

Abstract
Neutrophils and complement are key members of innate immunity. The alternative pathway (AP) of complement consists of C3, factor B, factor D and properdin, which amplifies AP activation. AP has been implicated in many neutrophil-mediated diseases, such as anti-neutrophil cytoplasmic antibody-associated vasculitis. The exact mechanism by which the AP and neutrophils interact remains largely unstudied. We investigated the ability of the AP to interact with neutrophil components which can be exposed and released upon activation. Our studies focused on neutrophil enzymes, including myeloperoxidase (MPO), proteinase 3 (PR3), azurocidin, elastase, lysozyme and cathepsin G. All enzymes except for azurocidin were able to bind properdin. However, only MPO could induce C3 activation. MPO mediated AP complement activation in the presence of MgEGTA compared to the EDTA control. This activation resulted in C3 deposition and required properdin to occur. Furthermore, we could show that MPO binds properdin directly, which then serves as a focus for AP activation. In summary, properdin can directly interact with neutrophil components. MPO demonstrates the ability to activate the AP which is dependent on properdin. Finally, MPO is capable of inducing properdin-initiated C3 and C5b-9 deposition in vitro.
Key Words: Alternative pathway, Complement, Myeloperoxidase, Neutrophils, Properdin
Introduction
The complement system consists of three pathways, the classical, lectin and alternative pathway (AP). The AP is composed of C3, factor B, factor D and properdin. The AP is unique in that it can become autoactivated, transforming C3 to C3H20 (C3b-like molecule). This activated C3 binds factor B, which is then cleaved by factor D forming the C3bBb (AP C3 convertase), which generates more C3b. The C3bBb is stabilized by properdin, promoting further C3b generation [1]. More recently, it was discovered that properdin has a pattern recognition capability allowing it to bind and become initiator of the AP. Properdin is then capable of binding C3b followed by factor B and factor D generating the stable AP C3 convertase [2, 3]. In short, properdin is the only known positive regulator in the complement system and can act as initiator of the AP.
Properdin can recognize various foreign structures such as Neisseria gonorrhoeae, Escherichia coli as well as lipopolysaccharide [4]. Properdin can also bind altered self, such as apoptotic and necrotic cells. Studies by Xu et al. [5] demonstrated that properdin can bind necrotic splenocytes from both wild-type and C3-deficient mice, excluding any role of C3b in the binding of properdin. Furthermore, the pattern recognition ability of properdin can promote phagocytosis of apoptotic cells [4]. Kemper et al. [6] demonstrated that properdin can bind apoptotic T lymphocytes which can then induce AP complement activation. Intriguingly, in their study, the apoptotic T cells attached to properdin were phagocytosed in the absence of any complement activation. Properdin can also bind to nonapoptotic/necrotic cells, such as the surface of viable stimulated neutrophils, allowing for complement activation, which demonstrates overlap between the AP and neutrophil-mediated processes [7].
The AP has been implicated in many neutrophil-mediated diseases, such as rheumatoid arthritis, ischemia-reperfusion injury and in particular anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis. With this in mind, exposure to various neutrophil constituents may facilitate interaction with the AP of complement. Human neutrophils are able to produce C3, while mouse neutrophils are able to produce factor B, both integral to the AP [8, 9]. Moreover, neutrophils store the only positive regulator of complement, properdin, in their secondary specific granules [10]. In contrast, the majority of ANCA autoantigens, including MPO, are stored in the primary azurophilic granules of neutrophils. Activation of neutrophils followed by degranulation enables an assortment of neutrophil enzymes to interact with complement factors. Various azurophil granule proteins and oxidants have the capacity to activate complement [11, 12]. MPO itself can activate human C5 to generate a hemolytic C5-9 [13]. Interestingly, neutrophils activated with anti-MPO or anti-PR3 IgG release factors capable of activating the complement system [14].
With this in mind, exposure to various neutrophil constituents may trigger the interaction with the AP of complement. Therefore, we investigated the ability of the AP including properdin to interact with these purified constituents in vitro. We show that various neutrophil enzymes can interact with properdin. In addition, MPO demonstrates a clear ability to induce properdin-dependent AP activation. Intriguingly, MPO illustrated an ability to anchor properdin, thus providing a platform for AP C3 and C5b-9 deposition.
Materials and Methods
Reagents
MPO and PR3 were isolated from human neutrophils [15]. Commercially available MPO from Athens Research (Athens, Ga., USA) and Cedarlane (Burlington, Ont., Canada) was also tested. Azurocidin, lysozyme and cathepsin G isolated from human neutrophils were from Athens Research. Native neutrophil elastase was from AbD Serotec (Oxford, UK). Human properdin was isolated from serum by anion exchange and size exclusion chromatography [5] or was obtained from a commercial source (Quidel, San Diego, Calif., USA).
The molecular characteristics of properdin were investigated by SDS-PAGE (fig. 1). Properdin-deficient serum was generated in house using zymosan depletion [16]. C3 was isolated from human serum and treated with trypsin for the generation of C3b [17]. The rabbit anti-human properdin digoxigenin (DIG) and mouse anti-human C3 DIG (RFK-22) are in-house reagents, and the mouse anti-human C5b-9 DIG was a kind gift from Dr. T.E. Mollnes (Bodo, Norway) [18]. Mouse anti-human factor P 2 was supplied by Quidel, while the goat anti-human factor P was from Complement Technology (Tyler, Tex., USA). Goat anti-mouse immunoglobulin horseradish peroxidase (HRP) and rabbit anti-goat immunoglobulin HRP were purchased from Dako (Heverlee, Belgium). Sheep anti-DIG peroxidase was from Roche (Mannheim, Germany). For use in ELISA, all antibodies were diluted in phosphate-buffered saline (PBS) containing 1% bovine serum albumin (BSA) and 0.05% Tween-20 (PTB).
Fig. 1.
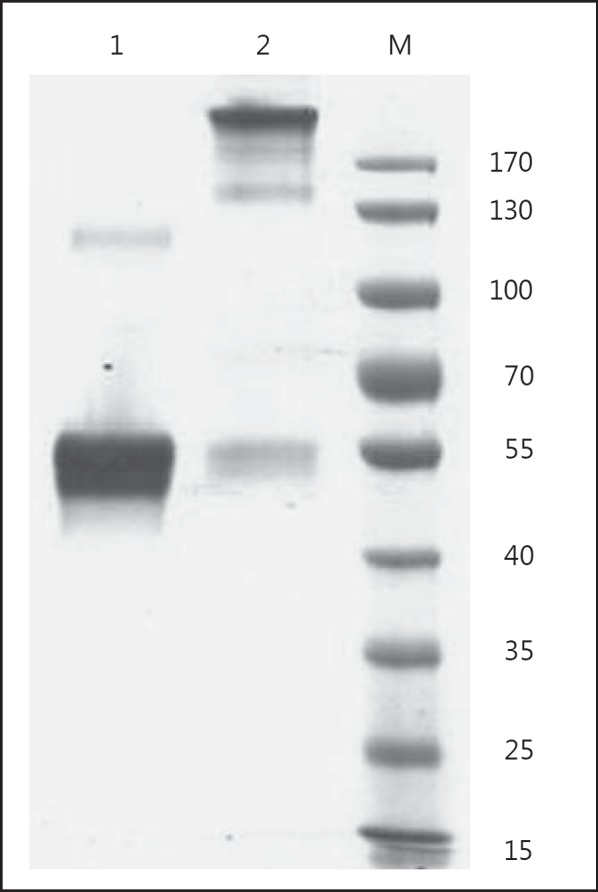
Molecular characteristics of properdin. Preparations of properdin were analyzed under nonreducing conditions in a 10% SDS-PAGE gel. Following electrophoresis, the gel was stained with Coomassie brilliant blue and destained using methanol. The commercial preparation (lane 1) showed dimeric properdin but no presence of aggregates, even when high concentrations are loaded (25 g), while in another preparation (lane 2, 1 g loaded), aggregates were formed following storage and this preparation was excluded from testing.
Properdin Binding to Neutrophil Components
The neutrophil enzymes MPO, PR3, elastase, azurocidin, lysozyme and cathepsin G or BSA were diluted in PBS and coated in a Nunc 96-well plate. The plate was incubated with 1% BSA/PBS as a blocking buffer for a minimum of 30 min at 37°C. Properdin was diluted in PTB and added to the wells for 1 h. Subsequently, properdin was detected using rabbit anti-human properdin-DIG for 1 h and sheep anti-DIG HRP for 1 h with development using 3-3,5,5-tetramethylbenzidine (TMB). The plate was washed in PBS Tween-20 (0.05%) between each step. Data are expressed as optical density values at 450 nm (OD450 nm). Furthermore, properdin binding was demonstrated using mouse anti-human factor P 2 and goat anti-human factor P, respectively. The primary antibodies were developed using the corresponding goat anti-mouse HRP and rabbit anti-goat HRP followed by TMB.
Complement Activation Assays
For the C3 activation assay, a variety of neutrophil constituents or BSA was coated on a 96-well plate using PBS. After coating, the plate was blocked using 1% BSA/PBS and incubated at 37°C for at least 30 min. The wells were exposed to normal human serum diluted in gelatin veronal buffer (GVB)++ MgEGTA (33.3 mM) or EDTA (20 mM) for 1 h at 37°C. The plate was washed in PBS Tween-20 (0.05%) with mouse anti-human C3 DIG (RFK-22) added thereafter for 1 h. Detection was completed using sheep anti-DIG HRP for 1 h and TMB. The plate was washed in PBS Tween-20 (0.05%) between each step.
The properdin deposition assays involving normal human serum (NHS) were performed similar those mentioned above; however, detection was performed using rabbit anti-human properdin DIG and sheep anti-DIG HRP using TMB as chromogen. All TMB readings were performed at 450 nm.
To show that AP activation is dependent upon properdin, we performed the following experiment. MPO was coated at 2.5 µg/ml using PBS on a 96-well plate. A blocking step of 1% BSA/PBS was added for a minimum of 30 min at 37°C following coating. The wells were exposed to a fixed concentration of properdin-deficient serum with or without properdin or just NHS in GVB++ MgEGTA (33.3 mM) or EDTA (20 mM) for 1 h. The plate was washed in PBS Tween-20 (0.05%). Mouse anti-human C3 DIG was diluted in PTB and added to all wells for 1 h followed by addition of sheep anti-DIG HRP in PTB for 1 h. The enzymatic reaction was developed using TMB and measured at OD450 nm. Studies testing the ability of properdin to initiate complement activation were performed on a 96-well plate coated with MPO and BSA, respectively. All MPO- and BSA-coated wells were blocked using 1% BSA/PBS for at least 30 min at 37°C. Increasing concentrations of properdin diluted in PTB were added to the individual wells. The wells were washed using PBS Tween-20 (0.05%). A fixed concentration of properdin-deficient human serum diluted in GVB−/− MgEGTA (33.3 mM) or GVB−/− EDTA (20 mM) was then added for 1 h. The plate was washed using PBS Tween-20 (0.05%) followed by addition of mouse anti-human C3 DIG or C5b-9 DIG; both were then detected using sheep anti-DIG HRP, as mentioned earlier. The assay was developed using TMB and read at 450 nm.
Properdin-C3b Assay
Based on previous studies demonstrating the ability of MPO to activate complement, we set out to determine the exact mechanism involved in the AP activation. We coated MPO or BSA using PBS onto a 96-well plate. The coated plate was incubated with 1% BSA/PBS as a blocking step for a minimum of 30 min at 37°C. Increasing concentrations of properdin were added to the individual wells for 1 h. The wells were washed using PBS Tween-20 (0.05%) and a fixed concentration of human C3b (30 µg/ml) was added to all wells for 1 h. C3b binding was detected using mouse anti-human C3 DIG (RFK-22) and sheep anti-DIG HRP, each incubated for 1 h, respectively. TMB was used as chromogen and the plate was read at 450 nm.
Results
Properdin Interacts with Various Components of Neutrophil Granules
It has been described that properdin may play a role in neutrophil-mediated diseases such as ANCA vasculitis [4, 19]. Properdin is present in secondary granules of neutrophils, and after degranulation it can act as a pattern recognition molecule [4, 19]. Therefore, we investigated the ability of properdin to interact with a selection of neutrophil components. We used an ELISA-based system and immobilized the following purified neutrophil factors: MPO, elastase, azurocidin, lysozyme, proteinase 3 and cathepsin G. Incubation with a fixed concentration of purified human properdin (5 µg/ml) resulted in a strong binding to MPO; intermediate binding to lysozyme, cathepsin G, proteinase 3 and elastase, and a weak binding to azurocidin or BSA as a control protein (fig. 2a).
Fig. 2.

The interaction between neutrophil components and the AP. a ELISA wells were coated with MPO, azurocidin, lysozyme, proteinase 3 and cathepsin G (all at 2 µg/ml, respectively). Furthermore, elastase was coated at 8 µg/ml with 1% BSA coated as negative control. The coated plate was washed and 1% BSA/PBS was added as a blocking step for at least 30 min at 37°C. The wells were washed and exposed to 5 µg/ml of properdin, followed by detection using rabbit anti-human properdin. b MPO, elastase, cathepsin G, proteinase 3 and lysozyme were coated at 2 µg/ml with 1% BSA coated as a control. Following coating, the plate was blocked using 1% BSA/PBS for a minimum of 30 min at 37°C. The wells were washed with 5% NHS diluted in GVB++ MgEGTA then added followed by detection using mouse anti-human C3. The values were measured in triplicate and plotted as means with SD.
As properdin is an instrumental protein in the AP of complement, we addressed whether under similar conditions immobilized neutrophil enzymes are capable of activating the AP of complement. After addition of a fixed concentration of 5% NHS in MgEGTA, to specifically allow only AP activation, complement activation was measured by the deposition of C3. MPO and to a lesser extent also cathepsin G were capable of activating the AP of complement (fig. 2b). Cathepsin G-induced complement activation has been demonstrated previously [20].
Properdin Binding to MPO
We further concentrated on the novel interaction between properdin and MPO. Although initial interaction of properdin with MPO was demonstrated with an in-house purified preparation [15], a similar binding was observed with two commercial preparations (fig. 3a). Binding of purified human properdin was demonstrated using a polyclonal antibody against properdin. Increasing the concentration of properdin showed dose-dependent binding to MPO, which was already clearly observed with 2.5 µg/ml of properdin, which is within the physiological range (fig. 3a). The binding of properdin was also dependent on the concentration of MPO immobilized on the plate when a fixed concentration of properdin was added (fig. 3b). Similar to figure 3a, dose-dependent binding of properdin could be demonstrated with either a mouse monoclonal antibody directed against properdin (fig. 3c) or with a goat anti-human properdin polyclonal antibody (fig. 3d). In all cases, only limited interaction was observed with the control protein BSA.
Fig. 3.

Properdin binding to MPO. a The various MPO preparations were coated to the ELISA wells at 0.5 µg/ml with 1% BSA as negative control. The coated plate was incubated at 37°C with 1% BSA/PBS for at least 30 min as a blocking step. Increasing concentrations of properdin were added to the wells followed by detection using rabbit anti-human properdin. b MPO coated at various concentrations or 1% BSA coated to the wells. All MPO- or BSA-coated wells had 1% BSA/PBS added for a minimum of 30 min at 37°C as a blocking agent. Properdin was added at a concentration of 0.25 µg/ml followed by detection using rabbit anti-human properdin. c MPO was coated at 2.5 µg/ml and 1% BSA as negative control, followed by a blocking step of all wells with 1% BSA/PBS for a minimum of 30 min at 37°C. Increasing concentrations of properdin were added to the wells and results were assessed using mouse anti-human properdin. d MPO was coated at 2.5 µg/ml and 1% BSA as negative control. The plate was blocked using 1% BSA/PBS and incubated at 37°C for at least 30 min. Increasing doses of properdin were added to the wells and results were assessed using goat anti-human properdin.
MPO Activates the AP of Complement Inducing C3 Deposition
It has previously been reported that MPO can interact directly with the human complement component C5 allowing for the generation of a hemolytic C5b-9 complex [13]. Therefore, we investigated the conditions required for MPO-mediated complement activation. MPO or BSA were exposed to 5% NHS diluted in either MgEGTA or EDTA, followed by detection of bound C3. MPO incubated with NHS in MgEGTA demonstrated a strong C3 deposition compared to its respective EDTA and BSA controls (fig. 4a). Furthermore, testing of a titration of NHS diluted in MgEGTA showed that the MPO-mediated C3 deposition is dose dependent compared to the control protein (fig. 4b). This suggests that MPO-mediated complement C3 deposition is supported under AP conditions.
Fig. 4.

MPO activates AP inducing C3 deposition. a ELISA wells were coated with MPO at 2.5 µg/ml and 1% BSA as negative control. All coated wells were incubated with 1% BSA/PBS as a blocking agent for a minimum of 30 min at 37°C. A fixed concentration of 5% NHS diluted in GVB++ MgEGTA or EDTA was added to the wells with detection by mouse anti-human C3 antibody. The values were measured in duplicate and expressed as means with SD. b MPO was coated at 2.5 µg/ml and 1% BSA as negative control to ELISA wells. The plate was blocked using 1% BSA/PBS at 37°C for at least 30 min. Increasing concentrations of NHS diluted in GVB++ MgEGTA were added to the wells followed by detection using mouse anti-human C3 antibody.
MPO-Mediated AP Activation Is Dependent on Properdin
Earlier studies have reported that properdin is integral in promoting AP complement activation [2]. To investigate whether properdin had a role in the MPO-mediated C3 activation, we added 10% NHS in MgEGTA or EDTA to immobilized MPO and detected properdin binding. The findings demonstrated significant properdin deposition on MPO in the presence of MgEGTA, but signals were diminished with EDTA. These findings were reaffirmed by comparing NHS and properdin-deficient serum under AP conditions (fig. 5a). The results illustrated a clear properdin binding in the NHS wells compared to the properdin-deficient serum, demonstrating the need for properdin activation in this setting (fig. 5a). Similar results were obtained with recombinant mouse MPO using normal mouse serum and properdin-deficient serum (data not shown).
Fig. 5.

MPO induces AP properdin (P) consumption. a MPO was coated at 2.5 µg/ml to ELISA wells. Following coating, all MPO-coated wells had 1% BSA/PBS added as a blocking step and were incubated at 37°C for a minimum of 30 min. The wells were washed and exposed to increasing concentrations of NHS or properdin-deficient serum diluted in GVB++ MgEGTA or EDTA followed by detection using the rabbit anti-human properdin antibody. b ELISA wells were coated with MPO at a concentration of 2.5 µg/ml. The wells were washed and incubated with 1% BSA/PBS as a blocking agent and incubated at 37°C for at least 30 min. A fixed concentration of 5% properdin-deficient serum was added alone or with 5 µg/ml of properdin diluted in GVB++ MgEGTA or EDTA. Also, 5% NHS was diluted in GVB++ MgEGTA or EDTA. Detection was performed using the monoclonal mouse anti-human C3 antibody. c MPO was coated at 2.5 µg/ml to ELISA wells. The wells were blocked using 1% BSA/PBS for a minimum of 30 min at 37°C. A fixed concentration of 5% properdin-deficient serum was added alone or with increasing concentrations of properdin diluted in GVB++ MgEGTA. The C3 deposited was detected using mouse anti-human C3 antibody. P = Properdin; P- = properdin deficient.
Considering the ability of MPO to activate the AP and induce both C3 and properdin deposition, we aimed to elucidate the role of properdin in this C3 activation. With this in mind, NHS or properdin-deficient serum with/without properdin was added to MPO. Properdin-deficient serum exhibited significantly less C3 bound compared to its NHS control; however, C3 deposition can be restored with the addition of properdin to the deficient serum (fig. 5b). To investigate the role of properdin further, we added increasing concentrations of properdin to 5% properdin-deficient serum (MgEGTA) which was then added to MPO. The C3 deposited on the well increases with the dose of properdin added to the deficient serum (fig. 5c). In short, these results indicate a significant role for properdin in MPO-mediated complement activation.
Properdin Initiates C3 Deposition on MPO
Recently published data have suggested that properdin may act as an initiator of the AP inducing C3 activation [3]. Therefore, we coated MPO or BSA and exposed it to increasing concentrations of properdin or buffer alone. Next, the plate was washed and properdin-deficient human serum diluted in MgEGTA or EDTA was added to all wells, followed by C3 detection. The results showed only background levels of C3 bound in BSA wells, with a slight increase in the MPO wells without supplemental properdin. MPO wells supplemented with properdin prior to the addition of properdin-deficient serum demonstrated a clear dose-dependent increase in C3 deposition compared to MPO wells unexposed to properdin (fig. 6). Similar results were obtained with a different properdin batch (data not shown). In summary, the results indicate that properdin may act as an initiator of the AP C3 deposition involving MPO.
Fig. 6.

Properdin initiated C3 deposition on MPO. ELISA wells were coated with MPO (1 µg/ml) or 1% BSA diluted in PBS. All coated wells were blocked using 1% BSA/PBS and incubated at 37°C for at least 30 min. The wells were exposed to increasing concentrations of properdin diluted in PTB. The wells were washed using PBS Tween and 5% properdin-deficient serum diluted in GVB−/− MgEGTA (33.3 mM) or EDTA (20 mM). C3 deposition was determined using mouse anti-human C3 DIG and sheep anti-DIG HRP.
Properdin-Mediated AP Generation of C5b-9
The results described above demonstrate a role for properdin in generating AP activation up to the C3 level. Therefore, we investigated whether this AP activation would lead to further complement activation and generation of C5b-9. This was performed similar to our assay for properdin-initiated C3 deposition on MPO, with detection performed using a mouse monoclonal antibody specific to human C5b-9. C5b-9 deposition on MPO was marked compared to BSA-coated wells which had received 5 μg/ml of properdin followed by serum diluted in MgEGTA. C5b-9 was generated in a complement-dependent manner as illustrated by its inhibition using EDTA.
Furthermore, the AP C5b-9 is generated in a dose-dependent manner when increasing concentrations of properdin were added before addition of properdin-deficient serum (fig. 7).
Fig. 7.

Properdin-mediated AP generation of C5b-9. Wells were coated with MPO at a concentration of 2.5 µg/ml or 1% BSA diluted in PBS. The coated plate was incubated with 1% BSA/PBS added as a blocking step at 37°C for a minimum of 30 min. Response to PTB-diluted properdin (up to 5 µg/ml) added to the wells followed by addition of 5% properdin-deficient serum diluted in GVB−/− MgEGTA (33.3 mM) or EDTA (20 mM) was assessed. C5b-9 was detected using a monoclonal antibody (AE-11) specific to the neoepitope of the MAC complex.
Properdin Directed C3b Deposition on MPO
Our results indicated a role for MPO in activating the AP of complement, with properdin remaining key for this activation. With a previous study demonstrating that MPO can induce complement activation [13], we examined the exact mechanism involved in our studies using purified components. MPO and BSA were coated followed by the addition of increasing concentrations of properdin. All wells were exposed to 30 µg/ml of C3b followed by the detection of C3 deposition. The results demonstrate a clear binding of C3b which is dependent on the presence of properdin for binding to MPO (fig. 8).
Fig. 8.

MPO binds directly to properdin allowing for C3b attachment. ELISA wells were coated with 0.5 µg/ml of MPO or 1% BSA diluted in PBS. All coated wells were incubated at 37°C with 1% BSA/PBS as a blocking step for at least 30 min. Increasing concentrations of properdin diluted in PTB were added to the wells. Next all wells were exposed to a fixed concentration of human C3b (30 µg/ml) diluted in PTB followed by detection using mouse anti-human C3.
Discussion
Several studies have demonstrated a role for the AP in neutrophil-mediated diseases. Activated neutrophils and the release of soluble mediators have increasingly become the focus of research in this field. In particular in ANCA, TNF-stimulated neutrophils, as well as neutrophil extracellular traps, have been postulated as sources of immune activation in the disease [21, 22]. Enzymes stored within the granules of neutrophils were shown to activate complement C5 with oxidants also illustrating similar capabilities [11, 12, 13]. Intriguingly, ANCA-activated neutrophils themselves release factors capable of activating the complement system [14]. This led us to address the question of whether neutrophil components may be involved in the AP activation, and by what possible mechanism it may become activated.
Initially, we focused on properdin and its interplay with neutrophil components, as it promotes AP activation. We found that properdin was capable of binding to MPO, PR3, elastase, cathepsin G and lysozyme but not azurocidin. With these results in mind, we wanted to assess the ability of these enzymes which bound properdin to activate AP C3 deposition. Interestingly, only MPO appeared capable of inducing significant C3 deposition. It is intriguing to see that not all properdin-binding enzymes, such as proteinase 3, elastase and lysozyme, could activate complement, suggesting a mode of possibly regulating properdin. To ensure our results were reproducible, we altered the MPO, properdin and antibody detection systems and attained similar results. After demonstrating that MPO can mediate AP C3 deposition, we focused on the exact conditions and whether the process is complement dependent. Our results showed clear complement activation in MgEGTA but not in EDTA conditions, indicating that activation is dependent on the AP of complement. In addition, we illustrated that during AP activation on MPO, properdin becomes deposited on the antigen in a dose-dependent manner. To address the importance of properdin in this AP-induced C3 deposition, we tested the ability of properdin-deficient serum to induce activation compared to NHS. The results show that properdin-deficient serum lacked the ability to induce AP activation compared to NHS. However, this activation could be restored by the addition of properdin to the properdin-deficient serum, and restoration of activation occurred in a dose-dependent manner. Properdin has two known distinct roles in the complement field. It can stabilize the AP C3 convertase and act as a pattern recognition molecule inducing AP activation [2, 4]. We evaluated the ability of properdin to interact with MPO initially, followed by exposure to properdin-deficient serum as a source of complement factors. Our results demonstrated that MPO was capable of inducing properdin-mediated C3 and C5b-9 deposition compared to the control.
These results indicate a new role for MPO in activating the complement system. In addition, these data demonstrate that MPO and the other neutrophil granule components demonstrate a pleiotropic range of capabilities in regulating complement. It would be interesting to take the role of autoantibodies to these autoantigens into account, e.g. whether they would enhance AP activation. Furthermore, MPO and other enzymes such as elastase are instrumental in the process of neutrophil extracellular traps, which may be able to control AP activation. A recent study by Camous et al. [7] reinforced the link between neutrophils and the AP. Their interesting findings showed that normal activated neutrophils could bind properdin released during activation and subsequently induced AP C3 convertase generation, indicating another link between the AP and neutrophils. The diverse mechanism of AP activation involving the activated neutrophil, acting as a source of properdin and a platform for AP activation using enzymes such as MPO, demonstrates the complex and varied interactions at work. Recent publications in the field of ANCA vasculitis demonstrated the presence of AP components at the site of injury, as well as AP activation in the circulation of patients with active ANCA vasculitis [23, 24]. These findings were further enforced by a mouse model of MPO ANCA vasculitis using various complement-deficient mice, which demonstrated that the AP is involved in the pathogenesis of the condition [14]. Our findings are all in vitro but are comparable with data in vivo (humans and mice in ANCA vasculitis); however, it will be important to test the principles in vivo in the setting of neutrophil immune and autoimmune-mediated diseases.
Disclosure Statement
The authors would like to state that there are no conflicts of interest.
Acknowledgments
The authors would like to thank Dr. D.E. Jenne for helpful discussions. This work was financially supported by the European Union (Marie Curie TranSVIR FP7-PEOPLE-ITN-2008 No. 238756).
References
- 1.Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058–1066. doi: 10.1056/NEJM200104053441406. [DOI] [PubMed] [Google Scholar]
- 2.Fearon DT, Austen KF. Properdin: binding to C3b and stabilization of the C3b-dependent C3 convertase. J Exp Med. 1975;142:856–863. doi: 10.1084/jem.142.4.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hourcade DE. The role of properdin in the assembly of the alternative pathway C3 convertases of complement. J Biol Chem. 2006;281:2128–2132. doi: 10.1074/jbc.M508928200. [DOI] [PubMed] [Google Scholar]
- 4.Kemper C, Atkinson JP, Hourcade DE. Properdin: emerging roles of a pattern-recognition molecule. Annu Rev Immunol. 2010;28:131–155. doi: 10.1146/annurev-immunol-030409-101250. [DOI] [PubMed] [Google Scholar]
- 5.Xu W, Berger SP, Trouw LA, de Boer HC, Schlagwein N, Mutsaers C, Daha MR, van Kooten C. Properdin binds to late apoptotic and necrotic cells independently of C3b and regulates alternative pathway complement activation. J Immunol. 2008;180:7613–7621. doi: 10.4049/jimmunol.180.11.7613. [DOI] [PubMed] [Google Scholar]
- 6.Kemper C, Mitchell LM, Zhang L, Hourcade DE. The complement protein properdin binds apoptotic T cells and promotes complement activation and phagocytosis. Proc Natl Acad Sci USA. 2008;105:9023–9028. doi: 10.1073/pnas.0801015105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camous L, Roumenina L, Bigot S, Brachemi S, Fremeaux-Bacchi V, Lesavre P, Halbwachs-Mecarelli L. Complement alternative pathway acts as a positive feedback amplification of neutrophil activation. Blood. 2011;117:1340–1349. doi: 10.1182/blood-2010-05-283564. [DOI] [PubMed] [Google Scholar]
- 8.Botto M, Lissandrini D, Sorio C, Walport MJ. Biosynthesis and secretion of complement component (C3) by activated human polymorphonuclear leukocytes. J Immunol. 1992;149:1348–1355. [PubMed] [Google Scholar]
- 9.Okuda T. Murine polymorphonuclear leukocytes synthesize and secrete the third component and factor B of complement. Int Immunol. 1991;3:293–296. doi: 10.1093/intimm/3.4.293. [DOI] [PubMed] [Google Scholar]
- 10.Wirthmueller U, Dewald B, Thelen M, Schafer MK, Stover C, Whaley K, North J, Eggleton P, Reid KB, Schwaeble WJ. Properdin, a positive regulator of complement activation, is released from secondary granules of stimulated peripheral blood neutrophils. J Immunol. 1997;158:4444–4451. [PubMed] [Google Scholar]
- 11.Venge P, Olsson I. Cationic proteins of human granulocytes. 6. Effects on the complement system and mediation of chemotactic activity. J Immunol. 1975;115:1505–1508. [PubMed] [Google Scholar]
- 12.Shingu M, Nonaka S, Nishimukai H, Nobunaga M, Kitamura H, Tomo-Oka K. Activation of complement in normal serum by hydrogen peroxide and hydrogen peroxide-related oxygen radicals produced by activated neutrophils. Clin Exp Immunol. 1992;90:72–78. doi: 10.1111/j.1365-2249.1992.tb05834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vogt W. Complement activation by myeloperoxidase products released from stimulated human polymorphonuclear leukocytes. Immunobiology. 1996;195:334–346. doi: 10.1016/S0171-2985(96)80050-7. [DOI] [PubMed] [Google Scholar]
- 14.Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170:52–64. doi: 10.2353/ajpath.2007.060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ballieux BE, Zondervan KT, Kievit P, Hagen EC, van Es LA, van der Woude FJ, Daha MR. Binding of proteinase 3 and myeloperoxidase to endothelial cells: ANCA-mediated endothelial damage through ADCC? Clin Exp Immunol. 1994;97:52–60. doi: 10.1111/j.1365-2249.1994.tb06579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bjornson AB, Michael JG. Factors in human serum promoting phagocytosis of Pseudomonas aeruginosa. 1. Interaction of opsonins with the bacterium. J Infect Dis. 1974;130((suppl)):S119–S126. doi: 10.1093/infdis/130.supplement.s119. [DOI] [PubMed] [Google Scholar]
- 17.Pepys MB, Butterworth AE. Inhibition by C3 fragments of C3-dependent rosette formation and antigen-induced lymphocyte transformation. Clin Exp Immunol. 1974;18:273–282. [PMC free article] [PubMed] [Google Scholar]
- 18.Gaarkeuken H, Siezenga MA, Zuidwijk K, van Kooten C, Rabelink TJ, Daha MR, Berger SP. Complement activation by tubular cells is mediated by properdin binding. Am J Physiol Renal Physiol. 2008;295:F1397–F1403. doi: 10.1152/ajprenal.90313.2008. [DOI] [PubMed] [Google Scholar]
- 19.Kallenberg CG, Heeringa P. Complement is crucial in the pathogenesis of ANCA-associated vasculitis. Kidney Int. 2013;83:16–18. doi: 10.1038/ki.2012.371. [DOI] [PubMed] [Google Scholar]
- 20.Maison CM, Villiers CL, Colomb MG. Proteolysis of C3 on U937 cell plasma membranes. Purification of cathepsin G. J Immunol. 1991;147:921–926. [PubMed] [Google Scholar]
- 21.Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, Grone HJ, Brinkmann V, Jenne DE. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009;15:623–625. doi: 10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hewins P, Morgan MD, Holden N, Neil D, Williams JM, Savage CO, Harper L. IL-18 is upregulated in the kidney and primes neutrophil responsiveness in ANCA-associated vasculitis. Kidney Int. 2006;69:605–615. doi: 10.1038/sj.ki.5000167. [DOI] [PubMed] [Google Scholar]
- 23.Xing GQ, Chen M, Liu G, Heeringa P, Zhang JJ, Zheng X, E J, Kallenberg CG, Zhao MH. Complement activation is involved in renal damage in human antineutrophil cytoplasmic autoantibody associated pauci-immune vasculitis. J Clin Immunol. 2009;29:282–291. doi: 10.1007/s10875-008-9268-2. [DOI] [PubMed] [Google Scholar]
- 24.Gou SJ, Yuan J, Chen M, Yu F, Zhao MH. Circulating complement activation in patients with anti-neutrophil cytoplasmic antibody-associated vasculitis. Kidney Int. 2013;83:129–137. doi: 10.1038/ki.2012.313. [DOI] [PubMed] [Google Scholar]