

Abstract
CRISPR/Cas9 technology enables the rapid generation of loss‐of‐function mutations in a targeted gene in mammalian cells. A single cell harboring those mutations can be used to establish a new cell line, thereby creating a CRISPR‐induced knockout clone. These clonal cell lines serve as crucial tools for exploring protein function, analyzing the consequences of gene loss, and investigating the specificity of biological reagents. However, the successful derivation of knockout clones can be technically challenging and may be complicated by multiple factors, including incomplete target ablation and interclonal heterogeneity. Here, we describe optimized protocols and plasmids for generating clonal knockouts in mammalian cell lines. We provide strategies for guide RNA design, CRISPR delivery, and knockout validation that facilitate the derivation of true knockout clones and are amenable to multiplexed gene targeting. These protocols will be broadly useful for researchers seeking to apply CRISPR to study gene function in mammalian cells. © 2019 The Authors.
Keywords: cell lines, CRISPR/Cas9, knockout, mammalian
INTRODUCTION
The CRISPR system initially evolved as a nucleic acid–targeting bacterial defense mechanism capable of conferring resistance to viral infection (Barrangou et al., 2007). It has since been co‐opted by scientists as a means to generate sequence‐specific double‐strand breaks (DSBs) and to induce other precise alterations in the genomes of cells and organisms (Cong et al., 2013). CRISPR has been particularly useful in the study of mammalian genetics and cell biology, as mammalian somatic cells have historically proven to be highly refractory to genetic modification (Komor, Badran, & Liu, 2017). By expressing the Cas9 nuclease and a suitable guide RNA (gRNA) in mammalian cells, a double‐strand break can be introduced at a locus of interest. The cell then has multiple options for repairing that break. If a suitable template is provided, the cell can use homology‐directed repair to integrate a novel allele or transgene at the targeted site (Ceasar, Rajan, Prykhozhij, Berman, & Ignacimuthu, 2016). Alternately, the cell can repair the lesion via nonhomologous end joining (NHEJ), an error‐prone process that commonly results in an insertion or deletion (indel) mutation at the DSB location (Brinkman et al., 2018). In this way, CRISPR can be used to introduce stable, nonrevertible alterations to mammalian genes. Below, we describe an efficient method to use CRISPR to generate knockout clones in mammalian somatic cell lines. The protocol is divided into five sections, as outlined in Figure 1:
-
1.
Choosing a knockout strategy;
-
2.
Selecting gRNA target sites and performing vector cloning;
-
3.
Introducing gRNAs by transfection or transduction;
-
4.
Isolation and expansion of single‐cell clones;
-
5.
Knockout verification by western blot analysis, PCR, and/or Sanger sequencing.
Figure 1.
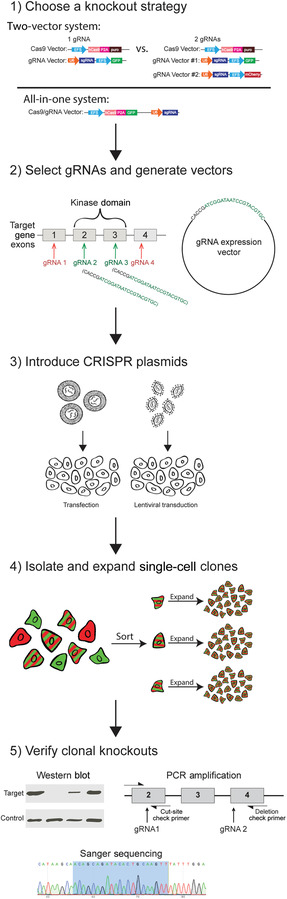
Schematic outline of the knockout process. The procedure includes (1) choosing a knockout strategy; (2) selecting gRNA target sites and performing vector cloning (Support Protocols 1–3, Basic Protocol 1); (3) introducing CRISPR plasmids by transfection or transduction (Basic Protocols 2–5); (4) isolation and expansion of single‐cell clones (Basic Protocol 6, Alternate Protocol 1); and (5) knockout verification by western blot analysis, PCR, and/or Sanger sequencing (Support Protocol 4, Basic Protocols 7–9).
STRATEGIC PLANNING
Choosing a Knockout Strategy
To knock out a gene of interest, the nuclease Cas9 and a target‐specifying guide RNA must be delivered to the cell. Various CRISPR platforms are suitable for generating knockouts, including “all‐in‐one” or one‐plasmid system (in which Cas9 and the guide RNA are encoded on the same vector), two‐plasmid systems (in which Cas9 and the guide RNA are encoded on different vectors), and direct ribonucleoprotein delivery. Ribonucleoprotein assembly and delivery have been described elsewhere (Farboud et al., 2018), and this protocol will focus on plasmid‐based knockout strategies.
The efficiency of each CRISPR system is limited by the efficacy with which it can be introduced into and expressed in the target cell. In the two‐vector approach, a Cas9‐expressing plasmid is first stably transduced using lentiviruses into the target cell line. Following selection for Cas9 expression, this line can be easily used to generate multiple knockout clones by expressing different guide RNAs. However, constitutive Cas9 expression may lead to higher levels of off‐target mutagenesis, and some cell lines have a tendency to silence virally expressed proteins (Ellis, 2005). In the one‐vector approach, the single Cas9/gRNA plasmid can be introduced into cells via either transfection or transduction. However, the size of the provirus generated from the all‐in‐one vector is near the lentiviral packaging limit, which may decrease subsequent viral titers (Kumar, Keller, Makalou, & Sutton, 2001). In general, if a researcher is seeking to analyze different knockouts in the same cell line, we recommend using the two‐vector approach and generating a stable Cas9‐expressing cell line. If a researcher is seeking to analyze a single knockout and to minimize off‐target effects, then transient transfection of the all‐in‐one vector may be superior. A number of labs have developed CRISPR plasmids with convenient drug‐resistance and fluorescent markers. In this protocol, we describe the use of plasmids that have been generated and validated by our own laboratory and by the Vakoc laboratory at Cold Spring Harbor Laboratory, though many of the other CRISPR vectors available from Addgene could be used in their place.
The Cas9 variant used in this protocol was obtained from the bacteria Streptococcus pyogenes and cuts 3 base pairs (bp) upstream of the prospacer adjacent motif (PAM) sequence, NGG. A number of tools to identify potent guide RNA sequences have been described (Graham & Root, 2015). In general, targeting Cas9 to exons that encode functional protein domains is more likely to ablate gene function than simply targeting a 5′ exon (Shi et al., 2015). Exon‐targeting guide RNA sequences can be identified using GUIDES (http://guides.sanjanalab.org/) (Meier, Zhang, & Sanjana, 2017). We also frequently design guide RNAs using Benchling (http://www.benchling.com/), which offers an easy‐to‐use graphical user interface for CRISPR experiments. Benchling does not currently support domain annotations, but these can be obtained by cross‐referencing guides found on Benchling with Ensembl or any other genome database. We often design three to five guides per target gene as some gRNA sequences may be more efficient than others; however, with the methods described in this protocol, we have found most gRNAs capable of cutting the target locus sufficiently well.
The number of gRNAs introduced into cell lines will vary depending on the experimental goals of the project and any unique characteristics of the gene of interest. The use of one gRNA is frequently sufficient to knock out the target gene within a portion of cells in a population (Popp & Maquat, 2016). However, as described in the Commentary below, alternative splicing, downstream translational initiation, and the production of functional protein fragments may occasionally allow cells to bypass single Cas9‐induced lesions (Lalonde et al., 2017; Sharpe & Cooper, 2017). To maximize the likelihood of knocking out a target gene, a two‐guide strategy can be employed. In this approach, two gRNAs, targeting two distinct exons, are introduced into the cells. This double cut can be repaired through either the generation of two independent mutations or the elimination of the DNA between the two targeted sites. It is important to note that the introduction of multiple guide RNAs into a cell may result in an increase in off‐target mutagenesis. Thus, it remains crucial to validate knockout phenotypes by using different combinations of guide RNAs and by analyzing multiple independent clones by western blotting, PCR, and sequencing.
Additionally, when generating KO cell lines, it is often useful to have control cell lines for downstream experimental comparisons. For this purpose, we often generate single‐cell‐derived clonal lines from cells harboring gRNAs targeting the safe harbor loci Rosa26 or AAVS1.
Guide RNA Cloning
To begin with, gRNAs targeting the gene of interest must first be cloned into the gRNA expression vector of choice.
Support Protocol 1. ISOLATION OF PLASMID DNA
In order to clone in gRNA sequences in later steps of the protocol (Basic Protocol 1), it is necessary to first purify sufficient quantities of the gRNA plasmid backbone (Table 1). These plasmid backbones can be obtained from Addgene.
Table 1.
CRISPR Plasmids
| Plasmid(s) | Addgene no. | Purpose |
|---|---|---|
| One‐plasmid knockout systems | ||
| Lenti‐Cas9‐gRNA‐GFP | 124770 | Coexpress Cas9, a gRNA, and GFP |
| Lenti‐Cas9‐gRNA‐TagBFP2 | 124774 | Coexpress Cas9, a gRNA, and TagBFP2 |
| Two‐plasmid knockout systems | ||
| LentiV_Cas9_puro | 108100 | Expresses Cas9 with puromycin resistance |
| LRG2.1 | 108098 | Expresses a gRNA with GFP |
| LRCherry2.1 | 108099 | Expresses a gRNA with mCherry |
| Lenti_sgRNA_EFS_BFP | 120577 | Expresses a gRNA with eBFP |
| LRG2.1‐mOrange | 124772 | Expresses a gRNA with mOrange |
| LRG2.1‐CyOFP1 | 124771 | Expresses a gRNA with CyOFP1 |
| LRG2.1‐TagBFP2 | 124773 | Expresses a gRNA with TagBFP2 |
Materials
Plasmid(s) of interest (Addgene)
Appropriate Escherichia coli strain
LB + 100 µg/ml ampicillin medium
50% (v/v) glycerol
GeneJET Plasmid Maxiprep Kit (Thermo Fisher Scientific; cat. no. K0491)
NanoDrop spectrophotometer or equivalent
1.5‐ml freezing tubes
1.5‐ml microcentrifuge tubes
-
1
Obtain the desired plasmid(s) from Addgene.
-
2
After receiving the stab culture of E. coli, inoculate the bacteria in 2 ml of LB + 100 µg/ml ampicillin medium overnight at 37°C.
-
3
The next day, prepare glycerol stocks by resuspending 500 µl of the bacterial culture in 500 µl of 50% glycerol and store at in 1.5‐ml freezing tubes −80°C. Additionally, transfer 1 ml of the overnight culture into 250 ml of LB + 100 µg/ml ampicillin medium and allow to grow for an additional 12‐18 hr at 37°C.
-
4
Isolate the plasmids from the E. coli stock using the GeneJET Plasmid Maxiprep Kit according to the manufacturer's instructions. Quantify the DNA concentration and purity using a NanoDrop spectrophotometer and store aliquots of the plasmid‐bearing stock in 1.5‐ml microcentrifuge tubes at −20°C for later use.
Support Protocol 2. PREPARATION OF COMPETENT STBL3 E. COLI FOR gRNA CLONING
The repetitive sequences in the lentiviral CRISPR plasmids may be unstable when the plasmids are propagated in E. coli. We therefore recommend transforming the cloned plasmids constructed in Basic Protocol 1 into the recombination‐deficient Stbl3 strain of E. coli to maximize vector stability (Al‐Allaf, Tolmachov, Zambetti, Tchetchelnitski, & Mehmet, 2013). Although purchasing premade competent Stbl3 is very expensive, we generate our own batches of competent Stbl3 cells using the Zymo Research Mix & Go Transformation Kit.
Materials
One Shot™ Stbl3™ Chemically Competent E. coli (Invitrogen; cat. no. C737303)
Mix & Go E. coli Transformation Kit and Buffer Set (Zymo Research; cat. no. T3001)
-
1
Prepare fresh batches of competent Stbl3 E. coli by following the Mix & Go Kit's instructions.
-
2
Store aliquots of cells at −80°C for later use.
Support Protocol 3. OBTAINING GUIDE RNA OLIGONUCLEOTIDES
Guide RNAs are cloned into expression vectors by BsmBI digestion. After a suitable 20‐bp guide RNA sequence is identified, BsmBI restriction sequences should be added to each strand of the gRNA sequence, as shown here:
Guide RNA post‐annealing schematic:
Forward oligo: 5′‐CACCG–20‐bp gRNA sequence‐3′
Reverse oligo: 3′‐C–20‐bp gRNA sequence–CAAA‐5′
Guide RNA oligo 5′‐3′ sequences:
Forward oligo: 5′‐CACCG–20 bp gRNA sequence‐3′
Reverse oligo: 5′‐AAAC–20 bp gRNA sequence–C‐3′
“CACCG” and “AAAC” are added to the oligos to ensure compatibility for cloning using a BsmBI digestion. The single C at the 3′ end of the reverse oligo is needed to anneal the sequence to the additional G added to the forward oligo. Guide RNAs are generally 25 bp long after these modifications are added to the sgRNA sequence.
Synthetic oligos of the designed guides can be purchased commercially from Integrated DNA Technologies (IDT; https://www.idtdna.com) or any commercial source. Generally, 25 nmol of DNA oligos from IDT prepared with standard desalting provides more than enough material for gRNA cloning (see Strategic Planning for guidelines for gRNA design). Although we have provided a list of gRNA‐expressing vectors that we commonly use in Table 1, a number of other gRNA vectors can be found on Addgene, and these plasmids may require the use of alternative restriction enzymes.
Basic Protocol 1. GUIDE RNA CLONING
The gRNA oligos can be cloned into either the gRNA vector (two‐vector system) or the “all‐in‐one” vector (one‐vector system) by restriction digestion and ligation. If the two‐gRNA approach is chosen, each gRNA can be cloned into a vector with different fluorescent markers to allow downstream sorting of cells that harbor both gRNAs. The gRNA cloning protocol consists of five overall steps: (1) linearization of the plasmid backbone, (2) 5′ phosphorylation and annealing of the oligonucleotides, (3) ligation of the annealed oligonucleotides into the linearized vector, (4) transformation of the ligation product into Stbl3 cells, and (5) sequence verification of the gRNA.
Materials
gRNA plasmid backbone or all‐in‐one vector (Support Protocol 1)
Competent Stbl3 cells (Support Protocol 2)
BsmBI restriction endonuclease and NEB buffer 3.1 (NEB; cat. no. R0580)
Nuclease‐free distilled, deionized water (ddH2O)
Alkaline phosphatase, calf intestinal (CIP) stock (10,000 U/ml; NEB; cat. no. M0290)
NucleoSpin® Gel and PCR clean‐up (Macherey‐Nagel; cat. no. 740609.50)
100 µM gRNA oligos (IDT or other DNA synthesis manufacturer; see Strategic Planning and Support Protocol 3)
T4 polynucleotide kinase (NEB; cat. no. M0201)
T4 DNA ligase and appropriate buffer (NEB; cat. no. M0202)
LB + 100 µg/ml ampicillin culture plates
LB + 100 µg/ml ampicillin medium
50% (v/v) glycerol
QIAprep Spin Miniprep Kit (Qiagen; cat. no. 27106)
U6 sequencing primer 5′‐GAGGGCCTATTTCCCATGATTCC‐3′ (IDT or other DNA provider)
1.5‐ml microcentrifuge tubes
Heat block
DNA NanoDrop spectrophotometer or equivalent
0.2‐ml thin‐walled PCR tubes
Thermocycler
1.5‐ml freezing tubes
Additional reagents and equipment for agarose gel electrophoresis (Current Protocols article: Voytas, 2001)
-
1To linearize the guide RNA plasmid backbone, mix the following in a 1.5‐ml microcentrifuge tube:
- 8 µg gRNA backbone
- 2 µl BsmBI enzyme
- 5 µl NEB buffer 3.1
- Nuclease‐free ddH2O to 50 µl final.
Incubate for 3 hr at 55°C.
-
2
After 3 hr of digestion, add 1 µl 10,000 U/ml CIP stock solution to each reaction and incubate 1 hr more at 37°C.
-
3
Run the digestion mixture on a 1% agarose gel. Two bands should be observed (bands around 11 and 2 kb for the all‐in‐one vector and bands around 7.5 and 2 kb for the vector backbone of the two‐plasmid system). Using the NucleoSpin® Gel and PCR Clean‐up Kit, cut out and extract the larger band. Determine the DNA concentration of the extracted vector using a NanoDrop spectrophotometer, and store the linearized vector at −20°C.
-
4To anneal and phosphorylate the 5′ end of the gRNA‐containing oligos, set up the following reaction in a 0.2‐ml PCR tube:
- 1 µl forward oligo (100 µM)
- 1 µl reverse oligo (100 µM)
- 1 µl 10× T4 ligase buffer
- 6.5 µl nuclease‐free ddH2O
- 0.5 µl T4 PNK enzyme.
-
5Place the reaction tube in a thermocycler and run the following program:
30 min 37°C 5 min 95°C 5 min 90°C 5 s 80°C (ramp 0.1°C/s) 30 s 70°C (ramp 0.1°C/s) 30 s 60°C (ramp 0.1°C/s) 1 min 50°C (ramp 0.1°C/s) 2 min 40°C (ramp 0.1°C/s) 2 min 30°C (ramp 0.1°C/s) Hold at 4°C. -
6
After the PCR program is completed, dilute the reaction 1:200 with nuclease‐free ddH2O and store at −20°C.
-
7To ligate the sgRNA into the linearized guide RNA backbone, set up the following reaction and incubate 30 min at room temperature. A reaction excluding the gRNA oligos from step 6 should be included the first time a new batch of digested vector is used to confirm the efficiency of the digestion. No colonies above background levels should be seen on Stbl3 cells transformed with only digested vector.
- X µl (25 ng) digested vector from step 3
- Y µl ddH2O to 10 µl total volume
- 1 µl phosphorylated and annealed oligo duplex from step 6
- 1 µl 10× T4 ligase buffer
- 1 µl T4 ligase.
-
8
To transform the sgRNA vectors into Stbl3 E. coli cells, add 5 µl of the ligation reaction from step 7 to 50 µl Stbl3 cells. Allow the mixture to rest on ice for 5 min.
-
9
Plate appropriate dilutions of the Stbl3 E. coli cells on LB + 100 µg/ml ampicillin plates and allow to grow overnight at 37°C.
-
10
Pick three to five colonies per gRNA and allow to grow in 3 ml LB + 100 µg/ml ampicillin medium overnight at 37°C.
-
11
After overnight growth, make 1 ml glycerol stocks of each of the colonies by mixing the LB culture 1:1 with 50% glycerol and storing at −80°C.
-
12
To verify the sequence of the sgRNA plasmid, generate a glycerol stock (see Support Protocol 1, step 3), purify the vectors from the bacteria in the liquid culture using the QIAprep Spin Miniprep Kit, and sequence the plasmids with the U6 primer.
The U6 primer will initiate sequencing at the promoter region upstream of the location where the sgRNA should be inserted.
CRISPR Delivery
The CRISPR system described in this protocol can be introduced into the cells of interest in a number of ways, including:
Transfection of the all‐in‐one CRISPR plasmid (Basic Protocol 5);
Transfection of a gRNA plasmid into a Cas9‐expressing cell line (cell line generation: Basic Protocols 2 and 3; transfection: Basic Protocol 5);
Transduction of the all‐in‐one CRISPR plasmid (Basic Protocol 2 and 4);
Transduction of a gRNA plasmid into a Cas9‐expressing cell line (cell line generation: Basic Protocols 2 and 3; transduction: Basic Protocol 2 and 4).
In general, transient expression of the CRISPR components may lead to fewer off‐target effects while also decreasing on‐target efficiency. Suitable strategies will depend on the researchers’ particular experimental goals and the system(s) in which they are working. Below, we describe various protocols for transfection and transduction to introduce the CRISPR plasmids into the cells of interest.
Basic Protocol 2. CALCIUM PHOSPHATE TRANSFECTION TO PRODUCE LENTIVIRUS
All lentiviruses used in this protocol (viral particles containing plasmids that express Cas9, a guide RNA, or the all‐in‐one vector) are generated in HEK293T cells via calcium phosphate transfection (Chang, Marran, Valentine, & Hannon, 2013). This transfection protocol involves the use of two viral packaging plasmids (PsPAX2.0 and VSVG). If the two‐vector approach is used, separate lentiviral pools should be generated for each vector.
Materials
HEK293T cells (ATCC CRL‐3216)
CRISPR plasmid DNA (“all‐in‐one” plasmid or gRNA/Cas9 plasmids from Basic Protocol 1)
psPAX2.0 plasmid (Addgene plasmid no. 12260)
VSVG plasmid (Addgene plasmid no. 12259)
2 M CaCl2
Solution B (2× HEPES‐buffered saline; see recipe).
100 mM chloroquine (Cayman Chemical; cat. no. 14194)
DMEM medium supplemented with 10% FBS, 1% Pen/Strep, and glutamine (Life Technologies; cat. no. 11995‐073)
10‐cm tissue culture plates with preferred culture medium for DMEM/10% FBS with 1% Pen/Strep and glutamine HEK293T cells
5‐ml polystyrene tubes
0.45‐µM filter
-
1
Prior to transduction, split HEK293T onto a 10‐cm plate to obtain cells that are 70‐80% confluent at the time of transfection.
-
2In one tube, prepare solution A (total volume 500 µl) by adding, in the following order:
- 20 µg CRISPR plasmid DNA
- 25 µg psPAX2.0 plasmid
- 4 µg VSVG plasmid
- ddH2O as required for a 500 µl total volume
- 62.5 µl 2 M CaCl2
-
3
Pipet 500 µl solution B into a separate tube.
-
4
Use a pipet to blow bubbles into solution B while adding solution A to solution B dropwise.
Bubbles will aid the even formation of precipitants, which is a crucial step in determining the success of a transfection.
-
5
Incubate this mixture 15 min room temperature.
-
6
During the incubation, add 2.5 µl 100 mM chloroquine to the 10‐cm plate of HEK293T cells from step 1.
-
7
After the 15‐min incubation is done, add the transfection mixture dropwise around the 10‐cm plate and then return the plate to a tissue culture incubator at 37°C.
-
8
At 8‐14 hr after transfection, replace the medium on the HEK293T cells with fresh 10% FBS DMEM medium.
-
9
Start collecting virus 24 hr after transfection. To harvest virus, collect all the old medium on the HEK293T cells using a syringe. Separate cell debris from viral supernatant using a 0.45‐µM filter, and either store the supernatant at −80°C or add it to a recipient cell line immediately. Typically, virus can be collected three times within the 24‐ to 72‐hr window after transfection. To perform multiple harvests, add an equal volume of the medium back to the cells and repeat the collection protocol the next day. The protocol can also be scaled up (to 15‐cm plates) or down (to 6‐ or 12‐well plates) as needed.
Several of the CRISPR plasmids described in this protocol contain a fluorescent marker and can be easily titered. To determine viral titer, add increasing volumes of virus to a recipient cell line using the transduction protocol listed below. Three days after transduction, quantify the fraction of fluorescent cells for each volume of virus to determine the number of infectious particles per microliter of solution. However, titration is often unnecessary, as the above transfection protocol is highly efficient. If titration is required for a nonfluorescent viral plasmid, a qPCR kit can be used instead (Clontech; cat. no. 631235).
Basic Protocol 3. GENERATING A CAS9‐EXPRESSING CELL LINE
When using the two‐vector system, a stable cell line that expresses Cas9 must first be generated. The cell line can then be reused in multiple knockout experiments. Alternatively, a number of vendors offer Cas9‐expressing cell lines, some of which have inducible Cas9 expression (https://www.genecopoeia.com).
Materials
Cas9 lentiviral supernatant from lentivirus generated using the calcium phosphate transfection protocol (Basic Protocol 2)
Recipient mammalian cell line
Polybrene (Santa Cruz Biotechnology; cat. no. sc‐134220)
Appropriate selective agent for the plasmid used
Additional reagents and equipment for cell culture and trypsinization (Current Protocols article: Phelan, 1996)
-
1
Trypsinize the target cells and plate them so that they will be ∼50% confluent.
-
2
Mix supernatant from the lentiviral culture into cells 1:1: e.g., add 5 ml viral supernatant to 5 ml cell culture.
-
3
Add polybrene to 8 µg/ml final.
-
4
Change medium 24 hr after transduction.
-
5
At 48 hr after transduction, select for Cas9 expression.
The Cas9 lentiviral plasmid most commonly used in our lab (Addgene plasmid no. 108100) harbors a puromycin‐resistance marker, and we select for Cas9‐expressing cells using 1‐4 µg/ml puromycin (Thermo Fisher Scientific; cat. no. A11138‐03). In some cell lines, sufficient Cas9 expression can be generated by a single round of transduction followed by 3‐5 days of selection. In other cell lines (particularly those that silence viral transgenes), optimal Cas9 expression requires multiple rounds of lentiviral transduction followed by 1‐2 weeks of drug selection. Cas9 expression can be checked in the population using qPCR, western blot analysis, or by transduction with a guide RNA with known biological activity.
Basic Protocol 4. TRANSDUCING GUIDE RNAs OR ALL‐IN‐ONE VECTOR INTO RECIPIENT CELLS
Guide RNA or all‐in‐one vectors can be permanently introduced into a recipient cell line by the transduction protocol described below.
Materials
Recipient mammalian cell line (use cell line from Basic Protocol 3 for two‐vector system)
Supernatant with lentiviruses carrying cloned gRNA vector or all‐in‐one vector (Basic Protocol 2)
Polybrene (Santa Cruz Biotechnology; cat. no. sc‐134220)
6‐well culture plates
Additional reagents and equipment for cell culture and trypsinization (Current Protocols article: Phelan, 1996)
-
1
Trypsinize the target cells and plate them in 6‐well plates so that they will be ∼50% confluent.
Typically, plating one well more than the number of transduction reaction is sufficient. The extra well can be used as an untransduced control when monitoring transduction efficiency.
-
2
Mix lentiviral supernatant into culture medium in 1:1 (or using an appropriate amount determined by titration).
-
3
Add polybrene to 8 µg/ml final.
-
4
Change medium 24 hr after transduction.
-
5
Monitor transduction efficiency based on the fluorescent marker present on the gRNA or all‐in‐one vector using a fluorescence microscope.
Basic Protocol 5. TRANSFECTING GUIDE RNAs OR THE ALL‐IN‐ONE VECTOR INTO RECIPIENT CELLS
Guide RNAs or all‐in‐one vectors can be temporarily introduced into a recipient cell line by a variety of transfection methods. We have found that transfection efficiency can vary greatly for different methods with different recipient cell lines. In general, we find that Lipofectamine 3000 is most likely to produce the highest transfection rates with the least amount of optimization required. However, in specific circumstances, other reagents (Lipofectamine 2000, TransIT‐LT1, etc.) may yield superior results. In general, we recommend starting with Lipofectamine 3000 and then testing other reagents if necessary.
To introduce CRISPR plasmids with Lipofectamine 3000, we use the following protocol (slightly modified from the manufacturer's protocol).
Materials
Recipient mammalian cell line (use the cell line from Basic Protocol 3 for the two‐vector system)
Lipofectamine 3000 (Thermo Fisher Scientific; cat. no. L3000001)
P3000 Reagent (supplied with Lipofectamine 3000)
Opti‐MEM Reduced Serum Media (Thermo Fisher Scientific; cat. no. 31985088)
Cloned gRNA vector or all‐in‐one vector
Pen/Strep‐free medium
5‐ml polystyrene tubes
Cell culture materials
6‐well tissue culture plates
Fluorescence microscope
-
1
Plate the target cell line so that it will be 60‐80% confluent in a 6‐well plate the next day.
-
2
Warm transfection reagents to room temperature.
-
3
Add 7.5 µl Lipofectamine 3000 reagent to 125 µl Opti‐MEM medium (called “tube A”).
-
4
Add 5 µg plasmid DNA (gRNA or all‐in‐one vector) and 10 µl P3000 Reagent to 125 µl medium (called “tube B”).
-
5
Mix the contents of tube A and tube B and incubate the mixture at room temperature for 5 min.
-
6
Change the medium on the recipient plate to Pen/Strep‐free medium.
-
7
Add the transfection mixture to the recipient cells dropwise.
-
8
Change the medium 24 hr after transfection.
-
9
Confirm transfection efficiency by assessing the fluorescent marker under a fluorescence microscope. Although higher transfection efficiencies will allow for easier knockout enrichment by FACS, even low transfection rates will allow you to proceed with the protocol.
Clonal Expansion of Knockout Cell Lines
Multiple approaches can be used to isolate potential knockout clonally derived populations.
Basic Protocol 6. SINGLE‐CELL EXPANSION BY DILUTION PLATING
After introduction of the gRNA(s) by transduction or transfection, single cells must be isolated in order to generate clonal lines that can be verified as complete knockouts. Two common methods for this are dilution cloning and fluorescence‐activated cell sorting (FACS) of single cells (Alternate Protocol 1). Although both approaches can yield single cell–derived clones, FACS sorting is often more effective given its ability to specifically isolate double‐positive cells. However, dilution cloning is cheaper and may cause less cellular stress than sorting.
Materials
Appropriate cell culture medium
-
Standard cell culture plates:
10‐cm plates (VWR: cat. no. 25382‐701)
96‐well flat‐bottom plates (VWR: cat. no. 29442‐056)
Microscope
Additional reagents and equipment for cell culture and trypsinization (Current Protocols article: Phelan, 1996)
-
1
Harvest the transduced cells by trypsinization.
-
2
Count the cells and dilute to a concentration of 20 cells per 100 µl.
-
3
Using a multichannel pipette, pipette 200 µl of the diluted cells into the first row of a 96‐well plate.
-
4
Pipette 100 µl of the appropriate culture medium into all remaining wells in the plate.
-
5
Take 100 µl from the first row of the plate containing cells and mix it with the 100 µl of appropriate medium in the row below. This will result in a two‐fold dilution of the cell concentration for the second row.
-
6
Repeat this process down the rows of the plate, resulting in a series of two‐fold dilution down the rows. This will result in some proportion of the wells containing a single cell.
-
7
Check by microscopy for wells that contain a single cell that are single or double‐positive for the gRNA marker(s). This can be done immediately following dilution or 12‐24 hr later, after the cells have settled. Circle these wells.
-
8
Wells with single cells should be expanded: allow the cells to grow to form microcolonies (∼50‐100 cells). Split the microcolony into a fresh well of a 96‐well plate. Once that well is near confluence, split to a well of a 48‐well plate. Continue expanding the line until a sufficient number of cells can be harvested for the validation assays described below.
Alternate Protocol 1. SINGLE‐CELL ISOLATION AND EXPANSION BY FACS
FACS can be used in multiple ways to isolate single cells. When using the two‐gRNA approach, FACS can be used to isolate cells that express both gRNAs by sorting for both fluorescent markers. Depending on your choice of system and the setup in your FACS facility, there are several approaches for this, listed below.
-
1
Double‐positive single cells can be sorted directly by FACS and plated into 96‐well plates.
-
2
Bulk populations of double‐positive cells can be isolated by FACS and plated into 96‐well plates by dilution plating, as described in Basic Protocol 6.
-
3Bulk populations of double positive cells can be isolated by FACS and plated on 10‐cm plates at a low concentration to allow single cells to form colonies:
- Plate 100‐200 cells from the FACS‐sorted sample of double‐positive cells onto a 10‐cm plate.
- Use a microscope to identify isolated microcolonies that begin to grow.
-
Use cloning cylinders (Chemglass; cat. no. CLS‐1777‐02) to isolate and trypsinize the individual microcolonies, and then move them to a 96‐well or 48‐well plate for clonal expansion.Clones can be expanded by plating to progressively larger plates until there are enough cells to freeze and take DNA and protein lysates for knockout verification.
Validation of Gene Knockout
The cornerstone of any strategy to verify that a protein of interest has been ablated is demonstrating its absence by western blot analysis. However, single western blots may be misleading, and characterizing the precise alterations caused by CRISPR at the DNA level may provide additional useful information. The options for knockout validation also differ slightly depending on whether the one‐guide or two‐guide approach was used. In particular, if a gene was targeted by two independent guide RNAs, a PCR‐based screening strategy can be applied to rapidly assess the knockout status of dozens of potential clones. Alternatively, the presence of indel mutations can be verified at each cut site, though this becomes more challenging if the resultant mutations are heterozygous. No single technique is perfect, and we recommend that researchers apply a combination of the methods described below to characterize the CRISPR‐induced lesions and identify valid knockout clones. Figures 2 and 3 show representative results of different validation experiments to confirm knockout of the MELK kinase from cancer cell lines (Giuliano, Lin, Smith, Palladino, & Sheltzer, 2018; Lin, Giuliano, Sayles, & Sheltzer, 2017).
Figure 2.
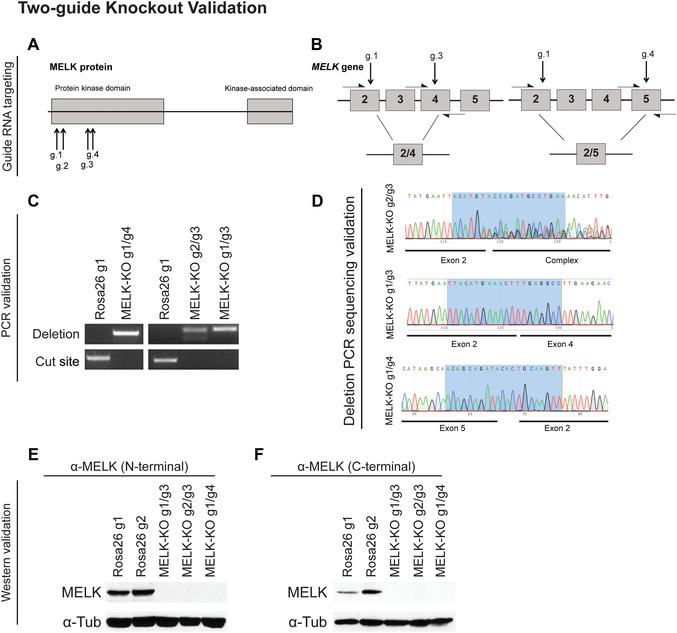
Two‐guide knockout validation. (A) Schematic illustrating the domain structure of MELK and sites targeted by guide RNAs. (B) Diagram showing the exons of MELK targeted by each gRNA. Half‐arrows indicate the deletion or cut‐site primers used to verify the presence of recombination events. (C) Sample results from PCR amplification verifying multi‐exon deletions in three independent MELK knockout (MELK‐KO) clones transduced with two guide RNAs. (D) Sample results from Sanger sequencing of deletion PCR products from three MELK‐KO clones. Clones transduced with the g1/g3 and g1/g4 combinations have homozygous deletions in MELK, whereas the clone transduced with the g2/g3 combination contain a heterozygous recombination event. If necessary, the heterozygous alleles could be further analyzed using TOPO cloning. The highlighted region in each chromatogram indicates the base pairs recognized by a guide RNA. (E and F) Sample results from validation of MELK knockout through western blotting using two antibodies that bind to two different epitopes. Clones transduced with guide RNAs targeting Rosa26, a nonessential and noncoding gene, were used as a control. (Reprinted from Lin et al., 2017, with permission of eLife.)
Figure 3.
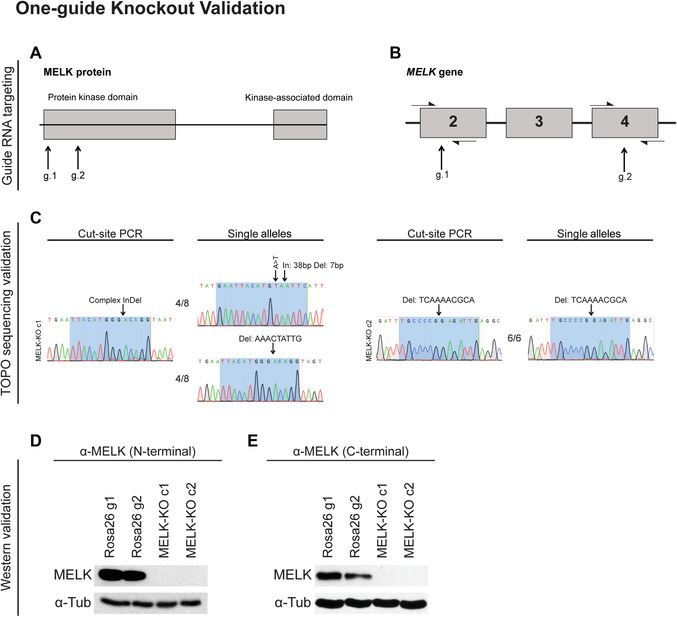
One‐guide knockout validation. (A) Schematic illustrating the domain structure of MELK and sites targeted by guide RNAs. (B) Diagram showing the exons of MELK targeted by each gRNA. Half‐arrows represent cut‐site primers used to verify the presence of indels. (C) Sample results from Sanger sequencing of cut‐site PCR products or single alleles isolated by TOPO cloning. MELK‐KO c1 has heterozygous cut‐site mutations, whereas MELK‐KO c2 has a homozygous mutation. Highlighted regions indicate base pairs recognized by each guide RNA. (D and E) Sample results from validation of MELK knockout through western blotting using two antibodies that bind to two different epitopes. Clones transduced with guide RNAs targeting Rosa26, a nonessential and noncoding gene, were used as a control. (Reprinted from Giuliano et al., 2018, with permission of eLife.)
Support Protocol 4. GENOMIC DNA EXTRACTION FROM POTENTIAL KNOCKOUT CELLS
To confirm gene knockout by PCR (Basic Protocol 7), mutation sequencing (Basic Protocol 8), or TOPO cloning (Basic Protocol 9), genomic DNA must first be prepared from control and potential knockout cell lines.
Materials
Potential knockout cells
DNeasy Blood & Tissue Kit (Qiagen; cat. no. 69506)
Cell culture materials
6‐well tissue culture plates
15‐ml tubes
1.5‐ml microcentrifuge tube
NanoDrop spectrophotometer or equivalent
-
1
Plate ∼300,000 cells in one well of a 6‐well plate for each putative knockout clone.
If the number of clones need to be screen is large and plating 300,000 cells per clone is not optimal, cells can be plated in smaller well plates and QuickExtract kits can be employed instead of DNeasy kits. However, for high‐quality genomic DNA and clean sequencing results in later steps, we do recommend plating ∼300,000 cells.
-
2
The next day, follow the manufacturer's protocol from the DNeasy Blood & Tissue Kit to isolate the genomic DNA.
-
3
Quantify the concentration and purity of the DNA with a NanoDrop spectrophotometer. A DNA yield of ∼1 µg is ideal.
Basic Protocol 7. GENOMIC DELETION AND CUT‐SITE PCR
To screen for genomic knockouts, design two sets of PCR primers: one that spans the putative deletion and one that spans either cut site. The primers should recognize a sequence that is ∼100‐200 bp from the CRISPR cut site, as larger sequences are more difficult to amplify. Suitable primers can be designed using NIH Primer Blast (https://www.ncbi.nlm.nih.gov/tools/primer‐blast/) and ordered from IDT (https://www.idtdna.com).
Materials
Genomic DNA (Support Protocol 4)
Primers
Taq 2× Master Mix (NEB; cat. no. M0270)
Nuclease‐free distilled, deionized water (ddH2O)
Thermocycler
0.2‐ml thin‐walled PCR tubes
Additional reagents and equipment for agarose gel electrophoresis (Current Protocols article: Voytas, 2001)
-
1
For each genomic DNA sample, set up one reaction with primers amplifying a single cut site and another reaction with primers spanning the deletion. As controls, set up a PCR reaction with genomic DNA from a wild‐type population and a PCR reaction with genomic DNA from a mixed population transduced/transfected with the two guides. Follow the manufacturer's instructions to perform PCR with the Taq 2× Master Mix.
-
2
After the PCR reactions are complete, run the PCR products on a 2% agarose gel.
If the DNA region between the cut sites was removed, then a PCR product would be expected in the reactions with deletion primers and not in the reactions with the cut‐site primers. The presence of a reaction product in the cut‐site primer lane indicates that not all copies of the intervening genetic material have been excised.
Basic Protocol 8. CHARACTERIZING CRISPR‐INDUCED MUTATIONS BY SEQUENCING
If the researcher chooses the single‐guide knockout approach, then demonstrating the presence of an indel mutation at the guide RNA cut site is strong evidence that the targeted gene has been knocked out. (Note that this approach is also appropriate if two gRNAs are used but no full deletions are detected.) Cut‐site analysis can be performed by PCR‐amplifying the region targeted by the guide RNA and then subjecting the amplicon to Sanger sequencing. In the event that the lesion is found to be heterozygous, then TOPO cloning can be applied to sequence individual alleles found within the clone.
For this approach, first isolate genomic DNA from each putative knockout clone and design PCR primers to amplify a ∼200‐ to 400‐bp region around the cut site, as described above. Then, perform PCR and sequence the amplicon:
Materials
Genomic DNA (Support Protocol 4)
Primers
2× Taq Master Mix (NEB; cat. no. M0270)
NucleoSpin® Gel and PCR Clean‐up (Macherey‐Nagel; cat. no. 740609.50)
Nuclease‐free water
Gel electrophoresis equipment
Thermocycler
0.2‐ml thin‐walled PCR tubes
1.5‐ml microcentrifuge tube
Additional reagents and equipment for agarose gel electrophoresis (Current Protocols article: Voytas, 2001)
-
1
To set up a PCR reaction, follow the manufacturer's protocol for PCR guidelines and thermocycler settings for the 2× Taq master mix. As a control, set up a PCR reaction using the genomic DNA from a wild‐type population.
-
2
Run the PCR product on a 2% gel. Verify that a band of the correct size is present.
-
3
Using the NucleoSpin® gel and PCR clean‐up, isolate the PCR fragments from the mixture.
-
4
Set up Sanger sequencing reactions according to the sequencing facility's guidelines. The forward and reverse primers used for PCR can also be applied for sequencing.
-
5
Compare sequences between the wild‐type population and each clone using standard chromatogram analysis software. Indels should be observed at or near the nuclease‐targeted site.
Basic Protocol 9. CHARACTERIZING HETEROZYGOUS MUTATIONS BY TOPO CLONING AND SEQUENCING
If Sanger sequencing reveals that the CRISPR‐induced lesion is heterozygous (as evidenced by the presence of overlapping bases in the chromatogram; Figure 3C), the alleles can be deconvolved by TOPO cloning and sequencing. First, the individual alleles are cloned into the TOPO vector and transformed into E. coli. Next, single colonies are picked and the plasmids are reisolated by miniprep and then sequenced individually. In this way, researchers can confirm the presence of indel mutations in each allele in a targeted cell line.
Note that the Taq polymerase used for PCR in this protocol leaves an adenine overhang at the 3′ end of the PCR product. As a result, the TOPO TA kit (Thermo Fisher Scientific; cat. no. 450071), in which the vector contains a T overhang, should be used for TOPO cloning of these amplicons. If a different polymerase with proofreading activity is used for PCR, then the TOPO blunt‐end kit (cat. no. 450159) should be used instead.
Materials
PCR products from cut‐site PCR reactions (Basic Protocol 8)
TOPO™ TA Cloning™ Kit for Sequencing, without competent cells (cat. no. 450071)
Competent Stbl3 E. coli cells (Support Protocol 2)
Nuclease‐free water
LB + 50 µg/ml kanamycin culture plates
LB + 50 µg/ml kanamycin medium
QIAprep Spin Miniprep Kit (Qiagen; cat. no. 27106)
0.2‐ml thin‐walled PCR tubes
Thermocycler
-
1
Ligate the products from cut‐site PCR into TOPO plasmids according to the manufacturer's guidelines.
-
2
Add 2‐3 µl of the ligation product to an aliquot of Stbl3 E. coli cells. Incubate the cells on ice for 5 min and then plate the E. coli cells on LB + kanamycin plates. Allow the plates to grow at 37°C overnight.
-
3
The next day, pick 15 colonies from the plates and grow each of them in 2 ml LB + kanamycin medium.
-
4
Isolate the plasmids from the E. coli cells using the QIAprep Spin Miniprep Kit.
-
5
Set up Sanger sequencing reactions according to the sequencing facility's guidelines. The forward primer or M13 forward primer from the TOPO cloning kit can be used to sequence the plasmids. Set up a sequencing reaction using the PCR product from the wild‐type population as well.
-
6
Analyze the resulting alleles using standard chromatogram analysis software. Indels should be observed at or near the nuclease targeted site.
REAGENTS AND SOLUTIONS
Solution B (HEPES‐buffered saline)
280 mM NaCl
1.5 mM anhydrous Na2HPO4
50 mM HEPES
12 mM dextrose or glucose
10 mM KCl
Dilute solution to 950 ml with ddH2O
Adjust pH to 7 with 1 M NaOH
Bring final volume up to 1 L with ddH2O
Adjust pH to 7 with 1 M NaOH
H2O to 1 liter
COMMENTARY
Background Information
A common approach to learn the function of a gene is to probe the phenotype of a cell or organism in which that gene has been eliminated. For instance, the generation and characterization of the Yeast Deletion Collection, a set of yeast strains among which every nonessential gene has been knocked out, has been a powerful tool for investigating gene function in a simple eukaryote (Giaever & Nislow, 2014). Until the development of CRISPR technology, applying this approach to mammalian cells was extremely difficult. Earlier methods for ablating gene activity included approaches using zinc‐finger nucleases (ZFNs) and transcription‐activator‐like effector nucleases (TALENs), both of which contain a DNA binding domain fused to the FokI nuclease. Although these approaches are capable of generating a DSB at a specific locus, both ZFNs and TALENs require complicated re‐engineering of the DNA‐binding domains to target different DNA sequences. Thus, their versatility and utility are extremely limited (Gaj, Gersbach, & Barbas, 2013).
The development of RNA interference (RNAi) technology in the early 2000s provided an additional approach for rapid analysis of gene function in mammalian cells (Kim & Rossi, 2008). With RNAi, small interfering RNAs (siRNAs) or short hairpin RNAs (shRNAs) could be easily generated and introduced into mammalian cells to knock down the expression of a targeted gene. RNAi technology was harnessed to conduct the first whole‐genome screens in human cells, and these methods greatly advanced the understanding of mammalian biology (Kittler & Buchholz, 2005; Kittler et al., 2004; Mukherji et al., 2006; Nagaoka‐Yasuda, Matsuo, Perkins, Limbaeck‐Stokin, & Mayford, 2007; Whitehurst et al., 2007). However, experiments conducted with RNAi are limited in multiple ways. First, RNAi represents a “partial,” rather than a “total,” loss‐of‐function (LOF) perturbation, as RNAi constructs decrease protein expression but cannot fully eliminate the targeted gene. This feature of RNAi may be especially problematic when studying enzymes or other proteins that can tolerate significant changes in gene expression while still maintaining wild‐type levels of activity. Secondly, careful experiments have revealed that RNAi constructs are highly prone to off‐target knockdowns that can confound the characterization of a gene of interest. Certain results obtained with RNAi have been called into question after further analysis revealed that the gene ostensibly targeted in an experiment was not the cause of the resultant phenotype (Huang et al., 2017; Hübner et al., 2009; Putzbach et al., 2017). It is important to note, as discussed below, that CRISPR may also result in off‐target activity, yet these off‐target activity appears to be less promiscuous than that of RNAi (Boettcher & McManus, 2015). Lastly, RNAi‐induced knockdowns are dependent on the continuous expression of the RNAi construct. shRNAs transduced into a cell population can be lost or epigenetically silenced, allowing target re‐expression and complicating the use of these cells in long‐term experiments (Ellis, 2005).
The CRISPR system provides several advantages for perturbing mammalian gene function compared to either FokI‐nuclease‐ or RNAi‐mediated approaches. Unlike ZFNs and TALENs, CRISPR is highly versatile, as targeting a new region in a cell line of interest requires only cloning and expressing a ∼20‐bp oligonucleotide in a guide RNA vector. Unlike RNAi, CRISPR has the potential to generate complete loss‐of‐function perturbations, bypassing the problems inherent in studying a partial‐LOF phenotype. Head‐to‐head comparisons have also revealed that CRISPR constructs exhibit fewer off‐target effects than RNAi constructs (Smith et al., 2017), a difference that may stem from the more stringent binding and cutting requirements for Cas9 (Boettcher & McManus, 2015). Finally, CRISPR has the potential to induce permanent and stable loss‐of‐function alterations, allowing long‐term analysis of a modified cell line.
CRISPR can be applied to perturb mammalian gene function in several ways. CRISPR‐mediated homology‐directed repair can be used to replace a wild‐type allele with a mutation of interest, as has been described elsewhere (Ran et al., 2013). Alternately, if the goal of an experiment is simply to eliminate gene function, this can be achieved by targeting Cas9 to that locus and relying on the cell's NHEJ pathway to repair the DSB with indel mutations. A population of cells can be transfected or transduced with a CRISPR construct at a high multiplicity of infection (MOI), leading to the development of mutations in a majority of cells. However, this approach is likely to generate a heterogeneous cell population: some cells may totally escape target modification, and certain mutations (e.g., in‐frame indels) may be tolerated without compromising protein function. In this protocol, we instead describe an approach to generate CRISPR‐modified clones derived from single cells. This strategy allows the construction of cell lines in which every individual cell harbors the same set of total LOF (or null) mutations in a gene of interest.
These CRISPR‐knockout cell lines are suitable for multiple downstream applications and can be used to study fundamental questions in cell biology, genetics, and cancer biology. In one recent study, for instance, CRISPR was used to generate isogenic human cell line models in which different DNA repair genes had been deleted (Zou et al., 2018). These lines were allowed to grow in culture for up to 1 month, and the resulting “mutational signatures” caused by each knockout were determined by whole‐genome sequencing. These patterns of hypermutation were then compared to the mutational signatures found in human tumors, allowing the researchers to experimentally verify the genetic underpinnings of these motifs. In another recent study, CRISPR was used to identify the transporter that imports the amino acid serine into mitochondria (Kory et al., 2018). These researchers first performed a CRISPR screen to identify guide RNAs that cause poor growth specifically in serine‐deficient media. The researchers then generated cell line knockouts of SFXN1, the screen's top hit, and discovered that these lines were deficient in mitochondrial serine import. These examples illustrate how CRISPR‐induced knockouts allow a precise interrogation of the relationship between genotype and phenotype, shedding light on the genetic architecture of mammalian cells.
CRISPR‐generated knockout cell lines are also useful for investigating the specificity of drugs, antibodies, and other biological reagents. For example, a series of experiments using RNAi suggested that multiple cancer types required the expression of the maternal embryonic leucine zipper kinase (MELK) in order to proliferate (Jiang & Zhang, 2013). Based on these preclinical results, one company developed a small‐molecule inhibitor of MELK and launched multiple clinical trials to treat cancer patients with this compound (Chung et al., 2012). However, subsequent work from our lab and the Gray lab at the Dana‐Farber Institute demonstrated that cancer cells tolerated CRISPR‐induced mutations in MELK with no loss in fitness (Giuliano et al., 2018; Huang et al., 2017; Lin et al., 2017). Thus CRISPR knockouts can be used to validate findings obtained using other protein‐depletion approaches. CRISPR knockouts have also been analyzed to test the specificity of MTH1 inhibitors (Kettle et al., 2016), CD95L‐targeting shRNAs (Putzbach et al., 2017), Sema4B‐targeting shRNAs (Peretz et al., 2018), and an increasing number of antibodies (Thermo Fisher, 2018; Origene, 2018; Abcam, 2018a). CRISPR knockouts are therefore a key laboratory tool for validating reagents and conducting reproducible biological research.
Nonetheless, there are multiple potential pitfalls that can interfere with the generation and analysis of CRISPR knockout cell lines. First, some CRISPR‐induced mutations can lead to incomplete target ablation via exon skipping and/or nonsense‐associated alternative splicing (NAS). In these poorly understood processes, the presence of a nonsense mutation causes cells to produce an alternative transcript that bypasses the CRISPR‐induced mutation. Although the resulting transcripts may differ from the dominant isoform expressed in a cell line, they may still be sufficient to carry out the function of the protein (Mou et al., 2017; Rodriguez‐Rodriguez et al., 2018). Secondly, while CRISPR‐mediated mutagenesis can effectively ablate the targeted locus, the resulting indel can itself have unintended consequences. Long‐read sequencing has recently shown that Cas9‐induced DSBs can cause large deletions and complex rearrangements that span multiple kilobases, potentially interfering with the expression or regulation of nearby genes (Kosicki, Tomberg, & Bradley, 2018). Finally, challenging a cell line to repopulate itself from a single progenitor cell represents a significant genetic bottleneck and may also select for certain genetic alterations. For instance, it has been reported that knocking out genes in a nontransformed cell line can select for p53 deficiencies, potentially confounding downstream analysis (Haapaniemi, Botla, Persson, Schmierer, & Taipale, 2018). It is important to be aware of these potential genetic compensatory mutations, especially when attempting to knock out an essential protein.
In this article, we describe a number of methods that can minimize—but not eliminate—these potential limitations of CRISPR‐mediated knockout generation. First, as a means to avoid incomplete protein ablation caused by exon skipping or NAS, we describe a two‐guide system to independently target two distinct exons. Secondly, we recommend designing guide RNAs to target conserved functional domains of the protein of interest, thereby increasing the likelihood that a mutation will compromise protein function (Shi et al., 2015). Thirdly, to address the potential problems caused by evolution and variability during single‐cell clonal expansion, we suggest the derivation of multiple independent knockout clones and the use of appropriate controls during experimental design.
Critical Parameters
The CRISPR/Cas9 system is a powerful tool for rapidly generating knockout cell lines. These lines have already proven instrumental in a number of recent publications by allowing researchers to determine the function of uncharacterized genes, analyze knockouts in isogenic backgrounds, and validate the activity of small‐molecule inhibitors and RNAi constructs (Giuliano et al., 2018; Huang et al., 2017; Kory et al., 2018; Lin et al., 2017; Zou et al., 2018). Nonetheless, there are several important considerations to take into account when generating and deploying knockout cell lines in experimental settings.
First, it is important to consider the genetic, epigenetic, and phenotypic heterogeneity that may be present within a population of cells prior to single‐cell isolation. Rederiving a cell line from a single cell represents a significant genetic bottleneck, and experiments may be confounded if a single cell–derived clone captures only a subset of the diversity present within the starting cell population. Indeed, some established cell lines are well known to exhibit significant interclonal heterogeneity; for instance, we and others have observed sizeable variation in proliferative capacity among clones derived from the widely used MDA‐MB‐231 breast cancer cell line (Giuliano et al., 2018; Khan, Kim, Shin, & Lee, 2017; Lin et al., 2017). Because of the possibility of parental heterogeneity, it is absolutely crucial that researchers analyze multiple independent knockout cell lines in order to ensure that any unexpected result is not a clonal artifact.
Secondly, challenging a single cell to proliferate to form a clonal population of millions of cells represents a significant selective pressure. This pressure could conceivably enrich for certain genetic and epigenetic alterations within the growing clone that promote proliferation. To account for this selective force, it is important to compare experimental knockouts with clonal “control” cell lines that were derived under similar conditions. These control cells should also harbor Cas9 and/or gRNAs targeting established nonessential loci to account for any nonspecific reaction to CRISPR‐induced DNA damage. In our experience, guides targeting confirmed nonessential loci have a slight but significant effect when compared to guides that lack any target sites in the genome. As a result, we recommend against using nontargeting guide RNAs as experimental controls, and instead suggest that control guide RNAs be designed to cut at an established safe‐harbor locus in the genome, such as AAVS1 or Rosa26 (Papapetrou & Schambach, 2016).
As an alternate approach to minimize the effects of cell cloning, or to verify an unexpected result, researchers may consider conducting bulk CRISPR assays in parallel to generating clonal knockout cell lines. In bulk assays, gRNAs can be introduced into the recipient cell line by transfection or transduction, as described above. If enough cells express the guide RNA of interest, an assay can be conducted directly with that population. Otherwise, FACS can be used to isolate a bulk population of cells harboring that guide, and that population can be subjected to genetic and phenotypic analysis. Rescue experiments—in which cells are generated that express a guide RNA‐resistant allele of the gene of interest—represent an alternate method to rule out clonal artifacts and to ensure that a phenotype results from on‐target activity. Importantly, through the use of CRISPR‐mediated HDR, gRNA‐resistance mutations can be introduced directly into the locus of interest, bypassing problems that could potentially result from the transgenic overexpression of resistant cDNA.
In sum, this protocol describes an effective strategy for generating knockout cell lines with CRISPR/Cas9. As the CRISPR system becomes increasingly ubiquitous, CRISPR‐generated knockout cell lines will represent an essential approach to characterizing gene function and exploring the activity of biological reagents.
Troubleshooting
In this section, we describe common problems, their likely causes, and appropriate corrective steps.
No plasmids with the correct gRNA sequence are recovered following cloning
In our experience, the most common cause of guide RNA cloning problems is incomplete excision of the BsmBI filler sequence. That is, ampicillin‐resistant colonies are recovered following transformation, but sequencing reveals that they contain undigested vector with no gRNA insert. If this occurs, the BsmBI digestion can be performed overnight to ensure complete spacer excision. Alternately, the use of a new stock of restriction enzyme may alleviate the problem. Additionally, the researchers should ensure that the agarose gel has been run long enough to separate the digested fragment from any undigested vector before performing the gel‐extraction step.
If no bacterial colonies are recovered after transformation, we recommend double‐checking the oligonucleotide sequences to ensure that the BsmBI cut‐site overhangs have been added correctly. Additionally, the ligation reaction can be extended from 30 min to 1 hr or longer.
Failure to generate lentivirus
If the lentiviral titer generated is low, several steps can be taken to increase virus yield. First, HEK293T cells that have been left in culture for too long or under suboptimal growth conditions may fail to produce sufficient lentivirus. Using freshly thawed low‐passage HEK293T cells can help increase viral titer. Second, the quality of transfection reagents can impact lentiviral titer: optimizing transfection by monitoring the expression of a GFP‐expressing plasmid, in the absence of lentiviral production, can help identify problems with transfection reagents. Remaking these solutions can often eliminate these problems. Finally, plasmid quality is highly important: researchers should ensure that the lentiviral and CRISPR plasmid preparations are pure, with high concentrations (>500 ng/µl), an A 260/A 280 ratio of ∼1.8, and an A 260/A 230 ratio of ∼2.0.
Cells fail to grow during single‐cell expansion
The plating of single cells can present a very challenging environment for an individual cell to grow. If the researchers are agnostic to cell line identity, they can simply choose to generate knockout clones in a cell line that exhibits consistently high clonogenicity (HeLa, HCT116, A375, DLD1, Cal51). Alternately, if cell line identity is important, there are a number of modifications that can greatly improve a cell line's clonogenic capacity. First, simply doubling the concentration of serum is sufficient to promote single‐cell growth in some cell lines. If this fails, cell line–conditioned medium can be used to supplement growth. To generate conditioned medium, seed ∼106 cells from the chosen cell line onto a 10‐cm plate. The next day, collect the supernatant, spin it down, pass it through a 0.45‐µm filter, and then use it to feed the plate on which the single cells will be deposited.
If each of the above methods have been tried and no successful knockout clones are identified, it is possible that the target gene is essential. We recommend using tools such as DepMap (https://depmap.org/portal/) to verify whether your target gene is required in a particular cell line. If the gene of interest is indeed necessary for cell survival, the generation of knockout clones may not be possible.
The protein of interest cannot be detected by western
If western blot analysis fails to detect any protein presence in control samples when tested with multiple antibodies, it is possible that the protein of interest is not actually expressed in the tested cell line. We recommend consulting the Cancer Cell Line Encyclopedia or using qRT‐PCR prior to single‐cell cloning to ensure that the target gene is expressed in the cell line of interest (Barretina et al., 2012).
No knockout clones are recovered
If western blot analysis fails to reveal any apparent knockout clones, this may simply indicate that the antibody used is nonspecific for its reported target. In this case, we would suggest that researchers investigate the DNA‐based validation approaches described above. Redoing the western blots with other antibodies (particularly antibodies that have previously been knockout‐validated) may also identify true knockout clones.
If DNA‐based validation indicates that the guide RNA cut site lacks any detectable mutations, this may indicate that the cell line did not express sufficient levels of Cas9 expression or that the guide RNA was not active. If this experiment was done in a stable, Cas9‐expressing cell line, then the researchers should test Cas9 expression (via qPCR or western blot) or confirm its activity using a validated guide RNA. Testing alternative guide RNAs may also allow successful knockout generation.
Finally, if multiple guide RNAs are tested and no knockout clones can be derived, it may be the case that the targeted gene is essential for cell survival and thus knockout generation is not possible. The derivation of many heterozygous clones may indicate that the CRISPR system is working effectively, but complete knockout of the gene of interest is lethal.
Knockout clones lack any detectable phenotype or display an unexpected phenotype
Single cell–derived clones can exhibit significant clonal variability, particularly in genomically unstable cell lines. It is always best to generate and analyze multiple knockout clones, if possible using different guide RNA combinations. In order to confirm that an unexpected phenotype is a true consequence of the loss of the targeted gene, rescue experiments can be performed by expressing guide RNA–resistant cDNAs in each clone. If no identifiable phenotype is detected, the researchers may wish to investigate the effects of ablating gene function without single‐cell cloning (for instance, by transducing high‐titer gRNA lentivirus and analyzing the population that results). Alternately, it is possible that the targeted gene is truly dispensable for the expected phenotype in the cell line being tested.
Understanding Results
Verified knockout clones should demonstrate gene knockout according to both gene evidence by PCR and Sanger sequencing and protein evidence by western blotting, as shown in Figures 2 and 3.
Western blot analysis remains the gold standard for verifying protein knockout. If an antibody detects the expression of a protein in a wild‐type or control clone but fails to detect the expression of that same protein in a putative knockout clone, that is strong evidence that the targeted protein has been ablated. However, a number of factors may confound this analysis. First, a CRISPR‐induced lesion may ablate the antigen specifically recognized by an antibody without eliminating all protein expression. This could lead to a false‐positive result, in which a western blot seemingly confirms the absence of a target although it remains present in the cell line. Secondly, alternative splicing or downstream translational initiation could lead to the production of functional protein fragments. These smaller isoforms may be dismissed as degradation products on a western blot or may not be detected if the blot is cut based on the expected size of the wild‐type gene product. Thirdly, many commercially available antibodies are nonspecific for their reported targets, potentially confounding western blot analysis (Berglund et al., 2008; Voskuil, 2014). Many of these problems can be overcome by targeting genes with multiple independent guide RNAs and validating knockouts with more than one antibody. For instance, by assessing protein expression with antibodies raised against different antigens present within the target, one can effectively rule out the possibility that the gene still produces functional protein fragments. It may require testing three to six antibodies to identify two to three that are suitable for western blotting, specific for the targeted protein, and that recognize unique antigens. These concerns aside, standard western blotting protocols, as published elsewhere, are generally sufficient for this analysis (Abcam, 2018b; JoVE Science Education Database, 2018; Current Protocols article: Gallagher et al., 2008).
If a cell line is transfected or transduced with two independent guide RNAs that target the same gene, recombination between these lesions will occasionally cause the deletion of the intervening genomic DNA. The frequency of this event will vary based on the distance between the cut sites, the local chromatin context, the identity of the specific cell line, and several other factors. Nonetheless, using two guides to eliminate multiple exons in a targeted gene is a good strategy to ensure complete target ablation. At the same time, these large deletions may be more likely to affect the expression of nearby genes and could also eliminate intronic microRNAs, transcription‐factor binding sites, or other important regulatory elements. As we describe, recombination between two distant cut sites can be easily detected by PCR using one primer that is upstream of the 5′ cut site and another primer that is downstream of the 3′ cut site. A second set of primers that recognize sequences between the cut sites can be used to differentiate between clones that are homozygous and heterozygous for the deletion event. This approach can be easily applied to screen a large number of clones and represents a reliable, antibody‐independent approach to identify knockouts.
Time Considerations
Generating the cloned plasmids will take 2 days, followed by another 2 to 3 days to verify the insert by sequencing. Time considerations for the introduction of the CRISPR system to the cell line of interest depend on the methodology chosen: 2 days are required to generate any lentivirus; generation of stable Cas9 expressing cell line may take 1 to 2 weeks depending on selection efficiency; transduction or transfection of a gRNA vector to a recipient cell line will take 2 to 3 days. Expansion of single‐cell clones is often the lengthiest part of the process, with single‐cell expansion rates ranging from 3 to 6 weeks depending on the proliferation rate of the cell line. Subsequent knockout validation assays (western blotting, PCR, Sanger sequencing) all take 1 to 3 days, assuming that the reagents have previously been optimized.
Acknowledgments
We thank Chris Vakoc (Cold Spring Harbor Laboratory) for sharing cell lines and plasmids used in this work. Research in the Sheltzer Laboratory is supported by an NIH Early Independence Award (1DP5OD021385), a Breast Cancer Alliance Young Investigator Award, a Damon Runyon–Rachleff Innovation Award, a Gates Foundation Innovative Technology Solutions grant, and a CSHL‐Northwell Translational Cancer Research Grant. This work was performed with assistance from the CSHL Flow Cytometry Shared Resource, which is supported by the CSHL Cancer Center Support Grant 5P30CA045508. C.J.G. and A.L. are supported by U.S. National Science Foundation Graduate Research Fellowships.
Giuliano, C. J. , Lin, A. , Girish, V. , & Sheltzer, J. M. (2019). Generating single cell–derived knockout clones in mammalian cells with CRISPR/Cas9. Current Protocols in Molecular Biology, 128, e100. doi: 10.1002/cpmb.100
Literature Cited
- Abcam . (2018a, accessed December 26). Knockout (KO) validation results. Retrieved from https://www.abcam.com/primary‐antibodies/knockout‐ko‐validation‐results.
- Abcam . (2018b, accessed December 31.) Western blot protocol. Retrieved from https://www.abcam.com/protocols/general‐western‐blot‐protocol.
- Al‐Allaf, F. A. , Tolmachov, O. E. , Zambetti, L. P. , Tchetchelnitski, V. , & Mehmet, H. (2013). Remarkable stability of an instability‐prone lentiviral vector plasmid in Escherichia coli Stbl3. 3 Biotech, 3, 61–70. doi: 10.1007/s13205-012-0070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou, R. , Fremaux, C. , Deveau, H. , Richards, M. , Boyaval, P. , Moineau, S. , … Horvath, P. (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science, 315, 1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Barretina, J. , Caponigro, G. , Stransky, N. , Venkatesan, K. , Margolin, A. A. , Kim, S. , … Garraway, L. A. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 483, 603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berglund, L. , Björling, E. , Oksvold, P. , Fagerberg, L. , Asplund, A. , Szigyarto, C. A.‐K. , … Uhlén, M. (2008). A genecentric human protein atlas for expression profiles based on antibodies. Molecular & Cellular Proteomics, 7, 2019–2027. [DOI] [PubMed] [Google Scholar]
- Boettcher, M. , & McManus, M. T. (2015). Choosing the right tool for the job: RNAi, TALEN or CRISPR. Molecular Cell, 58, 575–585. doi: 10.1016/j.molcel.2015.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman, E. K. , Chen, T. , de Haas, M. , Holland, H. A. , Akhtar, W. , & van Steensel, B. (2018). Kinetics and fidelity of the repair of Cas9‐induced double‐strand DNA breaks. Molecular Cell, 70, 801–813.e6. doi: 10.1016/j.molcel.2018.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceasar, S. A. , Rajan, V. , Prykhozhij, S. V. , Berman, J. N. , & Ignacimuthu, S. (2016). Insert, remove or replace: A highly advanced genome editing system using CRISPR/Cas9. Biochimica et Biophysica Acta Molecular Cell Research, 1863, 2333–2344. doi: 10.1016/j.bbamcr.2016.06.009. [DOI] [PubMed] [Google Scholar]
- Chang, K. , Marran, K. , Valentine, A. , & Hannon, G. J. (2013). Packaging shRNA retroviruses. Cold Spring Harbor Protocols, 2013, 734–737. doi: 10.1101/pdb.prot076448. [DOI] [PubMed] [Google Scholar]
- Chung, S. , Suzuki, H. , Miyamoto, T. , Takamatsu, N. , Tatsuguchi, A. , … Matsuo, Y. (2012). Development of an orally‐administrative MELK‐targeting inhibitor that suppresses the growth of various types of human cancer. Oncotarget, 3, 1629–1640. doi: 10.18632/oncotarget.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong, L. , Ran, F. A. , Cox, D. , Lin, S. , Barretto, R. , Habib, N. , … Zhang, F. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis, J. (2005). Silencing and variegation of gammaretrovirus and lentivirus vectors. Human Gene Therapy, 16, 1241–1246. doi: 10.1089/hum.2005.16.1241. [DOI] [PubMed] [Google Scholar]
- Farboud, B. , Jarvis, E. , Roth, T. L. , Shin, J. , Corn, J. E. , Marson, A. , … Hochstrasser, M. L. (2018). Enhanced genome editing with Cas9 ribonucleoprotein in diverse cells and organisms. Journal of Visualized Experiments, Retrieved from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6101420/. doi: 10.3791/57350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj, T. , Gersbach, C. A. , & Barbas, C. F. (2013). ZFN, TALEN and CRISPR/Cas‐based methods for genome engineering. Trends in Biotechnology, 31, 397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher, S. , Winston, S. E. , Fuller, S. A. , & Hurrell, J. G. (2008). Immunoblotting and immunodetection. Current Protocols in Molecular Biology, 83, 10.8.1–10.8.28. doi: 10.1002/0471142727.mb1008s83. [DOI] [PubMed] [Google Scholar]
- Giaever, G. , & Nislow, C. (2014). The yeast deletion collection: A decade of functional genomics. Genetics, 197, 451–465. doi: 10.1534/genetics.114.161620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliano, C. J. , Lin, A. , Smith, J. C. , Palladino, A. C. , & Sheltzer, J. M. (2018). MELK expression correlates with tumor mitotic activity but is not required for cancer growth. eLife, 7, e32838. doi: 10.7554/eLife.32838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, D. B. , & Root, D. E. (2015). Resources for the design of CRISPR gene editing experiments. Genome Biology, 16, 260. doi: 10.1186/s13059-015-0823-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapaniemi, E. , Botla, S. , Persson, J. , Schmierer, B. , & Taipale, J. (2018). CRISPR–Cas9 genome editing induces a p53‐mediated DNA damage response. Nature Medicine, 24, 927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- Huang, H.‐T. , Seo, H.‐S. , Zhang, T. , Wang, Y. , Jiang, B. , Li, Q. , … Gray, N. S. (2017). MELK is not necessary for the proliferation of basal‐like breast cancer cells. eLife, 6, e26693. doi: 10.7554/eLife.26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hübner, N. C. , Wang, L. H.‐C. , Kaulich, M. , Descombes, P. , Poser, I. , & Nigg, E. A. (2009). Re‐examination of siRNA specificity questions role of PICH and Tao1 in the spindle checkpoint and identifies Mad2 as a sensitive target for small RNAs. Chromosoma, 119, 149–165. doi: 10.1007/s00412-009-0244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, P. , & Zhang, D. (2013). Maternal embryonic leucine zipper kinase (MELK): A novel regulator in cell cycle control, embryonic development, and cancer. International Journal of Molecular Sciences, 14, 21551–21560. doi: 10.3390/ijms141121551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JoVE Science Education Database . (2018, accessed December 2018). The western blot. Retrieved from https://www.jove.com/science‐education/5065/the‐western‐blot.
- Kettle, J. G. , Alwan, H. , Bista, M. , Breed, J. , Davies, N. L. , Eckersley, K. , … Wylot, M. (2016). Potent and selective inhibitors of MTH1 probe its role in cancer cell survival. Journal of Medicinal Chemistry, 59, 2346–2361. doi: 10.1021/acs.jmedchem.5b01760. [DOI] [PubMed] [Google Scholar]
- Khan, G. N. , Kim, E. J. , Shin, T. S. , & Lee, S. H. (2017). Heterogeneous cell types in single‐cell‐derived clones of MCF7 and MDA‐MB‐231 cells. Anticancer Research, 37, 2343–2354. doi: 10.21873/anticanres.11572. [DOI] [PubMed] [Google Scholar]
- Kim, D. H. , & Rossi, J. J. (2008). RNAi mechanisms and applications. BioTechniques, 44, 613–616. doi: 10.2144/000112792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittler, R. , & Buchholz, F. (2005). Functional genomic analysis of cell division by endoribonuclease‐prepared siRNAs. Cell Cycle, 4, 564–567. doi: 10.4161/cc.4.4.1608. [DOI] [PubMed] [Google Scholar]
- Kittler, R. , Putz, G. , Pelletier, L. , Poser, I. , Heninger, A.‐K. , Drechsel, D. , … Buchholz, F. (2004). An endoribonuclease‐prepared siRNA screen in human cells identifies genes essential for cell division. Nature, 432, 1036–1040. doi: 10.1038/nature03159. [DOI] [PubMed] [Google Scholar]
- Komor, A. C. , Badran, A. H. , & Liu, D. R. (2017). CRISPR‐based technologies for the manipulation of eukaryotic genomes. Cell, 168, 20–36. doi: 10.1016/j.cell.2016.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kory, N. , Wyant, G. A. , Prakash, G. , uit de Bos, J. , Bottanelli, F. , Pacold, M. E. , … Sabatini, D. M. (2018). SFXN1 is a mitochondrial serine transporter required for one‐carbon metabolism. Science, 362, eaat9528. doi: 10.1126/science.aat9528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosicki, M. , Tomberg, K. , & Bradley, A. (2018). Repair of double‐strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nature Biotechnology, 36, 765–771. doi: 10.1038/nbt.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, M. , Keller, B. , Makalou, N. , & Sutton, R. E. (2001). Systematic determination of the packaging limit of lentiviral vectors. Human Gene Therapy, 12, 1893–1905. doi: 10.1089/104303401753153947. [DOI] [PubMed] [Google Scholar]
- Lalonde, S. , Stone, O. A. , Lessard, S. , Lavertu, A. , Desjardins, J. , Beaudoin, M. , … Lettre, G. (2017). Frameshift indels introduced by genome editing can lead to in‐frame exon skipping. PLOS One, 12, e0178700. doi: 10.1371/journal.pone.0178700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, A. , Giuliano, C. J. , Sayles, N. M. , & Sheltzer, J. M. (2017). CRISPR/Cas9 mutagenesis invalidates a putative cancer dependency targeted in on‐going clinical trials. eLife, 6, e24179. doi: 10.7554/eLife.24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier, J. A. , Zhang, F. , & Sanjana, N. E. (2017). GUIDES: SgRNA design for loss‐of‐function screens. Nature Methods, 14, 831–832. doi: 10.1038/nmeth.4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou, H. , Smith, J. L. , Peng, L. , Yin, H. , Moore, J. , Zhang, X.‐O. , … Xue, W. (2017). CRISPR/Cas9‐mediated genome editing induces exon skipping by alternative splicing or exon deletion. Genome Biology, 18, 108. doi: 10.1186/s13059-017-1237-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherji, M. , Bell, R. , Supekova, L. , Wang, Y. , Orth, A. P. , Batalov, S. , … Schultz, P. G. (2006). Genome‐wide functional analysis of human cell‐cycle regulators. Proceedings of the National Academy of Sciences of the United States of America, 103, 14819–14824. doi: 10.1073/pnas.0604320103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaoka‐Yasuda, R. , Matsuo, N. , Perkins, B. , Limbaeck‐Stokin, K. , & Mayford, M. (2007). An RNAi‐based genetic screen for oxidative stress resistance reveals retinol saturase as a mediator of stress resistance. Free Radical Biology & Medicine, 43, 781–788. [DOI] [PubMed] [Google Scholar]
- Origene . (2018, accessed December 26). Knockout (KO) validation. Retrieved from https://www.origene.com/products/antibodies/primary‐antibodies/ko‐validated‐antibodies.
- Papapetrou, E. P. , & Schambach, A. (2016). Gene insertion into genomic safe harbors for human gene therapy. Molecular Therapy, 24, 678–684. doi: 10.1038/mt.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz, L. , Besser, E. , Hajbi, R. , Casden, N. , Ziv, D. , Kronenberg, N. , … Behar, O. (2018). Combined shRNA over CRISPR/cas9 as a methodology to detect off‐target effects and a potential compensatory mechanism. Scientific Reports, 8, 93. doi: 10.1038/s41598-017-18551-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan, M. C. (1996). Technqiues for mammalian cell tissue culture. Current Protocols in Molecular Biology, 35, A.3F.1–A.3F.14. doi: 10.1002/0471142727.mba03fs35. [DOI] [PubMed] [Google Scholar]
- Popp, M. W. , & Maquat, L. E. (2016). Leveraging rules of nonsense‐mediated mRNA decay for genome engineering and personalized medicine. Cell, 165, 1319–1322. doi: 10.1016/j.cell.2016.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putzbach, W. , Gao, Q. Q. , Patel, M. , van Dongen, S. , Haluck‐Kangas, A. , Sarshad, A. A ., … Peter, M. E. (2017). Many si/shRNAs can kill cancer cells by targeting multiple survival genes through an off‐target mechanism. eLife, 6, e29702. doi: 10.7554/eLife.29702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, F. A. , Hsu, P. D. , Lin, C.‐Y. , Gootenberg, J. S. , Konermann, S. , Trevino, A. E. , … Zhang, F. (2013). Double nicking by RNA‐guided CRISPR Cas9 for enhanced genome editing specificity. Cell, 154, 1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez‐Rodriguez, J.‐A. , McKinley, K. L. , Sikirzhytski, V. , Corona, J. , Maciejowski, J. , Khodjakov, A. , … Jallepalli, P. (2018). The RZZ complex integrates spindle checkpoint maintenance with dynamic expansion of unattached kinetochores. bioRxiv, 297580 [Preprint]. Retrieved from https://www.biorxiv.org/content/10.1101/297580v1. [Google Scholar]
- Sharpe, J. J. , & Cooper, T. A. (2017). Unexpected consequences: Exon skipping caused by CRISPR‐generated mutations. Genome Biology, 18, 109. doi: 10.1186/s13059-017-1240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, J. , Wang, E. , Milazzo, J. P. , Wang, Z. , Kinney, J. B. , & Vakoc, C. R. (2015). Discovery of cancer drug targets by CRISPR‐Cas9 screening of protein domains. Nature Biotechnology, 33, 661–667. doi: 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, I. , Greenside, P. G. , Natoli, T. , Lahr, D. L. , Wadden, D. , Tirosh, I. , … Doench, J. G. (2017). Evaluation of RNAi and CRISPR technologies by large‐scale gene expression profiling in the Connectivity Map. PLOS Biology, 15, e2003213. doi: 10.1371/journal.pbio.2003213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thermo Fisher . (2018, accessed December 26). Knockout and knockdown antibody validation. Retrieved from https://www.thermofisher.com/us/en/home/life‐science/antibodies/invitrogen‐antibody‐validation/knockout‐knockdown‐antibody‐validation.html.
- Voskuil, J. (2014). Commercial antibodies and their validation. F1000Research, 3, 232. doi: 10.12688/f1000research.4966.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voytas, D. (2001). Agarose gel electrophoresis. Current Protocols in Molecular Biology, 51, 2.5A.1–2.5A.9. doi: 10.1002/0471142727.mb0205as51. [DOI] [PubMed] [Google Scholar]
- Whitehurst, A. W. , Bodemann, B. O. , Cardenas, J. , Ferguson, D. , Girard, L. , Peyton, M. , … White, M. A. (2007). Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature, 446, 815–819. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- Zou, X. , Owusu, M. , Harris, R. , Jackson, S. P. , Loizou, J. I. , & Nik‐Zainal, S. (2018). Validating the concept of mutational signatures with isogenic cell models. Nature Communications, 9, 1744. doi: 10.1038/s41467-018-04052-8. [DOI] [PMC free article] [PubMed] [Google Scholar]