

Abstract
Synaptic differentiation results from an exchange of informational molecules between synaptic partners during development. At the vertebrate neuromuscular junction, agrin is one molecule presented by the presynaptic motor neuron that plays an instructive role in postsynaptic differentiation of the muscle cell, most notably in aggregation of acetylcholine receptors (AChRs). Although agrin is the best-characterized synaptogenic molecule, its mechanism of action remains uncertain, but clearly, it requires the receptor tyrosine kinase MuSK (muscle-specific kinase), the intracellular protein rapsyn, an Src-like kinase, and cytoskeletal components. In addition, the transmembrane protein dystroglycan interacts with the cytoskeleton and is implicated in agrin responsiveness. This α–β heterodimer can bind agrin via its extracellular α subunit and associates with the membrane cytoskeleton via its β subunit. In this study, we demonstrate that overexpression of the β subunit of dystroglycan in cultured muscle cells inhibits agrin-induced AChR clustering. Deletion analysis and point mutagenesis demonstrate that the inhibition is mediated by an intracellular, juxtamembrane region composed of basic amino acids. Finally, the inhibition mediated by β-dystroglycan extends to the minimal agrin fragment required for AChR clustering, suggesting that dystroglycan plays an important role in postsynaptic differentiation in response to agrin.
Keywords: agrin, dystroglycan, juxtamembrane, acetylcholine receptor, dystrophin, neuromuscular junction
Introduction
Synapses represent extreme examples of spatially localized subcellular differentiation. Their extracellular, transmembrane, and cytosolic components are organized in precise registration between presynaptic and postsynaptic cells to enable, respectively, efficient release and response to neurotransmitters. The events underlying synapse formation are best characterized for the neuromuscular junction (NMJ) at which localized information exchange between synaptic partners drives structural differentiation of presynaptic and postsynaptic elements (Sanes and Lichtman, 2001). The nerve-derived information is best understood and includes agrin, the heparan sulfate proteoglycan. Neuron-specific isoforms of agrin are synthesized and released by motor neurons, and recombinant agrin can induce the aggregation of acetylcholine receptors (AChRs) when applied to cultured muscle cells or when expressed at nonsynaptic sites in vivo (Sanes and Lichtman, 2001). Furthermore, mice with a deletion of the neural agrin isoforms fail to form NMJs (Burgess et al., 1999). Thus, secreted neural isoforms of agrin are deposited into the extracellular matrix (ECM) during development and induce differentiation of the postsynaptic apparatus, as originally proposed by McMahan (1990). However, the postsynaptic events that mediate the response to agrin remain to be elucidated.
Two muscle cell proteins are required for agrin responsiveness: the receptor tyrosine kinase MuSK (muscle-specific kinase) (DeChiara et al., 1996), which is part of an agrin receptor complex, and the cytosolic protein rapsyn (Gautam et al., 1995), which has been genetically placed between activation of MuSK and clustering of postsynaptic molecules (Gautam et al., 1999). Although MuSK activation is necessary, it is not sufficient for agrin-mediated differentiation (Apel et al., 1997; Glass et al., 1997; but see Sander et al., 2001), and several candidate cytosolic mediators have been implicated in downstream signaling, including nitric oxide (Jones and Werle, 2000;Luck et al., 2000), calcium (Megeath and Fallon, 1998), small GTP-binding proteins (Weston et al., 2000), and an Src family tyrosine kinase (Fuhrer et al., 1997). The soluble kinase phosphorylates substrates that are required for aggregation of postsynaptic proteins, including MuSK (Mohamed et al., 2001) and the AChR itself (Qu and Huganir, 1994; Wallace, 1995; Ferns et al., 1996). Tyrosine-phosphorylated AChRs have enhanced association with the membrane cytoskeleton (Borges and Ferns, 2001) and restricted mobility, suggesting that agrin-stimulated phosphorylation can alter the interactions between AChRs and the postsynaptic membrane cytoskeleton. In addition, studies are consistent with agrin acting by organizing the synaptic cytoskeleton through activities of the small GTPases Cdc42 and Rac, at least in part (Weston et al., 2000), and the polymerization of actin (Dai et al., 2000).
Membrane cytoskeleton components at the NMJ include the dystrophin family proteins dystrophin and utrophin, which are present, respectively, at the troughs and crests of the postsynaptic junctional folds (Byers et al., 1991; Bewick et al., 1992). These proteins are components of the dystrophin glycoprotein complex (DCG)/utrophin glycoprotein complex (UGC) that also includes the dystroglycans, sarcoglycans, syntrophins, and dystrobrevins (Henry and Campbell, 1999). Dystroglycan is a transmembrane α–β heterodimer capable of binding ECM proteins (including laminin, perlecan, and agrin) via its extracellular α subunit and dystrophin/utrophin via its β subunit. Because dystrophin and utrophin interact directly with actin, the DGC/UGC can serve as a link between the cytoplasmic actin cytoskeleton and the ECM.
Direct binding of dystroglycan to agrin suggests that dystroglycan plays a role in agrin-induced postsynaptic differentiation (Campanelli et al., 1991; Gee et al., 1994; Sugiyama et al., 1994), a notion supported by studies in vivo and in vitro. First, dystroglycan−/− chimeric animals are deficient in NMJ synaptogenesis (Cote et al., 1999), and overexpression of dystroglycan decreases AChR accumulation at synaptic sites in developing Xenopus muscle (Heathcote et al., 2000). Second, in cultured muscle cells, antibodies against α-dystroglycan interfere with agrin-induced AChR cluster consolidation (Campanelli et al., 1994), antisense dystroglycan oligonucleotides inhibit agrin activity (Jacobson et al., 1998), and dystroglycan−/− myotubes form only immature AChR clusters in response to agrin stimulation (Grady et al., 2000). Together, these results are consistent with a role for dystroglycan in agrin-mediated AChR cluster formation, but the studies have not defined its mechanism of action.
In this study, we sought to define regions of β-dystroglycan that are functionally involved in agrin-induced postsynaptic differentiation. Our results demonstrate a dominant-negative effect of truncated dystroglycan expression on agrin-induced AChR clustering in cultured muscle cells, and localize an inhibitory activity to the cytoplasmic juxtamembrane domain of dystroglycan. The data suggest, importantly, that the basic mechanism of agrin-induced clustering involves dystroglycan.
Materials and Methods
Dystroglycan and CD8 constructs. Hemagglutinin (HA) epitope-tagged β-dystroglycan has been described previously (Chung and Campanelli, 1999). Truncated dystroglycan forms (DGΔP, DGtmO, DGtmI, and DGtm) and CD8tm mutants were amplified by PCR using oligonucleotide pairs (listed in Table 1) as follows: DGΔP, DG5 plus DGΔP3; DGtmO, DG5 plus tmO3; DGtmI, tmI5 plus DG3; DGtm, tmI5 plus tm3; CD8tm, CD85 plus CD83. All PCR products were ligated in-frame with the prolactin signal sequence and HA epitope as described for the β-dystroglycan construct (Chung and Campanelli, 1999). CD8tm was amplified from pBSCD8-GFP (Lee and Luo, 1999). CD8tmJ and CD8tmDGJ were created by ligating KSCD8tm (digested withNdeI and XbaI) with the appropriate complementary oligonucleotides (Table 1; CD8J5 plus CD8J3 or DGJ5 plus DGJ3, respectively). CD8tmDGJK was created from KSCD8tmDGJ (digested withNdeI and XbaI) with the oligonucleotides DGJK5 and DGJK3. CD8tmDGJ2RA, CD8tmDGJ3KA, and CD8tmDGJCYSF were created by ligating KSCD8tmJ (digested with NdeI and XbaI) with the appropriate complementary oligonucleotides (Table 1; 2RA3 plus 2RA5, 3KA3 plus 3KA5, or CYSF3 plus CYSF5). Constructs were moved into the pCMV expression vector using ClaI andXbaI. DGtmJ was obtained by a triple ligation of KSDGtmI digested with ClaI and PvuII (N terminus), KSDGtmO digested with PvuII and XbaI (C terminus), and pCMV digested with ClaI and XbaI. All subcloned constructs were sequenced (University of Illinois Biotechnology Center) to verify accurate PCR amplification and oligonucleotide synthesis. Schematics of mutant proteins are shown in Figure 1A (also see Fig. 8A).
Table 1.
Oligonucleotides used in this study
| DG5 | CCGGGGCATGCAGTCTATTGTG |
| DG3 | GAGCCTTCTCTTCCTGCAG |
| TMI5 | GACATGATAGCATGCGGAGCAGTGAGGATGACG |
| TMO3 | GGTCAGTCTAGATTAGCCCTCCT |
| CD85 | GAAGGGGAGCATGCTGGACTTC |
| CD83 | TTCGGCTCTAGATTACTTGCGGTAGCATATGAGAG |
| DGJ5 | TATGCTATAGAAAGAAGAGGAAGGGCTAAT |
| DGJ3 | CTAGATTAGCCCTTCCTCTTCTTTCTATAGCA |
| CD8J5 | TATGCTACCATCGATCGCGAAAGCGTTAAT |
| CD8J3 | CTAGATTAACGCTTTCGCGATCGATGGTAGCA |
| DGΔP3 | TCATGTTTCTAGATTAGGGACGGGA |
| DGJK5 | TATGCTATAGAAAGAAGAGGAAGGGCAAATAA |
| DGJK3 | CTAGTTATTTGCCCTTCCTCTTCTTTCTATAGCA |
| CYSF5 | TAAGCTTTAGAAAGAAGAGGAAGGGCAAATAAT |
| CYSF3 | CTAGATTATTTGCCCTTCCTCTTCTTTCTAAAGCT |
| 3KA5 | TATGCTATAGAGCTGCAAGGGCTGGCAAAATGAT |
| 3KA3 | CTAGATCATTTTGCCAGCCCTTGCAGCTCTATAGCA |
| 2RA5 | TATGCTATGCCAAGAAGGCAAAGGGCAAATAGAT |
| 2RA3 | TAGATCTATTTGCCCTTTGCCTTCTTGGCATAGCA |
Fig. 1.

Schematic and nomenclature of β-dystroglycan and CD8 variants. A, Full-length β-dystroglycan (S654–P895) is represented by DG. Variant nomenclature is as follows: ΔP, C-terminal deletion;tm, transmembrane domain; O, outside;I, inside; J, juxtamembrane. All mutants have an N-terminal HA epitope that is preceded in frame by the N-terminal-cleaved signal sequence derived from prolactin (data not shown). The C-terminal dystrophin/utrophin/Grb2-binding motif is indicated by single-letter amino acid code (PPXY). ecto, Ectodomain. B, Amino acid sequence alignment of dystroglycan (amino acids 746–782) and CD8 transmembrane (amino acids 189–225) and juxtamembrane deletion mutants. Alignment of CD8 variants with dystroglycan variants is based on the cytoplasmic juxtamembrane sequence (ICY). Dystroglycan residues are in bold.
Fig. 8.
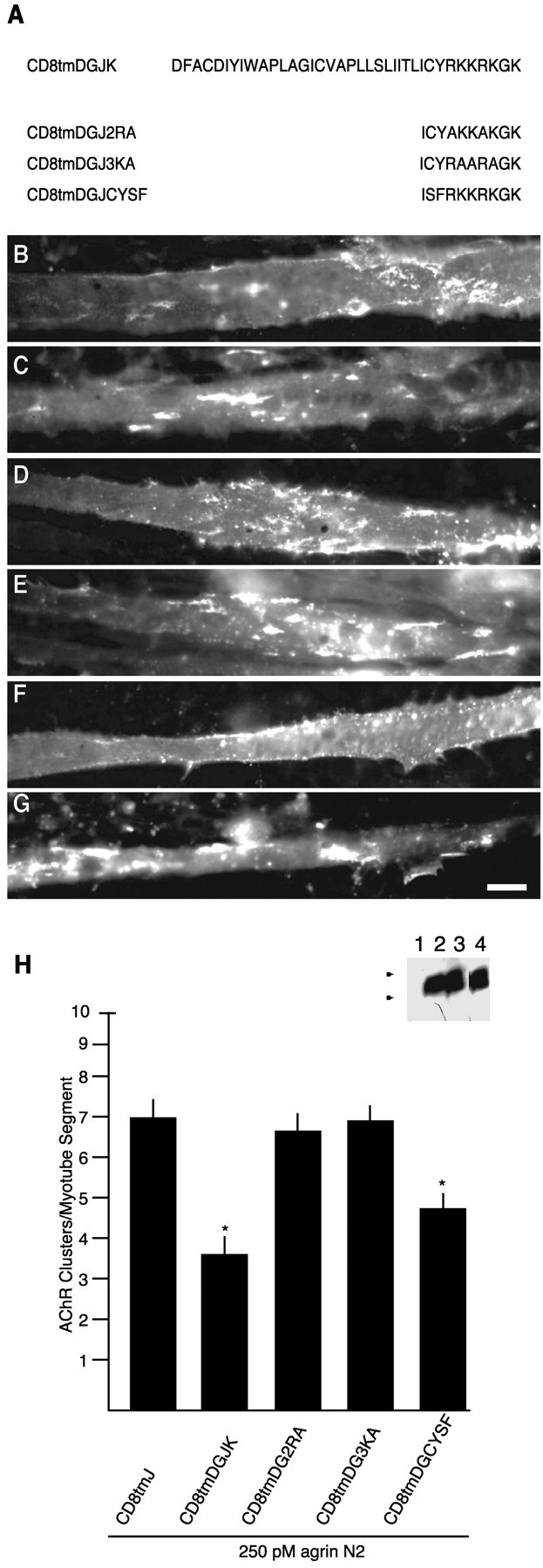
Intracellular juxtamembrane basic amino acids of β-dystroglycan are required for inhibition of AChR clustering in response to agrin. A, Schematic and nomenclature of CD8tmDGJ chimeras. Transfected myotubes were treated for 5 hr with 250 pm agrin N2(4,8). Cells were double-labeled for AChRs (Texas Red-conjugated α-bungarotoxin; C, E, G) and anti-HA immunoreactivity (FITC-conjugated anti-mouse IgG; B, D, F). B andC are a CD8tmDGJ2RA-transfected myotube,D and E are a CD8tmDGJ3KA-transfected myotube, and F and G are a CD8tmDGJCYSF-transfected myotube. H, Quantification of agrin-induced AChR clustering in myotubes transfected with CD8tmDGJ chimeras (mean ± SEM; n = 8). Mutation of either the arginine or lysine residues in the β-dystroglycan juxtamembrane region restores AChR clustering to control values. The chimeric mutant CD8tmDGJCYSF causes inhibition to the same extent as CD8tmDGJ. Asterisks indicate mean values that are statistically different from CD8tmJ (p < 0.05; Student's ttest). Inset, anti-HA Western blot of COS cells transiently transfected with no DNA (1), CD8tmDGJ2RA (2), CD8tmDGJ3KA (3), or CD8tmDGJCYSF (4). Samples were separated by 16% SDS-PAGE.Arrowheads indicate weight standards (×103): 6, 3. Scale bar, 50 μm.
COS cell transfection, immunostaining, and Western blotting.Purified pCMV plasmids (dystroglycan, CD8, and agrin variants) were transfected into COS cells as described previously (Cornish et al., 1999). Sham indicates a transfection with no DNA. Agrin used for C2 myotube stimulation [N2(4,8) or G3(8)] was collected, assayed, and applied at the indicated concentrations as conditioned medium diluted into fusion medium (Cornish et al., 1999). For immunofluorescence analysis, COS cells transfected in 12 well plates were replated onto coverslips at 12 hr after transfection and stained at 48 hr after transfection. Anti-HA immunostaining was performed on live cells at 4°C. Coverslips were rinsed with chilled growth medium, incubated at 4°C with mouse anti-HA.11 antibody (1:500; Covance, Richmond, CA) for 30 min, washed once with chilled growth medium and once with chilled PBS, and incubated for 20 min at 25°C with chilled fixative (4% paraformaldehyde, 4% sucrose in PBS). Cells were subsequently blocked, incubated with FITC-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA), and mounted for visualization. For Western blot analysis of dystroglycan and CD8 variants, COS cells were released from tissue culture dishes by divalent cation chelation in buffered saline and pelleted; whole-cell pellets were resuspended in SDS-PAGE loading buffer with 5% β-mercaptoethanol. Whole-cell lysates were separated by the indicated gel system [10, 12, or 16% SDS-PAGE (Laemmli, 1970)] and transferred to polyvinylidene fluoride membrane (Micron Sciences, Westborough, MA). Membranes were probed with anti-HA.11 and visualized with horseradish peroxidase-conjugated secondary antibodies (Zymed, South San Francisco, CA) and enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ) as described previously (Cornish et al., 1999). For control Western blots in Figure 2, COS cells in 12 well dishes were transfected with Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Whole-cell pellets were prepared as described above, and equal percentages of each transfection were analyzed by Western blotting.
Fig. 2.

β-Dystroglycan constructs express at similar levels when transfected into heterologous cells. A, Anti-HA immunoblot of COS cells transiently transfected with no DNA (1), DG (2), DGΔP (3), or DGtmO (4). Samples were separated by 10% SDS-PAGE. Arrows indicate weight standards (×103): 47, 34, 29, 20. B, Anti-HA immunoblot of COS cell transfected with no DNA (1), CD8tm (2), CD8tmJ (3), DGtm (4), DGtmJ (5), CD8tmDGJK (6), CD8tmDGJ3KA (7), CD8tmDGJ2RA (8), and CD8tmDGJCYSF (9). All of the β-dystroglycan constructs and CD8 constructs had similar levels of expression as determined by Western blotting. Samples were separated by 16% SDS-PAGE. Arrows indicate weight standards (×103): 6, 3.
C2-C12 cell culture, transfection, and clustering assay. C2–C12 myoblasts (4.3 × 105) were plated on nitric acid-washed 22 mm round coverslips (Fisher Scientific, Hanover Park, IL) coated with 0.1% gelatin followed by 2 μg/ml mouse collagen IV (Invitrogen). Cells were cultured until confluence in growth medium and subsequently maintained in fusion medium (Cornish et al., 1999). Forty-eight hours after myoblast fusion, cells were transfected with the Helios Gene Gun according to the manufacturer's instructions (Bio-Rad, Hercules, CA). After a 24 hr recovery period, cells were incubated for an additional 5 or 16 hr (as indicated) with recombinant soluble agrin. Cells were subsequently fixed in 2.5% paraformaldehyde for 15 min at room temperature and stained for either anti-HA or anti-α5-integrin followed by AChR labeling: HA.11, 1:1000; CD49e, 1:200 (PharMingen, San Diego, CA); fluorescein-conjugated goat anti-mouse, 1:200 (ICN Biomedicals, Costa Mesa, CA); or 83 nmTexas Red-α-bungarotoxin (Molecular Probes, Eugene, OR) without permeabilization (Cornish et al., 1999). For cell-surface-staining control experiments, C2 myotubes were transfected as above, fixed with paraformaldehyde, and stained with either anti-dystroglycan C-terminus antibody (43DAG1, 1:500; Novocastra, Newcastle on Tyne, UK), anti-light meromyosin, or anti-Ca2+-ATPase (MF20 or CaF2-5D2, 1:50 and 1:20, respectively; Developmental Studies Hybridoma Bank, Iowa City, IA). Images were acquired using a Zeiss (Thornwood, NY) Axioplan epifluorescence microscope (100 W HBO burner, 40× oil immersion objective; numerical aperture, 1.3; Plan Neofluar), a Photometrics Sensys CCD, and IP Lab software (Scanalytics, Fairfax, VA). Quantification of AChR clusters induced by agrin was performed in a blind manner. Anti-HA-reactive myotubes >200 μm in length and 10 μm in width were identified using fluorescein optics. On HA-positive myotubes, clusters of AChR >1 μm in their longest dimension were then counted manually using Texas Red optics. The average number of clusters per myotube segment was calculated for each coverslip (5–15 myotubes per coverslip), and n is the total number of coverslips (derived from at least three independent transfections). Microcluster analysis (Fig.3) was performed as described above with an additional tally of AChR puncta <1 μm recorded for each myotube segment analyzed. For cell-surface anti-HA fluorescence quantification, digital images of myotubes were normalized, the area and total fluorescence (arbitrary units) were then recorded for each immunopositive myotube, and the average fluorescence per pixel was calculated with IP Lab software. Data were analyzed by Student'st test.
Fig. 3.
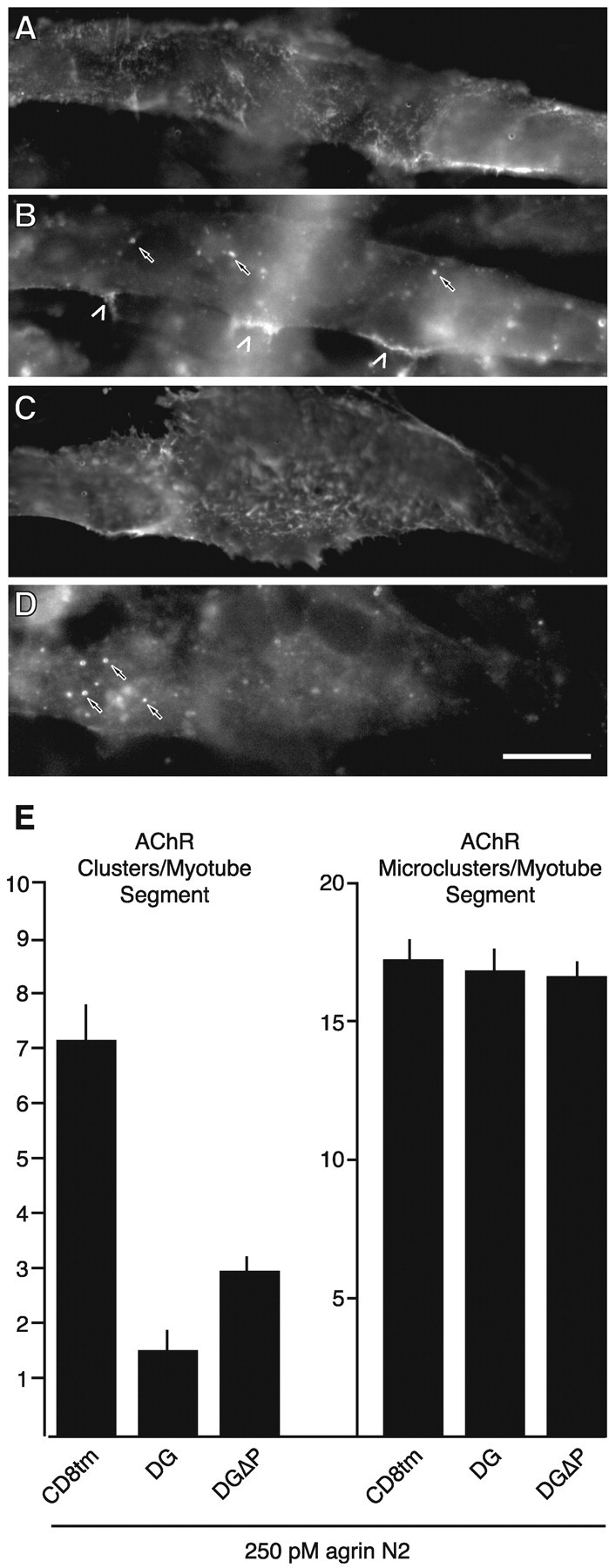
β-Dystroglycan expression inhibits agrin-induced formation of clusters without altering formation of microclusters. Transfected myotubes were incubated with 250 pm agrin N2(4,8) for 5 hr and double-labeled for AChRs (Texas Red-conjugated α-bungarotoxin; B, D) and anti-HA immunoreactivity (FITC-conjugated anti-mouse; A, C). A, B, A CD8tm-transfected myotube. C, D, A myotube transfected with DGΔP. In B andD, carets indicate large clusters, whereas small arrows point to a subset of the quantified microclusters (see Materials and Methods). E, Quantification of agrin-induced clusters (left) and microclusters (right). Agrin-induced formation of clusters is inhibited by dystroglycan and DGΔP relative to control CD8tm (p < 0.005; Student'st test; n = 4). In contrast, the numbers of microclusters per myotube segment were not different between myotubes transfected with the dystroglycan variants and controls. Scale bar, 20 μm.
Results
Dystroglycan is synthesized as a precursor protein that is subsequently cleaved into α and β subunits (Ibraghimov-Beskrovnaya et al., 1992). To assess the functional contribution of β-dystroglycan to the agrin responsiveness of muscle cells, we first determined whether β-dystroglycan could be expressed in the absence of the α subunit. The results of expressing β-dystroglycan in COS cells are shown in Figure4, and demonstrate synthesis of the appropriate size protein (Fig. 4D) that is present at the plasma membrane (Fig. 4B).
Fig. 4.
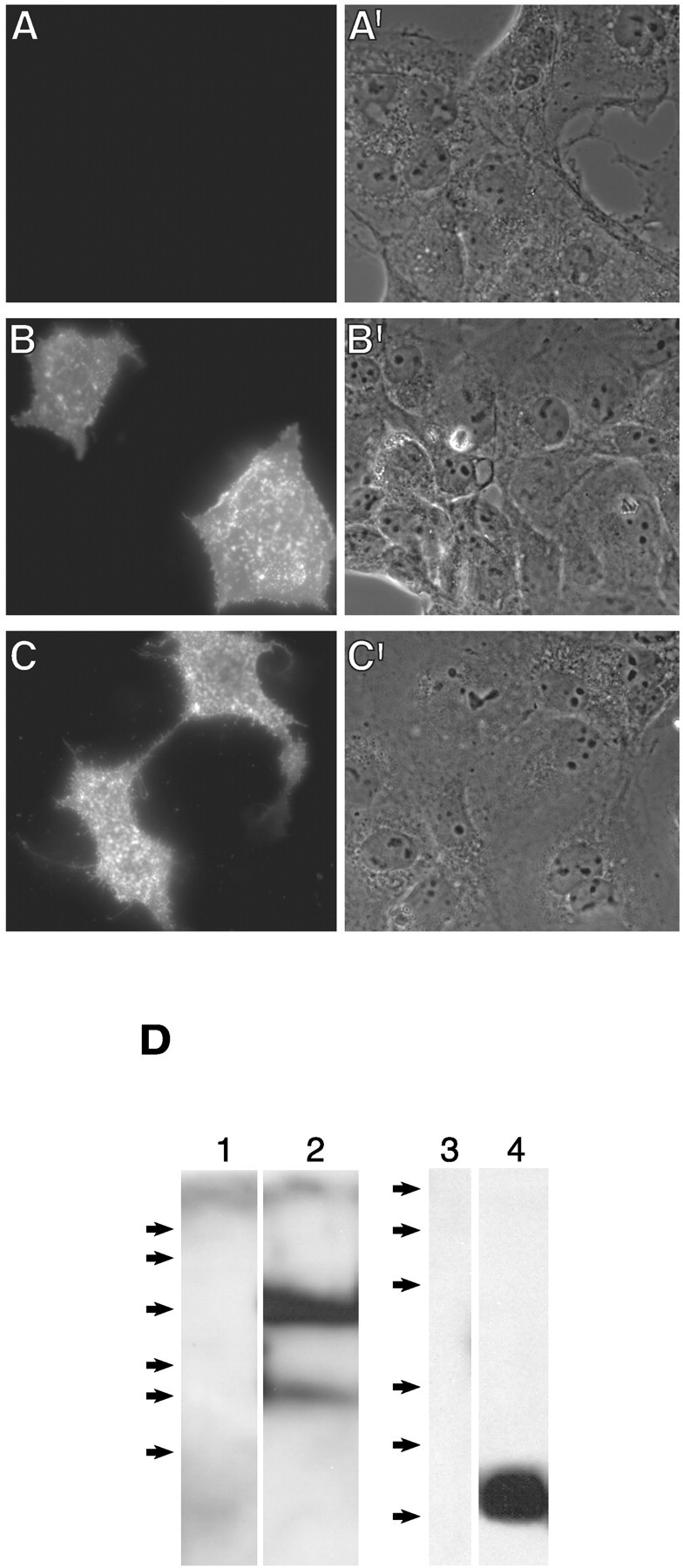
β-Dystroglycan is expressed at the cell surface in the absence of α-dystroglycan. Transiently transfected COS cells (sham, pCMVDG, or pCMVCD8tmJ; A–C, respectively) were stained live with anti-HA antibody to selectively detect extracellular epitopes (see Materials and Methods). A′–C′ are the corresponding phase-contrast images. Immunoreactivity observed inB and C demonstrates expression of the proteins at the cell surface with the extracellular N-terminal HA epitope. D, Immunoblot of transfected COS cell lysates probed with the anti-HA antibody. β-Dystroglycan and CD8tmJ migrate at an Mr of 43,000 and 5000, respectively, consistent with synthesis of full-length proteins. The lower-molecular-weight species observed in β-dystroglycan-transfected cells is a cleavage product of dystroglycan that has been documented by others (Marchand et al., 2001). Arrows indicate molecular weight standards (×103): 111, 77, 47, 34, 29, 20 (left, 12% SDS-PAGE); 43, 29, 18, 14, 6, 3 (right, 15% SDS-PAGE).
Dystroglycan colocalizes with agrin-induced AChR aggregates; binds ECM ligands via α-dystroglycan; and binds cytosolic proteins including dystrophin (Blake et al., 2002), utrophin (James et al., 2000), rapsyn (Cartaud et al., 1998), Grb2 (Yang et al., 1995; Cavaldesi et al., 1999; Russo et al., 2000), and caveolin (Sotgia et al., 2000) via cytoplasmic sequences of β-dystroglycan. We hypothesized that dystroglycan function involves coordinate binding of extracellular and intracellular partners, and that overexpression of only the β subunit would allow for cytoplasmic interactions but lack binding to extracellular ligands and therefore block function. To evaluate this, C2 myotubes were transfected with β-dystroglycan and challenged with agrin N2(4,8) for 16 hr to induce clustering of AChRs (see Materials and Methods). As shown in Figure 5, expression of β-dystroglycan resulted in a decrease in agrin-induced AChR clustering (Fig. 5B,D,G). In contrast, expression of soluble green fluorescent protein (GFP), a CD8GFP fusion protein, or an HA-tagged CD8 transmembrane domain (CD8tm; Fig.5E–G, data not shown) had no effect on agrin responsiveness.
Fig. 5.
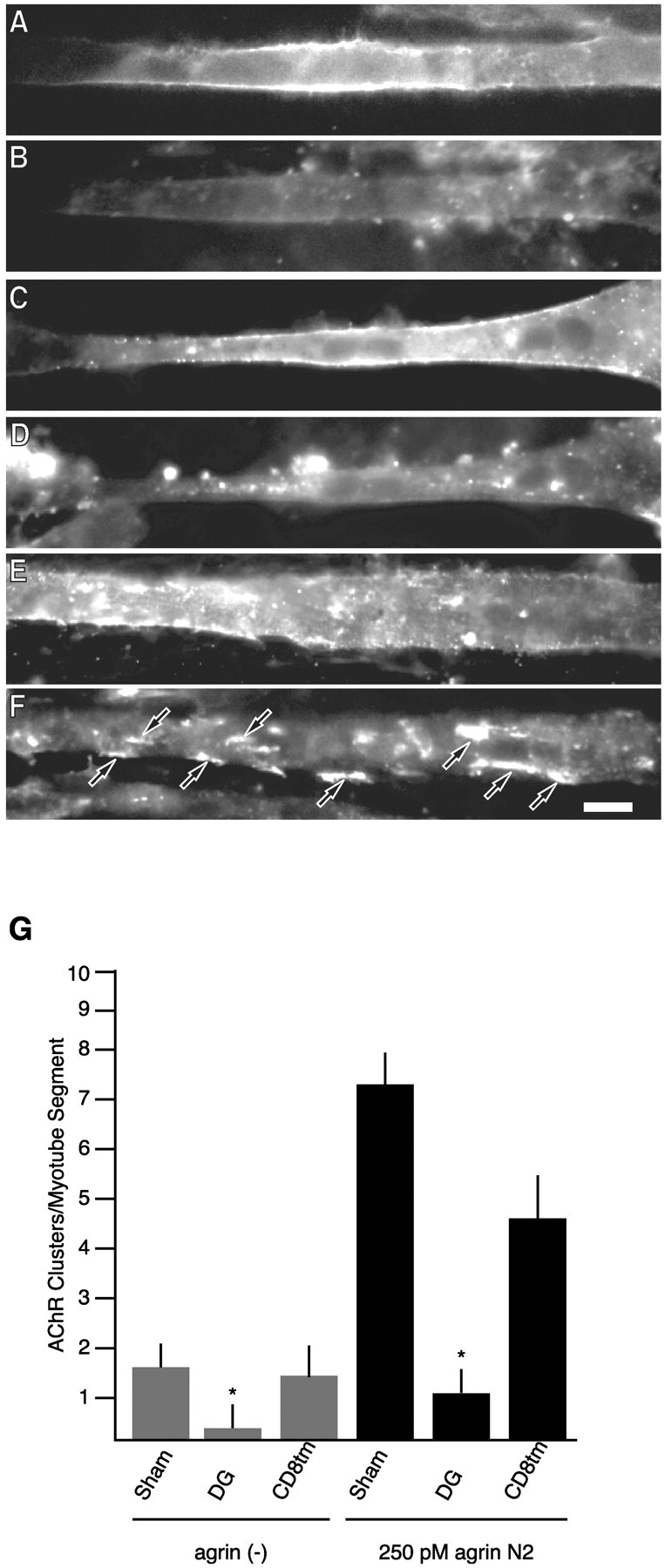
β-Dystroglycan expression inhibits agrin-induced AChR clustering. Biolistically transfected myotubes were treated for 16 hr with 250 pm agrin N2(4,8). Cells were double-labeled for AChRs (Texas Red-conjugated α-bungarotoxin; B, D, F) and anti-HA immunoreactivity (FITC-conjugated anti-mouse; A, C, E). A, B, A β-dystroglycan-transfected myotube in the absence of agrin. C, D, A β-dystroglycan-transfected myotube challenged with 250 pm agrin N2(4,8). E, F, An agrin-treated CD8tm-transfected myotube. Agrin-induced clusters are apparent in CD8tm (F) myotubes but not in the β-dystroglycan (D) myotube or in the control non-agrin-treated case (B). Arrows indicate AChR clusters. Scale bar, 20 μm. G, Quantification of the effects of exogenously expressed proteins on agrin-induced AChR clustering (mean ± SEM; n= 8). Expression of β-dystroglycan (DG) but not CD8tm inhibits agrin-induced clustering compared with untransfected myotubes. The difference between spontaneous clustering [sham vs DG-transfected myotubes; agrin(−)] is likely caused by experimental manipulations: spontaneous clustering in DG- and CD8tm-transfected myotubes is not statistically different (asterisks indicate that mean values are statistically different from sham; p < 0.05; Student'st test).
These results suggest that muscle-cell responsiveness to agrin can be abrogated by expression of β-dystroglycan. However, because recent evidence suggests that dystroglycan associates with AChRs in post-Golgi vesicles and that these proteins are cotransported to the plasma membrane (Marchand et al., 2001), one possibility is that inhibition reflects alterations in cell-surface delivery of these vesicles. Another possibility is that β-dystroglycan overexpression alters membrane protein distribution in a nonspecific manner. The following experiments were performed to address these possibilities. First, we evaluated the effects of β-dystroglycan overexpression on the distribution of cell-surface α5-integrin. No differences in α5-integrin expression could be detected between β-dystroglycan-transfected and untransfected myotubes on the same coverslip (Fig.6G,H) or between β-dystroglycan- and control-transfected myotubes (data not shown). Second, we evaluated our method of immunocytochemical staining for its ability to selectively detect cell-surface epitopes. β-Dystroglycan-transfected C2 myotubes were fixed and stained with antibodies against an extracellular epitope (anti-HA), antibodies against intracellular epitopes (anti-β-dystroglycan, anti-light meromyosin, or anti-Ca2+-ATPase), or secondary antibody alone. Expression of HA-tagged β-dystroglycan in C2 myotubes resulted in cell-surface localization indicated by the outline of HA immunoreactivity at the membrane (Fig. 6F). Staining for cytoplasmic antigens by this procedure resulted in myotubes that were indistinguishable from control myotubes (Fig.6B,D). Because AChRs and dystroglycan are cotransported, we conclude that cell-surface delivery of AChR proceeds in the presence of exogenous β-dystroglycan, and that the site of inhibitory action observed is at the plasma membrane. Therefore, this transient expression system enabled us to assess the effects of β-dystroglycan expression on agrin-induced AChR clustering.
Fig. 6.

Extracellular epitopes are specifically detected in transfected C2 myotubes. C2 myotubes were transfected with β-dystroglycan. Twenty-four hours later, cells were fixed and labeled for intracellular epitopes (anti-β-dystroglycan, anti-light meromyosin; B, D, respectively), extracellular epitopes (anti-HA; F) or secondary antibody only (data not shown). Corresponding phase-contrast images are shown inA, C, and E. Nonspecific effects of β-dystroglycan expression were analyzed by monitoring the distribution of α5-integrin in β-dystroglycan-transfected C2 myotubes. G, Surface expression of β-dystroglycan with anti-HA. H, The same field of myotubes probed for α5-integrin. α-5 Localization in the transfected myotube is indistinguishable from the untransfected cells in the field of view. Scale bar, 20 μm.
β-Dystroglycan has been shown to interact with dystrophin (Jung et al., 1995; Rentschler et al., 1999), Grb2 (Yang et al., 1995; Cavaldesi et al., 1999; Russo et al., 2000), and caveolin-3 (Sotgia et al., 2000) via the C-terminal PPxY-containing sequence, and to rapsyn (Cartaud et al., 1998) via cytoplasmic sequences closer to the membrane. We were interested in determining whether known binding domains of β-dystroglycan were involved in agrin-induced AChR clustering, so we sought to localize the region of β-dystroglycan that mediates the inhibitory effect; expression of β-dystroglycan with a deletion of the relevant binding site should no longer have resulted in inhibition. Deletion mutants of β-dystroglycan were expressed in C2 myotubes, and their effect on agrin-induced AChR clustering was quantified. In these and all subsequent experiments, the HA-tagged version of the CD8 transmembrane domain was used as the control for three reasons. First, in contrast to GFP and the CD8GFP fusion protein, which display significant intracellular signal, this protein displays plasma membrane localization similar to β-dystroglycan (Fig. 5C,E, data not shown). Second, because a common method of detection (i.e., the extracellular HA epitope) could be used for all expressed proteins, quantification could be performed in a blind manner on identically processed coverslips. Third, this construct is similar to our smallest inhibitory dystroglycan construct (see Fig. 1B and below). We first asked whether the observed inhibition could be abrogated by expressing a β-dystroglycan construct that lacked the C-terminal 15 aa harboring the dystrophin binding site (DGΔP) (Fig.1A). In this and subsequent experiments, agrin incubation was performed for 5 hr, a time that afforded greater cell viability and, as seen in Figure7I, yields potent β-dystroglycan inhibition of agrin activity. These results demonstrate that the effects of dystroglycan expression are apparent with short-duration agrin treatments (5 hr β-dystroglycan is the same as that observed after 16 hr), and that the effect is not dependent on the C-terminal 15 aa of β-dystroglycan (DGΔP) (Fig.7I).
Fig. 7.
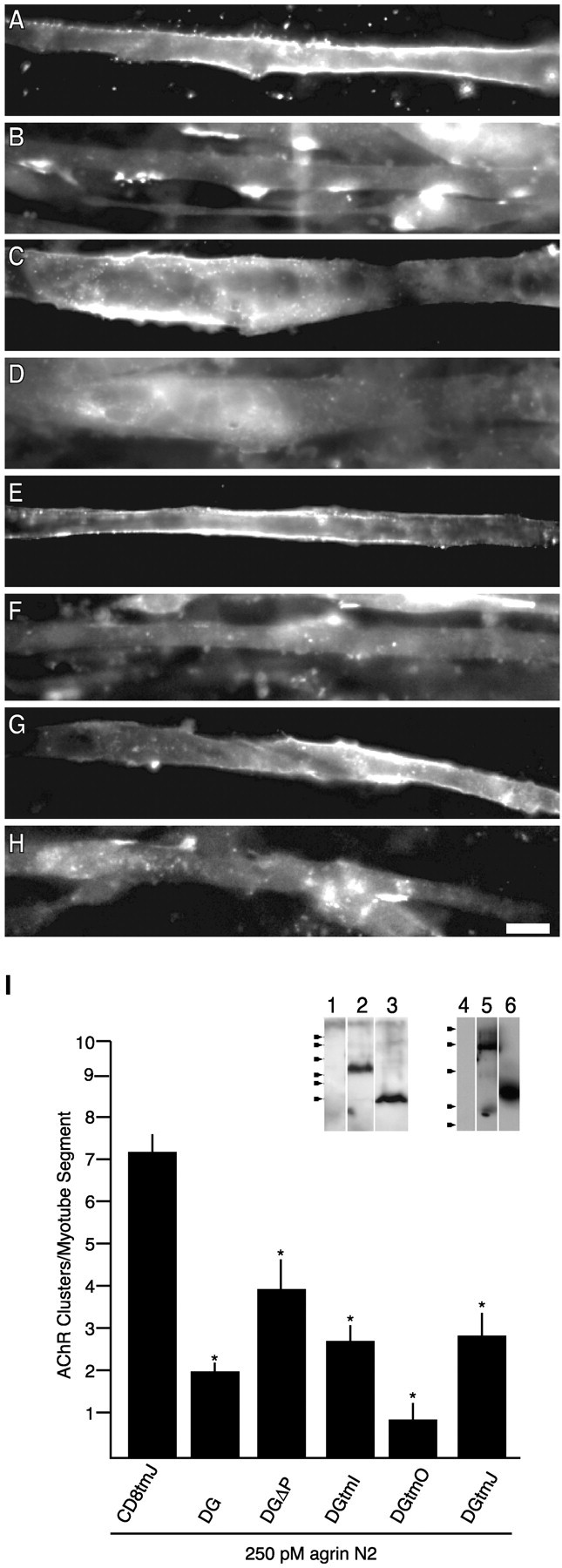
Deletion mutants of β-dystroglycan reveal an inhibitory motif within or near the transmembrane domain. Transfected myotubes were treated for 5 hr with 250 pm agrin N2(4,8) and double-labeled for AChRs (Texas Red-conjugated α-bungarotoxin; B, D, F, H) and anti-HA immunoreactivity (FITC-conjugated anti-mouse; A, C, E, G). Myotubes were transfected with the following constructs: DGΔP (A, B), DGtmI (C, D), DGtmO (E, F), and DGtmJ (G, H).I, Quantification of agrin-induced AChR clustering in transfected myotubes (mean ± SEM; n = 9). Significantly fewer clusters are observed in myotubes transfected with β-dystroglycan deletion constructs compared with CD8tmJ-transfected myotubes. Removal of the extracellular (DGtmI), cytoplasmic (DGtmO), or both extracellular and cytoplasmic (DGtmJ) regions as well as removal of the C-terminal 15 aa (DGΔP) of β-dystroglycan does not relieve inhibition. Asterisks indicate mean values that are statistically different from CD8tmJ (p < 0.05; Student's t test). Inset, COS cells were transiently transfected with no DNA (1, 4), DgtmI (2), DGtmJ (3), DGΔP (5), or DGtmO (6). Cell lysates were analyzed by anti-HA Western blot (12% SDS-PAGE). Arrowheads indicate molecular weight standards (×103): 43, 29, 18, 14, 6, 3 (left); 76, 49, 36, 29, 21 (right). Scale bar, 50 μm.
Previous reports suggested that the interaction of dystrophin family proteins with the C terminus of β-dystroglycan serves to stabilize large aggregates of microclusters (Phillips et al., 1993; Grady et al., 2000). Thus, our dystroglycan construct might be expected to result in an increase in microclusters by inhibiting dystroglycan–dystrophin interactions. We therefore quantified the frequency of agrin-induced clusters of <1 μm in control-transfected (CD8tm) and β-dystroglycan-transfected (with and without the C-terminal binding site; dystroglycan and DGΔP) myotubes. As shown in Figure 3, agrin-induced formation of clusters was inhibited by dystroglycan and DGΔP relative to control CD8tm, whereas the number of microclusters per myotube segment was not different between myotubes transfected with the dystroglycan variants and controls (Fig.3E). This experiment supports the notion that β-dystroglycan-mediated inhibition of agrin activity reflects more than dystrophin/utrophin binding to the C terminus of dystroglycan.
The inhibitory effects of β-dystroglycan were also probed by expressing variants of β-dystroglycan with larger deletions. All constructs were synthesized and expressed at the plasma membrane in COS cells as well as in C2 myotubes (Figs. 4, 7). Their apparent molecular weights, assessed by SDS-PAGE, were consistent with faithful synthesis of full-length proteins (Fig. 7I, inset). The mutants tested were gross deletions of the cytoplasmic domain, the ectodomain, or both the cytoplasmic and ectodomain of β-dystroglycan (DGtmO, DGtmI, and DGtmJ, respectively) (Fig. 1). As seen in Figure7I, all of these truncated proteins were effective in inhibiting agrin-induced AChR clustering. The inhibition by the DGtmJ variant is specific given the lack of effect produced by the equivalent CD8tmJ construct (Figs. 1B, 7I). Therefore, either the transmembrane or membrane-adjacent sequences of β-dystroglycan make functionally important contributions to agrin-induced AChR cluster formation.
Inspection of the C termini of the cytoplasmic deletions, DGtmO and DGtmJ, revealed that the juxtamembrane amino acid sequence, RKKRKG, was retained in these constructs (Fig. 1B). Charged juxtamembrane residues are believed to play a role in the orientation and targeting of transmembrane domains (Andrews et al., 1992; Wahlberg and Spiess, 1997) and were included for this reason. However, the juxtamembrane region of dystroglycan has been identified as a weak site of interaction with dystrophin (Rentschler et al., 1999) and utrophin (James et al., 2000) in a membrane-bound peptide binding assay, as well as DP116 in in vitro pull-down assays (Saito et al., 1999). The defined binding site corresponds to our juxtamembrane domain (Fig.1B) with an additional C-terminal lysine residue (Fig.8A). Therefore, additional deletions and a chimera were created to assess whether this sequence contributed to the inhibitory effect (Fig.1B and CD8tmDGJK in Fig. 8A). As seen in Figure 9, deletion of the KRKG sequence (DGtm) resulted in a loss of inhibition; the effect of DGtm expression is indistinguishable from control proteins (CD8tm and CD8tmJ). In contrast, replacement of the corresponding juxtamembrane region of CD8 with the β-dystroglycan juxtamembrane sequence (CD8tmDGJ and CD8tmDGJK) conferred inhibitory activity on the chimeric CD8 transmembrane construct (Fig. 9E–G). Together, these results demonstrate a functional activity of the membrane proximal amino acid sequence, ICYRKKRKGK, within the cytoplasmic domain of β-dystroglycan.
Fig. 9.
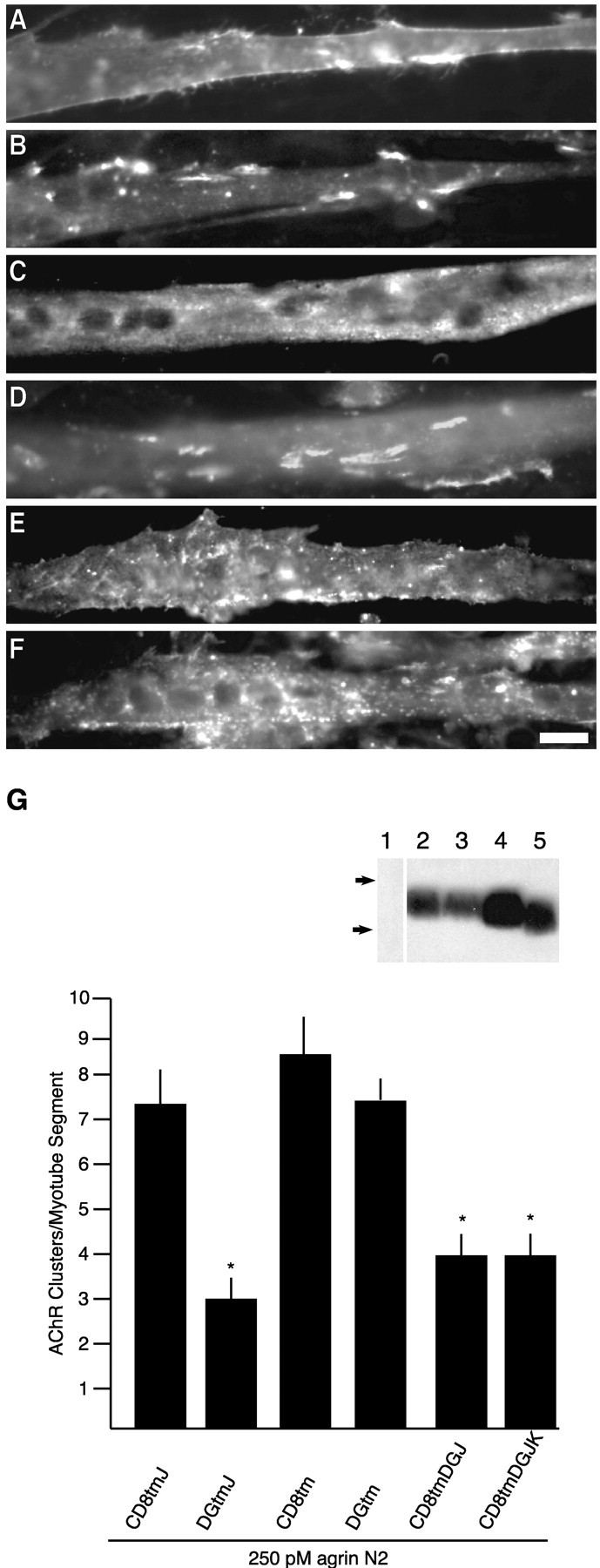
The intracellular juxtamembrane domain of β-dystroglycan contains an inhibitory element. Transfected myotubes were treated for 5 hr with 250 pm agrin N2(4,8) and double-labeled for AChRs (Texas Red-conjugated α-bungarotoxin;B, D, F) and anti-HA immunoreactivity (FITC-conjugated anti-mouse IgG; A, C, E).A and B are a CD8tm-transfected myotube.C and D are a DGtm-transfected myotube.E and F are a CD8tmDGJK-transfected myotube. G, Quantification of agrin- induced AChR clustering in myotubes transfected with β-dystroglycan and CD8 transmembrane constructs with and without the cytosolic juxtamembrane domain (mean ± SEM; n= 8). Removal of four amino acids (KRKG) from DGtmJ (DGtm) restores AChR clustering to control values. The corresponding CD8 mutants (CD8tmJ and CD8tm) are not statistically different, whereas the chimeras CD8tmDGJ and CD8tmDGJK cause inhibition.Asterisks indicate mean values that are statistically different from CD8tmJ (p < 0.05; Student's t test). Inset, Anti-HA Western blot of COS cells transiently transfected with no DNA (1), DGtm (2), CD8tm (3), CD8tmJ (4), or CD8tmDGJ (5). Samples were separated by 16% SDS-PAGE. Arrows indicate molecular weight standards (×103): 6, 3. Scale bar, 50 μm.
Juxtamembrane domains, either extracellular or intracellular, mediate a variety of protein–protein and protein–lipid interactions. For example, the juxtamembrane region of tyrosine kinase A has an Shc binding motif that is responsible for activation of Ras (Yoon et al., 1997); the juxtamembrane domain of the human epidermal growth factor receptor contains a positive basolateral-sorting determinant (Hobert and Carlin, 1995); and juxtamembrane regions of CD44, CD43, and intercellular adhesion molecule-2 (ICAM-2) harbor binding sites for ezrin/radixin/moesin (ERM) family proteins (Legg and Isacke, 1998; Yonemura et al., 1998). In addition, these short membrane proximal regions often contain amino acids that are capable of post-translational modification that can be functionally important (see Discussion). Our working hypothesis is that the juxtamembrane region of β-dystroglycan encodes localization information or confers specific molecular recognition that allows this domain to compete for function with endogenous dystroglycan because of either (1) post-translational modification of the cysteine or tyrosine residues or (2) protein–protein or protein–lipid interactions. As a step toward distinguishing among these possibilities, we generated point mutations to assess the contribution of individual amino acids in the juxtamembrane domain to inhibition of agrin-induced AChR clustering (Fig. 8A). As shown in Figure 8H, chimeric mutants with arginine or lysine residues replaced by alanines (CD8tmDGJ2RA and CD8tmDGJ3KA, respectively) resulted in a loss of inhibition and are statistically indistinguishable from the control CD8tmJ, whereas the mutant with cysteine and tyrosine replaced by serine and phenylalanine (CD8tmDGJCYSF) retained inhibitory activity. Thus, the inhibitory mechanism of β-dystroglycan partly depends on the basic amino acid residues RKKRK of the juxtamembrane region, whereas potential post-translational modifications of the cysteine and tyrosine residues are unlikely to be contributing factors.
The inhibitory effects of β-dystroglycan are presumed to reflect activity at the cell surface, because our assay detects only cell-surface exogenous protein. To rule out the possibility that some of our constructs act by inhibiting delivery of components to the cell surface, we compared the total levels of expression of our mutants as well as their level of cell-surface expression. All of the β-dystroglycan constructs and CD8 constructs had similar levels of expression as determined by Western blotting of transfected COS cells (Fig. 2 and see also Fig. 4D, insets, Figs. 7I, 8H, 9G). Furthermore, quantification of the anti-HA cell-surface labeling revealed no statistically significant differences among the proteins when expressed in myotubes (p > 0.05; Student's ttest; see Materials and Methods). Although we are unable to directly assess the total level of gene gun-mediated protein expression in myotubes because of the low frequency of transfection, the consistent levels of protein expression in COS cells, together with their equivalent levels of cell-surface expression in myotubes, argue against differences in protein trafficking between mutant proteins in myotubes.
The above experiments used the smallest agrin fragment with maximal AChR clustering activity; it contains four EGF-like domains and three laminin-like globular domains (G-domains). Deletion studies have demonstrated that the minimal agrin fragment that retains AChR clustering activity corresponds to the C-terminal (third) G-domain (Gesemann et al., 1995; Cornish et al., 1999). Because the first and second G-domains are believed to constitute the α-dystroglycan binding sites in agrin (Campanelli et al., 1996; Hopf and Hoch, 1996), we wondered whether the inhibitory effect of β-dystroglycan expression was specific to those agrin fragments that were capable of binding α-dystroglycan. Myotubes expressing β-dystroglycan or a control protein were challenged with the minimally active agrin fragment G3(8) (Cornish et al., 1999). As shown in Figure 10, β-dystroglycan expression results in inhibition of even this minimal, non-dystroglycan-binding agrin fragment. These results suggest that the cellular process inhibited by β-dystroglycan expression is common to the mechanism of clustering used by all neural agrin isoforms.
Fig. 10.
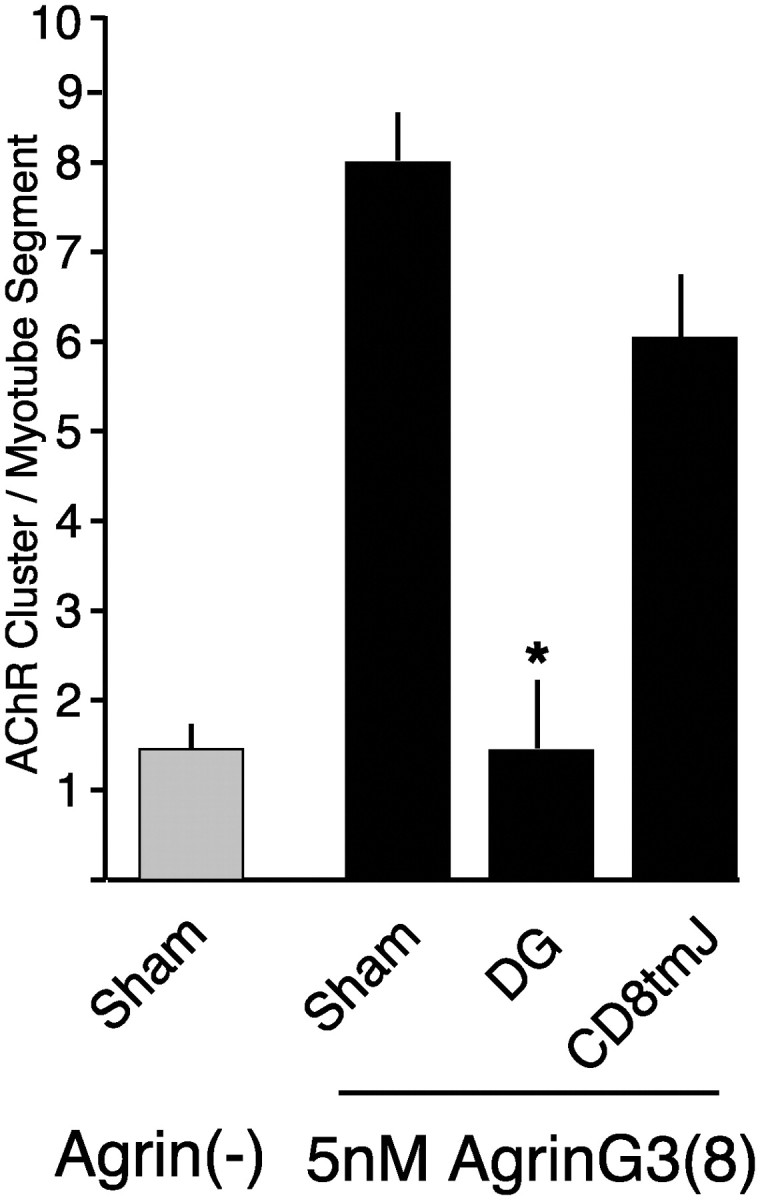
β-Dystroglycan inhibits activity of the minimally active domain of agrin. Transfected C2 myotubes were incubated with 5 nm agrin G3(8) for 16 hr. Expression of β-dystroglycan, but not CD8tmJ, inhibits agrin-induced clustering (mean ± SEM; n = 9). Theasterisk indicates mean value statistically different from sham (p < 0.05).
Discussion
We have demonstrated that β-dystroglycan can be expressed at the cell surface in the absence of coexpressed α-dystroglycan, and that expression of β-dystroglycan in cultured myotubes interferes with agrin-induced AChR clustering. Our results are consistent with previous findings that dystroglycan is involved in postsynaptic NMJ differentiation, but more importantly, our results define a region of β-dystroglycan that mediates interactions required for agrin-induced AChR clustering. We were surprised to find that deletion of the cytoplasmic dystrophin/utrophin/Grb2/caveolin binding sites did not relieve inhibition, which implies that these interactions are not required for dystroglycan function in AChR clustering. However, although our results demonstrate that the juxtamembrane region confers an inhibitory phenotype, the data cannot rule out the possibility that contributions are made by other cytoplasmic sequences; additional important interactions could be downstream or in parallel with juxtamembrane-mediated interactions. The potential nature of the juxtamembrane-mediated interaction as well as the relationship of our results to previous investigations of dystroglycan involvement in agrin activity is discussed below.
Experiments with myotubes derived from dystroglycan null cells reveal that although AChR clustering is profoundly altered, it can proceed in response to agrin (Grady et al., 2000). At least two possibilities could explain the apparent discrepancy between inhibition mediated by β-dystroglycan variants and cluster formation observed in dystroglycan null cells. First, most of our experiments have not included an analysis of clusters that are <1 μm in the largest dimension. The short time frame of the majority of our experiments (5 hr of agrin treatment) (Figs. 3, 7-9) may be insufficient for consolidation of microaggregates observed in dystroglycan null myotubes. However, even with longer incubations (16 hr of agrin treatment) (Figs. 5, 10), we still observed the inhibitory effect of β-dystroglycan expression, and our analysis of small AChR puncta (Fig. 3) reveals no alterations in microcluster numbers. These observations suggest that differences in analysis cannot explain the apparent contradiction of our results with previous work. Second, these experimental approaches differ significantly: removal of a protein versus introduction of truncated forms of a protein into the wild-type background. When a protein is deleted, it is possible for alternative pathways to perform the functions of the missing protein. When truncated forms are introduced, as in the present study, these variants reveal inhibition, because they engage only a subset of their binding partners that are required for function (i.e., cytosolic components but not ECM ligands). We favor this latter explanation: β-dystroglycan and its variants act in a dominant negative manner.
A previous study demonstrated that overexpression of full-length dystroglycan in Xenopus muscle in vivo resulted in a decrease in synaptic AChR density (Heathcote et al., 2000). The interpretation favored by these authors was that inhibition occurred by an indirect effect of α-dystroglycan binding to agrin, limiting its ability to interact productively, or at the correct location, with other important agrin-binding proteins. Our findings are consistent with this mechanism of inhibition for full-length dystroglycan. However, the inhibition demonstrated in the present work is mediated by β-dystroglycan sequences and is not limited to those fragments of agrin that bind α-dystroglycan. Therefore, we hypothesize that the juxtamembrane region of dystroglycan is involved in an interaction that is integral to the formation of postsynaptic membrane specializations in response to neural agrin.
What is the nature of the interaction mediated by the juxtamembrane basic region of β-dystroglycan? At least two scenarios are possible, and these are not mutually exclusive; this domain may mediate protein–protein or protein–lipid interactions. Previous findings are consistent with the notion that the juxtamembrane domain mediates protein–protein interactions. First, a membrane proximal interaction between β-dystroglycan and rapsyn has been documented. However, this interaction localizes to more C-terminal sequences in β-dystroglycan and is believed to involve a proline-rich region (Cartaud et al., 1998;Bartoli et al., 2001). Second, the juxtamembrane region of β-dystroglycan has been shown to bind dystrophin family proteins containing WW-hand and EF-hand domains [dystrophin, utrophin, and DP116 (Rentschler et al., 1999; Saito et al., 1999; James et al., 2000)]. Therefore, the dominant-negative effect may result from binding to one of these proteins in the absence of binding to ECM ligands. Alternatively, because the interaction between dystrophin family proteins and the juxtamembrane region of dystroglycan is less robust than the interaction of these proteins with the β-dystroglycan C-terminal PPxY motif, it is possible that additional proteins interact with the juxtamembrane region of dystroglycan. It is interesting that juxtamembrane regions of CD44, CD43, and ICAM-2 have been shown to harbor binding sites for ERM family proteins (Legg and Isacke, 1998; Yonemura et al., 1998) that are involved in cortical actin organization and interaction between F-actin and specific membrane proteins (Bretscher et al., 2000). The involvement of Rac and Cdc42 (Weston et al., 2000) as well as actin dynamics (Dai et al., 2000) in agrin-induced AChR clustering make an interaction between β-dystroglycan and F-actin linker proteins worthy of future investigation.
Several different observations suggest that the basic juxtamembrane domain of β-dystroglycan could mediate protein–lipid interactions. Basic amino acid sequences have been shown in several contexts to interact specifically with phosphatidylinositol head groups (Ben-Tal et al., 1996; Yonemura et al., 1998; Rohatgi et al., 2000). The interaction between phosphatidylinositol-3-phosphate and the FYVE membrane targeting domain of Vps27p is mediated by a basic pocket formed by an (R/K)(R/K) HHCR sequence (Misra and Hurley, 1999). Although this well characterized phospholipid binding domain is found in soluble proteins and is believed to enable recruitment of these proteins to the membrane, interactions of transmembrane proteins with phospholipids have been documented; syndecan-4 (Couchman and Woods, 1999) and ICAM-2 (Heiska et al., 1998) bind phosphatidylinositol-(4,5) bisphosphate via their juxtamembrane sequences. In these cases, functions other than membrane recruitment are likely, in that membrane protein–lipid interactions could serve to modulate the lipid environment or enable responses to changes in this environment. For example, specific phospholipids may concentrate proteins at sites of the cell membrane in response to local phopholipid kinase activation, as has been demonstrated in polarized pollen tube growth (Kost et al., 1999) and eukaryotic chemotaxis (Comer and Parent, 2002). Thus, the juxtamembrane sequence of dystroglycan could serve to localize dystroglycan to subdomains of the plasma membrane by interaction with specific phospholipids.
An alternative interaction between dystroglycan and specific lipids is suggested by the presence of a juxtamembrane cysteine residue. Several proteins with transmembrane domains have also been demonstrated to be acylated, including p75LNGFR (Barker et al., 1994), CD39 (Koziak et al., 2000), stomatin (Snyers et al., 1999), neurofascin (Ren and Bennett, 1998), and the transmembrane protein linker for activation of T cells (LAT) (Zhang et al., 1998, 1999; Harder and Kuhn, 2000). In p75 and neurofascin, the role of palmitoylation is unknown, although palmitoylation of stomatin and CD39 is proposed to play a role in oligomerization and targeting of these membrane proteins to caveolea. LAT acylation targets LAT to membrane microdomains (rafts), a localization that is suggested to be functionally important for T cell receptor signal transduction (Zhang et al., 1998, 1999; Harder and Kuhn, 2000). It has been suggested recently that agrin-induced AChR clustering may proceed through a mechanism that is shared with T cell function; it is interesting that both may involve agrin as well as the organization of signal transduction complexes in raft domains (Khan et al., 2001). The β-dystroglycan juxtamembrane domain could serve as a site of palmitoylation that would target dystroglycan to membrane domains involved in AChR cluster organization. Although our point mutagenesis data suggest that this type of modification is not absolutely required for agrin activity, we cannot rule out the possibility that dystroglycan becomes acylated while performing other cellular functions.
Available evidence is consistent with the following model for postsynaptic muscle-cell differentiation. Agrin acts through MuSK to initiates a series of events that include rapsyn and an Src-family kinase to prime cluster formation; dystroglycan-mediated events are then required for clusters to obtain or maintain their mature form. Our results leave open the question of how β-dystroglycan acts, but we favor the idea that the inhibitory effect of β-dystroglycan is attributable to the absence of an associated α-dystroglycan, the ECM binding subunit. However, although α-dystroglycan binds agrin directly, we found that activity of an agrin construct lacking dystroglycan binding domains is inhibited. This suggests that α-dystroglycan binding to ECM proteins in general, rather than to agrin specifically, is important for AChR clustering, and in addition, that these interactions must be coordinated with an intracellular interaction mediated by the juxtamembrane domain. It will now be informative to characterize the interactions mediated by this region of β-dystroglycan and to assess the contributions of the juxtamembrane domain to the other critical roles of dystroglycan outside the context of the synapse.
Footnotes
This work supported by grants from the Lucille P. Markey Charitable Trust and the Research Board of the University of Illinois to J.T.C. The monoclonal antibodies developed by D. Fischman (MF20) and D. Fambrough (CaF2-5D2) were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained by the Department of Biological Sciences of the University of Iowa.
Correspondence should be addressed to Dr. James T. Campanelli, Department of Biochemistry, MC 712, University of Illinois at Urbana-Champaign, 600 South Mathews Avenue, Urbana, IL 61801. E-mail:campanll@uiuc.edu.
References
- 1.Andrews DW, Young JC, Mirels LF, Czarnota GJ. The role of the N region in signal sequence and signal-anchor function. J Biol Chem. 1992;267:7761–7769. [PubMed] [Google Scholar]
- 2.Apel ED, Glass DJ, Moscoso LM, Yancopoulos GD, Sanes JR. Rapsyn is required for MuSK signaling and recruits synaptic components to a MuSK-containing scaffold. Neuron. 1997;18:623–635. doi: 10.1016/s0896-6273(00)80303-7. [DOI] [PubMed] [Google Scholar]
- 3.Barker PA, Barbee G, Misko TP, Shooter EM. The low affinity neurotrophin receptor, p75LNTR, is palmitoylated by thioester formation through cysteine 279. J Biol Chem. 1994;269:30645–30650. [PubMed] [Google Scholar]
- 4.Bartoli M, Ramarao MK, Cohen JB. Interactions of the rapsyn RING-H2 domain with dystroglycan. J Biol Chem. 2001;276:24911–24917. doi: 10.1074/jbc.M103258200. [DOI] [PubMed] [Google Scholar]
- 5.Ben-Tal N, Honig B, Peitzsch RM, Denisov G, McLaughlin S. Binding of small basic peptides to membranes containing acidic lipids: theoretical models and experimental results. Biophys J. 1996;71:561–575. doi: 10.1016/S0006-3495(96)79280-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bewick GS, Nicholson LV, Young C, O'Donnell E, Slater CR. Different distributions of dystrophin and related proteins at nerve-muscle junctions. NeuroReport. 1992;3:857–860. doi: 10.1097/00001756-199210000-00009. [DOI] [PubMed] [Google Scholar]
- 7.Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 8.Borges LS, Ferns M. Agrin-induced phosphorylation of the acetylcholine receptor regulates cytoskeletal anchoring and clustering. J Cell Biol. 2001;153:1–12. doi: 10.1083/jcb.153.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bretscher A, Chambers D, Nguyen R, Reczek D. ERM-merlin and EBP50 protein families in plasma membrane organization and function. Annu Rev Cell Dev Biol. 2000;16:113–143. doi: 10.1146/annurev.cellbio.16.1.113. [DOI] [PubMed] [Google Scholar]
- 10.Burgess RW, Nguyen QT, Son YJ, Lichtman JW, Sanes JR. Alternatively spliced isoforms of nerve- and muscle-derived agrin: their roles at the neuromuscular junction. Neuron. 1999;23:33–44. doi: 10.1016/s0896-6273(00)80751-5. [DOI] [PubMed] [Google Scholar]
- 11.Byers TJ, Kunkel LM, Watkins SC. The subcellular distribution of dystrophin in mouse skeletal, cardiac, and smooth muscle. J Cell Biol. 1991;115:411–421. doi: 10.1083/jcb.115.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Campanelli JT, Hoch W, Rupp F, Kreiner T, Scheller RH. Agrin mediates cell contact-induced acetylcholine receptor clustering. Cell. 1991;67:909–916. doi: 10.1016/0092-8674(91)90364-5. [DOI] [PubMed] [Google Scholar]
- 13.Campanelli JT, Roberds SL, Campbell KP, Scheller RH. A role for dystrophin-associated glycoproteins and utrophin in agrin-induced AChR clustering. Cell. 1994;77:663–674. doi: 10.1016/0092-8674(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 14.Campanelli JT, Gayer GG, Scheller RH. Alternative RNA splicing that determines agrin activity regulates binding to heparin and alpha-dystroglycan. Development. 1996;122:1663–1672. doi: 10.1242/dev.122.5.1663. [DOI] [PubMed] [Google Scholar]
- 15.Cartaud A, Coutant S, Petrucci TC, Cartaud J. Evidence for in situ and in vitro association between beta-dystroglycan and the subsynaptic 43K rapsyn protein. Consequence for acetylcholine receptor clustering at the synapse. J Biol Chem. 1998;273:11321–11326. doi: 10.1074/jbc.273.18.11321. [DOI] [PubMed] [Google Scholar]
- 16.Cavaldesi M, Macchia G, Barca S, Defilippi P, Tarone G, Petrucci TC. Association of the dystroglycan complex isolated from bovine brain synaptosomes with proteins involved in signal transduction. J Neurochem. 1999;72:1648–1655. doi: 10.1046/j.1471-4159.1999.721648.x. [DOI] [PubMed] [Google Scholar]
- 17.Chung W, Campanelli JT. WW and EF hand domains of dystrophin-family proteins mediate dystroglycan binding. Mol Cell Biol Res Commun. 1999;2:162–171. doi: 10.1006/mcbr.1999.0168. [DOI] [PubMed] [Google Scholar]
- 18.Comer FI, Parent CA. PI 3-kinase and PTEN: how opposites chemoattract. Cell. 2002;109:541–544. doi: 10.1016/s0092-8674(02)00765-1. [DOI] [PubMed] [Google Scholar]
- 19.Cornish T, Chi J, Johnson S, Lu Y, Campanelli JT. Globular domains of agrin are functional units that collaborate to induce acetylcholine receptor clustering. J Cell Sci. 1999;112:1213–1223. doi: 10.1242/jcs.112.8.1213. [DOI] [PubMed] [Google Scholar]
- 20.Cote PD, Moukhles H, Lindenbaum M, Carbonetto S. Chimaeric mice deficient in dystroglycans develop muscular dystrophy and have disrupted myoneural synapses. Nat Genet. 1999;23:338–342. doi: 10.1038/15519. [DOI] [PubMed] [Google Scholar]
- 21.Couchman JR, Woods A. Syndecan-4 and integrins: combinatorial signaling in cell adhesion. J Cell Sci. 1999;112:3415–3420. doi: 10.1242/jcs.112.20.3415. [DOI] [PubMed] [Google Scholar]
- 22.Dai Z, Luo X, Xie H, Peng HB. The actin-driven movement and formation of acetylcholine receptor clusters. J Cell Biol. 2000;150:1321–1334. doi: 10.1083/jcb.150.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- 24.Ferns M, Deiner M, Hall Z. Agrin-induced acetylcholine receptor clustering in mammalian muscle requires tyrosine phosphorylation. J Cell Biol. 1996;132:937–944. doi: 10.1083/jcb.132.5.937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fuhrer C, Sugiyama JE, Taylor RG, Hall ZW. Association of muscle-specific kinase MuSK with the acetylcholine receptor in mammalian muscle. EMBO J. 1997;16:4951–4960. doi: 10.1093/emboj/16.16.4951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gautam M, Noakes PG, Mudd J, Nichol M, Chu GC, Sanes JR, Merlie JP. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature. 1995;377:232–236. doi: 10.1038/377232a0. [DOI] [PubMed] [Google Scholar]
- 27.Gautam M, DeChiara TM, Glass DJ, Yancopoulos GD, Sanes JR. Distinct phenotypes of mutant mice lacking agrin, MuSK, or rapsyn. Brain Res Dev Brain Res. 1999;114:171–178. doi: 10.1016/s0165-3806(99)00013-9. [DOI] [PubMed] [Google Scholar]
- 28.Gee SH, Montanaro F, Lindenbaum MH, Carbonetto S. Dystroglycan-alpha, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell. 1994;77:675–686. doi: 10.1016/0092-8674(94)90052-3. [DOI] [PubMed] [Google Scholar]
- 29.Gesemann M, Denzer AJ, Ruegg MA. Acetylcholine receptor-aggregating activity of agrin isoforms and mapping of the active site. J Cell Biol. 1995;128:625–636. doi: 10.1083/jcb.128.4.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glass DJ, Apel ED, Shah S, Bowen DC, DeChiara TM, Stitt TN, Sanes JR, Yancopoulos GD. Kinase domain of the muscle-specific receptor tyrosine kinase (MuSK) is sufficient for phosphorylation but not clustering of acetylcholine receptors: required role for the MuSK ectodomain? Proc Natl Acad Sci USA. 1997;94:8848–8853. doi: 10.1073/pnas.94.16.8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grady RM, Zhou H, Cunningham JM, Henry MD, Campbell KP, Sanes JR. Maturation and maintenance of the neuromuscular synapse: genetic evidence for roles of the dystrophin–glycoprotein complex. Neuron. 2000;25:279–293. doi: 10.1016/s0896-6273(00)80894-6. [DOI] [PubMed] [Google Scholar]
- 32.Harder T, Kuhn M. Selective accumulation of raft-associated membrane protein LAT in T cell receptor signaling assemblies. J Cell Biol. 2000;151:199–208. doi: 10.1083/jcb.151.2.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heathcote RD, Ekman JM, Campbell KP, Godfrey EW. Dystroglycan overexpression in vivo alters acetylcholine receptor aggregation at the neuromuscular junction. Dev Biol. 2000;227:595–605. doi: 10.1006/dbio.2000.9906. [DOI] [PubMed] [Google Scholar]
- 34.Heiska L, Alfthan K, Gronholm M, Vilja P, Vaheri A, Carpen O. Association of ezrin with intercellular adhesion molecule-1 and -2 (ICAM-1 and ICAM-2). Regulation by phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 1998;273:21893–21900. doi: 10.1074/jbc.273.34.21893. [DOI] [PubMed] [Google Scholar]
- 35.Henry MD, Campbell KP. Dystroglycan inside and out. Curr Opin Cell Biol. 1999;11:602–607. doi: 10.1016/s0955-0674(99)00024-1. [DOI] [PubMed] [Google Scholar]
- 36.Hobert M, Carlin C. Cytoplasmic juxtamembrane domain of the human EGF receptor is required for basolateral localization in MDCK cells. J Cell Physiol. 1995;162:434–446. doi: 10.1002/jcp.1041620316. [DOI] [PubMed] [Google Scholar]
- 37.Hopf C, Hoch W. Agrin binding to alpha-dystroglycan. Domains of agrin necessary to induce acetylcholine receptor clustering are overlapping but not identical to the alpha-dystroglycan-binding region. J Biol Chem. 1996;271:5231–5236. doi: 10.1074/jbc.271.9.5231. [DOI] [PubMed] [Google Scholar]
- 38.Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355:696–702. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 39.Jacobson C, Montanaro F, Lindenbaum M, Carbonetto S, Ferns M. α-Dystroglycan functions in acetylcholine receptor aggregation but is not a coreceptor for agrin-MuSK signaling. J Neurosci. 1998;18:6340–6348. doi: 10.1523/JNEUROSCI.18-16-06340.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.James M, Nuttall A, Ilsley JL, Ottersbach K, Tinsley JM, Sudol M, Winder SJ. Adhesion-dependent tyrosine phosphorylation of (beta)-dystroglycan regulates its interaction with utrophin. J Cell Sci. 2000;113:1717–1726. doi: 10.1242/jcs.113.10.1717. [DOI] [PubMed] [Google Scholar]
- 41.Jones MA, Werle MJ. Nitric oxide is a downstream mediator of agrin-induced acetylcholine receptor aggregation. Mol Cell Neurosci. 2000;16:649–660. doi: 10.1006/mcne.2000.0901. [DOI] [PubMed] [Google Scholar]
- 42.Jung D, Yang B, Meyer J, Chamberlain JS, Campbell KP. Identification and characterization of the dystrophin anchoring site on beta-dystroglycan. J Biol Chem. 1995;270:27305–27310. doi: 10.1074/jbc.270.45.27305. [DOI] [PubMed] [Google Scholar]
- 43.Khan AA, Bose C, Yam LS, Soloski MJ, Rupp F. Physiological regulation of the immunological synapse by agrin. Science. 2001;292:1681–1686. doi: 10.1126/science.1056594. [DOI] [PubMed] [Google Scholar]
- 44.Kost B, Lemichez E, Spielhofer P, Hong Y, Tolias K, Carpenter C, Chua NH. Rac homologues and compartmentalized phosphatidylinositol 4,5-bisphosphate act in a common pathway to regulate polar pollen tube growth. J Cell Biol. 1999;145:317–330. doi: 10.1083/jcb.145.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koziak K, Kaczmarek E, Kittel A, Sevigny J, Blusztajn JK, Schulte Am Esch J, Imai M, Guckelberger O, Goepfert C, Qawi I, Robson SC. Palmitoylation targets CD39/endothelial ATP diphosphohydrolase to caveolae. J Biol Chem. 2000;275:2057–2062. doi: 10.1074/jbc.275.3.2057. [DOI] [PubMed] [Google Scholar]
- 46.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 47.Lee T, Luo L. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron. 1999;22:451–461. doi: 10.1016/s0896-6273(00)80701-1. [DOI] [PubMed] [Google Scholar]
- 48.Legg JW, Isacke CM. Identification and functional analysis of the ezrin-binding site in the hyaluronan receptor, CD44. Curr Biol. 1998;8:705–708. doi: 10.1016/s0960-9822(98)70277-5. [DOI] [PubMed] [Google Scholar]
- 49.Luck G, Hoch W, Hopf C, Blottner D. Nitric oxide synthase (NOS-1) coclustered with agrin-induced AChR-specializations on cultured skeletal myotubes. Mol Cell Neurosci. 2000;16:269–281. doi: 10.1006/mcne.2000.0873. [DOI] [PubMed] [Google Scholar]
- 50.Marchand S, Stetzkowski-Marden F, Cartaud J. Differential targeting of components of the dystrophin complex to the postsynaptic membrane. Eur J Neurosci. 2001;13:221–229. [PubMed] [Google Scholar]
- 51.McMahan UJ. The agrin hypothesis. Cold Spring Harb Symp Quant Biol. 1990;55:407–418. doi: 10.1101/sqb.1990.055.01.041. [DOI] [PubMed] [Google Scholar]
- 52.Megeath LJ, Fallon JR. Intracellular calcium regulates agrin-induced acetylcholine receptor clustering. J Neurosci. 1998;18:672–678. doi: 10.1523/JNEUROSCI.18-02-00672.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Misra S, Hurley JH. Crystal structure of a phosphatidylinositol 3-phosphate-specific membrane-targeting motif, the FYVE domain of Vps27p. Cell. 1999;97:657–666. doi: 10.1016/s0092-8674(00)80776-x. [DOI] [PubMed] [Google Scholar]
- 54.Mohamed AS, Rivas-Plata KA, Kraas JR, Saleh SM, Swope SL. Src-class kinases act within the agrin/MuSK pathway to regulate acetylcholine receptor phosphorylation, cytoskeletal anchoring, and clustering. J Neurosci. 2001;21:3806–3818. doi: 10.1523/JNEUROSCI.21-11-03806.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Phillips WD, Noakes PG, Roberds SL, Campbell KP, Merlie JP. Clustering and immobilization of acetylcholine receptors by the 43-kD protein: a possible role for dystrophin-related protein. J Cell Biol. 1993;123:729–740. doi: 10.1083/jcb.123.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qu Z, Huganir RL. Comparison of innervation and agrin-induced tyrosine phosphorylation of the nicotinic acetylcholine receptor. J Neurosci. 1994;14:6834–6841. doi: 10.1523/JNEUROSCI.14-11-06834.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ren Q, Bennett V. Palmitoylation of neurofascin at a site in the membrane-spanning domain highly conserved among the L1 family of cell adhesion molecules. J Neurochem. 1998;70:1839–1849. doi: 10.1046/j.1471-4159.1998.70051839.x. [DOI] [PubMed] [Google Scholar]
- 58.Rentschler S, Linn H, Deininger K, Bedford MT, Espanel X, Sudol M. The WW domain of dystrophin requires EF-hands region to interact with beta-dystroglycan. Biol Chem. 1999;380:431–442. doi: 10.1515/BC.1999.057. [DOI] [PubMed] [Google Scholar]
- 59.Rohatgi R, Ho HY, Kirschner MW. Mechanism of N-WASP activation by CDC42 and phosphatidylinositol 4,5-bisphosphate. J Cell Biol. 2000;150:1299–1310. doi: 10.1083/jcb.150.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Russo K, Di Stasio E, Macchia G, Rosa G, Brancaccio A, Petrucci TC. Characterization of the beta-dystroglycan-growth factor receptor 2 (Grb2) interaction. Biochem Biophys Res Commun. 2000;274:93–98. doi: 10.1006/bbrc.2000.3103. [DOI] [PubMed] [Google Scholar]
- 61.Saito F, Masaki T, Kamakura K, Anderson LV, Fujita S, Fukuta-Ohi H, Sunada Y, Shimizu T, Matsumura K. Characterization of the transmembrane molecular architecture of the dystroglycan complex in Schwann cells. J Biol Chem. 1999;274:8240–8246. doi: 10.1074/jbc.274.12.8240. [DOI] [PubMed] [Google Scholar]
- 62.Sander A, Hesser BA, Witzemann V. MuSK induces in vivo acetylcholine receptor clusters in a ligand-independent manner. J Cell Biol. 2001;155:1287–1296. doi: 10.1083/jcb.200105034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- 64.Snyers L, Umlauf E, Prohaska R. Cysteine 29 is the major palmitoylation site on stomatin. FEBS Lett. 1999;449:101–104. doi: 10.1016/s0014-5793(99)00417-2. [DOI] [PubMed] [Google Scholar]
- 65.Sotgia F, Lee JK, Das K, Bedford M, Petrucci TC, Macioce P, Sargiacomo M, Bricarelli FD, Minetti C, Sudol M, Lisanti MP. Caveolin-3 directly interacts with the C-terminal tail of beta-dystroglycan. Identification of a central WW-like domain within caveolin family members. J Biol Chem. 2000;275:38048–38058. doi: 10.1074/jbc.M005321200. [DOI] [PubMed] [Google Scholar]
- 66.Sugiyama J, Bowen DC, Hall ZW. Dystroglycan binds nerve and muscle agrin. Neuron. 1994;13:103–115. doi: 10.1016/0896-6273(94)90462-6. [DOI] [PubMed] [Google Scholar]
- 67.Wahlberg JM, Spiess M. Multiple determinants direct the orientation of signal-anchor proteins: the topogenic role of the hydrophobic signal domain. J Cell Biol. 1997;137:555–562. doi: 10.1083/jcb.137.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wallace BG. Regulation of the interaction of nicotinic acetylcholine receptors with the cytoskeleton by agrin-activated protein tyrosine kinase. J Cell Biol. 1995;128:1121–1129. doi: 10.1083/jcb.128.6.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weston C, Yee B, Hod E, Prives J. Agrin-induced acetylcholine receptor clustering is mediated by the small guanosine triphosphatases rac and cdc42. J Cell Biol. 2000;150:205–212. doi: 10.1083/jcb.150.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang B, Jung D, Motto D, Meyer J, Koretzky G, Campbell KP. SH3 domain-mediated interaction of dystroglycan and Grb2. J Biol Chem. 1995;270:11711–11714. doi: 10.1074/jbc.270.20.11711. [DOI] [PubMed] [Google Scholar]
- 71.Yonemura S, Hirao M, Doi Y, Takahashi N, Kondo T, Tsukita S. Ezrin/radixin/moesin (ERM) proteins bind to a positively charged amino acid cluster in the juxta-membrane cytoplasmic domain of CD44, CD43, and ICAM-2. J Cell Biol. 1998;140:885–895. doi: 10.1083/jcb.140.4.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoon SO, Soltoff SP, Chao MV. A dominant role of the juxtamembrane region of the TrkA nerve growth factor receptor during neuronal cell differentiation. J Biol Chem. 1997;272:23231–23238. doi: 10.1074/jbc.272.37.23231. [DOI] [PubMed] [Google Scholar]
- 73.Zhang W, Trible RP, Samelson LE. LAT palmitoylation: its essential role in membrane microdomain targeting and tyrosine phosphorylation during T cell activation. Immunity. 1998;9:239–246. doi: 10.1016/s1074-7613(00)80606-8. [DOI] [PubMed] [Google Scholar]
- 74.Zhang W, Irvin BJ, Trible RP, Abraham RT, Samelson LE. Functional analysis of LAT in TCR-mediated signaling pathways using a LAT-deficient Jurkat cell line. Int Immunol. 1999;11:943–950. doi: 10.1093/intimm/11.6.943. [DOI] [PubMed] [Google Scholar]