

Abstract
C1q has been shown to recognize apoptotic cells, to enhance their uptake and to modulate cytokine release by phagocytes and thus promote immune tolerance. Surface-exposed calreticulin (CRT), known as a C1q receptor, is also considered to be an early eat-me signal that enhances the phagocytosis of apoptotic cells and is capable of eliciting an immunogenic response. However, the molecular mechanisms that trigger these functions are not clear. We hypothesized that CRT and C1q might act together in these processes. We first showed, by means of fluorescence resonance energy transfer (FRET), that CRT interacts with the C1q globular region at the surface of early apoptotic cells. Next, we pointed out that knockdown of CRT on early apoptotic HeLa cells impairs the enhancement effect of C1q on their uptake by THP-1 monocyte-derived macrophages. Furthermore, a deficiency of CRT induces contrasting effects on cytokine release by THP-1 macrophages, increasing interleukin (IL)-6 and monocyte chemotactic protein 1/CCL2 and decreasing IL-8. Remarkably, these effects were greatly reduced when apoptotic cells were opsonized by C1q, which counterbalanced the effect of the CRT deficiency. These results demonstrate that CRT-C1q interaction is involved in the C1q bridging function and they highlight the particular ability of C1q to control the phagocyte inflammatory status, i.e. by integrating the molecular changes that could occur at the surface of dying cells.
Key Words: C1q, Calreticulin, Phagocytosis, Apoptotic cells, Interleukins
Introduction
The well-known complement component C1q [1, 2, 3, 4] was described in the last decade for its role in the recognition and removal of apoptotic cells. C1q acts as an opsonin and, by itself, facilitates the uptake of unwanted self-cells by phagocytes [5, 6, 7, 8, 9]. Numerous studies have also shown that C1q, which is predominantly synthesized by macrophages and dendritic cells, influences the phagocyte ‘status’ by modulating its differentiation and regulating its cytokine expression [5, 6, 10, 11, 12]. Accordingly, mutations in C1q and C1q deficiency are linked to systemic lupus erythematosus autoimmune disease. In addition, genetic variants of C1q are associated with the development of rheumatoid arthritis [9, 13, 14]. The binding of C1q to the apoptotic cell surface is a multimolecular event which has not yet been completely deciphered. We and others have characterized a number of the molecules that mark the apoptotic cell and serve as C1q ligands via interaction with its globular regions (C1qGR), such as phosphatidylserine (PS) [15], DNA [16, 17], annexins [18], GAPDH [19] and calreticulin (CRT) [20]. To add complexity, some of these molecules have been shown to interact, e.g. PS with CRT or annexins [20, 21, 22]. How these interactions modulate apoptotic cell uptake and/or the immune response initiated by the phagocyte is a major question that needs to be solved in order to understand the fragile balance between tolerance and immunogenicity. Recently characterized as an early eat-me signal that enhances the phagocytosis of apoptotic cells [23], surface-exposed CRT (ectoCRT) has been known for a long time to be associated with CD91 at the phagocyte surface and serves as a C1q collagen tail coreceptor, although the consequence of this interaction is still not clear [24, 25]. The way CRT plays out its functions on each side of the phagocytic synapse has still to be elucidated. Interestingly, recent studies have revealed the immunogenic consequences of CRT exposure at the surface of dying cells, and have demonstrated that CRT could trigger an immunogenic response in reaction to proapoptotic/anticancer drugs [26]. The apparent opposite effects of C1q viewed as tolerogenic correlated to the immunosuppressive behavior of apoptotic cells and of CRT mostly described as immunogenic are open to question. They prompted us to examine the combined effect of both proteins on the phagocyte response to apoptotic cells. In a previous study, we demonstrated that CRT binds with a high affinity to C1qGR but also to PS, which is well-recognized by C1q at the surface of early apoptotic cells. Notably, CRT knockdown obtained by specific siRNA, reducing exposure of CRT at the surface of HeLa cells, induces an increase of apoptotic cell binding to C1q [20].
In order to gain insight into the mutual effect of C1q and ectoCRT on the uptake of apoptotic cells, we examined the direct C1qGR-CRT interaction at the surface of apoptotic HeLa cells and measured how full-length C1q (used as an opsonin) and CRT impact the phagocytosis mediated by THP-1-derived macrophages and their cytokine profile.
Materials and Methods
Media, Reagents and Antibodies
Glutamax Dulbecco's modified Eagle's medium (DMEM), RPMI 1640, penicillin/streptomycin, trypsin-EDTA, phosphate-buffered saline (PBS) and carboxyfluorescein diacetate succinimidyl ester (CFSE) were purchased from Invitrogen. The fetal calf serum was obtained from Dutscher. Dimethyl sulfoxide (DMSO), phorbol 12-myristate 13-acetate (PMA), cytochalasin D and lipopolysaccharide (LPS; from Escherichia coli 055:B5) were purchased from Sigma-Aldrich. PA1-902A, a chicken polyclonal antibody directed against the N-terminus of CRT, was obtained from Affinity Bioreagents. SPA-601, a mouse monoclonal antibody against CRT, was from Stressgen and rabbit polyclonal antibodies directed against human C1q were from the Immune Response to Pathogens and Altered-Self Group (Institut de Biologie Structurale, Grenoble). Alexa-488-conjugated donkey anti-chicken IgY and cyanine-3-conjugated goat anti-rabbit IgG were from Jackson Immunoresearch. C1q and C1qGRs were purified from human serum and were prepared and quantified as described previously [27].
Cell Culture, Apoptosis Induction and THP-1 Differentiation
HeLa cells (American Type Culture collection, No. CCL2) were grown in Glutamax DMEM supplemented with 10% (v/v) fetal calf serum, penicillin (2.5 U/ml) and streptomycin (2.5 μg/ml) at 5% CO2. THP-1 cells (American Type Culture collection, No. TIB-202) were cultivated in RPMI supplemented with 10% (v/v) fetal calf serum, penicillin (2.5 U/ml) and streptomycin (2.5 μg/ml) at 5% CO2. The cells were regularly tested for Mycoplasma contamination (Mycoalert detection kit, Lonza). The apoptosis of HeLa cells cultured at 60–80% confluence was induced by UVB irradiation (1,000 mJ/cm2) at 312 nm in fresh DMEM as previously described [15]. Cells were then incubated for the indicated times at 37°C and 5% CO2. Measurement of early/late apoptosis was performed by flow cytometry using the Annexin V-FITC kit (MACS Miltenyi Biotec) according to the manufacturer's instructions. Up to 6 h after the UV irradiation, the cell population was considered to be early apoptotic as propidium iodide labeling had not increased significantly at this time point. Early apoptotic cells harvested 4 or 6 h after UV irradiation were used for the phagocytosis assay and the fluorescence resonance energy transfer (FRET) analysis, respectively. Late apoptotic cells were obtained 20 h after the UV irradiation. To induce the differentiation of THP-1 monocyte cells to macrophages, the cells were treated with 10 nM PMA for 72 h [28, 29]. Evaluation of cell phenotypes was performed by standard flow cytometry using fluorochrome-conjugated antibodies against the cell surface markers CD14, CD11b, CD11c, CD54 and HLA-DR. The appropriate fluorochrome-conjugated isotype control antibodies were used as negative controls. Fluorochrome-conjugated antibodies were obtained from eBioscience for anti-CD11c and anti-CD14. Anti-CD11b, anti-CD54 and anti-HLA-DR were from BD Pharmingen. Flow cytometry data were analyzed with a FACScan flow cytometer using CellQuest software (BD Biosciences) or Flowing software (Turku Center for Biotechnology, Finland).
RNA Interference
Cells were transfected with siRNA as described previously [20]. Briefly, HeLa cells were transfected with lipofectamine RNAiMAX (Invitrogen) by siRNA specific for CRT or by its complementary inverse sequence as a control at a final concentration of 10 nM. Forty-eight hours after each transfection, cells were systematically assessed for their CRT content by Western blotting of total cell lysate using mouse anti-CRT monoclonal antibody SPA-601 (1:1,000). As demonstrated previously [20], siRNA CRT treatment induces a decrease of 49–79% in surface-exposed CRT (i.e. it corresponds to a lower surface exposure of CRT of about 2- to 5-fold depending on the variability observed after transient cell transfections).
Confocal Microscopy and FRET Quantification
Cells were washed in PBS and fixed for 15 min with 4% paraformaldehyde at 37°C (EM Grade, Electron Microscopy Science). HeLa cells were then incubated with C1qGR or C1q (10 μg/ml) in PBS-1% BSA for 1 h at room temperature. CRT and bound C1qGR were then detected by indirect immunofluorescence with the PA1-902A chicken polyclonal anti-CRT antibody at 10 μg/ml and a rabbit polyclonal anti-C1q antibody was diluted 1:100. Bound antibodies were visualized with Alexa-488-conjugated donkey anti-chicken IgY and cyanine-3-conjugated goat anti-rabbit IgG; both secondary antibodies were diluted 1:200. Cells were mounted on glass slides with Vectashield HardSet solution (Vector Laboratories) and were visualized with a laser confocal fluorescence microscope LSM 710 (Zeiss) using a Plan-Apochromat 63×/1.4 oil with a 512 × 512 pixels scan. The efficiency of FRET is measured as a relative increase in donor fluorescence (Alexa-488) following the specific photobleaching of the acceptor fluorophore, which is cyanine-3. Several photobleaching steps with moderate laser power were performed and the data obtained were extrapolated by the linear regression fit to a zero-acceptor point in order to get more reliable estimation of FRET efficiency, independent of the degree of acceptor photobleaching or local variations of pixel intensities. The analysis of the fluorescence intensity of the fluorophore and the FRET efficiency were evaluated with the Volocity software (Perkin Elmer). Control FRET experiments using the antibodies (i.e. anti-C1q and anti-CRT plus the secondary-conjugated antibodies) in the absence of C1qGR showed no colocalization. In addition, the use of secondary antibodies alone in the presence of C1qGR indicated that none of the secondary antibodies interacted nonspecifically.
Uptake of Apoptotic Cells
The protocol was adapted from Fraser et al. [5.] HeLa cells were labeled with CFSE as follows: they were washed twice and then resuspended at 1 × 106 cells/ml in PBS and incubated with 5 µM CFSE at 37°C for 15 min. The remaining CFSE was quenched with DMEM-10% FCS, and the cells were then washed 3 times before the induction of apoptosis or siRNA transfection. Prior to phagocytosis, live or apoptotic HeLa cells were preincubated with 0 or 25 µg/ml C1q in RPMI 1640–10% FCS for 30 min at 37°C, washed and then added to THP-1-derived macrophages at a ratio of 1:1. The cells were then placed in contact for 1 h at 37°C and 5% CO2 after centrifugation for 5 min at 300 g. After incubation, cells were harvested with 0.25% trypsin/EDTA. The trypsin reaction was stopped with medium containing 10% FCS. Cells were pelleted by centrifugation at 300 g for 5 min, washed with PBS and resuspended in PBS-1% BSA for staining. THP-1 macrophages were stained with mouse anti-human CD11c-phycoerythrin (PE) or mouse IgG1-PE isotype control for 1 h on ice. The cells were then fixed with 4% paraformaldehyde for 15 min at room temperature, and then resuspended in PBS before FACS analysis. Phagocytosis was calculated as the percent of the double CFSE and CD11c-PE-labeled cells in the THP-1 macrophage population (CD11c-PE-positive cells). Phagocytosis negative controls performed at 4 or 37°C in the presence of 5 µM of cytochalasin (to block the uptake) showed ≤5% of the double CFSE and CD11c-PE-labeled cells.
Quantification of Cytokine Release
Cells were treated essentially according to the uptake assays described above, but unlabeled live or apoptotic HeLa cells were used. After the uptake of apoptotic cells, phagocytic cells were washed with PBS and resuspended in RPMI 1640–10% FCS. Supernatants were harvested for cytokine analysis 18 h after the phagocytosis assay (and in some conditions, after the addition of 30 ng/ml LPS), centrifuged to remove cellular debris and stored at −80°C until analysis. Cytokine production was analyzed in the same sample using a BD Cytometric Bead Array (BD Biosciences) for the detection of interleukin (IL)-1α, IL-1β, IL-6, IL-8, IL-10, IL-12p70, tumor necrosis factor-alpha (TNFα) and monocyte chemotactic protein 1 (MCP-1/CCL2) [30]. Analyses were performed at the Etablissement Français du Sang, Grenoble.
Results
The C1q Globular Regions and Calreticulin Interact at the Surface of Early Apoptotic Cells
We developed a FRET strategy to detect C1qGR interaction with endogenous ectoCRT. With our approach, exogenous C1qGR and cell-surface CRT were detected using specific antibodies. Analyses were performed on viable and UV-treated apoptotic HeLa cells. FRET was evaluated after acceptor photobleaching (Cy-3 dye, C1qGR labeling) by measuring donor fluorescence increase (Alexa-488 dye, CRT labeling). Representative images of C1qGR-CRT colocalization areas detected on early apoptotic cells (6 h after UV irradiation) and the corresponding FRET efficiency curves are shown in figure 1. On cells without visible membrane bleb, a significant FRET efficiency of about 6–12% was measured on all colocalization areas. The presence of a FRET signal, in spite of the spacing created by the antibodies, indicates the close proximity of the fluorophores and the direct interaction of the labeled proteins. An example of C1qGR and CRT colocalization and the corresponding FRET analysis (displaying a FRET efficiency of about 8%) is shown in figure 1a. Interestingly, for cells displaying the characteristic surface blebs (fig. 1b), which indicates that cells are in a more advanced stage of apoptosis, the result was heterogeneous. Indeed, some patches presented a FRET-positive signal similar to that observed previously (fig. 1a), but for most of the areas, no FRET signal could be detected (fig. 1b). This suggests that C1qGR-CRT interaction is mostly a feature of the earlier stages of apoptosis.
Fig. 1.

FRET study of C1qGR and CRT at the surface of apoptotic HeLa cells. FRET efficiency was estimated by photobleaching of the acceptor dye (Cy-3) on nonpermeabilized cells incubated with C1qGR and immunolabeled for CRT (Alexa-488) and C1qGR (Cy-3) as described in Materials and Methods. a Apoptotic cell without visible membrane bleb. b Apoptotic cell with characteristic membrane blebs. Regions used for the acceptor photobleaching and FRET analysis are shown. Bars: 5 µm. Middle: curves corresponding to the normalized fluorescence intensities of both dyes (Cy-3 in red and Alexa-488 in green) expressed as a percent of the signal measured before the gradual photobleaching started (blue arrow). Right: FRET efficiency (percent of acceptor fluorescence intensity increase) is expressed as a function of the percent of the normalized acceptor fluorescence intensity.
Downregulation of Calreticulin on Apoptotic Cells Impacts Their Uptake and the C1q Effect on Phagocytosis
In order to analyze the CRT effect on the uptake of apoptotic HeLa cells by macrophages, we measured the capacity of PMA-stimulated THP-1 cells to phagocyte siRNA-treated HeLa cells. siRNA-CRT treatment induced a very efficient knockdown of CRT (fig. 2), resulting in a decrease in the amount of surface CRT of about 2- to 5-fold when compared to control cells (as determined previously after biotinylation of cell-surface proteins [20]). Viable or apoptotic HeLa cells labeled with CFSE and partially depleted for CRT were submitted to differentiated THP-1. The percentage of CD11c-positive macrophages, which had engulfed HeLa cell(s), was determined by flow cytometry as described in Materials and Methods. To assess specifically the effect of C1q on phagocytosis, and because its globular regions alone do not promote cell-cell bridging, whole C1q (25 μg/ml) was used to opsonize apoptotic HeLa cells prior to the macrophage-target cell contact. As illustrated for a representative experiment (fig. 2), HeLa cells were efficiently engulfed by THP-1 macrophages and the phagocytosis increased with the development of apoptosis. CRT-deficient apoptotic cells (siRNA CRT) were significantly less engulfed than control siRNA-treated cells (siRNA ct), at either an early or a late stage of apoptosis (i.e. 4 or 20 h after UV irradiation). As expected from the published data [5], C1q only slightly but specifically enhanced the phagocytosis of early apoptotic cells. Remarkably, this was not observed for CRT-deficient cells. In our assays, C1q had no effect on the phagocytosis of late apoptotic cells.
Fig. 2.

Effect of CRT deficiency and of exogenous C1q on the uptake of early and late apoptotic cells. Early or late apoptotic HeLa cells labeled with CFSE, opsonized or not with C1q (25 µg/ml), were incubated with PMA-treated THP-1 cells at a ratio of 1:1 for 1 h at 37°C. THP-1 macrophages were labeled with an anti-CD11c-PE antibody just prior to flow cytometry analysis. a Dot-plot of a negative control phagocytosis performed at 4°C (negative control). b Dot-plot of the phagocytosis at 37°C (corresponding to late apoptotic CRT-deficient cells). a, b Inset shows macrophages that had engulfed HeLa cells (double-labeled cells). c Phagocytosis is expressed as the percent of the double-labeled cells in the macrophage population (i.e. CD11c-positive cells). Data are the mean ± SD of triplicates of a representative experiment of 5. * p ≤ 0.05, ** p ≤ 0.005, ANOVA 1-way test. d Representative SDS-PAGE analysis and immunoblotting of CRT expressed on siRNA-treated HeLa cells; 20 µg of soluble proteins were analyzed. The molecular mass markers (expressed in kilodaltons) are shown.
C1q and CRT Act Together in Regulating the Cytokine Profile
As it has already been shown that apoptotic cells, C1q and CRT have immunomodulatory effects on phagocytic cells, we next investigated IL-1α, IL-1β, IL-6, IL-8, IL-10, IL-12, MCP-1/CCL2 and TNFα cytokine production in our model. This analysis was performed under sterile conditions (i.e. in the absence of a microorganism component) to avoid the influence of other cytokine modulating factors. Under these conditions, IL-10 and TNFα were not detected, and IL-1α, IL-1β and IL-12 were only detected at a low level (data not shown). In contrast, the proinflammatory IL-6, IL-8 and MCP-1/CCL2 cytokines were efficiently produced and modulated by THP-1 macrophages after incubation with HeLa cells. As a result of the uptake of viable HeLa cells (fig. 2), THP-1 cells impressively enhanced the production of IL-6, MCP-1/CCL2 and IL-8 (fig. 3). When apoptotic cells were engulfed, these increases were significantly reduced. In addition, efficient CRT knockdown obtained by siRNA CRT treatment (fig. 2d) clearly reduced this anti-inflammatory effect by increasing the release of IL-6 and MCP-1/CCL2 but not IL-8 (fig. 3). Indeed, concentrations of IL-6 and MCP-1/CCL2 were 7.9- and 4.1-fold higher for siRNA CRT early apoptotic cells, compared to siRNA ct cells. Similarly, increases were also observed for late apoptotic cells (14.1- and 2.2-fold for IL-6 and MCP-1/CCL2, respectively). When early apoptotic cells were first opsonized by exogenous C1q, the differences between siRNA CRT and siRNA ct conditions were clearly of a lesser extent for both IL-6 and MCP-1/CCL2 cytokine levels. This suggests that the presence of C1q could compensate for the CRT deficiency. If IL-8 release was not affected by CRT on early apoptotic cells (fig. 3), it nevertheless significantly decreased following the uptake of late apo- ptotic CRT-deficient cells (i.e. late siRNA CRT vs. late siRNA ct cells). Interestingly, the level of IL-6 was differentially modulated by C1q, depending on the CRT expression on the early apoptotic cells (fig. 4a). The decrease of IL-6 was noticeably more marked for CRT-deficient cells than for control cells. No significant change was observed in response to the uptake of late apoptotic cells. To the contrary, the effect on IL-8 level was observed only for macrophages fed with late apoptotic cells, but, opposite to what we measured for IL-6, C1q affected the level of IL-8 in the phagocytosis of control cells more efficiently than that of CRT-deficient cells (fig. 4b). Finally, the emblematic IL-10 anti-inflammatory cytokine was only produced after LPS-stimulation of THP-1. Even under this condition, the level of IL-10 remained weak and appeared unaffected by CRT and C1q. The same observation was made for TNFα (data not shown).
Fig. 3.

Cytokines released from THP-1 macrophages during the uptake of CRT-deficient apoptotic cells. Cytokine production of PMA-treated THP-1 monocytes was analyzed using BDBiosciences Cytometric BeadArray in the supernatant of macrophages fed with early or late apoptotic cells deficient or not for CRT (i.e. siRNA CRT or siRNA ct cells). When indicated, early or late apoptotic cells were opsonized by C1q. Supernatants were collected 18 h after the phagocytosis assay. Data are the average concentrations ± SD from measurements of 3 independent experiments. * p ≤ 0.05, Student t test. Control cytokine levels produced by THP-1 alone or fed with viable cells are shown. Fold modulation of the cytokines released in the siRNA CRT versus siRNA ct conditions is reported when differences were statistically significant.
Fig. 4.
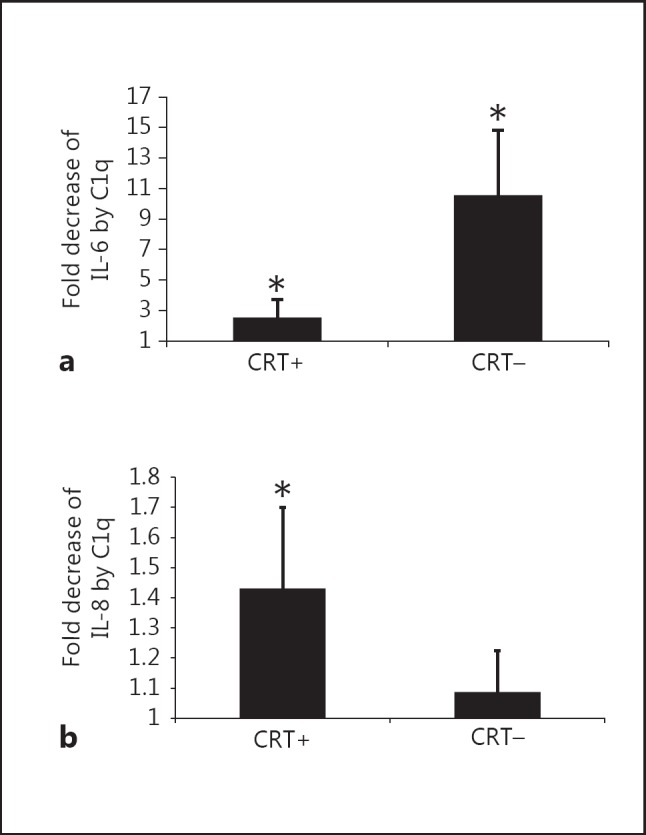
C1q modulation of IL-6 and IL-8 released from macrophages during the uptake of apoptotic cells. Cytokine levels were measured by analysis of the supernatant of differentiated THP-1 macrophages and fed with early (IL-6) or late (IL-8) apoptotic cells as described for figure 3. Selected results of figure 3 are expressed as fold difference in cytokine concentration (a IL-6 and b IL-8) compared to control levels from phagocytes, which had ingested apoptotic cells in the absence of C1q. Data are plotted as average fold decreases ± SD from 3 experiments. * p ≤ 0.05 (C1q-treated levels vs. control levels in the absence of C1q, Student t test). CRT+ = siRNA control-treated cells; CRT- = siRNA CRT-treated cells.
Discussion
The C1q globular region binds with high affinity to purified CRT and C1qGR partially colocalizes with ectoCRT at the surface of apoptotic cells; this raises the possibly that CRT and C1q act together to modulate the phagocytosis event by their interaction at the apoptotic cell surface. Accordingly, we have previously demonstrated that PS is a common C1q and CRT partner and that CRT knockdown increased C1q binding to apoptotic cell binding, possibly via its interaction with PS [20]. Here, we focused on C1q/CRT interactions that occur at the apoptotic cell surface and their effect on the phagocyte function. We first demonstrated by FRET that C1qGR binds directly to CRT exposed at the apoptotic cell surface, and our observation suggests that this interaction could be dependent on the apoptotic stage. Secondly, we analyzed the consequence of CRT deficiency on the uptake of the cells undergoing apoptosis by THP-1 macrophages. As expected from reports showing that CRT could be an early eat-me signal in the uptake of apoptotic cells, we have shown that CRT-deficient apoptotic HeLa cells were, in effect, less engulfed than control cells. We also observed that CRT deficiency abolished the C1q enhancement of the phagocytosis of early apoptotic cells. This result is logically supported by the direct CRT-C1q interaction and provides evidence that CRT is a C1q ligand involved in the C1q enhancement of apoptotic cell uptake at the early stage of apoptosis. However, together with our previous observation that reducing surface CRT exposure on early apoptotic cells induces a significant increase of C1q binding [20], this indicates that the presence of C1q at the cell surface is not sufficient for its bridging function. This also underlines that functions supported by C1q are dependent on the accessibility of its ligands on the apoptotic cell, and suggests that binding events could modulate responses triggered by C1q. To get an insight into C1q function in signaling and its possible partnership with ectoCRT in this process, we have quantified cytokines released from phagocytic THP-1 macrophages. Our findings indicated that, if apoptotic cell engulfment has anti-inflammatory consequences as IL-6, MCP-1/CCL2 and IL-8 levels are dramatically reduced, CRT deficiency on apoptotic cells induces contrasting effects on cytokine production, increasing IL-6 and MCP-1/CCL2 and decreasing IL-8. We conclude that CRT exposed at the apoptotic cell surface led to a downregulation of the expression of IL-6/MCP-1 and an upregulation of IL-8, thus suggesting that the global inflammatory response triggered by CRT is a complex equilibrium. Moreover, this should be dependent on the phagocyte differentiation. Interestingly, MCP-1/CCL2 and IL-6, which are among the most prevalent cytokines in the tumor microenvironment, have been shown to promote the survival of mononuclear cells and induce anti-inflammatory M2-type macrophage polarization [31]. Their downregulation mediated by CRT exposed at the surface of dying cells could be in favor of a more efficient immunogenic response. Remarkably, when apoptotic cells were opsonized by C1q, the CRT deficiency effects were greatly reduced, either with an IL-6 increase or an IL-8 decrease. Besides, we showed that C1q differentially modulated IL-6 and IL-8 releases by THP-1 macrophages during the phagocytosis of normal or CRT-deficient cells. Its anti-inflammatory effect is significantly higher on CRT-deficient cells with regard to the IL-6 release and is more efficient on normal cells for the IL-8 release (fig. 4), thus counterbalancing the effect of the CRT deficiency (fig. 3). It should be noted that C1q is also known to enhance IL-8 production by human umbilical vein endothelial cells in a CRT-dependent way [32]. Accordingly, we have observed that C1q enhances the production of IL-8 and IL-6 by HeLa cells (data not shown). This represents <1.5% of the amount of cytokines measured during phagocytosis and could thus not significantly influence our conclusion.
In this study, we focused on the C1q/CRT interaction at the apoptotic cell surface known to be mediated by the C1q globular region. We analyzed how this impacts on the phagocyte response. However, it should be kept in mind that CRT is also present at the macrophage surface, and it was proposed to act with CD91 as a coreceptor of the collagen tail of C1q. The functional consequence of this interaction is still unclear and needs further elucidation.
In our opinion, a key element is likely the C1q/PS/CRT partnership. Indeed, PS which is well-known for being linked to the anti-inflammatory effects triggered by apoptotic cells [33], interacts with both CRT and C1q. Our previous study suggested that surface CRT level could modulate C1q/PS binding, thus possibly modifying PS recognition by other phagocyte receptors and bridging molecules, and consequently affecting the downstream signaling events. To add complexity, other serum PS-binding molecules have been shown to regulate C1q function such as factor H and beta 2-glycoprotein 1 [34, 35], suggesting that they could also modulate interaction with CRT and the global inflammatory responses.
In conclusion, our observations highlight the crucial role of C1q in tissue homeostasis in controlling the inflammatory phagocyte status. Most notably, our data emphasize the dual role of C1q on uptake and on signaling events during the elimination of apoptotic cells. We are convinced that the versatile binding properties of C1q are a key element at the disposal of the phagocyte for integrating the subtle modifications which appear on its prey.
Acknowledgments
We thank David Laurin and Caroline Aspord from the Etablissement Français du Sang, Grenoble for their help on cytokine detection, Françoise Lacroix from the IBS platform of the Partnership for Structural Biology and the Institut de Biologie Structurale in Grenoble for assistance and access to the epifluorescence microscope facility and Sarah Ancelet for purification of C1q and its globular regions. We are grateful to Nicole Thielens for her critical reading of the manuscript and for her helpful assistance.
M.V. was supported by a PhD grant from University Joseph Fourier-Grenoble and R.A. and R.O. by PhD grants from provinces of South and North Lebanon, respectively. This work was supported in part by the French National Research Agency (ANR-09-PIRI-0021), Association Espoir and GEFLUC (Isère).
References
- 1.Gaboriaud C, Frachet P, Thielens NM, Arlaud GJ. The human C1q globular domain: structure and recognition of non-immune self ligands. Front Immunol. 2011;2:92. doi: 10.3389/fimmu.2011.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J Immunol. 1997;158:4525–4528. [PubMed] [Google Scholar]
- 3.Navratil JS, Watkins SC, Wisnieski JJ, Ahearn JM. The globular heads of C1q specifically recognize surface blebs of apoptotic vascular endothelial cells. J Immunol. 2001;166:3231–3239. doi: 10.4049/jimmunol.166.5.3231. [DOI] [PubMed] [Google Scholar]
- 4.Nayak A, Ferluga J, Tsolaki AG, Kishore U. The non-classical functions of the classical complement pathway recognition subcomponent C1q. Immunol Lett. 2010;131:139–150. doi: 10.1016/j.imlet.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 5.Fraser DA, Laust AK, Nelson EL, Tenner AJ. C1q differentially modulates phagocytosis and cytokine responses during ingestion of apoptotic cells by human monocytes, macrophages, and dendritic cells. J Immunol. 2009;183:6175–6185. doi: 10.4049/jimmunol.0902232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fraser DA, Pisalyaput K, Tenner AJ. C1q enhances microglial clearance of apoptotic neurons and neuronal blebs, and modulates subsequent inflammatory cytokine production. J Neurochem. 2010;112:733–743. doi: 10.1111/j.1471-4159.2009.06494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hochreiter-Hufford A, Ravichandran KS. Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol. 2013;5:a008748. doi: 10.1101/cshperspect.a008748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paidassi H, Tacnet-Delorme P, Arlaud GJ, Frachet P. How phagocytes track down and respond to apoptotic cells. Crit Rev Immunol. 2009;29:111–130. doi: 10.1615/critrevimmunol.v29.i2.20. [DOI] [PubMed] [Google Scholar]
- 9.Lu JH, Teh BK, Wang L, Wang YN, Tan YS, Lai MC, Reid KB. The classical and regulatory functions of C1q in immunity and autoimmunity. Cell Mol Immunol. 2008;5:9–21. doi: 10.1038/cmi.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castellano G, Woltman AM, Schlagwein N, Xu W, Schena FP, Daha MR, van Kooten C. Immune modulation of human dendritic cells by complement. Eur J Immunol. 2007;37:2803–2811. doi: 10.1002/eji.200636845. [DOI] [PubMed] [Google Scholar]
- 11.Benoit ME, Clarke EV, Morgado P, Fraser DA, Tenner AJ. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J Immunol. 2012;188:5682–5693. doi: 10.4049/jimmunol.1103760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghebrehiwet B, Hosszu KK, Valentino A, Peerschke EI. The C1q family of proteins: insights into the emerging non-traditional functions. Front Immunol. 2012;3:52. doi: 10.3389/fimmu.2012.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walport MJ, Davies KA, Botto M. C1q and systemic lupus erythematosus. Immunobiology. 1998;199:265–285. doi: 10.1016/S0171-2985(98)80032-6. [DOI] [PubMed] [Google Scholar]
- 14.Trouw LA, Daha N, Kurreeman FA, Bohringer S, Goulielmos GN, Westra HJ, Zhernakova A, Franke L, Stahl EA, Levarht EW, Stoeken-Rijsbergen G, Verduijn W, Roos A, Li Y, Houwing-Duistermaat JJ, Huizinga TW, Toes RE. Genetic variants in the region of the C1q genes are associated with rheumatoid arthritis. Clin Exp Immunol. 2013;173:76–83. doi: 10.1111/cei.12097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paidassi H, Tacnet-Delorme P, Garlatti V, Darnault C, Ghebrehiwet B, Gaboriaud C, Arlaud GJ, Frachet P. C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J Immunol. 2008;180:2329–2338. doi: 10.4049/jimmunol.180.4.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elward K, Griffiths M, Mizuno M, Harris CL, Neal JW, Morgan BP, Gasque P. CD46 plays a key role in tailoring innate immune recognition of apoptotic and necrotic cells. J Biol Chem. 2005;280:36342–36354. doi: 10.1074/jbc.M506579200. [DOI] [PubMed] [Google Scholar]
- 17.Paidassi H, Tacnet-Delorme P, Lunardi T, Arlaud GJ, Thielens NM, Frachet P. The lectin-like activity of human C1q and its implication in DNA and apoptotic cell recognition. FEBS Lett. 2008;582:3111–3116. doi: 10.1016/j.febslet.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 18.Martin M, Leffler J, Blom AM. Annexin A2 and A5 serve as new ligands for C1q on apoptotic cells. J Biol Chem. 2012;287:33733–33744. doi: 10.1074/jbc.M112.341339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Terrasse R, Tacnet-Delorme P, Moriscot C, Perard J, Schoehn G, Vernet T, Thielens NM, Di Guilmi AM, Frachet P. Human and pneumococcal cell surface glyceraldehyde-3-phosphate dehydrogenase (GAPDH) proteins are both ligands of human C1q protein. J Biol Chem. 2012;287:42620–42633. doi: 10.1074/jbc.M112.423731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paidassi H, Tacnet-Delorme P, Verneret M, Gaboriaud C, Houen G, Duus K, Ling WL, Arlaud GJ, Frachet P. Investigations on the C1q-calreticulin-phosphatidylserine interactions yield new insights into apoptotic cell recognition. J Mol Biol. 2011;408:277–290. doi: 10.1016/j.jmb.2011.02.029. [DOI] [PubMed] [Google Scholar]
- 21.Tarr JM, Young PJ, Morse R, Shaw DJ, Haigh R, Petrov PG, Johnson SJ, Winyard PG, Eggleton P. A mechanism of release of calreticulin from cells during apoptosis. J Mol Biol. 2010;401:799–812. doi: 10.1016/j.jmb.2010.06.064. [DOI] [PubMed] [Google Scholar]
- 22.Gerke V, Moss SE. Annexins: From structure to function. Physiol Rev. 2002;82:331–371. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- 23.Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 24.Ogden CA, deCathelineau A, Hoffmann PR, Bratton D, Ghebrehiwet B, Fadok VA, Henson PM. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med. 2001;194:781–795. doi: 10.1084/jem.194.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donnelly S, Roake W, Brown S, Young P, Naik H, Wordsworth P, Isenberg DA, Reid KB, Eggleton P. Impaired recognition of apoptotic neutrophils by the C1q/calreticulin and CD91 pathway in systemic lupus erythematosus. Arthritis Rheum. 2006;54:1543–1556. doi: 10.1002/art.21783. [DOI] [PubMed] [Google Scholar]
- 26.Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 27.Tacnet-Delorme P, Chevallier S, Arlaud GJ. Beta-amyloid fibrils activate the C1 complex of complement under physiological conditions: evidence for a binding site for A beta on the C1q globular regions. J Immunol. 2001;167:6374–6381. doi: 10.4049/jimmunol.167.11.6374. [DOI] [PubMed] [Google Scholar]
- 28.Schwende H, Fitzke E, Ambs P, Dieter P. Differences in the state of differentiation of THP-1 cells induced by phorbol ester and 1,25-dihydroxyvitamin D3. J Leukoc Biol. 1996;59:555–561. [PubMed] [Google Scholar]
- 29.Kurosaka K, Watanabe N, Kobayashi Y. Potentiation by human serum of anti-inflammatory cytokine production by human macrophages in response to apoptotic cells. J Leukoc Biol. 2002;71:950–956. [PubMed] [Google Scholar]
- 30.Varro R, Chen R, Sepulveda H, Apgar J. Bead-based multianalyte flow immunoassays: the cytometric bead array system. Methods Mol Biol. 2007;378:125–152. doi: 10.1007/978-1-59745-323-3_9. [DOI] [PubMed] [Google Scholar]
- 31.Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, Pienta KJ. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J Biol Chem. 2009;284:34342–34354. doi: 10.1074/jbc.M109.042671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van den Berg RH, Faber-Krol MC, van Wetering S, Hiemstra PS, Daha MR. Inhibition of activation of the classical pathway of complement by human neutrophil defensins. Blood. 1998;92:3898–3903. [PubMed] [Google Scholar]
- 33.Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tan LA, Yu B, Sim FC, Kishore U, Sim RB. Complement activation by phospholipids: the interplay of factor H and C1q. Protein Cell. 2010;1:1033–1049. doi: 10.1007/s13238-010-0125-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan LA, Yang AC, Kishore U, Sim RB. Interactions of complement proteins C1q and factor H with lipid A and Escherichia coli: further evidence that factor H regulates the classical complement pathway. Protein Cell. 2011;2:320–332. doi: 10.1007/s13238-011-1029-y. [DOI] [PMC free article] [PubMed] [Google Scholar]