

Abstract
Neuronal dystrophy is a pathological hallmark of Alzheimer's disease (AD) that is not observed in other neurodegenerative disorders that lack amyloid deposition. Treatment of cortical neurons with fibrillar amyloid β (Aβ) peptides induces progressive neuritic dystrophy accompanied by a marked loss of synaptophysin immunoreactivity (Grace et al., 2002). Here, we report that fibrillar Aβ-induced neuronal dystrophy is mediated by the activation of focal adhesion (FA) proteins and the formation of aberrant FA structures adjacent to Aβ deposits. In the AD brain, activated FA proteins are observed associated with the majority of senile plaques. Clustered integrin receptors and activated paxillin (phosphorylated at Tyr-31) and focal adhesion kinase (phosphorylated at Tyr-297) are mainly detected in dystrophic neurites surrounding Aβ plaque cores, where they colocalize with hyperphosphorylated tau. Deletion experiments demonstrated that the presence of the LIM domains in the paxillin C terminus and the recruitment of the protein-Tyr phosphatase (PTP)-PEST to the FA complex are required for Aβ-induced neuronal dystrophy. Therefore, both paxillin and PTP-PEST appear to be critical elements in the generation of the dystrophic response. Paxillin is a scaffolding protein to which other FA proteins bind, leading to the formation of the FA contact and initiation of signaling cascades. PTP-PEST plays a key role in the dynamic regulation of focal adhesion contacts in response to extracellular cues. Thus, in the AD brain, fibrillar Aβ may induce neuronal dystrophy by triggering a maladaptive plastic response mediated by FA protein activation and tau hyperphosphorylation.
Keywords: Alzheimer's disease, amyloid β, senile plaques, neuronal dystrophy, neurodegeneration, focal adhesion
Introduction
Alzheimer's disease (AD) neuropathology is characterized by the presence of neurofibrillar tangles (NFTs) and amyloid plaques in the brains of affected individuals. NFTs are intraneuronal bundles of paired helical filaments composed of hyperphosphorylated tau. Amyloid plaques are extracellular lesions composed primarily of a core of fibrillar amyloid β (Aβ) surrounded by reactive astrocytes, microglial cells, and dystrophic neurites (Selkoe, 2001). Previous work indicates that Aβ fibrils and protofibrils are neurotoxic (Yankner, 1996; Lashuel et al., 2002). Aβ fibrils induce neuronal dystrophy (Busciglio et al., 1992; Pike et al., 1992, Grace et al., 2002), tau hyperphosphorylation (Busciglio et al., 1995), and neuronal death in vitro (Yankner et al., 1990;Pike et al., 1991; Mattson et al., 1992) and in vivo (Geula et al., 1998). In this regard, neuronal dystrophy is specifically associated with AD and is not present in other neurodegenerative conditions that lack amyloid plaques (Benzing et al., 1993). In AD, the number of dystrophic neurites correlates with the degree of dementia (McKee et al., 1991), suggesting a close association between the development of neuronal dystrophy and the cognitive decline in AD patients. A pathological relationship between neuronal dystrophy and cognitive impairment is further suggested by studies of transgenic mice expressing the “Swedish” mutation of APP. These animals develop abundant amyloid deposits and neuronal dystrophy without significant cell death and exhibit behavioral deficits consistent with memory loss (Irizarry et al., 1997). In the AD brain, synaptic loss is observed in areas of aberrant neuronal sprouting (Geddes et al., 1986; Masliah et al., 1991), and in cortical cultures exposed to fibrillar Aβ, neuronal dystrophy is associated with synaptic loss (Grace et al., 2002). Thus, a pathological relationship among Aβ deposition, neuronal dystrophy, and synaptic loss may lead to cognitive impairment in AD.
The molecular mechanism by which neuronal dystrophy occurs is unclear. Neuronal dystrophy and cell death induced by fibrillar Aβ take place over different time courses and at different Aβ concentrations, raising the possibility that both events are mediated by separate molecular mechanisms (Grace et al., 2002). Extracellular signals producing alterations in the cytoskeleton are often transduced through adhesion proteins (Mueller et al., 1989). In this regard, fibrillar Aβ could promote dystrophy by aberrantly activating signal transduction cascades leading to cytoskeletal changes (Saitoh et al., 1993). Aβ binds to integrins (Kowalska and Badellino, 1994; Sabo et al., 1995; Goodwin et al., 1997) and activates the focal adhesion (FA) proteins paxillin and focal adhesion kinase (FAK), which are downstream of integrin receptors, suggesting that FA signaling cascades might be involved in Aβ-induced neuronal dystrophy, cell death, or both (Zhang et al., 1994, 1996; Berg et al., 1997; Williamson et al., 2002). To address the role of FA signaling in Aβ-induced neuronal dystrophy, we analyzed the expression and activity of FA proteins in the AD brain and in cultured neurons exposed to fibrillar Aβ. Our results indicate that the aberrant activation of FA proteins by Aβ may play a critical role in the development of neuronal dystrophy and AD neuropathology.
Materials and Methods
Neuronal cultures. Rat cortical cultures were established from embryonic day 17 fetuses as described previously (Busciglio et al., 1993). The cells were plated on laminin-coated (10 μg/ml) or poly-l-lysine-coated (250 μg/ml) dishes or glass coverslips at a density of 10,000 cells/cm2 in DMEM plus 10% calf serum for 2 hr. After cell attachment, the media were replaced with DMEM plus a 1:1 mixture of N2 and B27 supplements (Invitrogen, Grand Island, NY).
Aβ treatments. Peptide treatments were performed as described previously (Pigino et al., 2001; Grace et al., 2002). Synthetic Aβ peptides 1–40 and 25–35 (Bachem, King of Prussia, PA) were dissolved in double-distilled H2O to a concentration of 2 mm. The peptide stock solutions were incubated at 37°C for 3 d to allow fibril formation. These preparations are composed primarily of fibrillar Aβ, as shown by electron microscopy and Congo red birefringence (Lorenzo and Yankner, 1994). Peptide treatments were initiated at day 5 in culture. Fibrillar Aβ peptides were added directly to the media for a final concentration of 20 μm. Nonfibrillar Aβ peptides were added to the medium without preincubation.
Western blot and immunoprecipitation. Cultures were harvested in radioimmunoprecipitation assay (RIPA) buffer (1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS in PBS) plus protease inhibitors (Complete, Roche Molecular Biochemicals, Indianapolis, IN) and phosphatase inhibitors (Busciglio et al., 1993) at 4°C. The cell homogenates were centrifuged at 100,000 × g for 30 min. The supernatant was collected, and the protein content was determined using a commercial kit (Bio-Rad, Hercules, CA). Cytoskeletal proteins were obtained in a similar manner; however, before harvesting with RIPA buffer, the cells were placed in microtubule-stabilizing buffer (MSB; 0.13 m HEPES pH 6.9, 2 mm MgCl2, and 10 mm EGTA) at 37°C for 1 min, and then the cells were incubated with 0.2% Triton X-100 in MSB for 2 min at 37°C. The protein homogenates were separated on standard 10% polyacrylamide gels or precast 4–20% gradient gels and transferred to polyvinylidene difluoride membranes. After blocking with 5% milk or 5% bovine serum albumin in PBS, the membranes were incubated overnight at 4°C with mouse anti-paxillin (1:10,000; BioSource, Camarillo, CA) or mouse anti-phospho-Tyr (1:3000, 4G10; Upstate Biotechnology, Waltham, MA). To control for equal protein loading, membranes were incubated with mouse anti-tubulin class III (1:5000; Sigma, St. Louis, MO). Protein bands were visualized by chemiluminescence. Quantitative analysis was performed using a FastScan densitometer (Molecular Dynamics, Sunnyvale, CA) as described previously (Pigino et al., 2001). Volume analysis was performed on the appropriate bands using NIH Image software. The values were expressed as the percent change in protein levels in treated samples compared with control samples. Proteins were immunoprecipitated as described previously (Pigino et al., 2001). Briefly, whole-cell homogenates were incubated with primary antibody overnight at 4°C and immunoprecipitated with protein A-Sepharose beads (Amersham Biosciences, Piscataway, NJ) for 4 hr. The beads were washed and resuspended in sample buffer. The specificity of the immunoprecipitation was controlled by replacing the primary antibody with the corresponding nonimmune serum.
Transfection. Green fluorescent protein (GFP)-tagged full-length N- and C-terminal paxillin constructs were provided by Dr. R. Salgia (Dana-Faber Cancer Institute, Boston, MA). Paxillin constructs containing site-specific Cys-to-Ala mutations at amino acids 467, 470, and 523 and a double mutation at 467 and 470 were provided by Dr. M. L. Tremblay (McGill University, Montreal, Quebec, Canada). The FAK dominant negative construct FRNK was provided by Dr. L. Languino (Yale University, New Haven, CT). A protein-Tyr phosphatase (PTP)-PEST construct bearing a deletion in the Pro2 domain comprising amino acids 367–400 (PESTdl) was provided by Dr. M. D. Schaller (University of North Carolina, Chapel Hill, NC). A GFP expression vector (Clontech, Palo Alto, CA) was used as a control. Primary cortical neurons were transfected at day 5 using calcium phosphate as described previously (Pigino et al., 2001). Neurons were transfected in triplicate wells in 24-well plates. The cultures were processed 24 or 48 hr after transfection.
Immunocytochemistry. Cultured neurons were fixed in 4% paraformaldehyde and 0.12 m sucrose in PBS for 30 min at 37°C, permeabilized with 0.1% Triton X-100 in PBS, and blocked for 30 min in 5% BSA. The cells were then incubated overnight at 4°C with mouse anti-paxillin (1:100), rabbit anti-α5β1-integrin receptor (1:500; provided by Dr. Z. Zhan, The Burnham Institute, San Diego, CA), anti-PTP 4G10 (1:100), PY20 (1:100; Zymed, San Francisco, CA), and anti-PTP-PEST (1:500; provided by Dr. M. D. Schaller; Shen et al., 2000), followed by incubation in goat anti-mouse or rabbit secondary antibodies conjugated to Alexa fluoro-green (Molecular Probes, Eugene, OR). Double labeling was performed using phalloidin-Texas Red (Molecular Probes) to visualize microfilaments. Triple labeling was performed by sequential incubation with mouse anti-Aβ (1:500, clone 6F/3D; Dako, Glostrup, Denmark) followed by anti-mouse conjugated with Cascade blue (Molecular Probes).
Immunohistology. Brain tissue samples were obtained from the Harvard Brain Tissue Resource Center (McLean Hospital, Boston, MA). Bielchowski silver staining was performed on AD brain tissue as described previously (Carson, 1990). Paraffin-embedded tissue sections of AD brains and age-matched controls were processed as described previously (Busciglio et al., 1997). Briefly, the paraffin was removed, and the tissue was hydrated in H2O. Incubation with a boiling citrate solution (1.05 gm of citric acid in 450 ml of distilled H2O, pH 6.0) for 6 min was used to enhance epitope exposure. Double labeling was performed by sequential immunostaining. The following primary antibodies were used: anti-α5β1-integrin receptor (1:100), rabbit anti-β1-integrin subunit (1:40; Chemicon, Temecula, CA), PHF-1, which recognizes phosphorylated tau at Ser-396 and Ser-404 (1:500; Greenberg et al., 1992), mouse anti-Aβ 6E10 (1:500; Senetek, Napa, CA), rabbit anti-paxillin phosphorylated at residue 31 (1:100; Biosource), and rabbit anti-FAK phosphorylated at Tyr-397 (1:100; Biosource). The immunoreaction was visualized with the enzyme-linked fluorescence kit following the vendor's protocol (Molecular Probes). The second primary antibody was visualized by incubation with a secondary antibody conjugated with Cascade blue. The tissue was mounted and analyzed using a fluorescent Olympus Optical (Tokyo, Japan) IX-70 inverted microscope. Fluorescent images were captured with a CCD camera (Spot; Diagnostic Instruments, Sterling Heights, MI) driven by Spot acquisition software.
Viability assay. Neuronal viability was evaluated using a propidium iodide exclusion assay as described previously (Busciglio and Yankner, 1995; Grace et al., 2002). The number of propidium-positive nuclei (nonviable cells) in transfected neurons was expressed as a percentage of the number of nonviable neurons transfected with the GFP expression vector. More than 200 cells were analyzed per experimental condition in each individual experiment.
Morphometric analysis. Neurons were analyzed by fluorescent microscopy at a final magnification of 400×. Individual neurons were scored as nondystrophic or dystrophic according to defined morphological features (Pigino et al., 2001; Grace et al., 2002). Similar results scoring the number of dystrophic neurons on control and Aβ-treated cultures were obtained by three different examiners analyzing independent experiments. Individual neurons were characterized as dystrophic when neurites showed obvious aberrant morphology, such as acute angles and loops, or grew upward. The identity of the cultures was coded to exclude experimental bias. More than 250 cells were scored per experimental condition in each individual experiment. Neuronal dystrophy was expressed as a percentage of the number of dystrophic neurons transfected with GFP. Neuritic caliber was assessed by image analysis in control and dystrophic neurons. To standardize the point of caliber measurement between neurons, neurite caliber was measured in control and fibrillar Aβ-treated neurons immediately after the first branch point. This parameter was chosen instead of a fixed distance from the cell body to overcome the difficulty of measuring accurately the length of dystrophic neurites with tortuous morphology. In total, 57 neurons from three different experiments were analyzed. To evaluate the clustering of FA proteins, neurons were labeled with phalloidin, anti-integrin, anti-paxillin, or anti-tubulin antibodies, and the immunofluorescence intensity was measured at FA sites and at adjacent sites periodically localized along the neurite using NIH Image software. FA sites were identified by the increase in microfilament density compared with adjacent areas of similar size in the same neuronal process. In each case, FA sites were scored in at least 10 neurites per culture in triplicate cultures growing on laminin or poly-l-lysine.
Statistics. All experiments were performed in triplicate samples and replicated at least three times. Data are expressed as the mean ± SEM. Asterisks in the figures indicate significant statistical differences between groups as determined by Student'st test or ANOVA followed by the Student–Newman–Keulspost hoc test.
Results
Fibrillar Aβ-induced neuronal dystrophy, paxillin Tyr phosphorylation, and translocation to the cytoskeleton
During the assembly of FA structures, paxillin is phosphorylated at Tyr residues and translocates to the cytoskeleton, where it acts as a scaffolding protein binding integrin receptors, kinases such as FAK, phosphatases such as PTP-PEST, adapter proteins, and actin-binding proteins (Schaller et al., 1995; Turner, 2000). This FA complex mediates spatial and temporal interactions that are initiated on integrin receptor activation. The interplay between kinases and phosphatases leads to the initiation of multiple signal transduction cascades that promote local changes in the cytoskeleton, which result in modifications in cell morphology (Aplin et al., 1998; Turner, 2000). To establish the role of FA proteins in Aβ-induced neuronal dystrophy, cortical neurons grown on poly-l-lysine were treated with 20 μm fibrillar Aβ for 2 d. Under these treatment conditions, >80% of the neurons present in the culture remained viable, whereas >55% of them developed dystrophic morphological features (Grace et al., 2002), such as abnormal neuritic branches protruding from cell bodies and increased tortuosity of processes, including acute angles and loops (Fig.1A, arrows). A significant decrease in neuritic caliber also characterized dystrophic neurons (Fig. 1A). Neuritic caliber was measured in control and Aβ-treated neurons immediately after the first branch point to overcome the difficulty of measuring accurately the length of highly dystrophic neurites. The results showed average calibers of 1.09 ± 0.05 (SEM) μm for control neurons and 0.59 ± 0.04 μm for Aβ-treated neurons, indicating a significant decrease in neurite caliber in dystrophic neurons (p < 0.0001 by Student's t test). Similar dystrophic effects were observed with either Aβ1–40 or 25–35 peptides. Cultures treated with vehicle alone, reverse-sequence 35–25, or nonfibrillar Aβ1–40 did not show any morphological changes or decreased viability (Fig. 1A,control) (Grace et al., 2002).
Fig. 1.

Fibrillar Aβ induces neuronal dystrophy, Tyr phosphorylation, and paxillin activation in cortical neurons. Aβ was added at day 5, and the cultures were processed at day 7 in culture.A, Neurites appear smooth and healthy incontrol cultures. Fibrillar Aβ-induced aberrant neurite morphology, including decreased caliber and acute angles and loops (Aβ, arrows). Neurons were immunolabeled with anti-tubulin class III antibody. Scale bar, 10 μm.B, Western blot of whole-cell homogenates developed with anti-phospho-Tyr antibody 4G10. Note the increase in phospho-Tyr in several bands after Aβ treatment (arrowheads) and decreased Tyr phosphorylation in a band of ∼180 kDa (small arrow). Con, Control. C, Homogenates were immunoprecipitated (IP) with anti-paxillin (α-pax) antibody and immunostained with 4G10. Note the increase in paxillin Tyr phosphorylation in Aβ-treated neurons (Aβ). Immunoprecipitates with nonimmune (NI) serum were negative. Con, Control; WB, Western blot. D, Paxillin Tyr phosphorylation was quantified by densitometry in vehicle-treated samples (Con) and normalized as 100%. Note the significant increase (170 ± 30%) induced by fibrillar Aβ (Aβ). The membranes were reprobed for paxillin to confirm equal loading. Values are mean ± SEM;n = 4 independent experiments; *p < 0.05. E, Western blot analysis of whole-cell homogenates and cytoskeletal extracts. The blots were developed with anti-paxillin antibody (pax). Note the increase in paxillin (pax) in the cytoskeletal fraction (Cytosk) of Aβ-treated neurons (Aβ). Total paxillin levels in whole-cell homogenates did not change. Similar tubulin levels (tub) confirmed equal loading. Con, Control. F, Densitometric quantification of paxillin in the cytoskeleton revealed a 680 ± 200% increase in Aβ-treated neurons grown on poly-l-lysine and a 210 ± 30% increase in control (Con) neurons grown on laminin (Lam).n = 9 independent experiments; *p < 0.05 relative to control by Student'st test.
Western blot analysis of whole-cell homogenates revealed a marked increase in Tyr phosphorylation in several protein bands in Aβ-treated samples (Fig. 1B,arrowheads). We also detected a clear reduction in the level of phospho-Tyr in at least one band (Fig. 1B,arrow), consistent with the activation of Tyr phosphatases. Western blot analysis of whole-cell homogenates immunoprecipitated with an anti-paxillin antibody revealed a significant increase in paxillin Tyr phosphorylation in Aβ-treated cultures (Fig. 1C,D). This result is consistent with previous reports showing the activation of Tyr kinase activity and increased paxillin Tyr phosphorylation in neuronal cells treated with fibrillar Aβ (Berg et al., 1997;Williamson et al., 2002). Paxillin translocation to the cytoskeleton was assessed by Western blot of whole-cell homogenates and cytoskeletal preparations. Although there was no difference in the level of total paxillin in whole-cell homogenates of control and Aβ-treated samples (Fig. 1E), there was a dramatic increase in the level of paxillin associated with the cytoskeleton after Aβ treatment (Fig.1E,F). Neurons grown on laminin, which is an integrin ligand that activates FA assembly, also showed a significant increase in paxillin translocation to the cytoskeleton (Fig.1F). Thus, fibrillar Aβ induces neuronal dystrophy that correlates with increased paxillin Tyr phosphorylation and translocation to the cytoskeleton.
Fibrillar Aβ induces aberrant FA-like structures in dystrophic neurons
Ligand binding to the integrin receptor initiates a signaling cascade that initiates Tyr phosphorylation and recruits several proteins to the FA site, including paxillin, FAK, vinculin, and α-actinin, leading to a local increase in microfilament assembly (Aplin et al., 1998, Cukierman et al., 2001). The clustering and colocalization of two or more FA proteins is considered a hallmark for the morphological identification of FA contacts (Katoh et al., 1995;Dogic et al., 1998, Cukierman et al., 2001). To determine whether paxillin activation is associated with the formation of FA sites in dystrophic neurons, FA sites were identified as clusters of increased paxillin or integrin immunoreactivity associated with microfilament assembly. Neurons grown on poly-l-lysine substrate exhibited a homogeneous distribution of paxillin and integrin along the neurites, and the presence of microfilaments was mainly observed in the growth cone region (Fig.2A–D,arrows). This result is consistent with the absence of FA contacts in neurons growing on poly-l-lysine. In contrast, neurons grown on laminin, an integrin ligand that activates FA assembly, exhibited clusters of paxillin and integrin immunofluorescence associated with microfilaments that were localized periodically along the neurites, consistent with integrin activation and FA site formation (Fig. 2E,F, arrows). Double immunofluorescence with anti-integrin and anti-paxillin antibodies confirmed that both proteins colocalized at FA sites (data not shown). Quantitative image analysis revealed a significant increase in paxillin and integrin immunofluorescence intensity at FA sites compared with adjacent non-FA sites in the neurites (Fig.2O). This result is consistent with the formation of FA complexes in cortical neurons grown on laminin. In neurons grown on poly-l-lysine, treatment with fibrillar Aβ induced the appearance of aberrant structures in neurite areas close to Aβ deposits, which were reminiscent of FA sites (Fig.2G–N, arrows). Within these sites, there was a significant increase in paxillin and integrin immunofluorescence intensity that was comparable with the increase observed in neurons growing on laminin (Fig. 2O). In contrast, tubulin immunofluorescence intensity did not change along the neurites, ruling out a nonspecific protein clustering effect at these FA-like structures (data not shown). These results indicate that fibrillar Aβ promoted the clustering of FA proteins. In Aβ-induced FA sites, microfilaments extended outwardly in the middle of the neuritic shaft (Fig.2G,H, arrowheads), suggesting microfilament assembly at points of contact between Aβ fibrils and neuronal processes. In some instances, fibrillar Aβ appeared to actively induce microfilament extension. For example, in Figure 2, Iand J, a subset of filopodia from a growth cone appeared to reverse their orientation and extend toward the Aβ deposit (Fig.2I,J, arrowheads). Clusters of phosphorylated Tyr fluorescence colocalizing with filopodia and paxillin clusters were also observed in dystrophic neurites (Fig.2K–N), consistent with increased Tyr phosphorylation at FA-like structures. Thus, fibrillar Aβ promotes activation of FA proteins and the formation of aberrant FA-like structures.
Fig. 2.

Fibrillar Aβ induces FA-like structures in dystrophic neurons. Cortical neurons were treated with fibrillar Aβ at day 5, fixed at day 7, and stained with phalloidin-Texas Red, anti-Aβ, anti-paxillin (pax), and anti-integrin (int) antibodies. FAs were identified by colocalization of paxillin or integrin clusters with microfilaments.A–D, FA are absent in neurons grown on poly-l-lysine (control, PLL). Neuronal processes exhibit homogeneous distribution of paxillin and integrin, whereas microfilaments (MF, red) are primarily localized in growth cones (A, C, arrows). E, F, Neurons grown on laminin (control, Lam) exhibit paxillin and integrin clusters (arrows), associated with microfilaments periodically localized along the processes.G–J, Aβ-treated neurons on poly-l-lysine [Aβ (PLL)] develop FA-like structures proximal to Aβ fibrils (blue) that include clusters of paxillin and integrin (arrows). Microfilaments (MF, red) protruding from FA-like structures are evident in G and H (arrowheads). In some cases, growth cone filopodia appear to reverse orientation and extend toward Aβ deposits (I, J, arrowheads).K–N, Phospho-Tyr immunoreactivity (Tyr-P, green) colocalizes with paxillin (blue) and microfilaments (MF, red) in dystrophic processes (arrows). Scale bar: (in F)A–N, 5 μm. O, Quantification of integrin and paxillin clustering. Integrin receptor clustering increased 2.0 ± 0.2-fold on a laminin substrate and 1.8 ± 0.2-fold in neurons grown on poly-l-lysine after Aβ treatment. Paxillin clustering increased 3.0 ± 0.3-fold on a laminin substrate and 2.5 ± 0.3-fold in neurons grown on poly-l-lysine after Aβ treatment. Values are mean ± SEM; n = 3 independent experiments; >100 FA contacts were scored per condition; *p < 0.05 relative to control by ANOVA followed by the Student–Newman–Keulspost hoc test.
Expression of FA proteins in Alzheimer's brain
We then analyzed the presence of activated FA proteins in association with Aβ deposits in the AD brain. Silver staining of paraffin-embedded sections was used to observe the general features of senile plaques, including the presence of dystrophic neurites surrounding the Aβ core (Fig.3A). Double immunofluorescence with antibody PHF-1 and anti-Aβ antibodies showed the presence of hyperphosphorylated tau-positive dystrophic neurites surrounding the amyloid core (Fig. 3B). Double labeling with anti-integrin receptor α5β1 and anti-Aβ antibodies revealed strong integrin immunoreactivity in senile plaques in close apposition with the core of Aβ (Fig. 3C). Double labeling with anti-integrin and PHF-1 showed colocalization of integrin and hyperphosphorylated tau in senile plaques in dystrophic neurites and neuronal cell bodies (Fig.3D, arrows). Similar results were obtained with an antibody that recognizes specifically integrin β1 (data not shown). We also analyzed the presence of activated FA proteins in senile plaques. Paxillin Tyr phosphorylation was analyzed using an antibody that specifically recognizes paxillin phosphorylated at Tyr-31. Phosphorylation at this site is highly inducible on integrin activation and FA formation (Nakamura et al., 2000; Cukierman et al., 2001). Strong immunoreactivity for phosphorylated paxillin at Tyr-31 was observed in senile plaques in dystrophic neurites and cell bodies surrounding the plaque core (Fig. 3E). FAK, another major component of FA sites, undergoes autophosphorylation at Tyr-397 on binding to activated integrin receptors (Schaller and Parsons, 1994;Cukierman et al., 2001). Immunostaining with an antibody that recognizes specifically anti-FAK phosphorylated at Tyr-397 strongly labeled dystrophic neurites in senile plaques (Fig.3F). In nonlesioned AD brain areas and age-matched control sections, very low levels of diffuse integrin and Tyr-phosphorylated paxillin and FAK immunostaining were observed in the neuropil, whereas moderate immunoreactivity was associated with blood vessels (data not shown). To determine the frequency of integrin immunoreactivity in senile plaques, the number of plaques was quantified in adjacent sections after silver staining (Fig.3G) and integrin immunofluorescence (Fig3H), respectively. This analysis revealed that a very high percentage of silver-stained plaques was immunoreactive against anti-integrin antibodies (84 ± 6%) (Fig. 3I). A similar percentage of plaques stained positive for hyperphosphorylated tau (79 ± 14%) (Fig. 3I). Comparable results were obtained using sections from four different AD brains (three sporadic AD cases and one Down's syndrome–AD case). Thus, integrin clustering, activation of paxillin and FAK, and tau hyperphosphorylation are common features of dystrophic neurons surrounding amyloid deposits in the AD brain.
Fig. 3.
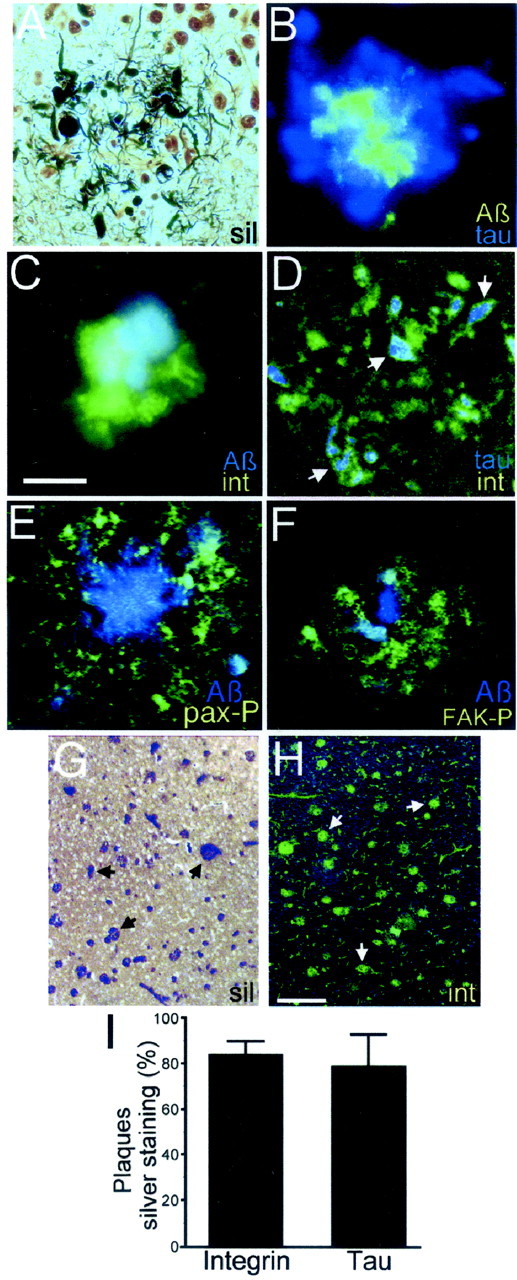
Expression of FA proteins in Alzheimer's brain. Paraffin-embedded brain sections of AD and age-matched control specimens were silver-stained or immunolabeled. A, In AD brains, silver staining revealed dystrophic neurites (black) in senile plaques, surrounding the core of Aβ (pale yellow). B, Immunofluorescence shows the Aβ core of the plaque (green) surrounded by dystrophic neurites immunostained with antibody PHF-1, which recognizes hyperphosphorylated tau (blue). C, Integrin receptor immunoreactivity in a senile plaque (green) surrounding the Aβ core (blue). D, Hyperphosphorylated tau (blue) and integrin receptors (green) colocalize in dystrophic neurites and cell bodies in a senile plaque (arrows).E, Phosphorylated paxillin (pax-P,green) in dystrophic neurites and cell bodies in a senile plaque. The plaque core is stained with anti-Aβ antibody (Aβ, blue). F, Dystrophic neurites in a senile plaque immunostained with anti-phosphorylated FAK antibody (FAK-P, green) surrounding the Aβ core (blue). Adjacent brain sections silver-stained (G) and immunostained with anti-integrin antibody (int,H) show similar plaque density.Arrows denote individual plaques. I, Quantification of integrin- and hyperphosphorylated tau-positive plaques shows that 84 ± 6% of silver-stained plaques were positive for integrin immunoreactivity, and 79 ± 14% were positive for hyperphosphorylated tau. Ten to 20 microscopic fields were analyzed in adjacent sections of four AD brain cases. At least 50 plaques were scored per silver-stained section. Scale bars:A–F, 50 μm; G, H, 250 μm.
Aβ-induced neuronal dystrophy requires paxillin LIM domains and the recruitment of PTP-PEST to the FA complex
To characterize the role of FA signaling in Aβ-induced neuronal dystrophy, we investigated the role of paxillin in this process. Paxillin is a scaffolding protein capable of binding to multiple D proteins. It contains four Src homology 2 (SH2)-binding domains, five Leu-rich LD domains, one Pro-rich SH3-binding domain, and four LIM domains (see Fig. 7) (Turner and Miller, 1994; Turner, 2000). We performed deletion experiments using GFP-tagged constructs containing the full-length, C-terminal, and N-terminal domains of paxillin (Fig.4A). All three chimeric proteins showed similar transfection efficiency and expression levels and a subcellular distribution similar to that of endogenous paxillin. After transfection, cortical neurons were treated with fibrillar Aβ, and the development of neuronal dystrophy was assessed by fluorescent microscopy. We found a significant reduction in Aβ-induced neuronal dystrophy in neurons expressing the N-terminal region of paxillin (N-pax), which lacks the C-terminal region of paxillin (Fig.4B), suggesting that the LIM domains of paxillin may be involved in Aβ-induced neuronal dystrophy. In contrast, no reduction in neuronal dystrophy was observed in neurons transfected with full-length paxillin (Fig. 4B,FL-pax) or the C-terminal domain of paxillin (Fig.4B, LIM-pax) compared with GFP-transfected cells. All three GFP-paxillin proteins retained the ability to translocate to the cytoskeleton (data not shown) and to cluster in FA-like structures after Aβ treatment (Fig. 4C–E) (Thomas et al., 1999). Thus, the decrease in neuronal dystrophy observed in N-pax-expressing neurons was not attributable to a reduced capacity of N-pax to assemble in FA contacts (Fig. 4D).
Fig. 7.

Model of the FA pathways involved in Aβ-induced neuronal dystrophy. Fibrillar Aβ binds to and induces the clustering of the integrin receptors, leading to the activation of paxillin and FAK and their translocation to the nascent FA complex. Paxillin binds to vinculin, which promotes microfilament stabilization at the FA site. PTP-PEST binds to paxillin, leading to dephosphorylation of several FA proteins, which prevents the stabilization of the FA contact, allowing the neuron to continuously respond to fibrillar Aβ stimuli. Alternatively, APP binds to Aβ fibrils, bringing them in contact with integrins, activating FA signaling through FE65, which binds to the C terminus of APP and associates with c-abl, which in turn binds and phosphorylates paxillin, or both. A pathway involving fyn, which is downstream of PTP-PEST, promotes GSK3β activity, whereas the interaction of cbl with c-abl increases CDK5 activity (Zukerberg et al., 2000). Both CDK5 and GSK3β hyperphosphorylate tau, leading to microtubular destabilization and neuronal dystrophy. An alternative pathway involving FAK activity leads to neuronal cell death but not neuronal dystrophy. Paxillin contains four SH2-binding domains (red), five Leu-rich LD domains (light blue), one Pro-rich SH3-binding domain (green) and four LIM domains (purple). MF, microfilaments;hyperphosp., hyperphosphorylated; P, phosphate groups.
Fig. 4.

Paxillin LIM domains are required for Aβ-induced neuronal dystrophy. A, Cortical neurons were transfected with GFP or GFP-paxillin expression vectors containing full-length paxillin (FL-pax) or deletions of the LIM domains (N-pax) or the LIM domains (LIM-pax), respectively. B, Cortical neurons were transfected at day 5 and treated with fibrillar Aβ. The number of dystrophic neurons was quantified 24 hr later and expressed as a percentage of the number of Aβ-induced dystrophic neurons expressing GFP (100%). Note the significant inhibition of neuronal dystrophy in neurons expressingN-pax (28.6 ± 15.1%). FL-pax and LIM-pax were not significantly different from GFP (FL-pax, 108 ± 12%; LIM-pax, 90.4 ± 21.9%). Values are the mean ± SEM; n = 4 independent experiments; 250 neurons were scored per experiment; *p < 0.05 relative to GFP-transfected cells by ANOVA. C–E, Neurons expressing the indicated GFP-tagged paxillin proteins were immunostained with anti-Aβ antibody (blue) and phalloidin-Texas Red. Note the formation of FA-like structures in neurons expressing FL-pax, N-pax, and LIM-pax (arrows).
The above results indicate that Aβ-induced neuronal dystrophy requires the presence of paxillin LIM domains (Fig.4A). Paxillin contains four LIM domains, each capable of protein binding through two zinc finger-like structures (Turner and Miller, 1994; Dawid et al., 1998). β1-Integrin binding is associated with a region of paxillin spanning LIM3, and PTP-PEST binds to paxillin in a region spanning LIM4 and part of LIM3 (Brown et al., 1996; Cote et al., 1999). β1-Integrin binding may be important for paxillin localization to FA sites (Schaller et al., 1995), whereas PTP-PEST is a phosphatase involved in the dynamic regulation of FA contacts (Angers-Loustau et al., 1999). To evaluate the functional role of integrin and PTP-PEST interaction with paxillin in Aβ-induced neuronal dystrophy, we used paxillin constructs containing Cys-to-Ala mutations at positions 467 and 470 in LIM3 and 523 in LIM4, which prevent the binding of the zinc molecule required for the formation of the finger-like structures (Fig.5A) (Dawid et al., 1998;Jurata and Gill, 1998). Cortical neurons were transfected with the indicated constructs, incubated with fibrillar Aβ, and analyzed for the development of neuronal dystrophy. The number of dystrophic neurons expressing paxillin point mutation C467A, which prevents its association with integrin and PTP-PEST, was similar to the number of dystrophic neurons in control GFP-transfected neurons (Fig.5B). Neurons transfected with constructs C470A and C467/470A, which suppress the ability of paxillin to either bind to PTP-PEST or associate with integrin, showed a clear reduction in Aβ-induced neuronal dystrophy that was statistically significant in the case of C467/470A compared with GFP-transfected neurons (Fig.5B). Neurons expressing paxillin C523A, which abrogates the ability of paxillin to bind to PTP-PEST, also significantly reduced Aβ-induced dystrophy. Similarly, neurons expressing a PTP-PEST construct bearing a deletion in the Pro2 domain comprising residues 367–400 (PESTdl), which prevents its binding to paxillin (Cote et al., 1999), showed a complete inhibition of neuronal dystrophy (Fig.5B). These results indicate that PTP-PEST binding to paxillin is a necessary step in the pathway mediating Aβ-induced neuronal dystrophy.
Fig. 5.

PTP-PEST binding to paxillin is required for Aβ-induced neuronal dystrophy. A, Aβ-induced neuronal dystrophy was assessed in neurons transfected with paxillin constructs bearing Cys-to-Ala point mutations, which disrupt the LIM domain tertiary structure, with FRNK, an FAK dominant negative construct, or with PESTdl, a PTP-PEST deletion construct.B, Mutations C470A, C467/470A, and C523A in paxillin, which prevent PTP-PEST binding and integrin association, significantly reduce Aβ-induced neuronal dystrophy (C470A, 62.6 ± 1.3%; C467/C470A, 21.2 ± 4.4%;C523A, 39.9 ± 18.5%). The mutationC467A, which also prevents PTP-PEST binding and integrin association, has no effect on Aβ-induced neuronal dystrophy (112.2 ± 36.1). Expression of PESTdl, which prevents PTP-PEST binding to paxillin, completely prevents neuronal dystrophy. Expression of FRNK, which contains the FAK focal adhesion targeting domain but lacks the kinase domain, has no effect on Aβ-induced neuronal dystrophy. The number of dystrophic neurons was quantified 24 hr after transfection and expressed as a percentage of the number of Aβ-induced dystrophic neurons expressing GFP (100%). Values are mean ± SEM; n = 3–7 individual experiments; 250 neurons were scored per condition in each experiment; *p < 0.05; **p < 0.01 relative to control (GFP) by ANOVA followed by the Student–Newman–Keuls post hoc test.
Previous work has demonstrated that fibrillar Aβ activates FAK in neuronal cells (Zhang et al., 1994, 1996; Williamson et al., 2002). To assess the involvement of FAK in Aβ-induced neuronal dystrophy, we used FRNK, a dominant negative form of FAK (Schaller et al., 1993;Zheng et al., 1999). FRNK contains the FA-targeting sequence of FAK and binds to paxillin but lacks the kinase domain (Fig. 5A). Cultures transfected with FRNK and treated with Aβ showed an increase in the number of dystrophic neurons similar to that in GFP-transfected cultures (Fig. 5B), indicating that the kinase activity of FAK is not required for Aβ-induced neuronal dystrophy.
FA proteins are involved in Aβ-induced neuronal death
Recent results suggest that Aβ-induced neuronal dystrophy and cell death are mediated by distinct molecular pathways (Grace et al., 2002). To evaluate the role of FA proteins in Aβ-induced neuronal death, neuronal viability was determined in cultures transfected with FL-pax, N-pax, PESTdl, and FRNK expression vectors and treated with fibrillar Aβ. Neurons expressing FL-pax showed a significant increase in cell death after Aβ treatment (Fig.6B). In contrast, neurons expressing FRNK became dystrophic but remained viable (Fig.6A,B). Interestingly, neurons expressing N-pax, which lacks paxillin LIM domains, exhibited significantly reduced neuronal dystrophy (Fig. 4B) and cell death (Fig.6B), indicating that paxillin LIM domains participate in both Aβ-induced neuronal dystrophy and cell death. Similarly, expression of PESTdl also reduced neuronal dystrophy and cell death (Fig. 6B). Thus, although FAK activity is not involved in neuronal dystrophy, it is involved in cell death, as is PTP-PEST, further suggesting that separate molecular pathways mediate Aβ-induced neuronal dystrophy and death.
Fig. 6.

PTP-PEST and FAK activity are involved in Aβ-induced neuronal death. A, Aβ treatment induces cell death in neurons expressing GFP (top panel, greenfluorescence). Nonviable cells show process retraction and disintegration and positive nuclear staining for propidium iodide (arrow). Neurons expressing FRNK (bottom panel, green fluorescence) became dystrophic but remained viable. Note the dystrophic appearance of neuritic processes and the absence of propidium nuclear staining in the neuron expressing FRNK.B, Quantification of cell death in transfected neurons treated with Aβ. A significant reduction in Aβ-induced neuronal death is observed in neurons expressing N-pax, which lacks paxillin LIM domains (20.4 ± 8%), andPESTdl, which lacks the paxillin-binding domain of PTP-PEST (13.3 ± 41.7%). FRNK completely prevents neuronal death (−15.3 ± 13%). Cell death is expressed as a percentage of Aβ-induced propidium-positive neurons transfected with GFP (100%). Values are mean ± SEM; n = 3 independent experiments; >200 neurons were scored per condition in each individual experiment; *p < 0.05; **p < 0.01 relative to control (GFP) by ANOVA followed by the Student–Newman–Keuls post hoc test. Cortical neurons were transfected at day 5, treated with fibrillar Aβ for 2 d, and processed for analysis. Before fixation, the nuclei of dead cells were labeled with propidium iodide.
Discussion
These experiments indicate that Aβ-induced neuronal dystrophy is mediated by the aberrant activation of FA proteins. FA sites are integrin-based structures that mediate cell–substrate adhesion and the bidirectional exchange of information between extracellular molecules and the cytoplasm. Fibrillar Aβ treatment induced integrin receptor clustering, paxillin Tyr phosphorylation, and translocation to the cytoskeleton and promoted the formation of aberrant FA-like structures, suggesting the activation of focal adhesion signaling cascades (Figs.1, 2). The presence of several antioxidants in the B27 supplement used to prepare our culture medium (see Materials and Methods) and use of calcium-deficient medium did not prevent the development of dystrophy (A. Deshpande, G. Pigino, and J. Busciglio, unpublished observations), suggesting that oxidative stress and intracellular calcium influx do not mediate Aβ-induced neuronal dystrophy. Previous studies indicate that Aβ peptides bind to integrin receptors (Sabo et al., 1995) and that integrins are expressed in the AD brain (Akiyama et al., 1991;Frohman et al., 1991; Eikelenboom et al., 1994; Van Gool et al., 1994), suggesting that Aβ-induced neuronal dystrophy may be mediated by integrin signaling. We observed high expression of integrin receptors in senile plaques. Interestingly, integrin β1, which is the subunit that directly binds to and recruits paxillin to the FA complex (Fig.7) (Schaller et al., 1995; Chen et al., 2000), was enriched in dystrophic neurites and cell bodies surrounding amyloid cores. Most importantly, we detected increased levels of Tyr-phosphorylated, activated paxillin and FAK in dystrophic neurites around plaque cores, consistent with the activation of FA signaling cascades in neuronal processes in contact with fibrillar Aβ. Quantitative analysis shows that most senile plaques exhibited strong integrin immunoreactivity, indicating that FA protein activation may be a common feature in the AD brain (Fig. 3).
Deletion experiments indicate that the LIM domains in the C terminus of paxillin mediate Aβ-induced neuronal dystrophy. Paxillin LIM domains are associated with its binding to β1-integrin (Fig. 7), suggesting that the absence of LIM domains could reduce Aβ-induced neuronal dystrophy by preventing paxillin recruitment to the FA site. Interestingly, although the deletion of the LIM domains suppressed Aβ-induced dystrophy, it did not prevent the recruitment of paxillin to FA sites, which may still occur via its N-terminal association with FAK (Hildebrand et al., 1995). Therefore, the inhibition of neuronal dystrophy was not produced by the inability of N-pax to bind to the FA complex (Fig. 4). Point mutations in paxillin LIM domains that prevent the binding of PTP-PEST significantly reduced Aβ-induced neuronal dystrophy, and a similar reduction was observed in neurons expressing PESTdl, which abrogates PTP-PEST binding to paxillin. Thus, the recruitment of PTP-PEST to the FA complex seems to be a critical step for the development of Aβ-induced neuronal dystrophy. PTP-PEST is required for the turnover of FA contacts (Angers-Loustau et al., 1999), suggesting that, in the absence of PTP-PEST, FA contacts may become stabilized, preventing further response to extracellular stimuli. In this regard, fibroblasts from PTP-PEST null mice show decreased motility and increased FA size (Angers-Loustau et al., 1999), similar to the large FA-like structures formed in Aβ-treated neurons expressing N-pax, which suppressed PTP-PEST binding and neuronal dystrophy (Fig. 4D). Current experiments are directed to characterize the substrates of PTP-PEST involved in the development of neuronal dystrophy. FRNK, a dominant negative form of FAK, did not alter Aβ-induced neuronal dystrophy, suggesting that although FAK is activated by fibrillar Aβ (Zhang et al., 1994, 1996; Williamson et al., 2002), it is not involved in the dystrophic response. Aβ-induced neuronal dystrophy and cell death occur over different Aβ concentrations and time courses, suggesting distinct signaling pathways (Grace et al., 2002). We observed Tyr phosphorylation and dephosphorylation of several proteins in response to Aβ, consistent with the activation of multiple signaling pathways (Fig.1B). Deletion experiments indicate that FAK and PTP-PEST activity are both involved in fibrillar Aβ-induced cell death, further suggesting that more than one pathway mediate Aβ-induced cell death (Fig. 7). Alternatively, because neurons expressing a dominant negative form of FAK became dystrophic but remained viable, dystrophy may lead to cell death over a significantly longer time course.
Recent results indicate that the RHDS sequence in Aβ, similar to the integrin ligand sequence RGDS, is not necessary for Aβ-induced neuronal dystrophy and cell death, but that the transition of Aβ to a fibrillar form is essential (Grace et al., 2002). Fibrillar Aβ has been shown to bind to cell surface amyloid precursor protein (Lorenzo et al., 2000), which colocalizes with integrins in neurons (Storey et al., 1996; Yamazaki et al., 1997) and participates in FA signaling (Sabo et al., 2001). We have found that APP is enriched in aberrant FA-like structures induced by Aβ (E. Grace, R. Lin, L. Heredia, A. Lorenzo, and J. Busciglio, unpublished results). APP may bring Aβ fibrils into physical contact with integrin receptors and may also activate paxillin through the adapter protein FE65 (Sabo et al., 2001), which is associated with c-abl, a nonreceptor Tyr kinase that phosphorylates paxillin (Salgia et al., 1995; Sabo et al., 2001) (Fig. 7). Interestingly, FE65 immunoreactivity colocalizes with neurofibrillary tangles (Delatour et al., 2001), and a polymorphism in the FE65 gene that reduces its binding to APP appears to confer resistance to late-onset AD (Hu et al., 1998; Lambert et al., 2000; Hu et al., 2002). Ongoing experiments are directed to establish the role of APP in the transduction of fibrillar Aβ-induced neuronal dystrophy.
Fibrillar Aβ induces aberrant morphological changes and tau hyperphosphorylation in primary neurons in culture and in vivo (Busciglio et al., 1992; Pike et al., 1992; Busciglio and Yankner, 1995; Geula et al., 1998), resembling dystrophic neurites in the AD brain. We found that FA proteins frequently colocalized with hyperphosphorylated tau in dystrophic neurites surrounding Aβ deposits in the AD brain (Fig. 3) and in culture (data not shown), suggesting that FA signaling induced by fibrillar Aβ may lead to deregulation of kinase and phosphatase activities responsible for tau hyperphosphorylation. In this regard, neurons transfected with N-pax, the paxillin construct lacking the LIM domains that significantly reduced Aβ-induced dystrophy, did not exhibit increased tau phosphorylation after Aβ treatment, further supporting the involvement of FA signaling in tau hyperphosphorylation (E. Grace and J. Busciglio, unpublished results). FA signaling leads to the activation of cyclin-dependent kinase 5 (CDK5) and glycogen synthase kinase 3β (GSK3β) (Fig. 7) (Bhat et al., 2000; Li et al., 2000), two kinases that phosphorylate tau at epitopes phosphorylated in neurofibrillary tangles (Mandelkow et al., 1992;Baumann et al., 1993; Flaherty et al., 2000), which have been implicated in neurofibrillary pathology (Yamaguchi et al., 1996; Pei et al., 1998; Flaherty et al., 2000). It is noteworthy that phosphorylation of tau by cdk5 may potentiate the phosphorylation of tau by GSK3β (Sengupta et al., 1997), suggesting a synergistic effect between these two kinases activated by the FA signaling cascade.
In summary, the aberrant activation of FA pathways appears to be critically involved in fibrillar Aβ-induced neuronal dystrophy. Neuronal dystrophy is associated with synaptic loss in culture (Grace et al., 2002) and in the AD brain (Masliah et al., 1991), and similarly to Aβ deposition, is a unique pathological feature of AD (Benzing et al., 1993). The ability of the neuron to respond dynamically to extracellular cues is reminiscent of plasticity mechanisms. In this regard, maladaptive neuronal plasticity may play a major role in AD (Cotman et al., 1998; Mesulam, 1999). Fibrillar Aβ has been shown to induce apoptotic cascades in neurites and synapses (Mattson and Duan, 1999). Thus, aberrant focal adhesion activation by Aβ may lead to the initiation of localized apoptotic cascades normally involved in adaptive plasticity in both neurites and synapses (Mattson and Duan, 1999). The alterations in the composition of the extracellular environment in the AD brain may stimulate aberrant cellular responses consistent with the dynamic regulation of FA contacts by PTP-PEST activity, which is required for Aβ-induced neuronal dystrophy. Brain regions with the highest plasticity are the most vulnerable in AD (Small, 1998), suggesting that, under pathological conditions such as presenilin mutations (Pigino et al., 2001) or Aβ deposition, neuronal plasticity may result in neuronal dysfunction. As such, the characterization of the molecular pathway(s) by which fibrillar Aβ induces neuronal dystrophy may lead to therapies directed to block maladaptive plasticity to preserve neuronal function and synaptic integrity.
Footnotes
This work was supported by grants from the University of Connecticut Health Center and the Alzheimer's Association and National Institutes of Health Grant HD38466 (J.B.). We are grateful to Dr. R. Salgia, Dr. M. L. Tremblay, Dr. L. Languino, Dr. Z. Zhan, and Dr. M. D. Schaller for generously providing reagents. We also thank Dr. Schaller for helpful discussions, Gustavo Pigino and Atul Deshpande for technical assistance, and Nancy Ryan for assistance with the silver-staining technique.
Correspondence should be addressed to Jorge Busciglio, Department of Neuroscience, University of Connecticut Health Center, 263 Farmington Avenue, Farmington, CT 06030. E-mail: busciglio@nso1.uchc.edu.
E. A. Grace's present address: Department of Pharmacology, Mount Sinai School of Medicine, New York, NY 10029.
References
- 1.Akiyama H, Kawamata T, Dedhar S, McGeer PL. Immunohistochemical localization of vitronectin, its receptor and beta-3 integrin in Alzheimer brain tissue. J Neuroimmunol. 1991;32:19–28. doi: 10.1016/0165-5728(91)90067-h. [DOI] [PubMed] [Google Scholar]
- 2.Angers-Loustau A, Cote JF, Charest A, Dowbenko D, Spencer S, Lasky LA, Tremblay ML. Protein tyrosine phosphatase-PEST regulates focal adhesion disassembly, migration, and cytokinesis in fibroblasts. J Cell Biol. 1999;144:1019–1031. doi: 10.1083/jcb.144.5.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aplin AE, Howe A, Alahari SK, Juliano RL. Signal transduction and signal modulation by cell adhesion receptors: the role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharmacol Rev. 1998;50:197–263. [PubMed] [Google Scholar]
- 4.Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 1993;336:417–424. doi: 10.1016/0014-5793(93)80849-p. [DOI] [PubMed] [Google Scholar]
- 5.Benzing WC, Mufson EJ, Armstrong DM. Alzheimer's disease-like dystrophic neurites characteristically associated with senile plaques are not found within other neurodegenerative diseases unless amyloid beta-protein deposition is present. Brain Res. 1993;606:10–18. doi: 10.1016/0006-8993(93)91563-8. [DOI] [PubMed] [Google Scholar]
- 6.Berg MM, Krafft GA, Klein WL. Rapid impact of beta-amyloid on paxillin in a neural cell line. J Neurosci Res. 1997;50:979–989. doi: 10.1002/(SICI)1097-4547(19971215)50:6<979::AID-JNR8>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 7.Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, Lee CM. Regulation and localization of tyrosine 216 phosphorylation of glycogen synthase kinase-3 beta in cellular and animal models of neuronal degeneration. Proc Natl Acad Sci USA. 2000;97:11074–11079. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown MC, Perrotta JA, Turner CE. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J Cell Biol. 1996;135:1109–1123. doi: 10.1083/jcb.135.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Busciglio J, Yankner BA. Apoptosis and increased generation of reactive oxygen species in Down's syndrome neurons in vitro. Nature. 1995;378:776–779. doi: 10.1038/378776a0. [DOI] [PubMed] [Google Scholar]
- 10.Busciglio J, Lorenzo A, Yankner BA. Methodological variables in the assessment of beta amyloid neurotoxicity. Neurobiol Aging. 1992;13:609–612. doi: 10.1016/0197-4580(92)90065-6. [DOI] [PubMed] [Google Scholar]
- 11.Busciglio J, Gabuzda DH, Matsudaira P, Yankner BA. Generation of beta-amyloid in the secretory pathway in neuronal and nonneuronal cells. Proc Natl Acad Sci USA. 1993;90:2092–2096. doi: 10.1073/pnas.90.5.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Busciglio J, Lorenzo A, Yeh J, Yankner BA. Beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14:879–888. doi: 10.1016/0896-6273(95)90232-5. [DOI] [PubMed] [Google Scholar]
- 13.Busciglio J, Hartmann H, Lorenzo A, Wong C, Baumann K, Sommer B, Staufenbiel M, Yankner BA. Neuronal localization of presenilin-1 and association with amyloid plaques and neurofibrillary tangles in Alzheimer's disease. J Neurosci. 1997;17:5101–5107. doi: 10.1523/JNEUROSCI.17-13-05101.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carson F. Histotechnology: a self-instructional text, pp 167–169. American Society for Clinical Pathology; Chicago: 1990. [Google Scholar]
- 15.Chen LM, Bailey D, Fernandez-Valle C. Association of beta 1 integrin with focal adhesion kinase and paxillin in differentiating Schwann cells. J Neurosci. 2000;20:3776–3784. doi: 10.1523/JNEUROSCI.20-10-03776.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cote JF, Turner CE, Tremblay ML. Intact LIM 3 and LIM 4 domains of paxillin are required for the association to a novel polyproline region (Pro 2) of protein-tyrosine phosphatase-PEST. J Biol Chem. 1999;274:20550–20560. doi: 10.1074/jbc.274.29.20550. [DOI] [PubMed] [Google Scholar]
- 17.Cotman CW, Hailer NP, Pfister KK, Soltesz I, Schachner M. Cell adhesion molecules in neural plasticity and pathology: similar mechanisms, distinct organizations? Prog Neurobiol. 1998;55:659–669. doi: 10.1016/s0301-0082(98)00025-2. [DOI] [PubMed] [Google Scholar]
- 18.Cukierman E, Pankov R, Stevens DR, Yamada KM. Taking cell-matrix adhesions to the third dimension. Science. 2001;294:1708–1712. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]
- 19.Dawid IB, Breen JJ, Toyama R. LIM domains: multiple roles as adapters and functional modifiers in protein interactions. Trends Genet. 1998;14:156–162. doi: 10.1016/s0168-9525(98)01424-3. [DOI] [PubMed] [Google Scholar]
- 20.Delatour B, Mercken L, El Hachimi KH, Colle MA, Pradier L, Duyckaerts C. FE65 in Alzheimer's disease: neuronal distribution and association with neurofibrillary tangles. Am J Pathol. 2001;158:1585–1591. doi: 10.1016/S0002-9440(10)64113-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dogic D, Rousselle P, Aumailley M. Cell adhesion to laminin 1 or 5 induces isoform-specific clustering of integrins and other focal adhesion components. J Cell Sci. 1998;111:793–802. doi: 10.1242/jcs.111.6.793. [DOI] [PubMed] [Google Scholar]
- 22.Eikelenboom P, Zhan SS, Kamphorst W, van der Valk P, Rozemuller JM. Cellular and substrate adhesion molecules (integrins) and their ligands in cerebral amyloid plaques in Alzheimer's disease. Virchows Arch. 1994;424:421–427. doi: 10.1007/BF00190565. [DOI] [PubMed] [Google Scholar]
- 23.Flaherty DB, Soria JP, Tomasiewicz HG, Wood JG. Phosphorylation of human tau protein by microtubule-associated kinases: GSK3 beta and cdk5 are key participants. J Neurosci Res. 2000;62:463–472. doi: 10.1002/1097-4547(20001101)62:3<463::AID-JNR16>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 24.Frohman EM, Frohman TC, Gupta S, de Fougerolles A, van den Noort S. Expression of intercellular adhesion molecule 1 (ICAM-1) in Alzheimer's disease. J Neurol Sci. 1991;106:105–111. doi: 10.1016/0022-510x(91)90202-i. [DOI] [PubMed] [Google Scholar]
- 25.Geddes JW, Anderson KJ, Cotman CW. Senile plaques as aberrant sprout-stimulating structures. Exp Neurol. 1986;94:767–776. doi: 10.1016/0014-4886(86)90254-2. [DOI] [PubMed] [Google Scholar]
- 26.Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA. Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity. Nat Med. 1998;4:827–831. doi: 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- 27.Goodwin JL, Kehrli ME, Jr, Uemura E. Integrin Mac-1 and beta-amyloid in microglial release of nitric oxide. Brain Res. 1997;768:279–286. doi: 10.1016/s0006-8993(97)00653-7. [DOI] [PubMed] [Google Scholar]
- 28.Grace E, Rabiner AC, Busciglio J. Characterization of neuronal dystrophy induced by fibrillar amyloid β: implications for Alzheimer's disease. Neuroscience. 2002;11:265–273. doi: 10.1016/s0306-4522(02)00241-5. [DOI] [PubMed] [Google Scholar]
- 29.Greenberg SG, Davies P, Schein JD, Binder LI. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem. 1992;267:564–569. [PubMed] [Google Scholar]
- 30.Hildebrand JD, Schaller MD, Parsons JT. Paxillin, a tyrosine phosphorylated focal adhesion-associated protein binds to the carboxyl terminal domain of focal adhesion kinase. Mol Biol Cell. 1995;6:637–647. doi: 10.1091/mbc.6.6.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hu Q, Kukull WA, Bressler SL, Gray MD, Cam JA, Larson EB, Martin GM, Deeb SS. The human FE65 gene: genomic structure and an intronic biallelic polymorphism associated with sporadic dementia of the Alzheimer type. Hum Genet. 1998;103:295–303. doi: 10.1007/s004390050820. [DOI] [PubMed] [Google Scholar]
- 32.Hu Q, Cool BH, Wang B, Hearn MG, Martin GM. A candidate molecular mechanism for the association of an intronic polymorphism of FE65 with resistance to very late onset dementia of the Alzheimer type. Hum Mol Genet. 2002;11:465–475. doi: 10.1093/hmg/11.4.465. [DOI] [PubMed] [Google Scholar]
- 33.Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol. 1997;56:965–973. doi: 10.1097/00005072-199709000-00002. [DOI] [PubMed] [Google Scholar]
- 34.Jurata LW, Gill GN. Structure and function of LIM domains. Curr Top Microbiol Immunol. 1998;228:75–113. doi: 10.1007/978-3-642-80481-6_4. [DOI] [PubMed] [Google Scholar]
- 35.Katoh K, Masuda M, Kano Y, Jinguji Y, Fujiwara K. Focal adhesion proteins associated with apical stress fibers of human fibroblasts. Cell Motil Cytoskeleton. 1995;31:177–195. doi: 10.1002/cm.970310302. [DOI] [PubMed] [Google Scholar]
- 36.Kowalska MA, Badellino K. Beta-amyloid protein induces platelet aggregation and supports platelet adhesion. Biochem Biophys Res Commun. 1994;205:1829–1835. doi: 10.1006/bbrc.1994.2883. [DOI] [PubMed] [Google Scholar]
- 37.Lambert JC, Mann D, Goumidi L, Harris J, Pasquier F, Frigard B, Cottel D, Lendon C, Iwatsubo T, Amouyel P, Chartier-Harlin MC. A FE65 polymorphism associated with risk of developing sporadic late-onset Alzheimer's disease but not with Abeta loading in brains. Neurosci Lett. 2000;293:29–32. doi: 10.1016/s0304-3940(00)01477-4. [DOI] [PubMed] [Google Scholar]
- 38.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT. Amyloid pores from pathogenic mutations. Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 39.Li BS, Zhang L, Gu J, Amin ND, Pant HC. Integrin α(1)β(1)-mediated activation of cyclin-dependent kinase 5 activity is involved in neurite outgrowth and human neurofilament protein H Lys-Ser-Pro tail domain phosphorylation. J Neurosci. 2000;20:6055–6062. doi: 10.1523/JNEUROSCI.20-16-06055.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lorenzo A, Yankner BA. Beta-amyloid neurotoxicity requires fibril formation and is inhibited by congo red. Proc Natl Acad Sci USA. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lorenzo A, Yuan M, Zhang Z, Paganetti PA, Sturchler-Pierrat C, Staufenbiel M, Mautino J, Vigo FS, Sommer B, Yankner BA. Amyloid beta interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer's disease. Nat Neurosci. 2000;3:460–464. doi: 10.1038/74833. [DOI] [PubMed] [Google Scholar]
- 42.Mandelkow EM, Drewes G, Biernat J, Gustke N, Van Lint J, Vandenheede JR, Mandelkow E. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992;314:315–321. doi: 10.1016/0014-5793(92)81496-9. [DOI] [PubMed] [Google Scholar]
- 43.Masliah E, Mallory M, Hansen L, Alford M, Albright T, DeTeresa R, Terry R, Baudier J, Saitoh T. Patterns of aberrant sprouting in Alzheimer's disease. Neuron. 1991;6:729–739. doi: 10.1016/0896-6273(91)90170-5. [DOI] [PubMed] [Google Scholar]
- 44.Mattson MP, Duan W. “Apoptotic” biochemical cascades in synaptic compartments: roles in adaptive plasticity and neurodegenerative disorders. J Neurosci Res. 1999;58:152–166. [PubMed] [Google Scholar]
- 45.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKee AC, Kosik KS, Kowall NW. Neuritic pathology and dementia in Alzheimer's disease. Ann Neurol. 1991;30:156–165. doi: 10.1002/ana.410300206. [DOI] [PubMed] [Google Scholar]
- 47.Mesulam MM. Neuroplasticity failure in Alzheimer's disease: bridging the gap between plaques and tangles. Neuron. 1999;24:521–529. doi: 10.1016/s0896-6273(00)81109-5. [DOI] [PubMed] [Google Scholar]
- 48.Mueller SC, Kelly T, Dai MZ, Dai HN, Chen WT. Dynamic cytoskeleton-integrin associations induced by cell binding to immobilized fibronectin. J Cell Biol. 1989;109:3455–3464. doi: 10.1083/jcb.109.6.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamura K, Yano H, Uchida H, Hashimoto S, Schaefer E, Sabe H. Tyrosine phosphorylation of paxillin α is involved in temporospatial regulation of paxillin-containing focal adhesion formation and F-actin organization in motile cells. J Biol Chem. 2000;275:27155–27164. doi: 10.1074/jbc.M000679200. [DOI] [PubMed] [Google Scholar]
- 50.Pei JJ, Grundke-Iqbal I, Iqbal K, Bogdanovic N, Winblad B, Cowburn RF. Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer's disease neurofibrillary degeneration. Brain Res. 1998;797:267–277. doi: 10.1016/s0006-8993(98)00296-0. [DOI] [PubMed] [Google Scholar]
- 51.Pigino G, Pelsman A, Mori H, Busciglio J. Presenilin-1 mutations reduce cytoskeletal association, deregulate neurite growth, and potentiate neuronal dystrophy and tau phosphorylation. J Neurosci. 2001;21:834–842. doi: 10.1523/JNEUROSCI.21-03-00834.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pike CJ, Walencewicz AJ, Glabe CG, Cotman CW. Aggregation-related toxicity of synthetic beta-amyloid protein in hippocampal cultures. Eur J Pharmacol. 1991;207:367–368. doi: 10.1016/0922-4106(91)90014-9. [DOI] [PubMed] [Google Scholar]
- 53.Pike CJ, Cummings BJ, Cotman CW. Beta-amyloid induces neuritic dystrophy in vitro: similarities with Alzheimer pathology. NeuroReport. 1992;3:769–772. doi: 10.1097/00001756-199209000-00012. [DOI] [PubMed] [Google Scholar]
- 54.Sabo S, Lambert MP, Kessey K, Wade W, Krafft G, Klein WL. Interaction of beta-amyloid peptides with integrins in a human nerve cell line. Neurosci Lett. 1995;184:25–28. doi: 10.1016/0304-3940(94)11159-g. [DOI] [PubMed] [Google Scholar]
- 55.Sabo SL, Ikin AF, Buxbaum JD, Greengard P. The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J Cell Biol. 2001;153:1403–1414. doi: 10.1083/jcb.153.7.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saitoh T, Horsburgh K, Masliah E. Hyperactivation of signal transduction systems in Alzheimer's disease. Ann NY Acad Sci. 1993;695:34–41. doi: 10.1111/j.1749-6632.1993.tb23023.x. [DOI] [PubMed] [Google Scholar]
- 57.Salgia R, Li JL, Lo SH, Brunkhorst B, Kansas GS, Sobhany ES, Sun Y, Pisick E, Hallek M, Ernst T, Tantravahi R, Chen LB, Griffin JD. Molecular cloning of human paxillin, a focal adhesion protein phosphorylated by P210BCR/ABL. J Biol Chem. 1995;270:5039–5047. doi: 10.1074/jbc.270.10.5039. [DOI] [PubMed] [Google Scholar]
- 58.Schaller MD, Parsons JT. Focal adhesion kinase and associated proteins. Curr Opin Cell Biol. 1994;6:705–710. doi: 10.1016/0955-0674(94)90097-3. [DOI] [PubMed] [Google Scholar]
- 59.Schaller MD, Borgman CA, Parsons JT. Autonomous expression of a noncatalytic domain of the focal adhesion-associated protein tyrosine kinase pp125FAK. Mol Cell Biol. 1993;13:785–791. doi: 10.1128/mcb.13.2.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schaller MD, Otey CA, Hildebrand JD, Parsons JT. Focal adhesion kinase and paxillin bind to peptides mimicking beta integrin cytoplasmic domains. J Cell Biol. 1995;130:1181–1187. doi: 10.1083/jcb.130.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 62.Sengupta A, Wu Q, Grundke-Iqbal I, Iqbal K, Singh TJ. Potentiation of GSK-3-catalyzed Alzheimer-like phosphorylation of human tau by cdk5. Mol Cell Biochem. 1997;167:99–105. doi: 10.1023/a:1006883924775. [DOI] [PubMed] [Google Scholar]
- 63.Shen Y, Lyons P, Cooley M, Davidson D, Veillette A, Salgia R, Griffin JD, Schaller MD. The noncatalytic domain of protein-tyrosine phosphatase-PEST targets paxillin for dephosphorylation in vivo. J Biol Chem. 2000;275:1405–1413. doi: 10.1074/jbc.275.2.1405. [DOI] [PubMed] [Google Scholar]
- 64.Small DH. The role of the amyloid protein precursor (APP) in Alzheimer's disease: does the normal function of APP explain the topography of neurodegeneration? Neurochem Res. 1998;23:795–806. doi: 10.1023/a:1022471729291. [DOI] [PubMed] [Google Scholar]
- 65.Storey E, Beyreuther K, Masters CL. Alzheimer's disease amyloid precursor protein on the surface of cortical neurons in primary culture co-localizes with adhesion patch components. Brain Res. 1996;735:217–231. doi: 10.1016/0006-8993(96)00608-7. [DOI] [PubMed] [Google Scholar]
- 66.Thomas JW, Cooley MA, Broome JM, Salgia R, Griffin JD, Lombardo CR, Schaller MD. The role of focal adhesion kinase binding in the regulation of tyrosine phosphorylation of paxillin. J Biol Chem. 1999;274:36684–36692. doi: 10.1074/jbc.274.51.36684. [DOI] [PubMed] [Google Scholar]
- 67.Turner CE. Paxillin interactions. J Cell Sci. 2000;113:4139–4140. doi: 10.1242/jcs.113.23.4139. [DOI] [PubMed] [Google Scholar]
- 68.Turner CE, Miller JT. Primary sequence of paxillin contains putative SH2 and SH3 domain binding motifs and multiple LIM domains: identification of a vinculin and pp125Fak-binding region. J Cell Sci. 1994;107:1583–1591. doi: 10.1242/jcs.107.6.1583. [DOI] [PubMed] [Google Scholar]
- 69.Van Gool D, Carmeliet G, Triau E, Cassiman JJ, Dom R. Appearance of localized immunoreactivity for the alpha 4 integrin subunit and for fibronectin in brains from Alzheimer's, Lewy body dementia patients and aged controls. Neurosci Lett. 1994;170:71–73. doi: 10.1016/0304-3940(94)90241-0. [DOI] [PubMed] [Google Scholar]
- 70.Williamson R, Scales T, Clark BR, Gibb G, Reynolds CH, Kellie S, Bird IN, Varndell IM, Sheppard PW, Everall I, Anderton BH. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-beta peptide exposure: involvement of Src family protein kinases. J Neurosci. 2002;22:10–20. doi: 10.1523/JNEUROSCI.22-01-00010.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamaguchi H, Ishiguro K, Uchida T, Takashima A, Lemere CA, Imahori K. Preferential labeling of Alzheimer neurofibrillary tangles with antisera for tau protein kinase (TPK) I/glycogen synthase kinase-3 beta and cyclin-dependent kinase 5, a component of TPK II. Acta Neuropathol (Berl) 1996;92:232–241. doi: 10.1007/s004010050513. [DOI] [PubMed] [Google Scholar]
- 72.Yamazaki T, Koo EH, Selkoe DJ. Cell surface amyloid beta-protein precursor colocalizes with beta 1 integrins at substrate contact sites in neural cells. J Neurosci. 1997;17:1004–1010. doi: 10.1523/JNEUROSCI.17-03-01004.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yankner BA. Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- 74.Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 75.Zhang C, Lambert MP, Bunch C, Barber K, Wade WS, Krafft GA, Klein WL. Focal adhesion kinase expressed by nerve cell lines shows increased tyrosine phosphorylation in response to Alzheimer's Aβ peptide. J Biol Chem. 1994;269:25247–25250. [PubMed] [Google Scholar]
- 76.Zhang C, Qiu HE, Krafft GA, Klein WL. A beta peptide enhances focal adhesion kinase/Fyn association in a rat CNS nerve cell line. Neurosci Lett. 1996;211:187–190. doi: 10.1016/0304-3940(96)12761-0. [DOI] [PubMed] [Google Scholar]
- 77.Zheng DQ, Woodard AS, Fornaro M, Tallini G, Languino LR. Prostatic carcinoma cell migration via α(v)β3 integrin is modulated by a focal adhesion kinase pathway. Cancer Res. 1999;59:1655–1664. [PubMed] [Google Scholar]
- 78.Zukerberg LR, Patrick GN, Nikolic M, Humbert S, Wu CL, Lanier LM, Gertler FB, Vidal M, Van Etten RA, Tsai LH. Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine phosphorylation, kinase upregulation, and neurite outgrowth. Neuron. 2000;26:633–646. doi: 10.1016/s0896-6273(00)81200-3. [DOI] [PubMed] [Google Scholar]