

Abstract
Purpose
Next-generation sequencing (NGS) for tumor molecular profiling can reveal secondary germline likely pathogenic and pathogenic variants (LPV/PV). The American College of Medical Genetics and Genomics (ACMG) recommends return of secondary results for a subset of 59 genes, but other genes with evidence of clinical utility are emerging. We previously reported that 4.3% of patients who underwent NGS of a targeted panel of 201 genes had LPV/PV on the basis of the ACMG list. We report the frequency of additional germline cancer-related gene variants and discuss their clinical utility.
Patients and Methods
Matched tumor and germline DNA NGS of a targeted panel of 201 genes was performed in a research laboratory on samples from 1,000 patients with advanced or metastatic solid tumors enrolled in a molecular testing protocol (ClinicalTrials.gov identifier: NCT01772771). The frequency of germline LPV/PV in 54 cancer-related genes, beyond the genes in ACMG list, were analyzed.
Results
Among 1,000 patients who underwent tumor/normal DNA sequencing, 46 (4.6%) were found to have a germline LPV/PV in the following genes: AR (n = 5), ATM (n = 4), BAP1 (n = 1), CDH1 (n = 1), CDKN2A (n = 1), CHEK1 (n = 2), CHEK2 (n = 10), EGFR (n = 1), ERCC3 (n = 4), ERCC5 (n = 1), HNF1B (n = 1), HRAS (n = 1), MITF (n = 4), MLL3 (n = 1), NF1 (n = 3), PKHD1 (n = 4), PTCH1 (n = 1), and SMARCA4 (n = 1). Thus, 8.7% of patients had an LPV/PV, with two patients having two concomitant germline LPV/PV. Five mutations in high-penetrance hereditary cancer predisposition genes were selected to be returned to patients or their representatives: BAP1, CDH1, CDKN2A, EGFR, and SMARCA4.
Conclusion
Broader genomic testing is likely to identify additional secondary pathogenic germline alterations, some with potential clinical utility for return to patients and their relatives. The recommended genes for which germline results should be returned are continually changing, which warrants continued study.
INTRODUCTION
With the exponential development of next-generation sequencing (NGS), specifically the ability to sequence larger panels of genes in more depth, molecular profiling increasingly is being integrated into oncology practice.1,2 The main goal of NGS is to identify actionable genomic alterations in the tumor to target by matched drugs in efforts to personalize treatment.3 In addition, NGS can be used for testing of prognostic biomarkers of disease progression and metastasis, testing of cancer predisposition genes, and cancer risk assessment for at-risk asymptomatic family members. Sequencing matched tumor and normal tissue samples from the same patient can assist in more-accurate calling of somatic variants.4-6 Often, germline variants are subtracted and ignored; however, analysis of secondary germline findings might identify variants associated with an increased susceptibility to develop cancer or other diseases. The returning of these results has implications for patients and their families, and some of these secondary germline findings could offer matched therapeutic opportunities.
The American College of Medical Genetics and Genomics (ACMG) has recommended that laboratories that perform clinical sequencing report germline pathogenic variants (PV) in 59 genes, if covered by the tested panel, regardless of the indication for which the clinical sequencing was ordered.7,8 ASCO endorses the return of medically relevant secondary germline findings and encourages physicians to solicit patients’ preference with regard to the return of pathogenic germline alterations before testing.9 However, the systematic return of germline alterations found during NGS requires implementation of bioinformatics programs for variant detection, curation, and annotation; such implementation increases the required resources, time, and costs.9,10
In this study, we sought to determine whether NGS of matched tumor and normal samples revealed secondary germline PV and likely pathogenic variants (LPV) in genes beyond those currently recommended by the ACMG. Accordingly, we assessed the prevalence of LPV/PV secondary germline findings in 54 additional cancer-related genes (having previously reported those recommended by ACMG11).
PATIENTS AND METHODS
Patients
Patients with locally advanced or metastatic solid tumors who exhausted standard treatment options were enrolled in the institutional review board–approved Clearinghouse protocol (ClinicalTrials.gov identifier: NCT01772771) using molecular profiling to assist with personalized cancer treatment at The University of Texas MD Anderson Cancer Center. Patients also were offered possible secondary germline mutation testing in a companion institutional review board–approved protocol.11 The patients’ relevant clinical characteristics were collected from electronic medical records and prospectively maintained institutional databases.
Matched Tumor/Normal DNA Sequencing
Paired tumor/normal DNA-targeted exome sequencing of 201 genes was performed in a research laboratory (Data Supplement).12 Tumor samples were acquired as formalin-fixed paraffin-embedded slides in which the tumor area was circled to facilitate macrodissection of the tumor-containing region. For normal DNA, blood or saliva was used. The key elements of NGS, including DNA extraction, library preparation, target enrichment, sequencing, and variant calling, were performed on tumor/normal tissue samples. In summary, genomic DNA was extracted using the QIAamp DNA FFPE Tissue Kit (QIAGEN, Hilden, Germany) according to the manufacturer’s protocol and quantified by Qubit assay (Invitrogen, Carlsbad, CA), and quality was assessed using Genomic DNA Screen Tape for the 2200 TapeStation (Agilent Technologies, Santa Clara, CA). The captured libraries were sequenced on a HiSeq 2000 sequencing system (Illumina, San Diego, CA). All regions were covered by more than 20 reads. Alignment of the sequenced data to human reference assembly hg19 and variant calling of the sequencing reads have been described previously.13-15 Single nucleotide variants and small indels were called using the Genome Analysis Tool Kit (Broad Institute, Cambridge, MA). We annotated the variants using variant effect predictor, annotate variation, sorting intolerant from tolerant, and polymorphism phenotyping. For each sample, we estimated an average allele fraction cutoff by averaging site-specific cutoffs over all the targeted sites. Loss of the normal allele in the tumor (loss of heterozygosity) was evaluated in the analyzed genes.
Selection of Genes for Analysis for Secondary Germline Findings
Our group previously published the germline LPV/PV results for 18 of 56 genes recommended by ACMG that were included in our 201-gene panel.11 PALB2 also was included in a previous secondary germline analysis because of its strong association with hereditary breast cancer.16 After a thorough literature search and evaluation of relevant databases,17-19 we selected 54 additional genes from our panel for which we analyzed germline LPV/PV. These genes were selected on the basis of the mode of inheritance and known penetrance for disease phenotype, including genes previously described as hereditary-cancer susceptibility; genes currently tested in commercially available hereditary panels; cancer-related genes often tested on matched normal/tumor panels, however with the mode of inheritance nonconsistent with phenotypic expression in patients; and noncancer-related genes with suggested familial inheritance (Data Supplement). On the basis of the mode of inheritance, known penetrance for a disease phenotype, and presence or absence of management guidelines, the identified LPV/PV were grouped into five categories (Data Supplement): (1) established hereditary cancer susceptibility genes (not in the ACMG recommended genes in 2015, which were previously reported by our group11) and SMAD4 (which was added to the recommended genes in 2016), (2) hereditary cancer susceptibility genes with moderate penetrance included in available genetic testing panels with suggested management guidelines (ATM and CHEK2), (3) genes wherein somatic variants are associated with cancer but germline mutations are associated with noncancer phenotypes, (4) other cancer-related genes with unknown clinical validity of pathogenic germline alterations, and (5) hereditary noncancer susceptibility genes with other possible clinical utility.
Clinical Significance Interpretation of Variants
Variant clinical significance classification was assigned according to ACMG guidelines.20,21 The online tools and databases used to classify the clinical significance of the remaining variants are detailed in Figure 1. The clinical significance annotation of all variants in the 54 additional genes analyzed in this study were independently analyzed using InterVar, a bioinformatics software tool that uses an annotated file generated from ANNOVAR22 and classifies each variant on the basis of the Association of Molecular Pathology/ACMG 2015 guidelines,20 and by a scientist with germline variants annotation expertise from the Institute for Personalized Cancer Therapy at The University of Texas MD Anderson Cancer Center. Per the 2017 ACMG recommendation for cancer somatic variant evaluation, we focused on tier I and II variants, which include LPV/PV.21 Furthermore, personal and family history of cancer was reviewed for all patients with identified LPV/PV.
FIG 1.
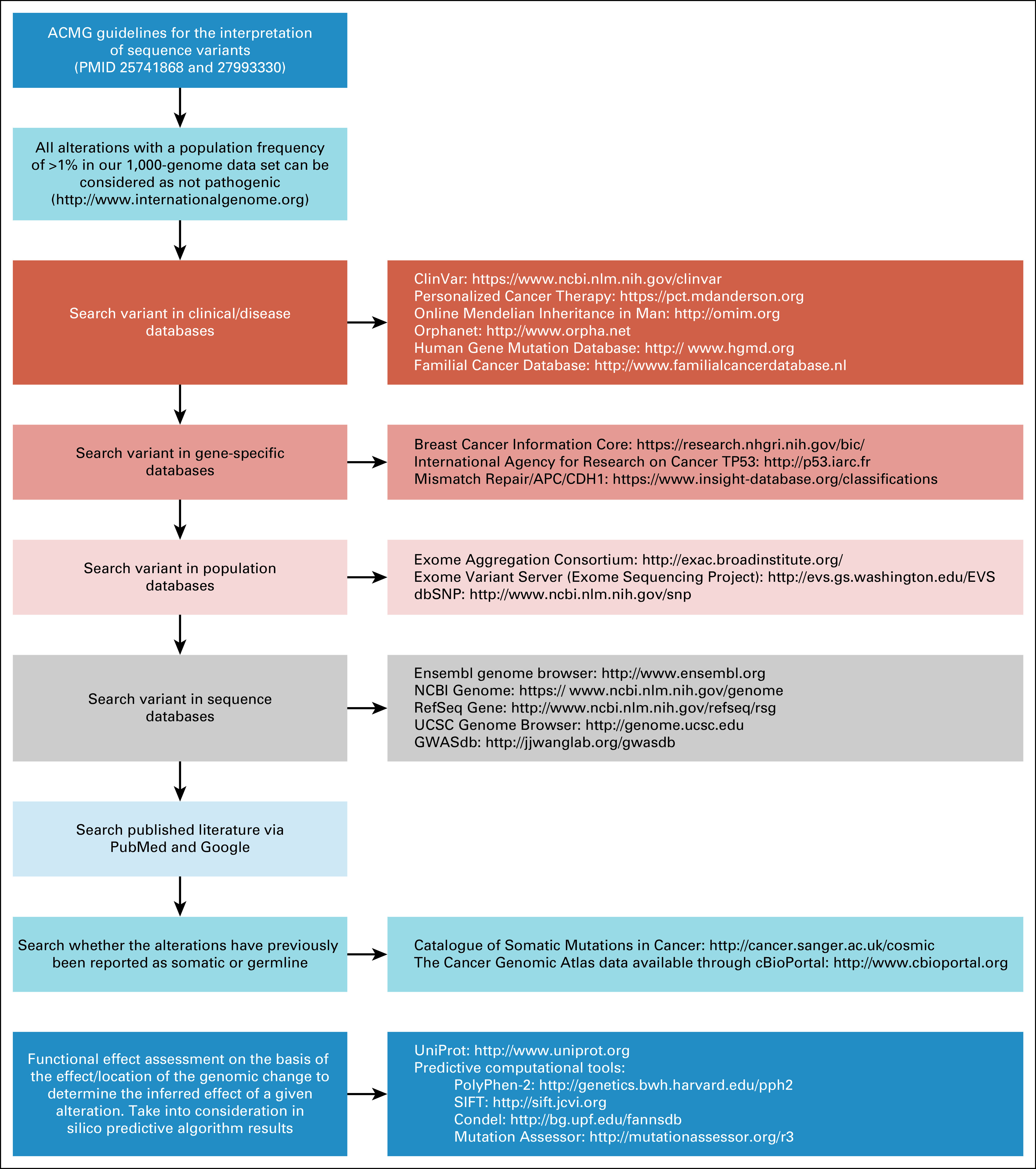
Databases and tools useful for interpreting the clinical significance of germline secondary findings. ACMG, American College of Medical Genetics and Genomics; Condel, consensus deleteriousness; dbSNP, Single Nucleotide Polymorphism Database; GWASdb, Genome-Wide Association Study Database; NCBI, National Center for Biotechnology Information; PMID, PubMed identifier; PolyPhen, polymorphism phenotyping; SIFT, sorting intolerant from tolerant; UCSC, University of California, Santa Cruz.
Determination of Which Results to Return to Patients
A committee of oncologists, a genetic counselor, molecular pathologists, an ethicist, and behavioral scientists developed criteria for return of results to patients. Any LPV/PV and the patient’s family and personal history were discussed in these committee meetings. The LPV/PV in well-established hereditary cancer predisposition genes with high penetrance for which management recommendations were available were recommended to be returned to patients who expressed interest in knowing about secondary germline findings. Variants selected to be returned to patients or their personal representative, were validated with an orthogonal assay in a Clinical Laboratory Improvement Amendments–certified laboratory using the same de-identified research specimen before formal genetic counseling and genetic testing.
RESULTS
Study Population
Among the 1,000 patients who underwent matched tumor/normal DNA sequencing for personalized cancer therapy in the Clearinghouse protocol, the most frequent tumor types were breast (25.3%) and colorectal (15.6%) cancers, glioblastoma (15.1%), melanoma (14.3%), and sarcoma (10.2%; Fig 2). At least one nonsynonymous germline variant in one of the additional 54 analyzed genes was found in 826 patients (82.6%). A median of 26 (range, 1 to 46) nonsynonymous germline variants per patient in these 54 genes was found. Median depth coverage of genes studied was 717 reads.
FIG 2.

Frequency of secondary germline likely pathogenic and pathogenic variants (LPV/PV). Data are presented as tumor type and number of patients enrolled in the study [eg, Breast (n = 253)]. GIST, GI stromal tumor; PNET, primary neuroectodermal tumor.
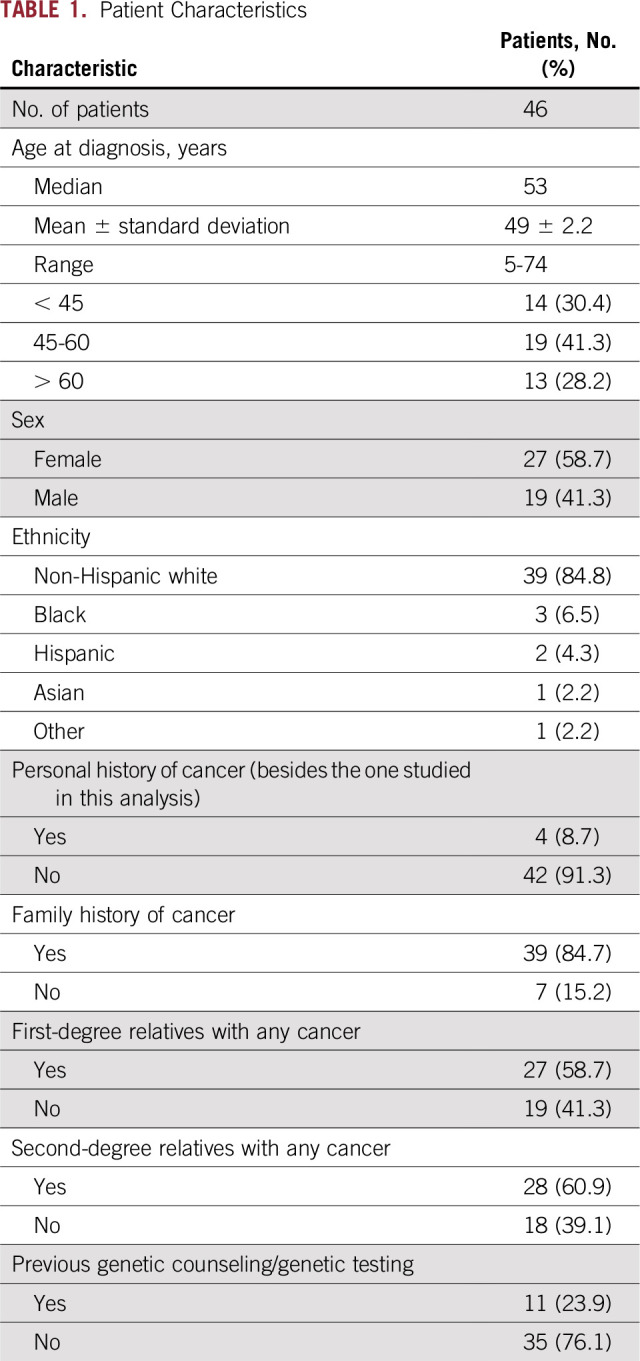
Most of these alterations were classified as benign, likely benign, or uncertain significance according to ACMG guidelines.7,8 However, we identified 46 new germline LPV/PV in 46 patients (4.6%), which added to the 4.3% previously identified. Thus, 8.7% of patients had a secondary germline LPV/PV, with two having two concomitant mutations. These 46 patients’ characteristics are listed in Table 1. The median age at diagnosis was 53 years (range, 5 to 74 years), and 58.7% were women. Four patients had a personal history of a previous malignancy (Data Supplement).
TABLE 1.
Patient Characteristics
We identified two patients who had two germline LPV/PV (one mutation from the ACMG-recommended return of results list that was found previously and one from the list of genes described here). One patient was a 60-year-old man who had metastatic melanoma with synchronous BRCA2 and CHEK2 germline mutations unknown before secondary germline results testing. He had a history of prostate cancer and family history of lung cancer in a sister and an unknown hematologic malignancy in a brother. The second patient was a 13-year-old girl who had a solid pseudopapillary neoplasm of the pancreas with two deleterious germline mutations (MSH6 and ERCC3), and although she had no relevant family history, previous genetic testing revealed a germline mutation in MSH6 suggestive of Lynch syndrome.
Of the 46 patients with LPV/PV, only 11 (24%) had been referred previously for genetic counseling. Of the 10 patients with breast cancer with LPV/PV identified in the current study, only three had previous genetic testing.
Germline LPV/PV Classification and Frequency
We identified germline LPV/PV in 18 (33%) of the 54 genes analyzed in this study. At least one variant of unknown significance was present in all analyzed genes. The most frequent LPV/PV were identified in the following genes: 10 CHEK2 mutations (1%), five AR mutations (0.5%), four ATM mutations (0.4%), four ERCC3 mutations (0.4%), four MITF mutations (0.4%), four PKHD1 mutations (0.4%), three NF1 mutations (0.3%), and two CHEK1 mutations (0.2%; Fig 3). On the basis of the currently available drugs (Food and Drug Administration approved or currently in clinical trials), 26 LPV/PV were found in eight potentially therapeutically actionable genes (CHEK2, AR, ATM, NF1, CDKN2A, EGFR, HRAS, PTCH1).
FIG 3.
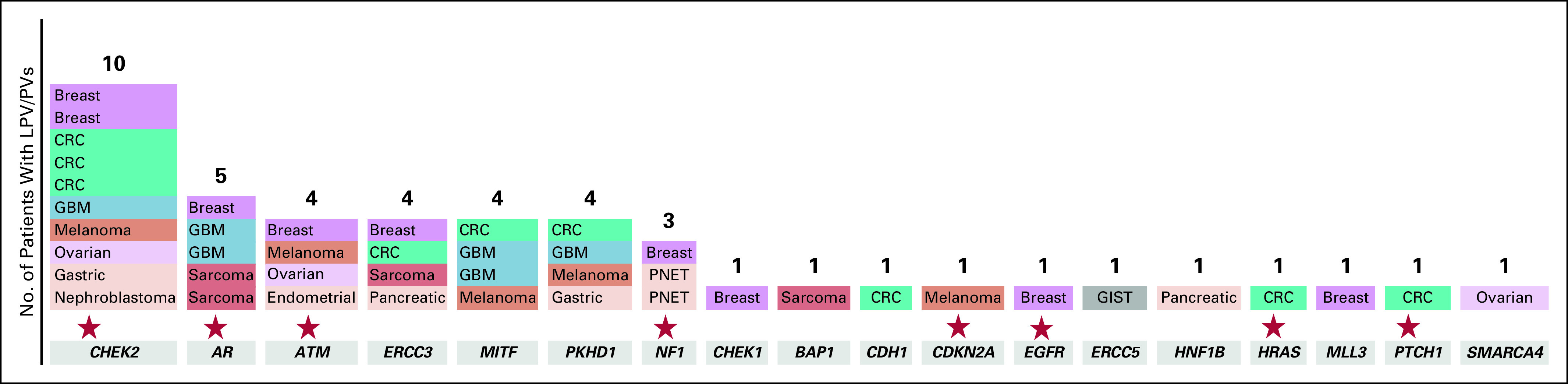
Frequency of secondary germline likely pathogenic and pathogenic variants (LPV/Ps) per genes analyzed. The stars indicate potentially therapeutically actionable genes. CRC, colorectal cancer; GBM, glioblastoma; GIST, GI stromal tumor; PNET, primary neuroectodermal tumor.
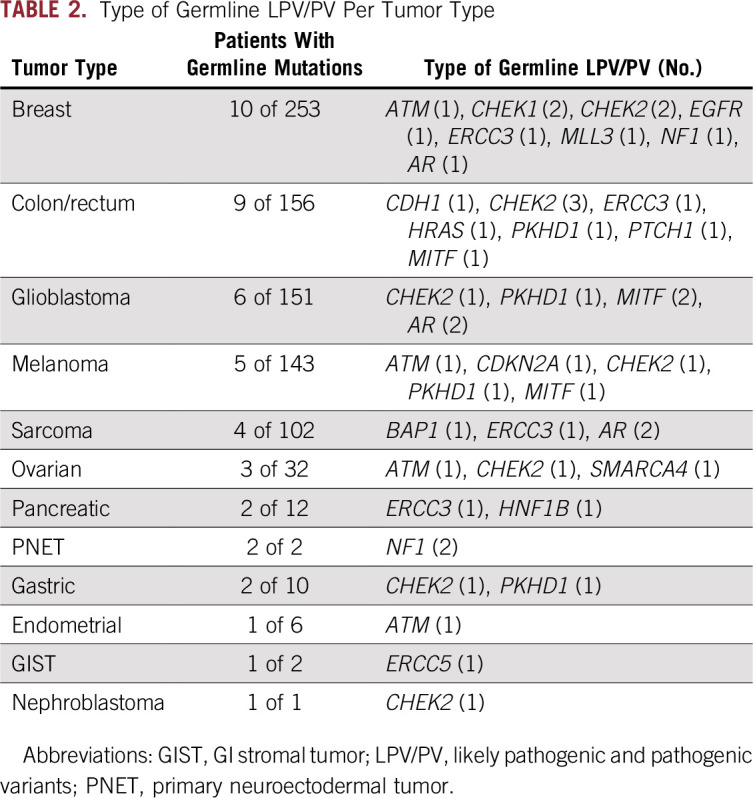
LPV/PV were found in several tumor types, most frequently breast cancer, colorectal cancer, glioblastoma, melanoma, sarcoma, and ovarian cancer (Table 2). Among the 10 patients with breast cancer with LPV/PV, 10 variants were observed in eight genes (Data Supplement).
TABLE 2.
Type of Germline LPV/PV Per Tumor Type
Of the 46 LPV/PV, 20 (44%) were known to be pathogenic and previously reported in ClinVar23 and related to an increased risk of cancer or other diseases. Twenty-six variants were identified as LPV on the basis of the effect/location of the genomic change on the protein function or the databases and online tools shown in Figure 1. The median allele fraction for the LPV/PV found in the current analysis was 46% (range, 11% to 55%). In cases of low median allele fraction, the results were rechecked and validated to be of germline origin. Four patients (8.7%) presented loss of heterozygosity in the same genes as the identified germline LPV/PV (two patients with NF1 mutation, one with ATM mutation, and one with SMARCA4 mutation).
Concordance of Manual and Automatic Variant Interpretation
Variant clinical significance was determined using databases and online tools (Fig 1). All variants also were annotated using InterVar.24 We compared manual annotations with the automatic annotation tool and observed concordance rates between 88% and 98%, which depended on the type of alteration analyzed (point v truncating mutations).
Return of Results to Patients or Their Personal Representative
The committee decided that germline LPV/PV in the following established hereditary cancer predisposition genes with high penetrance should be returned to patients: BAP1, CDH1, CDKN2A, EGFR, and SMARCA4 (Appendix Fig A1). All these results were previously unknown to the patient, and none of the five patients had been previously referred to genetic counseling or underwent prior genetic testing.
All five LPV/PV were confirmed using de-identified samples in a Clinical Laboratory Improvement Amendments–certified laboratory using a different platform (100% concordance obtained). At the time of writing this article, the results were returned to the patient with metastatic melanoma and a CDKN2A variant, who was alive. The patient underwent formal genetic testing and genetic counseling, and the CDKN2A p.G101W mutation was confirmed; she was enrolled in a pancreatic cancer screening program in addition to dermatologic surveillance because of her history of metastatic melanoma, which is currently without evidence of disease. The other four patients died before the results of this study, and we initiated the process of return of results to their personal representatives as recently described.25 Because the return of secondary germline findings to patients’ representatives in moderate penetrance genes such as ATM and CHEK2 in the absence of significant cancer family history have uncertainties and knowledge gaps, after discussion in the secondary germline review committee, these results were not returned.
DISCUSSION
Our findings confirm the feasibility of secondary germline analysis in genes beyond the ones currently recommended by the ACMG for patients with cancer who undergo molecular testing with therapeutic intent. In the current analysis of 1,000 patients who underwent tumor/normal DNA–targeted sequencing of 201 genes, 4.6% carried a germline LPV/PV in a gene linked to an inherited human disease other than those genes recommended for testing by ACMG, which were previously reported at a 4.3% frequency in the same cohort.11 Thus, 8.7% of our patients had an LPV/PV secondary germline finding on the basis of testing with a targeted NGS panel, with two patients having two concomitant germline LPV/PV.
The frequency of germline LPV/PV (between 4.3%26 and 17.5%27) likely depends on the number of genes analyzed, sequencing panel, tumor types and stages of patients enrolled, and annotation of clinical significance and could be higher in selected patients.28 In previous studies, germline mutations in 25 breast or ovarian cancer predisposition genes were found in 10.7% of patients,29 and 6% of patients with prostate cancer were reported to have a germline deleterious mutation in BRCA1/2.30 In the pediatric population, LPV/PV secondary germline findings were found in 5% to 8.5% of patients; many were unsuspected on the basis of family history or phenotypic presentation.31-33
Established guidelines exist for referral to genetic counseling, including germline testing criteria for patients with the BRCA1/2 mutations detected by tumor profiling34; however, carriers of deleterious germline mutations may not meet these criteria. One major advantage of reporting secondary germline findings is that the analysis is not constrained by family or personal history. Indeed, a recent study showed that more than one half of patients with deleterious secondary germline findings would not have been tested using current clinical guidelines.27
In the current study, from the additional genes tested, only the NF1 mutations had been identified before the matched normal/tumor DNA sequencing, which indicates that important cancer predisposition genes may be missed by current criteria for referral to a genetic counselor and genetic testing. Furthermore, in addition to colon, breast, and ovarian cancers, we identified LPV/PV in melanoma, glioblastoma, sarcoma, and pancreatic and gastric cancers.
The 2016 ACMG recommendations changed the terminology to secondary findings instead of incidental findings because the genes were intentionally analyzed. The ACMG list of recommended genes for secondary germline analysis likely will evolve continuously as more knowledge about hereditary syndromes is accumulated and as NGS is integrated into oncology practice, which thus will increase the likelihood of detecting secondary germline findings.8
The cancer risk predictions associated with our gene classifications is based on highly penetrant families. LPV/PV in hereditary cancer susceptibility genes also are present in the general population at a very low frequency, and whether these findings will have the same implications in patients without significant family histories is unknown. Although established public knowledgebases of LPV/PV, such as ClinVar,23 are helpful in determining the clinical significance of germline mutations, this might have conflicting results from different sources, and many variants are not yet described. To improve these databases, we need to share the results and establish guidelines for return of secondary germline findings.35
The potential effect of reporting secondary germline findings in cancer-related genes presents significant opportunities to assess and manage the risk of second primary cancers, assess familial risk, and provide targeted treatment options. Although the clinical utility of germline LPV/PV in some genes outside the ACMG list is still unknown, rapid advances of biologic knowledge and new drug development could aid in identifying new actionable alterations to serve as the basis for future clinical trial design and personalized cancer therapy.36,37
Increasing patient interest exists in knowing about secondary germline findings; however, which results should be returned remains controversial. In this study, a committee of oncologists, a genetic counselor, molecular pathologists, an ethicist, and behavioral scientists believed that in the absence of significant cancer family history, the clinical utility of pathogenic secondary germline findings in moderate penetrance genes like CHEK2 remains with many uncertainties and knowledge gaps; thus, moderate penetrance genes were not returned. This decision was made, in part, because the patients were deceased, which made it difficult to definitively confirm results because they were obtained in the research environment. However, emerging clinical screening guidelines are making a case for return of some moderate-penetrance genes. Evolving data exist for clinical implications of moderate-penetrance genes; thus, regular re-assessment is needed. Further development of guidelines is needed to determine clinical utility required for return of results. Whether clinical utility needs to be stronger for return of results obtained in the research environment and/or to patient representatives after a patient is deceased needs to be considered.
As biomarker-driven cancer therapy becomes a reality for more patients,15 integration of genetic counselors into molecular tumor boards and establishment of educational materials that differentiate between somatic and germline testing could enhance the return of secondary germline findings identified through molecular tumor profiling. Our process for identifying, confirming, and returning secondary germline findings is summarized in Figure 4.
FIG 4.
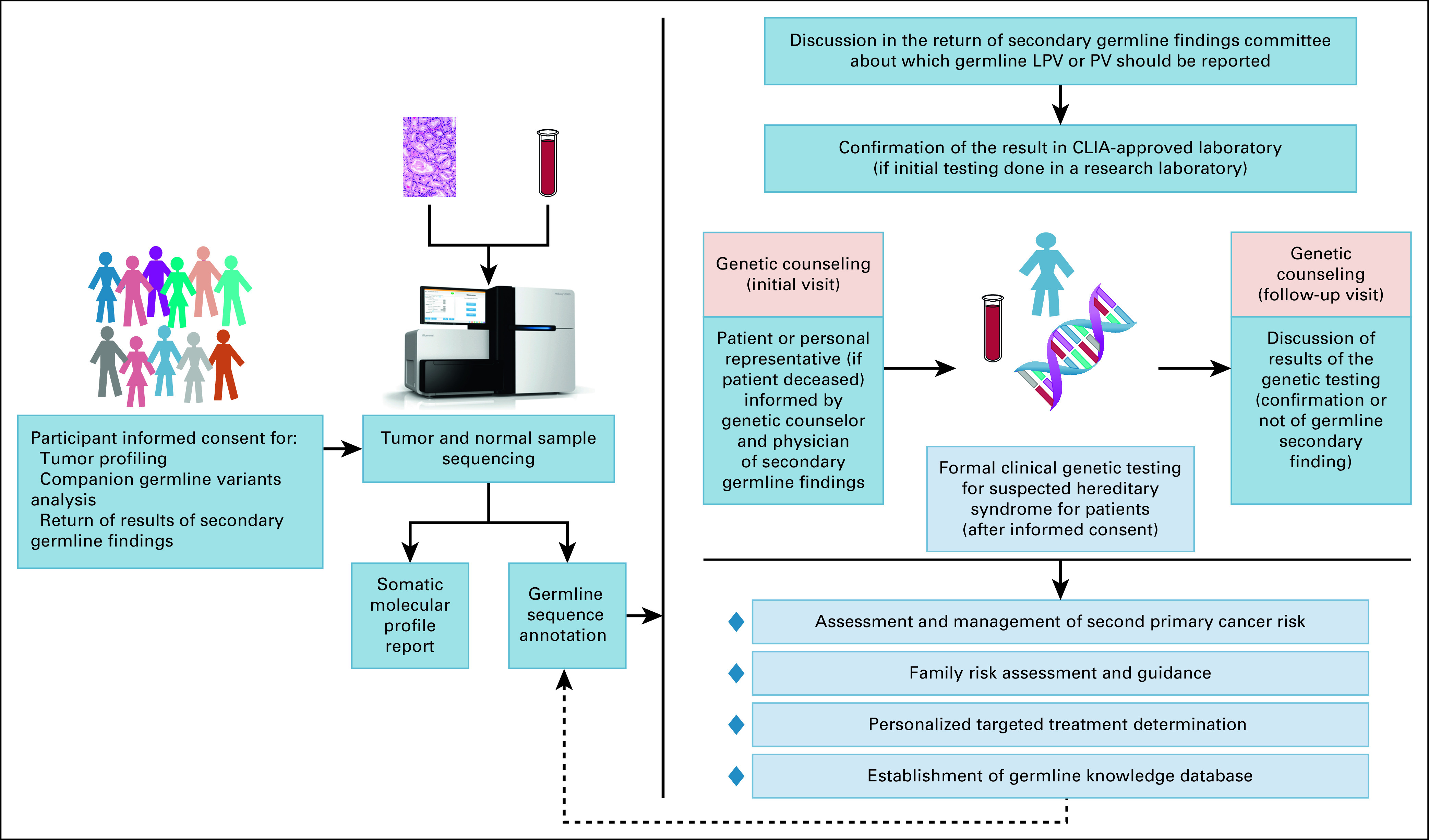
Clinical utility of analysis of germline secondary findings on tumor molecular sequencing using matched normal DNA. CLIA, Clinical Laboratory Improvement Amendments; LPV/PV, likely pathogenic and pathogenic variants.
One limitation of the current study is that the patient population was from an academic center where many patients were referred for consideration of clinical trials; this population may influence the overall results/detection of germline LPV/PV in particular tumor types. In addition, our return-of-results efforts did not include variants of unknown significance that might have clinical implications in the future with the rapidly evolving field.
Important barriers left to be overcome when disclosing germline deleterious variants are psychological outcomes and family communication barriers.38,39 In deceased patients, identification and contact of a personal representative and making the information on secondary germline findings available but allowing the right to decline represent other barriers in return of results.25 Currently, only 5% of genetic counselors feel prepared to handle tumor profiling results cases,40 but this might be overcome with the integration of new variant analysis tools and education programs to fill the knowledge gap among treating cancer care providers and patients.
In conclusion, secondary germline findings identified on tumor/normal DNA profiling could have implications in the assessment and management of second primary cancer risk; family risk assessment and guidance; and most importantly, personalized treatment determination. Most patients would like to know about these secondary germline findings for themselves and their families,41 but some barriers remain to be overcome, such as the determination of which results to disclose and how to disclose them as well as the burdens that this process would place on the cancer care program. A systematic analysis of germline variants could increase the cost and time involved in DNA sequencing and the interpretation of clinical significance. Education of cancer care providers and patients about possible secondary germline findings from tumor profiling is critical.
ACKNOWLEDGMENT
We thank Bryan Tutt in the Department of Scientific Publications, The University of Texas MD Anderson Cancer Center, for editorial assistance.
Appendix
FIG A1.
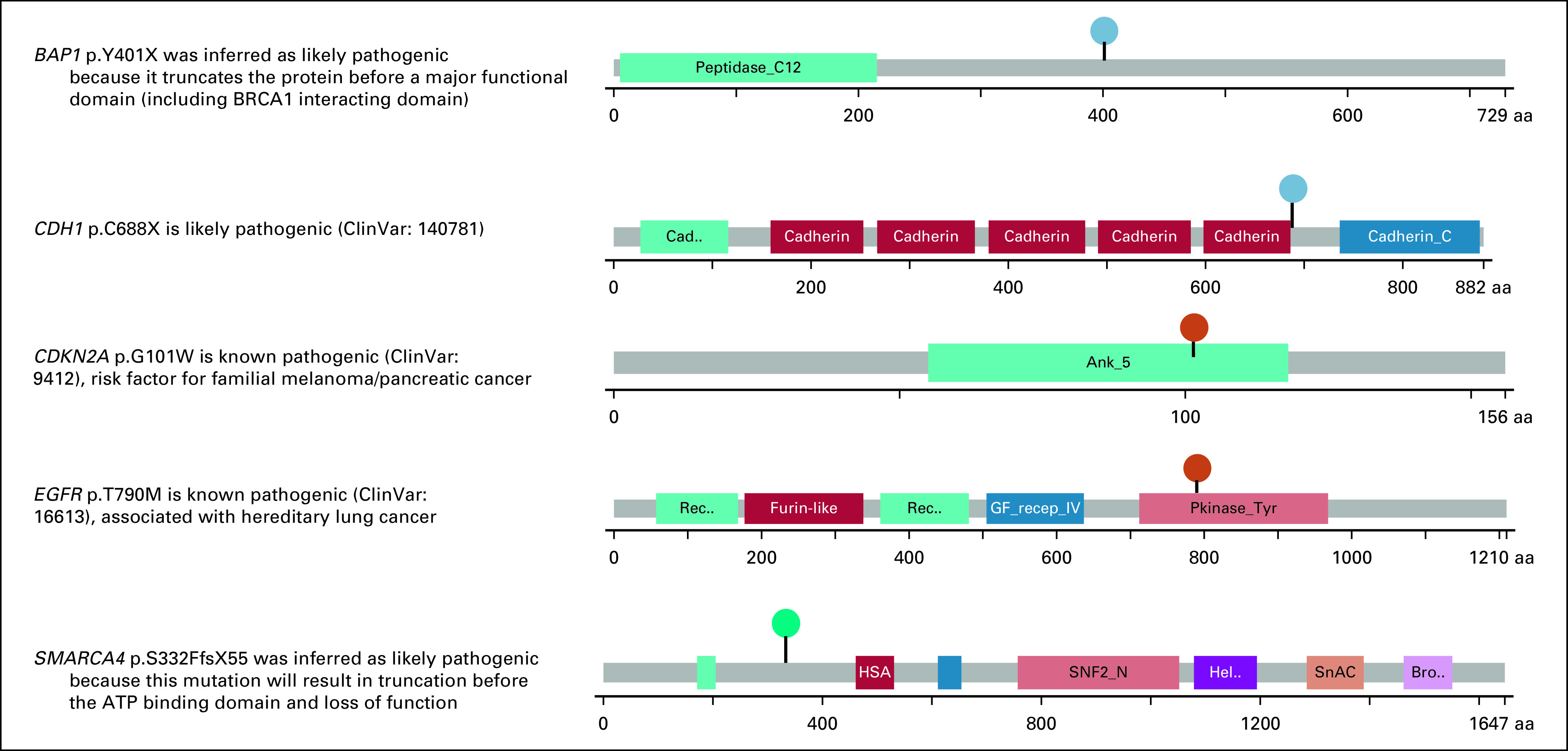
Germline likely pathogenic or pathogenic variants recommended by the secondary results committee to be returned to the patients or their personal representatives.
Footnotes
Supported by the Cancer Prevention Research Institute of Texas Precision Oncology Decision Support Core RP150535, Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy, National Cancer Institute Grant No. U01 CA180964, National Center for Advancing Translational Sciences Grant No. UL1 TR000371 (Center for Clinical and Translational Sciences), the Nellie B. Connally Breast Cancer Research Endowment, the Bosarge Foundation, and the MD Anderson Cancer Center support grant (National Cancer Institute Grant No. P30 CA016672).
See accompanying articles 10.1200/PO.18.00216,10.1200/PO.18.00258
AUTHOR CONTRIBUTIONS
Conception and design: Ecaterina Ileana Dumbrava, Lauren Brusco, Karen H. Lu, Louise C. Strong, Mark J. Routbort, John Mendelsohn, Gordon B. Mills, Funda Meric-Bernstam
Financial support: Funda Meric-Bernstam
Administrative support: Kenna R. Shaw, John Mendelsohn, Funda Meric-Bernstam
Provision of study material or patients: Chetna Wathoo, Mark J. Routbort, Sarina A. Piha-Paul, Vivek Subbiah, David S. Hong, Jordi Rodon, Scott Kopetz, Funda Meric-Bernstam
Collection and assembly of data: Ecaterina Ileana Dumbrava, Lauren Brusco, Chetna Wathoo, Kenna R. Shaw, Louise C. Strong, Jennifer Litton, A. Karina Eterovic, Mark J. Routbort, Keyur P. Patel, Vivek Subbiah, David S. Hong, Scott Kopetz, Funda Meric-Bernstam
Data analysis and interpretation: Ecaterina Ileana Dumbrava, Lauren Brusco, Molly S. Daniels, Chetna Wathoo, Xiaofeng Zheng, Louise C. Strong, Jennifer Litton, Banu K. Arun, Keyur P. Patel, Yuan Qi, Sarina A. Piha-Paul, Vivek Subbiah, Jordi Rodon, John Mendelsohn, Gordon B. Mills, Ken Chen, Funda Meric-Bernstam
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Lauren Brusco
Employment: Celgene
Jennifer Litton
Consulting or Advisory Role: Pfizer, AstraZeneca, Medivation
Speakers’ Bureau: Physicians’ Education Resource, UpToDate, Med Learning Group, Medscape
Research Funding: Novartis (Inst), Bristol-Myers Squibb (Inst), Genentech (Inst), Pfizer (Inst), EMD Serono (Inst), Jounce Therapeutics (Inst), GlaxoSmithKline (Inst), Medivation (Inst)
Patents, Royalties, Other Intellectual Property: UpToDate
Travel, Accommodations, Expenses: Physicians’ Education Resource, Med Learning Group, Medscape
Sarina A. Piha-Paul
Consulting or Advisory Role: Genentech
Research Funding: GlaxoSmithKline, Xuan Zhu, Puma Biotechnology, Novartis, Merck Sharp & Dohme, Curis, Principia Biopharma, Helix BioPharma, Bayer AG, AbbVie, Incyte, Five Prime Therapeutics, MedImmune, Medivation, BlueLink Pharmaceuticals, Pfizer, TESARO, Pieris Pharmaceuticals
Vivek Subbiah
Consulting or Advisory Role: MedImmune
Research Funding: Novartis (Inst), GlaxoSmithKline (Inst), NanoCarrier (Inst), Northwest Biotherapeutics (Inst), Genentech (Inst), Roche (Inst), BERG (Inst), Bayer AG (Inst), Incyte (Inst), Fujifilm (Inst), PharmaMar (Inst), D3 Oncology Solutions (Inst), Pfizer (Inst), Amgen (Inst), AbbVie (Inst), MultiVir (Inst), Blueprint Medicines (Inst), Loxo Oncology (Inst), Vegenics (Inst), Takeda Pharmaceuticals (Inst), Alfasigma (Inst), Agensys (Inst), Idera Pharmaceuticals (Inst), Boston Biomedical (Inst), InhibRx (Inst), Exelixis (Inst)
Travel, Accommodations, Expenses: PharmaMar, Bayer AG
David S. Hong
Stock and Other Ownership Interests: MolecularMatch, Oncorena
Honoraria: Adaptimmune, Baxter, Merrimack, Bayer AG
Consulting or Advisory Role: Baxter, Bayer AG, Guidepoint Global, Janssen Pharmaceuticals, Genentech, Eisai, GLG Pharma
Research Funding: Novartis, Genentech, Eisai, AstraZeneca, Pfizer, Mirna Therapeutics, Amgen, Daiichi Sankyo, Merck, Mirati Therapeutics, Eli Lilly, Adaptimmune, AbbVie, Bayer AG, Bristol-Myers Squibb, Genmab, Ignyta, Infinity Pharmaceuticals, Kite Pharma, Kyowa Hakko Kirin, Loxo Oncology, MedImmune, Molecular Templates, Takeda Pharmaceuticals, Seattle Genetics, Amgen
Travel, Accommodations, Expenses: Loxo Oncology, Mirna Therapeutics
Jordi Rodon
Consulting or Advisory Role: Novartis, Eli Lilly, ImClone Systems, SERVIER, Orion, Peptomyc, Kelun Pharmaceutical, Merck Sharp & Dohme, Spectrum Pharmaceuticals, Pfizer
Research Funding: Novartis, Bayer AG
Scott Kopetz
Stock and Other Ownership Interests: MolecularMatch, Navire Pharma
Consulting or Advisory Role: Roche, Genentech, EMD Serono, Merck, Karyopharm Therapeutics, Amal Therapeutics, Biocartis, Navire Pharma, Symphogen
Research Funding: Amgen (Inst), Sanofi (Inst), Biocartis (Inst), Guardant Health (Inst), Array BioPharma (Inst), Genentech (Inst), Roche (Inst), EMD Serono (Inst), MedImmune (Inst), Novartis (Inst)
John Mendelsohn
Stock and Other Ownership Interests: Merrimack
Patents, Royalties, Other Intellectual Property: Royalty payments from University of California, San Diego
Gordon B. Mills
Stock and Other Ownership Interests: Catena Pharmaceuticals, Spindletop Capital, Immunome, SignalChem, Tarveda Therapeutics
Honoraria: Nuevolution, AstraZeneca, Tarveda Therapeutics, TESARO, Symphogen, Immunome
Consulting or Advisory Role: AstraZeneca, Catena Pharmaceuticals, Critical Outcome Technologies, SignalChem, Tarveda Therapeutics, Symphogen, Takeda Pharmaceuticals, Millennium Pharmaceuticals, Ion Pharmaceuticals, Immunome
Research Funding: Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, AstraZeneca, NanoString Technologies, Breast Cancer Research Foundation, Karus Therapeutics, Immunome, Ion Pharmaceuticals, Komipharm, Ovarian Cancer Research Foundation, Pfizer, Prospect Creek Foundation, Tarveda Pharmaceuticals, Millennium Pharmaceuticals
Patents, Royalties, Other Intellectual Property: HRD assay to Myriad Genetics
Travel, Accommodations, Expenses: AstraZeneca, Pfizer, Immunome, Symphogen
Funda Meric-Bernstam
Honoraria: Sumitomo Group, Dialectica
Consulting or Advisory Role: Genentech, Inflection Biosciences, Pieris Pharmaceuticals, Clearlight Diagnostics, DarwinHealth, Samsung Bioepis, Spectrum Pharmaceuticals, Aduro Biotech, OrigiMed, Xencor, Debiopharm Group
Research Funding: Novartis, AstraZeneca, Taiho Pharmaceutical, Genentech, Calithera Biosciences, Debiopharm Group, Bayer AG, Aileron Therapeutics, Puma Biotechnology, CytomX Therapeutics, Jounce Therapeutics, Zymeworks, Curis, Pfizer, eFFECTOR Therapeutics, AbbVie, Boehringer Ingelheim (I)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Cummings CA, Peters E, Lacroix L, et al. The role of next-generation sequencing in enabling personalized oncology therapy. Clin Transl Sci. 2016;9:283–292. doi: 10.1111/cts.12429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11:685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 3.Meric-Bernstam F, Brusco L, Shaw K, et al. Feasibility of large-scale genomic testing to facilitate enrollment onto genomically matched clinical trials. J Clin Oncol. 2015;33:2753–2762. doi: 10.1200/JCO.2014.60.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jones S, Anagnostou V, Lytle K, et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med. 2015;7:283ra53. doi: 10.1126/scitranslmed.aaa7161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gray PN, Vuong H, Tsai P, et al. TumorNext: A comprehensive tumor profiling assay that incorporates high resolution copy number analysis and germline status to improve testing accuracy. Oncotarget. 2016;7:68206–68228. doi: 10.18632/oncotarget.11910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christoforides A, Carpten JD, Weiss GJ, et al. Identification of somatic mutations in cancer through Bayesian-based analysis of sequenced genome pairs. BMC Genomics. 2013;14:302. doi: 10.1186/1471-2164-14-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–574. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalia SS, Adelman K, Bale SJ, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–255. doi: 10.1038/gim.2016.190. [Erratum: Genet Med 19:484, 2017] [DOI] [PubMed] [Google Scholar]
- 9.Robson ME, Bradbury AR, Arun B, et al. American Society of Clinical Oncology policy statement update: Genetic and genomic testing for cancer susceptibility. J Clin Oncol. 2015;33:3660–3667. doi: 10.1200/JCO.2015.63.0996. [DOI] [PubMed] [Google Scholar]
- 10.Hegde M, Santani A, Mao R, et al. Development and validation of clinical whole-exome and whole-genome sequencing for detection of germline variants in inherited disease. Arch Pathol Lab Med. 2017;141:798–805. doi: 10.5858/arpa.2016-0622-RA. [DOI] [PubMed] [Google Scholar]
- 11.Meric-Bernstam F, Brusco L, Daniels M, et al. Incidental germline variants in 1000 advanced cancers on a prospective somatic genomic profiling protocol. Ann Oncol. 2016;27:795–800. doi: 10.1093/annonc/mdw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arango NP, Brusco L, Mills Shaw KR, et al. A feasibility study of returning clinically actionable somatic genomic alterations identified in a research laboratory. Oncotarget. 2017;8:41806–41814. doi: 10.18632/oncotarget.16018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen K, Meric-Bernstam F, Zhao H, et al. Clinical actionability enhanced through deep targeted sequencing of solid tumors. Clin Chem. 2015;61:544–553. doi: 10.1373/clinchem.2014.231100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bailey AM, Mao Y, Zeng J, et al. Implementation of biomarker-driven cancer therapy: Existing tools and remaining gaps. Discov Med. 2014;17:101–114. [PMC free article] [PubMed] [Google Scholar]
- 16.Antoniou AC, Casadei S, Heikkinen T, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371:497–506. doi: 10.1056/NEJMoa1400382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamosh A, Scott AF, Amberger JS, et al. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amberger JS, Hamosh A. Searching Online Mendelian Inheritance in Man (OMIM): A knowledgebase of human genes and genetic phenotypes. Curr Protoc Bioinforma. 2017;58:1.2.1–1.2.12. doi: 10.1002/cpbi.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rahman N. Realizing the promise of cancer predisposition genes. Nature. 2014;505:302–308. doi: 10.1038/nature12981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–423. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: A joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23. doi: 10.1016/j.jmoldx.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang K, Li M, Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Landrum MJ, Lee JM, Riley GR, et al. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Q, Wang K. InterVar: Clinical interpretation of genetic variants by the 2015 ACMG-AMP guidelines. Am J Hum Genet. 2017;100:267–280. doi: 10.1016/j.ajhg.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daniels M, Wathoo C, Brusco L, et al. Active disclosure of secondary germline findings to deceased research participants’ personal representatives: Process and outcomes. JCO Precis Oncol doi: doi: 10.1200/PO.17.00074. 10.1200/PO.17.00074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seifert BA, O’Daniel JM, Amin K, et al. Germline analysis from tumor-germline sequencing dyads to identify clinically actionable secondary findings. Clin Cancer Res. 2016;22:4087–4094. doi: 10.1158/1078-0432.CCR-16-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mandelker D, Zhang L, Kemel Y, et al. Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA. 2017;318:825–835. doi: 10.1001/jama.2017.11137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schrader KA, Cheng DT, Joseph V, et al. Germline variants in targeted tumor sequencing using matched normal DNA. JAMA Oncol. 2016;2:104–111. doi: 10.1001/jamaoncol.2015.5208. [Erratum: JAMA Oncol 2:279, 2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tung N, Lin NU, Kidd J, et al. Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J Clin Oncol. 2016;34:1460–1468. doi: 10.1200/JCO.2015.65.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–453. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kline CN, Joseph NM, Grenert JP, et al. Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro-oncol. 2017;19:699–709. doi: 10.1093/neuonc/now254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parsons DW, Roy A, Yang Y, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016;1200:1–9. doi: 10.1001/jamaoncol.2015.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373:2336–2346. doi: 10.1056/NEJMoa1508054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.NCCN Clinical Practice Guidelines in Oncology Genetic/familial high-risk assessment: Breast and ovarian. 2017 http://www.nccn.org/professionals/physician_gls
- 35.Slavin TP, Niell-Swiller M, Solomon I, et al. Clinical application of multigene panels: Challenges of next-generation counseling and cancer risk management. Front Oncol. 2015;5:208. doi: 10.3389/fonc.2015.00208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boland G, Piha-Paul S, Subbiah V, et al. Clinical next generation sequencing to identify actionable alterations in a phase I program. Oncotarget. 2015;6:20099–20110. doi: 10.18632/oncotarget.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan C, Pattabiraman N, Goecks J, et al. Impact of germline and somatic missense variations on drug binding sites. Pharmacogenomics J. 2017;17:128–136. doi: 10.1038/tpj.2015.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bombard Y, Robson M, Offit K. Revealing the incidentalome when targeting the tumor genome. JAMA. 2013;310:795–796. doi: 10.1001/jama.2013.276573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hofstatter E, Mehra K, Yushak M, et al. Tumor profiling and the incidentalome: Patient decisions and risks. Future Oncol. 2015;11:3299–3305. doi: 10.2217/fon.15.260. [DOI] [PubMed] [Google Scholar]
- 40.Goedde LN, Stupiansky NW, Lah M, et al. Cancer genetic counselors’ current practices and attitudes related to the use of tumor profiling. J Genet Couns. 2017;26:878–886. doi: 10.1007/s10897-017-0065-z. [DOI] [PubMed] [Google Scholar]
- 41.Daniels M, Wathoo C, Brusco L, et al. Active disclosure of secondary germline findings to deceased research participants’ personal representatives: Process and outcomes. JCO Precis Oncol 2017. doi:10.1200/PO17.00074 [DOI] [PMC free article] [PubMed] [Google Scholar]