirin and cancer risk: long-term observational follow-up of a randomized trial. Ann. Intern. Med 159, 77–85 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burn J et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomized controlled trial. Lancet 378, 2081–2087 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cunningham JM et al. The frequency of hereditary defective mismatch repair in a prospective series of unselected colorectal carcinomas. Am. J. Hum. Genet 69, 780–790 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fearon ER Molecular genetics of colorectal cancer. Annu. Rev. Pathol 6, 479–507 (2011). [DOI] [PubMed] [Google Scholar]
- 22.Eide TJ Natural history of adenomas. World J. Surg 15, 3–6 (1991). [DOI] [PubMed] [Google Scholar]
- 23.Fearon ER & Vogelstein B A genetic model for colorectal tumorigenesis. Cell 61, 759–767 (1990). [DOI] [PubMed] [Google Scholar]
- 24.Morson BC The evolution of colorectal carcinoma.Clin. Radiol 35, 425–431 (1984). [DOI] [PubMed] [Google Scholar]
- 25.Neugut AI, Johnsen CM, Forde KA& Treat MR Recurrence rates for colorectal polyps. Cancer 55, 1586–1589 (1985). [DOI] [PubMed] [Google Scholar]
- 26.Baron JA et al. A randomized trial of aspirin to prevent colorectal adenomas. N. Engl. J. Med 348, 891–899 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Sandler RS et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N. Engl. J. Med 348, 883–890 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Logan RF et al. Aspirin and folic acid for the prevention of recurrent colorectal adenomas. Gastroenterology 134, 29–38 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Benamouzig R et al. Daily soluble aspirin and prevention of colorectal adenoma recurrence: one-year results of the APACC trial. Gastroenterology 125, 328–336 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Ishikawa H et al. The preventive effects of low-dose enteric-coated aspirin tablets on the development of colorectal tumours in Asian patients a randomised trial. Gut 63, 1755–1759 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Drew DA et al. Colorectal polyp prevention by daily aspirin use is abrogated among active smokers. Cancer Causes Control 27, 93–103 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Pommergaard HC, Burcharth J, Rosenberg J & Raskov, H. Aspirin, calcitriol, and calcium do not prevent adenoma recurrence in a randomized controlled trial. Gastroenterology 150, 114–122 (2015). [DOI] [PubMed] [Google Scholar]
- 33.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT00501059 (2015).
- 34.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT00565708 (2015).
- 35.ISRCTN Registry. Aspirin in reducing events in the elderly. ISRCTN.org 10.1186/ISRCTN83772183 (2015). [DOI]
- 36.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02394769 (2015).
- 37.ISRCTN Registry. Finding the best dose of aspirin to prevent Lynch Syndrome cancers. ISRCTN.org 10.1186/ISRCTN16261285 (2015). [DOI]
- 38.ISRCTN Registry. The seAFOod (Systematic Evaluation of Aspirin and Fish Oil) polyp prevention trial. ISRCTN.org 10.1186/ISRCTN05926847 (2015). [DOI]
- 39.Whitlock EP, Williams SB, Burda BU,Feightner A & Beil T Aspirin Use in Adults: Cancer, All-Cause Mortality, and Harms: A Systematic Evidence Review for the U. S. Preventive Services Task Force. (Agency for Healthcare Research and Quality (US), 2015). [PubMed] [Google Scholar]
- 40.Lanas A, Wu P, Medin J & Mills EJ Low doses of acetylsalicylic acid increase risk of gastrointestinal bleeding in a meta-analysis. Clin. Gastroenterol. Hepatol 9, 762–768 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Serebruany VL et al. Analysis of risk of bleeding complications after different doses of aspirin in 192,036 patients enrolled in 31 randomized controlled trials. Am. J. Cardiol 95, 1218–1222 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Derry S & Loke YK Risk of gastrointestinal haemorrhage with long term use of aspirin: meta-analysis. BMJ 321, 1183–1187 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McQuaid KR & Laine L Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials. Am. J. Med 119, 624–638 (2006). [DOI] [PubMed] [Google Scholar]
- 44.The Dutch TIA Trial Study Group. A comparison of two doses of aspirin (30 mg versus 283 mg a day) in patients after a transient ischemic attack or minor ischemic stroke. N. Engl. J. Med 325, 1261–1266 (1991). [DOI] [PubMed] [Google Scholar]
- 45.Farrell B, Godwin J, Richards S & Warlow C The United Kingdom transient ischaemic attack (UK-TIA) aspirin trial: final results. J. Neurol. Neurosurg. Psychiatry 54, 1044–1054 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roderick PJ, Wilkes HC & Meade TW The gastrointestinal toxicity of aspirin: an overview of randomised controlled trials. Br. J. Clin. Pharmacol 35, 219–226 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weil J et al. Prophylactic aspirin and risk of peptic ulcer bleeding. BMJ 310, 827–830 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ulrich CM, Bigler J & Potter JD Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat. Rev. Cancer 6, 130–140 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Barry EL et al. CYP2C9 variants increase risk of colorectal adenoma recurrence and modify associations with smoking but not aspirin treatment. Cancer Causes Control 24, 47–54 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan AT, Hsu M, Zauber AG, Hawk ET & Bertagnolli MM The influence of UGT1A6 variants and aspirin use in a randomized trial of celecoxib for prevention of colorectal adenoma. Cancer Prev. Res. (Phila.) 5, 61–72 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chan AT et al. Cytochrome P450 2C9 variants influence response to celecoxib for prevention of colorectal adenoma. Gastroenterology 136, 2127–2136 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McGreavey LE et al. No evidence that polymorphisms in CYP2C8, CYP2C9, UGT1A6, PPARδ and PPARγ act as modifiers of the protective effect of regular NSAID use on the risk of colorectal carcinoma. Pharmacogenet. Genom 15, 713–721 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Roy HK et al. Spectral biomarkers for chemoprevention of colonic neoplasia: a placebo-controlled double-blinded trial with aspirin. Gut 10.1136/gutjnl-2015-309996 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomas SS et al. Tissue-specific patterns of gene expression in the epithelium and stroma of normal colon in healthy individuals in an aspirin intervention trial. BMC Med. Genet 16, 18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thompson CL et al. No association between cyclooxygenase-2 and uridine diphosphate glucuronosyltransferase 1A6 genetic polymorphisms and colon cancer risk. World J. Gastroenterol 15, 2240–2244 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Angstadt AY et al. The effect of UGT1A and UGT2B polymorphisms on colorectal cancer risk: haplotype associations and gene-environment interactions. Genes Chromosomes Cancer 53, 454–466(2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scherer D et al. Genetic variation in UGT genes modify the associations of NSAIDs with risk of colorectal cancer: colon cancer family registry. Genes Chromosomes Cancer 53, 568–578 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garcia-Albeniz X & Chan AT Aspirin for the prevention of colorectal cancer. Best Pract. Res. Clin. Gastroenterol 25, 461–472 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hull MA Cyclooxygenase-2: how good is it as a target for cancer chemoprevention? Eur. J. Cancer 41, 1854–1863 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Patrignani P & Patrono C Cyclooxygenase inhibitors: from pharmacology to clinical read-outs. Biochim. Biophys. Acta 1851, 422–432 (2015). [DOI] [PubMed] [Google Scholar]
- 61.Ranger GS Current concepts in colorectal cancer prevention with cyclooxygenase inhibitors. Anticancer Res. 34, 6277–6282 (2014). [PubMed] [Google Scholar]
- 62.Schror K Pharmacology and cellular/molecular mechanisms of action of aspirin and non-aspirin NSAIDs in colorectal cancer. Best Pract. Res. Clin. Gastroenterol 25, 473–484 (2011). [DOI] [PubMed] [Google Scholar]
- 63.Wang D & Dubois RN The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 29, 781–788 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dixon DA, Blanco FF, Bruno A & Patrignani P Mechanistic aspects of COX-2 expression in colorectal neoplasia. Recent Results Cancer Res. 191, 7–37 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fink SP et al. Aspirin and the risk of colorectal cancer in relation to the expression of 15-hydroxyprostaglandin dehydrogenase (HPGD). Sci. Transl Med 6, 233re2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chan AT, Ogino S & Fuchs CS Aspirin and the risk of colorectal cancer in relation to the expression of COX-2. N. Engl. J. Med 356, 2131–2142 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Elzagheid A et al. High cyclooxygenase-2 expression is associated with advanced stages in colorectal cancer. Anticancer Res. 33, 3137–3143 (2013). [PubMed] [Google Scholar]
- 68.Pugh S & Thomas GA Patients with adenomatous polyps and carcinomas have increased colonic mucosal prostaglandin E2. Gut 35, 675–678 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chulada PC et al. Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 60, 4705–4708 (2000). [PubMed] [Google Scholar]
- 70.Mutoh M et al. Involvement of prostaglandin E receptor subtype EP4 in colon carcinogenesis. Cancer Res. 62, 28–432 (2002). [PubMed] [Google Scholar]
- 71.Oshima M et al. Suppression of intestinal polyposis in ApcΔ716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 87, 803–809 (1996). [DOI] [PubMed] [Google Scholar]
- 72.Sonoshita M et al. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in ApcΔ716 knockout mice. Nat. Med 7, 1048–1051 (2001). [DOI] [PubMed] [Google Scholar]
- 73.Watanabe K et al. Role of the prostaglandin E receptor subtype EP1 in colon carcinogenesis. Cancer Res. 59, 5093–5096 (1999). [PubMed] [Google Scholar]
- 74.Nishihara R et al. Aspirin use and risk of colorectal cancer according to BRAF mutation status. JAMA 309, 2563–2571 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.I.Jspeert JE, Vermeulen L, Meijer GA& Dekker E Serrated neoplasia-role in colorectal carcinogenesis and clinical implications. Nat. Rev. Gastroenterol. Hepatol 12, 401–409 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Kedrin D & Gala MK Genetics of the serrated pathway to colorectal cancer. Clin. Transl Gastroenterol 6, e84(2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bettington M et al. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology 62, 367–386 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Myung SJ et al. 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc. Natl Acad. Sci. USA 103, 12098–12102 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yan M et al. 15-Hydroxyprostaglandin dehydrogenase inactivation as a mechanism of resistance to celecoxib chemoprevention of colon tumors. Proc. Natl Acad. Sci. USA 106, 9409–9413 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guda K et al. Inactivating mutation in the prostaglandin transporter gene, SLCO2A1, associated with familial digital clubbing, colon neoplasia, and NSAID resistance. Cancer Prev. Res. (Phila.) 7, 805–812 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Murphey LJ et al. Quantification of the major urinary metabolite of PGE2 by a liquid chromatographic/mass spectrometric assay: determination of cyclooxygenase-specific PGE2 synthesis in healthy humans and those with lung cancer. Anal. Biochem 334, 266–275 (2004). [DOI] [PubMed] [Google Scholar]
- 82.Wang D & DuBois RN Urinary PGE-M: a promising cancer biomarker. Cancer Prev. Res. (Phila.) 6, 507–510 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cai Q et al. Prospective study of urinary prostaglandin E2 metabolite and colorectal cancer risk. J. Clin. Oncol 24, 5010–5016 (2006). [DOI] [PubMed] [Google Scholar]
- 84.Shrubsole MJ et al. Urinary prostaglandin E2 metabolite and risk for colorectal adenoma. Cancer Prev. Res. (Phila.) 5, 336–342 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Johnson JC et al. Urine PGE-M: a metabolite of prostaglandin E2 as a potential biomarker of advanced colorectal neoplasia. Clin. Gastroenterol. Hepatol 4, 1358–1365 (2006). [DOI] [PubMed] [Google Scholar]
- 86.Bezawada N et al. Urinary PGE-M levels are associated with risk of colorectal adenomas and chemopreventive response to anti-inflammatory drugs. Cancer Prev. Res. (Phila.) 7, 758–765 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fedirko V et al. Urinary metabolites of prostanoids and risk of recurrent colorectal adenomas in the Aspirin/Folate Polyp Prevention Study (AFPPS). Cancer Prev. Res. (Phila.) 8, 1061–1068 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang D & Dubois RN Eicosanoids and cancer. Nat. Rev. Cancer 10, 181–193 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bigler J et al. Polymorphisms predicted to alter function in prostaglandin E2 synthase and prostaglandin E2 receptors. Pharmacogenet. Genom 17, 221–227 (2007). [DOI] [PubMed] [Google Scholar]
- 90.Liu W, Poole EM, Ulrich CM & Kulmacz RJ Decreased cyclooxygenase inhibition by aspirin in polymorphic variants of human prostaglandin H synthase-1. Pharmacogenet. Genom 22, 525–537 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hubner RA et al. Polymorphisms in PTGS1, PTGS2 and IL-10 do not influence colorectal adenoma recurrence in the context of a randomized aspirin intervention trial. Int. J. Cancer 121, 2001–2004 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Barry EL et al. Cyclooxygenase-2 polymorphisms, aspirin treatment, and risk for colorectal adenoma recurrence—data from a randomized clinical trial. Cancer Epidemiol. Biomarkers Prev. 18, 2726–2733 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Poole EM et al. Genetic variation in prostaglandin E2 synthesis and signaling, prostaglandin dehydrogenase, and the risk of colorectal adenoma. Cancer Epidemiol. Biomarkers Prev 19, 547–557 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Resler AJ et al. Genetic variation in prostaglandin synthesis and related pathways, NSAID use and colorectal cancer risk in the Colon Cancer Family Registry. Carcinogenesis 35, 2121–2126 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fresno Vara JA et al. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev 30, 193–204 (2004). [DOI] [PubMed] [Google Scholar]
- 96.Liao X et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N. Engl. J. Med 367, 1596–1606 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Prage EB et al. Location of inhibitor binding sites in the human inducible prostaglandin E synthase, MPGES1. Biochemistry 50, 7684–7693 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Morgenstern R, Zhang J & Johansson K Microsomal glutathione transferase 1: mechanism and functional roles. Drug Metab. Rev 43, 300–306(2011). [DOI] [PubMed] [Google Scholar]
- 99.Nakanishi M, Gokhale V, Meuillet EJ& Rosenberg DW mPGES-1 as a target for cancer suppression: a comprehensive invited review “Phospholipase A2 and lipid mediators”. Biochimie 92, 660–664 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kwon YJ et al. Genome-wide analysis of DNA methylation and the gene expression change in lung cancer. J. Thorac. Oncol 7, 20–33 (2012). [DOI] [PubMed] [Google Scholar]
- 101.Aoyama M et al. LMO3 interacts with neuronal transcription factor, HEN2, and acts as an oncogene in neuroblastoma. Cancer Res. 65, 4587–4597 (2005). [DOI] [PubMed] [Google Scholar]
- 102.MacDonald BT, Tamai K & He X Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bos CL et al. Effect of aspirin on the Wnt/β-catenin pathway is mediated via protein phosphatase 2A. Oncogene 25, 6447–6456 (2006). [DOI] [PubMed] [Google Scholar]
- 104.Gala MK & Chan AT Molecular pathways: aspirin and Wnt signaling—a molecularly targeted approach to cancer prevention and treatment. Clin. Cancer Res 21, 1543–1548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Castellone MD, Teramoto H, Williams BO, Druey KM & Gutkind JS Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-β-catenin signaling axis. Science 310, 1504–1510 (2005). [DOI] [PubMed] [Google Scholar]
- 106.Goessling W et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell 136, 1136–1147 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.He TC, Chan TA, Vogelstein B & Kinzler KW PPARδ is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 99, 335–345 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gupta RA et al. Prostacyclin-mediated activation of peroxisome proliferator-activated receptor delta in colorectal cancer. Proc. Natl Acad. Sci. USA 97,13275–13280 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gupta RA et al. Activation of nuclear hormone receptor peroxisome proliferator-activated receptor-δ accelerates intestinal adenoma growth. Nat. Med 10, 245–247 (2004). [DOI] [PubMed] [Google Scholar]
- 110.Wang D et al. Prostaglandin E2 promotes colorectal adenoma growth via transactivation of the nuclear peroxisome proliferator-activated receptor δ. Cancer Cell 6, 285–295 (2004). [DOI] [PubMed] [Google Scholar]
- 111.Ouyang N, Williams JL & Rigas B NO-donating aspirin isomers downregulate peroxisome proliferator-activated receptor (PPAR)δ expression in APCmin/+ mice proportionally to their tumor inhibitory effect: implications for the role of PPARδ in carcinogenesis. Carcinogenesis 27, 232–239 (2006). [DOI] [PubMed] [Google Scholar]
- 112.Tomlinson I et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat. Genet 39,984–988 (2007). [DOI] [PubMed] [Google Scholar]
- 113.Zanke BW et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on chromosome 8q24. Nat. Genet 39, 989–994 (2007). [DOI] [PubMed] [Google Scholar]
- 114.Pomerantz MM et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat. Genet 41, 882–884 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nan H et al. Aspirin use, 8q24 single nucleotide polymorphism rs6983267, and colorectal cancer according to CTNNB1 alterations. J. Natl Cancer Inst 105, 1852–1861 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Haiman CA et al. A common genetic risk factor for colorectal and prostate cancer. Nat. Genet 39, 954–956 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Montrose DC et al. The role of PGE2 in intestinal inflammation and tumorigenesis. Prostaglandins Other Lipid Mediat. 116–117, 26–36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ma X, Aoki T, Tsuruyama T & Narumiya S Definition of prostaglandin E2–EP2 signals in the colon tumor microenvironment that amplify inflammation and tumor growth. Cancer Res. 75, 2822–2832 (2015). [DOI] [PubMed] [Google Scholar]
- 119.Chan AT, Ogino S, Giovannucci EL& Fuchs CS Inflammatory markers are associated with risk of colorectal cancer and chemopreventative response to anti-inflammatory drugs. Gastroenterology 140, 799–808 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Song M et al. A prospective study of plasma inflammatory markers and risk of colorectal cancer in men. Br. J. Cancer 108, 1891–1898 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Heikkila K et al. Associations of circulating C-reactive protein and interleukin-6 with cancer risk: findings from two prospective cohorts and a meta-analysis. Cancer Causes Control 20, 15–26 (2009). [DOI] [PubMed] [Google Scholar]
- 122.Kakourou A et al. Interleukin-6 and risk of colorectal cancer: results from the CLUE II cohort and a meta-analysis of prospective studies. Cancer Causes Control 26, 1449–1460 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang X, Baek SJ & Eling TE The diverse roles of nonsteroidal anti-inflammatory drug activated gene (NAG-1/GDF15) in cancer. Biochem. Pharmacol 85, 597–606 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Breit SN et al. The TGF-β superfamily cytokine, MIC-1/GDF15: a pleotrophic cytokine with roles in inflammation, cancer and metabolism. Growth Factors 29, 187–195 (2011). [DOI] [PubMed] [Google Scholar]
- 125.Mehta RS et al. A prospective study of macrophage inhibitory cytokine-1 (MiC-1/GDF15) and risk of colorectal cancer. J. Natl Cancer Inst 106, dju016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Smyth E, Grosser T & FitzGerald G Goodman & Gillman’s The Pharmacological Basis of Therapeutics (McGraw-Hill, 2011). [Google Scholar]
- 127.Dixon DA et al. Expression of COX-2 in platelet-monocyte interactions occurs via combinatorial regulation involving adhesion and cytokine signaling. J. Clin. Invest 116, 2727–2738 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chan AT, Ogino S & Fuchs CS Aspirin use and survival after diagnosis of colorectal cancer. JAMA 302, 649–658 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Din FV et al. Effect of aspirin and NSAIDs on risk and survival from colorectal cancer. Gut 59, 1670–1679 (2010). [DOI] [PubMed] [Google Scholar]
- 130.Goh CH et al. Post-operative aspirin use and colorectal cancer-specific survival in patients with stage I–III colorectal cancer. Anticancer Res. 34, 7407–7414 (2014). [PubMed] [Google Scholar]
- 131.Li P et al. Aspirin use after diagnosis but not prediagnosis improves established colorectal cancer survival: a meta-analysis. Gut 64, 1419–1425 (2015). [DOI] [PubMed] [Google Scholar]
- 132.Ng K et al. Aspirin and COX-2 inhibitor use in patients with stage III colon cancer. J. Natl Cancer Inst. 107, 345 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Thun MJ, Namboodiri MM & Heath CW Jr. Aspirin use and reduced risk of fatal colon cancer. N. Engl. J. Med 325, 1593–1596 (1991). [DOI] [PubMed] [Google Scholar]
- 134.Algra AM & Rothwell PM Effects of regular aspirin on long-term cancer incidence and metastasis: a systematic comparison of evidence from observational studies versus randomised trials. Lancet Oncol. 13, 518–527 (2012). [DOI] [PubMed] [Google Scholar]
- 135.Rothwell PM et al. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet 379, 1591–1601 (2012). [DOI] [PubMed] [Google Scholar]
- 136.Tougeron D, Sha D, Manthravadi S& Sinicrope FA Aspirin and colorectal cancer: back to the future. Clin. Cancer Res 20, 1087–1094 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Domingo E et al. Evaluation of PIK3CA mutation as a predictor of benefit from nonsteroidal anti-inflammatory drug therapy in colorectal cancer. J. Clin. Oncol 31, 4297–4305 (2013). [DOI] [PubMed] [Google Scholar]
- 138.Reimers MS et al. Expression of HLA class I antigen, aspirin use, and survival after a diagnosis of colon cancer. JAMA Intern. Med 174, 732–739 (2014). [DOI] [PubMed] [Google Scholar]
- 139.Kothari N et al. Impact of regular aspirin use on overall and cancer-specific survival in patients with colorectal cancer harboring a PIK3CA mutation. Acta Oncol. 54, 487–492 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ye XF, Wang J, Shi WT & He J Relationship between aspirin use after diagnosis of colorectal cancer and patient survival: a meta-analysis of observational studies. Br. J. Cancer 111, 2172–2179 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.ISRCTN Registry. ADD-ASPIRIN: the effects of aspirin on disease recurrence and survival after primary therapy in common non-metastatic solid tumours. ISRCTN.org 10.1186/ISRCTN74358648 (2015). [DOI] [PMC free article] [PubMed]
- 142.Roy HK et al. Association between rectal optical signatures and colonic neoplasia: potential applications for screening. Cancer Res. 69, 4476–4483 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Radosevich AJ et al. Rectal optical markers for in vivo risk stratification of premalignant colorectal lesions. Clin. Cancer Res 21, 4347–4355 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liesenfeld DB et al. Aspirin reduces plasma concentrations of the oncometabolite 2-hydroxyglutarate: results of a randomized, double-blind, crossover trial. Cancer Epidemiol. Biomarkers Prev. 25, 180–187 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gerner EW & Meyskens FL Jr. Combination chemoprevention for colon cancer targeting polyamine synthesis and inflammation. Clin. Cancer Res. 15, 758–761 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Casero RA Jr & Marton, L. J. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov 6, 373–390 (2007). [DOI] [PubMed] [Google Scholar]
- 147.Gerner EW & Meyskens FL Jr. Polyamines and cancer: old molecules, new understanding. Nat. Rev. Cancer 4, 781–792 (2004). [DOI] [PubMed] [Google Scholar]
- 148.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT00983580 (2015).
- 149.Meyskens FL Jr et al. Difluoromethylornithine plus sulindac for the prevention of sporadic colorectal adenomas: a randomized placebo-controlled, double-blind trial. Cancer Prev. Res. (Phila.) 1, 32–38 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Martinez ME et al. Pronounced reduction in adenoma recurrence associated with aspirin use and a polymorphism in the ornithine decarboxylase gene. Proc. Natl Acad. Sci. USA 100, 7859–7864 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Barry EL et al. Ornithine decarboxylase polymorphism modification of response to aspirin treatment for colorectal adenoma prevention. J. Natl Cancer Inst 98, 1494–1500 (2006). [DOI] [PubMed] [Google Scholar]
- 152.Hubner RA et al. Ornithine decarboxylase G316A genotype is prognostic for colorectal adenoma recurrence and predicts efficacy of aspirin chemoprevention. Clin. Cancer Res 14, 2303–2309 (2008). [DOI] [PubMed] [Google Scholar]
- 153.Zell JA et al. Ornithine decarboxylase-1 polymorphism, chemoprevention with eflornithine and sulindac, and outcomes among colorectal adenoma patients. J. Natl Cancer Inst 102, 1513–1516 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Barry EL, Mott LA, Sandler RS, Ahnen DJ & Baron JA Variants downstream of the ornithine decarboxylase gene influence risk of colorectal adenoma and aspirin chemoprevention. Cancer Prev. Res. (Phila.) 4, 2072–2082 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wender RC Aspirin and NSAID chemoprevention, gene-environment interactions, and risk of colorectal cancer. JAMA 313, 1111–1112 (2015). [DOI] [PubMed] [Google Scholar]
- 156.Hull MA et al. A randomized controlled trial of eicosapentaenoic acid and/or aspirin for colorectal adenoma prevention during colonoscopic surveillance in the NHS Bowel Cancer Screening Programme (The seAFOod Polyp Prevention Trial): study protocol for a randomized controlled trial. Trials 14, 237 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Baron JA et al. Gastrointestinal adverse effects of short-term aspirin use: a meta-analysis of published randomized controlled trials. Drugs R D 13, 9–16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Abraham NS et al. ACCF/ACG/AHA 2010 expert consensus document on the concomitant use of proton pump inhibitors and thienopyridines: a focused update of the ACCF/ACG/AHA 2008 expert consensus document on reducing the gastrointestinal risks of antiplatelet therapy and NSAID use. Am. J. Gastroenterol 105, 2533–2549 (2010). [DOI] [PubMed] [Google Scholar]
- 159.Fuster V & Sweeny JM Aspirin: a historical and contemporary therapeutic overview. Circulation 123, 768–778 (2011). [DOI] [PubMed] [Google Scholar]
- 160.Vane JR Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol 231, 232–235 (1971). [DOI] [PubMed] [Google Scholar]
- 161.Kune GA, Kune S & Watson LF Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer Res. 48, 4399–4404 (1988). [PubMed] [Google Scholar]
- 162.Vogelstein B et al. Genetic alterations during colorectal-tumor development. N. Engl. J. Med 319, 525–532 (1988). [DOI] [PubMed] [Google Scholar]
- 163.Giovannucci E et al. Aspirin use and the risk for colorectal cancer and adenoma in male health professionals. Ann. Intern. Med 121, 241–246 (1994). [DOI] [PubMed] [Google Scholar]
- 164.Giovannucci E et al. Aspirin and the risk of colorectal cancer in women. N. Engl. J. Med 333, 609–614 (1995). [DOI] [PubMed] [Google Scholar]
- 165.Cole BF et al. Aspirin for the chemoprevention of colorectal adenomas: meta-analysis of the randomized trials. J. Natl Cancer Inst 101, 256–266 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]