

Abstract
The generation of properly functioning circuits during brain development requires precise timing of cell migration and differentiation. Disruptions in the developmental plan may lead to neurological and psychiatric disorders. Neocortical circuits rely on inhibitory GABAergic interneurons, the majority of which migrate from subcortical sources. We have shown that the pleiotropic molecule hepatocyte growth factor/scatter factor (HGF/SF) mediates interneuron migration. Mice with a targeted mutation of the gene encoding urokinase plasminogen activator receptor (uPAR), a key component in HGF/SF activation and function, have decreased levels of HGF/SF and a 50% reduction in neocortical GABAergic interneurons at embryonic and perinatal ages. Disruption of interneuron development leads to early lethality in most models. Thus, the long-term consequences of such perturbations are unknown. Mice of theuPAR−/− strain survive until adulthood, and behavior testing demonstrates that they have an increased anxiety state. TheuPAR−/− strain also exhibits spontaneous seizure activity and higher susceptibility to pharmacologically induced convulsions. The neocortex of the adultuPAR−/− mouse exhibits a dramatic region- and subtype-specific decrease in GABA-immunoreactive interneurons. Anterior cingulate and parietal cortical areas contain 50% fewer GABAergic interneurons compared with wild-type littermates. However, interneuron numbers in piriform and visual cortical areas do not differ from those of normal mice. Characterization of interneuron subpopulations reveals a near complete loss of the parvalbumin subtype, with other subclasses remaining intact. These data demonstrate that a single gene mutation can selectively alter the development of cortical interneurons in a region- and cell subtype-specific manner, with deficits leading to long-lasting changes in circuit organization and behavior.
Keywords: GABA, interneuron, epilepsy, anxiety, urokinase, plasminogen, calbindin, calretinin, somatostatin, parvalbumin, knock-out mouse, neocortex, uPAR
Introduction
Functional circuits in the CNS depend on a delicate balance of synaptic excitation and inhibition. Inherited disruption of these interactions in the cerebral cortex commonly leads to human neurological and psychiatric disorders such as epilepsy, anxiety, and depression (Sanacora et al., 2000;Sandford et al., 2000; Ballenger, 2001; Holmes and Ben-Ari, 2001), with a developmental onset during childhood or adolescence (Hauser, 1995;Heim and Nemeroff, 2001). Animal models of human disorders have been used to perturb circuit function by the mutation of genes encoding ion channels, neurotransmitter receptors, and developmentally essential molecules, or by environmental manipulations (Mehta and Ticku, 1999;Liu et al., 2000; Steinlein and Noebels, 2000; Anagnostopoulos et al., 2001). Several experimental approaches, including focal cortical freezing (Prince and Jacobs, 1998; Jacobs et al., 1999), methylazoxymethanol treatment (Chevassus-au-Louis et al., 1999; Baraban et al., 2000), and genetic disruption of critical developmental molecules such the transcription factor BETA2/NeuroD (Liu et al., 2000), lead to forebrain malformations and epileptic phenotypes. These studies also provide paradigms for studying adaptive changes that result from the developmental perturbations.
In the cerebral cortex, excitatory glutamatergic neurons arise from the proliferative zone of the neuroepithelium and migrate radially to form the cortical plate (Sidman and Rakic, 1973; Rakic, 1990; Tan et al., 1998). Simultaneously, the inhibitory GABAergic neurons relocate from their origin in the ganglionic eminence of the ventral telencephalon into the developing cortex by migrating tangentially before infiltrating the newly formed individual laminas of the cortex (Anderson et al., 1997; Tamamaki et al., 1997; Lavdas et al., 1999). Several molecules have been implicated in controlling interneuron migration, including the transcription factors DLX1 and DLX2 (Anderson et al., 1997; Sussel et al., 1999), the extracellular matrix molecule slit1 (Zhu et al., 1999), growth factors neurotrophin-4 (NT-4) (Brunstrom et al., 1997; Polleux et al., 2002) and hepatocyte growth factor/scatter factor (HGF/SF) (Powell et al., 2001), guidance molecules semaphorin 3A and semaphorin 3F, and the neuropilins (Marin et al., 2001). Disruption of GABAergic interneuron migration and differentiation could alter the balance of excitatory to inhibitory neurotransmission, leading to behavioral deficits. However, this hypothesis has not been directly tested, primarily because of the embryonic or perinatal lethality of many gene mutations that disrupt neural development more broadly than do GABAergic interneurons.
We have shown previously that HGF/SF mediates interneuron migration (Powell et al., 2001). Genetic deletion of HGF/SF results in midgestation lethality before the onset of interneuron migration. However, inactivation of the urokinase plasminogen activator receptor (uPAR) gene, which regulates HGF/SF activation (Blasi, 1993), leads to diminished levels of HGF/SF and a 50–65% reduction in cortical GABAergic interneurons beginning at embryonic day 16.5 (E16.5) (Powell et al., 2001). Because theuPAR−/− mouse survives to adulthood, it provides a unique opportunity for studying developmental perturbations of the cortical GABAergic interneurons in the mature brain. Here we demonstrate that theuPAR−/− mouse lacks specific subpopulations of cortical interneurons, displays increased anxiety, and exhibits a novel pattern of spontaneous myoclonic seizures. These deficits demonstrate the importance of the normal assembly of cortical interneurons in the establishment of functional circuits and the inability of the developing organism to induce adaptive responses that completely correct the defects.
Materials and Methods
Unless otherwise noted, all chemicals and reagents were purchased from Sigma (St. Louis MO).
Animals. C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME).uPAR−/− mice were a generous gift from P. Carmeliet (Center for Transgene Technology and Gene Therapy, Flanders Interuniversity Institute for Biotechnology, KU Leuven, Belgium). The mice were genotyped via PCR as described previously (Suh et al., 1994; Dewerchin et al., 1996) using the primer set 5′- GATGATAGAGAGCTGGAGGTGGTGAC-3′ (nucleotides 5054–5079) and 5′-CACCGGGTCTGGGCCTGTTGCAGAGGT-3′ (nucleotides 5201–5175). All mice used in the experiments were littermates that had been obtained from heterozygous matings. TheuPAR+/− mice were backcrossed onto the C57BL/6J background strain for >15 generations.
Behavior experiments. Mice were housed in a central animal facility with a 12 hr light cycle (from 7:00 A.M. to 7:00 P.M.) withad libitum access to food and water. Before all behavioral testing, mice were acclimated to the behavior facility for at least 1 hr. At least six male mice of each genotype were tested between the ages of postnatal day 90 (P90) and P120. Each animal was evaluated once for each test, beginning with open field, then light–dark anxiety assessment, and finally elevated plus maze. In all tests, the observer was blinded to the genotypes of the mice. Sample sizes are noted in figure legends.
Open-field exploration test. Mice were placed in the center of a 25 × 25 cm activity arena (TruScan; Colbourn Instruments, Allentown, PA) equipped with a computerized laser photobeam tracking system. Horizontal and vertical movements of the subject were automatically recorded. Activity of each mouse was assessed for 10 min in a brightly lit room between 9:00 A.M. to 12:00 P.M. Measurements of total distance traveled and time spent moving were calculated and analyzed with a Student's t test. Ethological parameters such as amount of defecation and time spent grooming were recorded manually.
Light–dark exploration test. The activity arena was partitioned into a darkened area with a black Plexiglas insert (Colbourn Instruments). The mouse was placed in the brightly lit area of the box at the start of the test and was allowed to explore both the light and dark sides for 10 min. The position of the subject was automatically recorded with the computerized laser photobeam tracking system. The time spent on each side was calculated and analyzed with a Student's t test.
Elevated plus maze exploration test. A standard elevated maze (San Diego Instruments, San Diego, CA) designed for mice was used. All arms were 50 cm long and 10 cm wide, with two arms enclosed by 40-cm-high opaque walls. The maze was raised 50 cm from the floor. The mouse was placed in the center of the maze and allowed to explore for 5 min. Each session was recorded on videotape and scored by at least two observers who were blinded to the genotype of the animals. Each mouse was allowed to explore the maze once; if the mouse fell off the maze, it was disqualified from the study. The position of the mouse in the maze was noted every 5 sec to calculate the percentage of time the mouse was located in the open arms, closed arms, or center of the maze. Statistical differences were evaluated with a two-way ANOVA followed by Student–Newman–Keuls post hoc analysis. Ethological parameters such as rearing and grooming also were recorded.
Electrocorticographic recordings. Adult mice were anesthetized with Avertin (0.02 ml/gm, i.p.) and Teflon-coated silver wire electrodes soldered to a microminiature connector positioned on the skull were implanted bilaterally into the subdural space. After several days of recovery, simultaneous digital EEG and video recordings of awake, freely moving mice were obtained during multiple 2–4 hr sessions taken over a 2 week period.
Pentylenetetrazol susceptibility. Pentylenetretrazol (PTZ) dissolved in buffered saline was administered subcutaneously at a dose of 50 mg/kg. Animals were monitored for up to 30 min after injection. Behavioral responses were scored using the following scale: 0, no signs of motor seizure; 1, isolated twitches; 2, tonic–clonic convulsions; 3, tonic extension or death (Erickson et al., 1996). The latency to the onset of the first seizure was recorded. Mice that did not develop seizures during the observation period were excluded from the latency analysis. The seizure severity data were analyzed for significance with an ANOVA test, whereas the seizure latency data were evaluated with a Student's t test.
Immunocytochemistry. Adult mice (>P90) were transcardially perfused using a fixative of 2% paraformaldehyde, 2% glutaraldehyde, and 0.2% picric acid in 0.1 m sodium phosphate, pH 7.2, and the brains were postfixed overnight at 4°C. Coronal sections were cut on a vibratome at 50 μm and stained using previously published protocols (Stanwood et al., 2001). Primary antibodies were used at the following dilutions: rat anti-GABA (1:1500; Protos Biotech, New York, NY), mouse anti-parvalbumin (PV) (1:1500; Sigma), rabbit anticalretinin (anti-CR) (1:2000; Sigma), and rabbit antisomatostatin (anti-SST) (1:5000; Bachem, Torrance, CA). Appropriate Cy3- or Cy2-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) were used at a 1:3000 dilution. Sections were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) to visualize cell nuclei and cytoarchitecture. Images were obtained with an Olympus Optical (Melville, NY) Provis confocal microscope (with Fluoview version 3.0 software; Olympus Optical) and extended views through 25 μm depth-of-section were shown.
Cell counting. Profiles of immunoreactive cells were counted in four areas of cortex: anterior cingulate, parietal (motor and somatosensory), piriform (at bregma levels +1.50, +0.50, −0.50, and −1.50 mm), and visual (at bregma levels −3.00, −3.50, −4.00, and −4.50 mm) using stereotaxic coordinates (Paxinos and Franklin, 2001). For each area, a 200 μm strip of cortex from the white–gray matter interface to the pial surface was counted on a Nikon (Tokyo, Japan) E800 microscope equipped with epifluorescent illumination. Brains from at least five different mice were counted for each genotype. All sections were coded such that the observer was blinded to the genotype. Statistical analysis was performed using a two-way ANOVA followed by a Student–Newman–Keuls post hocanalysis. Sample sizes are noted in the figure legends.
Results
uPAR−/− mice show increased anxiety
The uPAR−/− mouse has been reported to be grossly normal, without an obvious phenotype, thus enabling behavioral testing (Dewerchin et al., 1996; Crawley, 2000). Behavior of uPAR−/− mice and their wild-type (WT) littermates was assessed in a standard open-field paradigm. Both WT anduPAR−/− mice moved similar distances, 2004 ± 102 cm and 1950 ± 109 cm, respectively (Fig. 1A). The time spent moving was also equivalent: 410 ± 7 sec for WT mice compared with 417 ± 10 sec foruPAR−/− mice. Thus, theuPAR−/− mutation did not affect generalized motor activity. In addition, the percentage of time that mice spent at the margin of the arena (within 6 cm of the wall) compared with the center of the arena was calculated. WT mice occupied the margin for 66% of the time and the center for 34%, whereasuPAR−/− mice were found in the margin area for 76% of the time and the center 24% of the experiment time, which is significantly different from that of WT mice (p < 0.01). The decrease in time thatuPAR−/− mice spent in the center of the arena indicates decreased exploratory behavior and can be interpreted as an increase in anxiety (Crawley and Davis, 1982;Crawley, 1985; Crawley and Paylor, 1997).
Fig. 1.
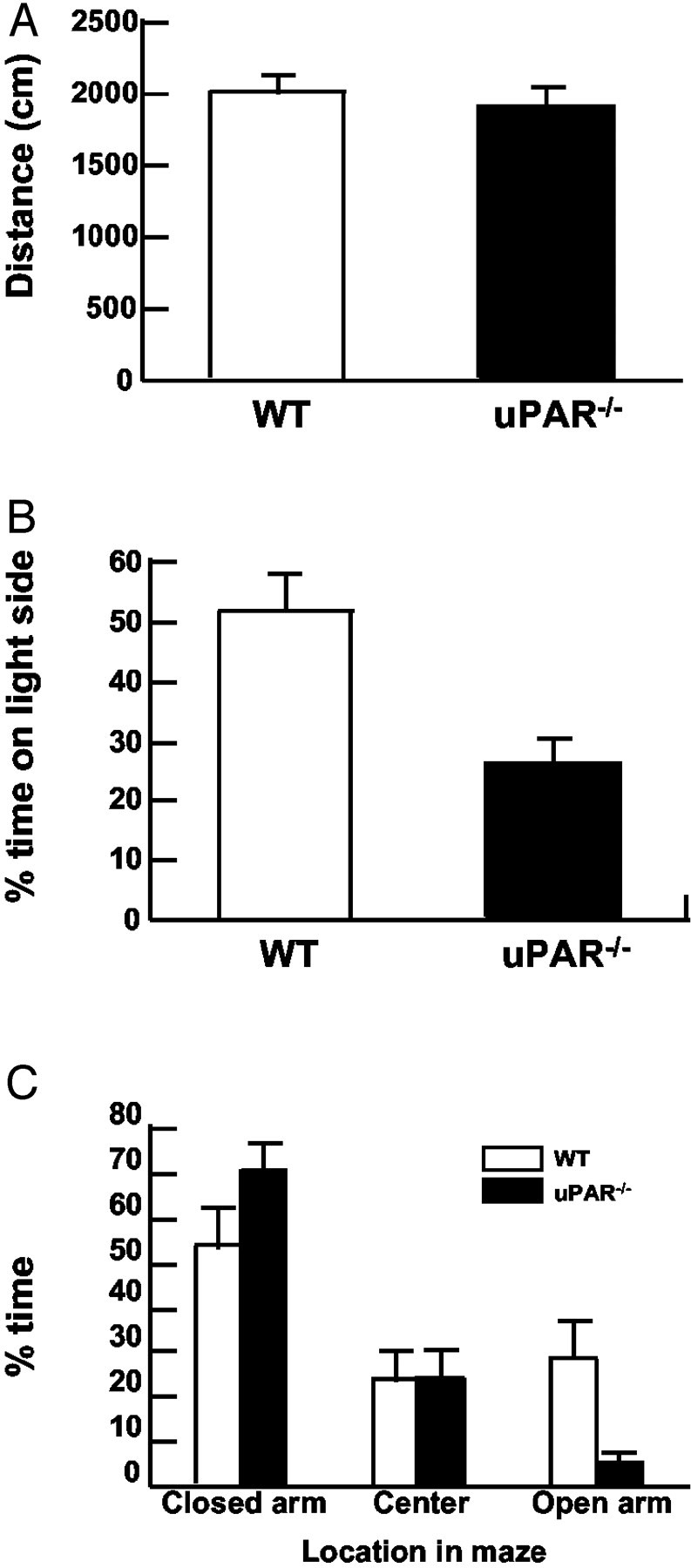
uPAR mutant mice display increased anxiety.A, Open-field test of WT anduPAR−/− animals demonstrates no significant difference in generalized motor activity. At least six mice were tested for 10 min, and the total distance moved was calculated with an automated photobeam tracking system.B,uPAR−/− mice do not remain on the light side of the light–dark avoidance box. WT mice spend ∼50% of the 10 min session exploring the lit side of the chamber, whereas uPAR−/−mice occupy the lit side for only 25% of the time. C,uPAR−/− mice show reduced exploration of the open arms on the elevated plus maze. Mice were placed in the center of the plus maze and allowed to explore the arena for 5 min. The percentage of time that the mice spent in each of three areas is shown. WT mice spent ∼35% of the time in the open arms, whereas uPAR−/−mice spent <5% of the time in the open arms.uPAR−/− mice spent >75% of the time in the closed arms. Both genotypes occupy the center of the maze ∼25% of the time. At least eight animals were tested for each genotype. Asterisks denote a significant difference (p < 0.05).
Anxiety was also assessed with the classic light–dark avoidance test and the elevated plus maze paradigms. In both assays, the test provides a conflict between the desire to explore an unknown area and the aversion of a brightly lit open space (Crawley and Davis, 1982; Zhuang et al., 1999; Crawley, 2000; Sibille and Hen, 2001). In the light–dark avoidance test, WT mice spent equivalent time (50%) between the light and dark sides of the open field. In contrast,uPAR−/− mice explored only the light part of the open field for 25% of the time, preferring the less aversive, darkened area (Fig. 1B), suggesting that uPAR−/− mice displayed a more anxious phenotype (Crawley and Davis, 1982).
The elevated plus maze serves as another measure of anxiety based on the same naturalistic conflict of exploring a novel, brightly lit, elevated region (open arms) versus the defined space of the closed arms (Pellow et al., 1985; Lister, 1987). Both WT anduPAR−/− mice actively traveled in the maze for the majority of the test period. Although WT mice explored the open arms for ∼25–30% of the test period, theuPAR−/− mice ventured into the open space <5% of the time (Fig. 1C), and none of theuPAR−/− mice explored the most distal end of the open arms. By all measures,uPAR−/− mice display behaviors consistent with increased anxiety compared with their WT littermates.
uPAR−/− mice display spontaneous seizures
Of >200 uPAR−/− mice observed, 5.9% were noted to exhibit overt convulsive activity, compared with none of their WT littermates. In a few instances the seizures were lethal. These observations were made during short (1–2 min), routine handling procedures. Although 24 hr surveillance of mice was not performed, the electroclinical correlation of these episodes was also explored with chronic video/electrographic recordings of freely moving uPAR−/− mice that had previously demonstrated convulsive activity. The baseline cortical activity of the mice showed periods of normal, low-amplitude desynchronized EEG with the frequent appearance of abnormal slow waves and interictal discharges (Fig.2A). A spontaneous bilateral tonic clonic seizure lasting ∼75 sec was captured, revealing an orderly buildup of periodic interictal discharges and the abrupt initiation of high-voltage rhythmic spike and spike-wave seizure discharges, terminating abruptly with no postictal depression (Fig.2B). During the electrographic seizure, mice maintained a tonic posture of flexion with an arched back and intermittent truncal myoclonus, some bilateral clonic forelimb movements accompanied by a Straub tail, followed by behavioral arrest until the seizure terminated.
Fig. 2.

Myoclonic seizure activity inuPAR−/− mutant mice.A, Electrocorticographic monitoring of control (WT) and adult uPAR−/− mice reveals abnormal bilaterally synchronous interictal discharges during waking behavior. In B, a spontaneous seizure episode displays a clear acceleration in the frequency of cortical discharges and a rapid progression from isolated to continuous spike and spike-wave rhythmic discharges, with abrupt return to normal cortical activity at the termination of the seizure. Representative stages of the seizure (76 sec in duration) are shown. At right, posture of a uPAR−/−mutant mouse during the final stage of a seizure. Calibration: 500 μV, 1 sec. Two mice of each genotype were recorded in separate sessions.
uPAR−/− mice have an increased susceptibility for PTZ-induced seizures
uPAR−/− mice were challenged with the convulsant PTZ, a GABA antagonist. After a single threshold dose (50 mg/kg, s.c.), all (n = 10) of theuPAR−/− mice displayed motor convulsions, compared with only 50% of their WT littermates (Fig.3A). The severity of the seizure activity was also characterized using a 0–3 rating system with 0 defined as no signs of motor seizure, 1 as isolated limb twitches, 2 as tonic–clonic convulsions, and 3 as tonic extension or death (Erickson et al., 1996). WT animals received scores of either 1 (50%) or 2 (50%), with no animals exhibiting tonic extension, whereas 90% of the uPAR−/− mice had seizures scored as tonic extension. The time to the first forelimb clonus was also recorded, with theuPAR−/− mice exhibiting a significantly decreased latency of 67% (456 ± 60 sec) compared with that of WT mice (681 ± 19 sec) (Fig. 3B).
Fig. 3.

uPAR−/− mice are more susceptible to PTZ-induced seizures. Mice were administered PTZ (50 mg/kg, i.p.) and observed for up to 30 min. A, The behavioral responses were scored (Erickson et al., 1996), with 0 indicating no motor seizure and 3 indicating tonic extension. The histogram shows the percentage of mice exhibiting each behavior. WT mice (n = 6) displayed isolated twitches or tonic–clonic convulsions, with none proceeding to tonic extension. The majority of uPAR−/− mice (n = 8) showed tonic extension. B, The latency time to seizure was defined as the time to the first tonic–clonic convulsion. In WT animals, the latency is shown for the 50% that displayed seizure activity. AlluPAR−/− mice seized and exhibited a shortened latency time. Asterisks denote a significant difference between the two genotypes (p < 0.05).
Neocortical GABAergic interneurons are decreased inuPAR−/− mice
Increased anxiety behavior and susceptibility to spontaneous and induced seizures suggested a defect in the balance between excitatory and inhibitory neurotransmission in the forebrain. Several transgenic mouse lines in which GABA modulation is altered, such as theGAD65 and NPY null mutants (Asada et al., 1996;Erickson et al., 1996; Kash et al., 1997), exhibit behavioral phenotypes similar to our observations of theuPAR−/− mouse. In addition, the uPAR−/− brain has a decrease in the number of neocortical GABAergic interneurons at birth (Powell et al., 2001). Gross anatomical observation of coronal brain sections with Nissl staining did not reveal any obvious morphological defects (Dewerchin et al., 1996) (Fig.4A,B). Nevertheless, immunocytochemical analysis of the cerebral cortex with anti-GABA antibodies demonstrated a substantial decrease in GABA-immunopositive cells in the adult uPAR−/−parietal cortex (Fig. 4D) compared with WT littermates (Fig. 4C). Although the decrease in GABA+ cells appeared uniform across all cortical laminas within the frontal cortex, regional variation in the reduction of GABA+ cells between frontal and visual cortices was evident.
Fig. 4.

GABAergic neurons are decreased in the neocortex of uPAR−/− mice.A, B, Thionin staining shows no obvious gross differences between cortices of WT anduPAR−/− mice. Confocal images of 50 μm coronal vibratome sections of adult parietal cortex show immunohistochemistry for GABA (C, D), PV (E, F), and SST (G, H). TheuPAR−/− mouse has fewer GABA-immunoreactive cells (compare C andD). The number of PV-immunoreactive cells is also decreased (E, F), whereas there is no difference in the SST-immunoreactive population (G, H).Roman numerals denote the cortical laminas. Scale bars:A, B, 400 μm; C–H, 100 μm. Five animals of each genotype were analyzed quantitatively.
The extent of GABA+ cell loss was quantified by counting 200 μm strips from the pial surface to the white–gray matter interface in several distinct cortical subregions. In the anterior cingulate and parietal regions, there was a reduction of ∼50% in GABA+ cells in theuPAR−/− mice compared with their WT littermates (Fig.5A). However, there were no significant differences in numbers of GABA+ cells in WT anduPAR−/− mice in either piriform or visual cortex (Fig. 5C,D). Thus, theuPAR mutation resulted in a selective deficit of GABA+ cells in distinct cortical areas, suggesting the possibility that specific interneuronal subtypes may be differentially impaired.
Fig. 5.

The numbers of GABA+ cells are decreased in specific regions of theuPAR−/− mouse cerebral cortex. For each area of cortex, a 200 μm profile of the pallial wall, from the pial to ventricular surface, was counted for five different brains from each genotype.uPAR−/− mice have fewer GABA+ cells in the anterior cingulate (A) and parietal cortical areas (B). However, the number of GABA+ cells in the piriform (C) and visual (D) cortex is not affected by theuPAR−/− mutation.Open bars represent WT; closed barsdenote uPAR−/−. Data are presented as mean ± SEM. Asterisks denote a significant difference between the WT anduPAR−/− brains (p < 0.05).
Cortical GABAergic interneurons can be identified using the molecular markers PV, CR, or SST (Kubota et al., 1994; Kawaguchi and Kubota, 1997). These subpopulations of interneurons possess distinct electrophysiological and neuroanatomical properties (Kawaguchi, 2001;Amitai et al., 2002; Pawelzik et al., 2002). Calbindin (CB) can be used as a marker for embryonic interneurons, but in adults, CB is expressed by both GABAergic and glutamatergic neurons, thus making it a poor marker for mature interneurons. The CB+interneuron subpopulation also overlaps with expression of PV and SST (Kubota et al., 1994). Immunocytochemical localization of either CR or SST showed no obvious difference in labeling between WT anduPAR−/− mice in any of the cortical areas examined (Fig. 4G,H and data not shown). However, the PV+ population was greatly diminished in the uPAR−/−parietal (Fig. 4E,F) and anterior cingulate cortex. Quantitative analysis demonstrated that the cingulate and parietal regions had a >90% decrease in the number of PV+ cells (Fig.6A,B). The PV+ subpopulation accounts for 45–50% of the GABAergic interneurons in the WT rodent cortex (Kawaguchi and Kubota, 1993; Kubota et al., 1994) (Table1). Thus, the near complete loss of the PV+ population may account for the missing 50% of the GABA+ cells in theuPAR−/− cingulate and parietal cortical regions.
Fig. 6.
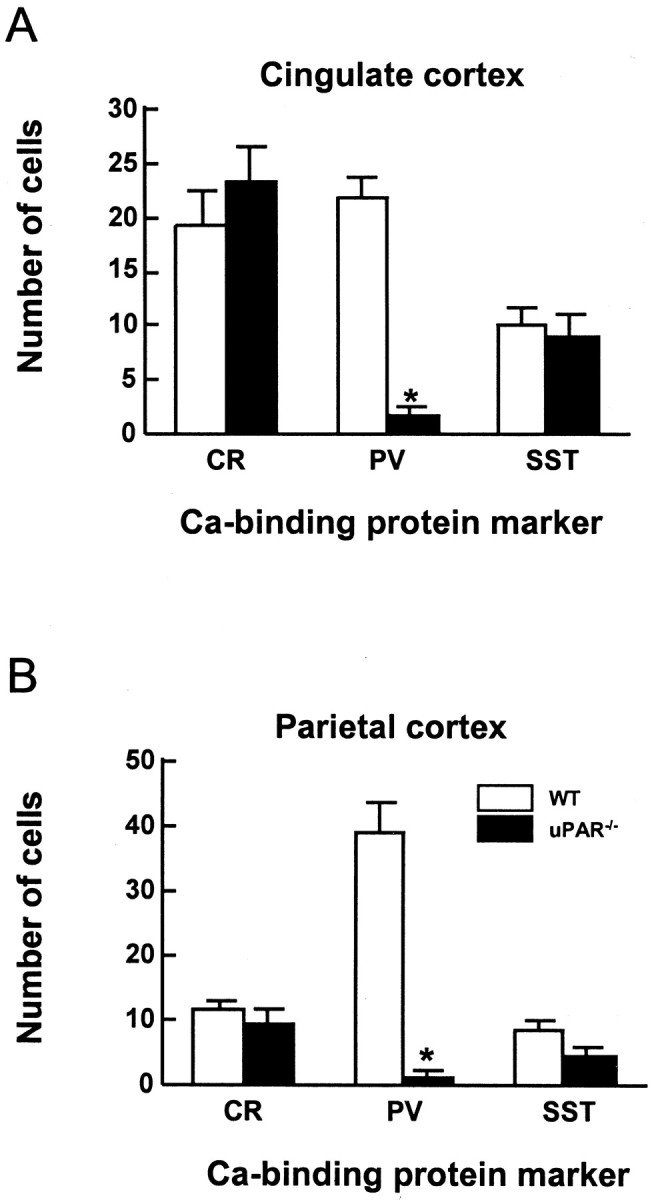
The PV-expressing subpopulation of GABAergic interneurons is decreased in theuPAR−/− mouse. The number of CR-, PV-, and SST-positive cells in a 200 μm strip of cortex was counted for anterior cingulate (A) and parietal (B) cortex. The PV subpopulation is significantly decreased in these regions of theuPAR−/− cortex. In contrast, there are no significant differences in the CR and SST populations. Data are presented as the mean ± SEM of the number of cells counted in five brains for each genotype.Asterisks denote a significant difference between genotypes (p < 0.05).
Table 1.
Comparison of calcium-binding protein subpopulations of GABAergic neurons in the parietal cortex of WT anduPAR−/− mice
| WT | uPAR−/− | |
|---|---|---|
| GABA+cells that are PV+ (%) | 46 ± 4.4 | 2 ± 0.9 |
| GABA+ cells that are CR+ (%) | 12 ± 1.4 | 7 ± 2.5 |
| GABA+ cells that are SST+ (%) | 11 ± 1.8 | 11 ± 1.9 |
| PV+ cells that are GABA+ (%) | 83 ± 2.8 | 11 ± 5.9 |
| CR+ cells that are GABA+ (%) | 51 ± 6.1 | 18 ± 5.0 |
| SST+ cells that are GABA+ (%) | 88 ± 3.5 | 45 ± 8.0 |
Values shown are means ± SEM.
To test the hypothesis that the absent GABA+ cells in theuPAR−/− cortex were comprised specifically of the PV+ subtype, we performed double-label immunohistochemistry and counted the number of cells that expressed combinations of GABA and PV, GABA and CR, and GABA and SST (Fig. 7 and Table 1). In WT mice, ∼46% of the GABA+ cells were also PV+, 12% were CR+, and 11% were SST+. InuPAR−/− mice, the PV+ subtype comprised only 2% of the GABA+ cells (Table 1). Because theuPAR−/− parietal cortex exhibited a 50% reduction in the total number of GABA+ cells but the same absolute number of CR+ and SST+ cells as that in WT mice, the percentage of GABA+ cells that also double-labeled with either CR or SST would be expected to double compared with that of WT mice. However, the percentage of GABA+/CR+cells decreased significantly from 12 to 7%. The GABA+/SST+fraction was not affected. Thus, it appears that some neurons that express calcium-binding protein markers do not produce detectable quantities of GABA in theuPAR−/− mouse.
Fig. 7.
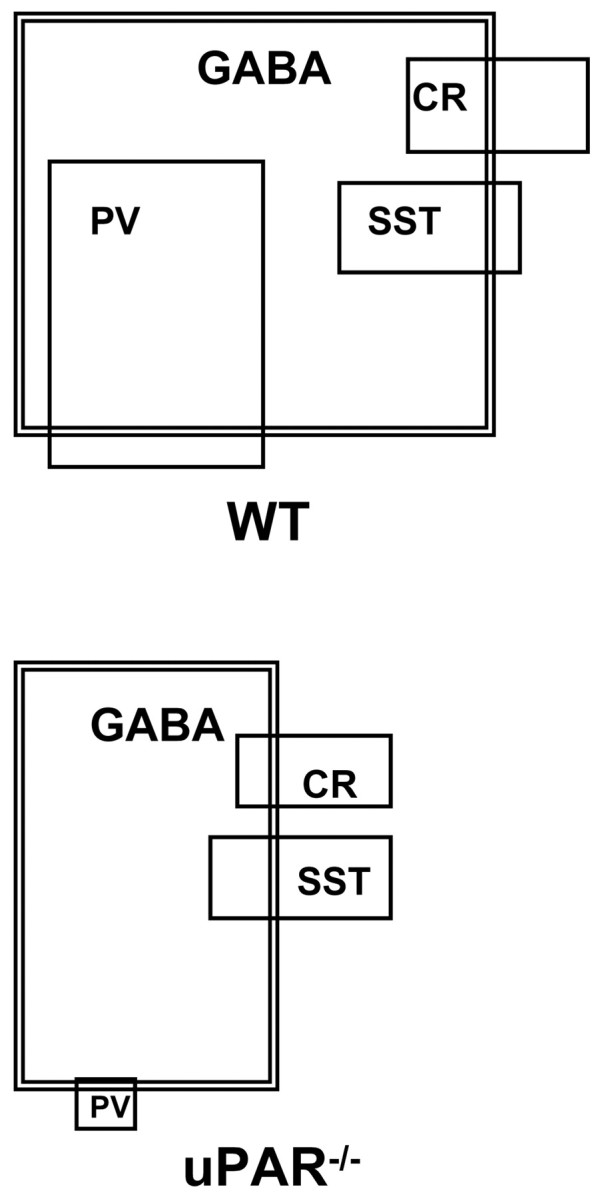
Venn diagrams illustrating the phenotypic profile of GABAergic interneurons in the parietal cortex of WT anduPAR−/− mice. Eachlarge box (outlined in a double line) represents the GABA+ population, with the box size proportional to the total number of cells. Cells expressing PV, SST, or CR are represented by smaller boxes (single outline), with overlapping boxes indicating colocalization of the markers. The percentage of cells in each population is shown in Table 1.
The double-labeled sections were counted to determine the number of cells that coexpressed PV, CR, or SST and GABA. In the WT parietal cortex, 83% of the PV+ cells and 88% of the SST+ cells were also GABA+, whereas only 51% of CR+ cells demonstrated GABA immunoreactivity. These values are in close agreement with previous studies in prefrontal areas of the rat (Kawaguchi and Kubota, 1993;Kubota et al., 1994). InuPAR−/− mice, the degree of colocalization of the calcium-binding marker proteins with GABA was decreased for all subtypes. Only 11% of the remaining PV+ cells expressed GABA, whereas 18% of CR+ and 45% of SST+ cells were GABA+ (Table 1). The shift in subtypes from GABA-expressing to GABA-nonexpressing is shown visually in Figure7. The data suggest that theuPAR−/− mouse parietal cortex has an overall decrease in GABA+interneurons, which is attributable both to a drastic reduction in the number of PV+ cells and to a decrease in detectable GABA immunoreactivity in the PV+, CR+, and SST+ subtypes.
Discussion
The present study provides behavioral and neuroanatomical evidence that deletion of the gene encoding uPAR causes developmental defects in interneuron maturation and long-term disruption of cortical circuit function. Initial characterization studies reported no obvious phenotype inuPAR−/− mice; however, slight deficits in immune responses were observed during challenges with infectious agents (Gyetko et al., 2000, 2001). Our analysis ofuPAR−/− mice demonstrated no gross anatomical abnormalities (Powell et al., 2001). Behavioral testing showed overall normal levels of motor activity, but increased anxiety in three paradigms: the open field, light–dark exploration, and the elevated plus maze.uPAR−/− mice also exhibited spontaneous myoclonic seizures and a greater susceptibility to pharmacologically induced convulsions. Morphological analyses uncovered a 50% deficit in GABA+ cells in both anterior cingulate and parietal cortical regions, with a highly preferential effect on the PV subtype, and a sparing of changes in visual and piriform areas. These data indicate that uPAR, in addition to being an active participant in the biochemical pathways mediating thrombolysis, angiogenesis, and metastasis, also plays a critical role during cortical development. The cortical interneuron deficit that we described previously inuPAR−/− mice at prenatal and perinatal periods (Powell et al., 2001) does not reflect merely a developmental delay, but rather a permanent absence of a specific interneuron population that is maintained in the adult and produces significant functional consequences.
Several studies report a critical role for the plasminogen system in brain development and function, including HGF/SF (also known as plasminogen-related growth factor 1) (Achim et al., 1997; Thewke and Seeds, 1999; Powell et al., 2001) and tissue-type plasminogen activator (tPA) (Baranes et al., 1998; Thewke and Seeds, 1999). The present study provides an additional role for the plasminogen system. Although uPA and tPA are both expressed in the developing forebrain (Seeds et al., 1992) and both can activate plasminogen and HGF/SF (Pepper et al., 1992), each system appears to have distinct roles. tPA has been shown to be important in the establishment of long-term potentiation in the hippocampus, whereas no role was found for uPA (Qian et al., 1993). The tPA null mouse is distinct in a number of ways from theuPAR−/− mouse. For example, the tPA mutant does not exhibit an anxious, seizure-prone behavioral phenotype, and it is less sensitive to pharmacologically induced hippocampal seizures, with reduced excitotoxic cell death and mossy fiber sprouting (Tsirka et al., 1995). In addition, thetPA null mouse does not appear to have a deficit in cortical GABAergic interneurons (Tsirka et al., 1995; Baranes et al., 1998). In contrast, uPA is the main ligand for uPAR, and uPA mRNA was increased after kainate-induced seizures (Masos and Miskin, 1997). These data demonstrate critical yet distinct roles for the tPA and uPA/uPAR systems in cortical development and function.
Role of uPAR in cerebral cortical interneuron development
We previously reported a migration defect of the interneurons in the uPAR−/− forebrain at embryonic and early postnatal ages (Powell et al., 2001). The number of GABAergic interneurons, assessed by calbindin immunoreactivity, was diminished by >50% in frontal and parietal neocortices at E16.5 and P0, whereas the cells appeared to differentiate and remain in or near the ganglionic eminence. uPAR can mediate cell migration in non-neuronal cells through either proteolytic- or nonproteolytic-mediated mechanisms (Blasi, 1993; Waltz et al., 2000). uPAR, via interactions with several α- and β-integrin subunits, increases cell migration in vitro and possibly in vivo during metastasis (Chapman et al., 1999). Lack of uPAR reduces neutrophil migration, a process normally mediated through uPAR–integrin interactions (Gyetko et al., 2000). uPAR, as the receptor for uPA, mediates the proteolytic activity of uPA and tPA, thus increasing the activated concentrations of the zymogen plasmin and several matrix metalloproteases, which can then digest extracellular matrix molecules to clear a pathway for migrating cells. In addition, zymogens can activate latent growth factors such as HGF/SF into their active forms (Naldini et al., 1992; Mars et al., 1993), which then can function as a motogenic or differentiation factor. HGF/SF is expressed in a pattern that is consistent with a role in directing interneurons from their subpallial origins in the ganglionic eminence into the neocortex (Powell et al., 2001). HGF/SF activation, by either uPA or tPA (Seeds et al., 1992), is more efficient in the presence of uPAR (Blasi, 1993). Thus, in the absence of uPAR during forebrain development, the levels of active HGF/SF are dramatically decreased, which we have suggested is a mechanism that leads to defects in prenatal interneuron migration (Powell et al., 2001).
In addition to its role in mediating migration, HGF/SF may also participate in neuronal differentiation. Postnatally, GABAergic interneurons complete migration into the cortical plate and undergo differentiation and synapse formation. HGF/SF and its cognate receptor c-met are expressed in a caudal–rostral (high-low) gradient at early postnatal ages (Thewke and Seeds, 1999). Thus, areas of anterior cingulate and parietal cortices exhibit lower expression of HGF/SF than the visual cortex. Expression also is robust in the piriform cortex (Achim et al., 1997; Powell et al., 2001) (data not shown). We suggest that the reduced transcript and protein levels of HGF/SF that we measured in theuPAR−/− mouse (Powell et al., 2001) may have their greatest effects on interneuron differentiation or survival in the cingulate and parietal cortex, because HGF/SF levels in these regions may drop below threshold.
Several in vivo and in vitro studies have indicated that forebrain interneuron differentiation is mediated by a variety of growth factors, including basic FGF (FGF-2), BDNF, NT-3, and HGF/SF (Pappas and Parnavelas, 1998; Fiumelli et al., 2000;Korhonen et al., 2000). In fact, combinatorial effects of these factors on neuronal differentiation have been reported. For example, HGF/SF, when present with BDNF or NT-3, has a synergistic effect and increases the expression of calbindin or calretinin, respectively, compared with either growth factor alone (Fiumelli et al., 2000). Although its role in GABA expression has not been documented previously, it is possible that the decreased level of HGF/SF inuPAR−/− mice could disrupt the maturation of GABAergic interneurons. Thus, HGF/SF could play a pleiotropic role by regulating both migration and differentiation.
Pathophysiology resulting from developmental defects
Functional defects in GABAergic transmission are widely believed to cause epilepsy in humans. Migration defects, most obviously in the form of ectopias, are commonly observed in a subgroup of patients with epileptic seizures (Porter et al., 2002). It is often the case, however, that the cause of the seizures is not known and alterations in cortical lamination and cellularity are subtle.
Decreases in cells that are immunoreactive for the calcium-binding proteins PV, CB, CR, and SST have been reported in human epileptic tissue (temporal lobe) and in rodent models of epilepsy (Gruber et al., 1994; Spreafico et al., 2000; Andre et al., 2001;Sundstrom et al., 2001). However, defects in subcortical structures, including the amygdala and striatum, may also contribute to the behavioral phenotype ofuPAR−/− mice. Initial cursory examination of the striatum and amygdala does not display the same dramatic interneuron defect (data not shown).
Similar to the human epileptic phenotype,uPAR−/− mice show a major decrease in the expression of calcium-binding proteins in the cerebral cortex. However, mice null for PV, CB, CR, or the combination of PV/CB or PV/CR show grossly normal forebrain morphology and do not exhibit spontaneous seizures or increased vulnerability to kainic acid-induced seizures (Bouilleret et al., 2000). These data suggest that the most likely cause of the seizures observed inuPAR−/− mice is the lack of appropriate GABAergic inhibitory innervation, rather than changes in the expression of calcium-binding proteins. The spontaneous seizure activity in uPAR mutant mice, however, might contribute secondarily to an altered neuropeptide expression profile (Scotti et al., 1997; Vogt Weisenhorn et al., 1998; Porter et al., 1999). This is consistent with reports that expression of PV by interneurons is sensitive to abnormal levels of activity (Andre et al., 2001; Gorter et al., 2001). Thus, in uPAR−/−mice, the early disruption of the GABA phenotype in the developing cerebral cortex may combine with later onset seizure activity to produce the calcium-binding protein profile of interneurons in the adult.
A variety of genetic mutations affecting GABAergic synaptic transmission, including GAD65; GABAA receptor subunits α3, β3, and γ2; and neuropeptide Y, result in epilepsy and abnormal anxiety in humans and in experimental animal models (Homanics et al., 1997; Crestani et al., 1999; Kash et al., 1999;Baulac et al., 2001; Wallace et al., 2001). Although the increased anxiety and spontaneous seizures that we observed in theuPAR−/− mouse are similar to neurobehavioral deficits reported in other mutations of the GABAergic system, the uPAR−/− mutant phenotype is unique in its regional and interneuronal subtype specificity. Thus, as in human neurological and psychiatric diseases, there are likely to be numerous molecular pathways leading to pathophysiological states that share common features (Mirnics et al., 2000). Conditional deletions of the HGF/SF and plasminogen signaling systems, both temporally and spatially, will provide additional opportunities to dissect the molecular pathways responsible for interneuron migration, subsequent differentiation, and adaptive capacity of cortical circuits.
Footnotes
This work was supported by National Institute of Mental Health Grant MH65299 (P.L.), by National Institute of Neurological Disorders and Stroke Grants 29709 (J.L.N.) and HD 97-003 (J.L.N.), and by National Research Service Award Grant MH12651 (E.M.P.). We are grateful to Dr. Kathie Eagleson, Dr. Frank Middleton, Kyoko Koshibu, and Kristine Roy for their insightful discussions.
Correspondence should be addressed to Dr. Elizabeth M. Powell, 408 South Biomedical Science Tower, Department of Pathology, University of Pittsburgh, School of Medicine, 200 Lothrop Street, Pittsburgh, PA 15261. E-mail: epowell@pitt.edu.
References
- 1.Achim CL, Katyal S, Wiley CA, Shiratori M, Wang G, Oshika E, Petersen BE, Li JM, Michalopoulos GK. Expression of HGF and cMet in the developing and adult brain. Brain Res Dev Brain Res. 1997;102:299–303. doi: 10.1016/s0165-3806(97)00108-9. [DOI] [PubMed] [Google Scholar]
- 2.Amitai Y, Gibson JR, Beierlein M, Patrick SL, Ho AM, Connors BW, Golomb D. The spatial dimensions of electrically coupled networks of interneurons in the neocortex. J Neurosci. 2002;22:4142–4152. doi: 10.1523/JNEUROSCI.22-10-04142.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anagnostopoulos AV, Mobraaten LE, Sharp JJ, Davisson MT. Transgenic and knockout databases: behavioral profiles of mouse mutants. Physiol Behav. 2001;73:675–689. doi: 10.1016/s0031-9384(01)00525-x. [DOI] [PubMed] [Google Scholar]
- 4.Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- 5.Andre V, Marescaux C, Nehlig A, Fritschy JM. Alterations of hippocampal GABAergic system contribute to development of spontaneous recurrent seizures in the rat lithium-pilocarpine model of temporal lobe epilepsy. Hippocampus. 2001;11:452–468. doi: 10.1002/hipo.1060. [DOI] [PubMed] [Google Scholar]
- 6.Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem Biophys Res Commun. 1996;229:891–895. doi: 10.1006/bbrc.1996.1898. [DOI] [PubMed] [Google Scholar]
- 7.Ballenger JC. Overview of different pharmacotherapies for attaining remission in generalized anxiety disorder. J Clin Psychiatry. 2001;62 [Suppl 19]:11–19. [PubMed] [Google Scholar]
- 8.Baraban SC, Wenzel HJ, Hochman DW, Schwartzkroin PA. Characterization of heterotopic cell clusters in the hippocampus of rats exposed to methylazoxymethanol in utero. Epilepsy Res. 2000;39:87–102. doi: 10.1016/s0920-1211(99)00104-7. [DOI] [PubMed] [Google Scholar]
- 9.Baranes D, Lederfein D, Huang YY, Chen M, Bailey CH, Kandel ER. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron. 1998;21:813–825. doi: 10.1016/s0896-6273(00)80597-8. [DOI] [PubMed] [Google Scholar]
- 10.Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud'homme JF, Baulac M, Brice A, Bruzzone R, LeGuern E. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet. 2001;28:46–48. doi: 10.1038/ng0501-46. [DOI] [PubMed] [Google Scholar]
- 11.Blasi F. Urokinase and urokinase receptor: a paracrine/autocrine system regulating cell migration and invasiveness. BioEssays. 1993;15:105–111. doi: 10.1002/bies.950150206. [DOI] [PubMed] [Google Scholar]
- 12.Bouilleret V, Schwaller B, Schurmans S, Celio MR, Fritschy JM. Neurodegenerative and morphogenic changes in a mouse model of temporal lobe epilepsy do not depend on the expression of the calcium-binding proteins parvalbumin, calbindin, or calretinin. Neuroscience. 2000;97:47–58. doi: 10.1016/s0306-4522(00)00017-8. [DOI] [PubMed] [Google Scholar]
- 13.Brunstrom JE, Gray-Swain MR, Osborne PA, Pearlman AL. Neuronal heterotopias in the developing cerebral cortex produced by neurotrophin-4. Neuron. 1997;18:505–517. doi: 10.1016/s0896-6273(00)81250-7. [DOI] [PubMed] [Google Scholar]
- 14.Chapman HA, Wei Y, Simon DI, Waltz DA. Role of urokinase receptor and caveolin in regulation of integrin signaling. Thromb Haemost. 1999;82:291–297. [PubMed] [Google Scholar]
- 15.Chevassus-au-Louis N, Baraban SC, Gaiarsa JL, Ben-Ari Y. Cortical malformations and epilepsy: new insights from animal models. Epilepsia. 1999;40:811–821. doi: 10.1111/j.1528-1157.1999.tb00786.x. [DOI] [PubMed] [Google Scholar]
- 16.Crawley JN. Exploratory behavior models of anxiety in mice. Neurosci Biobehav Rev. 1985;9:37–44. doi: 10.1016/0149-7634(85)90030-2. [DOI] [PubMed] [Google Scholar]
- 17.Crawley JN. What's wrong with my mouse? Behavioral phenotyping of transgenic and knockout mice. Wiley; New York: 2000. [Google Scholar]
- 18.Crawley JN, Davis LG. Baseline exploratory activity predicts anxiolytic responsiveness to diazepam in five mouse strains. Brain Res Bull. 1982;8:609–612. doi: 10.1016/0361-9230(82)90087-9. [DOI] [PubMed] [Google Scholar]
- 19.Crawley JN, Paylor R. A proposed test battery and constellations of specific behavioral paradigms to investigate the behavioral phenotypes of transgenic and knockout mice. Horm Behav. 1997;31:197–211. doi: 10.1006/hbeh.1997.1382. [DOI] [PubMed] [Google Scholar]
- 20.Crestani F, Lorez M, Baer K, Essrich C, Benke D, Laurent JP, Belzung C, Fritschy JM, Luscher B, Mohler H. Decreased GABAA-receptor clustering results in enhanced anxiety and a bias for threat cues. Nat Neurosci. 1999;2:833–839. doi: 10.1038/12207. [DOI] [PubMed] [Google Scholar]
- 21.Dewerchin M, Nuffelen AV, Wallays G, Bouche A, Moons L, Carmeliet P, Mulligan RC, Collen D. Generation and characterization of urokinase receptor-deficient mice. J Clin Invest. 1996;97:870–878. doi: 10.1172/JCI118489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erickson JC, Clegg KE, Palmiter RD. Sensitivity to leptin and susceptibility to seizures of mice lacking neuropeptide Y. Nature. 1996;381:415–421. doi: 10.1038/381415a0. [DOI] [PubMed] [Google Scholar]
- 23.Fiumelli H, Kiraly M, Ambrus A, Magistretti PJ, Martin JL. Opposite regulation of calbindin and calretinin expression by brain-derived neurotrophic factor in cortical neurons. J Neurochem. 2000;74:1870–1877. doi: 10.1046/j.1471-4159.2000.0741870.x. [DOI] [PubMed] [Google Scholar]
- 24.Gorter JA, van Vliet EA, Aronica E, Lopes da Silva FH. Progression of spontaneous seizures after status epilepticus is associated with mossy fibre sprouting and extensive bilateral loss of hilar parvalbumin and somatostatin-immunoreactive neurons. Eur J Neurosci. 2001;13:657–669. doi: 10.1046/j.1460-9568.2001.01428.x. [DOI] [PubMed] [Google Scholar]
- 25.Gruber B, Greber S, Rupp E, Sperk G. Differential NPY mRNA expression in granule cells and interneurons of the rat dentate gyrus after kainic acid injection. Hippocampus. 1994;4:474–482. doi: 10.1002/hipo.450040409. [DOI] [PubMed] [Google Scholar]
- 26.Gyetko MR, Sud S, Kendall T, Fuller JA, Newstead MW, Standiford TJ. Urokinase receptor-deficient mice have impaired neutrophil recruitment in response to pulmonary Pseudomonas aeruginosa infection. J Immunol. 2000;165:1513–1519. doi: 10.4049/jimmunol.165.3.1513. [DOI] [PubMed] [Google Scholar]
- 27.Gyetko MR, Sud S, Sonstein J, Polak T, Sud A, Curtis JL. Antigen-driven lymphocyte recruitment to the lung is diminished in the absence of urokinase-type plasminogen activator (uPA) receptor, but is independent of uPA. J Immunol. 2001;167:5539–5542. doi: 10.4049/jimmunol.167.10.5539. [DOI] [PubMed] [Google Scholar]
- 28.Hauser WA. Epidemiology of epilepsy in children. Neurosurg Clin N Am. 1995;6:419–429. [PubMed] [Google Scholar]
- 29.Heim C, Nemeroff CB. The role of childhood trauma in the neurobiology of mood and anxiety disorders: preclinical and clinical studies. Biol Psychiatry. 2001;49:1023–1039. doi: 10.1016/s0006-3223(01)01157-x. [DOI] [PubMed] [Google Scholar]
- 30.Holmes GL, Ben-Ari Y. The neurobiology and consequences of epilepsy in the developing brain. Pediatr Res. 2001;49:320–325. doi: 10.1203/00006450-200103000-00004. [DOI] [PubMed] [Google Scholar]
- 31.Homanics GE, DeLorey TM, Firestone LL, Quinlan JJ, Handforth A, Harrison NL, Krasowski MD, Rick CE, Korpi ER, Makela R, Brilliant MH, Hagiwara N, Ferguson C, Snyder K, Olsen RW. Mice devoid of gamma-aminobutyrate type A receptor beta3 subunit have epilepsy, cleft palate, and hypersensitive behavior. Proc Natl Acad Sci USA. 1997;94:4143–4148. doi: 10.1073/pnas.94.8.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacobs KM, Kharazia VN, Prince DA. Mechanisms underlying epileptogenesis in cortical malformations. Epilepsy Res. 1999;36:165–188. doi: 10.1016/s0920-1211(99)00050-9. [DOI] [PubMed] [Google Scholar]
- 33.Kash SF, Johnson RS, Tecott LH, Noebels JL, Mayfield RD, Hanahan D, Baekkeskov S. Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci USA. 1997;94:14060–14065. doi: 10.1073/pnas.94.25.14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kash SF, Tecott LH, Hodge C, Baekkeskov S. Increased anxiety and altered responses to anxiolytics in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci USA. 1999;96:1698–1703. doi: 10.1073/pnas.96.4.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawaguchi Y. Distinct firing patterns of neuronal subtypes in cortical synchronized activities. J Neurosci. 2001;21:7261–7272. doi: 10.1523/JNEUROSCI.21-18-07261.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawaguchi Y, Kubota Y. Correlation of physiological subgroupings of nonpyramidal cells with parvalbumin- and calbindinD28k-immunoreactive neurons in layer V of rat frontal cortex. J Neurophysiol. 1993;70:387–396. doi: 10.1152/jn.1993.70.1.387. [DOI] [PubMed] [Google Scholar]
- 37.Kawaguchi Y, Kubota Y. GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex. 1997;7:476–486. doi: 10.1093/cercor/7.6.476. [DOI] [PubMed] [Google Scholar]
- 38.Korhonen L, Sjoholm U, Takei N, Kern MA, Schirmacher P, Castren E, Lindholm D. Expression of c-Met in developing rat hippocampus: evidence for HGF as a neurotrophic factor for calbindin D-expressing neurons. Eur J Neurosci. 2000;12:3453–3461. doi: 10.1046/j.1460-9568.2000.00260.x. [DOI] [PubMed] [Google Scholar]
- 39.Kubota Y, Hattori R, Yui Y. Three distinct subpopulations of GABAergic neurons in rat frontal agranular cortex. Brain Res. 1994;649:159–173. doi: 10.1016/0006-8993(94)91060-x. [DOI] [PubMed] [Google Scholar]
- 40.Lavdas AA, Grigoriou M, Pachnis V, Parnavelas JG. The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci. 1999;19:7881–7888. doi: 10.1523/JNEUROSCI.19-18-07881.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lister RG. The use of a plus-maze to measure anxiety in the mouse. Pharmacol Biochem Behav. 1987;28:75–79. [Google Scholar]
- 42.Liu M, Pleasure SJ, Collins AE, Noebels JL, Naya FJ, Tsai MJ, Lowenstein DH. Loss of BETA2/NeuroD leads to malformation of the dentate gyrus and epilepsy. Proc Natl Acad Sci USA. 2000;97:865–870. doi: 10.1073/pnas.97.2.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marin O, Yaron A, Bagri A, Tessier-Lavigne M, Rubenstein JL. Sorting of striatal and cortical interneurons regulated by semaphorin-neuropilin interactions. Science. 2001;293:872–875. doi: 10.1126/science.1061891. [DOI] [PubMed] [Google Scholar]
- 44.Mars WM, Zarnegar R, Michalopoulos GK. Activation of hepatocyte growth factor by the plasminogen activators uPA and tPA. Am J Pathol. 1993;143:949–958. [PMC free article] [PubMed] [Google Scholar]
- 45.Masos T, Miskin R. mRNAs encoding urokinase-type plasminogen activator and plasminogen activator inhibitor-1 are elevated in the mouse brain following kainate-mediated excitation. Brain Res Mol Brain Res. 1997;47:157–169. doi: 10.1016/s0169-328x(97)00040-5. [DOI] [PubMed] [Google Scholar]
- 46.Mehta AK, Ticku MK. An update on GABAA receptors. Brain Res Brain Res Rev. 1999;29:196–217. doi: 10.1016/s0165-0173(98)00052-6. [DOI] [PubMed] [Google Scholar]
- 47.Mirnics K, Middleton FA, Marquez A, Lewis DA, Levitt P. Molecular characterization of schizophrenia viewed by microarray analysis of gene expression in prefrontal cortex. Neuron. 2000;28:53–67. doi: 10.1016/s0896-6273(00)00085-4. [DOI] [PubMed] [Google Scholar]
- 48.Naldini L, Tamagnone L, Vigna E, Sachs M, Hartmann G, Birchmeier W, Daikuhara Y, Tsubouchi H, Blasi F, Comoglio PM. Extracellular proteolytic cleavage by urokinase is required for activation of hepatocyte growth factor/scatter factor. EMBO J. 1992;11:4825–4833. doi: 10.1002/j.1460-2075.1992.tb05588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pappas IS, Parnavelas JG. Basic fibroblast growth factor promotes the generation and differentiation of calretinin neurons in the rat cerebral cortex in vitro. Eur J Neurosci. 1998;10:1436–1445. doi: 10.1046/j.1460-9568.1998.00147.x. [DOI] [PubMed] [Google Scholar]
- 50.Pawelzik H, Hughes DI, Thomson AM. Physiological and morphological diversity of immunocytochemically defined parvalbumin- and cholecystokinin-positive interneurones in CA1 of the adult rat hippocampus. J Comp Neurol. 2002;443:346–367. doi: 10.1002/cne.10118. [DOI] [PubMed] [Google Scholar]
- 51.Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates, Ed 2. Academic; San Diego: 2001. [Google Scholar]
- 52.Pellow S, Chopin P, File SE, Briley M. Validation of open:closed arm entries in an elevated plus-maze as a measure of anxiety in the rat. J Neurosci Methods. 1985;14:149–167. doi: 10.1016/0165-0270(85)90031-7. [DOI] [PubMed] [Google Scholar]
- 53.Pepper MS, Matsumoto K, Nakamura T, Orci L, Montesano R. Hepatocyte growth factor increases urokinase-type plasminogen activator (u-PA) and u-PA receptor expression in Madin-Darby canine kidney epithelial cells. J Biol Chem. 1992;267:20493–20496. [PubMed] [Google Scholar]
- 54.Polleux F, Whitford KL, Dijkhuizen PA, Vitalis T, Ghosh A. Control of cortical interneuron migration by neurotrophins and PI3-kinase signaling. Development. 2002;129:3147–3160. doi: 10.1242/dev.129.13.3147. [DOI] [PubMed] [Google Scholar]
- 55.Porter BE, Brooks-Kayal A, Golden JA. Disorders of cortical development and epilepsy. Arch Neurol. 2002;59:361–365. doi: 10.1001/archneur.59.3.361. [DOI] [PubMed] [Google Scholar]
- 56.Porter LL, Rizzo E, Hornung JP. Dopamine affects parvalbumin expression during cortical development in vitro. J Neurosci. 1999;19:8990–9003. doi: 10.1523/JNEUROSCI.19-20-08990.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Powell EM, Mars WM, Levitt P. Hepatocyte growth factor/scatter factor is a motogen for interneurons migrating from the ventral to dorsal telencephalon. Neuron. 2001;30:79–89. doi: 10.1016/s0896-6273(01)00264-1. [DOI] [PubMed] [Google Scholar]
- 58.Prince DA, Jacobs K. Inhibitory function in two models of chronic epileptogenesis. Epilepsy Res. 1998;32:83–92. doi: 10.1016/s0920-1211(98)00042-4. [DOI] [PubMed] [Google Scholar]
- 59.Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature. 1993;361:453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- 60.Rakic P. Principles of neural cell migration. Experientia. 1990;46:882–891. doi: 10.1007/BF01939380. [DOI] [PubMed] [Google Scholar]
- 61.Sanacora G, Mason GF, Krystal JH. Impairment of GABAergic transmission in depression: new insights from neuroimaging studies. Crit Rev Neurobiol. 2000;14:23–45. doi: 10.1615/critrevneurobiol.v14.i1.20. [DOI] [PubMed] [Google Scholar]
- 62.Sandford JJ, Argyropoulos SV, Nutt DJ. The psychobiology of anxiolytic drugs. Part 1: basic neurobiology. Pharmacol Ther. 2000;88:197–212. doi: 10.1016/s0163-7258(00)00082-6. [DOI] [PubMed] [Google Scholar]
- 63.Scotti AL, Kalt G, Bollag O, Nitsch C. Parvalbumin disappears from GABAergic CA1 neurons of the gerbil hippocampus with seizure onset while its presence persists in the perforant path. Brain Res. 1997;760:109–117. doi: 10.1016/s0006-8993(97)00309-0. [DOI] [PubMed] [Google Scholar]
- 64.Seeds NW, Verrall S, Friedman G, Hayden S, Gadotti D, Haffke S, Christensen K, Gardner B, McGuire P, Krystosek A. Plasminogen activators and plasminogen activator inhibitors in neural development. Ann NY Acad Sci. 1992;667:32–40. doi: 10.1111/j.1749-6632.1992.tb51592.x. [DOI] [PubMed] [Google Scholar]
- 65.Sibille E, Hen R. Combining genetic and genomic approaches to study mood disorders. Eur Neuropsychopharmacol. 2001;11:413–421. doi: 10.1016/s0924-977x(01)00118-3. [DOI] [PubMed] [Google Scholar]
- 66.Sidman RL, Rakic P. Neuronal migration, with special reference to developing human brain: a review. Brain Res. 1973;62:1–35. doi: 10.1016/0006-8993(73)90617-3. [DOI] [PubMed] [Google Scholar]
- 67.Spreafico R, Tassi L, Colombo N, Bramerio M, Galli C, Garbelli R, Ferrario A, Lo Russo G, Munari C. Inhibitory circuits in human dysplastic tissue. Epilepsia. 2000;41 [Suppl 6]:S168–S173. doi: 10.1111/j.1528-1157.2000.tb01576.x. [DOI] [PubMed] [Google Scholar]
- 68.Stanwood GD, Washington RA, Levitt P. Identification of a sensitive period of prenatal cocaine exposure that alters the development of the anterior cingulate cortex. Cereb Cortex. 2001;11:430–440. doi: 10.1093/cercor/11.5.430. [DOI] [PubMed] [Google Scholar]
- 69.Steinlein OK, Noebels JL. Ion channels and epilepsy in man and mouse. Curr Opin Genet Dev. 2000;10:286–291. doi: 10.1016/s0959-437x(00)00079-4. [DOI] [PubMed] [Google Scholar]
- 70.Suh TT, Nerlov C, Dano K, Degen JL. The murine urokinase-type plasminogen activator receptor gene. J Biol Chem. 1994;269:25992–25998. [PubMed] [Google Scholar]
- 71.Sundstrom LE, Brana C, Gatherer M, Mepham J, Rougier A. Somatostatin- and neuropeptide Y-synthesizing neurones in the fascia dentata of humans with temporal lobe epilepsy. Brain. 2001;124:688–697. doi: 10.1093/brain/124.4.688. [DOI] [PubMed] [Google Scholar]
- 72.Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- 73.Tamamaki N, Fujimori KE, Takauji R. Origin and route of tangentially migrating neurons in the developing neocortical intermediate zone. J Neurosci. 1997;17:8313–8323. doi: 10.1523/JNEUROSCI.17-21-08313.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tan SS, Kalloniatis M, Sturm K, Tam PP, Reese BE, Faulkner-Jones B. Separate progenitors for radial and tangential cell dispersion during development of the cerebral neocortex. Neuron. 1998;21:295–304. doi: 10.1016/s0896-6273(00)80539-5. [DOI] [PubMed] [Google Scholar]
- 75.Thewke DP, Seeds NW. The expression of mRNAs for hepatocyte growth factor/scatter factor, its receptor c-met, and one of its activators tissue-type plasminogen activator show a systematic relationship in the developing and adult cerebral cortex and hippocampus. Brain Res. 1999;821:356–367. doi: 10.1016/s0006-8993(99)01115-4. [DOI] [PubMed] [Google Scholar]
- 76.Tsirka SE, Gualandris A, Amaral DG, Strickland S. Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature. 1995;377:340–344. doi: 10.1038/377340a0. [DOI] [PubMed] [Google Scholar]
- 77.Vogt Weisenhorn DM, Celio MR, Rickmann M. The onset of parvalbumin-expression in interneurons of the rat parietal cortex depends upon extrinsic factor(s). Eur J Neurosci. 1998;10:1027–1036. doi: 10.1046/j.1460-9568.1998.00120.x. [DOI] [PubMed] [Google Scholar]
- 78.Wallace RH, Marini C, Petrou S, Harkin LA, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- 79.Waltz DA, Fujita RM, Yang X, Natkin L, Zhuo S, Gerard CJ, Rosenberg S, Chapman HA. Nonproteolytic role for the urokinase receptor in cellular migration in vivo. Am J Respir Cell Mol Biol. 2000;22:316–322. doi: 10.1165/ajrcmb.22.3.3713. [DOI] [PubMed] [Google Scholar]
- 80.Zhu Y, Li H, Zhou L, Wu JY, Rao Y. Cellular and molecular guidance of GABAergic neuronal migration from an extracortical origin to the neocortex. Neuron. 1999;23:473–485. doi: 10.1016/s0896-6273(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 81.Zhuang X, Gross C, Santarelli L, Compan V, Trillat AC, Hen R. Altered emotional states in knockout mice lacking 5-HT1A or 5-HT1B receptors. Neuropsychopharmacology. 1999;21:52S–60S. doi: 10.1016/S0893-133X(99)00047-0. [DOI] [PubMed] [Google Scholar]