

SUMMARY
Parkinson’s disease causes the most profound loss of the aldehyde dehydrogenase 1A1-positive (ALDH1A1+) nigrostriatal dopaminergic neuron (nDAN) subpopulation. The connectivity and functionality of ALDH1A1+ nDANs, however, remain poorly understood. Here, we show in rodent brains that ALDH1A1+ nDANs project predominantly to the rostral dorsal striatum, from which they also receive most monosynaptic inputs, indicating extensive reciprocal innervations with the striatal spiny projection neurons (SPNs). Functionally, genetic ablation of ALDH1A1+ nDANs causes severe impairments in motor skill learning, along with a reduction in high-speed walking. While dopamine replacement therapy accelerated walking speed, it failed to improve motor skill learning in ALDH1A1+ nDAN-ablated mice. Altogether, our study provides a comprehensive whole-brain connectivity map and reveals a key physiological function of ALDH1A1+ nDANs in motor skill acquisition, suggesting the motor learning processes require ALDH1A1+ nDANs to integrate diverse presynaptic inputs and supply dopamine with dynamic precision.
In Brief
Wu et al. show that aldehyde dehydrogenase 1A1-positive nigrostriatal dopaminergic neurons, which display the most profound degeneration in Parkinson’s disease, are essential in the acquisition of skilled movements in rodent models. The proper synaptic inputs to these neurons are critical for providing the timely dopamine release required in the learning process.
Graphical Abstract
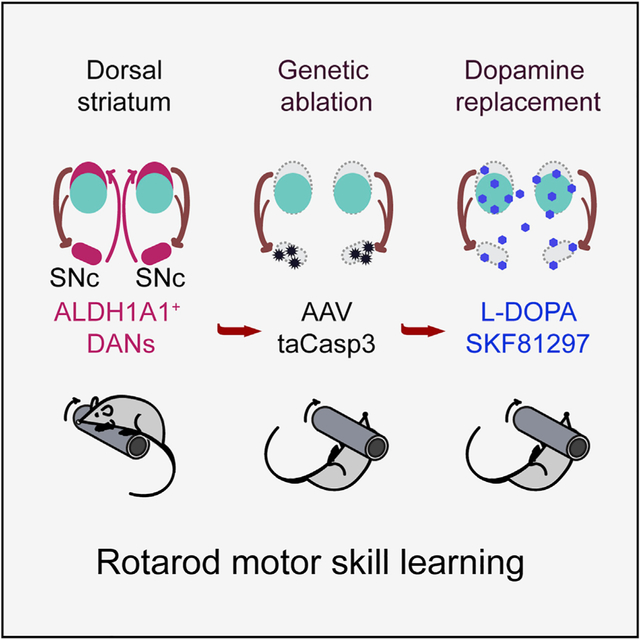
INTRODUCTION
The nigrostriatal dopaminergic neurons (nDANs) are heterogenous and can be categorized into subpopulations based on location, gene expression profiles, electrophysiological properties, morphology, projection pattern, physiological functions, and vulnerabilities to diseases (Beier et al., 2015; Lammel et al., 2011, 2012; Lerner et al., 2015; Menegas et al., 2015, 2018; Yang et al., 2013). Parkinson’s disease (PD), the most common degenerative movement disorder, particularly affects basal ganglia dopamine transmission (Sulzer and Surmeier, 2013; Vogt Weisenhorn et al., 2016). In the postmortem brains of PD patients, the most profound loss of nDANs was observed in the ventral tier of substantia nigra pars compacta (SNc) (Fearnley and Lees, 1991; Kordower et al., 2013). These ventral nDANs can be molecularly defined by the selective expression of aldehyde dehydrogenase 1A1 (ALDH1A1) (Cai et al., 2014; Liu et al., 2014). We referred to this distinct neuron subpopulation as the ALDH1A1-positive (ALDH1A1+) nDANs.
In DANs, ALDH1A1 oxidizes the highly reactive dopamine catabolic intermediate dopamine-3,4-dihydroxyphenylacetaldehyde (DOPAL) and protects ALDH1A1+ nDANs against DOPAL-induced cytotoxicity (Burke, 2010; Marchitti et al., 2007). The reduction of ALDH1A1 expression may contribute to the etiopathogenesis of the disease (Galter et al., 2003; Grünblatt et al., 2018; Mandel et al., 2007; Werner et al., 2008), whereas an increase of ALDH1A1 levels protects against dopaminergic neurodegeneration (Liu et al., 2014). Anatomically, the cell bodies of ALDH1A1+ nDANs display a topographically conserved distribution pattern in the ventral SNc of rodent brains (Liu et al., 2014; McCaffery and Dräger, 1994; Poulin et al., 2014), while the axons mainly innervate the rostral and dorsal portions of the dorsal striatum (DS), including both the dorsomedial striatum (DMS) and the dorsolateral striatum (DLS) (Pan et al., 2019; Poulin et al., 2018; Sgobio et al., 2017). However, little is known about which brain regions send afferents to ALDH1A1+ nDANs or about which specific motor or non-motor functions ALDH1A1+ nDANs regulate.
PD is known to cause motor control abnormalities, including impaired motor skill learning (Jankovic, 2008; Nieuwboer et al., 2009; Roncacci et al., 1996; Zhuang et al., 2013). Motor skill learning is essential for optimizing movements in every aspect of life (Kantak and Winstein, 2012). The cortico-basal gangliathalamus-cortex loops contribute to motor skill learning (Costa et al., 2004; Hélie et al., 2015; Hikosaka et al., 2002), within which the associated cortex-DMS and sensorimotor cortex-DLS circuits appear to function in parallel for the acquisition of skilled movements (Corbit et al., 2017; Kupferschmidt et al., 2017). Many studies have presented evidence that dopamine dynamically modulates synaptic weights of cortical and striatal neurons and serves as a reinforcement learning signal in the striatum (Valentin et al., 2016). Because both ALDH1A1+ and ALDH1A1-negative (ALDH1A1−) nDANs provide dopamine in the DS, how critical the ALDH1A1+ nDANs are in the acquisition of skilled movements remains to be determined.
Using a line of inducible Cre DNA recombinase (CreERT2) knockin (Aldh1a1-P2A-CreERT2) mice that selectively express CreERT2 in the ALDH1A1+ nDANs, we systematically examined the efferent projections from and the monosynaptic afferents onto ALDH1A1+ nDANs and produced a whole-brain connectivity map of ALDH1A1+ nDANs in the rodent brain. Our behavioral tests revealed an essential function of ALDH1A1+ nDANs in motor skill learning.
RESULTS
Uneven Distribution of ALDH1A1+ DANs in Different Midbrain Dopaminergic Subregions
Midbrain dopaminergic neurons can be divided into three main subgroups, retrorubral field (RRF, A8), SNc (A9), and ventral tegmental area (VTA, A10), in human and rodents (Bentivoglio and Morelli, 2005; Vogt Weisenhorn et al., 2016). The DANs in SNc and VTA are also called nDANs and mesocorticolimbic DANs, respectively (Fu et al., 2012). The ALDH1A1+ DANs accounted for 63.0% of SNc DANs, 31.9% of VTA DANs, and 5.1% of RRF DANs (Figures 1A and 1B). The mouse SNc and VTA DANs can be divided into five and seven subgroups, respectively (Fu et al., 2012). Among the SNc subgroups, SNc ventral (SNCV) and SNc medial (SNCM) were composed of more than 95% of ALDH1A1+ nDANs, while the largest subgroup, SNc dorsal (SNCD), contained around 54.6% of ALDH1A1+ nDANs (Figures 1C and 1D). In the VTA subgroups, interfascicular nucleus (IF) had the highest percentage (83.4%) of ALDH1A1+ mesocorticolimbic DANs (mDANs), while the largest subgroup, parabrachial pigmented nucleus (PBP), had about 22.1% of ALDH1A1+ mDANs (Figures 1C and 1D). Therefore, ALDH1A1+ nDANs and mDANs are distributed unevenly among different midbrain DAN subgroups.
Figure 1. Regional Distribution of ALDH1A1+ DANs in the Midbrain.

(A) ALDH1A1 and TH co-staining in sequential coronal midbrain sections of 3-month-old wild-type C57BL/6J mice. Hoechst was used to mark nuclei. Dashed lines and arrows indicate different SNc and VTA subregions. Scale bar: 200 μm.
(B–D) Percentages (B and C) and average numbers (D) of ALDH1A1+ DANs in each subregion.
N = 3.
Generation and Characterization of Aldh1a1-P2A-CreERT2 Knockin Mice
To facilitate the genetic manipulation of ALDH1A1+ DANs, we generated a new line of Aldh1a1-P2A-CreERT2 (Aldh1a1+/P2A-CreERT2) knockin (KI) mice, in which the inducible Cre recombinase CreERT2 is expressed under the control of the endogenous Aldh1a1 promoter (Figure S1). After crossing with a line of Cre-dependent tdTomato reporter mice (Ai9) (Madisen et al., 2010), we estimated that around 40% of ALDH1A1+ DANs in the Aldh1a1-P2A-CreERT2/Ai9 reporter bigenic mice showed tdTomato expression in the absence of Cre activity inducer 4-hydroxytamoxifen (4-OHT) (Figures S2A, S2B, S2D, and S2E). The exact mechanism of this leaky expression remains to be determined. However, we noticed that although ALDH1A1 proteins were mainly distributed in the cytosol, lower levels of ALDH1A1 proteins were present in the nuclei of DANs (Figures S2E and S2F). We suspect that some ALDH1A1-P2A-CreERT2 fusion proteins bypass the heat shock protein 90 (HSP90) chaperone activity and enter the nuclei in a 4-OHT-independent fashion.
Besides the midbrain DANs, we observed tdTomato-positive axon bundles in the optic tract (opt) underneath the lateral preoptic area of Aldh1a1-P2A-CreERT2/Ai9 reporter mice (open arrowhead, Figure S2A). We suspect those tdTomato-positive fibers came from the ALDH1A1+ ganglion cells in the retina, because ALDH1A1 is expressed by the retinal ganglion cells (Fan et al., 2003). In addition, when the Aldh1a1-P2A-CreERT2/Ai9 reporter mice were treated with a high dose (10 mg/kg of body weight) of 4-OHT, we observed several tdTomato-positive, glia-like cells randomly distributed in the brain sections (Figure S3). Those cells were negative for ALDH1A1, tyrosine hydroxylase (TH), NeuN, GFAP, Olig-2, and Iba1 immunostaining (Figure S3C). Such tdTomato-positive, glial-like cells were found in other 2A-CreERT2 KI mouse lines when crossbreeding with Ai9 reporter mice (Madisen et al., 2015). The nature of these tdTomato-positive, non-neuronal cells remains to be determined.
We next verified the uneven projection pattern of ALDH1A1+ nDAN axon fibers to the more rostral and dorsal portions of DS or caudate-putamen nuclei (CP) (Figures S2A, S2C, and S3B).
Uneven Projection of ALDH1A1+ nDANs in the DS
To selectively illustrate the projection patterns of ALDH1A1+ nDANs, we infected the Aldh1a1-P2A-CreERT2 KI mice with recombinant adeno-associated viruses (AAVs) by stereotactic injections targeted to the SNc to express Cre-dependent EGFP in one hemisphere and red fluorescent protein tdTomato in the other hemisphere (Figure S4A). The dual-color labeling allows study of the projections of two individually labeled neuronal clusters in the same animal. Co-staining with antibodies against ALDH1A1 confirmed the selective expression of EGFP and tdTomato in the cell bodies of ALDH1A1+ nDANs in the ventral SNc (Figure S4B). We then examined the EGFP- and tdTomato-positive axon fibers in sequential coronal sections across entire brains. We found the fluorescent protein-labeled fibers stayed in the same hemisphere and projected restrictively to the dorsal CP (Figure S4C). In line with previous reports (Poulin et al., 2018; Sgobio et al., 2017), we observed only a small fraction of those axons converged to the patch (also called striosome) compartments marked by staining with an antibody against μ-type opioid receptor 1 (MOR1) (Figure S4C). The administration of 4-OHT at 10 mg/kg of body weight did not induce fluorescent reporter-positive, glial-like cells, as observed in the Aldh1a1-P2A-CreERT2/Ai9 bigenic mice (Figure S3). Therefore, altering glial-like cells with injected AAVs was not a concern.
To delineate the projection patterns of ALDH1A1+ DANs in different SNc and VTA subgroups of Aldh1a1-P2A-CreERT2 KI mice, we labeled one small cluster of ALDH1A1+ DANs in each mouse brain by injecting an aliquot of diluted Cre-dependent tdTomato-expressing AAVs (Figure 2A). We managed to label a cluster of 10–100 ALDH1A1+ DANs in each mouse brain (Figure 2B). Most labeled neurons in clusters 1–4 were located in the SNc subregions, whereas neurons in clusters 5 and 6 were mostly in the VTA (Figures 2A and 2B). We found that ALDH1A1+ DANs in clusters 1–4 projected more restrictively to the DLS in the intermediate CP, whereas ALDH1A1+ DANs in cluster 5 and 6 projected more heavily to the rostral striatum, including both CP and ventral striatum (VS) (Figure 2C). Furthermore, ALDH1A1+ mDANs in the lateral VTA (PBP) represented by cluster 5 seemed to project to the DLS, while ALDH1A1+ mDANs in the medial VTA (IF, rostral linear nucleus [RLi], and caudal linear nucleus [CLi]) represented by cluster 6 projected more restrictively to the DMS (the bottom two rows, Figure 2C). Overall, it appears that ALDH1A1+ DANs arrange their projections to the CP in parallel along the medial to lateral axis corresponding to the locations of their cell bodies in the midbrain. In addition, ALDH1A1+ DANs in the more caudal midbrain regions seem to send their axons to the more rostral parts of CP.
Figure 2. Projection of Sparsely Labeled ALDH1A1+ DANs into the Striatum.

(A) Schematic brain atlas combines the cell bodies of tdTomato-expressing ALDH1A1+ DANs from six Aldh1a1-P2A-CreERT2 mice in 10 sequential midbrain coronal sections. Each ALDH1A1+ DAN cluster is marked by a different color.
(B) Distribution of labeled ALDH1A1+ DANs in different midbrain subregions from six individual ALDH1A1+ DAN clusters.
(C) Projections of tdTomato-positive axon terminals from the six ALDH1A1+ DAN clusters (A) in seven sequential striatal coronal sections. The axon terminals are marked by the same color as the cell bodies. Gray represents ALDH1A1 immunostaining in each section.
Whole-Brain Map of Monosynaptic Inputs of ALDH1A1+ DANs
To identify the neurons that directly innervate ALDH1A1+ DANs, we mapped monosynaptic inputs using a rabies-mediated retrograde labeling approach (Watabe-Uchida et al., 2012). We injected EGFP-tagged rabies tracers and supporting AAVs into the SNc or VTA of Aldh1a1-P2A-CreERT2 KI mice (Figure 3A). We analyzed five mice with the starter neurons mostly in the SNc and three mice with more starters in the VTA regions (Figure 3B). The starter neurons showed co-expression of EGFP, mCherry, TH, and ALDH1A1 in the midbrain (Figure 3C). We examined the distribution of EGFP-positive input neurons in sequential coronal sections across the entire brain (Figures 3D and 3E). We found that ALDH1A1+ DANs receive various inputs from several brain regions (Figure 3E; Figure S5). We marked the regions that contained more than 1% of total inputs on a schematic brain map (Figure 3E). It appeared that ALDH1A1+ nDANs and mDANs received inputs from overlapping brain regions, but ALDH1A1+ nDANs had more inputs from the CP (Figure 3E; Figure S5). We observed more ALDH1A1+ nDAN inputs distributed in the lateral CP (Figure 3F). However, the percentages of inputs from the patch compartments were comparable with the percentages of areas occupied by the patches in the dorsal CP (Figures 3D and 3G). These data suggest that ALDH1A1+ nDANs may not preferentially receive synaptic inputs from patch versus matrix compartments.
Figure 3. Whole-Brain Map of Monosynaptic Inputs of ALDH1A1+ DANs.

(A) Schematic brain atlas indicates the unilateral injections of AAVs and rabies in the midbrain regions of Aldh1a1-P2A-CreERT2 mice.
(B) Distribution of starter neurons in different midbrain regions of eight individual mice.
(C) Co-staining of EGFP, mCherry, ALDH1A1, and TH in a midbrain coronal section of mouse SNC#5. Arrows point to the starter neurons. Scale bar: 500 μm.
(D) Co-staining of EGFP, MOR1, and calbindin 1 (CALB1) in a striatal coronal section of mouse SNC#5. The bottom panels show the boxed area in the top panels. Scale bar: 500 μm.
(E) Schematic brain map highlights the locations of major input neurons (>1% total inputs) in the entire brain. The size of each circle indicates the percentage of total inputs.
(F) Distribution of ALDH1A1+ nDAN inputs in different CP subregions as described previously (Hintiryan et al., 2016). Rostral CP (CPr) is divided into subregions as medial (M), intermediate dorsal (D), intermediate ventral (Int. V), and lateral (L). Intermediate CP (CPi) is divided into subregions as dorsomedial (DM), dorsolateral (DL), ventromedial (VM), and ventrolateral (VL). Caudal CP (CPc) is divided into subregions as dorsal (D), intermediate (Int.), and ventral (V).
(G) Percentages of CP area occupied by patches and percentages of ALDH1A1+ nDAN inputs in the patches.
(H and I) ALDH1A1+ nDAN inputs from the cerebral cortex, pallidum, amygdala, and midbrain (H), as well as the hypothalamus and ventral striatum (I), compared with total nDANs.
We next compared the ALDH1A1+ nDANs’ input data with previously published input data that used dopamine transporter (Dat)-Cre mice to target all DANs (Watabe-Uchida et al., 2012). We found that ALDH1A1+ nDANs received fewer innervations from the cerebral cortex, pallidum, amygdala, and midbrain regions (Figure 3H; Figure S6) but more inputs from the VS and hypothalamus (Figure 3I; Figure S6). Differential distribution of afferent neurons was also observed in the ALDH1A1+ mDANs compared with the other mDANs (Figure S7). Altogether, we produced a comprehensive input and output circuit map of ALDH1A1+ DANs in the mouse brain.
Reciprocal Innervations between ALDH1A1+ nDANs and Dorsal CP SPNs
To elucidate the relationship between ALDH1A1+ nDANs and spiny projection neurons (SPNs), we compiled all starter ALDH1A1+ DANs and their corresponding striatal input neurons in a series of coronal sections sampled from the rostral to caudal midbrain and striatum (Figures 4A and 4B). We found that the striatal input neurons were more concentrated in the dorsal regions of the rostral and intermediate CP, an area also heavily innervated by the ALDH1A1+ nDANs (Figure 4C). In contrast, the distributions of striatal input neurons were more random in the caudal CP, which received fewer ALDH1A1+ nDAN projections (Figure 4C). We then generated heatmaps of ALDH1A1+ fibers, as well as the ALDH1A1+ nDAN and mDAN inputs in selective striatal sections (Figure 4D). The intensities of ALDH1A1+ nDAN projections showed stronger correlations with the inputs in the intermediate and rostral striatum compared with the caudal striatum (Figures 4E–4G). By contrast, the projections and inputs of ALDH1A1+ mDANs in different striatal regions were only weakly correlated (Figures 4F and 4G). Collectively, these results support a strong reciprocal innervation between subpopulations of ALDH1A1+ nDANs and SPNs in the dorsal CP.
Figure 4. Reciprocal Innervations between ALDH1A1+ nDANs and DS SPNs.

(A) Schematic brain atlas combines the starter ALDH1A1+ DANs from five Aldh1a1-P2A-CreERT2 mice in 6 sequential midbrain coronal sections. The starters from each mouse are indicated by different colors.
(B) Distribution of starter neurons in different midbrain subregions of five individual mice.
(C) Schematic brain atlas compiles the input neurons from the starters (A and B) in four sequential striatal coronal sections. The input neurons from each mouse are marked by the same color as the starters. Gray represents ALDH1A1 immunostaining in the respective striatal sections. Arrows point to the external globus pallidus (GPe).
(D) Density of combined ALDH1A1+ axon terminals (gray), SNc input neurons (blue, n = 5 mice), and VTA input neurons (red, n = 3 mice) in the selected CPr, CPi, and CPc sections.
(E and F) Correlations between the density of ALDH1A1 innervation in the CPi and the density of CPi input neurons to the SNc (E) and VTA (F), respectively. The plots were overlaid with lines derived from linear regression analyses.
(G) Comparisons of Pearson correlation coefficients (r) between the SNc and VTA starter neurons and their corresponding input neurons in different CP regions.
Genetic Ablation of ALDH1A1+ nDANs Led to Reduced Walking Speed
To elucidate the physiological functions of ALDH1A1+ nDANs, we genetically ablated these neurons in Aldh1a1-P2A-CreERT2 KI mice by bilateral stereotactic injection of SNc with AAVs expressing Cre-dependent apoptotic gene caspase-3 (taCasp3) (Yang et al., 2013). Selective losses of ALDH1A1+ nDAN cell bodies in the SNc and axon terminals in the dorsal CP were observed after viral infection for two to three weeks (Figures 5A and 5B). We carried out an unbiased stereology experiment to estimate the extent of ALDH1A1+ DAN loss in the SNc and VTA. We found an average of 80% loss of ALDH1A1+ nDANs in the taCasp3 AAV-injected mice (Figure 5C). We also obtained high-performance liquid chromatography (HPLC) data on the dopamine content in the DS and found around 60% reduction of dopamine concentration in the taCasp3 AAV-injected mice (Figure 5D).
Figure 5. Less Frequent High-Speed Walking of ALDH1A1+ nDAN-Ablated Mice.

(A) Co-staining of ALDH1A1 and TH in midbrain coronal sections of Aldh1a1-P2A-CreERT2 mice after injection with mCherry or taCasp3-expressing AAVs for three weeks. The arrow indicates the remaining ALDH1A1+ mDANs in the VTA regions. Scale bar: 500 μm.
(B) ALDH1A1, TH, and MOR1 co-staining in a striatal coronal section of the mouse in (A). The arrow indicates the remaining ALDH1A1+ axon terminals in the VS. Scale bar: 500 μm.
(C) Unbiased stereological estimation of remaining ALDH1A1+ DANs in the SNc and VTA of ALDH1A1+ nDAN-ablated mice (n = 3) and control mCherry mice (n = 3). One-way ANOVA with Sidak’s multiple comparison test, ****p < 0.0001.
(D) HPLC measurement of dopamine and 5-hydroxytryptamine (5-HT) content in the DS of ALDH1A1+ nDAN-ablated mice (n = 5) and control mCherry mice (n = 7). One-way ANOVA with Sidak’s multiple comparison test, ***p = 0.0006.
(E–G) Quantifications of the distance (E) (unpaired t test, p = 0.03), average velocity (F) (unpaired t test, p = 0.04), and numbers of movements (G) (unpaired t test, p = 0.38) of ALDH1A1+ nDAN-ablated mice (taCasp3, n = 10 males [M] and 10 females [F]) and age-matched control mice (mCherry, n = 9M, 9F).
(H) Velocity distribution of ALDH1A1+ nDAN-ablated and control mice in open-field tests. Arrows indicates the divergence at a speed > 20 cm/s. Wilcoxon matched-pairs signed rank test, p = 0.005.
(I and J) Comparison of the occurrence of high-speed walking (>20 cm/s) (I) (unpaired t test, p = 0.002) and low-speed walking (<5 cm/s) (J) (unpaired t test, p = 0.13) of ALDH1A1+ nDAN-ablated and control mice in open-field tests.
We used video tracking to monitor the voluntary movement of ALDH1A1+ nDAN-ablated mice in an open-field test four weeks after the viral infection. The ALDH1A1+ nDAN-ablated mice traveled less distance at a slower average walking speed during a session (Figures 5E and 5F). However, the numbers of movement bouts were comparable to the control mice (Figure 5G). We compared the velocity distribution between control and ALDH1A1+ nDAN-ablated mice and found that the ablation made the mice walk at higher speeds less often (Figure 5H). For example, the ALDH1A1+ nDAN-ablated mice walked less frequently at speeds faster than 20 cm/s (Figure 5I). However, the frequency of low-speed walks (<5 cm/s) was comparable between the control and the ablated mice (Figure 5J). To determine whether the lower walking speed resulted from a shorter step, we ran gait analyses in the free-moving animals. We found no apparent alterations in the stride length and the stance and swing time of the ALDH1A1+ nDAN-ablated mice (Figures S8A–S8C). These results demonstrate that the loss of ALDH1A1+ nDANs did not affect gait, or the initiation and termination of locomotion, but instead made the animals walk less often at higher speeds, supporting a role for ALDH1A1+ nDANs in regulating the vigor of locomotion.
We also carried out the pole test, another behavior paradigm, to examine potential nigrostriatal dopamine transmission deficiency in rodents (Ogawa et al., 1985). However, ALDH1A1+ nDAN-ablated mice showed no apparent impairments in the pole test (Figure S8D). ALDH1A1+ nDAN-ablated mice also did not display obvious alterations of anxiety-like responses in the open-field or light and dark box tests (Figures S8E and S8F). Collectively, these behavioral studies support a physiological function of ALDH1A1+ nDANs in regulating walking speed during locomotion.
Genetic Ablation of ALDH1A1+ nDANs Severely Disrupted Rotarod Motor Skill Learning
Because basal ganglion dopamine transmission is important in motor skill learning (Valentin et al., 2016), we applied a well-adopted repeated accelerating rotarod test (Costa et al., 2004; Yin et al., 2009) to evaluate the motor skill learning of ALDH1A1+ nDAN-ablated mice. We started the tests 28 days after the viral infection, ensuring a substantial ablation of taCasp3-expressing ALDH1A1+ nDANs (Figure 6A). The ALDH1A1+ nDAN-ablated mice performed similarly to the control mice in the first trial on the first day of the test (Figure 6B). However, their performance failed to improve in the subsequent trials over the 6-day training course compared with the controls (Figures 6B and 6C). These data revealed critical involvement of ALDH1A1+ nDANs in the acquisition of skilled movements.
Figure 6. Impaired Rotarod Motor Skill Learning of ALDH1A1+ nDAN-Ablated Mice.

(A) Timeline of viral injections and repeated accelerating rotarod tests after the ablation of ALDH1A1+ nDANs.
(B) Comparison of performance on the day 1, first 10 trials of ALDH1A1+ nDAN-ablated mice (Aldh1a1-P2A-CreERT2/Flex-taCasp3 [taCasp3], n = 6M, 12F) and two control cohorts (Aldh1a1+/+/Flex-taCasp3, n = 8M, 13F; Aldh1a1-P2A-CreERT2/Flex-mCherry [mCherry], n = 6M, 12F). Two-way ANOVA: performance, F(9, 504) = 8.86, p < 0.0001; treatment, F(2, 52) = 18.29, p < 0.0001. Tukey’s multiple comparison test: trial 1, mCherry versus taCasp3, p = 0.20; mCherry versus WT, p = 0.57; taCasp3 versus WT, p = 0.75. Trials 2–10, mCherry versus taCasp3, p < 0.01; mCherry versus WT, p = 0.51–0.98; taCasp3 versus WT, p < 0.01.
(C) Comparison of performance in 6-day repeated rotarod tests. Two-way ANOVA: performance, F(2, 260) = 29.04, p < 0.0001; treatment, F(2, 52) = 34.17, p < 0.0001. Tukey’s multiple comparison test: mCherry versus taCasp3, adjusted p < 0.0001; mCherry versus WT, p = 1.00; taCasp3 versus WT, p < 0.0001.
(D) Timeline of viral injections and repeated accelerating rotarod tests before ALDH1A1+ nDAN ablation.
(E) Comparison of performance in 27-day repeated rotarod tests of ALDH1A1+ nDAN-ablated mice (taCasp3, n = 6M, 4F) and controls (mCherry, n = 7M, 6F). Two-way ANOVA: performance, F(134, 1,340) = 5.78, p < 0.0001; treatment, F(1, 10) = 0.21, p = 0.411. Then, 6-day repeated rotarod tests. Two-way ANOVA: performance, F(29, 290) = 1.91, p = 0.0042; treatment, F(1, 10) = 1.36, p = 0.27.
(F) Post hoc histological study showing the percentages of ALDH1A1+ areas in the striatal sections of Aldh1a1-P2A-CreERT2 mice injected with mCherry AAVs (n6B = 2, n6E = 2) or taCasp3 AAVs (n6B = 4, n6E = 5). Two-way ANOVA: bregma, F(3, 40) = 22.91, p < 0.0001; treatment, F(2, 40) = 37.71, p < 0.0001. Tukey’s multiple comparison test: bregma 1.18, mCherry versus taCasp3-6B or taCasp3-6E, p < 0.0001; bregma 0.82, mCherry versus taCasp3-6B or taCasp3-6E, p = 0.02 or 0.04, respectively; bregma 0.5, mCherry versus taCasp3-6B or taCasp3-6E, p = 0.02 or 0.06, respectively; bregma −0.06, mCherry versus taCasp3-6B or taCasp3-6E, p = 0.003 or 0.02, respectively.
To investigate whether the ablation of ALDH1A1+ nDANs also affects the retention and maintenance of skilled movements, as well as overall performance, we started the rotarod tests 7 days after the viral infection and continued training the mice for the next 27 days (Figure 6D). We then rested the mice for 30 days before another 6-day trial series (Figure 6D). Because there was no apparent loss of ALDH1A1+ nDANs after viral infection for 7 days, the taCasp3-infected mice learned as well as the control mCherry-infected mice in the first 4 days of trials (Figure 6E). Moreover, the taCasp3-infected mice continued to perform well during trials after the ablation of ALDH1A1+ nDANs (Figure 6E). Post hoc histological analyses confirmed a similarly substantial loss (>70%) of ALDH1A1+ fibers in the dorsal CP of the taCasp3-infected mice randomly selected from the preceding two cohorts of mice after finishing the behavioral tests described in Figures 6B and 6E (Figure 6F). Altogether, our findings support a critical involvement of ALDH1A1+ nDANs in the acquisition phase of motor skill learning, but not in the retention or performance of learned motor skills.
Dopamine Replacement Therapy Improved Walking Speed, but Not Motor Skill Learning, in the ALDH1A1+ nDAN-Ablated Mice
Because dopamine precursor levodopa (L-DOPA) is widely applied to manage PD motor symptoms (Mhyre et al., 2012), we treated the ALDH1A1+ nDAN-ablated mice, which had already undergone a 6-day rotarod test, with L-DOPA 1 h before the motor behavioral tests. The administration of L-DOPA allowed the ALDH1A1+ nDAN-ablated mice to travel longer distances (Figure 7A), walk less often at lower speeds and walk more frequently at higher speeds during a session (Figures 7B–7D). However, L-DOPA failed to improve the motor skill learning of ALDH1A1+ nDAN-ablated mice in the repeated rotarod tests (Figure 7E). Considering that the extensive loss of ALDH1A1+ nDAN axon terminals might compromise the conversion of L-DOPA to dopamine in the dorsal CP of ALDH1A1+ nDAN-ablated mice, we treated the animals with dopamine receptor D1 (DRD1) agonist SKF81297, which directly stimulates the postsynaptic striatal neurons (Arnt et al., 1988). However, the administration of SKF81297 was snot effective in improving the performance of ALDH1A1+ nDAN-ablated mice in the rotarod tests (Figure 7E). In contrast, a dopamine receptor D2 (DRD2) agonist, quinpirole, severely impaired the mobility of both mCherry and tsCasp3 mice, which failed to move in the rotating rod (data not shown). These results demonstrate that while dopamine supplementation is effective in promoting walking speed, systemic injection of neither L-DOPA nor SKF81297 is sufficient to restore the function of ALDH1A1+ nDANs in motor skill learning.
Figure 7. Dopamine Replacement Therapy of ALDH1A1+ nDAN-Ablated Mice.

(A and B) Quantifications of the distance (A) (unpaired t test, two-tailed, mCherry+saline, n = 1M, 3F, versus mCherry+L-DOPA, n = 1M, 3F, p = 0.65; taCasp3+saline, n = 4M, 3F, versus taCasp3+L-DOPA, n = 5M, 2F, p = 0.0066), and velocity distribution of control and ALDH1A1+ nDAN-ablated mice treated with L-DOPA or saline in open-field tests (B). Arrows indicates the divergence at a speed > 15 cm/s. Wilcoxon matched-pairs signed rank test, p < 0.0001.
(C and D) Comparison of the occurrence of high-speed walking (D) (unpaired t test, p = 0.002) and low-speed walking (C) (unpaired t test, p = 0.13) of ALDH1A1+ nDAN-ablated and control mice in open-field tests.
(E) Comparison of the performance of control and ALDH1A1+ nDAN-ablated mice treated with saline, L-DOPA, or SKF81297 (mCherry+saline: n = 1M, 3F, mCherry+L-DOPA: n = 1M, 3F, mCherry+SKF81297: n = 3M, 5F; taCasp3+saline: n = 2M, 2F, taCasp3+L-DOPA: n = 2M, 3F, taCasp3+SKF81297: n = 4M, 5F) in 6-day repeated rotarod tests. Two-way ANOVA for the taCasp3 mice: performance, F(5, 84) = 0.44, p = 0.82; treatment, F(2, 84) = 7.44, p = 0.001. Tukey’s multiple comparison test: taCasp3+saline versus taCasp3+L-DOPA, p = 0.79; taCasp3+saline versus taCasp3–SKF81297, p = 0.12; taCasp3+L-DOPA versus taCasp3–SKF81297, p = 0.40.
Selective Deletion of Dopamine Transporter DAT in ALDH1A1+ nDANs Promoted Walking Speed, but Not Motor Skill Learning
Dopamine transporter DAT mediates the reuptake of extracellular dopamine in DANs, and suppression of DAT activity increases extracellular dopamine levels and promotes movement (Sulzer et al., 2016). We selectively deleted DAT in the ALDH1A1+ DANs of Aldh1a1+/P2A-CreERT2/Datfl/fl bigenic mice, which showed reduced DAT expression in the cell bodies and axon terminals of ALDH1A1+ nDAN (Figures S9A and S9B). Video tracking showed that Dat conditional knockout (KO) mice traveled longer distances with less frequent lower-speed and more frequent higher-speed walking even as they displayed comparable learning curves and performance in the rotarod tests compared with the controls (Figures S9C–S9G). Therefore, genetic deletion of DAT in ALDH1A1+ DANs promoted walking speed but did not affect motor skill learning in the Dat conditional KO mice.
Genetic Suppression of ALDH1A1 Expression in ALDH1A1+ nDANs Did Not Affect Motor Skill Learning
ALDH1A1 mediates the synthesis of inhibitory neurotransmitter γ-aminobutyric acid (GABA) in ALDH1A1+ DANs (Kim et al., 2015). Genetic suppression of ALDH1A1 in ALDH1A1+ DANs does not affect locomotion, but it renders the mice more susceptible to excessive consumption of alcohol (Kim et al., 2015; Pan et al., 2019). Genetic deletion of ALDH1A1 also leads to an increase of dopamine release in the DS (Sgobio et al., 2017). To examine whether the enzymatic activity of ALDH1A1 affects locomotion and motor skill learning, we suppressed the expression of ALDH1A1 in ALDH1A1+ nDANs by stereotactic injection of AAVs expressing both Aldh1a1 short hairpin RNA (shRNA) and GFP transgenes (AAV-Aldh1a1 shRNA/GFP) (Kim et al., 2015) in the SNc of 3-month-old wild-type C57BL/6J mice (Figure S10A). Subsequent behavioral tests demonstrated that while genetic suppression of ALDH1A1 expression in the ALDH1A1+ DANs slightly increased the occurrence of high-speed walking in the open-field tests, it did not affect motor skill learning in the rotarod tests (Figures S10B and S10C). These results demonstrate that ALDH1A1-mediated GABA synthesis is not required for motor skill learning.
DISCUSSION
We and others have shown a distinct projection pattern of ALDH1A1+ nDANs in the DS (Pan et al., 2019; Poulin et al., 2018; Sgobio et al., 2017). Here we extended the previous studies and provided a more detailed characterization of ALDH1A1+ DANs in different SNc and VTA subdivisions. We delineated the projection patterns of small numbers of ALDH1A1+ nDANs and mDANs clustered in overlapping midbrain subregions, both of which seem to arrange their axon fibers to the striatum along the medial to lateral axis in correlation with their cell body locations. Furthermore, ALDH1A1+ DANs in the more caudal midbrain regions tend to innervate the more rostral striatal regions. As expected, ALDH1A1+ nDANs project heavily to the DS, the striatal region that is also heavily innervated by the sensorimotor cortices (Hintiryan et al., 2016). In addition to innervating the VS, ALDH1A1+ mDANs in the lateral VTA (PBP) innervate part of the DLS and DMS, which is consistent with previous reports (Beier et al., 2015; Lammel et al., 2008, 2011). Moreover, the projections of ALDH1A1+ mDANs in the DS were more evident in rostral striatal regions, which also receive mixed innervations from both associative and sensorimotor cortices (Hintiryan et al., 2016). The convergence of diverse cortical glutamatergic and midbrain dopaminergic inputs in the rostral DS indicates the functional importance of this striatal region. In the current experimental setting, we cannot distinguish the ALDH1A1+ nDANs in the medial SNc and the ALDH1A1+ mDANs in the lateral VTA. With the accumulation of increasing numbers of single-cell RNA sequencing (RNA-seq) data, we expect additional genetic markers could be available to molecularly define different subpopulations of ALDH1A1+ nDANs and mDANs.
Regarding afferents, ALDH1A1+ nDANs receive differential monosynaptic inputs compared with the total nDAN populations (Watabe-Uchida et al., 2012). Specifically, ALDH1A1+ nDANs receive more innervations from VS and hypothalamus but less from cerebral cortices, pallidum, amygdala, and midbrain regions. Within cerebral cortices, primary and secondary motor cortices and somatosensory cortex provide the most inputs to the ALDH1A1+ nDANs, implying functional involvement of ALDH1A1+ nDANs in regulating sensorimotor activity. Furthermore, we revealed a strong correlation between distributions of ALDH1A1+ nDANs’ afferent neurons and axon fibers in the DS, supporting reciprocal innervation between subpopulations of ALDH1A1+ nDANs and SPNs in the dorsal DS. This finding is in line with previous observations that the DLS- and DMS-projecting nDANs receive reciprocal innervations from the DLS and DMS SPNs, respectively (Lerner et al., 2015). The DLS-projecting DANs are largely located in SNCD, while the DMS-projecting DANs are in SNCM. Most of those DLS- and DMS-projecting DANs are ALDH1A1+ nDANs. However, another study failed to establish such reciprocal connections between DANs and SPNs (Menegas et al., 2015). Because we did not observe substantial reciprocal connections between ALDH1A1+ mDANs and SPNs, we suspect that this discrepancy likely resulted from grouping DS-projecting nDANs and mDANs together in that study (Menegas et al., 2015). Reciprocal connections between ALDH1A1+ nDANs and SPNs may provide the anatomical framework for a feedback loop that enables the timely modulation of ALDH1A1+ nDANs in motor control by SPNs.
A previous study also described clusters of nDAN inputs located in the DS patch compartments marked by negative calbindin staining (Watabe-Uchida et al., 2012). In the present study, we used MOR1, a positive marker, to better define the boundary of patches compared with calbindin. We found some clusters of input neurons were located in the patch compartments. However, we observed other clusters of input neurons in the matrices. Because there were no apparently biased connections between ALDH1A1+ DANs and patch SPNs, our data did not support the notion that ALDH1A1+ nDANs preferentially receive monosynaptic inputs from the patch neurons in the DS. Instead, we provided evidence that both patch and matrix SPNs innervate ALDH1A1+ nDANs in the ventral SNc. It is still possible that a subpopulation of ALDH1A1+ nDANs forms strong reciprocal connections with patch SPNs.
As for the physiological function of nDANs, one study establishes a causal relationship between the activity of nDANs before movement initiation and the probability and vigor of future movements (da Silva et al., 2018). Accordingly, an inhibition of nDAN activity would make an animal walk less often and more slowly. We did observe a reduction in high-speed walking in the ALDH1A1+ nDAN-ablated mice in the open-field tests. However, we did not find obvious alterations in the numbers of movement bouts. We suspect that ALDH1A1+ nDANs are more engaged in regulating the vigor of movements than in regulating the initiation. Because the earlier study combined both ALDH1A1+ and ALDH1A1− nDANs in the SNc (da Silva et al., 2018), ALDH1A1− nDANs may play a more important role in regulating the initiation of movements. Live imaging of neural activity in behaving animals by miniature microscope or fiber photometry will be employed to directly correlate the activity of different DAN subtypes with specific motor behavioral phenotypes.
Compared with a modest reduction in high-speed walking, the ALDH1A1+ nDAN-ablated mice showed a more severe impairment in rotarod motor skill leaning. Unlike control animals, ALDH1A1+ nDAN-ablated mice essentially failed to improve their performance during the course of rotarod tests. The repeated rotarod test is a well-adopted motor training paradigm to examine the motor skill learning in rodents (Sommer et al., 2014), and it includes both the initial acquisition phase to optimize the foot placement on the rotating rod and the later retention phase to maintain the optimal stepping practice (Cao et al., 2015). When the ALDH1A1+ nDANs were ablated after the mice had reached maximal performance, the ablation no longer affected the test results, supporting an essential function of ALDH1A1+ nDANs in the acquisition of skilled movements. These findings are in line with the theory that nigrostriatal dopamine serves as the key feedback cue for reinforcement learning mediated by the cortico-striatal networks (Valentin et al., 2016). With the disruption of dopamine transmission in both DLS and DMS in ALDH1A1+ nDAN-ablated mice, our results are also consistent with the notion that the associate cortex-DMS and sensorimotor-DLS circuits function in parallel during motor skill training (Kupferschmidt et al., 2017). Therefore, our findings unravel an essential physiological function of ALDH1A1+ nDANs in the acquisition of skilled movements.
High temporal resolution of dopamine inputs in the DS is proposed as a key requirement for reinforcement learning by cortico-striatal circuits, in which dopamine release would be temporally paired with a correct response, while the residual extracellular dopamine should be removed quickly to avoid coupling with a potentially wrong response (Hélie et al., 2015). This theory may explain how the low temporal resolution of dopamine replacement therapy failed to rescue the motor learning impairments in the ALDH1A1+ nDAN-ablated mice. Furthermore, our circuit mapping study revealed that ALDH1A1+ nDANs receive diverse presynaptic inputs from various brain regions, including the striatum and cerebral cortex. We provide evidence that ALDH1A1+ nDANs form reciprocal innervations with the SPNs in the DS. These results imply that the activity of ALDH1A1+ nDANs can be dynamically regulated by afferent striatal neurons in response to different behavioral paradigms. Systematic administration of L-DOPA or dopamine receptor agonists apparently cannot mimic the dynamic release of dopamine from ALDH1A1+ nDANs under normal circuit control during the motor learning process. In support of this notion, dopamine replacement therapy is less effective in treating learning and memory deficiency in PD patients (Emre, 2003; Heremans et al., 2016). However, L-DOPA was shown to enable a rescue of rotarod motor skill learning in a line of paired-like homeodomain 3 (Pitx3) spontaneous KO mice, which fail to develop the nDANs in the embryonic stages (Beeler et al., 2010). We suspect that a more robust adaptation to the lack of dopamine inputs during early development likely causes the Pitx3 KO mice to apply a different mechanism to achieve motor learning and the resulting, adapted neural circuits are somehow more responsive to the L-DOPA treatment.
ALDH1A1+ nDANs also possess some special intrinsic properties compared with the other DANs. ALDH1A1+ nDANs contain a higher level of dopamine transporter DAT (Sgobio et al., 2017), which would enhance the reuptake of dopamine from the extra-cellular space and improve the temporal resolution of dopamine signaling. However, our observations from the DAT conditional KO mice did not support critical involvement of DAT activity in ALDH1A1+ nDAN-mediated motor skill learning. A lack of DAT might trigger as-yet-unspecified compensatory pathways for the removal of dopamine from the extracellular space. ALDH1A1+ nDANs can also produce and release the inhibitory neurotransmitter GABA in the striatum (Kim et al., 2015). Genetic deletion of Aldh1a1 in ALDH1A1+ DANs blocks GABA synthesis and makes the animals more susceptible to excessive alcohol drinking (Kim et al., 2015). By contrast, ALDH1A1 deficiency did not affect the performance of rotarod tests, suggesting that ALDH1A1-mediated GABA synthesis and release is not required for the motor skill learning.
The observed failure of dopamine replacement therapy to rescue motor learning in this study may portend a weakness in cell therapy approaches for PD. Fetus and induced pluripotent stem cell (iPSC)-derived dopaminergic progenitor cells had been successfully transplanted into the putamen of PD patients and primate PD models. The precursor cells appear to be able to differentiate into mature DANs, release dopamine, and mitigate many PD-related motor symptoms (Björklund and Lindvall, 2017; Kikuchi et al., 2017). However, the grafted DANs were unlikely to fully re-establish complex, natural connections with the rest of the brain. As we demonstrated in this study, for a more demanding task such as rotarod motor skill learning, the proper synaptic inputs to ALDH1A1+ nDANs are critical for providing the timely dopamine release required in the learning process. Therefore, more challenging motor skill learning tests would be useful to critically evaluate how effectively the grafted DANs are integrated into the neural circuits that govern complexed behaviors.
In summary, this study establishes an essential physiological function of ALDH1A1+ nDANs in rotarod motor skill learning and illustrates the utility of rotarod testing to reveal a complex motor phenotype in PD-related rodent models (Shiotsuki et al., 2010). Future studies will be needed to understand how ALDH1A1+ nDANs regulate motor skill learning at both cellular and circuit levels. It is also of interest to examine the roles of ALDH1A1+ nDANs in other forms of learning and memory, as well as the connectivity and functionality of ALDH1A1− DANs. Therefore, our work represents an initial attempt to understand specific physiological functions and underlying circuit mechanisms of molecularly defined midbrain DAN subpopulations in regulating various behavioral phenotypes.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Mouse lines generated in this study will be deposited to the Jackson Laboratory. Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Huaibin Cai (caih@mail.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All mouse studies were in accordance with the guidelines approved by Institutional Animal Care and Use Committees (IACUC) of the National Institute on Aging (NIA), NIH. Mice were housed in NIH mouse facility in a twelve-hour-light /twelve-hour-dark cycle and were fed water and regular diet ad libitum. All the behavioral tasks were performed during the light cycles. The gender and age of mice used for experiments were indicated in the figure legends.
Generation of Aldh1a1-P2A-CreERT2 knock-in mouse line
An ~8.0 kb genomic fragment containing the Aldh1a1 locus was digested from BAC clone B6Ng01-066I24 (RIKEN BRC) by KpnI and AvrII, and subcloned into modified pBlueScript II SK (+) vector (Agilent Technologies) for further modifications. Peptide P2A DNA fused with CreERT2 (P2A-CreERT2) was generated by PCR using plasmid pCAG-CreERT2 (Addgene) as the template and was then inserted between the last coding codon and the stop codon of Aldh1a1 gene. The 5′ recombination arm together with P2A-CreERT2 and the 3′ recombination arm were separately subcloned into pDNDF-7 backbone (Shim et al., 2012) to generate the targeting construct pDNDF-7-DTA-5′ arm-P2A-CreERT2-STOP-Frt-Neo-Frt-3′ arm-DTA. The AvrII and SphI enzyme sites for 5′ and 3′ Southern blots were introduced into the construct near the Frt-Neo-Frt cassette during subcloning. All PCR fragments were validated by sequencing. Targeting construct pDNDF-7-DTA-5′ arm-P2A-CreERT2-STOP-Frt-Neo-Frt-3′ arm-DTA was linearized by BsiWI enzyme and then was electroporated (Gene Pulser II, Bio-Rad) into male C57BL/6N JM8A3 Embryonic Stem Cells (ESCs) (UCDAVIS KOMP Repository). The JM8A3 ESCs were cultured in 2i Medium [KnockOut DMSM (GIBCO), KnockOut Serum Replacement (GIBCO), Insulin (Sigma-Aldrich), LIF (Millipore), MEK inhibitor PD0325901 (Tocris) and GSK3 inhibitor CHIR99021 (Tocris)] as described in details on the website (https://www.komp.org) (Pettitt et al., 2009). ESC colonies were selected by G418 (Sigma-Aldrich) for 7 days. Successful recombination was validated by Southern blot. For Southern blot, the 5′ probe and 3′ probe were amplified by PCR (KAPA HiFi PCR Kit) from BAC clone B6Ng01-066I24 and were radioactively labeled using Amersham Rediprime II DNA Labeling System (GE Healthcare) with dCTP, [α−32P] (PerkinElmer). Two positive ESC clones were microinjected into blastocysts in NCI (National Cancer Institute) Transgenic Mouse Facility. Male chimeric mice of agouti coat more than 50% were bred with congenic C57BL/6N homozygous ACTBFLPe female (The Jackson Laboratory). The resulting Aldh1a1-P2A-CerERT2 mice were genotyped by multiplex PCR with primers Ald1-wt-F, Ald1-Cre-F and Ald1-wt-R. The sizes of wild-type allele and knock-in allele are 394 bp and 349 bp respectively. The deletion of the Neo-Frt cassette and the residual Frt was confirmed by genotyping with primers Ald1-delNeo-F and Ald1-delNeo-R. The sizes of wild-type allele and Frt allele are 241 bp and 323 bp respectively. The Aldh1a1-P2A-CerERT2/ACTBFLPe mice were backcrossed with wild-type C57BL/6J mice (The Jackson Laboratory, JAX) to breed out ACTBFLPe transgene. Aldh1a1-P2A-CerERT2 mice were further backcrossed with C57BL6/J mice for at least two generations for the later experiments.
The Aldh1a1-P2A-CerERT2/Ai9 reporter mouse line
The Aldh1a1-P2A-CerERT2 mice were bred with congenic C57BL6/J homozygous Ai9 mice (Madisen et al., 2010) from JAX to generate the Aldh1a1-P2A-CerERT2/Ai9 reporter mouse line. 4-Hydroxytamoxifen (4-OHT) (Sigma-Aldrich) was first dissolved in ethanol (Sigma-Aldrich) at a concentration of 20 mg/ml and was then emulsified in two-fold volume of corn oil (Sigma-Aldrich). Ethanol was carefully vaporized using a speed vacuum (Eppendorf) and 4-OHT solution was diluted in corn oil to make 1 mg/ml solution. The 4-OHT solution was filtered with 0.2 μm membrane (Corning), aliquoted and stored in −40°C freezer. Aldh1a1-P2A-Cer-ERT2/Ai9 reporter mice were injected intraperitoneally (i.p.) of 4-OHT at 2 months of age with the doses varying from 0 – 10 mg per kilogram (mg/kg) bodyweight and were sacrificed at ages of 3-month-old.
Generation of Datfl/fl mouse line
C57BL/6N Slc6a3tm1a(KOMP)Wtsi Embryonic Stem Cells (UCDAVIS KOMP Repository) were expanded and the microinjection of ESC clones into blastocysts was performed in NCI Transgenic Mouse Facility. Male chimeric mice of agouti coat more than 50% were crossbred with female homozygous ACTFLPe mice to generate Slc6a3fl/+ mice. Slc6a3fl/+ mice were crossbred with Aldh1a1+/P2A-CreERT2 mice to obtain Aldh1a1+/P2A-CreERT2/Slc6a3+/fl and Slc6a3+/fl mice. Then Aldh1a1+/P2A-CreERT2/Slc6a3+/fl mice and Slc6a3+/fl mice were crossbred to produce Aldh1a1+/P2A-CreERT2/Slc6a3fl/fl mice and Slc6a3fl/fl mice. All the Aldh1a1+/P2A-CreERT2/Slc6a3fl/fl (also known as Aldh1a1+/P2A-CreERT2/Datfl/fl) and Slc6a3fl/fl (also known as Aldh1a1+/+/Datfl/fl, or Datfl/fl) cohorts were expanded by further crossbreeding. Genotyping of Dat+/fl and Datfl/fl mice were performed by PCR with primers CSD-Slc6a3-F and CSD-Slc6a3-ttR. The sizes of wild-type allele and floxed allele were 483 bp and 680 bp respectively. Both Aldh1a1+/P2A-CreERT2/Datfl/fl and Datfl/fl cohorts were i.p. injected with 4-OHT at 2 months of age with the dose of 10 mg/kg bodyweight.
METHOD DETAILS
Immunohistochemistry and light microscopy
Anesthetized mice were first perfused with PBS and then with 4% paraformaldehyde (PFA) (Electron Microscopy Sciences). Dissected mouse brains were post-fixed in 4% PFA overnight and dehydrated in 30% sucrose. 40 μm coronal or sagittal brain slices were sectioned using Leica cryostat CM3050S (Leica Biosystems) unless specific thickness of slices was indicated. Mouse brain sections were rinsed in 0.3 M Glycine in PBS for 20 minutes at room temperature and then were incubated in PBS + 0.3% Triton X-100 + 10% Donkey serum (Sigma-Aldrich) at 4°C overnight. Primary antibodies specific to ALDH1A1 (Rabbit 1:1000, Sigma-Aldrich; Goat 15 μg/mL, R&D Systems), TH (Mouse 1:1000, ImmunoStar; Rabbit 1:1000, Pel-Freez Biologicals; Chicken 1:500, Aves Labs), RFP (Rabbit 1:1000, Rockland), GFP (Chicken 1:500, Aves Labs), MOR1 (Rabbit 1:3000, ImmunoStar), Calbindin (Mouse 1:500, Abcam), and DAT (Rat 1:1000, Millipore; Rabbit 1:1000, Abcam) were freshly diluted in PBS + 0.3% Triton X-100 + 1% Donkey serum and were used in combinations indicated in the figure legends. Brain sections were incubated in primary antibody solutions with continuous gentle shaking at 4°C overnight. After extensive washes, Alexa Fluor (ThermoFisher Scientific) or Dylight (Jackson ImmunoResearch) 405-, 488-, 546-, or 647-conjugated secondary antibodies against the specific hosts of primary antibodies were used as suggested by manufacturers to visualize the staining. Optionally, Hoechst 33342 (Sigma-Aldrich) of 1:10,000 dilution was used to counterstain the cellular nuclei when applicable. Fluorescence images were acquired using a laser scanning confocal microscope LSM 780 (Zeiss). 10 × objective lens and 10 μm z-intervals were used to capture whole brain or whole hemisphere section images, 20 × objective lens and 8 μm z-intervals were used to capture whole striatal section images, 40 × objective lens and 4 μm z-intervals were used to capture whole SNC or VTA region images, whereas 63 × objective lens and 0.5–1 μm z-intervals were used to capture neuronal projection terminals. All the tiling images were acquired with 10% overlapping. The resolution of 10 × images were 512 pixels by 512 pixels, and all the other images were at the resolution of 1024 pixels by 1024 pixels. Paired samples used in comparison were collected and processed at the same time in the same way, and images were captured using the same laser intensities, gains and offsets. Also, post-acquisition image processing was applied to all paired images uniformly. The images were presented either as maximum intensity projections of z series stacks or as single optic layers.
Quantification of ALDH1A1-positive neurons
Three whole brains of 3-month-old male C57BL/6J mice were sectioned coronally from rostral to caudal regions. Every two brain slices were co-stained with anti-ALDH1A1 and anti-TH, and counterstained with Hoechst 33342 (Sigma-Aldrich). The whole hemisphere images of each mouse were captured using LSM 780 confocal microscopy with 10 × objective lens. The ALDH1A1+/TH+ neurons, ALDH1A1−/TH+ neurons and almost neglectable ALDH1A1+/TH− neurons from different subregions of midbrains were registered and summarized using Neurolucida (MBF Bioscience). TH+ neurons outside midbrain regions such as locus coeruleus (LC) were not registered since there were no identifiable ALDH1A1+ neurons in those regions.
Stereotactic injection
Stereotactic survival surgery was performed with aseptic technique and in accordance with the ARAC guidelines for Survival Rodent Surgery. Anesthetized mice were mounted on a stereotaxic frame (Stoelting), and the skulls of the mice were exposed by a scalp cut. Bregma and Lambda coordinates were used to adjust the position of the mouse brains. Small holes in mouse skulls were drilled by a small 0.6 mm drill bit at the chosen coordinates. The coordinates for SNc are AP +1.0 mm (based on Lambda coordinate), ML +1.5 mm or −1.5 mm (bilateral: ± 1.5 mm), DV −4.2 mm; and the coordinates for VTA are AP +0.9 mm (from Lambda coordinate), ML +0.5 mm or −0.5 mm (bilateral: ± 0.5 mm), DV −3.8 mm. 50 nL to 1 μL of AAVs titers from 1012 to 1013 g.c. /mL were loaded into 2 μL Neuros Syringes (Hamilton) and were injected into brain areas at chosen coordinates. The infusion of AAVs was controlled by a motorized stereotaxic injector (Stoelting) at a speed of 75 nL/min. Then the needle of the syringe stood for one more minute and was withdrawn slowly to allow the virus to diffuse extensively. After all injections, Vetbond tissue adhesive (3M) was applied to the wound for a quick and painless closure. Mice were subcutaneously injected with ketoprofen in lactated Ringers solution at a dose of 5 mg/kg body weight and re-dosed every 24 hours for three more days. Every two postoperative mice were housed in single cage with a transparent divider. Both young and old mice, with bodyweight greater than 18 g were used for surgery. However, for surgery mice involved in behavioral tasks, 8- to 11-week old littermates were randomly assigned to control groups and experimental groups, and the genders of the mice were distributed roughly evenly. Mice were i.p. injected with 4-OHT of 10 mg/kg body weight one week after surgery unless specified.
Sparse labeling
AAV1-CAG-flex-tdTomato with titer of 1012 g.c. /mL was diluted to 1:50 and 50 – 100 nL of diluted AAV was injected to SNc or VTA of Aldh1a1-P2A-CreERT2 mice unilaterally. Mouse brain sections were sectioned from rostral to caudal and were co-stained with anti-ALDH1A1, anti-TH and anti-RFP antibodies, the latter of which was used to boost the intensity of fluorescent protein tdTomato. Every midbrain section from rostral and caudal was imaged and all tdTomato-positive (tdTomato+) neurons from different midbrain sub-regions were registered and summarized using Neurolucida (MBF Bioscience). The tdTomato+ neurons with only partial soma on the boundaries of slices were not counted. In six of the labeled mouse brains, 95% of all tdTomato+ cells were also ALDH1A1+/TH+ neurons, and these were subsequently used for further analyses. For the same mice, every other striatal section from rostral to caudal was co-stained to guarantee that the seven representative images of ALDH1A1 staining could be aligned well among all six subjects.
Rabies labeling
AAV5-CAG-flex-TCB (-TVA-mCherry with more brightness) and AAV8-CAG-flex-RG with titers of 1013 g.c. /mL were mixed 1:1 in volume and were injected into SNc or VTA of Aldh1a1-P2A-CreERT2 mice unilaterally. The wound was sutured due to the need for the second injection. One week later, 10 mg/kg bodyweight of 4-OHT was administrated through i.p. and 3 weeks after surgery, 0.3 – 0.5 μL of EnvA G-deleted Rabies-eGFP with titers of 107 – 108 TU/mL was injected into the same site as the previous AAV injections. Mice were housed in Biosafety Level 2 facility and brain tissues were collected six days after the second injection. Specifically, 100 or 50 μm coronal slices were sectioned from rostral to caudal. All 100 μm sections were counterstained with NeuroTrace 435/455 Blue (ThermoFisher Scientific) for counting of the monosynaptic input neurons. 50 μm midbrain sections were co-stained with anti-TH and anti-ALDH1A1 antibodies to define the starter neurons, which were also co-labeled with EGFP and mCherry. Starter neurons from different midbrain subregions were registered and quantified using Neurolucida (MBF Bioscience). 50 μm striatal sections were co-stained with striosome marker anti-MOR1 (ImmunoSTAR) and matrix marker anti-Calbindin (Abcam) antibodies to calculate the occupancy ratios of monosynaptic input neurons in the striosomes and matrixes. Five and three mouse brains were analyzed for SNc and VTA labeling, respectively.
Images analyses
Neuron counting – hemisphere image series of rabies labeled input neurons were analyzed using MATLAB scripts to count the number of input neurons from each brain region. Confocal images were aligned and projected onto a corresponding color-coded Allen Mouse Brain volumetric atlas image (https://scalablebrainatlas.incf.org) (Bakker et al., 2015). The script allowed the user to manually outline a brain region, and subsequently the script would determine the number of neurons along with the brain region name. Neuron soma recognition was achieved with built-in filtering, thresholding, and segmentation functions. Brain region name was determined based on the pixel color value of the center of mass of the outlined region of interest. The pixel color would then map to a corresponding brain region name. This process would repeat until each brain region and input neuron was accounted for. The definition of different subregions of striatum was based on a recently published resource (Hintiryan et al., 2016). CPr denotes rostral CP, including medial (M), intermediate dorsal (D), intermediate ventral (Int.V) and lateral (L) striatum; CPi denotes intermediate CP, including dorsal medial (DM), dorsolateral (DL), ventromedial (VM) and ventrolateral (VL) striatum; and CPc denotes caudate CP, including dorsal (D), intermediate (Int.) and ventral (V) striatum.
Soma and axon terminal mapping – The hemispheric image series from each mouse were assigned a different color. They were then aligned to corresponding Bregmas. For each of the seven Bregmas (1.42mm, 1.15mm, 0.95mm, 0.62mm, 0.25mm, −0.15mm, and −0.56mm), we obtained a representative ALDH1A1 staining and converted it to grayscale to serve as the template. Each image was cropped and resized to its corresponding ALDH1A1 template. The program recognized pixel signal intensity above a certain threshold and converted the fluorescence signal to its assigned color. Pixel color values were added together across the color-coded individual images to create the combined images. The images were then color inverted and slightly dilated to produce the results in the figure shown.
Heatmap – the heatmaps of the distribution of ALDH1A1+ nDAN inputs were created from images in Figure 4C. Three Bregmas were chosen to represent the rostral striatum, intermediate striatum, and the caudal striatum. Each ALDH1A1 grayscale template was divided into 50 × 50 partitions. The pixels values that fell within a partition were summed together for the partition value. For each color-coded brain, we combined corresponding relative partition values to create an average heatmap for SNc inputs at a certain bregma. The results were visualized using MATLAB’s ‘heatmap’ function. The same process was repeated for ALDH1A1+ mDAN inputs.
Quantification of ALDH1A1+ nDAN axon terminal ablation in striatum – Sections corresponding to Bregma 1.18mm, 0.82mm, 0.50mm and −0.06mm of taCasp3 or mCherry-infected mice were co-stained with anti-TH and anti-ALDH1A1 antibodies. Images were taken by LSM 780 confocal microscope under 10 × objective lens (Zeiss). Boundary grids were corrected by using shading correction (Zeiss). The ALDH1A1+ area in the dorsal striatum was outlined by using a threshold to differentiate specific staining from background (ImageJ). The percentages of ALDH1A1+ area from the total striatum area of each Bregma were calculated.
Open-field test
Mice were acclimated for at least an hour in the behavioral room, in which 20 W phosphorescent lamps were shielded in a light box and placed in the dark opposite to the testing area to provid a diffuse source of light. Then the mice were placed in a 60 cm × 60 cm × 40 cm (length × width × height) white opaque chamber, and their activities were recorded by a digital camera (PointGrey) above for 5 minutes. The sampling frequency was 52 frames per second. Before and after the test of each mouse, the chamber was rinsed first with 70% alcohol once and then with sterile water twice. Videos of mice in the open field were analyzed using MATLAB scripts (Barbera et al., 2016). Videos were converted to grayscale so the mouse’s position could be determined by a threshold between the mouse’s black fur and the white chamber. The mouse’s position was registered for each frame, and the velocity was subsequently calculated. The velocity was then smoothed using a Butterworth filter. With the mouse position and velocity for every recorded frame, further analysis could be conducted such as calculating distance traveled, velocity distributions, and time spent in border (8cm from the edge) versus center. A simple bout analysis was implemented to see the number of instances each mouse surpassed a set velocity threshold. The results were similar for multiple thresholds, so 4cm/s was set as the threshold.
Gait analysis
The FreeWalkScan system (CleverSys Inc) was used for gait analysis. Mice were allowed to move freely in a 40 cm × 40 cm × 30 cm (length × width × height) chamber. A high-speed camera below a clear bottom plate was used captured mouse movement for 5 min in the red light. The surface was cleaned with 70% ethanol solution after each test. Videos were analyzed using FreewalkScanTM2.0 software (CleverSys Inc) for various characteristic parameters of gait including stride length and stance/swing time of each paw.
Rotarod task
The Rotarod apparatus (Model 3375-R4, TSE Systems) with grooved rod of 70-mm diameter was used in the task (Beeler et al., 2010). As the test started, the rod accelerated form 0 rpm to 40 rpm in 5min. The duration that each mouse was able to stay on the rotating rod in each trial was recorded as the latency to fall. The standard motor learning task was performed as ten trials per day for six consecutive days (Yin et al., 2009). The long-term motor learning task was performed as five trials per day for 27 consecutive days. Mice were allowed to take a rest for 1 minute between trials. During the test, the rod was kept dry and clean by wiping the mouse urine and feces off.
Pole test
Shortly after videotaping started, the mouse was placed head upward on the upper end of a 50 cm long and 1 cm diameter vertical pole with a cardboard barrier to prevent traversal from the top (Balkaya et al., 2013). When placed on the pole, the mouse turned downward and descended on the floor of the home cage. The time for each mouse to turn around completely and land on the floor of the home cage was recorded, respectively.
Light/dark box test
Light/dark box testing was conducted in a plexiglass chamber with 10cm wide passage between light and dark areas (18 cm × 29 cm each). The light area had white walls and was illuminated by white light (500 lux); the dark area had black walls and was illuminated by red light (perceived to be dark by mice; 40 lux). Mice were acclimated to the dark behavioral room 30 min prior to testing. Each mouse started in the light area and their movements between light and dark areas were recorded for 10 minutes. Less time spent in the light area would suggest elevated anxiety. Chamber surfaces were wiped down with ethanol and water between mice. TopScan 3.0 (CleverSys Inc) was used for animal tracking.
Levodopa, R (+)-SKF81297 and Quinpirole administration
All injections were administrated at 10 μL/g body weight using Monoject™ Ultra Comfort Syringes and mice were returned back to their home cages after injections. 25 mg/kg body weight of levodopa (Sigma-Aldrich) with 12.5 mg/kg body weight of Benserazide hydrochloride (Sigma-Aldrich) was i.p. injected into mice 1 hour prior to behavioral task. 2 mg/kg body weight of SKF81297 (Sigma-Aldrich) was i.p. injected into mice 30 min prior to the behavioral task. 1.5 mg/kg body weight of Quinpirole ((Sigma-Aldrich) was i.p. injected into mice 30 min prior to the behavioral task.
Biogenic amine analyses
Biogenic amines were analyzed in the Vanderbilt University Neurochemistry Core.
Tissue Extraction
The brain sections were homogenized, using a tissue dismembrator, in 100–750 μL of 0.1M TCA, which contains 10−2 M sodium acetate, 10−4 M EDTA, and 10.5% methanol (pH 3.8). Ten microliters of homogenate were used for protein assay. Protein concentration was determined using the BSA Protein Assay Kit (Thermo Scientific). Then samples were spun in a microcentrifuge at 10,000 × g for 20 minutes. The supernatant was removed for biogenic monoamines analysis.
Biogenic Amine Analysis using HPLC-ECD
Biogenic amine concentrations were determined utilizing an Antec Decade II (oxidation: 0.65) electrochemical detector operated at 33°C. Twenty microliter samples of the supernatant were injected using a Water 2707 autosampler onto a Phenomenex Kintex C18 HPLC column (100 × 4.60 mm, 2.6 μm). Biogenic amines are eluted with a mobile phase consisting of 89.5% 0.1M TCA, 10−2 M sodium acetate, 10−4 M EDTA and 10.5% methanol (pH 3.8). Solvent was delivered at 0.6 ml/min using a Waters 515 HPLC pump. Using this HPLC solvent the following biogenic amines were eluted in the following order: Norepinephrine, Epinephrine, DOPAC, Dopamine, 5-HIAA, HVA, 5-HT, and 3-MT. Data acquisition was managed by Empower software (Waters Corporation, Milford, MA USA). Isoproterenol (5 ng/mL) was included in the homogenization buffer for use as an internal standard to quantify the biogenic amines of interest.
Stereological quantification of ALDH1A1+ DANs in the SNc and VTA
A series of coronal sections (40 μm per section, every forth section from Bregma −2.46mm to −4.16mm) were selected and stained with anti-TH and anti-ALDH1A1 antibodies and DAPI for the quantification. The entire midbrain regions were scanned under 10 × objective lens (LSM 780, Zeiss). Nine to 10 sections were scanned per mice. The numbers of ALDH1A1+ DANs in the SNc and VTA were assessed by Stereo Investigator (MBF Bioscience).
QUANTIFICATION AND STATISTICAL ANALYSIS
All the data were analyzed by Prism 7 software (Graphpad). Data were presented as mean ± SEM. N represents animal numbers and is indicated in the figure legends. Statistical significance was determined by comparing means of different groups using t test or ANOVA followed by post hoc tests.
DATA AND CODE AVAILABILITY
All data and custom MATLAB scripts used in analysis are available upon request.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat polyclonal anti-Aldehyde Dehydrogenase 1-A1 (Aldh1a1) | R&D Systems | Cat# AF5869; RRID: AB_2044597 |
| Rabbit polyclonal anti-Aldh1a1 | Sigma-Aldrich | Cat# HPA002123; RRID: AB_1844722 |
| Rabbit polyclonal anti-RFP Pre-adsorbed | Rockland | Cat# 600-401-379; RRID: AB_2209751 |
| Mouse monoclonal anti-Tyrosine Hydroxylase (TH) | ImmunoStar | Cat# 22941; RRID: AB_572268 |
| Rabbit polyclonal anti-TH | Pel-Freez Biologicals | Cat# P40101–150; RRID: AB_2313713 |
| Chicken polyclonal anti-TH | Aves Labs | Cat# TYH; RRID: AB_10013440 |
| Rabbit antiserum anti-Opioid Receptor-Mu (MOR1) | ImmunoStar | Cat# 24216; RRID: AB_572251 |
| Mouse monoclonal [AF2E5] anti-Calbindin | Abcam | Cat# ab75524; RRID: AB_1310017 |
| Chicken polyclonal anti-GFP | Aves Labs | Cat# GFP-1020; RRID: AB_10000240 |
| Rat monoclonal anti-Dopamine Transporter (DAT), N terminus, clone DAT-Nt | Millipore | Cat# MAB369; RRID: AB_2190413 |
| Rabbit monoclonal [EPR19695] anti-Dopamine Transporter (DAT), C terminus | Abcam | Cat# ab184451 |
| Mouse monoclonal anti-NeuN, clone A60 | Millipore | Cat# MAB377; RRID: AB_2298772 |
| Rabbit polyclonal anti-Olig-2 | Millipore | Cat# AB9610; RRID: AB_570666 |
| Rabbit antisera anti-Iba1 | Wako | Cat# 016-20001 |
| Goat polyclonal anti-GFAP | Abcam | Cat# ab53554; RRID: AB_880202 |
| Bacterial and Virus Strains | ||
| Mouse: MSM/Ms BAC clone B6Ng01-066I24 | RIKEN BRC | Mouse BAC clone: B6Ng01-066I24 |
| AAV1-CAG-Flex-tdTomato | Penn Vector Core | Cat# AV-1-ALL864 |
| AAV9-CAG-Flex-EGFP | Penn Vector Core | Cat# AV-9-ALL854 |
| AAV1-EF1a-DIO-eYFP | Penn Vector Core | Cat# AV-1-27056 |
| EnvA G-deleted Rabies-eGFP | Salk Institute | Cat# 32635 |
| AAV5-CAG-Flex-TCB (TVA-mCherry) | Vigene Biosciences | Custom; N/A |
| AAV8-CAG-Flex-RG | Vigene Biosciences | Custom; N/A |
| AAV5-EF1a-Flex-mCherry | Vigene Biosciences | Custom; N/A |
| AAV1-EF1a-Flex-taCasp3-TEVp | Vigene Biosciences | Custom; N/A |
| AAV/DJ-Aldh1a1 shRNA/EGFP | Vigene Biosciences | Custom; N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 4-Hydroxytamoxifen (4-OHT) | Sigma-Aldrich | Cat# H7904; CAS: 68047-06-3 |
| Levodopa (L-DOPA) | Sigma-Aldrich | Cat# 1361009; CAS: 59-92-7 |
| Benserazide hydrochloride | Sigma-Aldrich | Cat# B7283; CAS: 14919-77-8 |
| R(+)-SKF-81297 hydrobromide | Sigma-Aldrich | Cat# S179; CAS: 253446-15-0 |
| Critical Commercial Assays | ||
| dCTP, [α−32P]- 800Ci/mmol 10mCi/ml EasyTide Lead, 250 μCi | PerkinElmer | Cat# NEG513A250UC |
| KAPA HiFi PCR Kit | KAPA Biosystems | Cat# KK2101 |
| Amersham Rediprime II DNA Labeling System | GE Healthcare Life Sciences | Cat# RPN1633 |
| Rapid-hyb Buffers | GE Healthcare Life Sciences | Cat# RPN1636 |
| AvrII Enzyme | New England BioLabs | Cat# R0714L |
| SphI-HF Enzyme | New England BioLabs | Cat# R3182L |
| BsiWI Enzyme | New England BioLabs | Cat# R3553L |
| KpnI-HF Enzyme | New England BioLabs | Cat# R3142L |
| NeuroTrace 435/455 Blue Fluorescent Nissl Stain | ThermoFisher Scientific | Cat# N21479 |
| Experimental Models: Cell Lines | ||
| Mouse: JM8A3 Embryonic Stem Cell line | UCDAVIS KOMP Repository | C57BI76N JM8A3 (RRID: CVCL_J959) |
| Mouse: Slc6a3tm1a(KOMP)Wtsi Embryonic Stem Cell line | UCDAVIS KOMP Repository | Cat# KOMP: CSD46925-1a-Wtsi; RRID: IMSR_KOMP: CSD46925-1a-Wtsi |
| Experimental Models: Organisms/Strains | ||
| Mouse: ACTBFLPe: B6N.Cg-Tg(ACTFLPe)9205Dym/CjDswJ | The Jackson Laboratory | Cat# 019100 RRID: IMSR_JAX:019100 |
| Mouse: Ai9: B6. Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J | The Jackson Laboratory | Cat# 007909 RRID: IMSR_JAX:007909 |
| Mouse: C57BL/6J | The Jackson Laboratory | Cat# 000664; RRID: IMSR_JAX:000664 |
| Oligonucleotides | ||
| P2A sequence: 5′-ggatccggagccacgaacttctctctgttaaagcaagcaggagacgtggaagaaaaccccggtcct-3′ | Szymczak-Workman et al., 2012 | N/A |
| Primers for 5′ Southern Probe 5′-Pb-F: 5′-agagttcaacagccccataaaa-3′ | This paper | Custom; N/A |
| Primers for 5′ Southern Probe 5′-Pb-R: 5′-gtgtgccatgtcaagaagtgtt-3′ | This paper | Custom; N/A |
| Primers for 3′ Southern Probe 3′-Pb-F: 5′-gatggcaagcttcctcataatc-3′ | This paper | Custom; N/A |
| Primers for 3′ Southern Probe 3′-Pb-R: 5′-tacagggttccatgagtgagtg-3′ | This paper | Custom; N/A |
| Genotyping primers for Aldh1a1+/P2A-CreERT2 mice Ald1-wt-F: 5′-cttcaaggagtcactgagggac-3′ | This paper | Custom; N/A |
| Genotyping primers for Aldh1a1+/P2A-CreERT2 mice Ald1-Cre-F: 5′-gcgcgcctgaagatatagaaga-3′ | This paper | Custom; N/A |
| Genotyping primers for Aldh1a1+/P2A-CreERT2 mice Ald1-wt-R: 5′-gctcaagacagtcgcaatgaag-3′ | This paper | Custom; N/A |
| Genotyping primers for Neo-Frt deletion Ald1-delNeo-F: 5′-agtagctacaatggaaagcacct-3′ | This paper | Custom; N/A |
| Genotyping primers for Neo-Frt deletion Ald1-delNeo-R: 5′-acacatctcctatagtaaccagca-3′ | This paper | Custom; N/A |
| Genotyping primers for Dat+/fl (Slc6a3+/fl) mice CSD-Slc6a3-F: 5′-gctgtggtggctactttcactatgc-3′ | UCDAVIS KOMP Repository | N/A |
| Genotyping primers for Dat+/fl (Slc6a3+/fl) mice CSD-Slc6a3-ttR: 5′-aacctgaagaaggtcagcactaccc-3′ | UCDAVIS KOMP Repository | N/A |
| Recombinant DNA | ||
| Plasmid: pDNDF-7 vector | A gift from Hoon Shim | N/A |
| Plasmid: pBlueScript II SK (+) | Aglient Technologies | Cat# 212205 |
| Plasmid: pCAG-CreERT2 | A gift from Connie Cepko | Addgene Plasmid #14797 |
| Plasmid: pAAV-CAG-Flex-TCB | A gift from Liqun Luo | Addgene Plasmid #48332 |
| Plasmid: pAAV-CAG-Flex-RG | A gift from Liqun Luo | Addgene Plasmid #48333 |
| Plasmid: pAAV-EF1a-Flex-taCasp3-TEVp | A gift from Nirao Shah & Jim Wells | Addgene Plasmid #45580 |
| Plasmid: pAAV-Aldh1a1 shRNA/EGFP | A gift from Lu Chen | N/A |
| Software and Algorithms | ||
| Neurolucida | MBF Bioscience | RRID:SCR_001775 |
| Stereo Investigator | MBF Bioscience | RRID:SCR_002526 |
| Prism 7 | GraphPad | RRID:SCR_002798 |
| ZEN Digital Imaging for Light Microscopy | Zeiss | RRID:SCR_013672 |
| Stereo Investigator | MBF Bioscience | RRID:SCR_002526 |
| MATLAB Script for Open-field test | Barbera et al., 2016 | N/A |
Highlights.
Whole-brain connectivity map of ALDH1A1+ nigrostriatal dopaminergic neurons (nDANs)
Reciprocal innervations between ALDH1A1+ nDANs and dorsal striatal neurons
ALDH1A1+ nDANs are essential for rotarod motor skill acquisition
Dopamine therapy fails to restore motor learning in the ALDH1A1+ nDAN-ablated mice
ACKNOWLEDGMENTS
This work was supported in part by the intramural research programs of National Institute on Aging, NIH (AG000928 to H.C.), National Institute of Neurological Disorders and Stroke (NS003135 to Z.K.), National Institute on Drug Abuse, and National Natural Science Foundation of China (81430021 to W.L. and 81430023 to B.T.). We thank members of the Cai lab for their suggestions and technical assistance; Drs. Watabe-Uchida and Uchida of Harvard University for sharing their original monosynaptic input data; Drs. Jun Ding and Lu Chen of Stanford University for providing the Aldh1a1 shRNA AAV vectors; Dr. Hoon Shim for providing the pDNDF-7 gene targeting vector; Drs. David Lovinger and Armando Salinas of NIAAA for inputs in functional studies; the NIMH Rodent Behavioral Core for assisting with various mouse behavioral tests; the NCI Transgenic Mouse Core for blastocyst injections of mouse embryonic stem cells (ESCs); and the Vanderbilt University Neurochemistry Core (supported by the Vanderbilt Kennedy Center on Developmental Disabilities grant 90DD0595) for biogenic amine analyses.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.095.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Arnt J, Bøgesø KP, Hyttel J, and Meier E (1988). Relative dopamine D1 and D2 receptor affinity and efficacy determine whether dopamine agonists induce hyperactivity or oral stereotypy in rats. Pharmacol. Toxicol 62, 121–130. [DOI] [PubMed] [Google Scholar]
- Bakker R, Tiesinga P, and Kötter R (2015). The Scalable Brain Atlas: Instant Web-Based Access to Public Brain Atlases and Related Content. Neuroinformatics 13, 353–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkaya M, Kröber JM, Rex A, and Endres M (2013). Assessing post-stroke behavior in mouse models of focal ischemia. J. Cereb. Blood Flow Metab 33, 330–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbera G, Liang B, Zhang L, Gerfen CR, Culurciello E, Chen R, Li Y, and Lin DT (2016). Spatially Compact Neural Clusters in the Dorsal Striatum Encode Locomotion Relevant Information. Neuron 92, 202–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beeler JA, Cao ZF, Kheirbek MA, Ding Y, Koranda J, Murakami M, Kang UJ, and Zhuang X (2010). Dopamine-dependent motor learning: insight into levodopa’s long-duration response. Ann. Neurol 67, 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier KT, Steinberg EE, DeLoach KE, Xie S, Miyamichi K, Schwarz L, Gao XJ, Kremer EJ, Malenka RC, and Luo L (2015). Circuit Architecture of VTA Dopamine Neurons Revealed by Systematic Input-Output Mapping. Cell 162, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentivoglio M, and Morelli M (2005). The organization and circuits of mesencephalic dopaminergic neurons and the distribution of dopamine receptors in the brain In Handbook of Chemical Neuroanatomy, Dopamine SB, Dunnett M, Bentivoglio A, Björklund., and Hökfelt T, eds. (Elsevier; ), pp. 1–107. [Google Scholar]
- Björklund A, and Lindvall O (2017). Replacing Dopamine Neurons in Parkinson’s Disease: How did it happen? J. Parkinsons Dis 7 (s1), S21–S31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE (2010). Intracellular signalling pathways in dopamine cell death and axonal degeneration. Prog. Brain Res 183, 79–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Liu G, Sun L, and Ding J (2014). Aldehyde Dehydrogenase 1 making molecular inroads into the differential vulnerability of nigrostriatal dopaminergic neuron subtypes in Parkinson’s disease. Transl. Neurodegener 3, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao VY, Ye Y, Mastwal S, Ren M, Coon M, Liu Q, Costa RM, and Wang KH (2015). Motor Learning Consolidates Arc-Expressing Neuronal Ensembles in Secondary Motor Cortex. Neuron 86, 1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbit VL, Ahmari SE, and Gittis AH (2017). A Corticostriatal Balancing Act Supports Skill Learning. Neuron 96, 253–255. [DOI] [PubMed] [Google Scholar]
- Costa RM, Cohen D, and Nicolelis MA (2004). Differential corticostriatal plasticity during fast and slow motor skill learning in mice. Curr. Biol 14, 1124–1134. [DOI] [PubMed] [Google Scholar]
- da Silva JA, Tecuapetla F, Paixão V, and Costa RM (2018). Dopamine neuron activity before action initiation gates and invigorates future movements. Nature 554, 244–248. [DOI] [PubMed] [Google Scholar]
- Emre M (2003). Dementia associated with Parkinson’s disease. Lancet Neurol 2, 229–237. [DOI] [PubMed] [Google Scholar]
- Fan X, Molotkov A, Manabe S, Donmoyer CM, Deltour L, Foglio MH, Cuenca AE, Blaner WS, Lipton SA, and Duester G (2003). Targeted disruption of Aldh1a1 (Raldh1) provides evidence for a complex mechanism of retinoic acid synthesis in the developing retina. Mol. Cell. Biol 23, 4637–4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnley JM, and Lees AJ (1991). Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain 114, 2283–2301. [DOI] [PubMed] [Google Scholar]
- Fu Y, Yuan Y, Halliday G, Rusznák Z, Watson C, and Paxinos G (2012). A cytoarchitectonic and chemoarchitectonic analysis of the dopamine cell groups in the substantia nigra, ventral tegmental area, and retrorubral field in the mouse. Brain Struct. Funct 217, 591–612. [DOI] [PubMed] [Google Scholar]
- Galter D, Buervenich S, Carmine A, Anvret M, and Olson L (2003). ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol. Dis 14, 637–647. [DOI] [PubMed] [Google Scholar]
- Grünblatt E, Ruder J, Monoranu CM, Riederer P, Youdim MB, and Mandel SA (2018). Differential Alterations in Metabolism and Proteolysis-Related Proteins in Human Parkinson’s Disease Substantia Nigra. Neurotox. Res 33, 560–568. [DOI] [PubMed] [Google Scholar]
- Hélie S, Ell SW, and Ashby FG (2015). Learning robust cortico-cortical associations with the basal ganglia: an integrative review. Cortex 64, 123–135. [DOI] [PubMed] [Google Scholar]
- Heremans E, Nackaerts E, Vervoort G, Broeder S, Swinnen SP, and Nieuwboer A (2016). Impaired Retention of Motor Learning of Writing Skills in Patients with Parkinson’s Disease with Freezing of Gait. PLoS ONE 11, e0148933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikosaka O, Nakamura K, Sakai K, and Nakahara H (2002). Central mechanisms of motor skill learning. Curr. Opin. Neurobiol 12, 217–222. [DOI] [PubMed] [Google Scholar]
- Hintiryan H, Foster NN, Bowman I, Bay M, Song MY, Gou L, Yamashita S, Bienkowski MS, Zingg B, Zhu M, et al. (2016). The mouse cortico-striatal projectome. Nat. Neurosci 19, 1100–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic J (2008). Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 79, 368–376. [DOI] [PubMed] [Google Scholar]
- Kantak SS, and Winstein CJ (2012). Learning-performance distinction and memory processes for motor skills: a focused review and perspective. Behav. Brain Res 228, 219–231. [DOI] [PubMed] [Google Scholar]
- Kikuchi T, Morizane A, Doi D, Magotani H, Onoe H, Hayashi T, Mizuma H, Takara S, Takahashi R, Inoue H, et al. (2017). Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 548, 592–596. [DOI] [PubMed] [Google Scholar]
- Kim JI, Ganesan S, Luo SX, Wu YW, Park E, Huang EJ, Chen L, and Ding JB (2015). Aldehyde dehydrogenase 1a1 mediates a GABA synthesis pathway in midbrain dopaminergic neurons. Science 350, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordower JH, Olanow CW, Dodiya HB, Chu Y, Beach TG, Adler CH, Halliday GM, and Bartus RT (2013). Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136, 2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupferschmidt DA, Juczewski K, Cui G, Johnson KA, and Lovinger DM (2017). Parallel, but Dissociable, Processing in Discrete Corticostriatal Inputs Encodes Skill Learning. Neuron 96, 476–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Hetzel A, Häckel O, Jones I, Liss B, and Roeper J (2008). Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron 57, 760–773. [DOI] [PubMed] [Google Scholar]
- Lammel S, Ion DI, Roeper J, and Malenka RC (2011). Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron 70, 855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, Deisseroth K, and Malenka RC (2012). Input-specific control of reward and aversion in the ventral tegmental area. Nature 49, 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner TN, Shilyansky C, Davidson TJ, Evans KE, Beier KT, Zalocusky KA, Crow AK, Malenka RC, Luo L, Tomer R, and Deisseroth K (2015). Intact-Brain Analyses Reveal Distinct Information Carried by SNc Dopamine Subcircuits. Cell 162, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Yu J, Ding J, Xie C, Sun L, Rudenko I, Zheng W, Sastry N, Luo J, Rudow G, et al. (2014). Aldehyde dehydrogenase 1 defines and protects a nigrostriatal dopaminergic neuron subpopulation. J. Clin. Invest 124, 3032–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Garner AR, Shimaoka D, Chuong AS, Klapoetke NC, Li L, van der Bourg A, Niino Y, Egolf L, Monetti C, et al. (2015). Transgenic mice for intersectional targeting of neural sensors and effectors with high specificity and performance. Neuron 85, 942–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel SA, Fishman T, and Youdim MB (2007). Gene and protein signatures in sporadic Parkinson’s disease and a novel genetic model of PD. Parkinsonism Relat. Disord 13 (Suppl 3), S242–S247. [DOI] [PubMed] [Google Scholar]
- Marchitti SA, Deitrich RA, and Vasiliou V (2007). Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol. Rev 59, 125–150. [DOI] [PubMed] [Google Scholar]
- McCaffery P, and Dräger UC (1994). High levels of a retinoic acid-generating dehydrogenase in the meso-telencephalic dopamine system. Proc. Natl. Acad. Sci. USA 91, 7772–7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegas W, Bergan JF, Ogawa SK, Isogai Y, Umadevi Venkataraju K, Osten P, Uchida N, and Watabe-Uchida M (2015). Dopamine neurons projecting to the posterior striatum form an anatomically distinct subclass. eLife 4, e10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegas W, Akiti K, Amo R, Uchida N, and Watabe-Uchida M (2018). Dopamine neurons projecting to the posterior striatum reinforce avoidance of threatening stimuli. Nat. Neurosci 21, 1421–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mhyre TR, Boyd JT, Hamill RW, and Maguire-Zeiss KA (2012). Parkinson’s disease. Subcell. Biochem 65, 389–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwboer A, Rochester L, Müncks L, and Swinnen SP (2009). Motor learning in Parkinson’s disease: limitations and potential for rehabilitation. Parkinsonism Relat. Disord 15 (Suppl 3), S53–S58. [DOI] [PubMed] [Google Scholar]
- Ogawa N, Hirose Y, Ohara S, Ono T, and Watanabe Y (1985). A simple quantitative bradykinesia test in MPTP-treated mice. Res. Commun. Chem. Pathol. Pharmacol 50, 435–441. [PubMed] [Google Scholar]
- Pan J, Yu J, Sun L, Xie C, Chang L, Wu J, Hawes S, Saez-Atienzar S, Zheng W, Kung J, et al. (2019). ALDH1A1 regulates postsynaptic μ-opioid receptor expression in dorsal striatal projection neurons and mitigates dyskinesia through transsynaptic retinoic acid signaling. Sci. Rep 9, 3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettitt SJ, Liang Q, Rairdan XY, Moran JL, Prosser HM, Beier DR, Lloyd KC, Bradley A, and Skarnes WC (2009). Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nat. Methods 6, 493–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin JF, Zou J, Drouin-Ouellet J, Kim KY, Cicchetti F, and Awatramani RB (2014). Defining midbrain dopaminergic neuron diversity by single-cell gene expression profiling. Cell Rep 9, 930–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin JF, Caronia G, Hofer C, Cui Q, Helm B, Ramakrishnan C, Chan CS, Dombeck DA, Deisseroth K, and Awatramani R (2018). Mapping projections of molecularly defined dopamine neuron subtypes using intersectional genetic approaches. Nat. Neurosci 21, 1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncacci S, Troisi E, Carlesimo GA, Nocentini U, and Caltagirone C (1996). Implicit memory in parkinsonian patients: evidence for deficient skill learning. Eur. Neurol 36, 154–159. [DOI] [PubMed] [Google Scholar]
- Sgobio C, Wu J, Zheng W, Chen X, Pan J, Salinas AG, Davis MI, Lovinger DM, and Cai H (2017). Aldehyde dehydrogenase 1-positive nigrostriatal dopaminergic fibers exhibit distinct projection pattern and dopamine release dynamics at mouse dorsal striatum. Sci. Rep 7, 5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim H, Wang CT, Chen YL, Chau VQ, Fu KG, Yang J, McQuiston AR, Fisher RA, and Chen CK (2012). Defective retinal depolarizing bipolar cells in regulators of G protein signaling (RGS) 7 and 11 double null mice. J. Biol. Chem 287, 14873–14879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiotsuki H, Yoshimi K, Shimo Y, Funayama M, Takamatsu Y, Ikeda K, Takahashi R, Kitazawa S, and Hattori N (2010). A rotarod test for evaluation of motor skill learning. J. Neurosci. Methods 189, 180–185. [DOI] [PubMed] [Google Scholar]
- Sommer WH, Costa RM, and Hansson AC (2014). Dopamine systems adaptation during acquisition and consolidation of a skill. Front. Integr. Nuerosci 8, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, and Surmeier DJ (2013). Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord 28, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Cragg SJ, and Rice ME (2016). Striatal dopamine neurotransmission: regulation of release and uptake. Basal Ganglia 6, 123–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szymczak-Workman AL, Vignali KM, and Vignali DA (2012). Design and construction of 2A peptide-linked multicistronic vectors. Cold Spring Harb. Protoc 2012, 199–204. [DOI] [PubMed] [Google Scholar]
- Valentin VV, Maddox WT, and Ashby FG (2016). Dopamine dependence in aggregate feedback learning: A computational cognitive neuroscience approach. Brain Cogn 109, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt Weisenhorn DM, Giesert F, and Wurst W (2016). Diversity matters—heterogeneity of dopaminergic neurons in the ventral mesencephalon and its relation to Parkinson’s Disease. J. Neurochem 139 (Suppl 1), 8–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, and Uchida N (2012). Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron 74, 858–873. [DOI] [PubMed] [Google Scholar]
- Werner CJ, Heyny-von Haussen R, Mall G, and Wolf S (2008). Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci 6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CF, Chiang MC, Gray DC, Prabhakaran M, Alvarado M, Juntti SA, Unger EK, Wells JA, and Shah NM (2013). Sexually dimorphic neurons in the ventromedial hypothalamus govern mating in both sexes and aggression in males. Cell 153, 896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin HH, Mulcare SP, Hilário MR, Clouse E, Holloway T, Davis MI, Hansson AC, Lovinger DM, and Costa RM (2009). Dynamic reorganization of striatal circuits during the acquisition and consolidation of a skill. Nat. Neurosci 12, 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang X, Mazzoni P, and Kang UJ (2013). The role of neuroplasticity in dopaminergic therapy for Parkinson disease. Nat. Rev. Neurol 9, 248–256. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and custom MATLAB scripts used in analysis are available upon request.