

Abstract
BETA2/NeuroD1 is a basic helix–loop–helix transcription factor that is expressed widely throughout the developing nervous system. Previous studies have shown that BETA2/NeuroD1 influences the fate of retinal cells in culture. To analyze the effect of BETA2/NeuroD1 on the structure and function of the retina, we examined a line of BETA2/NeuroD1 knock-out mice that survives until adulthood. At 2–3 months of age, homozygous null mice showed a 50% reduction in rod-driven electroretinograms (ERGs) and a 65% reduction in cone-driven ERGs. ERGs measured from knock-out mice that were >9 months of age were undetectable. At 2–3 months, the number of photoreceptors in the outer nuclear layer was reduced by 50%. In addition, electron microscopy showed that the surviving photoreceptors had shortened outer segments. The number of cones labeled by peanut agglutinin was decreased 50–60%. By 18 months, retinas from null mice were completely devoid of photoreceptors. There appeared to be few changes in the inner retina, although BETA2/NeuroD1 is expressed in this area. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining revealed a dramatic increase in cell death, peaking at approximately postnatal day 3 and continuing into adulthood. No defects in cell birth were detected using bromodeoxyuridine staining. Our results reveal that BETA2/NeuroD1 not only plays an important role in terminal differentiation of photoreceptors but also serves as a potential survival factor. Loss of BETA2/NeuroD1 results in an age-related degeneration of both rods and cones.
Keywords: retina, BETA2/NeuroD1, BETA2, NeuroD, NeuroD1, photoreceptor, degeneration, development, bHLH, electroretinogram, differentiation
Introduction
The retina is the first neural tissue that processes visual information; defects in retinal neurons, such as the photoreceptors, lead to several blinding eye disorders. Because of its accessibility, well studied anatomy and physiology, and known function, the retina serves as an excellent model to study how genetic defects might disturb the delicate balance between developmental, molecular, and physiological systems. Recently, transcription factors, such as cone–rod homebox (CRX) and neural retina leucine zipper (NRL), have been added to the growing list of genes mutated in retinal disease (Phelan and Bok, 2000). The basic helix–loop–helix (bHLH) transcription factors are involved in both cell determination and differentiation throughout the body, including the retina (Jan and Jan, 1993; Cepko, 1999). Because of their importance during photoreceptor development, bHLH factors are candidate genes for photoreceptor degeneration.
BETA2, a bHLH transcription factor, was cloned as an important regulator for insulin gene expression (Naya et al., 1995). It was also isolated from embryos and referred to as NeuroD because it could convert epidermal cell fate into neuronal cell fate (Lee et al., 1995). BETA2/NeuroD1 is expressed in the endocrine cells of the pancreas; its deletion results in neonatal diabetes secondary to islet defects (Naya et al., 1997). BETA2/NeuroD1 is also expressed widely throughout the developing CNS and the auditory and vestibular systems. Loss of BETA2/NeuroD1 leads to severe defects in these systems (Miyata et al., 1999; Schwab et al., 2000; Liu et al., 2000a,b; Kim et al., 2001). In this study, we investigated how BETA2/NeuroD1 regulates development and maintenance in the visual system and whether this transcription factor plays a role in retinal degeneration.
Studies from cultured retinal cells showed that loss of BETA2/NeuroD1 resulted in the death of a subset of rod photoreceptors, increased bipolar cell population, delayed amacrine differentiation, and increased gliogenesis (Morrow et al., 1999). However, these studies in cell culture systems could not address the question of how BETA2/NeuroD1 affects neuronal function, cell morphology, and synaptic connectivity in living intact retinas. It is also impossible to analyze light-evoked responses of photoreceptors in culture, because they do not form outer segments. Furthermore, because retinal explants do not survive long enough in a culture environment, it is difficult to examine whether BETA2/NeuroD1 regulates age-related degenerative processes in the retina. To test how BETA2/NeuroD1 affects the retina in an in vivo model, we examined the retinal function in BETA2/NeuroD knock-out mice bred onto a 129SvJ background (Liu et al., 2000b). These mice have a higher survival rate, allowing both electrophysiological and structural studies of the photoreceptors at prenatal, postnatal, and adult stages. The absence of visual evoked cortical potentials in these mice suggested a defect in the visual pathway (J. L. Noebels, personal communication). To test retinal function, we measured both rod- and cone-driven electroretinograms (ERGs). ERGs revealed a previously unnoted role of BETA2/NeuroD1 in cone survival. Morphological analysis using light and electron microscopy and immunohistochemistry revealed numerous signs of photoreceptor degeneration in the knock-out mice. Our evidence suggests that BETA2/NeuroD1 not only plays a role in photoreceptor differentiation but also is an important factor in the long-term maintenance and survival of photoreceptors.
Materials and Methods
Animals. BETA2/NeuroD1 null mice were generated in the 129SvJ background as described previously (Liu et al., 2000a,b). All animals were handled in accordance with the policies on the treatment of laboratory animals of Baylor College of Medicine.
Electroretinograms. Before testing, mice were allowed to adapt to the dark overnight. Under dim red light, mice were anesthetized with a solution of ketamine (95 mg/ml) and xylazine (5 mg/ml). The pupils were dilated with a single drop of 0.5% Mydryacil and 2.5% phenylephrine. A drop of 0.5% proparacaine hydrochloride was applied for corneal anesthesia. Mice were placed on a heating pad maintained at 39°C, inside a Ganzfeld dome coated with highly reflective white paint (Munsell Paint, New Windsor, NY). A small amount of 2.5% methylcellulose gel was applied to the eye, and a platinum electrode was placed in contact with the center of the cornea. Similar platinum reference and ground electrodes were placed in the forehead and tail, respectively. After placement in the dome, mice were allowed to remain in complete darkness for 5 min. Signals were amplified with a Grass P122 amplifier (bandpass 0.1 Hz-1000 Hz; Grass Instruments, West Warwick, RI). Data were acquired with a National Instruments Lab personal computer Data Acquisition board (sampling rate 10,000 Hz; National Instruments, Austin, TX). Traces were averaged and analyzed with custom software written in Matlab (Mathworks, Natick, MA).
Flashes were calibrated in a manner similar to that described byLyubarsky and Pugh (1996); they have been described in detail previously (Howes et al., 2002). Flashes for scotopic measurements were generated by a Grass PS-33+ photostimulator. Light was spectrally filtered with a 500 nm interference filter (Edmund Industrial Optics, Barrington, NJ). A series of metal plates with holes of varying diameters and glass neutral density filters were used to attenuate the flash. As the intensity of the flash increased, the number of trials was decreased and the time between each flash was increased. To remove oscillatory potentials before fitting, the scotopic b wave was digitally filtered using the filtfilt function in Matlab (low-pass filter; Fc = 60 Hz). The relationship between b-wave amplitude and flash intensity can be described by a saturating hyperbolic function (Naka–Rushton) with the form:b = (bmax,scot ×I)/(bmax,scot +I0.5), wherebmax,scot is the saturated scotopic b-wave amplitude andI0.5 is the intensity that provides half saturation. The baseline and peak of each filtered trace were measured, and the data for multiple intensities were fitted to the above equation.
For analysis of the a wave and cone function, 1500 W Novatron (Dallas, TX) xenon flash lamps provided intense illumination. To analyze the rod function, we used the following equation (Lamb–Pugh model) to fit a series of a waves at increasing intensities:
where a(t) is the a wave,amax is its saturating amplitude, φ is the number of photoisomerizations per rod produced by the flash,A is the amplification factor, andteff is a brief delay. This equation assumes normal outer segment length. Because histology indicated that the outer segments were shorter in null mice, we substituted φ =I × k into the equation and fit for the parameter k × A (Hood and Birch, 1994). I is the intensity of the flash in scotopic cd-sec/m2, and k is a variable with units φ/(scotopic cd-sec/m2). Cone-driven responses were recorded using the paired flash technique. The first flash drove the rods temporarily into saturation. The second flash, delivered 2.5 sec later, allowed the cones to recover but not the rods. To remove oscillatory potentials before measurement, the cone-driven b wave was digitally filtered using the filtfilt function in Matlab (low-pass filter; Fc = 30 Hz).
Light and electron microscopy. Mice were killed by asphyxiation with CO2. The eyes were enucleated and placed in 4% paraformaldehyde containing PBS. The cornea and lens were removed after 15 min, and the eyecup was allowed to fix overnight at 4°C. The eyecups were dehydrated, oriented, and embedded in JB-4 (Electron Microscopy Sciences, Fort Washington, PA) for cutting. Sections were cut until the optic nerve was visualized. Images were taken from a region just lateral to the optic nerve head. Sections were stained with hematoxylin and eosin. For electron microscopy (EM), the eyecups were fixed in 3% glutaraldehyde buffered to pH 7.2 with 0.01 m PIPES for at least 24 hr. The tissue was then rinsed in buffer and postfixed in PIPES-buffered osmium tetroxide, pH 7.2, for 1 hr at room temperature. The tissue was rinsed in several changes of distilled water and dehydrated through a graded series of ethanol solutions. The dehydrated tissue was incubated in two 1.5 hr changes of propylene oxide followed by a 1:1 mixture of propylene oxide and Spurr's resin overnight. The tissue was then incubated in pure resin for 1.5 hr and transferred to fresh resin in block molds. Sections (1 μm thick) were cut and stained with toluidine blue and basic fuchsin. The areas of interest were trimmed, and 60 nm sections were cut, mounted on copper grids, and stained with uranyl acetate and lead citrate. Specimens were examined in a Jeol (Peabody, MA) 100C Temscan electron microscope. Electron micrographs were made on Kodak (Rochester, NY) 4489 EM film.
Immunohistochemistry. Immunohistochemistry was performed using the indirect antibody method. For immunolabeling experiments, whole-mount retina or free-floating 40 mm vibratome sections were blocked with 3% normal donkey serum in PBS with 0.5% Triton X-100 and 0.1% sodium azide for 2 hr to overnight to reduce nonspecific labeling. The tissues were incubated in primary antibodies in the presence of 1% donkey serum and PBS with 0.5% Triton X-100 and 0.1% sodium azide for 1–5 d at 4°C. Tissues were then washed with PBS containing 0.5% Triton X-100 and 0.1% sodium azide; tissue immunoreactivity was visualized by incubating with fluorescent secondary antibodies overnight. After extensive rinsing, the tissue was mounted with Vectashield (Vector Laboratories, Burlingame, CA) and observed with a confocal laser scanning microscope (LSM) with a krypton–argon laser (LSM 510; Zeiss, Thornwood, NY). Images were acquired using 40× and 63× oil immersion objectives and processed with Zeiss LSM personal computer software and Adobe Photoshop 6.0 (Adobe Systems, San Jose, CA). For cell-counting studies, 352 × 352 μm fields were taken at evenly spaced intervals across the retina. Counters were blinded to the genotype of the mice. The antibodies used were GFAP (1:1000; Novus Biologicals, Littleton, CO), PKCα (1:200; Transduction Laboratories, Lexington, KY), Goα (1:1000;Chemicon, Temecula, CA), glutamine synthetase (1:1000; Transduction Laboratories), kinesin II (1:100, 30 min fixation only; Covance Research Products, Princeton, NJ), and peanut agglutinin (PNA) (1:20 dilution; Vector Laboratories).
Detection of apoptotic cells in the retina. Apoptotic cell death was detected by the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay (Gavrieli et al., 1992), with the following modifications. Briefly, the eyes were dissected out from the embryos [embryonic day 16.5 (E16.5) to E18.5], postnatal mice [postnatal day 0 (P0) to P5], and adult mice. Samples were fixed in 10% neutral buffered formalin (NBF) at 4°C for 16 hr. NBF-fixed eyes were embedded in paraffin and sectioned at 7 μm. Sections were deparaffinized and rehydrated in a series of graded ethanol solutions and incubated in PBS for 30 min. Sections were permeabilized with 20 μg/ml proteinase K in 10 mm Tris, pH 8.0, for 20 min at room temperature and were incubated in 3% H2O2 for 10 min at room temperature. After a brief rinse, slides were placed in the TUNEL buffer (in mm: 30 Tris, pH 7.2, 140 cacodalate, and 1 CoCl2) for 10 min and incubated for 1 hr in TUNEL buffer containing 1 nmol of biotin-16-deoxyUTP (Roche Products, Hertfordshire, UK) and 50 U/ml of terminal transferase (Invitrogen, San Diego, CA) at 37°C for 1 hr. Enzyme reactions were terminated by adding high-salt buffer (in mm: 300 NaCl and 30 sodium citrate). Biotin-labeled cells were visualized by the ABC detection system (Vector Laboratories) and counterstained with methyl-green.
5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside activity staining. Eyes were fixed in 2% PFA for 2 hr, rinsed three times with PBS, and incubated in 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) solution [2 mmMgCl2, 0.01% deoxycholate, 0.02% NP-40, 100 mm PBS, pH 7.4, 5 mmpotassium ferrocyanide (K4Fe(CN)6), 5 mm potassium ferricyanide (K3Fe(CN)6), and 1 mg/ml X-gal] for 8–16 hr. X-gal-stained eyes were postfixed with 2% PFA and embedded in paraffin. Sections were deparaffinized, rehydrated, and counterstained with nuclear fast red.
Results
Expression of BETA2/NeuroD1 in the retina
The LacZ reporter gene was introduced into the mouse BETA2/NeuroD1 locus to study the expression pattern of BETA2/NeuroD1 in the retina. We performed X-gal staining on developing and adult mice. The expression of BETA2/NeuroD1 was first detected at E10.5 in scattered cells in the central retina (Fig.1A,G). By E13, the pattern of expression expanded from the central to the peripheral retina. (Fig. 1B,H) At E16.5, the expression in the outer neuroblastic layer increased in intensity, and punctate staining was observed in the ganglion cell layer (GCL) (Fig.1C,I). BETA2/NeuroD1 expression peaked at approximately E18.5, with the heaviest staining present in the outer half of the retina and scattered staining present in the GCL (Fig.1D,J). There appear to be two populations of cells labeled in the outer neuroblastic layer. The outermost cells likely represent developing photoreceptors, whereas the labeled cells at the inner margin could be amacrine cells. At P3, most of the BETA2/NeuroD1 expression was restricted to the outer nuclear layer (ONL), with slight expression in the developing region of the inner nuclear layer (INL) (Fig. 1E,K). Moderate levels of BETA2/NeuroD1 expression in the ONL and INL persisted throughout early postnatal development and remained at a stable level in the adult retina (Fig. 1F,L). In addition, expression was observed in a subset of cells (possibly displaced amacrine cells) in the GCL. The pattern of expression for BETA2/NeuroD1 in our line of mice was similar to that reported previously (Brown et al., 1998; Morrow et al., 1999). We did not observe a central-to-peripheral gradient of expression in the adult mouse. This minor discrepancy could have arisen because of the differences between the backgrounds of the mice used (Miyata et al., 1999; Liu et al., 2000b).
Fig. 1.
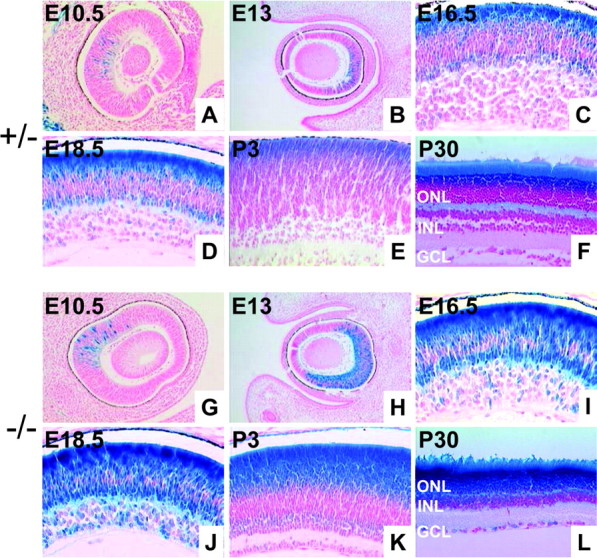
Expression of BETA2/NeuroD1 in the developing and adult retina. A–F, X-gal staining from +/− mice at E10.5, E13, E16.5, E18.5, P3, and P30. G–L, X-gal staining from −/− mice at the same time points. The pattern of expression is similar for +/− and −/− mice all time points. However, null mice display a higher level of expression because of the presence of two copies of the β-gal gene. At earlier time points (A, B and G, H) BETA2/NeuroD1 is expressed central to the peripheral pattern in both the inner and outer retina. High levels of expression are seen in the outer layers of the retina, and punctate staining is seen in the ganglion cell layer.
Adult BETA2/NeuroD1 null mice have functional retinal defects in both rod- and cone-driven systems
The ERG is an effective tool for analyzing retinal function. Under scotopic conditions, the ERG detects responses from rod-driven circuitry, whereas under photopic conditions it detects responses from cone-driven circuitry. We measured scotopic b waves, a waves, and cone-driven b waves from both young (1- to 3-month-old) and old (10- to 18-month-old) mice. The scotopic b wave is the extracellular field potential that primarily arises from rod bipolar cells in response to dim flashes of light (Pugh et al., 1998). Figure2A–D shows scotopic b-wave responses to increasing intensities of flashed light. The relationship between scotopic b-wave amplitude and intensity can be modeled using a hyperbolic saturation function (see Materials and Methods). This model yields two parameters,bmax,scot andI0.5, representing the maximum b-wave amplitude and the intensity that provides half saturation. In wild-type mice bmax,scot measured ∼650 μV and I0.5 = 16.3 scotopic cd-sec/m2 (Table1). Heterozygous mice were indistinguishable from wild-type mice for this and other tests of visual function (Fig. 2B,F,J,N). Homozygous null mice had a severe decrease in the scotopic b wave, withbmax,scot measuring on average 300 μV and I0.5 = 120.3 scotopic cd-sec/m2. To determine whether the decrease in the ERG worsened with age, we tested several mice that were >9 months of age (Fig. 2D,H,L,P). Neither rod-driven nor cone-driven responses were detectable from null mice at these ages. ERGs from corresponding control mice were only slightly decreased, consistent with normal aging (data not shown). To determine the earliest point at which decreased function was detectable, we measured ERGs from mice at P21. Mutant mice at this age already showed a significant decrease in the ERG (data not shown). Recordings from younger mutant mice were not possible because of their inability to survive the anesthesia.
Fig. 2.

ERGs from BETA2/NeuroD1 mice. Representative ERGs from 1- to 3-month-old (first three columns) and 10-month-old (fourth column) BETA2/NeuroD1 mice.A–D, Measurement of scotopic b waves. Traces were obtained using a series of 500 nm flashes with intensities ranging from −3.86 to −1.76 log scotopic cd-sec/m2 (estimated to produce between 0.13 and 16.1 photoisomerizations per rod in photoreceptors with normal outer segments). Each trace represents the average of at least five flashes. E–H, ERG response to an intense flash (2.97 log scotopic cd-sec/m2 or an estimated 800,000 photoisomerizations per rod). The initial negative deflection is the saturated a wave. I–L, Close-up of multiple a-wave responses to flashes varying in intensity from −0.51 to 2.97 log scotopic cd-sec/m2. The traces have been fitted using the Lamb–Pugh model (dotted lines) as described in Materials and Methods. M–P, Cone-isolated ERGs using the double-flash method. Only the response to the second flash is displayed.
Table 1.
Summary of ERG parameters
| Genotype | bmax, scot(μV) | I0.5 (scotopic cd-sec/m2) | amax (μV) | k × A (sec−2 (scotopic cd-sec m−2)−1) | bmax, phot(μV) |
|---|---|---|---|---|---|
| +/+ | 650 ± 18 (n = 4) | (16.3 ± 2.3) × 10−4 | 580 ± 130 | 3340 ± 950 | 180 ± 30 (n = 5) |
| +/− | 675 ± 11 (n = 5) | (17.6 ± 1.3) × 10−4 | 620 ± 60 | 4040 ± 1030 | 200 ± 70 (n = 4) |
| −/− | 335 ± 8 (n = 5) | (120.3 ± 10.5) × 10−4 | 285 ± 60 | 3100 ± 375 | 65 ± 25 (n = 5) |
| −/−, 9 months | 0 (n = 3) | 0 | 0 |
The first column indicates the genotype of the BETA2/NeuroD mice. The parameter bmax,scot represents the saturating amplitude of the scotopic b wave, andI0.5 is the intensity, measured as scotopic cd-sec/m2, which produces one-half ofbmax,scot (see Materials and Methods). The value shown in the table represents the best fit, and the second number is the error of the fit. For the remaining parameters, the first value represents the mean and the second value represents the SE. The parameter amax is the amplitude of the a wave in response to an intense, saturating flash measured to produce 2.97 log scotopic cd-sec/m2. The parameter k × Awas obtained by fitting the a wave using the model developed byLamb and Pugh (1992) and modified by Hood and Birch (1994). Isolated cone responses were measured using the paired-flash technique (see experimental procedures in Lyubarsky et al., 1999). bmax,phot measures the peak amplitude of the filtered b wave in response to a flash that produces 143 photopic cd-sec/m2.
To characterize rod photoreceptor function more directly, we measured the ERG a wave, which in the mouse arises almost exclusively from the rod photoreceptors (Pugh et al., 1998). Figure 2E–Hshows the response to an intense flash, which saturated the rod photoreceptors. Wild-type mice produced a saturated a wave, oramax, measuring ∼600 μV (Table 1). In null mice, the saturated a wave was reduced by almost half, to 300 μV. To analyze the amplification of the rod transduction cascade, a series of a waves to different intensities were fitted (Lamb and Pugh, 1992; Hood and Birch, 1994), and the parameter K ×A was derived (Fig. 2I–L, Table 1). There was no significant difference between wild-type and heterozygous mice. In null mice, the amplification constant was reduced, but the difference was not significant for the number of mice tested.
To determine whether degeneration extended to cones, we used the double-flash technique, which isolated responses from cone photoreceptors (Lyubarsky et al., 1999). Figure 2M–Pshows the ERG response to a white flash, which stimulated both S- and M-cones. The cone-driven b wave in wild-type mice measures ∼200 μV (Table 1). This signal is reduced almost 65% in the null mice. The cone-driven a wave in mice is small, because cones make up only ∼3% of the total photoreceptor population (Carter-Dawson and LaVail, 1979). Wild-type mice had a cone a wave measuring ∼30 μV, whereas the cone a wave in null mice was undetectable. The reduced cone-driven a and b waves provide evidence for cone dysfunction.
Structural defects in BETA2/NeuroD1 retinas
To elucidate the cause of decreased ERGs in null mice, we examined retinal structure using light microscopy and EM and immunohistochemistry. Figure3A,B shows light micrographs taken from BETA2/NeuroD1 control and null mice at 2 months of age. Overall, the retina of null mice was decreased in thickness. The ONL, which contains the photoreceptors, normally measures 10–12 cells thick (Carter-Dawson and LaVail, 1979). In null mice, the thickness of the ONL was reduced to five to six cells. There also appeared to be a thinning of the outer plexiform layer (OPL) and a shortening of the outer segments. There was no change in the thickness or appearance of the INL, inner plexiform layer (IPL), or GCL in null mice. Light microscopy of 18-month-old mice revealed that null mice are completely devoid of photoreceptors (Fig. 3C). To study the structural properties of the photoreceptor outer segment more closely, we used transmission electron microscopy (TEM). Figure 3D,Eshows TEM images of the outer and inner segments from control and null mice. Both sections were taken from similar regions in the posterior pole of the retina. These images reveal that the length of both the outer and inner segments was decreased in null mice.
Fig. 3.
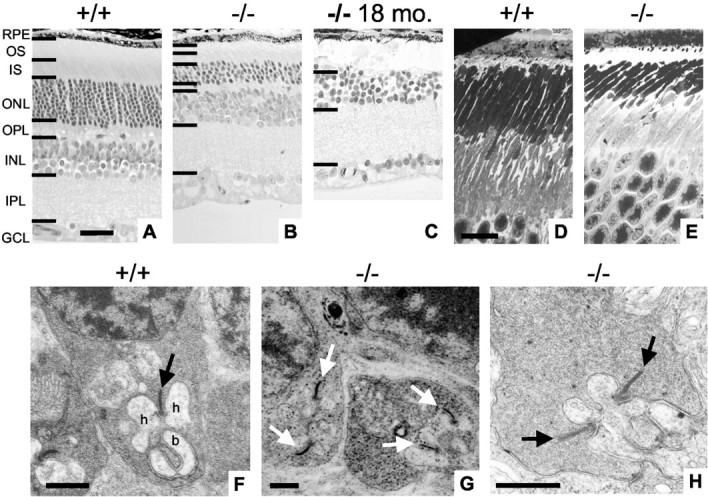
Light microscopy and EM of the retina in BETA2/NeuroD1 mice. Light microscopy of retinal slices shows an area taken from the posterior retina of 2-month-old +/+ mice (A), 2-month-old −/− mice (B), and 18-month-old −/− mice (C). In 2-month-old null mice, the ONL thickness is reduced to four to five cells. Null mice also demonstrate decreased thickness of the outer segments (OS), inner segments (IS), and OPL. In 18-month-old −/− mice, the ONL has completely degenerated. Scale bar, D (forA–C), 20 μm. EM for 2-month-old +/+ (D) and −/− (E) mice focusing on the OS is shown. In null mice, both outer and inner segments are shorter. Scale bar, F (for Dand E), 10 μm. F, EM of a rod synapse in a +/+ mouse. Note the presence of a synaptic ribbon (arrow), two postsynaptic horizontal cell processes (h), and a bipolar cell dendrite (b) forming a classic synaptic triad.G, OPL taken from a −/− mouse. Note the presence of multiple ribbons in rod terminals (arrows).H, Higher magnification taken from a −/− mouse showing multiple synaptic ribbons. RPE, Retinal pigment epithelial. Scale bar, 0.5 μm forF–H.
The decrease in photoreceptor population is not limited to the rods. Figure 4A,B shows flat-mount retinas stained with fluorescent PNA. Figure4C–E shows retinal slices doubled-stained with PNA (green) and kinesin (red). PNA stains the cone photoreceptor-associated matrix and the synaptic terminals (Fig. 4C–E, arrowheads) (Blanks and Johnson, 1983). The number of labeled cone matrices and synapses were decreased ∼60% in the null mice. We examined the thinning of the OPL using immunohistochemistry and EM. Figure 4F,G shows flat-mount retinas stained with anti-kinesin II, which labels synaptic ribbons (Muresan et al., 1999). There was a clear decrease in the total number of synaptic ribbons in null mice (Fig. 4C–E,arrows, F,G). To determine whether the remaining synapses had normal morphology, we imaged them at the EM level. Figure3F demonstrates a classic synaptic triad in a wild-type mouse composed of a single presynaptic ribbon complex, two horizontal cell processes, and one to two bipolar cell dendrites. In null mice, it is still possible to find rod synapses that contain both presynaptic and postsynaptic elements. However, many of these synapses are enlarged and have multiple ribbons (Fig. 3G,H). These morphological changes have been reported in several different models of retinal degeneration (Jansen et al., 1997). In addition, immunohistochemical labeling showed retracted bipolar cell processes and extension of horizontal cell processes into the ONL (data not shown). Similar changes in the INL have been reported in the retinal degeneration mouse (Strettoi and Pignatelli, 2000). The changes found in the OPL and INL of BETA2/NeuroD null are mice are consistent with other models of retinal degeneration.
Fig. 4.
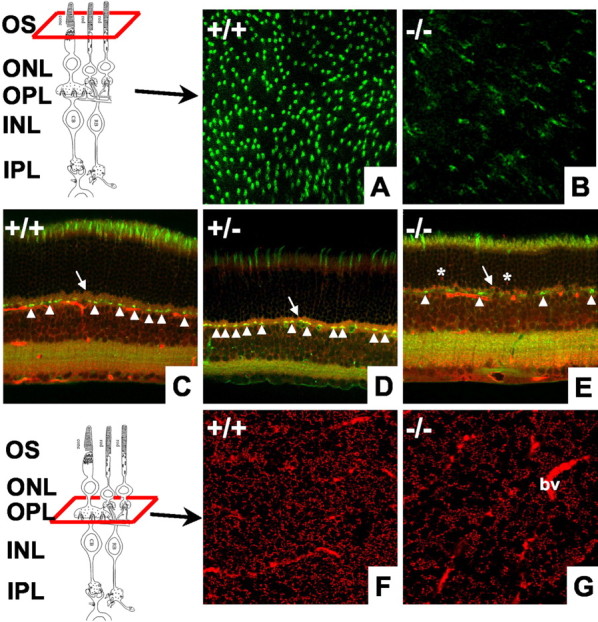
Analysis of cone photoreceptors and synaptic ribbons. A, B, Flat-mount retinas from +/+ and −/− mice stained with PNA to label cone photoreceptors. Labeled cones are reduced ∼60% in null mice. C–E, Retinal slices from +/+, +/−, and −/− mice double labeled with PNA (green) and anti-kinesin II (red). Null mice have decreased numbers of cone synapses (arrowheads), decreased thickness of anti-kinesin-labeled ribbons in the OPL (arrow), and aberrant ribbons present in the ONL (asterisks).F, G, Stacked images from flat-mount retinas taken at the level of the OPL and stained with anti-kinesin II. Labeled synaptic ribbons are significantly decreased in null mice. Some blood vessels (bv) are also labeled. Drawings of the retina are modified from Dowling and Boycott (1966). CB, Cone cell body; RB, rod cell body.
Putative photoreceptors undergo apoptosis in null mice
Previous work has shown that BETA2/NeuroD1 is involved in influencing the balance of cell populations in the retina (Morrow et al., 1999). To determine whether the loss of photoreceptors in our mice resulted from decreased cell birth or increased cell death, we used bromodeoxyuridine (BrdU) and TUNEL staining. There were no differences observed in BrdU staining between wild-type and null mice (data not shown). Figure 5 demonstrates the results for TUNEL staining from ages E16.5 to P30. In control mice, scattered TUNEL-positive cells are observed between E16.5 and P3 (Fig.5A–C). After P3, few if any TUNEL-positive cells are observed in the wild-type mice (Fig. 5D–F). At E18, null mice show a higher proportion of TUNEL-positive cells located primarily in the outer half of the neuroblastic layer (Fig.5H). Cell death peaks at P3 and begins to subside by P5 (Fig. 5I,J). Higher magnification revealed that most of the TUNEL-positive cells had no cytoplasm, were rounded or oval in shape, and had a condensed nucleus (data not shown). In addition, immunohistochemistry from null P3 mice revealed an increase in staining for caspase-3, a marker of apoptosis (data not shown). By P10, once the retina has laminated, one can observe that most of the cell death is present in the ONL (Fig. 5K). Cell death continues in adult null mice, as evidenced by occasional TUNEL-positive cells in the ONL (Fig. 5L).
Fig. 5.
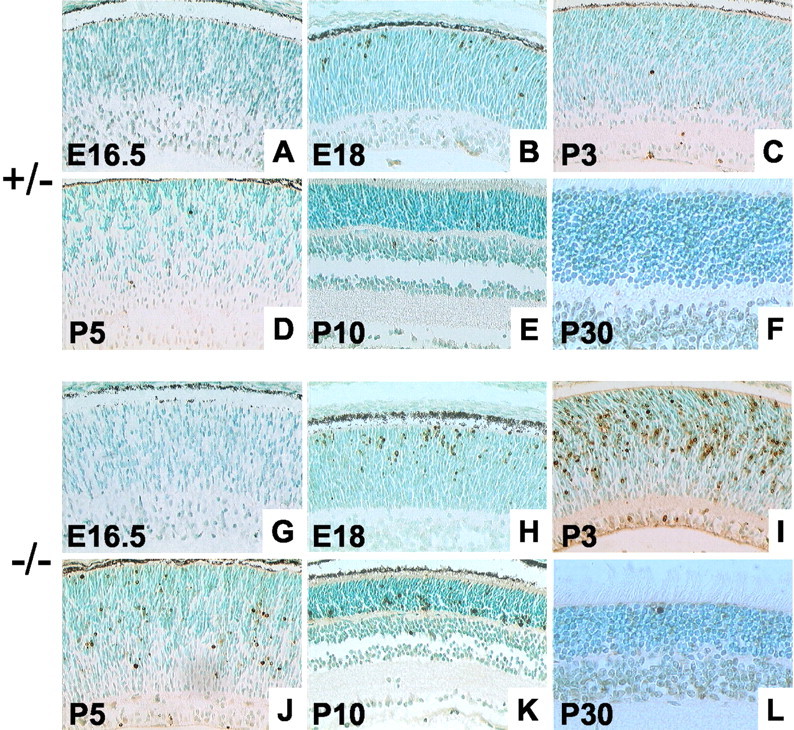
Detection of apoptosis in BETA2/NeuroD1 mice.A–F, TUNEL staining from (+/−) mice at E16.5, E18, P3, P5, P10, and P30. G–L, TUNEL staining from −/− mice at the same time points. In the control mice, some TUNEL-positive cells are observed from E18 to P3. In adult mice, few if any TUNEL-positive cells are observed. In contrast, null mice show a higher number of TUNEL-positive cells at E18, and the count increases at P3. The number of dying cells seems to decrease after P3, but cell death is present even in the adult retina. Most of the cell death seems to be occurring in the ONL, the location of developing photoreceptors.
Effect of BETA2/NeuroD on inner retinal cells
Previous studies have reported that BETA2/NeuroD1 plays a role in modulating the ratio of glial to neural cells. Specifically, a loss of BETA2/NeuroD1 leads to a threefold to fourfold increase in Müller cells and a twofold to threefold increase in bipolar cells (Morrow et al., 1999). However, a second study, also performed using retinal cultures, reported only a slight increase in Müller glial cells (Inoue et al., 2002). We observed no alteration in the density of glial cells stained by glutamine synthetase, suggesting little change in the number of Müller cells in null mice (Fig.6A–C). However, we did observe a remarkable increase in GFAP staining in these cells. Figure6D–F shows that GFAP is normally found only in the foot processes of the Müller cells in wild-type and heterozygous mice. GFAP expression is markedly increased in the retinas from null mice. Reactive gliosis, such as that seen here, is a generalized response to photoreceptor degeneration and is characterized by the upregulation of GFAP in the Müller cells (Tuccari et al., 1986;Semple-Rowland, 1991; de Raad et al., 1996).
Fig. 6.

Gliosis in BETA2/NeuroD1 null mice.A–C, Immunohistochemical staining with anti-glutamine synthetase. There is little difference in the number Müller glia in the different lines of mice. D–F, Immunohistochemical staining with anti-GFAP in +/+, +/−, and −/− mice. In wild-type and heterozygous mice, anti-GFAP stains only astrocytic processes near the inner limiting membrane. In null mice, anti-GFAP staining demonstrates a reactive gliosis in Müller glia.
Light microscopy does not reveal a change in the thickness of the INL. We used immunohistochemistry to more closely analyze the cell populations in the INL (Dhingra et al., 2000). Figure7A–C shows retinal slices stained with anti-Goα antibody, which labeled both rod and ON-cone bipolar cells. There was little difference between control mice and null mice. Figures 7D–F demonstrates labeling with anti-PKCα antibody, which labeled the rod bipolar cells (Greferath et al., 1990). Once again, little difference is seen in the number of cells between wild-type and null mice. To assess the bipolar cell population in a more quantitative way, we counted the number of PKC-labeled bipolar cell processes using flat mounts (Fig. 7G,H). There was no statistically significant difference between control and null mice. It is possible that the discrepancy between our model and previous work arises because of the difference in the genetic backgrounds of mice used (129SvJ in our experiments). Another possibility might result from the different experimental conditions: in vitro retinal explants (Morrow et al., 1999; Inoue et al., 2002) versus in vivo retinas from postnatal mice (this study).
Fig. 7.
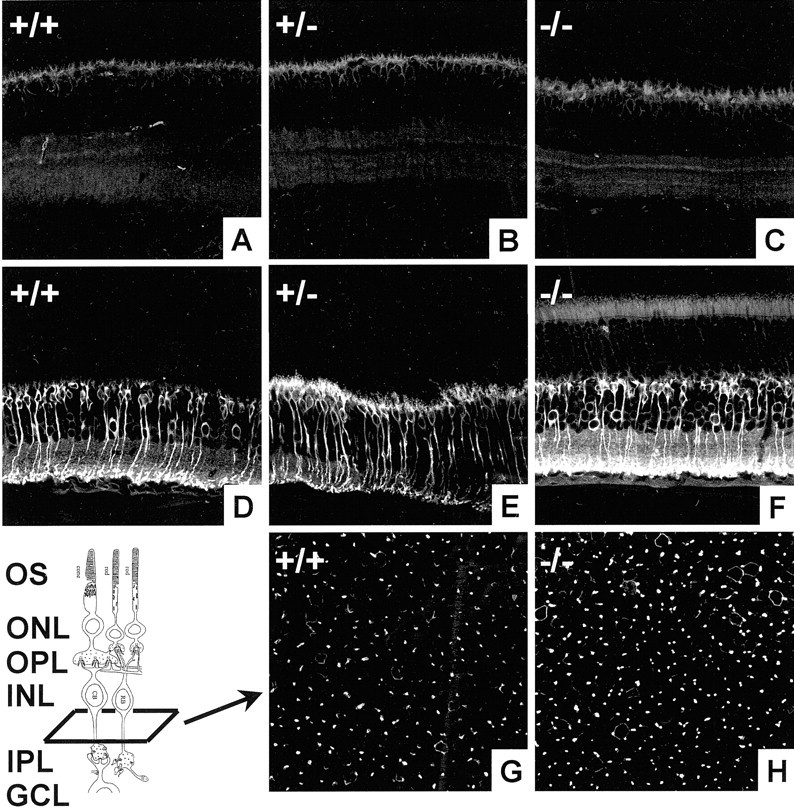
Analysis of bipolar cells in BETA2/NeuroD mice.A–C, Immunohistochemical staining using anti-Goα in +/+, +/−, and −/− mice. Anti-Goα stains rod bipolar and ON-cone bipolar cells. There is little difference between wild-type, heterozygous, and null mice. D–F, Immunohistochemical staining using anti-PKC, which labels rod bipolar cells. G, H, Cross sections taken from anti-PKC-stained flat-mount retinas. There is no significant difference between the number of labeled cells. OS, Outer segment.
Discussion
In this study, we have examined the functional role of BETA2/NeuroD1 using an in vivo model. BETA2/NeuroD1 is essential for photoreceptor survival; its loss leads to severe functional defects of both rod- and cone-mediated vision. BETA2/NeuroD1 is required for the maintenance of photoreceptor cells by preventing apoptosis. Photoreceptor degeneration occurs in an age-dependent manner only when both copies of the gene are missing. Our data suggest that BETA2/NeuroD1 null mice can be used as an excellent animal model for photoreceptor degeneration mediated by transcription factor loss.
Both functional and histological evidence demonstrate that loss of BETA2/NeuroD leads to photoreceptor degeneration
ERGs demonstrated several defects in null mice. First, at 2–3 months of age there was a 50% reduction in both the scotopic b wave and the rod a wave. The scotopic b wave arises primarily from the rod bipolar cells (Pugh et al., 1998). Thus, a decrease could arise from a defect in the bipolar cells, the input from the rod photoreceptors, or the synaptic connections between the two. There was no evidence for a decrease in the number of bipolar cells in null mice. Although the total number of synaptic ribbons was decreased in null mice, this seems to be secondary to cell death of the photoreceptors. The OPL is disorganized and shows signs of synaptic remodeling, but the remaining photoreceptors have synaptic terminals with ribbons and postsynaptic processes. Although, we cannot rule out synaptic defects, the most likely cause of the diminished b wave is decreased input to the bipolar cell attributable to the loss of photoreceptors. Analysis of the saturated rod a wave lends evidence to support this hypothesis. The amplitude of the saturated a wave is proportional to the number of photoreceptors (ONL count) times the length of the outer segment (Machida et al., 2000). Light microscopy demonstrated that the ONL count in 2- to 3-month-old null mice was reduced to five to six cells per column, compared with 10–12 cells per column in control mice. In addition, EM showed that the lengths of the outer segments in null mice were shortened. Together, these two findings can account for the 50% reduction measured by the ERGs.Morrow et al. (1999) found no difference in the number of cone photoreceptors at P0. Our data indicate that the cone pathway also appears to degenerate in null mice. In 3-month-old null mice, the number of cone sheaths labeled by PNA was diminished by 50–65%; cone-driven ERGs were reduced almost 65%.
The exact cause of cell death in null mice is not clear. TUNEL staining suggested two phases of cell death: an early phase peaking at approximately P3, when the photoreceptors started dying, and a late phase that continues through adulthood and was characterized by a slower rate of cell death. The early phase of cell death could result from a failure of these cells to differentiate properly. The loss of BETA2/NeuroD1 has been linked to the downregulation of developmental markers and defects in differentiation. For example, BETA2/NeuroD1 is necessary for the expression of insulin in β cells of the pancreas and proper formation of the islets (Mutoh et al., 1997; Qiu et al., 1998). In the hippocampus, the loss of BETA2/NeuroD1 is associated with the loss of the marker Prox-1 and deformations in the dentate gyrus (Liu et al., 2000a; Schwab et al., 2000). In the auditory system, the loss of BETA2/NeuroD1 resulted in the downregulation of tyrosine receptor kinase B and C in the cochlear–vestibular ganglion (Liu et al., 2000b; Kim et al., 2001). BETA2/NeuroD1 may play a similar role in the retina by inducing and maintaining the expression of photoreceptor-specific genes. Mutations in photoreceptor-specific transcription factors such as CRX and NRL have been associated with both defects in photoreceptor development and subsequent degeneration (Freund et al., 1997; Sohocki et al., 1998; Bessant et al., 1999). CRX and NRL have been shown to synergistically regulate rhodopsin expression by binding to sites in the rhodopsin proximal promoter region (RPPR) (Mitton et al., 2000). In the rat, BETA2/NeuroD1 can bind the Eopsin-1 element in the RPPR, and overexpression of BETA2/NeuroD1 increases the proportion of opsin-positive cells (Ahmad et al., 1998). A similar relationship likely exists in the mouse, because the rat BETA2/NeuroD1 sequence is identical to the mouse sequence at the amino acid level (Acharya et al., 1997). The Eopsin-1 element is distinct from the binding sites for CRX and NRL, suggesting that BETA2/NeuroD1 may represent an additional mediator of rhodopsin expression. BETA2/NeuroD1 null mice show reduced expression of rhodopsin at P3, which correlates with the peak time of cell death (data not shown). Approximately one-half of the photoreceptors survive this early round of death and are able to form shortened outer segments. This decrease in length may be secondary to a reduced expression of rhodopsin in these cells. Western blots from 2- to 3-month-old mice show that rhodopsin levels are decreased even after cell death has been accounted for (data not shown). Continued degeneration of the photoreceptors may result from decreased rhodopsin expression.
Although rhodopsin expression was decreased in null mice, the ratio of other phototransduction proteins is likely normal. An analysis of the rod a wave using the Lamb–Pugh model to calculate the amplification constant of phototransduction showed that there was no significant difference in null mice. This parameter reflects the gain of the phototransduction cascade and would be decreased if transducin or phosphodiesterase levels were down. There have yet to be any reports of BETA2/NeuroD1 regulating the cone-specific genes in the mouse. However, in the chicken, the forced expression of BETA2/NeuroD1 has been shown to induce the expression of cone pigment genes in cultured retinal pigment epithelial cells (Yan and Wang, 2000a,b). Cone degeneration may be the result of the downregulation of cone-specific genes. Alternatively, cone death may be secondary to rod death, as is seen in other degenerative diseases, such as retinitis pigmentosa (Phelan and Bok, 2000).
At the time of the writing of this paper, no visual disease in humans has been linked to the BETA2/NeuroD1 locus. BETA2/NeuroD1 maps close to, but does not overlap with, the locus for Bardet–Biedl syndrome (BBS) type 5. Knock-out mice manifest two primary symptoms of BBS (retinal degeneration and learning disabilities) and two secondary features (ataxia and diabetes mellitus) (Katsanis et al., 2001). However, these mice do not exhibit obesity, dysmorphic extremities, or renal defects and thus do not pass the minimum criteria for the diagnosis of BBS. Heterozygous mutations in BETA2/NeuroD1 are associated with the development of both type 1 and type 2 diabetes mellitus in humans (Iwata et al., 1999; Malecki et al., 1999). Diabetic patients often have visual loss secondary to complications of their disease. Although visual function was not analyzed in these patients, our data suggest that photoreceptor degeneration such as that seen in our model would result only from the loss of both copies of BETA2/NeuroD1. No homozygous mutations in BETA2/NeuroD1 have been reported in humans. Because of the severity of the phenotype seen in mice, it is possible that a homozygous mutation would be lethal in humans. It remains possible that heterozygous mutations in BETA2/NeuroD1 will be a risk factor for retinal degeneration, especially the mutations generating nonfunctional proteins that interfere with the function of the BETA2/NeuroD1 from the normal copy.
Previous work has proposed that BETA2/NeuroD1 plays an important role in the terminal differentiation of cells in the retina (Morrow et al., 1999; Inoue et al., 2002). Our results support this hypothesis and also demonstrate that BETA2/NeuroD1 is necessary for photoreceptor survival and maintenance. Additional studies will be needed to elucidate the genes that are regulated by BETA2/NeuroD1. In conclusion, our results demonstrate that BETA2/NeuroD1 is necessary for proper photoreceptor function and should be included in future screening for mutations of retinal diseases.
M.E.P. and J.-H.C. contributed equally to this work.
This work was supported by National Institutes of Health (NIH) Grants EY04446 and HD17379, by NIH Vision Core Grant EY02520, by the Retina Research Foundation of Houston, by Research to Prevent Blindness, Inc., and by International Retina Research Foundation, Inc. M.E.P. was supported by Baylor Research Advocates for Student Scientists and Medical Scientist Training Program Training Grant GM07330. We thank Drs. Wolfgang Baehr, Roy Jacoby, Kartik Pappu, and Mayra Mori for critical reading of this manuscript and Susan Robbins, Khoi Chu, and Emily Hui for technical assistance. We thank Min Liu and Sophia Tsai for helpful discussions.
Correspondence should be addressed to either of the following: Dr. Ming-Jer Tsai, Department of Molecular and Cellular Biology, Baylor College of Medicine, One Baylor Plaza, M734, Houston, TX 77030, E-mail:mtsai@bcm.tmc.edu; or Dr. Samuel M. Wu, Department of Ophthalmology, Baylor College of Medicine, One Baylor Plaza, NC416, Houston, TX 77030, E-mail: swu@bcm.tmc.edu.
References
- 1.Acharya HR, Dooley CM, Thoreson WB, Ahmad I. cDNA cloning and expression analysis of NeuroD mRNA in human retina. Biochem Biophys Res Commun. 1997;233:459–463. doi: 10.1006/bbrc.1997.6483. [DOI] [PubMed] [Google Scholar]
- 2.Ahmad I, Acharya HR, Rogers JA, Shibata A, Smithgall TE, Dooley CM. The role of NeuroD as a differentiation factor in the mammalian retina. J Mol Neurosci. 1998;11:165–178. doi: 10.1385/JMN:11:2:165. [DOI] [PubMed] [Google Scholar]
- 3.Bessant DA, Payne AM, Mitton KP, Wang QL, Swain PK, Plant C, Bird AC, Zack DJ, Swaroop A, Bhattacharya SS. A mutation in NRL is associated with autosomal dominant retinitis pigmentosa. Nat Genet. 1999;21:355–356. doi: 10.1038/7678. [DOI] [PubMed] [Google Scholar]
- 4.Blanks JC, Johnson LV. Selective lectin binding of the developing mouse retina. J Comp Neurol. 1983;221:31–41. doi: 10.1002/cne.902210103. [DOI] [PubMed] [Google Scholar]
- 5.Brown NL, Kanekar S, Vetter ML, Tucker PK, Gemza DL, Glaser T. Math5 encodes a murine basic helix-loop-helix transcription factor expressed during early stages of retinal neurogenesis. Development. 1998;125:4821–4833. doi: 10.1242/dev.125.23.4821. [DOI] [PubMed] [Google Scholar]
- 6.Carter-Dawson LD, LaVail MM. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J Comp Neurol. 1979;188:245–262. doi: 10.1002/cne.901880204. [DOI] [PubMed] [Google Scholar]
- 7.Cepko CL. The roles of intrinsic and extrinsic cues and bHLH genes in the determination of retinal cell fates. Curr Opin Neurobiol. 1999;9:37–46. doi: 10.1016/s0959-4388(99)80005-1. [DOI] [PubMed] [Google Scholar]
- 8.de Raad S, Szczesny PJ, Munz K, Reme CE. Light damage in the rat retina: glial fibrillary acidic protein accumulates in Muller cells in correlation with photoreceptor damage. Ophthal Res. 1996;28:99–107. doi: 10.1159/000267881. [DOI] [PubMed] [Google Scholar]
- 9.Dhingra A, Lyubarsky A, Jiang M, Pugh EN, Birnbaumer L, Sterling P, Vardi N. The light response of ON bipolar neurons requires G[alpha]o. J Neurosci. 2000;20:9053–9058. doi: 10.1523/JNEUROSCI.20-24-09053.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dowling JE, Boycott BB. Organization of the primate retina: electron microscopy. Proc R Soc Lond B Biol Sci. 1966;166:80–111. doi: 10.1098/rspb.1966.0086. [DOI] [PubMed] [Google Scholar]
- 11.Freund CL, Gregory-Evans CY, Furukawa T, Papaioannou M, Looser J, Ploder L, Bellingham J, Ng D, Herbrick JA, Duncan A, Scherer SW, Tsui LC, Loutradis-Anagnostou A, Jacobson SG, Cepko CL, Bhattacharya SS, McInnes RR. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell. 1997;91:543–553. doi: 10.1016/s0092-8674(00)80440-7. [DOI] [PubMed] [Google Scholar]
- 12.Gavrieli Y, Sherman Y, Ben Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greferath U, Grunert U, Wassle H. Rod bipolar cells in the mammalian retina show protein kinase C-like immunoreactivity. J Comp Neurol. 1990;301:433–442. doi: 10.1002/cne.903010308. [DOI] [PubMed] [Google Scholar]
- 14.Hood DC, Birch DG. Rod phototransduction in retinitis pigmentosa: estimation and interpretation of parameters derived from the rod a-wave. Invest Ophthalmol Vis Sci. 1994;35:2948–2961. [PubMed] [Google Scholar]
- 15.Howes KA, Pennesi ME, Sokal I, Church-Kopish J, Schmidt B, Margolis D, Frederick JM, Rieke F, Palczewski K, Wu SM, Detwiler PB, Baehr W. GCAP1 rescues rod photoreceptor response in GCAP1/GCAP2 knockout mice. EMBO J. 2002;21:1545–1554. doi: 10.1093/emboj/21.7.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoue T, Hojo M, Bessho Y, Tano Y, Lee JE, Kageyama R. Math3 and NeuroD regulate amacrine cell fate specification in the retina. Development. 2002;129:831–842. doi: 10.1242/dev.129.4.831. [DOI] [PubMed] [Google Scholar]
- 17.Iwata I, Nagafuchi S, Nakashima H, Kondo S, Koga T, Yokogawa Y, Akashi T, Shibuya T, Umeno Y, Okeda T, Shibata S, Kono S, Yasunami M, Ohkubo H, Niho Y. Association of polymorphism in the NeuroD/BETA2 gene with type 1 diabetes in the Japanese. Diabetes. 1999;48:416–419. doi: 10.2337/diabetes.48.2.416. [DOI] [PubMed] [Google Scholar]
- 18.Jan YN, Jan LY. HLH proteins, fly neurogenesis, and vertebrate myogenesis. Cell. 1993;75:827–830. doi: 10.1016/0092-8674(93)90525-u. [DOI] [PubMed] [Google Scholar]
- 19.Jansen HG, Hawkins RK, Sanyal S. Synaptic growth in the rod terminals of mice after partial photoreceptor cell loss: a three-dimensional ultrastructural study. Microsc Res Tech. 1997;36:96–105. doi: 10.1002/(SICI)1097-0029(19970115)36:2<96::AID-JEMT3>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 20.Katsanis N, Lupski JR, Beales PL. Exploring the molecular basis of Bardet-Biedl syndrome. Hum Mol Genet. 2001;10:2293–2299. doi: 10.1093/hmg/10.20.2293. [DOI] [PubMed] [Google Scholar]
- 21.Kim WY, Fritzsch B, Serls A, Bakel LA, Huang EJ, Reichardt LF, Barth DS, Lee JE. NeuroD-null mice are deaf due to a severe loss of the inner ear sensory neurons during development. Development. 2001;128:417–426. doi: 10.1242/dev.128.3.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamb TD, Pugh EN. A quantitative account of the activation steps involved in phototransduction in amphibian photoreceptors. J Physiol (Lond) 1992;449:719–758. doi: 10.1113/jphysiol.1992.sp019111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix-loop-helix protein. Science. 1995;268:836–844. doi: 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- 24.Liu M, Pleasure SJ, Collins AE, Noebels JL, Naya FJ, Tsai MJ, Lowenstein DH. Loss of BETA2/NeuroD leads to malformation of the dentate gyrus and epilepsy. Proc Natl Acad Sci USA. 2000a;97:865–870. doi: 10.1073/pnas.97.2.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu M, Pereira FA, Price SD, Chu M, Shope C, Himes D, Eatock RA, Brownell WE, Lysakowski A, Tsai MJ. Essential role of BETA2/NeuroD1 in development of the vestibular and auditory systems. Genes Dev. 2000b;14:2839–2854. doi: 10.1101/gad.840500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyubarsky AL, Falsini B, Pennesi ME, Valentini P, Pugh EN. UV- and midwave-sensitive cone-driven retinal responses of the mouse: a possible phenotype for coexpression of cone photopigments. J Neurosci. 1999;19:442–455. doi: 10.1523/JNEUROSCI.19-01-00442.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lyubarsky AL, Pugh EN. Recovery phase of the murine rod photoresponse reconstructed from electroretinographic recordings. J Neurosci. 1996;16:563–571. doi: 10.1523/JNEUROSCI.16-02-00563.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Machida S, Kondo M, Jamison JA, Khan NW, Kononen LT, Sugawara T, Bush RA, Sieving PA. P23H rhodopsin transgenic rat: correlation of retinal function with histopathology. Invest Ophthalmol Vis Sci. 2000;41:3200–3209. [PubMed] [Google Scholar]
- 29.Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, Saad M, Warram JH, Montminy M, Krolewski AS. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. 1999;23:323–328. doi: 10.1038/15500. [DOI] [PubMed] [Google Scholar]
- 30.Mitton KP, Swain PK, Chen S, Xu S, Zack DJ, Swaroop A. The leucine zipper of NRL interacts with the CRX homeodomain: a possible mechanism of transcriptional synergy in rhodopsin regulation. J Biol Chem. 2000;275:29794–29799. doi: 10.1074/jbc.M003658200. [DOI] [PubMed] [Google Scholar]
- 31.Miyata T, Maeda T, Lee JE. NeuroD is required for differentiation of the granule cells in the cerebellum and hippocampus. Genes Dev. 1999;13:1647–1652. doi: 10.1101/gad.13.13.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morrow EM, Furukawa T, Lee JE, Cepko CL. NeuroD regulates multiple functions in the developing neural retina in rodent. Development. 1999;126:23–36. doi: 10.1242/dev.126.1.23. [DOI] [PubMed] [Google Scholar]
- 33.Muresan V, Lyass A, Schnapp BJ. The kinesin motor KIF3A is a component of the presynaptic ribbon in vertebrate photoreceptors. J Neurosci. 1999;19:1027–1037. doi: 10.1523/JNEUROSCI.19-03-01027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mutoh H, Fung BP, Naya FJ, Tsai MJ, Nishitani J, Leiter AB. The basic helix-loop-helix transcription factor BETA2/NeuroD is expressed in mammalian enteroendocrine cells and activates secretin gene expression. Proc Natl Acad Sci USA. 1997;94:3560–3564. doi: 10.1073/pnas.94.8.3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naya FJ, Stellrecht CM, Tsai MJ. Tissue-specific regulation of the insulin gene by a novel basic helix-loop-helix transcription factor. Genes Dev. 1995;9:1009–1019. doi: 10.1101/gad.9.8.1009. [DOI] [PubMed] [Google Scholar]
- 36.Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, Tsai MJ. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997;11:2323–2334. doi: 10.1101/gad.11.18.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Phelan JK, Bok D. A brief review of retinitis pigmentosa and the identified retinitis pigmentosa genes. Mol Vis. 2000;6:116–124. [PubMed] [Google Scholar]
- 38.Pugh EN, Jr, Falsini B, Lyubarsky AL. The origin of the major rod- and cone-driven components of the rodent electroretinogram, and the effect of age and light rearing history on the magnitudes of these components. In: Williams TP, Thistle AB, editors. Photostasis and related topics. Plenum; New York: 1998. pp. 93–128. [Google Scholar]
- 39.Qiu Y, Sharma A, Stein R. p300 mediates transcriptional stimulation by the basic helix-loop-helix activators of the insulin gene. Mol Cell Biol. 1998;18:2957–2964. doi: 10.1128/mcb.18.5.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwab MH, Bartholomae A, Heimrich B, Feldmeyer D, Druffel-Augustin S, Goebbels S, Naya FJ, Zhao S, Frotscher M, Tsai MJ, Nave KA. Neuronal basic helix-loop-helix proteins (NEX and BETA2/Neuro D) regulate terminal granule cell differentiation in the hippocampus. J Neurosci. 2000;20:3714–3724. doi: 10.1523/JNEUROSCI.20-10-03714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Semple-Rowland SL. Expression of glial fibrillary acidic protein by Muller cells in rod chick retina. J Comp Neurol. 1991;305:582–590. doi: 10.1002/cne.903050405. [DOI] [PubMed] [Google Scholar]
- 42.Sohocki MM, Sullivan LS, Mintz-Hittner HA, Birch D, Heckenlively JR, Freund CL, McInnes RR, Daiger SP. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am J Hum Genet. 1998;63:1307–1315. doi: 10.1086/302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strettoi E, Pignatelli V. Modifications of retinal neurons in a mouse model of retinitis pigmentosa. Proc Natl Acad Sci USA. 2000;97:11020–11025. doi: 10.1073/pnas.190291097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tuccari G, Trombetta C, Giardinelli MM, Arena F, Barresi G. Distribution of glial fibrillary acidic protein in normal and gliotic human retina. Basic Appl Histochem. 1986;30:425–432. [PubMed] [Google Scholar]
- 45.Yan RT, Wang SZ. Differential induction of gene expression by basic fibroblast growth factor and neuroD in cultured retinal pigment epithelial cells. Vis Neurosci. 2000a;17:157–164. doi: 10.1017/s0952523800171172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yan RT, Wang SZ. Expression of an array of photoreceptor genes in chick embryonic retinal pigment epithelium cell cultures under the induction of neuroD. Neurosci Lett. 2000b;280:83–86. doi: 10.1016/s0304-3940(99)01003-4. [DOI] [PubMed] [Google Scholar]