

Abstract
Type 1 inositol (1,4,5)-trisphosphate receptors (InsP3R1s) play a major role in neuronal calcium (Ca2+) signaling. The InsP3R1s are phosphorylated by protein kinase A (PKA), but the functional consequences of InsP3R1 phosphorylation and the mechanisms that control the phosphorylated state of neuronal InsP3R1s are poorly understood. In a yeast two-hybrid screen of rat brain cDNA library with the InsP3R1-specific bait, we isolated the protein phosphatase 1α (PP1α). In biochemical experiments, we confirmed the specificity of the InsP3R1–PP1α association and immunoprecipitated the InsP3R1–PP1 complex from rat brain synaptosomes and from the neostriatal lysate. We also established that the association with PP1 facilitates dephosphorylation of PKA-phosphorylated InsP3R1 by the endogenous neostriatal PP1 and by the recombinant PP1α. We demonstrated that exposure of neostriatal slices to 8-bromo-cAMP, dopamine, calyculin A, or cyclosporine A, but not to 10 nm okadaic acid, promotes the phosphorylation of neostriatal InsP3R1 by PKA in vivo. We discovered that PKA activates and PP1α inhibits the activity of recombinant InsP3R1 reconstituted into planar lipid bilayers. We found that phosphorylation of InsP3R1 by PKA induces at least a fourfold increase in the sensitivity of InsP3R1 to activation by InsP3 without shifting the peak of InsP3R1 bell-shaped Ca2+dependence. Based on these data, we suggest that InsP3R1 may participate in cross talk between cAMP and Ca2+signaling in the neostriatum and possibly in other regions of the brain.
Keywords: inositol trisphosphate receptor, calcium signaling, dopamine, protein phosphorylation, yeast two-hybrid, planar lipid bilayers
Introduction
Calcium ions (Ca2+) are universal second messengers. Changes in cytosolic Ca2+ concentration influence most fundamental cellular processes in neuronal and non-neuronal cells (Berridge, 1993, 1998). Inositol 1,4,5-trisphosphate (InsP3), a soluble compound generated by enzymatic cleavage of the lipid phosphatidylinositol 4,5-bisphosphate after activation of phospholipase C (PLC), is a second messenger used by many cell types to stimulate Ca2+ release from intracellular Ca2+ stores. InsP3-induced Ca2+release in these cells is supported by a highly specialized Ca2+ channel, the inositol (1,4,5)-trisphosphate receptor (InsP3R). Three mammalian isoforms of InsP3R have been identified, each with the unique expression pattern (for review, seeFuruichi et al., 1994; Taylor et al., 1999). The three mammalian InsP3R isoforms share 60–70% amino acid homology, but the differences in their functional properties are poorly understood (for review, see Thrower et al., 2001). The type 1 InsP3R (InsP3R1) is a predominant isoform in the CNS. Targeted deletion ofInsP3R1 gene in mice induces ataxia and epileptic seizures, followed by a premature death (Matsumoto et al., 1996), highlighting the importance of InsP3R1 for brain function.
InsP3R1s are subjected to multiple levels of regulation in cells (Bezprozvanny and Ehrlich, 1995). Binding of InsP3 triggers the InsP3R1 channel opening. The activity of InsP3R1 is under feedback control by cytosolic Ca2+; at low Ca2+ concentrations, Ca2+ acts as a coactivator of the InsP3R1, and at high Ca2+ concentrations, the InsP3R1 is inhibited by Ca2+ (Iino, 1990; Bezprozvanny et al., 1991; Finch et al., 1991; Kaznacheyeva et al., 1998). The activity of InsP3R1 is allosterically potentiated by ATP (Ferris et al., 1990; Iino, 1991; Bezprozvanny and Ehrlich, 1993). The InsP3R1 is also one of the major substrates of protein kinase A (PKA) phosphorylation in the brain (Walaas et al., 1986; Supattapone et al., 1988; Maeda et al., 1990; Danoff et al., 1991; Ferris et al., 1991a; Haug et al., 1999; Pieper et al., 2001). PKA can phosphorylate InsP3R1 at two sites, S1589 and S1755 (Danoff et al., 1991; Ferris et al., 1991a; Haug et al., 1999; Pieper et al., 2001). Both sites are located in the coupling domain of the InsP3R1 (Furuichi et al., 1994), and PKA phosphorylation is likely to affect InsP3R1 function. However, functional consequences of neuronal InsP3R1 phosphorylation by PKA remain controversial. An activation (Volpe and Alderson-Lang, 1990; Nakade et al., 1994; Wojcikiewicz and Luo, 1998) or an inhibition (Supattapone et al., 1988; Cameron et al., 1995) of InsP3R1 by PKA was observed using Ca2+ flux measurements.
What are the mechanisms that control phosphorylation of InsP3R1 by PKA in the brain? What are the functional consequences of InsP3R1 phosphorylation by PKA? Is modulation of neuronal InsP3R1 function by PKA physiologically relevant? Here we address some of these questions. In a yeast two-hybrid screen of rat brain cDNA library with the InsP3R1-specific bait, we isolated a cDNA of protein phosphatase 1α (PP1α). In a series of biochemical and electrophysiological in vitro experiments, we analyzed the importance of InsP3R1–PP1α association for control of InsP3R1 phosphorylation by PKA and modulation of InsP3R1 activity. In addition, we characterized the phosphorylation of InsP3R1 by PKA during neostriatal dopaminergic signaling in vivo. Our results suggest that InsP3R1 may play a role in the cross talk between cAMP and Ca2+signaling pathways in the neostriatum (Greengard et al., 1999) and possibly in other regions of the brain.
Materials and Methods
Yeast two-hybrid methods. The C-terminal regions of rat InsP3R1 (Mignery et al., 1990) (amino acids Q2714-A2749), rat InsP3R2 (Sudhof et al., 1991) (amino acids Q2666-H2701), and rat InsP3R3 (Blondel et al., 1993) (amino acids Q2641-R2670) were amplified by PCR and cloned into pLexN vector to yield IC1, IC2, and IC3 baits. Mutant and truncated versions of IC1 bait were generated by PCR and verified by sequencing. The yeast two-hybrid screen of rat brain cDNA library in pVp16-3 vector (3 × 105 independent clones; gift from Dr T. Südhof, University of Texas Southwestern Medical Center, Howard Hughes Medical Institute, Dallas, TX) with IC1 bait was performed according to published procedures (Hata et al., 1996). Coding sequences of mouse PP1β and human PP1γ were amplified by PCR from expressed sequence tags (ESTs) (GenBank accession numbers BF179322 and BG389563) and subcloned into pVp16-3 prey vector. The liquid yeast two-hybrid assays were performed as described previously (Maximov et al., 1999).
In vitro binding assay. RIGLLGHPPHMNVNPQQPA (RIGL-V1, 2731–2749 of rat InsP3R1), RLGFLGSNTPHENHHMPPH (RLGF-V2, 2683–2701 of rat InsP3R2), and RLGFVDVQNCMSR (RLGF-V3, 2658–2670 of rat InsP3R3) peptides were synthesized and coupled toN-hydroxysuccinimide (NHS)-activated Sepharose according to the manufacturer's (Amersham Biosciences, Uppsala, Sweden) instructions. The rat PP1α was cloned into hemagglutinin (HA)-pCMV5 vector (Maximov et al., 1999), expressed in COS7 cells by DEAE–dextran transient transfection, and solubilized in the extraction buffer A [1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 137 mm NaCl, 2.7 mm KCl, 4.3 mmNa2HPO4, 1.4 mmKH2PO4, 5 mm EDTA, 5 mm EGTA, and protease inhibitors]. The HA–PP1α-containing extract was clarified by 20 min of centrifugation (100,000 × g in TL-100) and incubated with RIGL-V1, RLGF-V2, and RLGF-V3 Sepharose beads for 16 hr at 4°C. Beads were washed with 40 bead volumes of the extraction buffer A, and attached proteins were sequentially eluted with 1 bead volume of 1 m NaCl and then 1 bead volume of 1% SDS. Samples were resolved by SDS-PAGE and analyzed by immunoblotting with anti-HA antibodies.
Immunoprecipitations. The RT1 baculovirus encoding the SI(−)/SII(+) splice variant of rat InsP3R1 (Mignery et al., 1990) has been described previously (Tu et al., 2002). The RT1ΔC baculovirus encoding rat InsP3R1 truncated at position G2736 was generated using the Bac-to-Bac system (Invitrogen, San Diego, CA) as described previously (Tu et al., 2002). High Five (Invitrogen) or Sf9 (American Type Culture Collection, Manassas, VA) insect cells were infected with high titer (>108 colony-forming units/ml) stocks of RT1 and RT1ΔC baculoviruses as described previously (Tu et al., 2002). At 48 hr after infection, the insect cells were solubilized in the extraction buffer A. Rat PP1α, mouse PP1β, and human PP1γ were amplified by PCR from ESTs, cloned into HA–pCMV vector (Maximov et al., 1999), expressed in COS7 cells by DEAE–dextran transient transfection, and solubilized in the extraction buffer A. Extracts from Sf9 cells and COS7 cells were clarified by centrifugation (100,000 × g in TL-100), mixed together, and immunoprecipitated for 2 hr at 4°C with anti-HA monoclonal antibodies (mAbs) attached to protein G-agarose beads. mAb against InsP3R1 was used as a positive control. The amount of precipitated InsP3R1 was quantified by [3H]InsP3 binding as described previously (Kaznacheyeva et al., 1998). GlutathioneS-transferase (GST), GST–IC1, GST–IC2, and GST–IC3 fusion proteins (in pGEX-KG; Amersham Biosciences) were expressed in BL21 cells, purified on glutathione beads as described previously (Maximov et al., 1999), and added to immunoprecipitation reactions at a concentration of 200 μg/ml. Cortex rat brain synaptosomes and rat neostriatum homogenates were prepared according to published procedures (Jones and Matus, 1974; Nishi et al., 1997; Maximov et al., 1999) and verified by Western blotting with anti-postsynaptic density 95 (PSD95) and anti-dopamine and cAMP-regulated phosphoprotein (DARPP)-32 polyclonal antibodies, respectively. The synaptosomes and neostriatum homogenates were solubilized in extraction buffer A, clarified by centrifugation (100,000 × g in TL-100), and immunoprecipitated with anti-InsP3R1 T443 polyclonal antibodies attached to protein A-Sepharose beads. The precipitate was analyzed by Western blotting with mAbs against PP1.
In vitro dephosphorylation assay. Recombinant RT1 and RT1ΔC were precipitated from insect cell extracts with the anti-InsP3R1 polyclonal antibodies (T443 or cytl3b2, respectively) attached to protein A-Sepharose beads and phosphorylated as described previously (Wojcikiewicz and Luo, 1998). Briefly, precipitated RT1 or RT1ΔC was washed three times with the ice-cold phosphorylation buffer (120 mm KCl, 50 mm Tris, pH 7.2, 0.3 mmMgCl2, 0.1% Triton X-100) and resuspended in the phosphorylation buffer. The phosphorylation reaction was initiated by addition of 5 μCi [γ-32P]ATP, 5 μm ATP, and 10 U of PKA bovine heart catalytic subunit in 200 μl volume; continued for 1 hr at 30°C; and stopped by addition of 1.3 ml of ice-cold phosphorylation buffer containing 2 mm ATP. The beads were pelleted, washed two times with the dephosphorylation buffer I (50 mm NaCl, 50 mm Tris, pH 7.2, 0.7 mg/ml BSA, 3.3 mm caffeine, 0.15 mmMnCl2, 1.0 mm DTT), and resuspended in 100 μl of the dephosphorylation buffer I. The dephosphorylation reactions were initiated by addition of 0.1 U of rabbit recombinant PP1α or 2 × 10−4 U of human recombinant PP1γ (both from Calbiochem, La Jolla, CA), incubated at 30°C for 0–40 min, and stopped by addition of 5 mm EDTA.
The rat neostriatum homogenate prepared as described previously (Nishi et al., 1999) was used as a source of endogenous PP1 activity (nsPP1). The dephosphorylation reactions with nsPP1 were performed in dephosphorylation buffer II (50 mm Tris-Cl, 50 mm NaCl, pH 7.2, 0.1 mm EGTA, 1 nmokadaic acid, 1 mm DTT, 0.7 mg/ml BSA, 3.3 mmcaffeine). The nsPP1 dephosphorylation reactions were initiated by addition of 4 μg of striatal homogenate and stopped by addition of 1.4 ml of ice-cold dephosphorylation buffer II, brief centrifugation, and rapid (within 3 min) addition of equal volume of 2× SDS-gel loading buffer. Resulting samples were boiled for 5 min, separated by SDS-electrophoresis on 8% polyacrylamide gel, and analyzed by phosphoimaging (Bio-Rad, Richmond, CA). GST/GST–IC1 (200 μg/ml), DARPP-32/pDARPP-32 (0.2 μm), or PP1 inhibitor-2 (Inh2) (0.2 μm) were added to dephosphorylation reactions as indicated in Results.
Neostriatal InsP3R1 back-phosphorylation. The neostriatum of adult rats was dissected (Nishi et al., 1997), chopped into small slices (∼1–2 × 1–2 mm) in ice-cold, oxygenated (95%O2–5% CO2) Krebs–HCO3− buffer, aliquoted, washed, and preincubated in 5 ml of fresh Krebs–HCO3− buffer at 30°C under constant oxygenation for 60 min, with a single change of medium. The neostriatum slices were then placed into fresh Krebs–HCO3− buffer containing 20 μm IBMX and treated with 8-bromo-cAMP (8-Br-cAMP), dopamine, cyclosporine A, calyculin A, or okadaic acid as indicated in Results. After the drug treatment, the pieces were collected, homogenized, and solubilized in the extraction buffer A containing 0.5 mmNa3VO4. The extracts were clarified by centrifugation (100,000 × g in TL-100), and protein concentration in lysates was determined by Bio-Rad assay. The equal amounts of protein from each lysate were used for immunoprecipitation with anti-InsP3R1 T443 polyclonal antibodies attached to protein A-Sepharose beads. The precipitated InsP3R1s were phosphorylatedin vitro by the catalytic subunit of PKA in the presence of [γ-32P]ATP and analyzed by phosphoimaging as described above. When the neostriatal lysate was dephosphorylated by PP1α before in vitro phosphorylation by PKA, the measured content of the32P-InsP3R1 band (32PPP1α) was interpreted as total InsP3R1 in the neostriatal sample. To calculate the fraction of InsP3R1 in the PKA-phosphorylated state, the 32P content of the InsP3R1 band at each data point (32P-InsP3R1) was normalized to the total InsP3R1 content, as follows: pInsP3R1 = (32PPP1α −32P-InsP3R1)/32PPP1α.
Planar lipid bilayer experiments. Single-channel recordings of recombinant RT1 or RT1ΔC activity were performed as described previously (Tu et al., 2002) at 0 mV transmembrane potential using 50 mm Ba2+dissolved in HEPES, pH 7.35, in the trans (intraluminal) side as a charge carrier. The cis (cytosolic) chamber contained 110 mm Tris dissolved in HEPES, pH 7.35, −log ([Ca2+]) (pCa) 6.7 (0.2 mm EGTA plus 0.14 mmCaCl2) (Bezprozvanny et al., 1991), and 3% sucrose. InsP3R1s were activated by addition of 2 μm InsP3 (Alexis) to thecis chamber. The cis chamber contained 0.5 mm MgATP or 0.3 mmMgCl2 plus 0.1 mmLi4ATPγS as indicated in Results. PKA bovine heart catalytic subunit was diluted in 110 mm Tris/HEPES, pH 7.35, containing 0.2 mm ruthenium red to 2 U/μl. Rabbit recombinant PP1α was diluted in 110 mm Tris/HEPES, pH 7.35, containing 0.2 mm ruthenium red and 0.2 mm MnCl2 to 1 U/μl. One microliter of PKA or PP1α stocks was added directly to the bilayer without stirring. The phosphorylation/dephosphorylation reactions were stopped 1 min after PKA/PP1α addition by stirring the solution in thecis chamber for 30 sec. Stirring resulted in a 3000-fold reduction of PKA/PP1α concentration (1 μl in 3 ml dilution), greatly reducing the rate of InsP3R1 phosphorylation/dephosphorylation in the bilayer. In Ca2+-dependence experiments, the free Ca2+ concentration in thecis-chamber was controlled in the range of 10 nm (pCa 8) to 10 μm (pCa 5) by a mixture of 1 mm EGTA, 1 mm HEDTA, and variable concentrations of CaCl2. The resulting free Ca2+ concentration was calculated by using a program described by Fabiato (1988). InsP3dependence was measured by consecutive addition of InsP3 to the cis chamber from 1 mm stock. All additions (InsP3, ATP, CaCl2) were to the cis chamber from the concentrated stocks, with at least 30 sec of stirring of solutions in both chambers. The InsP3R1 single-channel currents were amplified (OC-725; Warner Instruments, Hamden, CT), filtered at 1 kHz with a low-pass eight pole Bessel filter, digitized at 5 kHz (Digidata 1200;Axon Instruments, Foster City, CA), and stored on computer hard drive and recordable optical disks.
For off-line computer analysis (pClamp 6; Axon Instruments) single-channel data were filtered digitally at 500 Hz; for presentation of the current traces, data were filtered at 200 Hz. Evidence for the presence of two to three functional channels in the bilayer was obtained in the majority of experiments. The number of active channels in the bilayer was estimated as a maximal number of simultaneously open channels during the course of an experiment (Horn, 1991). The open probability of closed level and first and second open levels was determined by using half-threshold crossing criteria (t≥ 2 msec) from the records lasting at least 2.5 min. The single-channel open probability (Po) for one channel was calculated using the binomial distribution for the levels 0, 1, and 2, assuming that the channels were identical and independent (Colquhoun and Hawkes, 1983). To construct InsP3 and Ca2+dependence curves for the InsP3R1 in control and PKA-phosphorylated states, the determined values ofPo were averaged across several independent experiments at each InsP3 or Ca2+ concentration. For InsP3-dependence experiments, the averaged values of Po are presented as mean ± SE (n = number of independent experiments) and fit by the following equation: Po(InsP3) =Pmax(InsP3)n/[(InsP3)n+ kInsP3n)], modified from Lupu et al. (1998), wherePmax is a maximalPo value, n is a Hill coefficient, and kInsP3 is the apparent affinity of InsP3R1 for InsP3. For Ca2+-dependence experiments, the averaged values of Po are presented as mean ± SE (n = number of independent experiments) and fit by the following bell-shaped equation:Po(Ca2+) = 4Pmkn(Ca2+)n/[(kn+ [Ca2+]n)(Kn+ [Ca2+]n)], modified from Bezprozvanny et al. (1991), wherePm is a parameter proportional to the maximal Po value, n is a Hill coefficient, k is the apparent affinity of the Ca2+ activating site, and K is the apparent affinity of the Ca2+inhibitory site. The fitting procedure used in this study differs from the procedure used in our previous studies (Bezprozvanny et al., 1991;Kaznacheyeva et al., 1998; Lupu et al., 1998; Nosyreva et al., 2002; Tu et al., 2002) in that Po values in the present study were not normalized to the maximalPo before averaging and fitting. Because Po values were not normalized,Pm is equal to maximalPo when k =K. If k ≠ K, Pm is proportional (and higher) than maximalPo.
Materials. The following mAbs were used: anti-HA for HA.11 (Covance), anti-InsP3R1 (Calbiochem), and anti-PP1 mAb (Transduction Laboratories, Lexington, KY). The following polyclonal antibodies were used: C-terminal anti-InsP3R1 T443 (Kaznacheyeva et al., 1998), N-terminal anti-InsP3R1 cytl3b2 (gift from J. Parys, Ku Leuven, Belgium) (Sipma et al., 1999), anti-PSD95 (gift from T. Südhof), and anti-DARPP-32 (Cell Signaling Technologies). Protein G-agarose beads were supplied by Santa Cruz Biotechnology (Santa Cruz, CA); protein A-Sepharose beads and [γ-32P]ATP were obtained from Amersham Biosciences; rabbit recombinant PP1α, human recombinant PP1γ, DARPP-32, pDARPP-32, PP1 inhibitor-2, calyculin A, and okadaic acid were obtained from Calbiochem, and InsP3 was supplied by Alexis. PKA bovine heart catalytic subunit, Li4ATPγS, and all other reagents are from Sigma (St. Louis, MO).
Results
InsP3R1 specifically binds PP1α
Each of three mammalian InsP3R isoforms contains a unique cytosolic C-terminal tail preceded by a highly conserved region (Fig. 1a). To search for the InsP3R1-specific neuronal binding partners, we performed a yeast two-hybrid screen of rat brain cDNA library with the IC1 bait (amino acids Q2714-A2749 of rat InsP3R1) (Fig. 1a) and isolated the full-length clone of PP1α. When the corresponding regions of InsP3R2 (IC2) and InsP3R3 (IC3) (Fig. 1a) were tested in a liquid yeast two-hybrid assay, we found that PP1α did not bind IC2 and only weakly associated with IC3 (Fig. 1b). Three isoforms of PP1 are expressed in mammalian brain, each with a unique expression pattern (da Cruz e Silva et al., 1995). In a yeast two-hybrid assay, IC1 associated with PP1α but not with PP1β or PP1γ (Fig. 1b). No interaction of IC2 or IC3 baits with PP1β or PP1γ was detected in our yeast two-hybrid experiments (Fig. 1b). Thus, the association appears to be specific for the InsP3R1–PP1α pair.
Fig. 1.

InsP3R1 specifically binds PP1α in a yeast two-hybrid assay. a, Alignment of C-terminal regions of the InsP3R1, InsP3R2, and InsP3R3. The IC1 fragment was used as a bait in the yeast two-hybrid screen. The position of the2731RIGL2734 motif in the IC1 bait is indicated by a bar above the sequence. The domain structure of the InsP3R C-terminal region [constant (C)-RXGX-variable (V)] is shown below the alignment.b, Specificity of InsP3R–PP1 interactions. IC1, IC2, and IC3 baits were tested for the strength of interactions with PP1α, PP1β, and PP1γ preys in liquid yeast two-hybrid assays. The data are normalized to the strength of interaction for the IC1–PP1α pair and are shown as mean ± SEM (n ≥ 3). β-gal, β-galactosidase.
The PP1-targeting proteins share the R/K-V/I-X-F docking motif (Greengard et al., 1999). A similar RIGL motif is present within IC1 sequence (Fig. 1a, indicated by a bar). However, a similar RLGF motif is also present in IC2 and IC3 sequences (Fig.1a), which are not strong PP1α-binding partners (Fig.1b). Where is a specific PP1α-binding site in the IC1 sequence? To address this question, we performed a systematic analysis of PP1α binding specificity by liquid yeast two-hybrid assay. From sequence alignment of InsP3R isoforms, we reasoned that the InsP3R C-terminal sequence could be divided into a conserved (C) domain, RXGX motif, and variable (V) regions (Fig. 1a). A deletion of conserved domain had no effect on IC1 association with PP1α (Fig.2a), indicating that the RIGL motif and V1 variable domain (RIGL-V1) is sufficient for association with PP1α. In contrast, the corresponding regions of IC2 and IC3 baits (RLGF-V2 and RLGF-V3) did not bind PP1α (Fig. 2a), confirming a specificity of the interaction. To determine a role for the RIGL motif in specific association with PP1α, we generated a series of IC1 bait point mutants and tested them in a yeast two-hybrid assay with PP1α prey. We found that in the context of the IC1 bait, mutations of RIGL motif to RIGA or RAGL had no apparent effect on the strength of interactions with PP1α (Fig. 2a, bold indicates mutated residues). In fact, mutations of the RIGL motif to the RLGFmotif present in IC2 and IC3 baits or to the RIGF motif corresponding to the “canonical” PP1 docking motif (Greengard et al., 1999) resulted in approximately a twofold increase in the strength of interaction with PP1α (Fig. 2a). Thus, presence of the RIGL motif does not explain PP1α specificity for the IC1 bait. Interestingly, the V1 variable region of IC1 bait alone is not sufficient for association with PP1α (Fig. 2a). From these results, we concluded that the association with PP1α requires the RIGL motif and V1 variable region of IC1 (RIGL-V1, R2731-A2749), with the specificity for IC1 conferred by the variable region. To further confirm these findings, we coupled the peptides corresponding to the RXGX motif and variable sequence of InsP3R1, InsP3R2, and InsP3R3 to NHS-Sepharose and performed pull-down experiments with HA-tagged PP1α transiently expressed in COS cells (Fig.3b). We found that the InsP3R1-specific peptide (RIGL-V1) but not the InsP3R2- or the InsP3R3-specific peptides (RLGF-V2 and RLGF-V3) formed a salt-sensitive complex with HA–PP1α (Fig.2b).
Fig. 2.
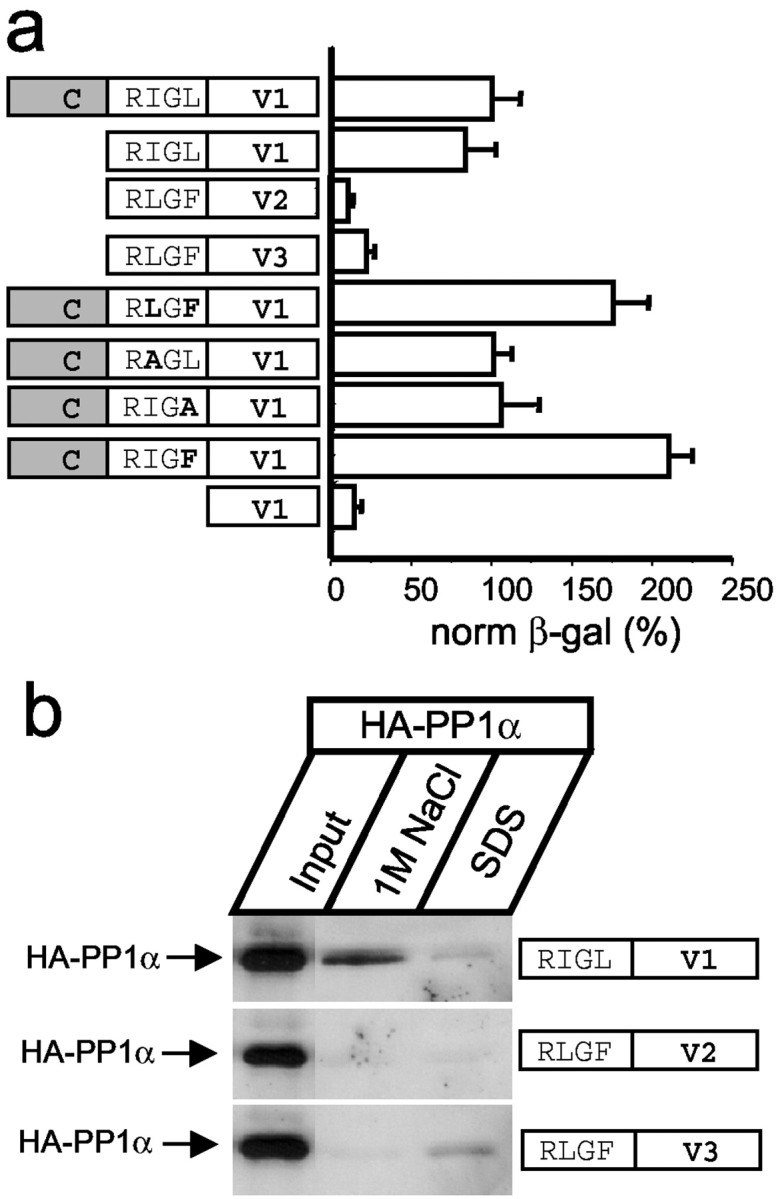
PP1α-binding motif in the InsP3R1 sequence. a, Analysis of PP1α binding specificity. IC1 (C-RIGL-V1, 2714–2749 of InsP3R1), RIGL-V1 (2731–2749 of InsP3R1), RLGF-V2 (2683–2701 of InsP3R2), RLGF-V3 (2658–2670 of InsP3R3), IC1 point mutants in the RIGL motif (indicated in bold), and V1 (2736–2749 of InsP3R1) baits were tested with PP1α prey in liquid yeast two-hybrid assays. The data are normalized to the strength of interaction for the IC1–PP1α pair and are shown as mean ± SEM (n ≥ 3). β-gal, β-galactosidase. b, HA–PP1α pull-down experiments with RIGL-V1 (2731–2749 of InsP3R1), RLGF-V2 (2683–2701 of InsP3R2), and RLGF-V3 (2658–2670 of InsP3R3) peptides. Fractions eluted from the beads by 1m NaCl and SDS were analyzed by Western blotting with anti-HA mAbs. The input lane on all three panelscontains th of the COS cell lysate used for pull-downs.
Fig. 3.
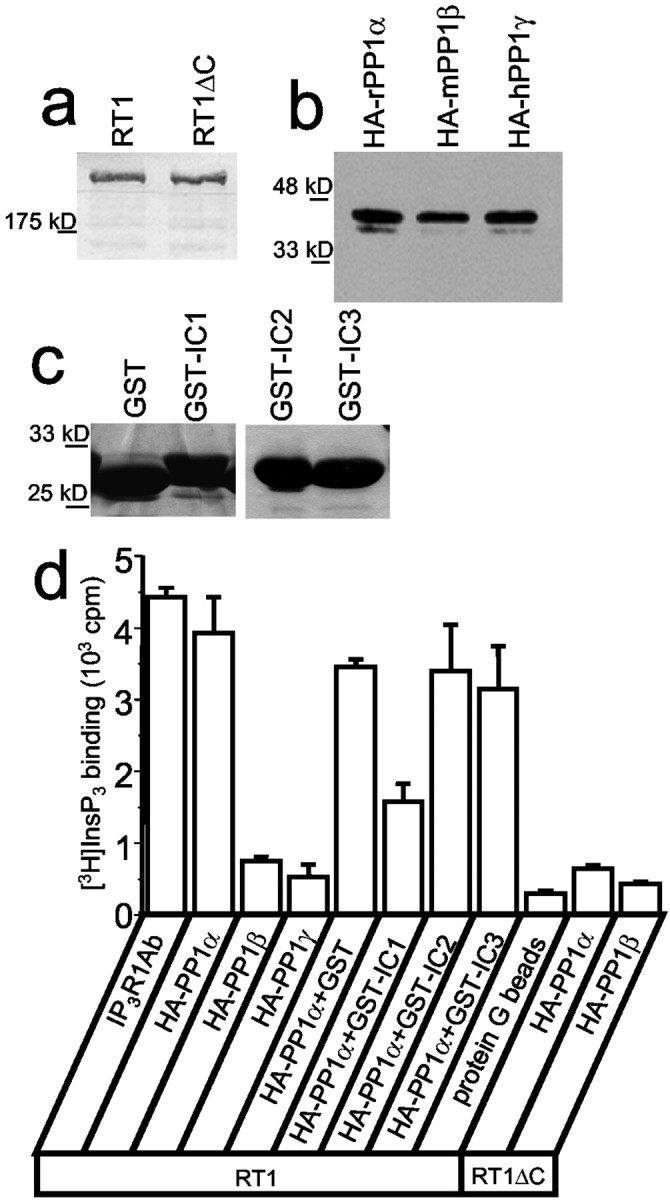
InsP3R1 binds PP1α in vitro. a, Expression of full-length (RT1) or truncated (RT1ΔC) recombinant InsP3R1 in Sf9 cells was analyzed by Western blotting with polyclonal antibodies directed against the InsP3R1 N-terminal region (cytl3b2).b, Expression of HA-tagged α, β, and γ PP1 isoforms in COS cells was analyzed by Western blotting with anti-HA mAbs. c, GST, GST–IC1, GST–IC2, and GST–IC3 proteins were expressed in BL21 Escherichia coli was purified on glutathione beads and analyzed by Coomassie staining. d, Analysis of InsP3R1–PP1 association in vitro by immunoprecipitation. HA-tagged α, β, and γ PP1 isoforms were mixed with solubilized full-length (RT1) or truncated (RT1ΔC) recombinant InsP3R1 and precipitated with anti-HA mAbs. GST, GST–IC1, GST–IC2, and GST–IC3 proteins were included in the immunoprecipitation reactions at a 200 μg/ml concentration as indicated. The amount of precipitated InsP3R1 was quantified by [3H]InsP3 binding. Anti-InsP3R1 mAbs (IP3R1Ab) were used as a positive control; empty beads (protein G beads) were used as a negative control.
To further confirm a specific association between InsP3R1 and PP1α, we performed a series ofin vitro binding experiments. For these experiments, full-length (RT1) and truncated (RT1ΔC) rat InsP3R1 were expressed in insect cells by baculovirus infection (Fig. 3a) and solubilized in CHAPS. The HA-tagged PP1α, PP1β, and PP1γ were transiently expressed in COS cells (Fig. 3b), solubilized in CHAPS, mixed with the InsP3R1-containing lysates, and precipitated with anti-HA antibodies. The amount of immunoprecipitated InsP3R1 was quantified by [3H]InsP3 binding assay. We found that HA–PP1α, but not HA–PP1β or HA–PP1γ, efficiently precipitated the InsP3R1 (Fig.3d). The ability of HA–PP1α to precipitate InsP3R1 critically depended on the InsP3R1 C-terminal region, because HA–PP1α did not precipitate RT1ΔC protein (Fig. 3d). In complementary experiments, we found that GST–IC1, but not GST alone, GST–IC2, or GST–IC3 proteins (Fig. 3c), effectively interfered with the InsP3R1 precipitation by HA–PP1α (Fig.3d). Thus, the most C-terminal region of the InsP3R1 is both necessary and sufficient for specific association with PP1α.
Do InsP3R1 and PP1α associate in vivo? In the brain, PP1α is concentrated in postsynaptic spines (Ouimet et al., 1995). The InsP3R1 is also present in postsynaptic terminals (Sharp et al., 1993a,b). To establish whether InsP3R1–PP1α complexes form in synaptic locations, we isolated cortical rat brain synaptosomes, extracted the obtained material in CHAPS, precipitated with the anti-InsP3R1 polyclonal antibody, and blotted with the anti-PP1 mAb. We found that PP1 was precipitated by anti-InsP3R1 antibodies but not by the preimmune sera (Fig. 4a). In the brain, the PP1α isoform is most enriched in the neostriatum region (da Cruz e Silva et al., 1995). Are InsP3R1–PP1α complexes formed in the neostriatum? By following published procedures (Nishi et al., 1997), we isolated the neostriatum region of the adult rat brain and performed immunoprecipitation experiments. Similar to experiments with the synaptosomes, PP1 was precipitated from the neostriatum by anti-InsP3R1 antibodies but not by the preimmune sera (Fig. 4b). Thus, InsP3R1–PP1 complexes exist in synaptic locations and in the neostriatum region of the brain. The PP1 mAbs available to us do not discriminate between different PP1 isoforms, but based on the specificity of InsP3R1 interactionsin vitro (Figs. 1-3), it is likely that the observed complexes correspond to InsP3R1–PP1α.
Fig. 4.

InsP3R1 binds PP1α in vivo. The InsP3R1 forms complexes with PP1 in brain synaptosomes (a) and in the neostriatum (b). The samples were precipitated with anti-InsP3R1 polyclonal antibodies (T443) and blotted with anti-PP1 mAbs. Preimmune sera (P/S) were used as a negative control. The input lane on a andb contains th of the lysate used for immunoprecipitation. Quantification of PP1 band intensity suggests that 3.8% (synaptosomes) and 3.4% (neostriatum) of total PP1 is associated with the InsP3R1.
PP1α dephosphorylates PKA-phosphorylated InsP3R1in vitro
The InsP3R1 is one of the major substrates of PKA phosphorylation in the brain (Supattapone et al., 1988; Danoff et al., 1991; Ferris et al., 1991a; Haug et al., 1999; Pieper et al., 2001). In the neostriatum, PP1 and PKA play an antagonistic role (Greengard et al., 1999). Can neostriatal PP1 dephosphorylate InsP3R1? To answer this question, we performed a series of in vitro dephosphorylation experiments. For these experiments, InsP3R1 (RT1) was expressed in insect cells by baculovirus infection (Fig. 3a), immunoprecipitated, and phosphorylated in vitro by a catalytic subunit of PKA in the presence of [γ-32P]ATP. The32P-InsP3R1 was incubated for a variable amount of time with the rat neostriatal homogenate. Rapid dephosphorylation of32P-InsP3R1 by neostriatal homogenate was observed (Fig.5a). The dephosphorylation assay was performed in the presence of 0.1 mmEGTA and 1 nm okadaic acid to inhibit PP2A, PP2B, and PP2C activities (Nishi et al., 1999). Under these conditions, dephosphorylation of InsP3R1 by neostriatal homogenate was almost completely inhibited by Inh2 (Fig.5c), confirming that the observed phosphatase activity corresponds to the activity of endogenous nsPP1.
Fig. 5.
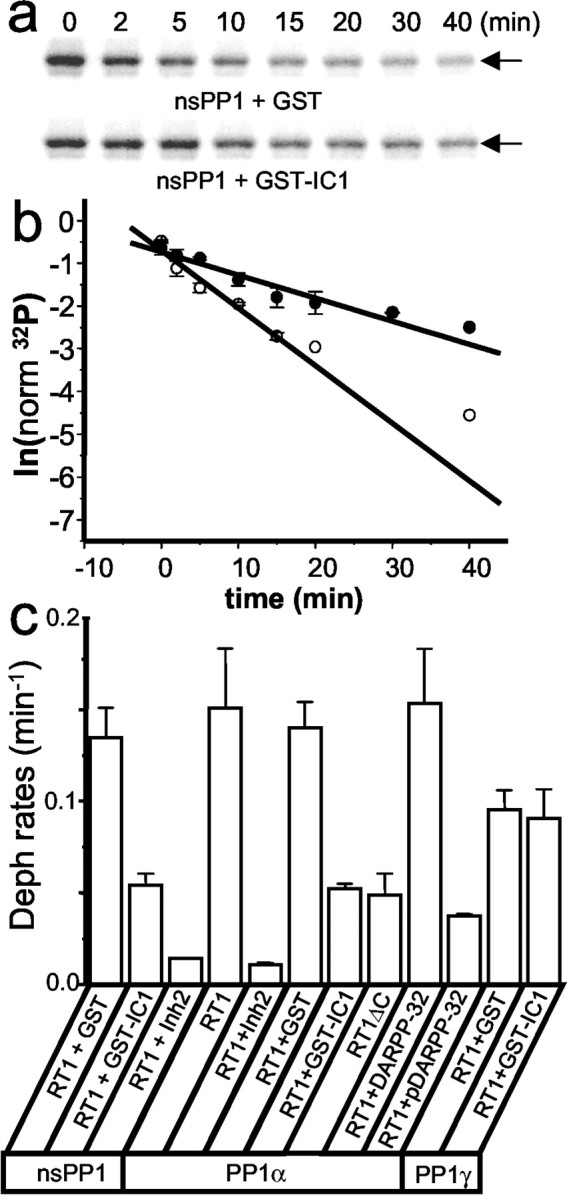
PP1 dephosphorylates the InsP3R1in vitro. a, Effects of GST (top) and GST–IC1 (bottom) on the32P-InsP3R1 dephosphorylation by endogenous nsPP1 in vitro. For each sample, the time of incubation with nsPP1 is indicated above the autoradiogram. The experiment was repeated three times with similar results. b, The normalized data from several independent experiments were averaged together and plotted in semilogarithmic coordinates as mean ± SE (n = 3) for experiments in the presence of GST (open circles) and GST–IC1 (filled circles). The rate of the dephosphorylation reaction is determined from the slope of the straight line used to fit the data. c, Summary of the32P-InsP3R1 in vitrodephosphorylation experiments. The rates of32P-InsP3R1 dephosphorylation (Deph) reactions determined as described forb are shown as mean ± SE (n ≥ 3). DARPP-32 and pDARPP-32 indicate recombinant unphosphorylated and PKA-phosphorylated forms of DARPP-32, respectively.
The substrate specificity of PP1 is primarily determined by its targeting subunits, such as spinophilin, neurabin, and GM (Greengard et al., 1999). Is it possible that the identified InsP3R1–PP1α association (Figs.1-4) facilitates the InsP3R1 dephosphorylation by PP1? To test this hypothesis, we compared the rates of32P-InsP3R1 dephosphorylation by nsPP1 in the presence of GST and GST–IC1 proteins (Fig. 3c). We found that dephosphorylation of32P-InsP3R1 by nsPP1 was significantly faster in the presence of GST than in the presence of GST–IC1 (Fig. 5a). To quantify these data, the content of the 32P-InsP3R1 band at each time point was quantified by phosphoimaging (Fig.5a) and normalized to time 0. When the normalized32P-InsP3R1 content was plotted versus time of incubation with nsPP1 in semilogarithmic coordinates, the measured values could be fitted by the straight line (Fig. 5b), the slope of which corresponds to the rate of the32P-InsP3R1 dephosphorylation. On average, for our experimental conditions the nsPP1 dephosphorylated32P-InsP3R1 at a rate of 0.13 ± 0.02 (n = 3) min−1 in the presence of GST and at a rate of 0.054 ± 0.006 (n = 3) min−1 in the presence of GST–IC1 (Fig.5c).
To further analyze the specificity of InsP3R1 dephosphorylation by PP1, we performed a series of in vitrodephosphorylation experiments with recombinant PP1. Similar to results with endogenous nsPP1, we found that recombinant PP1α rapidly dephosphorylates32P-InsP3R1 (Fig.5c). As with nsPP1, dephosphorylation of32P-InsP3R1 by PP1α was abolished by PP1 inhibitor 2 (Fig. 5c). Similar to nsPP1, we observed a threefold reduction in the rate of32P-InsP3R1 dephosphorylation by PP1α in the presence of GST–IC1 but not in the presence of GST (Fig. 5c). A similar effect was caused by truncation of the InsP3R1 C-terminal in RT1ΔC mutant (Fig. 5c). Similar to PP1α, the PP1γ isoform was also able to dephosphorylate32P-InsP3R1 in vitro (Fig. 5c). The absolute rates of PP1α- and PP1γ-mediated dephosphorylation of32P-InsP3R1 are not comparable, because different amounts of phosphatase activity were added to the dephosphorylation reactions. Importantly, in contrast to experiments with PP1α, GST–IC1 had no effect on the rate of32P-InsP3R1 dephosphorylation by PP1γ (Fig. 5c). This result agrees with the inability of InsP3R1 and PP1γ to form a complex in yeast two-hybrid and biochemical assays (Figs.1b, 3d). From the results shown on Figure5c, we concluded that direct association of PP1α with the InsP3R1 C-terminal enables efficient dephosphorylation of32P-InsP3R1 by PP1α. In the neostriatum, DARPP-32 plays a predominant role in control of PP1 activity (Greengard et al., 1999). We found that the PKA-phosphorylated form pDARPP-32, but not DARPP-32 itself, inhibited32P-InsP3R1 dephosphorylation by PP1α (Fig. 5c).
PKA phosphorylation of neostriatal InsP3R1in vivo
In the neostriatum, stimulation of D1 dopamine receptors causes an increase in cAMP levels (Greengard et al., 1999). Are neostriatal InsP3R1s phosphorylated by PKA when cAMP is elevated? To answer this question, we used the PKA back-phosphorylation method to determine the fraction of neostriatal InsP3R1 in the phosphorylated state (pInsP3R1). By following published procedures (Nishi et al., 1997), we isolated neostriatal slices from adult rat brains and incubated them in the oxygenated Krebs media. For back-phosphorylation experiments, the neostriatal InsP3R1 was solubilized in CHAPS in the presence of phosphatase inhibitors, immunoprecipitated with anti-InsP3R1 antibodies, phosphorylated in vitro by the catalytic subunit of PKA in the presence of [γ-32P]ATP, separated by electrophoresis, and analyzed by phosphoimaging. When the sample was incubated with PP1α before in vitro phosphorylation by PKA, the content of the32P-InsP3R1 band was the greatest (Fig. 6a,PP1α lane). This value was interpreted as total InsP3R1 in the neostriatal sample (32PPP1α). Without preincubation with PP1α, the content of the32P-InsP3R1 band was reduced by ∼30% (Fig. 6a, Ctr lane). By using the normalization procedure described in Materials and Methods, we determined that 37 ± 3% (n = 4) of InsP3R1s in the neostriatum are in the PKA-phosphorylated state in control conditions (Fig. 6b). When neostriatal slices were incubated with 1 mm8-Br-cAMP or 100 μm dopamine for 10 min, we observed a drastic reduction in the content of the32P-InsP3R1 band (Fig. 6a, cAMP and Dop lanes). We estimated that 8-Br-cAMP and dopamine increased the fraction of PKA-phosphorylated InsP3R1 in the neostriatum to 86 ± 6% (n = 4) and 85 ± 8% (n = 4), respectively (Fig. 6b). Preincubation of neostriatal slices for 60 min with 5 μm cyclosporine A, a calcineurin inhibitor, or 400 μm calyculin A, a PP1/PP2A inhibitor, increased the fraction of PKA-phosphorylated InsP3R1 to 71 ± 4% (n = 4) and 68 ± 3% (n = 4), respectively (Fig.6a,b, CsA and CalA lanes). In contrast, preincubation of neostriatal slices with 10 nm okadaic acid, a specific inhibitor of PP2A at this concentration, had only a minor effect on the PKA-phosphorylated state of InsP3R1 when compared with control conditions (Fig. 6a,b, OA lane).
Fig. 6.

PKA phosphorylation of neostriatal InsP3R1 in vivo. PKA back-phosphorylation of neostriatal InsP3R1 after pretreatment with PP1α (PP1α); in controls (Ctr); after 10 min of incubation of neostriatal slices with 1 mm 8-Br-cAMP (cAMP) or 100 μm dopamine (Dop); or after 60 min of treatment with 5 μm cyclosporine A (CsA), 400 μm calyculin A (CalA), or 10 nm okadaic acid (OA). a, Autoradiogram of a representative experiment. The data for calyculin A and okadaic acid are taken from the different experiments.b, Summary of neostriatal InsP3R1 back-phosphorylation experiments. The estimated fraction of PKA-phosphorylated InsP3R1 in neostriatal slices (see Materials and Methods) is shown as mean ± SE (n = 4).
To determine a dynamic of dopamine-induced InsP3R1 phosphorylation in the neostriatal slices, we used the PKA back-phosphorylation method at different time points after application of 100 μmdopamine. We found that the fraction of PKA-phosphorylated InsP3R1 peaks 10–15 min after dopamine application and returns to prestimulation levels within 30 min (Fig.7a). Thus, similar to DARPP-32 (Nishi et al., 1997; Greengard et al., 1999), the dopamine-induced phosphorylation of neostriatal InsP3R1 by PKA is transient, although with a slower time course. The dopamine-induced changes in the InsP3R1 phosphorylated state were abolished by preincubation of neostriatal slices with 400 μm calyculin A, a PP1/PP2A inhibitor (Fig.7b). In contrast, preincubation of neostriatal slices with 10 nm okadaic acid, a specific inhibitor of PP2A at this concentration, had only a minimal effect on dopamine-induced changes in neostriatal InsP3R1 phosphorylated state (Fig. 7c). From the obtained pharmacological profile (Figs. 6, 7), we concluded that the PKA-phosphorylated state of neostriatal InsP3R1 in the resting state and in response to stimulation with dopamine is determined by the activity of PP1 and PP2B phosphatases but not by the activity of PP2A phosphatase.
Fig. 7.

Dopamine induces transient phosphorylation of neostriatal InsP3R1 by PKA. a, Time course of changes in neostriatal InsP3R1 PKA-phosphorylated state in response to application of 100 μm dopamine. Dopamine was applied to neostriatal slices at time 0. At each time point, the fraction of neostriatal InsP3R1 in the PKA-phosphorylated state is shown as mean ± SE (n = 3) (filled circles). b, c, The same experiment as in a performed with slices exposed to 400 μm calyculin A (CalA, b) (open circles) or 10 nm okadaic acid (OA, c) (filled squares) for 60 min before the application of dopamine.
PKA activates and PP1α inhibits InsP3R1
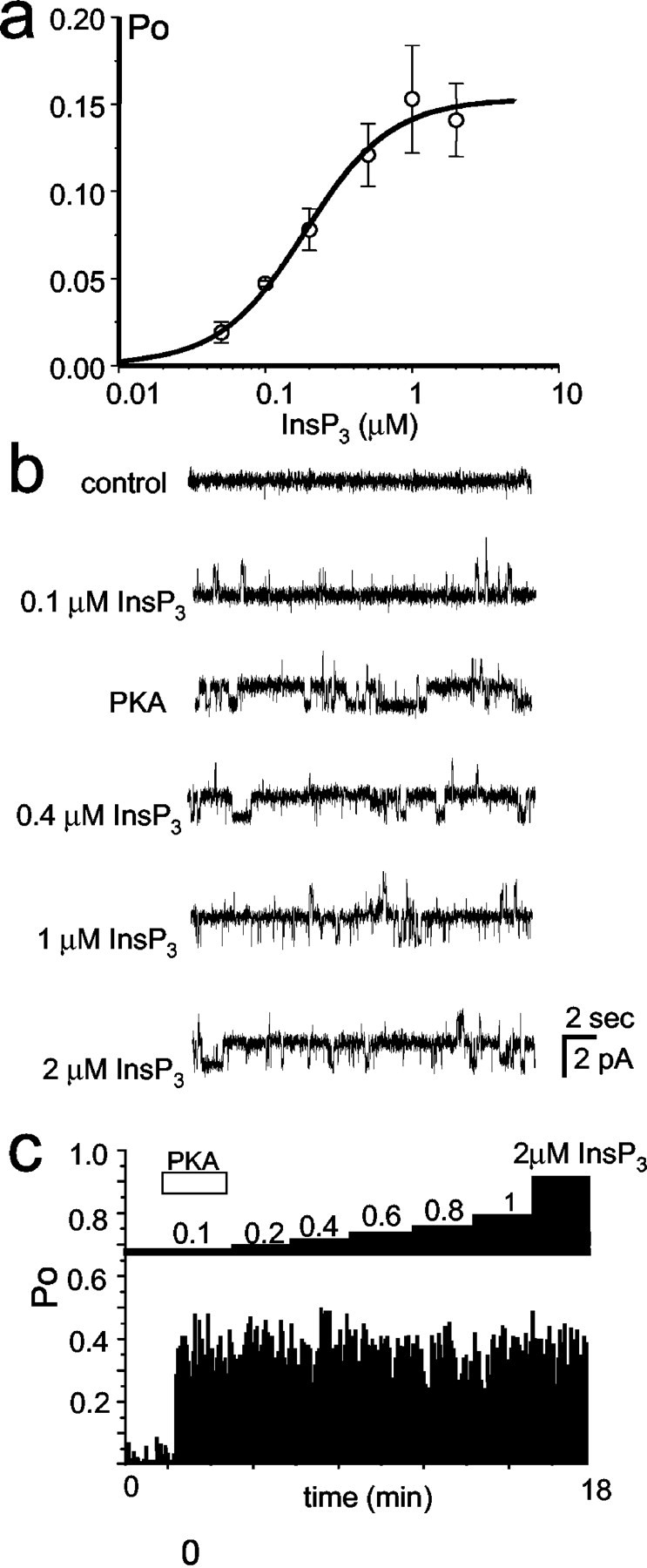
What are the functional consequences of InsP3R1 phosphorylation by PKA? Ca2+ flux measurements used previously to address this question provided conflicting answers (Supattapone et al., 1988; Nakade et al., 1994; Cameron et al., 1995; Wojcikiewicz and Luo, 1998). To study modulation of the InsP3R1 by PKA phosphorylation, we incorporated recombinant InsP3R1 expressed in insect cells into planar lipid bilayers by microsomal fusion (Tu et al., 2002). Addition of 2 μm InsP3 to the cytosolic (cis) chamber induced InsP3R1 activity (Fig. 8a, second trace), but the Po was only 5–10% (Fig. 8b). In the presence of 0.5 mm MgATP in the cis chamber, the application of PKA catalytic subunit directly to the bilayer induced immediate facilitation in channel activity (Fig. 8a,third trace), with a Po of phosphorylated channels in the range of 30–40% (Fig. 8b). Application of PP1α to the bilayer resulted in almost complete inhibition of channel activity (Fig. 8a, fourth trace, b). Inactivation of InsP3R1 by PP1α could be reversed by a second application of PKA catalytic subunit (Fig. 8a,fifth trace, b), which in turn could be counteracted by the second application of PP1α (Fig. 8a,sixth trace, b). Results similar to the experiment shown in Figure 8a,b were obtained in three independent experiments. No effect was observed if the catalytic subunit of PKA was boiled before addition to the bilayer (n = 5) or if 0.5 mmNa2ATP was present in the cis chamber instead of MgATP (n = 3). As an additional control, we performed experiments with a nonhydrolysable ATP analog, ATPγS. If 100 μm Mg-ATPγS was present in thecis chamber, the application of a catalytic subunit of PKA to the bilayer resulted in InsP3R1 activation that could no longer be reversed by PP1α or affected by a second application of PKA (Fig. 8c). From our experiments, we concluded that under identical experimental conditions PP1α-dephosphorylated InsP3R1s have lowPo (<2–3%), and PKA-phosphorylated InsP3R1s have much higherPo (30–40%).
Fig. 8.

PKA and PP1α modulate InsP3R1 activity in planar lipid bilayers. a, PKA activates and PP1α inhibits the recombinant InsP3R1 reconstituted into planar lipid bilayers. Each trace corresponds to 10 sec of current recordings from the same experiment. The experiment is performed in the presence of pCa 6.7 and 0.5 mm MgATP in the cis chamber. Additions of 2 μmInsP3 to the cis chamber and PKA/PP1α directly to the bilayer are indicated. Similar results were obtained in three independent experiments. b, The average InsP3R1 Po is calculated for a 5 sec window of time and plotted for the duration of an experiment. The times of InsP3, PKA, and PP1α additions are shown above the Po plot. The same experiment was used to generate a and b.c, The InsP3R1 Poplot for the experiment performed in the presence of 100 μm Mg-ATPγS in the cis chamber. The times of InsP3, PKA, and PP1α additions are shown above the Po plot. Similar results were obtained in three independent experiments.
To test the importance of InsP3R1–PP1α association for InsP3R1 modulation by PKA phosphorylation, we performed planar lipid bilayer experiments with RT1ΔC mutants expressed in Sf9 cells (Fig. 3a). We found that RT1ΔC mutants formed functional InsP3-gated channels, which were modulated by PKA and PP1α in a manner similar to the wild-type InsP3R1 (data not shown). To explain these results, we reasoned that because of the high concentration of PP1α added to the bilayer, the C-terminal PP1α-docking site in the InsP3R1 sequence is not important for functional regulation of InsP3R1 in our in vitroexperiments. However, in vivo the concentration of PP1α is much lower, and InsP3R1–PP1α association is likely to play an important role in control of the InsP3R1 PKA-phosphorylated state.
Mechanism of InsP3R1 activation by PKA
To obtain mechanistic insights into InsP3R1 activation by PKA, we evaluated effects of PKA phosphorylation on Ca2+ and InsP3dependence of recombinant InsP3R1 reconstituted into planar lipid bilayers. In the first series of experiments, the activity of InsP3R1 was recorded at variable Ca2+ concentrations in the presence of 2 μm InsP3 and 0.5 mmMg-ATP. With addition of InsP3, we observed two distinct populations of InsP3R1. In some (6 of 14) experiments, the initial activity of InsP3R1 was low, with Po ≤10% (“low-activity” channels). In other experiments (8 of 14), the activity of InsP3R1 was much higher, withPo ∼30% (“high-activity” channels). As described in the previous section, addition of PKA to low-activity channels increased theirPo to 30–40% (Fig. 8). Addition of PKA to high-activity channels had very little or no effect on theirPo, but addition of PP1α reduced their Po to levels of <10% (data not shown). In in vitro back-phosphorylation experiments, we determined that ∼20% of recombinant InsP3R1 in microsomes isolated from Sf9 cells are in the PKA-phosphorylated state (data not shown), presumably because of activity of endogenous PKA present in Sf9 cells. Thus, we reasoned that high-activity channels are likely to correspond to partially phosphorylated InsP3R1, and low-activity channels correspond to unphosphorylated InsP3R1.
The experiments in the previous section (Fig. 8) were performed with low-activity channels. In Ca2+-dependence experiments, we compared the behavior of low-activity, high-activity, and PKA-phosphorylated channels. In agreement with our previous findings (Nosyreva et al., 2002; Tu et al., 2002), recombinant high-activity InsP3R1 displayed bell-shaped dependence on cytosolic Ca2+ with the peak at pCa 6.65 (Fig. 9, open circles). The parameters of the optimal fit (Pm, n, k,K) for each series of Ca2+-dependence experiments are presented in Table 1. Fit to the data using the modified bell-shaped equation (see Materials and Methods) yielded the affinity of activating site equal to 0.22 μmCa2+, the affinity of inhibitory site equal to 0.21 μmCa2+, and the cooperativity coefficient of 1.31 (Fig. 9, smooth curve; Table 1). Recombinant low-activity InsP3R1 displayed similar bell-shaped Ca2+ dependence with the peak at pCa 6.55 (Fig. 9, open triangles). Fit to low-activity data set yielded the affinity of activating site equal to 0.10 μm Ca2+, the affinity of inhibitory site equal to 0.72 μmCa2+, and the cooperativity coefficient of 2.09 (Fig. 9, smooth curve; Table 1). When the same experiment was performed with the InsP3R1 phosphorylated by PKA in bilayers (initially displaying low activity), we found that the PKA-phosphorylated InsP3R1 also displayed bell-shaped Ca2+ dependence that peaked at pCa 6.65 (Fig. 9, filled circles). For PKA-phosphorylated InsP3R1, the fit yielded the affinity of activating site equal to 0.24 μmCa2+, the affinity of inhibitory site equal to 0.21 μmCa2+, and the cooperativity coefficient of 1.32 (Fig. 9, smooth curve; Table 1). From these experiments, we concluded that PKA phosphorylation induces only minor changes in bell-shaped Ca2+ dependence of the InsP3R1.
Fig. 9.

Effect of PKA on InsP3R1 Ca2+ dependence. The Poof recombinant InsP3R1 was determined in the presence of 2 μm InsP3 and 0.5 mm MgATP atcis (cytosolic) Ca2+ concentrations in the range between 10 nm and 5 μmCa2+. Po values measured in several independent experiments were averaged together at each Ca2+ concentration as described in Materials and Methods and shown as mean ± SE for low-activity InsP3R1 (n = 2; open triangles), high-activity InsP3R1 (n = 3; open circles), and PKA-phosphorylated InsP3R1 (n = 3;filled circles). The averaged data were fitted by the bell-shaped equation modified from Bezprozvanny et al. (1991), as explained in Materials and Methods. The parameters of the optimal fits (smooth curves) are shown in Table 1.
Table 1.
Parameters of the bell-shaped fit to the Ca2+-dependence data obtained with control (low and high activity) and PKA-phosphorylated InsP3R1
| InsP3R1 | Pm | Hill coefficient (n) | Affinity of the activating site k(μm) | Affinity of the inhibitory site K(μm) | Peak of Ca2+ dependence (pCa) |
|---|---|---|---|---|---|
| Low activity | 2.25 | 2.10 | 0.10 | 0.72 | 6.55 |
| High activity | 0.3 | 1.31 | 0.22 | 0.21 | 6.65 |
| PKA | 0.38 | 1.32 | 0.24 | 0.21 | 6.65 |
In the next series of experiments, we analyzed effects of PKA on InsP3 dependence of InsP3R1. These experiments were performed in the presence of 300 nm Ca2+ (pCa 6.7) and 0.5 mm MgATP on the cytosolic side of the bilayer. By adding increasing amounts of InsP3 to thecis chamber, we determined that the apparent affinity of high-activity InsP3R1 for InsP3 (kInsP3) is equal to 0.19 μm InsP3(Fig. 10a, open circles). The apparent affinity of low-activity InsP3R1 could not be reliably determined because of the extremely low Po of these channels at low InsP3 concentrations (data not shown). To determine the effect of PKA on the InsP3 dependence of InsP3R1, we started an experiment by addition of 100 nm InsP3. The InsP3R1 activity at this concentration of InsP3 was very low (Fig. 10b,second trace), with a Po of 1–2% (Fig. 10c). Addition of PKA to the bilayer resulted in dramatic activation of InsP3R1 (Fig.10b, third trace), with thePo increased to 30–40% (Fig.10c). Increasing the InsP3concentration from 100 nm to 2 μm did not result in additional InsP3R1 activation (Fig. 10b,traces 4–6, and c). Thus, PKA-phosphorylated InsP3R1s are maximally activated by 100 nm InsP3, indicating that the kInsP3 value for PKA-phosphorylated InsP3R1 must be <50 nm InsP3. This estimate is in contrast to the values measured for high-activity InsP3R1 in the absence of PKA treatment (Fig.10a). From these experiments, we concluded that PKA phosphorylation causes at least a fourfold increase in InsP3R1 sensitivity to activation by InsP3.
Fig. 10.
Apparent affinity of InsP3R for InsP3 is increased by PKA phosphorylation.a, The InsP3 dependence of InsP3R1 in control conditions. The InsP3R1Po values measured in several independent experiments were averaged together at each InsP3concentration as described in Materials and Methods and shown as mean ± SE for high-activity InsP3R1 (n = 3; open circles). The averaged data were fitted by the equation modified from Lupu et al. (1998), as explained in Materials and Methods. The parameters of optimal fit (smooth curve) yieldedkInsP3 = 0.19 μm;n = 1.47; Pmax = 0.154. b, c, Effect of PKA on the InsP3R1 InsP3 dependence. b, Each tracecorresponds to 20 sec of recombinant InsP3R1 current recordings from the same experiment. The experiment was performed in the presence of pCa 6.7 and 0.5 mm MgATP in thecis (cytosolic) chamber at InsP3 concentrations from 100 nm to 2 μm. Addition of PKA directly to the bilayer increased the InsP3R1 activity (third trace). The filter frequency is 200 Hz for alltraces shown. c, The InsP3R1Po was calculated for a 5 sec window of time and was plotted for the duration of an experiment. Changes in InsP3 concentration (from 100 nm to 2 μm as indicated) in the cis chamber and the time of PKA addition to the bilayer are shown by the bar diagram. The data shown in b andc are from the same experiment.
Discussion
Modulation of InsP3R1 by PKA and PP1α was investigated in this study. The main conclusions of our study are as follows: (1) the InsP3R1 specifically associates with PP1α via the C-terminal region; (2) association with PP1α facilitates dephosphorylation of PKA-phosphorylated InsP3R1; (3) the neostriatal InsP3R1s are phosphorylated by PKA after exposure of neostriatal slices to 8-Br-cAMP, cyclosporine A, calyculin A, but not to 10 nm okadaic acid; (4) the neostriatal InsP3R1s are transiently phosphorylated by PKA after application of dopamine; (5) the dopamine-induced PKA phosphorylation of neostriatal InsP3R1 is affected by cyclosporine A but not by 10 nm okadaic acid; (6) the InsP3R1s reconstituted into planar lipid bilayers are activated by PKA and inhibited by PP1α; (7) phosphorylation of InsP3R1 by PKA does not shift the peak of InsP3R1 bell-shaped Ca2+ dependence; (8) phosphorylation of InsP3R1 by PKA induces at least a fourfold increase in the sensitivity of InsP3R1 to activation by InsP3. Implications of these findings for InsP3R1 function and dopaminergic signaling in the neostriatum are briefly discussed below.
Modulation of InsP3R1 activity by PKA
A number of previous biochemical studies analyzed phosphorylation of InsP3R1 by PKA. The neuronal InsP3R1 is one of the best-known substrates for both endogenous and exogenous PKA (Walaas et al., 1986; Supattapone et al., 1988; Maeda et al., 1990; Danoff et al., 1991; Ferris et al., 1991b; Wojcikiewicz and Luo, 1998; Haug et al., 1999; Pieper et al., 2001). Two putative PKA phosphorylation sites (S1756 and S1589) are present in the coupling domain of the InsP3R1 (Danoff et al., 1991; Ferris et al., 1991a; Haug et al., 1999). In the cerebellum, Ser-1756 is the primary site of phosphorylation by PKA, whereas much higher PKA activity is required to phosphorylate Ser-1589 (Ferris et al., 1991b; but see Haug et al., 1999). Interestingly, the region of InsP3R1 between these two phosphorylation sites is alternatively spliced in a tissue-specific manner, with the stretch of 39 aa residues deleted in the non-neuronal isoform of the receptor (Danoff et al., 1991). This splicing event appears to change the pattern of phosphorylation by PKA, because InsP3R purified from vas deferens (short, non-neuronal isoform) is phosphorylated by PKA almost exclusively on Ser-1589 (Danoff et al., 1991). Potentially, this difference may form a basis for a tissue-specific regulation of InsP3R1 function by cAMP-mediated signaling pathways.
Despite this wealth of biochemical information, the functional consequences of InsP3R1 phosphorylation by PKA are poorly understood. The potency of InsP3 to release Ca2+ from cerebellar microsomes was reduced 10-fold because of PKA phosphorylation (Supattapone et al., 1988; Cameron et al., 1995), indicating that the link between InsP3 binding and channel opening is impaired in PKA-phosphorylated InsP3R1. To the contrary, data from other groups, obtained with cerebellar microsomes (Volpe and Alderson-Lang, 1990), with the proteoliposomes containing purified cerebellar InsP3R1 (Nakade et al., 1994), or with permeabilized SH–SY5Y neuroblastoma cells (Wojcikiewicz and Luo, 1998), indicated that InsP3-mediated Ca2+ release is facilitated by PKA phosphorylation. The reasons for these conflicting results are not clear and more importantly, interpretation of these data are obscured by changes in the rate of Ca2+ uptake into the stores, known to be affected by PKA phosphorylation.
In this study, the effects of PKA on InsP3R1 function were evaluated using the planar lipid bilayer reconstitution technique. This technique has been used previously to analyze effects of PKA on skeletal and cardiac ryanodine receptors (RyanRs) (Hain et al., 1995; Marx et al., 2000). The planar lipid bilayer reconstitution method offers a number of advantages compared with previously used Ca2+ flux measurements. In planar lipid bilayer experiments, we were able to describe the functional effects of InsP3R1 phosphorylation by PKA in well defined experimental conditions, such as, for example, different cytosolic Ca2+ (Fig. 9) and InsP3 (Fig. 10) concentrations. Most importantly, in planar lipid bilayer experiments, effects of PKA on InsP3R1 can be studied in isolation from effects on Ca2+-ATPase and other signaling proteins. From our experiments, we concluded that PKA activates and PP1α inhibits the activity of InsP3R1 (Fig. 8). Interestingly, similar functional effects of PKA and PP1 on skeletal and cardiac RyanRs has been described previously (Hain et al., 1995;Marx et al., 2000), suggesting that both families of intracellular Ca2+ release channels are subject to similar modulation by PKA/PP1. Activation of InsP3R1 by PKA resulted from an increase in InsP3R1 sensitivity to InsP3 activation (Fig. 10), with minimal effect on InsP3R1 Ca2+dependence (Fig. 9). The recombinant InsP3R1 used in our studies corresponds to neuronal InsP3R1 isoform (Mignery et al., 1990). Future studies will be required to test the effects of PKA on non-neuronal InsP3R1 isoform. These experiments will be enabled by functional expression of both InsP3R1 isoforms in Sf9 cells (Tu et al., 2002). Additional experiments will also be needed to evaluate the role of two PKA phosphorylation sites in control of InsP3R1 activity. These studies will require generation of InsP3R1 point mutations in S1755 and S1589 PKA phosphorylation sites.
InsP3R1 as a core of macromolecular signaling complex
An emerging theme in signal transduction is the association of signaling molecules in macromolecular signaling complexes. Direct association of upstream and downstream signaling components increases the speed, efficiency, and specificity of signal transduction. Association between signaling molecules is mediated frequently by adaptor proteins. For example, recent data suggested that cardiac RyanR2 forms a complex with cAMP-dependent kinase-anchoring protein 6 (AKAP6)/PKA, spinophilin/PP1, and PR130/PP2A, and that cardiac RyanR2 is activated by PKA phosphorylation and inhibited by PP1 dephosphorylation (Marx et al., 2000, 2001). Association of AKAP6, spinophilin, and PR130 with RyanR2 is mediated via noncanonical leucine–isoleucine zipper (LIZ) motifs (Marx et al., 2001). Our sequence analysis (data not shown) reveals that putative AKAP-binding LIZ motif is also present in the InsP3R1 sequence, but that the spinophilin and PR130-binding LIZ motifs are absent. Future studies will be needed to clarify the role of putative AKAP-binding LIZ motif in the InsP3R1 sequence.
In this study, we discovered direct and specific association of the C-terminal portion of InsP3R1 with PP1α (Figs.1-3) and show that this association facilitates dephosphorylation of PKA-phosphorylated InsP3R1 (Fig. 5). The association of InsP3R1 with FKBP12/calcineurin has been reported previously (Cameron et al., 1995, 1997; but seeBultynck et al., 2001a,b). The N-terminal of InsP3R1 binds to the adaptor protein Homer (Tu et al., 1998) and to Ca2+-binding protein caldendrin, which affects InsP3R1 gating (Yang et al., 2002). The middle coupling domain of InsP3R1 binds to calmodulin (Yamada et al., 1995). Future experiments will likely lead to identification of additional InsP3R1-binding partners. Nevertheless, it is becoming apparent that InsP3R1 forms a core of macromolecular signaling complex that includes a number of associated signaling proteins, some of which are able to modulate the InsP3R1 activity.
Potential role of InsP3R1 in cross talk between Ca2+ and cAMP signaling in neostriatum
A cross talk between cAMP and Ca2+-signaling pathways plays an important role in dopaminergic signaling in the neostriatum (Greengard et al., 1999). From our results, we hypothesize that InsP3R1 may participate in this process (Fig.11). We reason that because of direct association between InsP3R1 and PP1α (Figs.1-4), a fraction of PKA-phosphorylated InsP3R1 in the neostriatum is kept below 40% (Fig. 6b). The effect of calyculin A and cyclosporine A on the InsP3R1 phosphorylated state (Fig. 6) indicates that even under resting conditions, the phosphorylated state of neostriatal InsP3R1 is determined by a balance between competing activities of kinases and phosphatases. A similar conclusion has been reached previously regarding the levels of DARPP-32 phosphorylation (Nishi et al., 1997). The PKA-phosphorylated state of neostriatal InsP3R1 appears to depend on PP1 and PP2B activity but not on PP2A activity, because 10 nm okadaic acid had only a minor effect on the InsP3R1 phosphorylated state in our experiments (Figs. 6, 7c). The effect of calyculin A on InsP3R1 phosphorylation is likely attributable to direct inhibition of PP1 phosphatase, but the effect of cyclosporine A is likely mediated by inhibition of PP2B phosphatase, which controls the phosphorylation state of DARPP-32 (Nishi et al., 1999) (Fig.11).
Fig. 11.

Model of InsP3R1 participation in cross talk between cAMP and Ca2+ signaling pathways during dopaminergic signaling in the neostriatum. The model drawing is adapted from Greengard et al. (1999). Arrows depict activating influence, whereas blocking arrows (⊣) depict inhibitory influence. The points of cyclosporine A (CsA) and calyculin A (CalA) interference with the phosphorylated state of neostriatal InsP3R1 in our experiments are indicated. See Discussion. NMDAR, NMDA receptor; AMPAR, AMPA receptor; VDCaC, Voltage-gated Ca2+ channels.
We propose that after release of dopamine and stimulation of D1 receptors, an increase in the cAMP level leads to activation of PKA and transient phosphorylation of the InsP3R1 (Fig.7a) and DARPP-32 (Nishi et al., 1997; Greengard et al., 1999) proteins. Phosphorylation of InsP3R1 by PKA promotes InsP3R1 activation (Fig. 8) and release of Ca2+ from intracellular stores. An increase in intracellular Ca2+ leads to activation of calcineurin (PP2B), which dephosphorylates DARPP-32 protein and closes the negative feedback loop (Nishi et al., 1997;Greengard et al., 1999). After dephosphorylation of DARPP-32, the InsP3R1-associated PP1α is able to dephosphorylate neostriatal InsP3R1, returning it to the initial state (Fig. 7a). A similar negative feedback mechanism has been proposed previously to involve Ca2+ influx via NMDA receptors (Blank et al., 1997; Cepeda et al., 1998; Snyder et al., 1998), AMPA receptors (Yan et al., 1999), and voltage-gated Ca2+channels (Surmeier et al., 1995; Cepeda et al., 1998). Influx of Ca2+ via plasma membrane channels may have an additive effect with Ca2+ released via InsP3R1 by directly activating calcineurin, or it may have a synergistic effect caused by the potentiating effect of Ca2+ on the InsP3R1 (Bezprozvanny et al., 1991) (Fig. 11). The proposed model may also help to explain the antagonism between D2 and D1 dopamine receptors coexpressed in a subpopulation of neostriatal medium spiny neurons (Surmeier et al., 1996; Nishi et al., 1997; Lindskog et al., 1999). The activation of PLCβ via D2 receptors (Vallar et al., 1990;Hernandez-Lopez et al., 2000) causes an increase in InsP3 levels that can boost the InsP3R1–Ca2+–PP2B negative feedback loop (Fig. 11). In this study, we focused on cross talk between dopamine/cAMP and Ca2+signaling systems in the neostriatum. A similar model may also be relevant in the context of synaptic plasticity in the hippocampus and in other regions of the brain, with the PP1-inhibitor 1 (Allen et al., 2000) playing the role analogous to the role of DARPP-32 in the neostriatum.
Note added on proof. While this paper was prepared for publication, association of InsP3R1 with PKA, PP1, and PP2A was reported by deSouza et al. (2002).
T.-S.T. and H.T. contributed equally to this work
This work was supported by the Welch Foundation and National Institutes of Health Grant R01 NS38082 (I.B.). The initial yeast two-hybrid screen was performed by Dale Elmer and Anton Maximov. We are grateful to them for their effort. We are thankful to Thomas C. Südhof, Anton Maximov, and Elena Nosyreva for many helpful discussions and valuable experimental advice and to Phyllis Foley for expert administrative assistance. We thank Thomas C. Südhof for the rat InsP3R1 clone and the rat brain cDNA library, Gregory Mignery for the rat InsP3R2 clone, Graeme Bell for the rat InsP3R3 clone, and Jan Parys for the cytl3b2 antibody.
Correspondence should be addressed to Dr. Ilya Bezprozvanny, Department of Physiology, K4.112, University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX 75390-9040. E-mail:Ilya.Bezprozvanny@UTSouthwestern.edu.
References
- 1.Allen PB, Hvalby O, Jensen V, Errington ML, Ramsay M, Chaudhry FA, Bliss TV, Storm-Mathisen J, Morris RG, Andersen P, Greengard P. Protein phosphatase-1 regulation in the induction of long-term potentiation: heterogeneous molecular mechanisms. J Neurosci. 2000;20:3537–3543. doi: 10.1523/JNEUROSCI.20-10-03537.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- 3.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 4.Bezprozvanny I, Ehrlich BE. ATP modulates the function of inositol 1,4,5-trisphosphate-gated channels at two sites. Neuron. 1993;10:1175–1184. doi: 10.1016/0896-6273(93)90065-y. [DOI] [PubMed] [Google Scholar]
- 5.Bezprozvanny I, Ehrlich BE. The inositol 1,4,5-trisphosphate (InsP3) receptor. J Membr Biol. 1995;145:205–216. doi: 10.1007/BF00232713. [DOI] [PubMed] [Google Scholar]
- 6.Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- 7.Blank T, Nijholt I, Teichert U, Kugler H, Behrsing H, Fienberg A, Greengard P, Spiess J. The phosphoprotein DARPP-32 mediates cAMP-dependent potentiation of striatal N-methyl-d-aspartate responses. Proc Natl Acad Sci USA. 1997;94:14859–14864. doi: 10.1073/pnas.94.26.14859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blondel O, Takeda J, Janssen H, Seino S, Bell GI. Sequence and functional characterization of a third inositol trisphosphate receptor subtype, IP3R-3, expressed in pancreatic islets, gastrointestinal tract, and other tissues. J Biol Chem. 1993;268:11356–11363. [PubMed] [Google Scholar]
- 9.Bultynck G, Rossi D, Callewaert G, Missiaen L, Sorrentino V, Parys JB, De Smedt H. The conserved sites for the FK506-binding proteins in ryanodine receptors and inositol 1,4,5-trisphosphate receptors are structurally and functionally different. J Biol Chem. 2001a;276:47715–47724. doi: 10.1074/jbc.M106573200. [DOI] [PubMed] [Google Scholar]
- 10.Bultynck G, De Smet P, Rossi D, Callewaert G, Missiaen L, Sorrentino V, De Smedt H, Parys JB. Characterization and mapping of the 12 kDa FK506-binding protein (FKBP12)-binding site on different isoforms of the ryanodine receptor and of the inositol 1,4,5-trisphosphate receptor. Biochem J. 2001b;354:413–422. doi: 10.1042/0264-6021:3540413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cameron AM, Steiner JP, Roskams AJ, Ali SM, Ronnett GV, Snyder SH. Calcineurin associated with the inositol 1,4,5-trisphosphate receptor-FKBP12 complex modulates Ca2+ flux. Cell. 1995;83:463–472. doi: 10.1016/0092-8674(95)90124-8. [DOI] [PubMed] [Google Scholar]
- 12.Cameron AM, Nucifora FC, Jr, Fung ET, Livingston DJ, Aldape RA, Ross CA, Snyder SH. FKBP12 binds the inositol 1,4,5-trisphosphate receptor at leucine-proline (1400–1401) and anchors calcineurin to this FK506-like domain. J Biol Chem. 1997;272:27582–27588. doi: 10.1074/jbc.272.44.27582. [DOI] [PubMed] [Google Scholar]
- 13.Cepeda C, Colwell CS, Itri JN, Chandler SH, Levine MS. Dopaminergic modulation of NMDA-induced whole cell currents in neostriatal neurons in slices: contribution of calcium conductances. J Neurophysiol. 1998;79:82–94. doi: 10.1152/jn.1998.79.1.82. [DOI] [PubMed] [Google Scholar]
- 14.Colquhoun D, Hawkes AG. The principles of stochastic interpretation of ion-channel mechanisms. In: Sakmann B, Neher E, editors. Single-channel recording. Plenum; New York: 1983. pp. 135–174. [Google Scholar]
- 15.da Cruz e Silva EF, Fox CA, Ouimet CC, Gustafson E, Watson SJ, Greengard P. Differential expression of protein phosphatase 1 isoforms in mammalian brain. J Neurosci. 1995;15:3375–3389. doi: 10.1523/JNEUROSCI.15-05-03375.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Danoff SK, Ferris CD, Donath C, Fischer GA, Munemitsu S, Ullrich A, Snyder SH, Ross CA. Inositol 1,4,5-trisphosphate receptors: distinct neuronal and nonneuronal forms derived by alternative splicing differ in phosphorylation. Proc Natl Acad Sci USA. 1991;88:2951–2955. doi: 10.1073/pnas.88.7.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeSouza N, Reiken S, Ondrias K, Yang YM, Matkovich S, Marks AR. Protein kinase A and two phosphatases are components of the inositol 1,4,5-trisphosphate receptor macromolecular signaling complex. J Biol Chem. 2002;277:39397–39400. doi: 10.1074/jbc.M207059200. [DOI] [PubMed] [Google Scholar]
- 18.Fabiato A. Computer programs for calculating total from specified free or free from specified total ionic concentrations in aqueous solutions containing multiple metals and ligands. Methods Enzymol. 1988;157:378–417. doi: 10.1016/0076-6879(88)57093-3. [DOI] [PubMed] [Google Scholar]
- 19.Ferris CD, Huganir RL, Snyder SH. Calcium flux mediated by purified inositol 1,4,5-trisphosphate receptor in reconstituted lipid vesicles is allosterically regulated by adenine nucleotides. Proc Natl Acad Sci USA. 1990;87:2147–2151. doi: 10.1073/pnas.87.6.2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferris CD, Cameron AM, Bredt DS, Huganir RL, Snyder SS. Inositol 1,4,5-trisphosphate receptor is phosphorylated by cyclic AMP-dependent protein kinase at serines 1755 and 1589. Biochem Biophys Res Commun. 1991a;175:192–198. doi: 10.1016/s0006-291x(05)81219-7. [DOI] [PubMed] [Google Scholar]
- 21.Ferris CD, Huganir RL, Bredt DS, Cameron AM, Snyder SH. Inositol trisphosphate receptor-phosphorylation by protein kinase-C and calcium calmodulin-dependent protein kinases in reconstituted lipid vesicles. Proc Natl Acad Sci USA. 1991b;88:2232–2235. doi: 10.1073/pnas.88.6.2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finch EA, Turner TJ, Goldin SM. Calcium as a coagonist of inositol 1,4,5-trisphosphate-induced calcium release. Science. 1991;252:443–446. doi: 10.1126/science.2017683. [DOI] [PubMed] [Google Scholar]
- 23.Furuichi T, Kohda K, Miyawaki A, Mikoshiba K. Intracellular channels. Curr Opin Neurobiol. 1994;4:294–303. doi: 10.1016/0959-4388(94)90089-2. [DOI] [PubMed] [Google Scholar]
- 24.Greengard P, Allen PB, Nairn AC. Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron. 1999;23:435–447. doi: 10.1016/s0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- 25.Hain J, Onoue H, Mayrleitner M, Fleischer S, Schindler H. Phosphorylation modulates the function of the calcium release channel of sarcoplasmic reticulum from cardiac muscle. J Biol Chem. 1995;270:2074–2081. doi: 10.1074/jbc.270.5.2074. [DOI] [PubMed] [Google Scholar]
- 26.Hata Y, Butz S, Sudhof TC. CASK: a novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J Neurosci. 1996;16:2488–2494. doi: 10.1523/JNEUROSCI.16-08-02488.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haug LS, Jensen V, Hvalby O, Walaas SI, Ostvold AC. Phosphorylation of the inositol 1,4,5-trisphosphate receptor by cyclic nucleotide-dependent kinases in vitro and in rat cerebellar slices in situ. J Biol Chem. 1999;274:7467–7473. doi: 10.1074/jbc.274.11.7467. [DOI] [PubMed] [Google Scholar]
- 28.Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, Surmeier DJ. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[β]1-IP3-calcineurin-signaling cascade. J Neurosci. 2000;20:8987–8995. doi: 10.1523/JNEUROSCI.20-24-08987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horn R. Estimating the number of channels in patch recordings. Biophys J. 1991;60:433–439. doi: 10.1016/S0006-3495(91)82069-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iino M. Biphasic Ca2+ dependence of inositol 1,4,5-trisphosphate-induced Ca release in smooth muscle cells of the guinea pig taenia caeci. J Gen Physiol. 1990;95:1103–1122. doi: 10.1085/jgp.95.6.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iino M. Effects of adenine nucleotides on inositol 1,4,5-trisphosphate-induced calcium release in vascular smooth muscle cells. J Gen Physiol. 1991;98:681–698. doi: 10.1085/jgp.98.4.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones DH, Matus AI. Isolation of synaptic plasma membrane from brain by combined flotation-sedimentation density gradient centrifugation. Biochim Biophys Acta. 1974;356:276–287. doi: 10.1016/0005-2736(74)90268-5. [DOI] [PubMed] [Google Scholar]
- 33.Kaznacheyeva E, Lupu VD, Bezprozvanny I. Single-channel properties of inositol (1,4,5)-trisphosphate receptor heterologously expressed in HEK-293 cells. J Gen Physiol. 1998;111:847–856. doi: 10.1085/jgp.111.6.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindskog M, Svenningsson P, Fredholm BB, Greengard P, Fisone G. Activation of dopamine D2 receptors decreases DARPP-32 phosphorylation in striatonigral and striatopallidal projection neurons via different mechanisms. Neuroscience. 1999;88:1005–1008. doi: 10.1016/s0306-4522(98)00411-4. [DOI] [PubMed] [Google Scholar]
- 35.Lupu VD, Kaznacheyeva E, Krishna UM, Falck JR, Bezprozvanny I. Functional coupling of phosphatidylinositol 4,5-bisphosphate to inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1998;273:14067–14070. doi: 10.1074/jbc.273.23.14067. [DOI] [PubMed] [Google Scholar]
- 36.Maeda N, Niinobe M, Mikoshiba K. A cerebellar Purkinje cell marker P400 protein is an inositol 1,4,5-trisphosphate (InsP3) receptor protein. Purification and characterization of InsP3 receptor complex. EMBO J. 1990;9:61–67. doi: 10.1002/j.1460-2075.1990.tb08080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 38.Marx SO, Reiken S, Hisamatsu Y, Gaburjakova M, Gaburjakova J, Yang YM, Rosemblit N, Marks AR. Phosphorylation-dependent regulation of ryanodine receptors: a novel role for leucine/isoleucine zippers. J Cell Biol. 2001;153:699–708. doi: 10.1083/jcb.153.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsumoto M, Nakagawa T, Inoue T, Nagata E, Tanaka K, Takano H, Minowa O, Kuno J, Sakakibara S, Yamada M, Yoneshima H, Miyawaki A, Fukuuchi Y, Furuichi T, Okano H, Mikoshiba K, Noda T. Ataxia and epileptic seizures in mice lacking type 1 inositol 1,4,5-trisphosphate receptor. Nature. 1996;379:168–171. doi: 10.1038/379168a0. [DOI] [PubMed] [Google Scholar]
- 40.Maximov A, Sudhof TC, Bezprozvanny I. Association of neuronal calcium channels with modular adaptor proteins. J Biol Chem. 1999;274:24453–24456. doi: 10.1074/jbc.274.35.24453. [DOI] [PubMed] [Google Scholar]
- 41.Mignery GA, Newton CL, Archer BT, Sudhof TC. Structure and expression of the rat inositol 1,4,5-trisphosphate receptor. J Biol Chem. 1990;265:12679–12685. [PubMed] [Google Scholar]
- 42.Nakade S, Rhee SK, Hamanaka H, Mikoshiba K. Cyclic AMP-dependent phosphorylation of an immunoaffinity-purified homotetrameric inositol 1,4,5-trisphosphate receptor (type I) increases Ca2+ flux in reconstituted vesicles. J Biol Chem. 1994;269:6735–6742. [PubMed] [Google Scholar]
- 43.Nishi A, Snyder GL, Greengard P. Bidirectional regulation of DARPP-32 phosphorylation by dopamine. J Neurosci. 1997;17:8147–8155. doi: 10.1523/JNEUROSCI.17-21-08147.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishi A, Snyder GL, Nairn AC, Greengard P. Role of calcineurin and protein phosphatase-2A in the regulation of DARPP-32 dephosphorylation in neostriatal neurons. J Neurochem. 1999;72:2015–2021. doi: 10.1046/j.1471-4159.1999.0722015.x. [DOI] [PubMed] [Google Scholar]
- 45.Nosyreva E, Miyakawa T, Wang Z, Glouchankova L, Mizushima A, Iino M, Bezprozvanny I. The high affinity calcium-calmodulin-binding site does not play a role in modulation of type 1 inositol (1,4,5)-trisphosphate receptor function by calcium and calmodulin. Biochem J. 2002;365:659–667. doi: 10.1042/BJ20011789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ouimet CC, da Cruz e Silva EF, Greengard P. The α and γ1 isoforms of protein phosphatase 1 are highly and specifically concentrated in dendritic spines. Proc Natl Acad Sci USA. 1995;92:3396–3400. doi: 10.1073/pnas.92.8.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pieper AA, Brat DJ, O'Hearn E, Krug DK, Kaplin AI, Takahashi K, Greenberg JH, Ginty D, Molliver ME, Snyder SH. Differential neuronal localizations and dynamics of phosphorylated and unphosphorylated type 1 inositol 1,4,5-trisphosphate receptors. Neuroscience. 2001;102:433–444. doi: 10.1016/s0306-4522(00)00470-x. [DOI] [PubMed] [Google Scholar]
- 48.Sharp AH, Dawson TM, Ross CA, Fotuhi M, Mourey RJ, Snyder SH. Inositol 1,4,5-trisphosphate receptors: immunohistochemical localization to discrete areas of rat central nervous system. Neuroscience. 1993a;53:927–942. doi: 10.1016/0306-4522(93)90478-x. [DOI] [PubMed] [Google Scholar]
- 49.Sharp AH, McPherson PS, Dawson TM, Aoki C, Campbell KP, Snyder SH. Differential immunohistochemical localization of inositol 1,4,5-trisphosphate- and ryanodine-sensitive Ca2+ release channels in rat brain. J Neurosci. 1993b;13:3051–3063. doi: 10.1523/JNEUROSCI.13-07-03051.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sipma H, De Smet P, Sienaert I, Vanlingen S, Missiaen L, Parys JB, De Smedt H. Modulation of inositol 1,4,5-trisphosphate binding to the recombinant ligand-binding site of the type-1 inositol 1,4,5-trisphosphate receptor by Ca2+ and calmodulin. J Biol Chem. 1999;274:12157–12162. doi: 10.1074/jbc.274.17.12157. [DOI] [PubMed] [Google Scholar]
- 51.Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. 1998;18:10297–10303. doi: 10.1523/JNEUROSCI.18-24-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sudhof TC, Newton CL, Archer BT, Ushkaryov YA, Mignery GA. Structure of a novel InsP3 receptor. EMBO J. 1991;10:3199–3206. doi: 10.1002/j.1460-2075.1991.tb04882.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Supattapone S, Danoff SK, Theibert A, Joseph SK, Steiner J, Snyder SH. Cyclic AMP-dependent phosphorylation of a brain inositol trisphosphate receptor decreases its release of calcium. Proc Natl Acad Sci USA. 1988;85:8747–8750. doi: 10.1073/pnas.85.22.8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Surmeier DJ, Bargas J, Hemmings HC, Jr, Nairn AC, Greengard P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. 1995;14:385–397. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- 55.Surmeier DJ, Song WJ, Yan Z. Coordinated expression of dopamine receptors in neostriatal medium spiny neurons. J Neurosci. 1996;16:6579–6591. doi: 10.1523/JNEUROSCI.16-20-06579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Taylor CW, Genazzani AA, Morris SA. Expression of inositol trisphosphate receptors. Cell Calcium. 1999;26:237–251. doi: 10.1054/ceca.1999.0090. [DOI] [PubMed] [Google Scholar]
- 57.Thrower EC, Hagar RE, Ehrlich BE. Regulation of Ins(1,4,5)P3 receptor isoforms by endogenous modulators. Trends Pharmacol Sci. 2001;22:580–586. doi: 10.1016/s0165-6147(00)01809-5. [DOI] [PubMed] [Google Scholar]
- 58.Tu H, Miyakawa T, Wang Z, Glouchankova L, Iino M, Bezprozvanny I. Functional characterization of the type 1 inositol 1,4,5-trisphosphate receptor coupling domain SII(+/−) splice variants and the opisthotonos mutant form. Biophys J. 2002;82:1995–2004. doi: 10.1016/S0006-3495(02)75548-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tu JC, Xiao B, Yuan JP, Lanahan AA, Leoffert K, Li M, Linden DJ, Worley PF. Homer binds a novel proline-rich motif and links group 1 metabotropic glutamate receptors with IP3 receptors. Neuron. 1998;21:717–726. doi: 10.1016/s0896-6273(00)80589-9. [DOI] [PubMed] [Google Scholar]
- 60.Vallar L, Muca C, Magni M, Albert P, Bunzow J, Meldolesi J, Civelli O. Differential coupling of dopaminergic D2 receptors expressed in different cell types. Stimulation of phosphatidylinositol 4,5-bisphosphate hydrolysis in LtK-fibroblasts, hyperpolarization, and cytosolic-free Ca2+ concentration decrease in GH4C1 cells. J Biol Chem. 1990;265:10320–10326. [PubMed] [Google Scholar]
- 61.Volpe P, Alderson-Lang BH. Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release. II. Effect of cAMP-dependent protein kinase. Am J Physiol. 1990;258:C1086–C1091. doi: 10.1152/ajpcell.1990.258.6.C1086. [DOI] [PubMed] [Google Scholar]
- 62.Walaas SI, Nairn AC, Greengard P. PCPP-260, a Purkinje cell-specific cyclic AMP-regulated membrane phosphoprotein of Mr 260,000. J Neurosci. 1986;6:954–961. doi: 10.1523/JNEUROSCI.06-04-00954.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wojcikiewicz RJ, Luo SG. Phosphorylation of inositol 1,4,5-trisphosphate receptors by cAMP-dependent protein kinase. Type I, II, and III receptors are differentially susceptible to phosphorylation and are phosphorylated in intact cells. J Biol Chem. 1998;273:5670–5677. doi: 10.1074/jbc.273.10.5670. [DOI] [PubMed] [Google Scholar]
- 64.Yamada M, Miyawaki A, Saito K, Nakajima T, Yamamoto-Hino M, Ryo Y, Furuichi T, Mikoshiba K. The calmodulin-binding domain in the mouse type 1 inositol 1,4,5-trisphosphate receptor. Biochem J. 1995;308:83–88. doi: 10.1042/bj3080083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yan Z, Hsieh-Wilson L, Feng J, Tomizawa K, Allen PB, Fienberg AA, Nairn AC, Greengard P. Protein phosphatase 1 modulation of neostriatal AMPA channels: regulation by DARPP-32 and spinophilin. Nat Neurosci. 1999;2:13–17. doi: 10.1038/4516. [DOI] [PubMed] [Google Scholar]
- 66.Yang J, McBride S, Mak DO, Vardi N, Palczewski K, Haeseleer F, Foskett JK. Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca2+ release channels. Proc Natl Acad Sci USA. 2002;7:7–12. doi: 10.1073/pnas.102006299. [DOI] [PMC free article] [PubMed] [Google Scholar]