

Abstract
Particulate systems in the form of liposomes, polymeric micelles, polymeric nano- and microparticles, and many others offer a rational approach for selective delivery of therapeutic agents to the macrophage from different physiological portals of entry. Particulate targeting of macrophages and intracellular drug release processes can be optimized through modifications of the drug carrier physicochemical properties, which include hydrodynamic size, shape, composition and surface characteristics. Through such modifications together with understanding of macrophage cell biology, targeting may be aimed at a particular subset of macrophages. Advances in basic and therapeutic concepts of particulate targeting of macrophages and related nanotechnology approaches for immune cell modifications are discussed.
Key Words: Drug delivery, Macrophage diseases, Nanomedicine, Nanoparticles, Phagocytosis
Introduction
The macrophage is the major differentiated cell of the mononuclear phagocyte system. Macrophages are widely distributed and strategically placed in many tissues of the body not only to combat infection but also to perform a vast array of other immunological, physiological and homeostatic functions and therefore play important roles in disease control and progression [1]. Some of these functions include antigen processing and presentation as well as coordination of the adaptive immune responses, clearance of senescent or damaged cells, autoimmunity and hyperinflammation, chemotaxis, tissue remodeling and repair, placental development and lipid and iron metabolism. Since phagocytosis is a vital determinant of host defense against microbial intruders and injury, there have been numerous attempts towards development of colloidal particles of variable size and dimensions for macrophage-specific targeting [2,3]. Indeed, the propensity of macrophages for phagocytic clearance (often through pattern recognition receptors including the family of scavenger receptors, Dectins and mannose receptor, Fc and complement receptors) [4] offers a viable approach for the design and engineering of particulate carrier systems to combat diseases and disorders where macrophages play a central role. This strategy is achievable since the surfaces of particulate drug carriers, such as liposomes, polymeric nano- and microparticles, nanocapsules and polymer micelles (fig. 1), are not only prone to opsonization events in the blood and other body fluids, but also amenable to modification with macrophage/monocyte receptor ligands (fig. 2). Indeed, various nanoparticles have been used for delivery of therapeutic and diagnostic agents to different macrophages in the body to combat persistent infections, treat macrophage genetic disorders, modulate macrophage accessory functions, induce macrophage death and detect local pathologies (e.g. cancer) [2,3]. Encapsulation or incorporation of drug molecules into the aforementioned particles affords protection against drug degradation or inactivation en route to the macrophages. This may even result in a reduction of the amount of therapeutic agent needed to obtain a clinical effect, and may effectively reduce drug-induced toxicity and other side effects. However, possible adverse effects of nanoparticles must also be considered. These concepts are reviewed in this article and the potential application of nanotechnology to the understanding of macrophage heterogeneity and future development of personalized immune-cell therapies is also discussed. There are cells other than macrophages that may participate in the uptake and clearance of nano- and microparticles, but this depends on physicochemical characteristics of the particles in question and their mode of entry into the body. These include immune cells such as neutrophils, dendritic cells (DCs), B cells, T cell subtypes and M cells, as well as nonimmune cells (e.g. platelets, hepatocytes, endothelial cells, epithelial cells, enterocytes and tumor cells) [2,3]. A detailed discussion of these aspects is beyond the scope of this article.
Fig. 1.

Examples of particulate drug carriers and functional nanoparticles for macrophage/monocyte targeting. Transmission electron microscopy (TEM) image of a multilamellar vesicle consisting of several lipid bilayers separated from one another by aqueous spaces (a) and a cryo-TEM image of small unilamellar vesicles consisting of a single bilayer surrounding the entrapped aqueous space (b). Drug molecules can be either entrapped in the aqueous space or intercalated into the lipid bilayer, depending on the physicochemical characteristics of the drug. The micrograph (c) is the scanning electron microscope image of polymeric nanospheres. These entities can be assembled from a variety of preformed synthetic polymers of different architecture (e.g. linear, di- and tri-block, cross-linked, dendronized) or by polymerization of monomers. Polymers of natural origin (e.g. albumin, gelatin, chitosan, hyaluronic acid) and pseudosynthetic polymers [e.g. poly (amino acids)] have also been used for nanoparticle construction. Polymeric nanoparticles are usually classified as either nanospheres or nanocapsules. In nanospheres, drugs or contrast agents are dispersed throughout the structure, whereas nanocapsules are composed of an oily or an aqueous drug-containing core surrounded by a polymeric membrane. d, e TEM images of two different types of polymeric micelles, respectively. Micelles are formed in solution as aggregates in which the amphiphilic component molecules are generally arranged in a spheroidal structure with hydrophobic cores shielded from water by a mantle of hydrophilic groups. These entities are used for solubilization of water-insoluble drugs. f Atomic force microscope image of single-walled carbon nanotubes (CNTs). CNTs are basically graphene sheets rolled up into hollow cylinders that can be many microns in length and typically with a small diameter. They consist of carbon atoms arranged in a hexagonal lattice, the physical properties of CNTs depend greatly on the diameter of the tubes and the orientation of the hexagons relative to the central CNT axis, which can be adjusted according to the purpose. CNTs can be divided in two groups, namely single-walled CNTs and multiwalled CNTs. Both exhibit interesting physical properties, such as high thermal stability, unique electronic characteristics that are very sensitive to their geometric structure as well as high mechanical strength combined with an ultra-low weight and large aspect ratio. These properties make CNTs attractive candidates for disease diagnosis and treatment, but are challenged by high toxicity.
Fig. 2.

Schematic representation of chemical strategies for coupling of biological ligands to the phospholipid headgroups on the surface of preformed liposomes. Procedures represent amine (a), carboxylic acid (b), aldehyde (c), hydrazine (d), maleimide (e), thiol (f), thiol (disulfide bond formation; g), bromoacetyl (h), cysteine (i), cyanur (j), p-nitrophenylcarbonyl (k), alkyne (l) and triphosphine (m) functionalization. Detailed chemical reactions are not shown. Modified with permission [117].
Basic Concepts
Anatomical Considerations: Reaching the Macrophage
Intravenous Route
Similar to microbial pathogens, particulate drug carriers and functional nanoparticles can reach macrophages from different physiological portals of entry. Intravenously injected particulate drug carriers are rapidly intercepted by hepatic macrophages (Kupffer cells), macrophages in the marginal zone and the red-pulp regions of the sinusoidal spleens (fig. 3) as well as persinal macrophages in the bone marrow (species-dependent; fig. 4), since these macrophages are in direct, open contact with the bloodstream [2,3]. Blood monocytes are also capable of internalizing particulate matters. In many mammals, including humans, rat and mouse, Kupffer cells are the largest population of macrophages in direct contact with the blood, and the bulk of intravenously injected particles are therefore predominantly localized to the liver. Periportal Kupffer cells (approximately comprising 45% of total liver macrophages) are the main particle scavengers; this is also reflected in their larger size and higher lysosomal enzyme activity (on a per cell basis), compared with Kupffer cells located in the hepatic midzonal and perivenous regions [5]. In contrast to Kupffer cells, pulmonary intravascular macrophages are the main scavengers in calves, pigs, cats, goats and sheep [6]. These macrophages are junctionally adherent to the capillary endothelium of lungs and morphologically similar to the Kupffer cells. However, differentiated pulmonary intravascular macrophages are rare in newborn pigs, whereas monocytes are closely associated with the pulmonary capillary endothelium.
Fig. 3.

Capture of intravenously injected polymeric nanoparticles by rat Kupffer cells and splenic red-pulp macrophages. a Fluorescent light microscopy of rat liver after administration of fluorescently labeled polystyrene nanoparticles (60 nm in size). The fluorescence representing nanoparticles is localized in large quantities in Kupffer cells (arrow heads). b An electron micrograph of a rat Kupffer cell with ingested polystyrene nanoparticles (200 nm). c A micrograph of a Kupffer cell lysosome packed with 60 nm particles (scale bar = 100 nm). d An electron micrograph of the rat spleen red-pulp region showing accumulation of poloxamine 908-coated nanospheres (220 nm in size) in macrophages. These nanoparticles are resistant to ingestion by Kupffer cells. The micrograph also shows a venous sinus. Particles are filtered first at interendothelial cell slits before macrophage engulfment (nanoparticle filtration still proceeds after destruction of red-pulp macrophages by clodronate-containing liposomes). e An enlarged view of a red-pulp macrophage with ingested poloxamine 908-coated nanoparticles. Modified with permission [33,103].
Fig. 4.

Schematic representation of hemopoietic bone marrow showing characteristic cellular associations in hemopoiesis and sinus structure (a) and electron micrograph of a rabbit bone marrow sinus endothelial cell (E) with ingested poloxamer 407-coated polystyrene nanoparticles (b). In a the sinus consists of endothelium (end), basement membrane and adventitial cells (adv). Apertures are present in the endothelium (diaphragmed), and hematopoietic cells, en route to the circulation, are passing through them. The sketch also shows a macrophage (mΦ), which extends a process into the lumen of the vascular sinus (a persinal macrophage) as well as megakaryocytes (meg), stromal cells (str), an erythroblastic islet (erb islet) and a granulocyte islet (gran islet). Macrophages are also associated with both islets. In b nanoparticles are 150 nm in size. Modified with permission [8,116].
Access to stromal or hematopoietic macrophages of the bone marrow is typically through nanoparticle passages across the bone marrow sinus endothelium [7,8]. The bone marrow sinus endothelium is capable of removing particles by both transcellular and intercellular routes (fig. 4). The former route occurs through the diaphragmed fenestrae of endothelial walls and the latter route is associated with the formation of bristle-coated pits on the luminal surface of endothelium, where internalized particles are often directed to dense bodies [7].
The intravenous route also offers a gateway for particle access (depending on their size and surface characteristics) to interstitial macrophages at locations where the endothelial barrier is perturbed by inflammatory processes or by dysregulated angiogenesis (e.g. arthritis, atherosclerotic lesions and solid tumors) [2,3]. However, there is marked variability in endothelial permeability at pathological sites that may control the extent of particle extravasation and hence access to local macrophages. For example, variability in endothelial function and permeability has been widely documented in different tumor types, different vessels within the same tumor and during tumor growth, regression and relapse [9,10]. Pore frequency and cutoff size (200–1,200 nm) in tumors may be controlled by microenvironmental factors and may increase with the histological grade and malignant potential of tumors.
Intraperitoneal Route
Intraperitoneal injection will present particles first to macrophages in the peritoneal cavity, but some particles may ultimately reach macrophages in the lymph nodes through lymphatics in the diaphragm and, subsequently, many could reach the bloodstream by this route [11]. Indeed, stomata, which are exclusive to the diaphragm, serve as the main drainage channels for absorption from the peritoneal cavity where the fluid enters subperitoneal lymphatic lacunae. The principal extrinsic lymphatic drainage is via parasternal lymphatic trunks that carry lymph (and hence the suspended particles) to the parasternal/mediastinal lymph nodes and, ultimately, to the right lymphatic or upper terminal thoracic duct.
Pleural Cavity
The lymphatic system also returns fluid from the pleural space surrounding the lungs through peribronchial and subpleural networks. The peribronchial network follows the bronchial tree, draining lymph from the bronchi and most of the lungs, whereas the subpleural network collects lymph from the peripheral lung and visceral pleura. These lymphatic networks provide routes for particle migration out of the pleural space. For instance, injection of carbon particles into the pleural cavity of Japanese monkeys demonstrated particle accumulation in the subpleural lymphatic lacunae through the mesothelial cells, which are present on the parietal pleura [12]. Communication between abdominal lymph nodes and pleural space has also been demonstrated [12].
Subcutaneous Route
Subcutaneously injected particles will be challenged first by interstitial macrophages (and to some extent by local DCs), but a significant fraction of particles will eventually drain into the initial lymphatic system by penetrating the thin-walled and fenestrated lymphatic microvessels. From here, they and are subsequently conveyed to the regional draining lymph nodes via the afferent lymph (fig. 5) [13,14]. In the lymph nodes, macrophages of the subcapsular floor and medullary sinuses are the key scavengers responsible for elimination of some drained particles. There are also indications that lymph node sinus-resident immature DCs could participate in the capture of nanoparticles [15]. This process may have important implications in vaccination strategies with nanoparticulate adjuvants. The role of DC types (e.g. interstitial and dermal DCs, and Langerhans cells) and their endocytic receptors in nanoparticle recognition, uptake and transport processes is beyond the scope of this article and is discussed elsewhere [16]. Drained particles that by-pass lymph node retention eventually reach the bloodstream through the thoracic duct and the right lymphatic duct. The former duct begins in the abdomen, passes along the vertebral column and opens into the venous system at the junction of the left jugular vein and subclavian veins. The latter duct receives lymph only from the upper-right portion of the body; the lymph is next emptied into the right brachiocephalic vein.
Fig. 5.

The fate of polymeric nanoparticles injected into an intradermal region of the rat footpad. The top panel is a schematic diagram showing the location of lymph nodes draining the footpad region of the rat. Popliteal node (1) drains the footpad, foot and hind leg through lymph vessels running with greater and lesser saphenous veins. The efferent popliteal trunk follows the femoral vein to a retroperitoneal lymphatic plexus dorsal to the iliac vessels and the main trunk continues centrally to the iliac node (3), while smaller tributaries travel with the superficial epigastric vessels to the inguinal nodes (2). The position of the caudal node (4) is also shown. Following intradermal injection some nanoparticles (60 nm in this study) may aggregate (a); particles individually or in aggregated form (arrow and arrowheads) are prone to phagocytic clearance by the local macrophages (b). Particles also drain from the injection site into the initial lymphatic vessels; a blind-ended lymphatic vessel is shown in c. In lymphatic capillaries, numerous endothelial cells overlap extensively at their margin. d Following intradermal injection, many of the overlapped endothelial cells are separated and passageways, known as patent junctions, are provided between the interstitium and the lymphatic lumen. e A phagocyte of the subcapsular sinus from a regional draining node with captured drained nanoparticles (60 nm in diameter); note the presence of extracellular nanoparticle aggregates. Modified with permission [14]. Scale bar = 250 nm in a and 500 nm in b, d and e.
The possibility of nanoparticle extravasation from afferent lymphatic branches into perinodal adipose tissue has not received much attention, but DCs are also found in abundance in perinodal adipose tissue [17,18]. This plausible route may account for DC uptake of some interstitially injected nanoparticles and eventual DC migration into the lymph node.
Intratracheal Route
Intratracheally administered particles can readily reach alveolar macrophages but not interstitial macrophages that are separated from the alveolar space by an epithelial barrier. There are reports that have shown the presence of asbestos fibers in the pleural space after intratracheal injection [19]. The process of fiber transfer to the pleura after inhalation is unclear, but presumably is through lymphatic spread and/or dependent on particle morphology (rod shape) and spatial orientation.
Other Routes
Finally, macrophages in organs such as brain, gut, eye, testis and the synovial cavity of joints are mainly accessible by local administration but it is cumbersome to reach the majority of local macrophages in these locations. For instance, only a small fraction of orally administered particles gain access to macrophages through M cells at the Peyer's patches; the M cell apical membranes bind and take up particles from the gut lumen and transport them to underlying macrophages and DCs [20]. However, some macrophages are able to send processes through the epithelium from the lamina propria and may be able to directly sample nanoparticles in the intestinal lumen [20].
Physicochemical Considerations of Particles Controlling Macrophage/Monocyte Clearance
Colloidal targeting of macrophages/monocytes is controlled by the physicochemical parameters of particles such as size, shape and surface characteristics; these issues are reviewed in detail elsewhere [2,3,21]. Collectively, these parameters affect the extent of particle attachment to macrophages/monocytes and their subsequent internalization and trafficking processes. Here, we discuss the key and most recent concepts.
Particle Surface Characteristics and Size
The physicochemical parameters of particulate drug carriers are often changed once in contact with biological fluids, such as the blood and lymph [2,21,22,23,24]. These alterations may further impose significant effects on macrophage/monocyte recognition and clearance rate of drug carriers. For instance, an important consequence of particle contact with blood is adsorption and deposition of a range of plasma proteins on the particle surface [2,23]. The composition of the plasma protein coat differs considerably in amount and heterogeneity and in a time-dependent manner, depending on the physicochemical properties of particles. Some of these proteins act as opsonic factors. Examples of key opsonic proteins include various subclasses of antibodies that are capable of mediating particle recognition by different macrophage Fc receptors. They also include complement activation products, such as C3b and iC3b, which also prime the particle surface for interaction with a plethora of macrophage complement receptors. The binding of different opsonic proteins and/or dynamic changes in surface opsonization processes may determine which subpopulation of macrophages will host the particles. Therefore, dynamic opsonization processes could indicate a recognition hierarchy phenomenon, wherein a specific macrophage receptor (or population) might recognize the earliest surface changes and other receptors (or other subpopulations) might engage later [21]. Understanding of differential opsonization processes may further allow means for design of drug carriers with high specificity towards only one macrophage subset in a typical organ.
A recent study demonstrated that intravenously injected liposomes containing the anionic lipid succinate predominantly localizes to the bone marrow macrophages in rhesus monkey, rabbit and hamster, but not in rats or mice where these liposomes are sequestered by both the hepatic and the splenic macrophage [25]. Whether these observations are related to blood opsonization processes and/or to differences in macrophage recognition of succinic acid still remains to be elucidated. In contrast to this attempt, an earlier study demonstrated exclusive localization of some intravenously injected engineered nanoparticles to sinus endothelial cells of the rabbit bone marrow [8].
During the past 2 decades we have witnessed rapid developments in stealth technologies, resulting in the engineering of particulate delivery systems that remain resistant to rapid clearance by macrophages in contact with the blood [2]. Stealth characteristics were initially achieved through either covalent attachment of polyethylene glycol 2000 (PEG2000) molecules to the nanoparticle surface (fig. 6) or by surface adsorption of polyoxypropylene-polyoxyethylene linear or star-shaped block copolymers (e.g. poloxamer 407 and poloxamine 908, respectively) on hydrophobic nanoparticles. Today, there are many other polymers that can confer longevity to nanoparticles following surface grafting or adsorption and these are discussed elsewhere [2,3,21,26]. Typically, these particles are below 150 nm in size and the stealth characteristics are conferred through increased surface hydrophilicity, where the surface projected polymer chains strongly associate with neighboring water molecules. Therefore, surface-projected PEG or polyethylene oxide chains provide stability to the particle suspension by repulsion through a steric mechanism of stabilization involving both enthalpic and entropic contributions [27]. Due to its hydrophilicity, the steric barrier dramatically suppresses protein adsorption, but this may not necessarily prevent opsonization processes [27,28,29]. For instance, complement activation and fixation still proceeds with some surface-engineered particles (fig. 6), where the extent of complement activation and fixation depends on surface density and conformation of the projected polymers [23,30]. The surface polymer conformational changes have further been shown to switch complement activation from one pathway to another [23]. The steric barrier on the nanoparticle surface, however, can interfere with the binding of C3b and/or iC3b to their corresponding macrophage receptors [29]. As a result of their macrophage-evading property, stealth nanoparticles have widely been used for both drug delivery to solid tumors and tumor detection using different imaging modalities [2,3,21,26].
Fig. 6.

Stealth liposomes. Schematic diagram of a stealth liposome is shown in the left panel. The stealth property arises from covalent attachment of methoxypolyethylene glycol 2000 (mPEG2000) to phospholipids. A typical stealth vesicle usually contains 5 mol% of mPEG-phospholipid. PEGylated liposomes can still activate the complement system (upper right). Despite complement activation these liposomes exhibit prolonged circulation times in the blood. Scintigraphic images of 2 rats at 4 h postinjection of 99mTcO–4 labeled stealth liposomes are shown in the lower right panel. The images show strong activity in the heart region (representing the rat blood pool), but poor activity in both the liver and spleen. At 4 h, approximately 70% of the injected liposomes were still in the circulatory blood pool. Hepatic and splenic sequestration accounted for 14 and 2.5% of the injected dose, respectively.
Through rational design and understanding of intrasplenic microcirculation pathways, a range of splenotropic nanoparticles have also been designed that mainly localize to the marginal zone and the red-pulp macrophages with minimum sequestration by Kupffer cells [31,32]. Morphologically, splenotropic nanoparticles are rigid nondeformable entities of 220–250 nm in diameter exhibiting stealth characteristics to the hepatic Kupffer cell recognition (e.g. through surface coating with poloxamine 908). As a result of poor recognition and clearance by Kupffer cells, nanoparticle concentration in the blood remains high. Therefore, more nanoparticles are exposed to the spleen in time (the rate of blood flow to the spleen is substantially lower than the liver). It is the combined particle size and nondeformable characteristics that contribute to particle filtration at interendothelial cell slits located at splenic venous sinuses, since the width of interendothelial cell slits is below 220 nm. Remarkably, macrophages in the vicinity of cell slits eventually take up filtered nanoparticles, but the mechanism of recognition remains unknown (fig. 3) [33]. In our studies, we have eliminated the role for the splenic macrophage complement receptors in sequestration of splenotropic nanoparticles. The whole process of splenotropic particle filtration and eventual macrophage uptake resembles erythrocytes containing rigid inclusions (e.g. Heinz bodies or malarial plasmodia) of 200––300 nm. Such erythrocytes are thought to be ‘pitted’ of their inclusions at interendothelial cell slits, which are eventually cleared by the red-pulp macrophages. Nevertheless, these splenotropic particles may have important applications with respect to examining splenic microcirculatory pathways in addition to selective delivery of antimicrobials and other therapeutic molecules to marginal zone and red-pulp macrophages [34].
Similarly, following subcutaneous injection, size and surface characteristics of particles can also control their clearance rates and the extent of macrophage sequestration [13,14,35,36,37]. Generally, small particles (often below 20 nm) can leak from interstitial spaces into the blood capillaries, whereas the majority of larger particles (20–150 nm) drain into the initial lymphatic capillaries. However, if particles are too large (150 nm or above) they move slowly from the site of injection and drainage into the lymphatic vessels may take a period of days. The slow drainage can induce inflammation and renders particles susceptible to ingestion by interstitial macrophages as well as local and dermal DCs [13]. Surface properties may further induce particle aggregation at the injection site and control the extent of particle interaction with the amorphous ground substance of the interstitium [35]. Numerous attempts have addressed these concepts through surface engineering of polymeric particles and liposomes with polymers [35,36,37]. In one study, the surfaces of 45 nm polystyrene nanoparticles were modified by adsorption of the block copolymer poloxamer 407. Adjusting the adlayer thickness of poloxamer 407 on the surface of polystyrene nanoparticles to 1.5–2.5 nm not only reduced the extent of nanoparticle aggregation at interstitial spaces, compared with uncoated nanoparticles, but also accelerated the rate of nanoparticle drainage into the initial lymphatic vessels (fig. 7) [35]. Furthermore, macrophages in the draining lymph nodes were able to recognize and ingest the flowing poloxamer-coated nanoparticles (fig. 7). On the other hand, increasing the adlayer thickness of the poloxamer to above 4 nm accelerated particle clearance from the interstitium further, but the drained particles were poorly recognizable by the lymph node macrophage [35]. Thus by precision surface engineering one can simultaneously control both the extent of the nanoparticle drainage rate from interstitium and fate within the lymphatic system. These approaches have laid the foundation for the design and engineering of advanced contrast agents for both lymph node and lymphatic vessel imaging in different pathologies [38].
Fig. 7.

The effect of poloxamer 407 adlayer thickness (δ) on lymphatic distribution of polystyrene nanoparticles at 6 h postinterstitial injection into rat footpads. Nanoparticles were 45.5 nm in size before coating with poloxamer 407. Extent of nanoparticle retention at the injection site (a) and distribution of drained nanoparticles among popliteal and iliac nodes (b). c Structure of poloxamer 407 and scanning electron micrographs of representative uncoated and poloxamer-coated nanoparticles. Nanoparticle packing arrangement is most regular when poloxamerδ ≥3.9 nm. The upper part is a schematic representation of a poloxamer molecule on a nanoparticle surface. At low poloxamer concentrations the surface is partly covered by poloxamer molecules. At such concentrations the ethylene oxide chains will be close to the nanoparticle surface (mushroom configuration) due to a large available surface area per adsorbed poloxamer molecule. Ethylene oxide chains will assume a ‘brush-like’ configuration at high concentrations of poloxamer. This is due to a smaller available surface area per adsorbed poloxamer molecule and repulsive forces arising from the ether oxygen atoms of projecting ethylene oxide chains (larger δ values). Modified with permission [35].
Particle Shape
A recent study has shown that particles of variable shapes are capable of initiating phagocytosis in at least one orientation [39]. The particle shape at the point of initial contact could control whether macrophages proceed with phagocytosis or rest over particles, but the particle size primarily affected the completion of phagocytosis in cases where particle volume exceeded the cell volume. Similarly, shape-switching particles (engineered from stimulus-responsive polymers) also exhibit unique interactions with macrophages; elliptical disk-shaped particles that are not phagocytosed by macrophages were made to internalize through shape switching into spherical particles [40]. These observations are of particular interest for the design and engineering of drug carriers for pulmonary administration and targeting of alveolar macrophages, particularly for treatment of tuberculosis.
Another interesting case is intravenously injected amino-functionalized single-walled carbon nanotubes with average lengths of 200–300 nm that by-pass efficient macrophage sequestration, but undergo rapid renal clearance by glomerular filtration. This process involves partial tubular reabsorption and transient translocation into the proximal tubular cell nuclei, but does not involve active secretion via specific transporters [41]. Longer entities such as filomicelles (2–18 µm), however, exhibit prolonged circulation half-lives, often in the order of days [42]. Hydrodynamic shears tend to stretch and flow-align the cylindrical filomicelles in most blood vessels, which minimizes filomicelle interactions with macrophages by pulling off the filomicelles as they come into contact with the phagocytes. However, following cell contact, a ‘nanofragment’ of a filomicelle might break off and be taken up by macrophages in the liver and the spleen [42].
Surface Engineering with Macrophage Ligands
There are continuous efforts that target particles to specific macrophage receptors through surface grafting with receptor-specific ligands [2,3]. One such example is tuftsin, a natural macrophage activator peptide that can stimulate accessory functions to combat infections [43]. Another candidate macrophage receptor, which has been exploited in particle surface engineering and macrophage targeting, is the hemoglobin scavenger receptor CD163. This receptor is overexpressed in tissue-resident macrophages of the M2 phenotype and in macrophages in sites of inflammation and tumor growth [44,45,46]. Ligands of mannose receptor carbohydrate recognition domains modulate macrophage function [47,48] and have also been used for nanoparticle decoration and macrophage (and DC) targeting [2,3]. Heparin-binding epidermal growth factor is expressed on leukocytes and particularly by the monocytes. A recent attempt has targeted heparin-binding epidermal growth factor expressed by monocytes and conventional DCs with cross-reacting material 197 (a nontoxic variant of diphtheria toxin) functionalized liposomes [49].
The rate of drainage and lymph node capture of interstitially injected PEGylated (or stealth) liposomes of 100 nm in size was also improved by grafting of nonspecific IgG to the distal end of surface-projected PEG chains [36,37,50]. The amount of IgG-PEG-liposome deposition in the regional lymph nodes was further improved following an adjacent subcutaneous injection of pentameric IgM against the surface-grafted IgG components without compromising the vesicle drainage rate from the interstitium [37]. The mechanism of action was suggested to arise from the formation of large immune-aggregates within the lymphatic vessels with subsequent transport to and trapping by macrophages at the floor of subcapsular sinuses. Rapidly drained IgM molecules may also bind lymph node macrophage Fc receptors. This in turn could generate platforms for the subsequent trapping of drained IgG-decorated liposomes or their aggregates. The antibody attachment procedures in these studies have generated immune-vesicles displaying IgG molecules in random orientation. Accordingly, random antibody conjugation through the Fab region may expose the Fc region, thereby facilitating Fc receptor recognition of immunoliposomes. IgG antibodies are also glycoproteins with N-linked carbohydrate in the Fc domain. The Fc glycan plays an important role on antibody effector functions, but plays a minimal role on antibody half-life or its antigen binding. The Fc glycan portion of IgG can be oxidized for coupling to PEGylated liposomes (e.g. by means of hydrazine-PEG-phospholipid). This mode of IgG coupling reaction, however, diminished Fc segment exposure to macrophages, but macrophages recognized and internalized immunovesicles predominantly via the scavenger receptor class A-I/II [50].
Selected Therapeutic Concepts
Lysosomal and Cytoplasmic Targeting: Antimicrobial, Nucleic Acid and Antigen Delivery
Macrophages serve as sites of proliferation of certain pathogens during some or all of the infection process. Notable examples include infections associated with developing countries such as visceral leishmaniasis, malaria and tuberculosis, where most of the conventionally used dosage forms of antimicrobial treatments have shortcomings of severe toxic side effects and emergence of drug resistance [3]. Passive targeting of colloidal carriers with encapsulated antimicrobial agents to infected macrophages, particularly via intravenous and pulmonary routes of administration, serve as logical strategies to effective microbial killing, while minimizing adverse drug related effects [51,52]. This may result in a further reduction of the amount of the active agent needed to obtain therapeutic efficacy since the phagocytic/endocytic pathways will direct the carrier to lysosomes where persistent pathogens are often resident, and may effectively reduce antimicrobial-induced adverse effects. Degradation of the carrier by hydrolytic lysosomal enzymes releases the drug into the organelle or into the cytoplasm. The latter is either by diffusion or by specific transporters and can challenge cytoplasmic infections. A regulatory-approved liposomal formulation of amphotericin B is now available for treatment of visceral leishmaniasis or confirmed infections caused by various fungal species [2,3]. This formulation has significantly reduced nephrotoxicity associated with amphotericin B as well as the release of proinflammatory cytokines, while achieving therapeutic drug concentrations in the infected macrophages. A wide range of liposomal formulations containing immunomodulators have also been used to successfully combat infections as well as for inducing macrophage-mediated destruction of metastases [2,3].
Advances in material design and engineering have further provided new opportunities for assembly of stimuli-responsive drug carriers, thus offering better control over the release of encapsulated cargo in to the cytoplasm [3,21,53,54,55]. Notable examples include particulate carriers composed of pH-sensitive polymers, pH-sensitive and charge-reversible liposomes, and fusion-competent vesicles that release their encapsulated drugs in late endosomes due to low microenvironmental pH. This also results in efficient cytosolic cargo delivery due to carrier-mediated endosomal destabilization and is important for combating cytoplasmic infections as well as for nucleic acid release and antigen presentation to the major histocompatibility complex class I pathway [3,53]. There are ongoing efforts that combine such carries in conjunction with ligands against macrophage plasma membrane receptors, such as mannose receptor and Dectins, as well as endosomal toll-like receptors for efficient antigen transport and processing [16]. On the other hand, some polymers, when coupled to molecules carrying a delocalized positive charge, can exclusively localize to mitochondrion and by-pass the usual endolysosomal routes (fig. 8). These polymers may be components of polymer micelles, but mitochondrial localization is exclusive to the monomer.
Fig. 8.

Live-cell microscopy of rat peritoneal macrophages treated with a newly engineered PEGylated polymer micelle for 1 h at 37°C. Polymer micelles are dynamic structures and are in equilibrium with monomers with the concentration of monomers being equal to the critical micelle concentration. The results demonstrate translocation of internalized monomer chains (covalently labeled with rhodamine B) to the mitochondrial network (a) and mitochondria stained with Mitotracker Green (b). c Merged images of a and b together with H33342 nuclear stain, confirming mitochondrial translocation of micelles. d Merged image showing that monomer chains do not colocalize with lysosome (lysosomes were stained with LysoTracker Blue). Neither the monomer nor micelle internalization occurred at 4°C. Imaging was performed on a Leica AF6000LX microscope using a 63×/1.47 oil objective with a 1.6 magnification and filters GFP (excitation band pass 475/40, emission band pass 530/50 nm), Cy3 (excitation band pass 555/25, emission band pass 605/52) and A4 (excitation band pass 360/40, emission band pass 470/40).
There are concerns regarding the cytotoxicity of therapeutic nanoparticles, or some of their constituents (e.g. polycations), as they may perturb endolysosomal membranes, form pores and affect organelle integrity [56,57,58,59,60,61,62,63,64,65]. Some cathepsins are stationed in endosomes and their release through nanomaterial-mediated endosomal damage may induce apoptosis through Bid cleavage. Likewise, leakage of lysosomal proteases into cytosol is known to occur when lysosomal integrity is compromised, even only moderately, as in the case of antibiotic treatments such as gentamicin [66]. Lysosomal cathepsins are also endowed with the capacity to cleave Bid as well as induce cell death through other complex intracellular processes [67,68]. The polycationic constituent of some nanoparticles (e.g. as in nucleic acid delivery systems) may further form complexes with highly anionic macromolecules such as F-actin or even interfere with actin polymerization, thereby inducing membrane damage as well as affecting internalization modes and subsequent responses. Polymeric and/or surfactant constituents of nanoparticles may even modulate global gene expression, as in activation of nuclear factor-ĸβ and hence cytokine production. Another concern is macrophage interception of nanoparticles carrying cytotoxic cargos to solid tumors, which may also induce macrophage destruction through the release of encapsulated cytotoxic drugs. In the case of Kupffer cell destruction, full restoration of the Kupffer cell population often takes 2 weeks [69]; this may increase the risk of bacteremia during the period of macrophage deficiency in cancer patients. Such issues must be seriously considered within the context of a therapeutic benefit-to-risk ratio, paying particular attention to the frequency of nanoparticle dosing, macrophage turnover and heterogeneity.
Gaucher's Disease and Crossing the Blood-Brain Barrier
The ultimate lysosomal destination of colloidal particles in macrophages has opened a viable approach for lysosomal enzyme replacement [70,71]. A typical example is the treatment of Gaucher's disease, which is an autosomal recessive condition with its highest prevalence in Ashkenazi Jews [72]. This typical lysosomal storage disease is caused by a deficiency in glucocerebrosidase, responsible for a step in the cleavage of glucose from ceramide, resulting in glucocerebroside substrate accumulation within macrophages [73]. There are three types of Gaucher's disease. Type I is a noncerebral form of the storage disease in adults, with some residual enzymatic activity, which is where colloidal delivery of the enzyme has demonstrated significant value and therapeutic efficacy at the experimental level. Type II is infantile and is an acute disease dominated by cerebral accumulation of substrate with hepatosplenic involvement, where no enzyme activity is usually detected. Type III is a juvenile disorder intermediate in signs and symptoms from other Gaucher's disease types.
Since the blood-brain barrier poses a formidable barrier to particle delivery to the brain, ex vivo modulation of monocytes with drug carriers may offer future opportunities to combat type II Gaucher's disease (e.g. through gene delivery) and other local conditions such as cerebral HIV infection. It is now known that some monocytes emigrate from the systemic circulation and develop into macrophages in different tissues [74]. Many stimuli also elicit increased recruitment of some monocytes to peripheral sites, where differentiation into macrophages and DCs occurs [75]. These processes contribute to the host defense and tissue repair. The brain contains two sets of monocyte-macrophage cells [76]. These are parenchymal microglial cells and blood-borne monocytes. The parenchymal microglial cells are mesodermally derived cells and are believed to enter the brain early in gestation. They are highly stable and comprise approximately 12% of cells in the brain and are more abundant in the gray matter. They are also replenished from bone marrow cells at a very low rate. The blood-borne brain monocytes are predominantly located in the perivascular spaces, the leptomeninges and the choroid plexus. In steady-state, microglia does not arise from monocytes; the earliest macrophages following brain injury arise from parenchymal microglia [76]. However, within a few days most macrophages arise from perivascular monocytes [76]. A recent report, however, has shown that Ly-6ChiCCR2+ monocytes are preferentially recruited to the lesioned brain where they differentiate into microglia [77]. These cells could potentially be used for therapeutic interventions in the lesioned brain following nanoparticle challenge. Others have demonstrated the potential brain targeting ability of intravenously injected ex vivo loaded monocyte-derived macrophages with indinavir-encapsulated nanoparticles, which resulted in reduced HIV-1 replication in HIV-1 encephalitis brain regions [78].
Macrophage Silencing and Destruction
Macrophages are attractive targets for RNA interference (siRNA) therapy because of their involvement in chronic conditions such as atherosclerosis, rheumatoid arthritis, diabetes and inflammatory bowel disease. Recently, the ability of β1,3-D-glucan-encapsulated siRNA particles as efficient oral delivery vehicles that potentially silence Map4k4 (mitogen-activated protein kinase kinase kinase kinase 4) gene in mouse macrophages was demonstrated [79]. This approach protected animals from lipopolysaccharide-induced lethality by inhibiting TNF-α and interleukin-1β production. Others have focused on using nanoparticles to manipulate phagocytic cell numbers. Examples include liposomal formulations of the bisphosphonate drugs for macrophage destruction in conditions such as autoimmune blood disorders, spinal cord injury, rheumatoid arthritis, T cell-mediated autoimmune diabetes and restenosis [80]. Selective compartmental macrophage elimination, as in synovial lining of murine knee joints, has also been achieved with boron-10-containing liposomes in combination with neutron irradiation, following local administration [81]. These macrophage ‘suicide’ approaches have further enabled the study of macrophage recruitment in a particular organ as well as many functions of these cells in relation to microenvironmental conditions.
Selective macrophage destruction may further benefit from recent advances in nanotechnology and nanoparticle engineering, such as the development of composite metal nanoshells [82,83]. These entities consist of a spherical dielectric core of 20–80 nm made from silica surrounded by a thin metal shell of 5–20 nm (e.g. gold). The design takes advantage of the plasmon resonance property of the metal shell. Consequently, the type of plasmon that exists on the surface of a metallic nanoshell is directly related to the shape and curvature of the particle. Therefore, by controlling the relative thickness of the core and shell layers, the plasmon resonance and resultant optical absorption properties can be adjusted from near-UV to mid infrared [82]. In addition to these properties, drugs can also be incorporated within the nanoshell core. The plasmon resonance property of near infrared-responsive gold nanoshells could be worthy of exploitation for photothermal ablation of macrophages under magnetic resonance guidance. This approach, however, has proved to be highly effective for the destruction of glioblastoma and astrocytoma [83].
Macrophage Imaging
Colloidal targeting of macrophages has many applications in diagnostic medicine with different modalities including positron emission tomography (fig. 9), MRI and spectral fluorescence imaging [3,14,38,84,85,86]. For example, particulate contrast agents such as superparamagnetic iron oxide nanocrystals are widely used for detection of cancer as well as atherosclerosis by MRI following an intravenous route of administration [3,85]. Here, through macrophage loading, the surrounding parenchyma will change in intensity. Likewise, subcutaneous injection of radiolabelled nanoparticles has allowed for better cancer detection, including demonstration of the route of lymphatic drainage from the tumor and identifying lymph nodes at risk of metastases, thus affording surgical guidance and better treatment planning (e.g. localizing regional lymph nodes for lymphadenectomy and radiation therapy) [49]. An interesting development is the application of quantum dots for sentinel lymph node mapping and particularly in multicolor wavelength-resolved spectral fluorescence lymphangiography [86]. Quantum dots are nanoscale crystalline structures composed of different compounds (e.g. cadmium selenide) that can transform the spectrum of radiative emission [3]. These entities absorb white light and then reemit it a couple of nanoseconds later at a specific wavelength. Therefore, by varying the size and composition of quantum dots, the emission wavelength can be tuned from blue to near infrared (fig. 10). Quantum dot toxicity, however, is of concern [87,88,89,90]. Cadmium selenide quantum dots are lethal to cells under UV irradiation and when their protective coating is lost intracellularly.
Fig. 9.

Positron emission tomography images of a mouse receiving an intravenous dose of stealth liposome. 64Cu radionuclide was used to label the liposomes and biodistribution was followed for 24 h. Initially the aorta, the main veins and the heart region gave the highest signal intensity (representing liposome presence in the systemic circulation). However, splenic and hepatic signal intensities increased at later time points. The images further illustrate that the spleen receives the highest liposome dose g–1 tissue followed by the liver, where resident macrophages (hepatic Kupffer cells and splenic marginal zone and red-pulp macrophages) are responsible for liposome clearance. Modified with permission [84].
Fig. 10.

The relationship between size and color of cadmium selenide quantum dots irradiated with a UV light. Quantum dots are semiconductors with electronic characteristics that are controlled by size and shape of individual crystals. By increasing the size, the emission wavelength of quantum dots may be tuned from blue to near infrared due to quantum confinement. Generally, the smaller the size and the larger the band gap, the greater the difference in energy between the highest valence band and the lowest conduction band becomes. As a result, more energy is required to excite the quantum dot and therefore more energy is released when the dot returns to its resting state. Quantum dots are not soluble in aqueous environments. For biological applications their surface must be modified (e.g. with polymer layers) to make them water dispersible. These approaches will dramatically increase the hydrodynamic size of the quantum dots (often in the order of 30–50 nm).
Future Directions
Targeting Macrophage Heterogeneity
Macrophage exhibit extensive receptor repertoire and versatile functions; these make them highly responsive to environmental microheterogeneity and also demonstrable in vitro [4,5,75,91,92,93,94,95,96,97]. For instance, ovarian cancer cells have been shown to switch cocultured macrophages to a phenotype similar to that found in ovarian tumors [97]. Macrophage populations display remarkable heterogeneity even within the same tissue. For instance, there are at least five different subpopulations of macrophages in the mouse spleen [98]. Functional and phenotypic heterogeneity are further evident in rat and human Kupffer cell populations [5,99,100]. Marked heterogeneity in the turnover and variable lifespan of macrophages has also been indicated [75]. It is well known that circulating monocytes may give rise to mature macrophages, but monocytes are also heterogeneous themselves and monocyte heterogeneity is well conserved in humans and mice [75]. The different monocyte subsets may reflect developmental stages with distinct physiological roles. From a pharmacological perspective, these findings are important for the development of particulate-based strategies that target a particular subpopulation of monocytes or subsets of macrophages (as in primed/activated Kupffer cells, infected macrophages, tumor-resident macrophages, or by optimizing delivery to newly recruited monocytes versus local resident macrophages). In support of this notion, there are attempts which have demonstrated the ability of newly recruited liver macrophages (as well as activated macrophages) to sequester nanoparticles with stealth characteristics (particles resistant to recognition by the quiescent resident Kupffer cells, which consequently exhibit prolonged circulation half-lives) [101,102,103]. Similarly, sex hormones can profoundly influence the phagocytic activity of the host defense system and modulate drug carrier distribution among different macrophage subsets. For instance, treatment of mice with the synthetic estrogen diethylstilbestrol indicated that liposome recognition via the complement receptor Mac-1 (CD11b/CD18) in the liver was exclusive to the newly recruited monocytes [104].
Understanding of Endo-Lysosomal Events in DCs and Macrophages
DCs are the only antigen-presenting cells having cross-presentation capacity, which is an important part of modulating the immune response. Interestingly, DCs lose the ability of cross-presentation in the late stage of maturation even though they retain their ability of internalization [105]. There are strong indications that this is associated with a change in the acidification of endosomes which results in differences in transfer to the cytosol in early and late stages of maturation. The fate of antigens presented on nanoparticles also seem to differ depending on particle size and acidification of the endosomes; however, precise knowledge of how this is correlated to DC maturation is not known [106,107]. Thus, there is a need to investigate differences in DC maturation and endosome acidification in relation to antigen presentation by particle-based delivery systems and particle size and hydrophobicity seem highly important parameters and need special attention. Some of these issues can perhaps be studied and resolved through the recent development of nanoparticle-based time-resolved pH measurements in endosomes and lysosomes (fig. 11) [108,109]. Examples include nanoparticle sensors with a pH measurement range of 3.2–7.0, which was synthesized by adding three fluorophores, of which two were pH-sensitive [108].
Fig. 11.
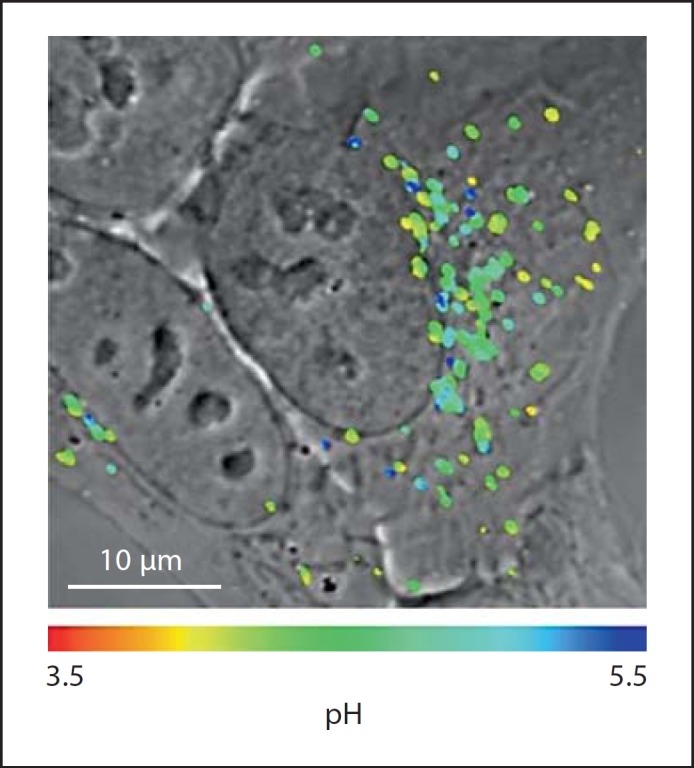
Image of internalized nanoparticle pH sensors in HepG2 cells. Image was obtained by confocal microscopy 24 h postnanoparticle challenge. The original image is converted into a color-coded illustration based on the pH gradient using a calibration curve and overlaid on the differential interference contrast image. The nanoparticle sensors are located in endosomes and lysosomes. Modified with permission [108].
Translational Mechanical and Mechanosensitivity Processes
Understanding of monocyte/macrophage mechanical and mechanosensitivity processes that control particle binding and internalization is also important for better design and surface modification of phagocyte-specific carriers that operate under shear flow conditions or at interstitial sites. An interesting example is the catch-bond concept. Catch bonds are bonds between a ligand and its receptor that are enhanced by a mechanical force that pulls apart the ligand-receptor complex [110]. Here, current evidence suggests that force-induced structural alterations in the receptor protein are allosterically linked to a high-affinity conformation of its ligand-binding pocket [110]. Catch bonds are widespread among adhesive proteins such as Escherichia coli adhesin FimH, which mediates catch bond formation with the target P- and L-selectins.
Translation of Molecular Adhesion Processes
Advantage must also be taken from an understanding of cellular and molecular adhesion mechanisms that are used by migrating monocytes to leave vasculature. Translation of these events to nanoparticle engineering may result in the design of entities with endowed ability to cross biological barriers, such as the blood-brain barrier for therapeutic interventions.
Understanding of Macrophage Contribution to Infusion-Related Reactions
The available clinical evidence suggests that intravenous infusion of therapeutic liposomes and nanoparticles (including stealth systems) may trigger adverse acute allergic-like reactions in some individuals and are often associated with flushing and circulatory disturbances [24,111,112,113]. These reactions are not initiated or mediated by immunoglobulin E antibodies and remain unpredictable by the standard allergy tests. There is strong evidence that complement activation may be a contributing, but not a rate-limiting factor in eliciting acute allergic-like reactions in sensitive individuals [24,112,113]. In addition to this, nanoparticle infusion-mediated symptomatic responses, reaction severity and duration may have further links to the state of macrophage responsiveness and receptor functionality. Acute allergic-like reactions to the same nanoparticles as used in humans are very common in pigs [112,113]. Hemodynamic responses in pigs include a massive rise in pulmonary arterial pressure and a decline in systemic arterial pressure, cardiac output and left ventricular end-diastolic pressure [112,113]. Depending on nanoparticle composition and dose transient electrocardiogram alterations, including tachycardia, bradycardia, ST segment depression and T wave changes, ventricular fibrillation or cardiac arrest may occur. However, prior destruction of pulmonary intravascular macrophages with clodronate liposomes can prevent these events. Therefore, a clear understanding of the macrophage role (and other immune cells such as mast cells) in infusion-related adverse reactions remains pivotal to the future development of nanoparticle-based therapeutics.
Beyond State of the Art Nanoparticle Engineering
There are many limitations with current approaches in particulate drug delivery systems for macrophage targeting. An endemic problem is poor drug loading in polymeric drug carriers. One example is poor loading efficiency of ampicillin in polyalkylcyanoacrylate nanoparticles, which were used for treatment of Salmonella typhimurium- and Listeria monocytogenes-infected peritoneal macrophages [60,61]. Accordingly, higher amounts of nanoparticles are required to deliver sufficient amounts of antibiotic for microbial killing, but this can also induce cytotoxicity due to intracellular overloading of the cyanoacrylic polymer.
The levels of sophistication in design and nanoengineering are also generating more heterogeneous populations of nanoparticles [21]. Consequently, these may cause problems in the targeting of macrophage subsets through different portals of entry and further intracellular processing.
There have been recent breakthroughs in the nanofabrication of polymeric particles that address some of these issues, which are expected to dramatically improve drug loading, macrophage targeting, intracellular drug delivery and controlled compartmental drug release. One remarkable approach is the Particle Replication in Nonwetting Templates (PRINT), which is based on the exploitation of the low surface energy of novel fluoropolymeric molds to produce a variety of organic particles with well-defined controllable physicochemical parameters [114]. Indeed, PRINT allows for precise control over particle size (from tens of nanometers to several microns), geometry (spheres, cylinders, discs and toroids), porosity and texture and yield monodisperse particles. Additionally, the concentration of drug cargos in the particles can be exactly chosen to meet specific needs since this technology does not rely on kinetic drug trapping as in the case with liposomes and micelles.
Phagocytosis Resistant ‘Backpacks’
While ex vivo modification and loading of monocytes/macrophages with therapeutic nanoparticles have shown promising results in different therapeutic scenarios after reinjection, the technique suffers from the fact that the drug carriers are sequestered in the phagolysosomal compartments. This reduces the drug release rate at the pathological sites and may even degrade or inactivate the drug. This limitation was recently addressed through design innovations allowing particles to be attached to macrophages without being internalized [115]. These entities were fabricated using a standard photolithography lift-off technique of layer-by-layer and spray-deposited film. Surface attachment was through a hyaluronic acid-CD44-mediated adhesion process. Remarkably, these procedures did not interfere with macrophage functions, including phagocytosis, and the ‘backpacks’ were capable of releasing an entrapped model protein in a controlled and sustained manner in vitro. Such approaches may hold great promise for the treatment of cancer and other inflammatory processes through monocyte/macrophage homing; however, detailed experiments are still necessary to assess their safety at different levels.
A ‘Patient-Centric’ Translational Approach
Finally, current advances in nanotechnology are expected to contribute enormously to a ‘patient-centric’ translational research approach with monocytes and macrophages. Potential examples include micro- and nanotechnology fabrication methodologies for immune cell confinement in two-dimensional (based on microfluidic and micropatterning with cell-specific ligand or nanoparticles on conducting polymers) or three-dimensional (hydrogel-based) scaffolds. These approaches could potentially allow parallel individual processing and selection of millions of monocytes, macrophages and even DCs, not only for understanding of cellular heterogeneity, but perhaps for planning and implementation of personalized immune-cell therapies for combating difficult diseases in the future [21].
Acknowledgments
Financial support from the Danish Agency for Science, Technology and Innovation (Det Strategiske Forskningsråd), reference 2106-08-0081, is gratefully acknowledged. P.P.W. is a recipient of a PhD Scholarship Award from the Faculty of Health and Medical Sciences, University of Copenhagen.
References
- 1.Gordon S. Vol. 158. Berlin: Springer; 2003. The Macrophage as Therapeutic Target. [Google Scholar]
- 2.Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53:283–318. [PubMed] [Google Scholar]
- 3.Moghimi SM, Hunter AC, Murray JC. Nanomedicine: current progress and future prospects. FASEB J. 2005;19:311–330. doi: 10.1096/fj.04-2747rev. [DOI] [PubMed] [Google Scholar]
- 4.Taylor PR, Martinez-Pomarez L, Stacey M. Macrophage receptors and immune recognition. Ann Rev Immunol. 2005;23:901–944. doi: 10.1146/annurev.immunol.23.021704.115816. [DOI] [PubMed] [Google Scholar]
- 5.Sleyster EC, Knook DL. Relation between localization and function of rat liver Kupffer cells. Lab Invest. 1982;47:484–490. [PubMed] [Google Scholar]
- 6.Winkler GC. Pulmonary intravascular macrophages in domestic animal species: review of structural and functional properties. Am J Anat. 1988;181:217–234. doi: 10.1002/aja.1001810302. [DOI] [PubMed] [Google Scholar]
- 7.Moghimi SM. Exploiting bone marrow microvascular structure for drug delivery and future therapies. Adv Drug Deliv Rev. 1995;17:61–73. [Google Scholar]
- 8.Porter CJH, Moghimi SM, Illum L. The polyoxyethylene/polyoxypropylene block co- polymer poloxamer-407 selectively redirects intravenously injected microspheres to sinusoidal endothelial cells of rabbit bone marrow. FEBS Lett. 1992;305:62–66. doi: 10.1016/0014-5793(92)80655-z. [DOI] [PubMed] [Google Scholar]
- 9.Hobbs SK, Monsky WL, Yuan F. Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci USA. 1998;95:4604–4612. doi: 10.1073/pnas.95.8.4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Munn LL. Aberrant vascular architecture in tumors and its importance in drug-based therapies. Drug Discov Today. 2003;8:396–403. doi: 10.1016/s1359-6446(03)02686-2. [DOI] [PubMed] [Google Scholar]
- 11.Abu-Hijleh MF, Habbal OA, Moqattash ST. The role of the diaphragm in lymphatic absorption from the peritoneal cavity. J Anat. 1995;186:453–467. [PMC free article] [PubMed] [Google Scholar]
- 12.Miura T, Shimada T, Tanaka K. Lymphatic drainage of carbon particles injected into the pleural cavity of the monkey as studied by video-assisted thoracoscopy and electron microscopy. J Thorac Cardiovasc Surg. 2000;120:437–447. doi: 10.1067/mtc.2000.108906. [DOI] [PubMed] [Google Scholar]
- 13.Moghimi SM. Nanoparticle engineering for the lymphatic system and lymph node targeting. In: Broz P, Polymer-Based Nanostructures: Medical Applications, editor. Cambridge: Royal Society of Chemistry; 2010. pp. pp 81–97. [Google Scholar]
- 14.Moghimi SM, Rajabi-Siahboomi AR. Advanced colloid-based systems for efficient delivery of drugs and diagnostic agents to the lymphatic tissues. Prog Biophys Mol Biol. 1996;65:221–249. doi: 10.1016/s0079-6107(96)00012-0. [DOI] [PubMed] [Google Scholar]
- 15.Reddy ST, van der Vlies AJ, Simenoi E. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25:1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 16.Moghimi SM, The innate immune responses, adjuvants and delivery systems . Delivery Technologies for Biopharmaceuticals: Peptides, Proteins, Nucleic Acids and Vaccines. In: Jorgensen L, Nielsen HM, editors. Chichester: John Wiley & Sons; 2009. pp. pp 113–127. [Google Scholar]
- 17.Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol. 2006;6:772–783. doi: 10.1038/nri1937. [DOI] [PubMed] [Google Scholar]
- 18.Knight SC. Specialized perinodal fat fuels and fashions immunity. Immunity. 2008;28:135–138. doi: 10.1016/j.immuni.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 19.Viallat RT, Raybuad F, Passarel M. Pleural migration of chrysotile fibers after intratracheal injection in rats. Arch Environ Health. 1986;41:282–286. doi: 10.1080/00039896.1986.9936697. [DOI] [PubMed] [Google Scholar]
- 20.Mowat AM, Bain CC. Mucosal macrophages in intestinal homeostasis and inflammation. J Innate Immun. 2011;3:550–564. doi: 10.1159/000329099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moghimi SM, Hunter AC, Andresen TL. Factors controlling nanoparticle pharmacokinetics: an integrated approach and perspective. Annu Rev Pharmacol Toxicol. 2012;52:481–503. doi: 10.1146/annurev-pharmtox-010611-134623. [DOI] [PubMed] [Google Scholar]
- 22.Monopoli MP, Walczyk D, Campbell A. Physical-chemical aspects of protein corona: relevance to in vitro and in vivo biological impacts of nanoparticles. J Am Chem Soc. 2011;133:2525–2534. doi: 10.1021/ja107583h. [DOI] [PubMed] [Google Scholar]
- 23.Hamad I, Al-Hanbali O, Hunter AC. Distinct polymer architecture mediates switching of complement activation pathways at the nanosphere-serum interface: implications for stealth nanoparticle engineering. ACS Nano. 2010;4:6629–6638. doi: 10.1021/nn101990a. [DOI] [PubMed] [Google Scholar]
- 24.Moghimi SM, Andersen AJ, Ahmadvand D. Material properties in complement activation. Adv Drug Deliv Rev. 2011;63:1000–1007. doi: 10.1016/j.addr.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 25.Sou K, Goins B, Takeoka S. Selective uptake of surface-modified vesicles by bone marrow macrophages in vivo. Biomaterials. 2006;28:2655–2666. doi: 10.1016/j.biomaterials.2007.01.041. [DOI] [PubMed] [Google Scholar]
- 26.Moghimi SM. Recent developments in polymeric nanopartricle engineering and their applications in experimental and clinical oncology. Anticancer Agent Med Chem. 2006;6:553–561. doi: 10.2174/187152006778699130. [DOI] [PubMed] [Google Scholar]
- 27.Moghimi SM, Muir IS, Illum L. Coating particles with a block co-polymer (poloxamine-908) suppresses opsonization but permits the activity of dysopsonins in the serum. Biochim Biophys Acta-Mol Cell Res. 1993;1179:157–165. doi: 10.1016/0167-4889(93)90137-e. [DOI] [PubMed] [Google Scholar]
- 28.Moghimi SM, Szebeni J. Stealth liposomes and long circulating nanoparticles: critical issues in pharmacokinetics, opsonization and protein-binding properties. Prog Lipid Res. 2003;42:463–478. doi: 10.1016/s0163-7827(03)00033-x. [DOI] [PubMed] [Google Scholar]
- 29.Moghimi SM, Hamad I, Andresen TL. Methylation of the phosphate oxygen moiety of phospholipid-methoxy(polyethylene glycol) conjugate prevents PEGylated liposome-mediated complement activation and anaphylatoxin production. FASEB J. 2006;20:2591–2593. doi: 10.1096/fj.06-6186fje. [DOI] [PubMed] [Google Scholar]
- 30.Moghimi SM, Hunter AC. Complement monitoring of carbon nanotubes. Nat Nanotechnol. 2010;5:382. doi: 10.1038/nnano.2010.109. [DOI] [PubMed] [Google Scholar]
- 31.Moghimi SM, Porter CJH, Muir IS. Non-phagocytic uptake of intravenously injected microspheres in rat spleen: influence of particle size and hydrophilic coating. Biochem Biophys Res Commun. 1991;177:861–866. doi: 10.1016/0006-291x(91)91869-e. [DOI] [PubMed] [Google Scholar]
- 32.Moghimi SM. Mechanisms of splenic clearance of blood cells and particles: towards development of new splenotropic agents. Adv Drug Deliv Rev. 1995;17:103–115. [Google Scholar]
- 33.Moghimi SM, Hedeman IS, Muir IS. An investigation of the filtration capacity and the fate of large filtered sterically-stabilized microspheres in rat spleen. Biochim Biophys Acta-Mol Cell Res. 1993;1157:233–240. doi: 10.1016/0304-4165(93)90105-h. [DOI] [PubMed] [Google Scholar]
- 34.Moghimi SM, Hedeman H, Illum L. Effect of splenic congestion associated with haemolytic anaemia on filtration of ‘spleen-homing' microspheres. Clin Sci (Lond) 1993;84:605–609. doi: 10.1042/cs0840605. [DOI] [PubMed] [Google Scholar]
- 35.Moghimi SM. Modulation of lymphatic distribution of subcutaneously injected poloxamer 407-coated nanospheres: the effect of the ethylene oxide chain configuration. FEBS Lett. 2003;540:241–244. doi: 10.1016/s0014-5793(03)00273-4. [DOI] [PubMed] [Google Scholar]
- 36.Moghimi SM. The effect of methoxy-PEG chain length and molecular architecture on lymph node targeting of immuno-PEG liposomes. Biomaterials. 2006;27:136–144. doi: 10.1016/j.biomaterials.2005.05.082. [DOI] [PubMed] [Google Scholar]
- 37.Moghimi SM, Moghimi M. Enhanced lymph node retention of subcutaneously injected IgG1-PEG2000-liposomes through pentameric IgM antibody-mediated vesicular aggregation. Biochim Biophys Acta-Biomemb. 2008;1778:51–55. doi: 10.1016/j.bbamem.2007.08.033. [DOI] [PubMed] [Google Scholar]
- 38.Jain R, Dandekar P, Patravale V. Diagnostic nanocarriers for sentinel lymph node imaging. J Control Release. 2009;138:90–102. doi: 10.1016/j.jconrel.2009.05.010. [DOI] [PubMed] [Google Scholar]
- 39.Champion JA, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci USA. 2006;103:4930–4934. doi: 10.1073/pnas.0600997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoo JW, Mitragotri S. Polymer particles that switch shape in response to a stimulus. Proc Natl Acad Sci USA. 2010;107:11205–11210. doi: 10.1073/pnas.1000346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ruggiero A, Villa CH, Bander E. Paradoxical glomerular filtration of carbon nanotubes. Proc Natl Acad Sci USA. 2010;107:12369–12374. doi: 10.1073/pnas.0913667107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geng Y, Dalhaimer P, Cai S. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat Nanotechnol. 2007;2:249–255. doi: 10.1038/nnano.2007.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Agrawal AK, Gupta CM. Tuftsin-bearing liposomes in treatment of macrophage-based infections. Adv Drug Deliv Rev. 2000;41:135–146. doi: 10.1016/s0169-409x(99)00061-7. [DOI] [PubMed] [Google Scholar]
- 44.Kristiansen M, Graversen JH, Jacobsen C. Identification of the hemoglobin scavenger receptor. Nature. 2002;409:198–201. doi: 10.1038/35051594. [DOI] [PubMed] [Google Scholar]
- 45.Buechler C, Ritter M, Orso E. Regulation of the scavenger receptor CD163 expression in human monocytes and macrophages by pro- and anti-inflammatory stimuli. J Leukoc Biol. 2000;67:97–103. [PubMed] [Google Scholar]
- 46.Etzerodt A, Maniecki MB, Graversen JH. Efficient intracellular drug-targeting of macrophages using stealth liposomes directed to the hemoglobin scavenger receptor CD163. J Control Release. 2012 doi: 10.1016/j.jconrel.2012.01.034. DOI: 10.1016/j.jconrel.2012.01.034. [DOI] [PubMed] [Google Scholar]
- 47.Royer PJ, Emara M, Yang C. The mannose receptor mediates the uptake of diverse native allergens by dendritic cells and determines allergen-induced T cell polarization through modulation of IDO activity. J Immunol. 2010;185:1522–1531. doi: 10.4049/jimmunol.1000774. [DOI] [PubMed] [Google Scholar]
- 48.Gazi U, Martinez-Pomares L. Influence of the mannose receptor in host immune responses. Immunobiol. 2009;214:554–561. doi: 10.1016/j.imbio.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 49.Schenk GJ, Haasnoot PCJ, Centlivre M. Efficient CRM197-mediated drug targeting to monocytes. J Control Release. 2012;158:139–147. doi: 10.1016/j.jconrel.2011.09.091. [DOI] [PubMed] [Google Scholar]
- 50.Moghimi M, Moghimi SM. Lymphatic targeting of immuno-PEG-liposomes: evaluation of antibody-coupling procedures on lymph node macrophage uptake. J Drug Target. 2008;16:586–590. doi: 10.1080/10611860802228905. [DOI] [PubMed] [Google Scholar]
- 51.Gupta S, Pal A, Vyas SP. Drug delivery strategies for therapy of visceral leishmaniasis. Expt Opin Drug Deliv. 2010;7:371–402. doi: 10.1517/17425240903548232. [DOI] [PubMed] [Google Scholar]
- 52.Griffiths G, Nystroem B, Sable SB. Nanobead-based interventions for the treatment and prevention of tuberculosis. Nat Rev Microbiol. 2010;8:827–834. doi: 10.1038/nrmicro2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Drummond DC, Zignani M, Leroux JC. Current status of pH-sensitive liposomes in drug delivery. Prog Lipid Res. 2000;39:409–460. doi: 10.1016/s0163-7827(00)00011-4. [DOI] [PubMed] [Google Scholar]
- 54.Yoo JW, Irvine DJ, Discher DE. Bio-inspired bioengineered and biomimetic drug delivery carriers. Nat Rev Drug Discov. 2011;10:521–535. doi: 10.1038/nrd3499. [DOI] [PubMed] [Google Scholar]
- 55.Heffernan MJ, Kasturi SP, Yang SC. The stimulation of CD8+ T cells by dendritic cells pulsed with polyketal microparticles containing ion-paired protein antigens and poly(inosinic acid)-poly(cytidylic acid) Biomaterials. 2009;30:910–918. doi: 10.1016/j.biomaterials.2008.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gibaud S, Weingarten C, Andreux JP. Targeting bone marrow with the help of polyalkylcyanoacrylate nanoparticles. Ann Pharm Fr. 1999;57:324–331. [PubMed] [Google Scholar]
- 57.Scholer N, Hahn H, Muller RH. Effect of lipid matrix and size of solid lipid nanoparticles (SLN) on the viability and cytokine production of macrophages. Int J Pharm. 2002;231:167–176. doi: 10.1016/s0378-5173(01)00882-1. [DOI] [PubMed] [Google Scholar]
- 58.Elferink JG. Cytolytic effect of polylysine on rabbit polymorphonuclear leukocytes. Inflammation. 1985;9:321–331. doi: 10.1007/BF00916280. [DOI] [PubMed] [Google Scholar]
- 59.Chellat F, Merhi Y, Moreau A. Therapeutic potential of nanoparticulate systems for macrophage targeting. Biomaterials. 2005;26:7260–7275. doi: 10.1016/j.biomaterials.2005.05.044. [DOI] [PubMed] [Google Scholar]
- 60.Pinto-Alphandary H, Balland O, Laurent M. Intracellular visualization of ampicillin-loaded nanoparticles in peritoneal macrophages infected in vitro with Salmonella typhimurium. Pharm Res. 1994;11:38–46. doi: 10.1023/a:1018985308984. [DOI] [PubMed] [Google Scholar]
- 61.Forestier F, Gerrier P, Chaumard C. Effect of nanoparticle-bound ampicillin on the survival of Listeria monocytogenes in mouse peritoneal macrophages. J Antimicrob Chemother. 1992;30:173–179. doi: 10.1093/jac/30.2.173. [DOI] [PubMed] [Google Scholar]
- 62.Symonds P, Murray C, Hunter AC. Low and high molecular weight poly(L-lysine)s/poly(L-lysine)-DNA complexes initiate mitochondrial-mediated apoptosis differently. FEBS Lett. 2005;579:6191–6198. doi: 10.1016/j.febslet.2005.09.092. [DOI] [PubMed] [Google Scholar]
- 63.Moghimi SM, Symonds P, Murray JC. A two-staged poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol Ther. 2005;11:990–995. doi: 10.1016/j.ymthe.2005.02.010. [DOI] [PubMed] [Google Scholar]
- 64.Moghimi SM, Hunter AC, Murray JC. Cellular distribution of micelles. Science. 2004;303:626–627. doi: 10.1126/science.303.5658.626. [DOI] [PubMed] [Google Scholar]
- 65.Parhamifar L, Larsen AK, Hunter AC. Polycation cytotoxicity: a delicate matter for nucleic acid therapy-Focus on polyethylenimine. Soft Matt. 2010;6:4001–4009. [Google Scholar]
- 66.Kirkeggard T, Jaattela M. Lysosomal involvement in cell death and cancer. Biochim Biophys Acta-Mol Cell Res. 2009;1793:746–754. doi: 10.1016/j.bbamcr.2008.09.008. [DOI] [PubMed] [Google Scholar]
- 67.Stoka V, Turk B, Schendel SL. Lysosomal protease pathway to apoptosis: cleavage of bid, not pro-caspases, is the most likely route. J Biol Chem. 2001;276:3149–3157. doi: 10.1074/jbc.M008944200. [DOI] [PubMed] [Google Scholar]
- 68.Stoka V, Turk V, Turk B. Lysosomal cysteine cathepsins: signalling pathways in apoptosis. Biol Chem. 2007;388:555–560. doi: 10.1515/BC.2007.064. [DOI] [PubMed] [Google Scholar]
- 69.Daemen T, Hofstede G, Kate MTT. Liposomal doxorubicin-induced toxicity: depletion and impairment of phagocytic activity of liver macrophages. Int J Cancer. 1995;61:716–721. doi: 10.1002/ijc.2910610520. [DOI] [PubMed] [Google Scholar]
- 70.Dale GL, Kuhl W, Beutler E. Incorporation of glucocerebrosidase into Gaucher's disease monocytes in vitro. Proc Natl Acad Sci USA. 1979;76:473–475. doi: 10.1073/pnas.76.1.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Belchetz PE, Crawley JC, Braidman IP. Treatment of Guacher's disease with liposome entrapped glucocerebroside beta-glucosidase. Lancet. 1977;2:116–117. doi: 10.1016/s0140-6736(77)90123-4. [DOI] [PubMed] [Google Scholar]
- 72.Balicki D, Beutler E. Gene therapy of human disease. Medicine. 2002;81:69–86. doi: 10.1097/00005792-200201000-00005. [DOI] [PubMed] [Google Scholar]
- 73.Inoue K, Lupski JR. Molecular mechanisms for genomic disorders. Ann Rev Genom Hum Genet. 2002;3:199–242. doi: 10.1146/annurev.genom.3.032802.120023. [DOI] [PubMed] [Google Scholar]
- 74.Ebert RH, Florey HW. The extravascular development of the monocyte observed in vivo. Brit J Exp Pathol. 1939;20:342–356. [Google Scholar]
- 75.Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 76.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 77.Mildner A, Schmidt H, Nitsche M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nature Neurosci. 2007;10:1544–1553. doi: 10.1038/nn2015. [DOI] [PubMed] [Google Scholar]
- 78.Dou HY, Grotepas CB, McMillan JM. Macrophage delivery of nanoformulated antiretroviral drug to the brain in a murine model of neuroAIDS. J Immunol. 2009;183:661–669. doi: 10.4049/jimmunol.0900274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aouadi M, Tesz GJ, Nicoloro SM. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature. 2009;458:1180–1184. doi: 10.1038/nature07774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kraal G, van Rooijen N, Reaching the macrophage: routes of delivery . The Macrophage as Therapeutic Target. In: Gordon S, editor. Vol. 158. Berlin: Springer; 2003. pp. pp 55–65. [Google Scholar]
- 81.van Lent PLEM, Krijger GC, Hofkens W. Selectively induced death of macrophages in the synovial lining of murine knee joints using B-10-liposomes and boron neutron capture synovectomy. Int J Rad Biol. 2009;85:860–871. doi: 10.1080/09553000903072454. [DOI] [PubMed] [Google Scholar]
- 82.Hirsch LR, Gobin AM, Lowery AR. Metal nanoshells. Ann Biomed Engineering. 2006;34:15–22. doi: 10.1007/s10439-005-9001-8. [DOI] [PubMed] [Google Scholar]
- 83.Hirsch LR, Stafford RJ, Bankson JA. Nanoshell-mediated near-infrared thermal therapy of tumors under magnetic resonance guidance. Proc Natl Acad Sci USA. 2003;100:13549–13554. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Petersen AL, Binderup T, Rasmussen P. 64Cu loaded liposomes as positron emission tomography imaging agents. Biomaterials. 2011;32:2334–2341. doi: 10.1016/j.biomaterials.2010.11.059. [DOI] [PubMed] [Google Scholar]
- 85.Moghimi SM, Bonnemain B. Subcutaneous and intravenous delivery of diagnostic agents to the lymphatic system: aplications in lymphoscintigraphy and indirect lymphography. Adv Drug Deliv Rev. 1999;37:295–312. doi: 10.1016/s0169-409x(98)00099-4. [DOI] [PubMed] [Google Scholar]
- 86.Kim S, Lim YT, Soltesz Near-infrared fluorescent type II quantum dots for sentinel lymph node mapping. Nat Biotechnol. 2004;22:93–97. doi: 10.1038/nbt920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maysinger D, Lovric J, Eisenberg A. Fate of micelles and quantum dots in cells. Eur J Pharm Biopharm. 2007;65:270–281. doi: 10.1016/j.ejpb.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 88.Derfus AM, Chan WCW, Bhatia SN. Probing the cytotoxicity of semiconductor quantum dots. Nano Lett. 2004;4:11–18. doi: 10.1021/nl0347334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Al-Hajaj, Moquin A, Neibert KD. Short ligands affect modes of QD uptake and elimination in human cells. ACS Nano. 2011;5:4909–4918. doi: 10.1021/nn201009w. [DOI] [PubMed] [Google Scholar]
- 90.Neibert KD, Maysinger D. Mechanisms of cellular adaptation to quantum dots – the role of glutathione and transcription factor EB. Nanotoxicology. 2012;6:249–262. doi: 10.3109/17435390.2011.572195. [DOI] [PubMed] [Google Scholar]
- 91.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 92.Wilson HM. Macrophage heterogeneity in atherosclerosis – implications for therapy. J Cell Mol Med. 2010;14:2055–2065. doi: 10.1111/j.1582-4934.2010.01121.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stoger JL, Goossens P, de Winther MPJ. Macrophage heterogeneity: relevance and functional implications in atherosclerosis. Curr Vascular Pharmacol. 2010;8:233–248. doi: 10.2174/157016110790886983. [DOI] [PubMed] [Google Scholar]
- 94.Kim WK, Sun Y, Do H. Monocyte heterogeneity underlying phenotypic changes in monocyte according to SIV disease stage. J Leukoc Biol. 2010;87:557–567. doi: 10.1189/jlb.0209082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ghosn EEB, Cassado AA, Govoni GR. Two physically, functionally, and developmentally distinct peritoneal macrophage subsets. Proc Natl Acad Sci USA. 2010;107:2568–2573. doi: 10.1073/pnas.0915000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Martinez FO, Gordon S, Locati M. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;176:7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 97.Hagemann T, Wilson J, Burke F. Ovarian cancer cells polarize macrophage towards a tumor-associated phenotype. J Immunol. 2006;176:5023–5032. doi: 10.4049/jimmunol.176.8.5023. [DOI] [PubMed] [Google Scholar]
- 98.Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol. 2005;5:606–616. doi: 10.1038/nri1669. [DOI] [PubMed] [Google Scholar]
- 99.Tomita M, Yamamoto K, Kobashi H. Immunohistochemical phenotyping of liver macrophages in normal and diseased human liver. Hepatology. 1994;20:317–325. [PubMed] [Google Scholar]
- 100.Hoedemakers RMJ, Morselt HWM, Scherp- hof GL. Secretion pattern of the rat-liver macrophage population following activation with liposomal muramyl dipeptide in-vivo and in-vitro. J Immunother. 1994;15:265–272. doi: 10.1097/00002371-199405000-00005. [DOI] [PubMed] [Google Scholar]
- 101.Moghimi SM, Hedeman H, Christy NM. Enhanced hepatic clearance of intravenously administered sterically stabilized microspheres in zymosan-stimulated rats. J Leukoc Biol. 1993;54:513–517. doi: 10.1002/jlb.54.6.513. [DOI] [PubMed] [Google Scholar]
- 102.Moghimi SM, Murray JC. Poloxamer-188 revisited: a potentially valuable immune modulator? J Natl Cancer Inst. 1996;88:766–768. doi: 10.1093/jnci/88.11.766. [DOI] [PubMed] [Google Scholar]
- 103.Moghimi SM, Gray T. A single dose of intravenously injected poloxamine-coated long-circulating particles triggers macrophage clearance of subsequent doses in rats. Clin Sci (Lond) 1997;93:371–379. doi: 10.1042/cs0930371. [DOI] [PubMed] [Google Scholar]
- 104.Moghimi SM, Patel HM. Modulation of murine liver macrophage clearance of liposomes by diethylstilbestrol: the effect of vesicle surface charge and a role for the complement receptor Mac-1 (CD11b/CD18) of newly recruited macrophages in liposome recognition. J Control Release. 2002;78:55–65. doi: 10.1016/s0168-3659(01)00481-3. [DOI] [PubMed] [Google Scholar]
- 105.Hotta C, Fujimaki H, Yoshinari M. The delivery of an antigen from the endocytic compartment into the cytosol for cross-presentation is restricted to early immature dendritic cells. Immunology. 2006;117:97–107. doi: 10.1111/j.1365-2567.2005.02270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tran KK, Shon H. The role of phagosomal pH on the size-dependent efficiency of cross-presentation by dendritic cells. Biomaterials. 2009;7:1356–1362. doi: 10.1016/j.biomaterials.2008.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kubo M, Hanada T, Yoshimura A. Suppressors of cytokine signaling and immunity. Nat Immunol. 2003;4:1169–1176. doi: 10.1038/ni1012. [DOI] [PubMed] [Google Scholar]
- 108.Benjaminsen RV, Sun H, Henriksen JR. Evaluating nanoparticle sensor design for intracellular pH measurement. ACS Nano. 2011;5:5864–5873. doi: 10.1021/nn201643f. [DOI] [PubMed] [Google Scholar]
- 109.Pramod Kumar EK, Almdal K, Andresen TL. Synthesis and characterization of ratiomateric nanosensors for pH quantification: a mixed micelle approach. Chem Commun (Camb) 2012;18:4776–4778. doi: 10.1039/c2cc17213e. [DOI] [PubMed] [Google Scholar]
- 110.Sokurenko EV, Vogel V, Thomas WE. Catch-bond mechanism of force-induced adhesion: counterintuitive, elusive, but widespread? Cell Host Microbe. 2008;16:314–323. doi: 10.1016/j.chom.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Moghimi SM, Hunter AC, Dadswell CM. Causative factors behind poloxamer 188 (Pluronic F68, Flocor™)-induced complement activation in human sera: a protective role against poloxamer-mediated complement activation by elevated levels of lipoproteins. Biochim Biophys Acta. 2004;1689:103–113. doi: 10.1016/j.bbadis.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 112.Moghimi SM, Andersen AJ, Hashemi SH. Complement activation cascade triggered by PEG-PL engineered nanomedicines and carbon nanotubes: the challenges ahead. J Control Release. 2010;146:175–181. doi: 10.1016/j.jconrel.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 113.Szebeni J, Muggia F, Gabizon A. Activation of complement by therapeutic liposomes and other lipid excipient-based therapeutic products: prediction and prevention. Adv Drug Deliv Rev. 2011;63:1020–1030. doi: 10.1016/j.addr.2011.06.017. [DOI] [PubMed] [Google Scholar]
- 114.Gratton SEA, Pohlhaus PD, Lee J. Nanofabricated particles for engineered drug therapies: a preliminary biodistribution study of PRINT™ nanoparticles. J Control Release. 2007;121:10–18. doi: 10.1016/j.jconrel.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Doshi N, Swiston AJ, Gilbert JB. Cell-based drug delivery devices using phagocytosis-resistant backpacks. Adv Mat. 2011;23:H105–H109. doi: 10.1002/adma.201004074. [DOI] [PubMed] [Google Scholar]
- 116.Moghimi SM, Illum L, Davis SS. Physiopathological and physicochemical considerations in targeting of colloids and drug carriers to the bone marrow. Crit Rev Ther Drug Carr Syst. 1990;7:187–209. [PubMed] [Google Scholar]
- 117.Jolk RI, Feldborg LN, Andersen S. Engineering liposomes and nanoparticles for biological targeting. Adv Biochem Engin Biotechnol. 2011;125:251–280. doi: 10.1007/10_2010_92. [DOI] [PubMed] [Google Scholar]