

Abstract
Mirror-image allodynia is a mysterious phenomenon that occurs in association with many clinical pain syndromes. Allodynia refers to pain in response to light touch/pressure stimuli, which normally are perceived as innocuous. Mirror-image allodynia arises from the healthy body region contralateral to the actual site of trauma/inflammation. Virtually nothing is known about the mechanisms underlying such pain. A recently developed animal model of inflammatory neuropathy reliably produces mirror-image allodynia, thus allowing this pain phenomenon to be analyzed. In this sciatic inflammatory neuropathy (SIN) model, decreased response threshold to tactile stimuli (mechanical allodynia) develops in rats after microinjection of immune activators around one healthy sciatic nerve at mid-thigh level. Low level immune activation produces unilateral allodynia ipsilateral to the site of sciatic inflammation; more intense immune activation produces bilateral (ipsilateral + mirror image) allodynia. The present studies demonstrate that both ipsilateral and mirror-image SIN-induced allodynias are (1) reversed by intrathecal (peri-spinal) delivery of fluorocitrate, a glial metabolic inhibitor; (2) prevented and reversed by intrathecal CNI-1493, an inhibitor of p38 mitogen-activated kinases implicated in proinflammatory cytokine production and signaling; and (3) prevented or reversed by intrathecal proinflammatory cytokine antagonists specific for interleukin-1, tumor necrosis factor, or interleukin-6. Reversal of ipsilateral and mirror-image allodynias was rapid and complete even when SIN was maintained constantly for 2 weeks before proinflammatory cytokine antagonist administration. These results provide the first evidence that ipsilateral and mirror-image inflammatory neuropathy pain are created both acutely and chronically through glial and proinflammatory cytokine actions.
Keywords: microglia, astrocyte, interleukin-1, tumor necrosis factor, interleukin-6, allodynia, p38 MAP kinase
Introduction
Trauma and inflammation of peripheral nerves induce pathological pain, referred to as neuropathic pain (Zimmermann, 2001). Although neuropathic pain is perceived to arise from the skin innervated by the damaged/inflamed nerve, pathological pain can also arise from sites contralateral to (the mirror image of) the site of pathology (Watkins and Maier, 2002). Mirror-image pain occurs in chronic pain conditions, including reflex sympathetic dystrophy (Maleki et al., 2000), causalgia (Shir and Seltzer, 1991), atypical facial pain (Woda and Pionchon, 2000), idiopathic facial arthromyalgia (Woda and Pionchon, 2000), and stomatodynia (Woda and Pionchon, 2000). It is typically characterized by mechanical allodynia (Moriwaki and Yuge, 1999; Baron, 2000). That is, mirror-image pain is perceived in response to light touch/pressure stimuli such as clothing and bed sheets (Slart et al., 1997).
How mirror-image pain is created is unknown. Although neuropathic pain from the area of nerve trauma can be accounted for, in part, by ectopic action potentials and hyperexcitability (Woolf and Salter, 2000), no abnormal activity has been reported in the healthy contralateral nerve. Hence abnormal contralateral peripheral nerve responsivity cannot account for mirror-image pain. Rather, mirror-image pain likely arises from altered spinal processing of incoming sensory information (Koltzenburg et al., 1999; Watkins and Maier, 2002). Various neurocircuits have been proposed for how altered contralateral neural processing of pain may occur (Koltzenburg et al., 1999; Ossipov et al., 2000), but whether such neurocircuits adequately account for mirror-image allodynia is unknown. Furthermore, virtually nothing is known regarding the neurochemical bases of this pain phenomenon.
The ability to study mirror-image allodynia has recently been facilitated by the development of the sciatic inflammatory neuropathy (SIN) model (Chacur et al., 2001; Gazda et al., 2001). This animal model creates neuropathic pain as a result of localized inflammation of one healthy sciatic nerve. Inflammation is induced by perisciatic microinjection of an immune activator (yeast cell walls; zymosan). This procedure creates rapid, robust, low-threshold mechanical allodynia. Low levels of immune activation create a unilateral mechanical allodynia ipsilateral to the inflamed sciatic nerve. Stronger immune activation creates bilateral allodynia; that is, allodynia is observed both ipsilateral and contralateral (mirror-image) to the nerve inflammation (Chacur et al., 2001; Gazda et al., 2001). Mirror-image allodynia produced by SIN cannot be accounted for by systemic spread of the immune activator (Chacur et al., 2001). Rather, expression of contralateral allodynia is correlated with well defined immunological and anatomical changes in and around the inflamed sciatic nerve (Gazda et al., 2001).
The purpose of the present series of experiments is to provide an initial investigation of spinal cord mechanisms underlying mirror-image allodynia. It also provides the first investigation of spinal mediators of inflammatory neuropathy pain. In contrast to the wealth of studies focused on the spinal neurochemistry of pain from traumatic neuropathy (Zimmermann, 2001), the spinal mediators of inflammatory neuropathy pain have not been identified. Specifically, these experiments examine whether spinal cord glia are involved in SIN-induced ipsilateral and contralateral pain changes and, if so, which glially derived substance(s) is critically involved in creating or maintaining such pathological pain.
Materials and Methods
Subjects
Pathogen-free adult male Sprague Dawley rats (300–450 gm; Harlan Labs, Madison, WI) were used in all experiments. Rats were housed in temperature-controlled (23 ± 3°C) and light-controlled (12 hr light/dark; lights on at 7:00 A.M.) rooms with standard rodent chow and water available ad libitum. Behavioral testing was performed during the light cycle. There were five to six rats per in every experiment. All procedures were approved by the Institutional Animal Care and Use Committee of the University of Colorado at Boulder.
Drugs
Zymosan (yeast cell walls; Sigma, St. Louis, MO) was made fresh daily by suspension in a vehicle of incomplete Freund's adjuvant (Sigma) to final concentrations of 0.08 μg/μl and 3.2 μg/μl. The glial metabolic inhibitor fluorocitrate (Sigma) (Paulsen et al., 1987; Hassel et al., 1992) was dissolved initially in 2 m HCl and then diluted in sterile, endotoxin-free 10 mm PBS (Invitrogen, Gaithersburg, MD) to attain a final concentration of 1 nmol fluorocitrate per microliter, pH 6.0. This solution was aliquoted and stored at −70°C. The vehicle (0.3% 2 m HCl in PBS, pH 6.0) was aliquoted and stored at 4°C. The p38 mitogen-activated protein (MAP) kinase inhibitor CNI-1493 (Denham et al., 2000) was synthesized at North Shore University Hospital as described previously (Bianchi et al., 1995). CNI-1493 was dissolved in endotoxin-free sterile distilled water (9 μg/μl), aliquoted, and stored at −70°C. Endotoxin-free solutions of recombinant met-human interleukin-1 receptor antagonist (IL1ra; 100 μg/μl; lot number 2010316L6; Amgen, Thousand Oaks, CA) and IL1ra vehicle (lot number 0210306L6; Amgen) were stored at 4°C. Lyophilized tumor necrosis factor binding protein (TNFbp; endotoxin-free polyethylene glycol recombinant human soluble TNF receptor type I; lot number 36000D8; Amgen) was reconstituted at 30 μg/μl (its limit of solubility) in endotoxin-free sterile distilled water, aliquoted on ice, and stored at −75°C. TNFbp vehicle (lot number 1105208E8; Amgen) was stored at 4°C. Lyophilized affinity-purified sheep anti-rat interleukin-6 (IL6) IgG (National Institute for Biological Standards and Control, Potters Bar, UK) was reconstituted at 1.3 μg/μl in endotoxin-free sterile distilled water, aliquoted, and stored at −70°C. At the time of testing, a thawed aliquot of anti-rat IL6 was diluted in sterile 0.9% saline to a final concentration of 0.065 μg/μl. Affinity-purified normal sheep IgG (control; lot number 31K9105; Sigma) was reconstituted to 1.3 μg/μl, aliquoted, stored, and diluted at the time of test to 0.065 μg/μl in an identical manner.
Behavioral measures
The von Frey test (Chaplan et al., 1994) was performed within the sciatic innervation area of the hindpaws as described previously in detail (Milligan et al., 2000, 2001b; Chacur et al., 2001; Gazda et al., 2001). Briefly, a logarithmic series of 10 calibrated Semmes-Weinstein monofilaments (von Frey hairs; Stoelting, Wood Dale, IL) was applied randomly to the left and right hindpaws to determine the stimulus intensity threshold stiffness required to elicit a paw withdrawal response. Log stiffness of the hairs is determined by log10 (milligrams × 10). The 10 stimuli had the following log-stiffness values (value in grams is given in parentheses): 3.61 (407 mg), 3.84 (692 mg), 4.08 (1202 mg), 4.17 (1479 mg), 4.31 (2041 mg), 4.56 (3630 mg), 4.74 (5495 mg), 4.93 (8511 mg), 5.07 (11749 mg), and 5.18 (15136 mg). The range of monofilaments used in these experiments (0.407–15.136 gm) produces a logarithmically graded slope when interpolating a 50% response threshold of stimulus intensity (expressed as log10 (milligrams × 10)) (Chaplan et al., 1994). Assessments were made before (baseline) and at specific times after perisciatic and intrathecal drug administration, as detailed below for each experiment. Behavioral testing was performed blind with respect to drug administration. The behavioral responses were used to calculate the 50% paw withdrawal threshold (absolute threshold), by fitting a Gaussian integral psychometric function using a maximum-likelihood fitting method (Harvey, 1986; Treutwein and Strasburger, 1999), as described in detail previously (Milligan et al., 2000, 2001b). This fitting method allows parametric statistical analyses (Milligan et al., 2000, 2001b).
Surgery and microinjections
Chronic intrathecal catheters. Lumbosacral intrathecal catheters were constructed and implanted by lumbar approach as described previously in detail (Milligan et al., 1999). The indwelling catheters were used to microinject drugs into the CSF space surrounding the lumbosacral spinal cord. Only one intrathecal injection was made per animal. All intrathecal microinjections were performed as detailed previously, using an 8 μl void volume to ensure complete drug delivery (Milligan et al., 1999). All catheter placements were verified after completion of behavioral testing by visual inspection. Data were only analyzed from animals with catheters verified as having the catheter tip at the lumbosacral spinal level.
Chronic perisciatic catheters. Perisciatic catheters were constructed and implanted at mid-thigh level of the left hindleg as described previously in detail (Chacur et al., 2001; Gazda et al., 2001; Milligan et al., 2003). This method allowed multiday recovery of the animal from isofluorane anesthesia before microinjection of an immune activator around the sciatic nerve. This avoids the deleterious effects of anesthetics on the function of both immune (Lockwood et al., 1993; Sato et al., 1995; Miller et al., 1996) and glial cells (Tas et al., 1987; Mantz et al., 1993; Miyazaki et al., 1997; Feinstein et al., 2001). In addition, this indwelling catheter method allowed perisciatic immune activation to be either acute (single injection of an immune activator) or chronic (repeated injections across weeks) (Milligan et al., 2003). Both methods were used in the present experiments in awake unrestrained rats. These acute and chronic perisciatic microinjections over the left sciatic nerve were performed as detailed previously (Milligan et al., 1999; Chacur et al., 2001). For all experiments, catheters were verified by visual inspection at the time animals were killed. Data were analyzed only from confirmed sites.
Data analysis
All statistical comparisons were computed using Statview 5.0.1 for the Macintosh. Data from the von Frey test were analyzed as the interpolated 50% threshold (absolute threshold) in log base 10 of stimulus intensity (monofilament stiffness in milligrams × 10). Pre-drug baseline measures were analyzed by one-way ANOVA. Post-drug time course measures were analyzed by repeated measures ANOVAs followed by Fisher's protected least significant difference post hoccomparisons, where appropriate.
Experiment 1: effect of intrathecal fluorocitrate on sciatic inflammatory neuropathy-induced allodynia: blockade of allodynia
Fluorocitrate selectively inhibits aconitase, an enzyme that is in the Krebs' energy cycle of glia, but not neurons (Paulsen et al., 1987; Hassel et al., 1992). Although prolonged glial disruption can indirectly lead to altered neuronal function (e.g., by altering glially regulated uptake of excitatory amino acids; for discussion, seeMilligan et al. (2000)), effects observed at short post-drug times after 1 nmol fluorocitrate reflect glial inactivation (Paulsen et al., 1987; Hassel et al., 1992). Blockade of the initial development of exaggerated pain states by 1 nmol intrathecal fluorocitrate has provided supportive evidence that spinal cord glia are involved in enhanced pain induced by peripheral inflammation (Meller et al., 1994;Watkins et al., 1997) and direct spinal immune activation (Milligan et al., 2000). Thus, intrathecal fluorocitrate was tested here to assess whether spinal cord glia may participate in the development of ipsilateral or mirror-image SIN-induced low-threshold mechanical allodynias.
After baseline behavioral assessments, the glial metabolic inhibitor fluorocitrate (1 nmol) (Paulsen et al., 1987; Hassel et al., 1992) or equivolume vehicle (1 μl) was microinjected intrathecally. This intrathecal dose has previously been documented to block enhanced pain states produced by peripheral inflammation or spinal glial activation (Meller et al., 1994; Watkins et al., 1997; Milligan et al., 2000;Chacur et al., 2001). Thirty minutes after the intrathecal injection, rats received perisciatic microinjections of 0, 4, or 160 μg zymosan in 50 μl of vehicle. The 4 and 160 μg zymosan doses were chosen for this and all subsequent experiments on the basis of their effectiveness in producing ipsilateral (relative to the site of injection) and bilateral allodynia, respectively, in intrathecal catheterized rats (Milligan et al., 2003). Behavior was reassessed 1, 2, and 3 hr later. Behavioral testing was restricted to these early postinjection times to avoid potential nonselective effects of fluorocitrate on neuronal function that may occur at later time points (for discussion seeMilligan et al. (2000)).
Experiment 2: effect of intrathecal CNI-1493 on sciatic inflammatory neuropathy-induced allodynia: prevention of allodynia
CNI-1493 is a p38 MAP kinase inhibitor (Denham et al., 2000). p38 MAP kinase participates in one of the major intracellular signaling cascades leading to the production and release of proinflammatory cytokines (TNF, IL1, IL6) in glia and immune cells (Lee et al., 2000). In addition, p38 MAP kinase is part of the intracellular signaling cascade activated by proinflammatory cytokines binding to their receptors (Lee et al., 2000). Thus, p38 MAP kinase inhibitors can disrupt both production of and signaling by these proteins. As an initial screen for potential proinflammatory cytokine mediation of SIN-induced allodynias, intrathecal CNI-1493 was administered before induction of SIN.
After baseline behavioral assessments, the p38 MAP kinase inhibitor CNI-1493 (9 μg) (Denham et al., 2000) or equivolume saline (1 μl) was microinjected intrathecally. p38 MAP kinase is implicated in the intracellular signaling cascades (1) activated in response to proinflammatory cytokines binding their receptors (Raingeaud et al., 1995; Ridley et al., 1997) and (2) which lead to the production of proinflammatory cytokines (Lee et al., 1994, 2000). The CNI-1493 dose used here has previously been documented to block enhanced pain states produced by direct spinal glial activation (Watkins et al., 1997;Milligan et al., 2000). Thirty minutes after intrathecal injection, each rat received a perisciatic microinjection of 0, 4, or 160 μg zymosan, performed as above. Behavior was reassessed 1, 1.5, 2, 3, and 24 hr later.
Experiment 3: effect of intrathecal CNI-1493 on sciatic inflammatory neuropathy-induced allodynia: reversal of allodynia
After baseline behavioral assessments, perisciatic microinjections of 0, 4, or 160 μg zymosan were performed as above. Behavior was reassessed 13 hr later to confirm the effectiveness of the perisciatic injections before intrathecal drug delivery. At 14.5 hr after perisciatic injection, each rat received an intrathecal injection of either CNI-1493 (9 μg) or equivolume saline (1 μl). Behavior was then assessed 0.5, 2.5, and 4.5 hr later (that is, 15, 17, and 19 hr after the perisciatic injection).
Experiment 4: effect of intrathecal tumor necrosis factor binding protein on sciatic inflammatory neuropathy-induced allodynia: blockade of allodynia
It should be noted here that CNI-1493 has recently been demonstrated to be capable of crossing the blood–brain barrier into spinal cord after systemic administration (Milligan et al., 2001a). Whether it could cross the blood–brain barrier from lumbar cerebrospinal fluid to reach the dorsal root ganglia is unknown. However, if this were possible, the CNI-1493 effects in experiments 2 and 3 might be accounted for by alterations in dorsal root ganglia proinflammatory cytokine function. Because dorsal root ganglia proinflammatory cytokines are upregulated in at least some pathological pain states (Watkins and Maier, 2002), this is an intriguing possibility. Testing TNF, IL6, and IL1 antagonists in this and subsequent experiments will clarify whether spinal proinflammatory cytokines are involved because these antagonists are all large proteins that do not diffuse across the blood–brain barrier; however, this does leave the question of potential dorsal root ganglia proinflammatory cytokine involvement open for future studies.
After baseline behavioral assessments, and 60 min before perisciatic injections, the TNF antagonist TNFbp (TNF soluble receptor; 300 μg) or equivolume vehicle (10 μl) was microinjected intrathecally. The TNFbp dose and timing of its administration were based on previous studies in which TNFbp blocked enhanced pain states produced by direct spinal glial activation (Milligan et al., 2001b). Behavior was reassessed 1, 3, and 24 hr after perisciatic microinjection of 0, 4, or 160 μg zymosan.
Experiment 5: effect of intrathecal anti-rat interleukin-6 on sciatic inflammatory neuropathy-induced allodynia: reversal of allodynia 1 d later
After baseline behavioral assessments, perisciatic microinjections of 0, 4, or 160 μg zymosan were performed as above. Behavior was reassessed 13 hr later to confirm the effectiveness of the perisciatic injections before intrathecal drug delivery. At 14.5 hr after perisciatic injection, each rat received an intrathecal injection of either affinity-purified sheep anti-rat IL6 IgG (0.065 μg in 5 μl) or affinity-purified normal sheep IgG (0.065 μg in 5 μl). Behavior was then assessed 0.5, 2.5, and 4.5 hr later (that is, 15, 17, and 19 hr after the perisciatic injection).
Experiment 6: effect of intrathecal interleukin-1 receptor antagonist on sciatic inflammatory neuropathy-induced allodynia: reversal of allodynia 1 d later
After baseline behavioral assessments, perisciatic microinjections of 0, 4, or 160 μg zymosan were performed as above. Behavior was reassessed 13 hr later to confirm the effectiveness of the perisciatic injections before intrathecal drug delivery. At 14.5 hr after perisciatic injection, each rat received an intrathecal injection of either IL1ra (100 μg) or equivolume vehicle (1 μl). Behavior was then assessed 0.5, 2.5, and 4.5 hr later (that is, 15, 17, and 19 hr after the perisciatic injection).
Experiment 7: effect of intrathecal interleukin-1 receptor antagonist on sciatic inflammatory neuropathy-induced allodynia: reversal of allodynia 2 weeks later
Behavioral assessments were recorded before (baseline) and at 1, 4, 8, 10, 12, and 14 d after baseline. In half of the animals, a perisciatic microinjection of 160 μg zymosan was delivered immediately after baseline (day 0) and 3, 5, 7, 9, 11, and 13 d later. This injection schedule was based on pilot studies aimed at maintaining robust allodynia across days. The remaining animals were identically implanted with indwelling perisciatic catheters and injected with equivolume saline on corresponding days. On day 14, after an initial behavioral assessment (day 14 baseline), all animals received either IL1ra (100 μg) or equivolume (1 μl) vehicle intrathecally. Behavior was reassessed 0.5, 1, 1.5, 2 and 2.5 hr later.
Results
Experiment 1: effect of intrathecal fluorocitrate on sciatic inflammatory neuropathy-induced allodynia: blockade of allodynia
Immunohistochemical evidence of bilateral astrocyte and microglial activation has been observed after traumatic neuropathies (Colburn et al., 1999; Winkelstein et al., 2001). Whether glial activation is the consequence of nerve trauma or nerve inflammation in these models is unknown. However, intense sciatic nerve inflammation has recently been reported to also induce immunohistochemical evidence of bilateral glial activation (Herzberg and Sagen, 2001). This suggests that glial activation may occur in response to SIN. Thus the purpose of the present experiment was to determine whether fluorocitrate, a glial metabolic inhibitor, could block pathological pain induced by inflammatory neuropathy.
As in our previous studies (Chacur et al., 2001; Gazda et al., 2001), low-dose zymosan induced a unilateral allodynia (Fig.1A), whereas higher dose zymosan induced a bilateral allodynia (Fig. 1B), compared with vehicle controls (Fig. 1A). Pretreatment with intrathecal fluorocitrate prevented the development of SIN-induced pain changes (Fig.1A,B).
Fig. 1.

Blockade of perisciatic SIN-induced mechanical allodynias by intrathecal fluorocitrate, a glial metabolic inhibitor. Rats were assessed for low-threshold mechanical sensitivity (von Frey test) both before (baseline) and 1, 2, and 3 hr after completion of intrathecal drug administration. Replicating our earlier studies (Chacur et al., 2001; Gazda et al., 2001), low-dose zymosan induced a unilateral allodynia (A), whereas high-dose zymosan induced a bilateral allodynia (B). Although fluorocitrate had no effect in the absence of perisciatic zymosan (A), it greatly reduced both unilateral (A) and bilateral (B) allodynias induced by perisciatic zymosan. The 10 stimuli tested had the following log-stiffness values (value in grams is given in parentheses): 3.61 (407 mg), 3.84 (692 mg), 4.08 (1202 mg), 4.17 (1479 mg), 4.31 (2041 mg), 4.56 (3630 mg), 4.74 (5495 mg), 4.93 (8511 mg), 5.07 (11,749 mg), and 5.18 (15,136 mg). i.t., Intrathecal; Inj, injection; Veh, vehicle; perisci, perisciatic; Lo Zym, low-dose zymosan; Hi Zym, high-dose zymosan. Abbreviations apply to Figures 1-7.
These observations were supported by statistical analyses. ANOVA revealed reliable main effects of zymosan dose (F(2,210) = 214.125; p< 0.0001), intrathecal fluorocitrate (F(2,62) = 182.756; p < 0.0001), laterality (F(1,62 = 88.069; p < 0.0001), and time (F(2,124) = 33.601;p < 0.0001), and interactions between intrathecal fluorocitrate and zymosan dose (F(2,62) = 70.867; p< 0.0001), zymosan dose and laterality (F(2,62) = 31.775; p< 0.0001), intrathecal fluorocitrate, zymosan dose, and laterality (F(2,62) = 12.070; p< 0.0001) and time, intrathecal fluorocitrate, zymosan dose, and laterality (F(4,124) = 2.692;p < 0.05).
Post hoc means comparison revealed several important points. After 4 μg zymosan (Fig. 1A), mechanical allodynia was observed in the left (ipsilateral) hindpaw compared with the right (contralateral) hindpaw (p < 0.0001). Mechanical responses of the right hindpaw after 4 μg zymosan did not differ from that after perisciatic vehicle (Fig. 1A), supporting the conclusion that 4 μg zymosan induced only a unilateral allodynia ipsilateral to the site of injection. Fluorocitrate greatly reduced the allodynic effects of 4 μg zymosan (p < 0.0001 comparing the ipsilateral paw of rats receiving 4 μg zymosan with versus without intrathecal fluorocitrate) (Fig. 1A), because mild allodynia was observed only at 1 hr after perisciatic zymosan (p < 0.001). Intrathecal fluorocitrate, in the absence of perisciatic zymosan, had no effect on paw withdrawal thresholds, compared with intrathecal vehicle controls (p > 0.5) (Fig. 1A).Post hoc means comparison also revealed that bilateral mechanical allodynia occurred in response to 160 μg zymosan. That is, the thresholds of the left and right hindpaws did not differ (p > 0.05 comparing the ipsilateral and contralateral paws of rats receiving 160 μg zymosan but no fluorocitrate) (Fig. 1B), but the thresholds for both the left and right paws for all of these groups were reliably different from those of the vehicle controls (p < 0.0001 and p < 0.0001, respectively) (Fig.1A). Fluorocitrate greatly reduced the perisciatic allodynia in both the ipsilateral (p < 0.0001) and contralateral (p < 0.0001) paws, compared with 160 μg zymosan-injected rats receiving vehicle intrathecally (Fig. 1B). Indeed, only mild, transient allodynia was observed in the fluorocitrate-treated animals at 1 hr after perisciatic zymosan (p < 0.05).
Experiment 2: effect of intrathecal CNI-1493 on sciatic inflammatory neuropathy-induced allodynia: prevention of allodynia
Experiment 1 provided initial evidence that spinal cord glia may be involved in the mediation of both ipsilateral and mirror-image SIN-induced allodynias. This is the first evidence that pain induced by inflammation around healthy peripheral nerves likely involves spinal cord glia. This suggests that SIN-induced pain changes would be mediated by pain-enhancing substances known to be released by activated glia. Although various substances are released by activated glia, proinflammatory cytokines have recently been implicated as mediators of diverse exaggerated pain states (Watkins et al., 2001). Hence, CNI-1493, a global inhibitor of proinflammatory cytokine function, was tested.
As in our previous studies (Chacur et al., 2001; Gazda et al., 2001), low-dose zymosan induced a unilateral allodynia (Fig.2A), whereas higher dose zymosan induced a bilateral allodynia (Fig. 2B) compared with vehicle controls (Fig. 2A). Pretreatment with intrathecal CNI-1493 abolished these SIN-induced pain changes through 3 hr after perisciatic injection (Fig.2A,B). Allodynia recovered by 24 hr, in accordance with known pharmacokinetics for this compound (Cerami et al., 1996).
Fig. 2.

Blockade of perisciatic SIN-induced mechanical allodynias by intrathecal CNI-1493, a p38 mitogen-activated kinase inhibitor. Rats were assessed for low-threshold mechanical sensitivity (von Frey test) both before (baseline) and 1, 1.5, 2, 3, and 24 hr after completion of intrathecal drug administration. Replicating our earlier studies (Chacur et al., 2001; Gazda et al., 2001), low-dose zymosan induced a unilateral allodynia (A), whereas high-dose zymosan induced a bilateral allodynia (B). Although CNI-1493 had no effect in the absence of perisciatic zymosan (A), it abolished unilateral allodynia (A) and greatly reduced bilateral allodynia (B) induced through 3 hr by perisciatic zymosan. Both unilateral and bilateral allodynias returned by 24 hr (A, B).
These observations were supported by statistical analyses. ANOVA revealed reliable main effects of zymosan dose (F(2,120) = 30.742; p< 0.0001), intrathecal CNI-1493 (F(1,120) = 132.044; p< 0.0001), and laterality (F(1,120) = 46.807; p < 0.0001), and interactions between intrathecal CNI-1493 and zymosan dose (F(2,60) = 28.656; p< 0.0001), zymosan dose and laterality(F(2,60) = 7.481;p < 0.01), and intrathecal CNI-1493, zymosan dose, and laterality (F(2,60) = 11.843;p < 0.0001). Allodynia recovered by 24 hr. Both ipsilateral allodynia (Fig. 2A) and bilateral allodynia (Fig. 2B) were fully restored by this time. At 24 hr, ANOVA revealed reliable main effects of zymosan dose (F(1,60) = 13.925; p< 0.0001) and laterality (F(1,60) = 21.370; p < 0.0001), and interactions between zymosan dose and laterality (F(1,60) = 9.864;p < 0.001). As in experiment 1, 4 μg zymosan (Fig.2A) produced mechanical allodynia in the left (ipsilateral) hindpaw compared with the right (contralateral) hindpaw (p < 0.0001). Also as in experiment 1, mechanical responses of the right hindpaw after 4 μg zymosan did not differ from that after perisciatic vehicle (p > 0.2) (Fig. 2A), indicating that 4 μg zymosan induced only a unilateral allodynia ipsilateral to the site of injection. CNI-1493 abolished the allodynic effects of 4 μg zymosan through 3 hr (p < 0.0001) comparing the ipsilateral paw of rats receiving 4 μg zymosan + intrathecal CNI-1493 with the ipsilateral paw of rats receiving no zymosan + intrathecal CNI-1493 (Fig.2A). Intrathecal CNI-1493, in the absence of perisciatic zymosan, had no effect on paw withdrawal thresholds, compared with intrathecal vehicle controls (p> 0.5) (Fig. 2A).
Post hoc means comparison also revealed that bilateral mechanical allodynia occurred in response to 160 μg zymosan. That is, the thresholds of the left and right hindpaws did not differ, except at the 1 hr time point (p < 0.001) (Fig.2B). The thresholds for both the left and right paws for all of these groups were reliably different from those of the appropriate vehicle controls (p < 0.0001) (Fig.2A). CNI-1493 greatly reduced the perisciatic allodynia in both the ipsilateral (p < 0.0001) and contralateral (p < 0.0001) paws through 3 hr, compared with 160 μg zymosan-injected rats receiving vehicle intrathecally (Fig. 2A,B).
Experiment 3: effect of intrathecal CNI-1493 on sciatic inflammatory neuropathy-induced allodynia: reversal of allodynia
Experiment 2 provided initial evidence that p38 MAP kinase pathways are involved in the initiation of both ipsilateral and mirror-image SIN-induced allodynias. The present experiment tests whether p38 MAP kinases may be involved in the maintenance of these ipsilateral and mirror-image allodynias as well. This is a key issue for pathological pain states, because drug manipulations can often prevent but not reverse pathological pain once it develops (Traub, 1996; Bianchi and Panerai, 1997). Thus, CNI-1493 was administered the day after the allodynias were fully developed to determine whether either allodynia would be reversed by this drug.
Compared with vehicle controls (Fig.3A), low-dose perisciatic zymosan induced a unilateral allodynia (Fig. 3A), whereas higher dose perisciatic zymosan induced a bilateral allodynia (Fig.3B), measured 13 hr later. CNI-1493 injected intrathecally at 14.5 hr abolished these SIN-induced pain changes within 2.5–4.5 hr (that is, 17–19 hr after perisciatic injection) (Fig.3B).
Fig. 3.

Reversal of perisciatic SIN-induced mechanical allodynias by intrathecal CNI-1493, a p38 mitogen-activated kinase inhibitor. Rats were assessed for low-threshold mechanical sensitivity (von Frey test) both before (baseline) and 13, 15, 17, and 19 hr after completion of perisciatic drug administration. Replicating and extending our earlier studies (Chacur et al., 2001; Gazda et al., 2001), low-dose zymosan induced a unilateral allodynia (A), whereas high-dose zymosan induced a bilateral allodynia (B) at 13 hr after injection. CNI-1493 reversed both of these allodynias, although it had no effect on behavior in the absence of perisciatic zymosan (A).
These observations were supported by statistical analyses. ANOVA revealed reliable main effects of zymosan dose (F(2,66) = 125.279; p< 0.0001) and laterality (F(1,66) = 80.804; p < 0.0001), and interactions between zymosan dose and laterality (F(2,66) = 30.638;p < 0.0001). Post hoc means comparison revealed that 4 μg zymosan (Fig. 3A) induced mechanical allodynia in the left (ipsilateral) hindpaw compared with the right (contralateral) hindpaw (p < 0.0001). Mechanical responses of the right hindpaw after 4 μg perisciatic zymosan did not differ from that after perisciatic vehicle (Fig.3A), showing that 4 μg zymosan induced only a unilateral allodynia ipsilateral to the site of injection. In addition, post hoc analyses showed that bilateral mechanical allodynia occurred in response to 160 μg perisciatic zymosan. That is, the thresholds of the left and right hindpaws did not differ (Fig. 3B). The thresholds for both the left and right paws for all of these groups were reliably different from those of the vehicle controls (p < 0.0001) (Fig. 3A). At 14.5 hr, either CNI-1493 or vehicle was injected intrathecally. Behavior was recorded 0.5, 2.5, and 4.5 hr later (that is, 15, 17, and 19 hr after perisciatic drug administration). Although CNI-1493 had no effect in the absence of perisciatic zymosan (Fig. 3A), it reversed both unilateral (Fig. 3A) and bilateral (Fig. 3B) allodynias induced by perisciatic zymosan. ANOVA revealed reliable main effects of zymosan dose (F(2,60) = 111.593; p < 0.0001), intrathecal CNI-1493 (F(1,60) = 162.991; p< 0.0001), and laterality (F(1,60) = 61.422; p < 0.0001), and interactions between intrathecal CNI-1493 and zymosan dose (F(2,60) = 84.320; p< 0.0001), and intrathecal CNI-1493, zymosan dose, and laterality (F(2,60) = 15.675; p< 0.0001).
Post hoc means comparisons showed that CNI-1493 reversed the allodynic effects of 4 μg zymosan by 17–19 hr (p < 00001 comparing the ipsilateral paw of rats receiving 4 μg zymosan with vs without intrathecal CNI-1493) (Fig. 3A). Indeed, the response thresholds of rats receiving 4 μg zymosan + CNI-1493 were not different from vehicle controls at this time (p > 0.3) (Fig. 3A). This drug reversed the bilateral allodynic effects of 160 μg perisciatic zymosan by 17–19 hr as well (p < 0.0001 comparing the ipsilateral paw of rats receiving 160 μg zymosan with vs without intrathecal CNI-1493; p < 0.0001 comparing the contralateral paw of rats receiving 160 μg zymosan with vs without intrathecal CNI-1493) (Fig.3A,B). Again, the response thresholds of rats receiving 160 μg zymosan + CNI-1493 were not different from vehicle controls at this time (p> 0.1) (Fig. 3A). Intrathecal CNI-1493, in the absence of perisciatic zymosan, had no effect on paw withdrawal thresholds, compared with intrathecal vehicle controls (Fig. 3A).
Experiment 4: effect of intrathecal tumor necrosis factor binding protein (soluble receptors) on sciatic inflammatory neuropathy-induced allodynia: blockade of allodynia
Experiments 2 and 3 demonstrate that p38 MAP kinase is a key mediator in the intracellular signaling leading to SIN-induced allodynias. Given that p38 MAP kinases are strongly associated with proinflammatory cytokine production and signaling (Lee et al., 2000), this suggests that disruption of SIN-induced allodynias by CNI-1493 may result from disruption of proinflammatory cytokine function. There are three proinflammatory cytokines known to be affected by p38 MAP kinase inhibitors: TNF, IL1, and IL6 (Lee et al., 2000). All three have been implicated in the spinal mediation of pain arising from traumatic neuropathies (Arruda et al., 2000; Sweitzer et al., 2001; Winkelstein et al., 2001). Whether it is the trauma or associated inflammation of the peripheral nerve that recruits spinal proinflammatory cytokine involvement in these models is unknown. The present experiment tested whether disruption of TNF signaling with TNF soluble receptors (TNFbp) would mimic the effects of CNI-1493, that is, prevent the development of SIN-induced allodynias. The experiments that follow examine IL1 and IL6 in turn.
As before, low-dose zymosan again induced a unilateral allodynia (Fig.4A), whereas higher dose zymosan induced a bilateral allodynia (Fig. 4B) through 24 hr, compared with vehicle controls (Fig.4A). Pretreatment with intrathecal TNFbp prevented these SIN-induced pain changes through 24 hr, in keeping with the prolonged half-life of this compound relative to IL1ra (Bendele et al., 1998; Edwards, 1999) (Fig.4A,B).
Fig. 4.

Blockade of perisciatic SIN-induced mechanical allodynias by intrathecal TNFbp (TNF-soluble receptors), a TNF antagonist. Rats were assessed for low-threshold mechanical sensitivity (von Frey test) both before (baseline) and 1, 3, and 24 hr after completion of intrathecal drug administration. Replicating our earlier studies (Chacur et al., 2001; Gazda et al., 2001), low-dose zymosan induced a unilateral allodynia (A), whereas high-dose zymosan induced a bilateral allodynia (B). Although TNFbp had no effect in the absence of perisciatic zymosan (A), it abolished both unilateral (A) and bilateral allodynia (B) induced through 3 hr by perisciatic zymosan. There was no evident return of allodynia by 24 hr, in accord with its prolonged half-life (A, B).
These observations were supported by statistical analyses. ANOVA revealed reliable main effects of intrathecal TNFbp (F(1,58) = 35.604; p< 0.0001) and laterality (F(1,58) = 23.061; p < 0.0001) and time (F(2,116) = 6.141; p< 0.005), and interactions between intrathecal TNFbp and zymosan dose (F(2,58) = 11.509;p < 0.0001), zymosan dose and laterality (F(2,58) = 6.736; p < 0.01), intrathecal TNFbp and zymosan dose and laterality (F(2,58) = 7.331; p < 0.005), time, and zymosan dose (F(4,116) = 3.739; p< 0.01), and time, intrathecal TNFbp, zymosan dose, and laterality (F(4,116) = 3.681; p< 0.01). Post hoc means comparison revealed that 4 μg zymosan induced a unilateral mechanical allodynia observed in the left (ipsilateral) hindpaw compared with the right (contralateral) hindpaw (p < 0.0001) (Fig.4A). Mechanical responses of the right hindpaw after 4 μg zymosan did not differ from that after perisciatic vehicle (p > 0.05) (Fig. 4A), showing that 4 μg zymosan induced only a unilateral allodynia ipsilateral to the site of injection. TNFbp prevented the allodynic effects of 4 μg zymosan (p < 0.0001 comparing the ipsilateral paw of rats receiving 4 μg zymosan with vs without intrathecal TNFbp;p > 0.25 comparing the ipsilateral paw of rats receiving 4 μg zymosan + intrathecal TNFbp vs control groups receiving perisciatic vehicle) (Fig. 4A). Intrathecal TNFbp, in the absence of perisciatic zymosan, had no effect on paw withdrawal thresholds, compared with intrathecal vehicle controls (p > 0.05) (Fig. 4A).
Post hoc means comparison also revealed that bilateral mechanical allodynia occurred in response to 160 μg zymosan. That is, the thresholds of the left and right hindpaws did not differ (p > 0.05) (Fig. 4B), but the thresholds for both the left and right paws for all of these groups were reliably different from those of the vehicle controls (p < 0.0001 and p < 0.0001 for ipsilateral and contralateral comparisons, respectively) (Fig.4A). TNFbp prevented the perisciatic 160 μg zymosan-induced allodynia in both the ipsilateral and contralateral paws compared with 160 μg zymosan-injected rats receiving vehicle intrathecally (p < 0.0001 and p< 0.0001 for ipsilateral and contralateral comparisons, respectively) and compared with control groups receiving perisciatic vehicle (p > 0.05 and p > 0.05 for ipsilateral and contralateral comparisons, respectively) (Fig.4A,B).
Experiment 5: effect of intrathecal anti-rat interleukin-6 on sciatic inflammatory neuropathy-induced allodynia: reversal of allodynia 1 d later
Experiment 4 provided evidence that the proinflammatory cytokine TNF is a key mediator of SIN-induced allodynias. Because TNF can both induce the release of IL6 (Benveniste et al., 1990) and synergize with IL6 (Dinarello, 1997), involvement of TNF in SIN-induced effects does not exclude the possibility that IL6 may also be involved. Thus the present experiment sought to extend the findings of experiment 4 by determining whether anti-IL6 could reverse both ipsilateral and mirror-image pain states. Thus, this experiment with anti-IL6 paralleled the design of experiment 3 (reversal of SIN-induced allodynias by CNI-1493).
As before, low-dose perisciatic zymosan induced a unilateral allodynia (Fig. 5A), whereas higher dose perisciatic zymosan induced a bilateral allodynia (Fig. 5B), measured 13 hr later. Anti-IL6 injected intrathecally at 14.5 hr reversed these SIN-induced pain changes within 2.5–4.5 hr (that is, 17–19 hr after perisciatic injection) (Fig.5A,B).
Fig. 5.

Reversal of perisciatic SIN-induced mechanical allodynias by intrathecal anti-rat IL6, 1 d later. Rats were assessed for low-threshold mechanical sensitivity (von Frey test) both before (baseline) and 13, 15, 17, and 19 hr after completion of perisciatic drug administration. Replicating experiment 3 and extending our earlier studies (Chacur et al., 2001; Gazda et al., 2001), low-dose zymosan induced a unilateral allodynia (A), whereas high-dose zymosan induced a bilateral allodynia (B) at 13 hr after injection. Both were reversed by intrathecal anti-rat IL6 at this time, whereas anti-IL6 had no effect on behavior in the absence of perisciatic zymosan (A).
These observations were supported by statistical analyses. Before intrathecal anti-IL6 administration, ANOVA revealed reliable main effects of zymosan dose (F(2,60) = 94.296; p < 0.0001) and laterality (F(1,60) = 23.977; p< 0.0001), and interactions between zymosan dose and laterality (F(2,60) = 21.085; p< 0.0001). Post hoc means comparison revealed that 4 μg zymosan (Fig. 5A) induced mechanical allodynia in the left (ipsilateral) hindpaw compared with the right (contralateral) hindpaw (p < 0.0001). Mechanical responses of the right hindpaw after 4 μg perisciatic zymosan did not differ from that after perisciatic vehicle (p > 0.5) (Fig.5A), showing that 4 μg zymosan induced only a unilateral allodynia ipsilateral to the site of injection. In addition, post hoc analyses showed that bilateral mechanical allodynia occurred in response to 160 μg perisciatic zymosan. That is, the thresholds of the left and right hindpaws did not differ (p > 0.5) (Fig. 5B). The thresholds for both the left and right paws for all of these groups were reliably different from those of the vehicle controls (p < 0.0001) (Fig.5A). At 14.5 hr, either affinity-purified sheep anti-rat IL6 IgG or affinity-purified normal sheep IgG (control) was injected intrathecally. Behavior was recorded 0.5, 2.5, and 4.5 hr later (that is, 15, 17, and 19 hr after perisciatic drug administration). Although anti-IL6 had no effect in the absence of perisciatic zymosan (Fig.5A), it completely reversed unilateral allodynia (Fig.5A) and greatly reduced bilateral allodynia (Fig.5B) induced by perisciatic zymosan. ANOVA revealed reliable main effects of zymosan dose (F(2,60)= 40.371; p < 0.0001), intrathecal anti-IL6 (F(1,60) = 29.745; p< 0.0001), and laterality (F(1,60) = 16.992; p < 0.0001), and interactions between zymosan dose and intrathecal anti-IL6 (F(2,60)= 18.022; p < 0.0001), zymosan dose and laterality (F(2,60) = 5.186; p < 0.01), and zymosan dose and intrathecal anti-IL6 and laterality (F(2,60) = 5.849; p < 0.005).
Post hoc means comparisons showed that anti-IL6 reversed the allodynic effects of 4 μg zymosan by 17–19 hr (p < 0.0001 comparing the ipsilateral paw of rats receiving 4 μg zymosan with vs without intrathecal anti-IL6;p > 0.2 comparing the ipsilateral paw of rats receiving 4 μg zymosan + intrathecal anti-IL6 versus perisciatic vehicle controls) (Fig. 5A). This antiserum reversed the bilateral allodynic effects of 160 μg perisciatic zymosan by 17–19 hr as well (p < 0.001 comparing the ipsilateral paw of rats receiving 160 μg zymosan with vs without intrathecal anti-IL6; p < 0.001 comparing the contralateral paw of rats receiving 160 μg zymosan with vs without intrathecal anti-IL6;p > 0.1 and p > 0.1 comparing the ipsilateral and contralateral paw of rats receiving 160 μg zymosan + intrathecal anti-IL6 vs perisciatic vehicle controls) (Fig.5A). Intrathecal anti-IL6, in the absence of perisciatic zymosan, had no effect on paw withdrawal thresholds, compared with intrathecal IgG controls (p > 0.09) (Fig.5A).
Experiment 6: effect of intrathecal interleukin-1 receptor antagonist on sciatic inflammatory neuropathy-induced allodynia: reversal of allodynia 1 d later
Experiments 4 and 5 provided evidence that the proinflammatory cytokines TNF and IL6 are key mediators of SIN-induced allodynias. Because TNF and IL6 can both (1) induce the release of IL1 (Watkins et al., 1999; Milligan et al., 2001b) and (2) synergize with IL1 (Dinarello, 1997), involvement of TNF and IL6 in SIN-induced effects does not exclude the possibility that IL1 may also be involved. Thus the present experiment sought to extend the findings of experiments 4 and 5 by examining whether IL1 receptor antagonist could reverse SIN-induced pain states.
As before, low-dose perisciatic zymosan induced a unilateral allodynia (Fig. 6A), whereas higher dose perisciatic zymosan induced a bilateral allodynia (Fig.6B), measured 13 hr later. IL1ra injected intrathecally at 14.5 hr greatly reduced these SIN-induced pain changes within 2.5–4.5 hr (that is, 17–19 hr after perisciatic injection) (Fig. 6A,B).
Fig. 6.

Reversal of perisciatic SIN-induced mechanical allodynias by intrathecal IL1ra, an IL1 receptor antagonist, 1 d later. Rats were assessed for low-threshold mechanical sensitivity (von Frey test) both before (baseline) and 13, 15, 17, and 19 hr after completion of perisciatic drug administration. Replicating experiments 3 and 5 and extending our earlier studies (Chacur et al., 2001; Gazda et al., 2001), low-dose zymosan induced a unilateral allodynia (A), whereas high-dose zymosan induced a bilateral allodynia (B) at 13 hr after injection. Both were reversed by intrathecal IL1ra at this time, whereas IL1ra had no effect on behavior in the absence of perisciatic zymosan (A).
These observations were supported by statistical analyses. ANOVA revealed reliable main effects of zymosan dose (F(2,68) = 39.635; p< 0.0001) and laterality (F(1,68) = 28.972; p < 0.0001), and interactions between zymosan dose and laterality (F(2,68) = 12.846;p < 0.0001). Post hoc means comparison revealed that 4 μg zymosan (Fig. 6A) induced mechanical allodynia in the left (ipsilateral) hindpaw compared with the right (contralateral) hindpaw (p < 0.0001). Mechanical responses of the right hindpaw after 4 μg perisciatic zymosan did not differ from that after perisciatic vehicle (p > 0.30) (Fig. 6A), showing that 4 μg zymosan induced only a unilateral allodynia ipsilateral to the site of injection. In addition, post hoc analyses showed that bilateral mechanical allodynia occurred in response to 160 μg perisciatic zymosan, that is, the thresholds of the left and right hindpaws did not differ (p > 0.4) (Fig.6B). The thresholds for both the left and right paws for all of these groups were reliably different from those of the vehicle controls (p < 0.0001) (Fig.6A). At 14.5 hr, either IL1ra or vehicle was injected intrathecally. Behavior was recorded 0.5, 2.5, and 4.5 hr later (that is, 15, 17, and 19 hr after perisciatic drug administration). Although IL1ra had no effect in the absence of perisciatic zymosan (Fig.6A), it completely reversed unilateral allodynia (Fig. 6A) and greatly reduced bilateral allodynia (Fig. 6B) induced by perisciatic zymosan. ANOVA revealed reliable main effects of zymosan dose (F(2,68) = 33.459; p< 0.0001), intrathecal IL1ra (F(1,68)= 24.257; p < 0.0001), and laterality (F(1,68) = 7.489; p < 0.01), and time (F(1,68) = 4.543; p< 0.05), and interactions between zymosan dose and intrathecal IL1ra (F(2,68) = 26.404; p< 0.0001), zymosan dose and laterality (F(2,68) = 5.114; p < 0.01), and time, intrathecal IL1ra, and zymosan dose (F(2,68) = 7.412; p < 0.01).
Post hoc means comparisons showed that IL1ra reversed the allodynic effects of 4 μg zymosan by 19 hr (p < 0.0001 at 19 hr, comparing the ipsilateral paw of rats receiving 4 μg zymosan with or without intrathecal IL1ra;p > 0.9 at 19 hr, comparing the ipsilateral paw of rats receiving 0 or 4 μg zymosan with intrathecal IL1ra) (Fig.6A). This drug greatly reduced the bilateral allodynic effects of 160 μg perisciatic zymosan by 19 hr as well (p < 0.0001 at 19 hr, comparing the ipsilateral paw of rats receiving 160 μg zymosan with or without intrathecal IL1ra; p < 0.0001 at 19 hr, comparing the contralateral paw of rats receiving 160 μg zymosan with vs without intrathecal IL1ra) (Fig. 6B). Intrathecal IL1ra in the absence of perisciatic zymosan had no effect on paw withdrawal thresholds, compared with intrathecal vehicle controls (p > 0.05) (Fig. 6A).
Experiment 7: effect of intrathecal interleukin-1 receptor antagonist on sciatic inflammatory neuropathy-induced allodynia: reversal of allodynia 2 weeks later
Experiment 6 provided evidence that the proinflammatory cytokine IL1 is a key mediator of SIN-induced allodynias and is involved in the maintenance of allodynia. Because chronic pain can persist for long periods of time, it was of interest to determine whether IL1 would still be a key mediator of allodynia once pain enhancement was maintained for weeks. If so, this would support the idea that IL1 in particular, and proinflammatory cytokines in general, may be clinically relevant targets for pain control. Thus, this last experiment tested whether IL1ra would still be able to reverse long-standing (2 weeks) SIN-induced allodynias.
Figure 7, A andB (left panels), presents the behavioral results of chronic perisciatic injections, with no intrathecal injections made at any time. That is, all rats in any given left panel are identical in terms of the drugs administered to them through the day 14 time point. Thus the small differences between groups within any givenleft panel reflect random variability. On day 14, the group designations within each panel of Figure 7 become meaningful because it is on day 14 that the single intrathecal injection of IL1ra or vehicle was administered. The day 14 data shown in the left panelssimultaneously serve as the baseline measure for the intrathecal IL1ra versus vehicle test presented in the right panels.
Fig. 7.
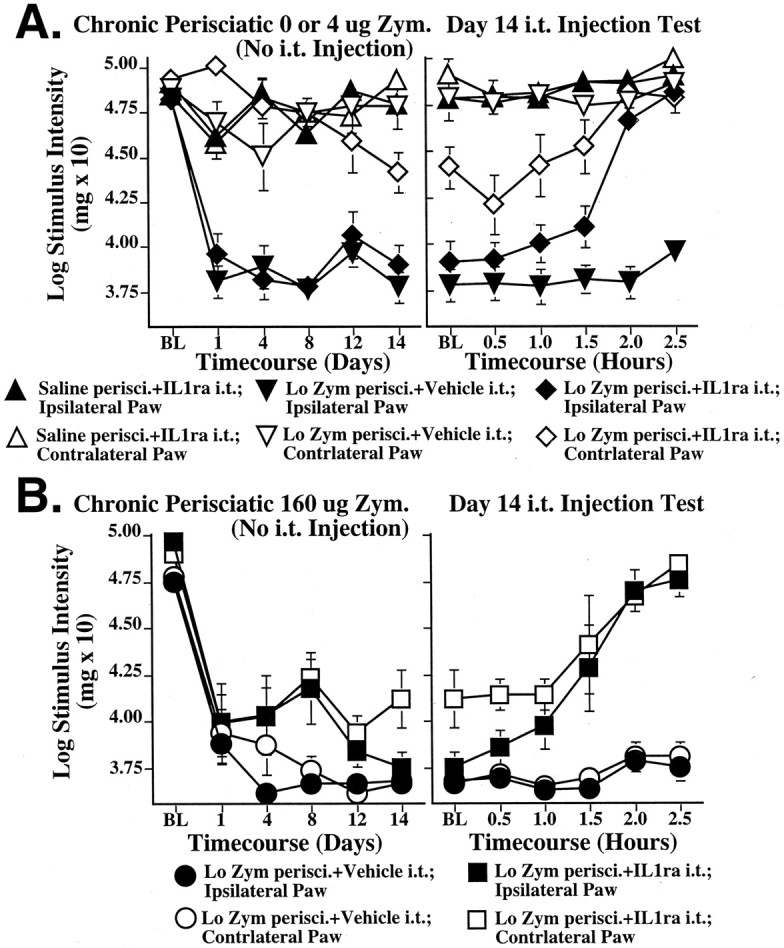
Reversal of perisciatic SIN-induced mechanical allodynias by intrathecal IL1ra, an IL1 receptor antagonist, 2 weeks later. Rats were assessed for low-threshold mechanical sensitivity (von Frey test) both before (baseline) and across 2 weeks after completion of perisciatic drug administration. Extending our earlier studies (Chacur et al., 2001; Gazda et al., 2001), low-dose zymosan induced a unilateral allodynia (A), whereas high-dose zymosan induced a bilateral allodynia (B) by 1 d after injection and stably maintained for 2 weeks. Both were reversed by intrathecal IL1ra at this time, whereas IL1ra had no effect on behavior in the absence of perisciatic zymosan (A,B, right panels).
Low dose perisciatic zymosan induced a unilateral allodynia by 1 d and was stably maintained across the 2 week observation period (Fig.7A, left panel). There was a trend toward mild allodynia developing in the contralateral paw with chronic zymosan, but there was still clearly a marked allodynia in the ipsilateral paw compared with the contralateral paw (Fig.7A, left panel). Higher dose perisciatic zymosan also induced a bilateral allodynia by 1 d and was also stably maintained across the 2 week observation period (Fig.7B, left panel). This is the first demonstration that stable allodynias can be chronically produced by this SIN model. IL1ra injected intrathecally on day 14 reversed these SIN-induced pain changes within 2.5 hr (Fig.7A,B, right panels).
These observations were supported by statistical analyses. ANOVA revealed reliable main effects of zymosan dose (F(2,36) = 46.844; p< 0.0001) and laterality (F(1,36) = 18.455; p < 0.0001), and interactions between sciatic treatment and laterality (F(2,36) = 8.550; p < 0.0001). Post hoc means comparison revealed that 4 μg zymosan (Fig. 7A) induced mechanical allodynia in the left (ipsilateral) hindpaw compared with the right (contralateral) hindpaw (p < 0.0001). Mechanical responses of the right hindpaw after 4 μg perisciatic zymosan did not differ from that after perisciatic vehicle, showing that 4 μg zymosan induced only a unilateral allodynia ipsilateral to the site of injection. In addition, post hoc analyses showed that bilateral mechanical allodynia occurred in response to 160 μg perisciatic zymosan (Fig. 7B). The thresholds for both the left and right paws for all of these groups were reliably different from those of the vehicle controls (p < 0.0001). At 14 d, either IL1ra or vehicle was injected intrathecally. Behavior was then recorded for 2.5 hr. Although IL1ra had no effect in the absence of perisciatic zymosan, it completely reversed unilateral allodynia (Fig. 7A, right) and greatly reduced bilateral allodynia (Fig. 7B,right) induced by perisciatic zymosan. ANOVA revealed reliable main effects of intrathecal IL1ra (F(4,42) = 91.950; p< 0.0001), and laterality (F(1,42) = 53.390; p < 0.0001), and time (F(4,168) = 48.251; p< 0.0001), and interactions between intrathecal IL1ra and laterality (F(4,42) = 22.964; p< 0.0001) and time and intrathecal IL1ra (F(16,168) = 8.799; p< 0.0001).
Post hoc means comparisons showed that IL1ra abolished the allodynic effects of 4 μg zymosan by 2.5 hr on day 14 (p < 0.0001 comparing the ipsilateral paw of rats receiving 0 or 4 μg zymosan with intrathecal IL1ra) (Fig.7A, right). This drug greatly reduced the bilateral allodynic effects of 160 μg perisciatic zymosan by 2.5 hr on day 14 as well (p < 0.0001 comparing the ipsilateral paw of rats receiving 160 μg zymosan with or without intrathecal anti-IL6; p < 0.0001 comparing the contralateral paw of rats receiving 160 μg zymosan with vs without intrathecal IL1ra) (Fig. 7B, right). Intrathecal IL1ra, in the absence of perisciatic zymosan, had no effect on paw withdrawal thresholds, compared with intrathecal vehicle controls.
Discussion
These experiments provide the first identification of spinal mediators of mirror-image low-threshold mechanical allodynia. They also provide the first identification of neurochemical bases of inflammatory neuropathy-induced pain. These data support the conclusion that spinal cord glia and proinflammatory cytokines (TNF, IL1, IL6) are key mediators of these pathological pain phenomena. These experiments thus support and extend the recent report that spinal proinflammatory cytokines (IL1) induce allodynia and hyperexcitability of pain transmission neurons (Reeve et al., 2000). As in previous studies of SIN, low-level sciatic inflammation created ipsilateral allodynia, whereas higher level sciatic inflammation created both ipsilateral and contralateral (mirror-image) allodynias. All allodynias were (1) blocked by intrathecal fluorocitrate, a glial metabolic inhibitor (Paulsen et al., 1987; Hassel et al., 1992), (2) blocked by an inhibitor of the p38 MAP kinase pathway (intrathecal CNI-1493) implicated in both proinflammatory cytokine production and intracellular signaling cascades (Lee et al., 2000), (3) reversed by this inhibitor after the allodynias were fully developed, (4) blocked by an intrathecal TNF antagonist (TNFbp), (5) reversed by an intrathecal IL6 antagonist after the allodynias were fully developed (anti-IL6), and (6) reversed by an intrathecal IL1ra after allodynia was maintained for either 1 d or 2 weeks. This last experiment also provides the first demonstration that the SIN model can be used to study pain chronically maintained by repeated immune activation. The fact that IL1ra reversed ipsilateral and mirror-image pain after 2 weeks of sciatic inflammation strongly indicates that proinflammatory cytokines are not important simply for the creation of pathological pain; rather, spinal proinflammatory cytokines are critical for maintenance of pathological pain as well. Last, given that nerve trauma always results in inflammation, these data have implications for all neuropathic pain regardless of whether pain arises from traumatic or nontraumatic etiologies.
SIN is not alone in creating mirror-image effects. As noted above, numerous clinical pain syndromes are associated with mirror-image pain, primarily allodynic in nature. Mirror-image thermal hyperalgesia and mechanical allodynia have also been observed in diverse animal models of pathological pain (Seltzer et al., 1990; Coderre and Melzack, 1991;Aloisi et al., 1993; Tal and Bennett, 1994; Rees et al., 1996;Takahashi et al., 1996; Sinnott et al., 1999; Hunt et al., 2001). Few studies have examined the mechanisms involved. Mirror-image thermal hyperalgesia is mediated, at least in part, by substance P, NMDA receptors, non-NMDA receptors, and dynorphin (Coderre and Melzack, 1991; Chen et al., 2000; Malan et al., 2000). Mirror-image allodynia is distinct from mirror-image thermal hyperalgesia because it is not mediated by NMDA or dynorphin (Malan et al., 2000). Until now, no spinal mediators of mirror-image allodynia had been identified.
The involvement of glia in creating mirror-image effects is intriguing. Glia are well suited for creating expansions of the body region from which pain is perceived, for two reasons. First, proinflammatory cytokines act in a paracrine manner to excite distant cells (Watkins et al., 1999). This would potentially allow the proinflammatory cytokines to reach spinal terminations of neighboring nerves, causing hyperexcitability of the pain transmission neurons (Reeve et al., 2000). Second, glia are organized as widespread networks via gap junctions and propagated calcium waves (Haydon, 2001). Excitation of glia at one site can activate distant glia, causing them to release pain-enhancing substances as well (Hassinger et al., 1995;Innocenti et al., 2000; Parri et al., 2001). If the spread of excitation were able to reach the contralateral dorsal horns, mirror-image pain might be anticipated to occur through release of proinflammatory cytokines, glutamate, nitric oxide, or other products released by the newly excited glia. Indeed SIN-induced mirror-image pain is abolished by inhibiting the function of astrocyte gap junctions (Watkins et al., 2003).
Although the present data support a role for glial activation in mediating the allodynic effects of sciatic nerve inflammation, they do not indicate how nerve inflammation leads to glial activation. There are three obvious possibilities. The first is that glia are directly activated by neurotransmitters released in the dorsal horn by the inflamed sensory neurons. Indeed astrocytes and microglia may be activated by “pain” neurotransmitters, including substance P, ATP, calcitonin gene-related peptide (CGRP), and glutamate. Spinal cord astrocytes are activated by substance P-binding neurokinin-1 (NK-1) receptors (Palma et al., 1997). Microglia express nonclassical NK-1 receptors as well (Martin et al., 1993). Substance P synergizes with IL1 and TNF, enhancing release of IL6 and prostaglandin from human spinal cord glia (Palma et al., 1997). It also synergizes with lipopolysaccharide, enhancing IL1 release (Martin et al., 1993). Furthermore, substance P releases IL6 and prostaglandins from astrocytes (Marriott et al., 1991; Cadman et al., 1994; Gitter et al., 1994). Indeed, astrocytes in spinal cord, but not astrocytes isolated from various brain regions, release prostaglandins in response to substance P (Marriott et al., 1991), suggesting that spinal glia are uniquely responsive to neurotransmitters in dorsal horn. Extracellular ATP and ATP metabolites also stimulate astrocytes to release prostaglandins (Marriott et al., 1991) and microglia to release TNF (Hide et al., 2000), IL1 (Chakfe et al., 2002), and IL6 (Shigemoto-Mogami et al., 2001). CGRP and glutamate stimulate IL6 release as well (Kiriyama et al., 1997; Wu et al., 1997). Whether such responses occur in spinal astrocytes and microglia is not known; however, it is likely that rat spinal cord glia will respond to at least ATP and glutamate because they express ATP, NMDA, AMPA, and kainate receptors (Agrawal and Fehlings, 1997; Aicher et al., 1997; Fam et al., 2000).
The second possibility is that sciatic inflammation activates spinal glia indirectly. Dorsal horn neurons are strongly activated in response to SIN, and strong neuronal activation can release fractalkine from the neuronal external surface (Chapman et al., 2000). Fractalkine is a member of the immune-related family of proteins called chemokines (Broxmeyer et al., 1999). These are proinflammatory, causing activation of immune and glial cells (Kuby, 1992). Indeed, fractalkine receptors are expressed by microglia in dorsal horn (Verge et al., 2002), and blocking these receptors abolishes ipsilateral and mirror-image SIN-induced pain (Milligan et al., 2002a). Furthermore, intrathecal fractalkine induces mechanical allodynia, which is blocked by intrathecal IL1ra (Milligan et al., 2002a). Thus, SIN-driven dorsal horn neuronal activation appears to activate glia and release proinflammatory cytokines, at least in part, by release of fractalkine as a neuron-to-glia signal.
The third possibility is that signals arising from nerve inflammation activate a spinal cord–brain–spinal cord loop (Watkins and Maier, 1997), which in turn activates glia. Certainly, exaggerated pain arising from peripheral tissue inflammation activates such a circuit (Wiertelak et al., 1994). Notably, the resultant exaggerated pain state is mediated by spinal cord glial activation and proinflammatory cytokines (Watkins et al., 1997). Furthermore, pain from traumatic neuropathies are created by spinal cord–brain–spinal cord loops (Ossipov et al., 2000) and have also been linked to spinal cord glial activation (Colburn et al., 1999; Winkelstein et al., 2001) and proinflammatory cytokines (Arruda et al., 2000; Sweitzer et al., 2001; Winkelstein et al., 2001). Thus, although such a loop circuit may indeed exist for SIN in general and for mirror-image pain in particular, present evidence is that such a pathway would ultimately lead to spinal cord glial activation, most likely via centrifugal axonal release of substance P and excitatory amino acids (Watkins et al., 1997; Watkins and Maier, 2000).
Regardless of how glia ultimately become activated, the implications for neuropathic pain treatment are clear. Neuropathic pain, regardless of whether it arises from traumatic or inflammatory insults, is essentially uncontrolled by currently available drug therapies. Indeed, it is common for these pharmacological treatments to provide no pain relief for 60–80% of patients and only partial relief for the rest (McQuay et al., 1995, 1996; Sindrup and Jensen, 1999). The present data suggest that these therapies may fail because they target neurons rather than glia. As such, this suggests that targeting spinal cord glial and proinflammatory cytokine function may provide a novel approach for pain control. It is notable along these lines that CNI-1493, the p38 MAP kinase inhibitor used here to block and reverse ipsilateral and mirror-image allodynias, has recently been shown to be effective systemically for controlling pathological pain arising from spinal glial activation (Milligan et al., 2001a). A novel and highly effective gene therapy approach has also been developed recently to disrupt spinal proinflammatory cytokine function (Milligan et al., 2002b; Watkins, 2002; Watkins et al., 2003). Here, the intrathecal injection of a viral vector that encodes for interleukin-10 (IL10) (Moore et al., 2001) blocked chronic pain. IL10 is an anti-inflammatory cytokine that suppresses all levels of proinflammatory cytokine expression from transcription through protein release, downregulates proinflammatory cytokine production, and upregulates production of endogenous proinflammatory cytokine antagonists (Moore et al., 2001). To date, this new gene therapy approach has been shown to block and reverse pathological pain arising from spinal cord inflammation, SIN, and partial nerve injury (Milligan et al., 2002b; Watkins, 2002). Thus, controlling pathological pain via targeting of spinal proinflammatory cytokines is an exciting possibility for clinical pain control.
Footnotes
This work was supported by National Institutes of Health Grants MH01558, MH00314, MH45045, and NS38020. We thank Amgen (Thousand Oaks, CA) for their gift of IL1ra, TNFbp, and vehicles appropriate for each. Affinity-purified anti-rat IL6 was provided by Dr. Stephen Poole (National Institute for Biological Standards and Control, UK). This antibody was raised as part of the European Community-funded Concerted Action Program Biomed I “Cytokines in the Brain” (PL931450).
Correspondence should be addressed to Linda R. Watkins, Department of Psychology, Campus Box 345, University of Colorado at Boulder, Boulder, CO 80309-0345. E-mail: lwatkins@psych.colorado.edu.
References
- 1.Agrawal SK, Fehlings MG. Role of NMDA and non-NMDA ionotropic glutamate receptors in traumatic spinal cord axonal injury. J Neurosci. 1997;17:1055–1063. doi: 10.1523/JNEUROSCI.17-03-01055.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aicher SA, Sharma S, Cheng PY, Pickel VM. The N-methyl-d-aspartate (NMDA) receptor is postsynaptic to substance P-containing axon terminals in the rat superficial dorsal horn. Brain Res. 1997;772:71–81. doi: 10.1016/s0006-8993(97)00637-9. [DOI] [PubMed] [Google Scholar]
- 3.Aloisi AM, Porro CA, Cavazzuti M, Baraldi P, Carli G. “Mirror pain” in the formalin test: behavioral and 2-deoxyglucose studies. Pain. 1993;55:267–273. doi: 10.1016/0304-3959(93)90156-J. [DOI] [PubMed] [Google Scholar]
- 4.Arruda JL, Rutkowski MD, Sweitzer SM, DeLeo JA. Antibody and IgG attenuates mechanical allodynia in a mononeuropathy model in the rat: potential role of immune modulation in neuropathic pain. Brain Res. 2000;879:216–225. doi: 10.1016/s0006-8993(00)02807-9. [DOI] [PubMed] [Google Scholar]
- 5.Baron R. Peripheral neuropathic pain: from mechanisms to symptoms. Clin J Pain. 2000;16(Suppl 2):S12–20. doi: 10.1097/00002508-200006001-00004. [DOI] [PubMed] [Google Scholar]
- 6.Bendele A, McAbee T, Woodward M, Scherrer J, Collins D, Frazier J, Chlipala E, McCabe D. Effects of interleukin-1 receptor antagonist in a slow-release hylan vehicle on rat type II collagen arthritis. Pharmacol Res. 1998;15:1557–1561. doi: 10.1023/a:1011903100188. [DOI] [PubMed] [Google Scholar]
- 7.Benveniste EN, Sparacio SM, Norris JG, Grennett HE, Fuller GM. Induction and regulation of interleukin-6 gene expression in rat astrocytes. J Neuroimmunol. 1990;30:201–212. doi: 10.1016/0165-5728(90)90104-u. [DOI] [PubMed] [Google Scholar]
- 8.Bianchi M, Panerai AE. Formalin injection in the tail facilitates hindpaw withdrawal reflexes induced by thermal stimulation in the rat: effect of paracetamol. Neurosci Lett. 1997;237:89–92. doi: 10.1016/s0304-3940(97)00819-7. [DOI] [PubMed] [Google Scholar]
- 9.Bianchi M, Ulrich P, Bloom O, Meistrell MR, Zimmerman GA, Zimmerman G, Schmidmayerova H, Bukrinsky M, Donnelley T, Bucala R, Sherry B, Manogue K, Tortolani A, Cerami A, Tracey K. An inhibitor of macrophage arginine transport and nitric oxide production (CNI-1493) prevents acute inflammation and endotoxin lethality. Mol Med. 1995;1:254–266. [PMC free article] [PubMed] [Google Scholar]
- 10.Broxmeyer HE, Kim CH, Cooper SH, Hangoc G, Hromas R, Pelus LM. Effects of CC, CXC, C, and CX3C chemokines on proliferation of myeloid progenitor cells, and insights into SDF-1-induced chemotaxis of progenitors. Ann NY Acad Sci. 1999;872:142–162. doi: 10.1111/j.1749-6632.1999.tb08460.x. [DOI] [PubMed] [Google Scholar]
- 11.Cadman ED, Witrte DG, Lee CM. Regulation of the release of interleukin-6 from human astrocytoma cells. J Neurochem. 1994;63:980–987. doi: 10.1046/j.1471-4159.1994.63030980.x. [DOI] [PubMed] [Google Scholar]
- 12.Cerami C, Zhang X, Ulrich P, Bianchi M, Tracey KJ, Berger BJ. High-performance liquid chromatographic method for guanylhydrazone compounds. J Chromatogr B Biomed Appl. 1996;675:71–75. doi: 10.1016/0378-4347(95)00346-0. [DOI] [PubMed] [Google Scholar]
- 13.Chacur M, Milligan ED, Gazda LS, Armstrong C, Wang H, Tracey KJ, Maier SF, Watkins LR. A new model of sciatic inflammatory neuritis (SIN): induction of unilateral and bilateral mechanical allodynia following acute unilateral peri-sciatic immune activation in rats. Pain. 2001;94:231–244. doi: 10.1016/S0304-3959(01)00354-2. [DOI] [PubMed] [Google Scholar]
- 14.Chakfe Y, Seguin R, Antel JP, Morissete C, Malo D, Henderson D, Seguela P. ADP and AMP induce interleukin-1β release from microglial cells through activation of ATP-primed P2X7 receptor channel. J Neurosci. 2002;22:3061–3069. doi: 10.1523/JNEUROSCI.22-08-03061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 16.Chapman GA, Moores K, Harrison D, Campbell CA, Steward BR, Strijbos PJLM. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci. 2000;20:1–5. doi: 10.1523/JNEUROSCI.20-15-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen HS, Chen J, Sun YY. Contralateral heat hyperalgesia induced by unilaterally intraplantar bee venom injection is produced by central changes: a behavioral study in the conscious rat. Neurosci Lett. 2000;284:45–48. doi: 10.1016/s0304-3940(00)00955-1. [DOI] [PubMed] [Google Scholar]
- 18.Coderre TJ, Melzack R. Central neural mediators of secondary hyperalgesia following heat injury in rats: neuropeptides and excitatory amino acids. Neurosci Lett. 1991;131:71–74. doi: 10.1016/0304-3940(91)90339-u. [DOI] [PubMed] [Google Scholar]
- 19.Colburn RW, Rickman AJ, DeLeo JA. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp Neurol. 1999;157:289–304. doi: 10.1006/exnr.1999.7065. [DOI] [PubMed] [Google Scholar]
- 20.Denham W, Yang J, Wang H, Botchkina G, Tracey KJ, Norman J. Inhibition of p38 mitogen activate kinase attenuates the severity of pancreatitis-induced adult respiratory distress syndrome. Crit Care Med. 2000;28:2567–2572. doi: 10.1097/00003246-200007000-00064. [DOI] [PubMed] [Google Scholar]
- 21.Dinarello CA. Role of pro- and anti-inflammatory cytokines during inflammation: experimental and clinical findings. J Biol Regul Homeost Agents. 1997;11:91–103. [PubMed] [Google Scholar]
- 22.Edwards CK, Edwards CK., III PEGylated recombinant human soluble tumour necrosis factor receptor type I (r-Hu-sTNF-RI): novel high affinity TNF receptor designed for chronic inflammatory diseases. Ann Rheum Dis. 1999;58(Suppl 1):173–181. doi: 10.1136/ard.58.2008.i73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fam SR, Gallagher CJ, Salter MW. P2Y(1) purinoceptor-mediated Ca(2+) signaling and Ca(2+) wave propagation in dorsal spinal cord astrocytes. J Neurosci. 2000;20:2800–2808. doi: 10.1523/JNEUROSCI.20-08-02800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feinstein DL, Murphy P, Sharp A, Galea E, Gavrilyuk V, Weinberg G. Local anesthetics potentiate nitric oxide synthase type 2 expression in rat glial cells. J Neurosurg Anesthesiol. 2001;13:99–105. doi: 10.1097/00008506-200104000-00006. [DOI] [PubMed] [Google Scholar]
- 25.Gazda LS, Milligan ED, Hansen MK, Twining CM, Paulos N, Chacur M, O'Connor KA, Armstrong C, Maier SF. Sciatic inflammatory neuritis (SIN): behavioral allodynia is paralleled by peri-sciatic proinflammatory cytokine and superoxide production. J Peripheral Nerv Sys. 2001;6:111–129. doi: 10.1046/j.1529-8027.2001.006001111.x. [DOI] [PubMed] [Google Scholar]
- 26.Gitter BD, Regoli D, Howbert JJ, Glasebrook AL, Waters DC. Interleukin-6 secretion from human astrocytoma cells induced by substance P. J Neuroimmunol. 1994;51:101–108. doi: 10.1016/0165-5728(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 27.Harvey LOJ. Efficient estimation of sensory thresholds. Behav Res Methods Instrum Comput. 1986;18:623–632. [Google Scholar]
- 28.Hassel B, Paulsen RE, Johnson A, Fonnum F. Selective inhibition of glial cell metabolism by fluorocitrate. Brain Res. 1992;249:120–124. doi: 10.1016/0006-8993(92)90616-h. [DOI] [PubMed] [Google Scholar]
- 29.Hassinger TD, Atkinson PB, Strecker GJ, Whalen LR, Dudek FE, Kossel AH, Kater SB. Evidence for glutamate-mediated activation of hippocampal neurons by glial calcium waves. J Neurobiol. 1995;28:159–170. doi: 10.1002/neu.480280204. [DOI] [PubMed] [Google Scholar]
- 30.Haydon PG. Glia: listening and talking to the synapse. Nat Rev Neurosci. 2001;2:185–193. doi: 10.1038/35058528. [DOI] [PubMed] [Google Scholar]
- 31.Herzberg U, Sagen J. Peripheral nerve exposure to HIV viral envelope protein gp120 induces neuropathic pain and spinal gliosis. J Neuroimmunol. 2001;116:29–39. doi: 10.1016/s0165-5728(01)00288-0. [DOI] [PubMed] [Google Scholar]
- 32.Hide I, Tanaka M, Inoue A, Nakajima K, Kohsaka S, Inoue K, Nakata Y. Extracellular ATP triggers tumor necrosis factor-alpha release from rat microglia. J Neurochem. 2000;2000:965–972. doi: 10.1046/j.1471-4159.2000.0750965.x. [DOI] [PubMed] [Google Scholar]
- 33.Hunt JL, Winkelstein BA, Rutkowski MD, Weinstein JN, DeLeo JA. Repeated injury to the lumbar nerve roots produces enhanced mechanical allodynia and persistent spinal neuroinflammation. Spine. 2001;26:2073–2079. doi: 10.1097/00007632-200110010-00005. [DOI] [PubMed] [Google Scholar]
- 34.Innocenti B, Parpura V, Haydon PG. Imaging extracellular waves of glutamate during calcium signaling in cultured astrocytes. J Neurosci. 2000;2000:1800–1808. doi: 10.1523/JNEUROSCI.20-05-01800.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kiriyama Y, Murayama T, Tokumisu Y, Nomura Y. Protein kinase A-dependent IL6 production induced by calcitonin in human glioblastoma A172 cells. J Neuroimmunol. 1997;76:139–144. doi: 10.1016/s0165-5728(97)00044-1. [DOI] [PubMed] [Google Scholar]
- 36.Koltzenburg M, Wall PD, McMahon SB. Does the right side know what the left is doing? Trends Neurosci. 1999;22:122–127. doi: 10.1016/s0166-2236(98)01302-2. [DOI] [PubMed] [Google Scholar]
- 37.Kuby J. Immunology. W. H. Freeman; New York: 1992. [Google Scholar]
- 38.Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- 39.Lee JC, Kumar S, Griwsold DE, Underwood DC, Votta BJ, Adams JL. Inhibition of p38 MAP kinase as a therapeutic strategy. Immunopharmacology. 2000;47:185–201. doi: 10.1016/s0162-3109(00)00206-x. [DOI] [PubMed] [Google Scholar]
- 40.Lockwood LL, Silbert LH, Laudenslager ML, Watkins LR, Maier SF. Anesthesia-induced modulation of in vivo antibody levels: a study of pentobarbital, chloral hydrate, methoxyflurane, halothane, and ketamine/xylazine. Anesthes Analg. 1993;77:769–774. doi: 10.1213/00000539-199310000-00020. [DOI] [PubMed] [Google Scholar]
- 41.Malan TP, Ossipov MH, Gardell LR, Ibrahim M, Bian D, Lai J, Porreca F. Extraterritorial neuropathic pain correlates with multisegmental elevation of spinal dynorphin in nerve-injured rats. Pain. 2000;86:185–194. doi: 10.1016/s0304-3959(00)00243-8. [DOI] [PubMed] [Google Scholar]
- 42.Maleki J, LeBel AA, Bennett GJ, Schwartzman RJ. Patterns of spread in complex regional pain syndrome, type I (reflex sympathetic dystrophy). Pain. 2000;88:259–266. doi: 10.1016/S0304-3959(00)00332-8. [DOI] [PubMed] [Google Scholar]
- 43.Mantz J, Cordier J, Giaume C. Effects of general anesthetics on intercellular communications mediated by gap junctions between astrocytes in primary culture. Anesthesiology. 1993;78:892–901. doi: 10.1097/00000542-199305000-00014. [DOI] [PubMed] [Google Scholar]
- 44.Marriott D, Wilkin GP, Coote PR, Wood JN. Eicosanoid synthesis by spinal cord astrocytes is evoked by substance P; possible implications for nociception and pain. Adv Prostaglandin Thromboxane Leukot Res. 1991;21B:739–741. [PubMed] [Google Scholar]
- 45.Martin FC, Anton PA, Gornbein JA, Shanahan F, Merrill JE. Production of interleukin-1 by microglia in response to substance P: role for a non-classical NK-1 receptor. J Neuroimmunol. 1993;42:53–60. doi: 10.1016/0165-5728(93)90212-h. [DOI] [PubMed] [Google Scholar]
- 46.McQuay H, Carroll D, Jadad AR, Wiffen P, Moore A. Anticonvulsant drugs for management of pain: a systematic review. Br Med J. 1995;311:1047–1052. doi: 10.1136/bmj.311.7012.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McQuay HJ, Tramer M, Nye BA, Carroll D, Wiffen PJ, Moore RA. A systematic review of antidepressants in neuropathic pain. Pain. 1996;68:217–227. doi: 10.1016/s0304-3959(96)03140-5. [DOI] [PubMed] [Google Scholar]
- 48.Meller S, Dykstra C, Grzbycki D, Murphy S, Gebhart G. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 49.Miller LS, Morita Y, Rangan U, Kondo S, Clemens MG, Bulkley GB. Suppression of cytokine-induced neutrophil accumulation in rat mesenteric venules in vivo by general anesthesia. Int J Microcirc Clin Exp. 1996;16:147–154. doi: 10.1159/000179165. [DOI] [PubMed] [Google Scholar]
- 50.Milligan ED, Hinde JL, Mehmert KK, Maier SF, Watkins LR. A method for increasing the viability of the external portion of the lumbar catheters placed in the spinal subarachnoid space of rats. J Neurosci Methods. 1999;90:81–86. doi: 10.1016/s0165-0270(99)00075-8. [DOI] [PubMed] [Google Scholar]
- 51.Milligan ED, Mehmert KK, Hinde JL, Harvey LOJ, Martin D, Tracey KJ, Maier SF, Watkins LR. Thermal hyperalgesia and mechanical allodynia produced by intrathecal administration of the human immunodeficiency virus-1 (HIV-1) envelope glycoprotein, gp120. Brain Res. 2000;861:105–116. doi: 10.1016/s0006-8993(00)02050-3. [DOI] [PubMed] [Google Scholar]
- 52.Milligan ED, O'Connor KA, Armstrong CB, Martin D, Tracey KJ, Maier SF, Watkins LR. Systemic administration of CNI-1493, a p38 MAP kinase inhibitor, blocks HIV-1 gp120-induced enhanced pain states in rats. J Pain. 2001a;6:326–333. doi: 10.1054/jpai.2001.26174. [DOI] [PubMed] [Google Scholar]
- 53.Milligan ED, O'Connor KA, Nguyen KT, Armstrong CB, Twining C, Gaykema R, Holguin A, Martin D, Maier SF, Watkins LR. Intrathecal HIV-1 envelope glycoprotein gp120 enhanced pain states mediated by spinal cord proinflammatory cytokines. J Neurosci. 2001b;21:2808–2819. doi: 10.1523/JNEUROSCI.21-08-02808.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Milligan ED, Verge G, Twining C, Chapman G, Maier SF, Naeve G, Watkins L. The potential role of fractalkine, a neural chemokine, in creating spinally mediated exaggerated pain states. J Pain (Suppl) 2002a;1:30. [Google Scholar]
- 55.Milligan ED, O'Connor KA, Hammack SE, Wiesler-Frank JL, Langer SJ, Leinwand LA, Maier SF, Watkins LR. A novel gene therapy approach for controlling pathological pain states: intrathecal (i.t.) delivery in rats of viral vectors encoding the anti-inflammatory cytokine, interleukin-10, p 124. 10th World Congress on Pain Abstracts. IASP; Seattle: 2002b. [Google Scholar]
- 56. Milligan ED, Maier SF, Watkins LR. Sciatic inflammatory neuropathy: a new model for studying neuropathic pain of inflammatory origin. Pain research methods and protocols: methods of molecular medicine Luo D. 2003. Humana; New York, in press. [Google Scholar]
- 57.Miyazaki H, Nakamura Y, Arai T, Kataoka K. Increase of glutamate uptake in astrocytes: a possible mechanism of action of volatile anesthetics. Anesthesiology. 1997;86:1359–1366. doi: 10.1097/00000542-199706000-00018. [DOI] [PubMed] [Google Scholar]
- 58.Moore KW, de Waal-Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 59.Moriwaki K, Yuge O. Topographical features of cutaneous tactile hypoesthetic and hyperesthetic abnormalities in chronic pain. Pain. 1999;81:1–6. doi: 10.1016/s0304-3959(98)00257-7. [DOI] [PubMed] [Google Scholar]
- 60.Ossipov MH, Lai J, Malan TP, Porreca F. Spinal and supraspinal mechanisms of neuropathic pain. Ann NY Acad Sci. 2000;909:12–24. doi: 10.1111/j.1749-6632.2000.tb06673.x. [DOI] [PubMed] [Google Scholar]
- 61.Palma C, Minghetti L, Astolfi M, Ambrosini E, Silberstein FC, Manzini S, Levi G, Aloisi F. Functional characterization of substance P receptors on cultured human spinal cord astrocytes: synergism of substance P with cytokines in inducing interleukin-6 and prostaglandin E2 production. Glia. 1997;21:183–193. doi: 10.1002/(sici)1098-1136(199710)21:2<183::aid-glia2>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 62.Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci. 2001;4:803–812. doi: 10.1038/90507. [DOI] [PubMed] [Google Scholar]
- 63.Paulsen RE, Contestabile A, Villani L, Fonnum F. An in vivo model for studying function of brain tissue temporarily devoid of glial cell metabolism: the use of fluorocitrate. J Neurochem. 1987;48:1377–1385. doi: 10.1111/j.1471-4159.1987.tb05674.x. [DOI] [PubMed] [Google Scholar]
- 64.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Proinflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 65.Rees H, Sluka KA, Lu Y, Westlund KN, Willis WD. Dorsal root reflexes in articular afferents occur bilaterally in a chronic model of arthritis in rats. J Neurophysiol. 1996;76:4190–4193. doi: 10.1152/jn.1996.76.6.4190. [DOI] [PubMed] [Google Scholar]
- 66.Reeve AJ, Patel S, Fox A, Walker K, Urban L. Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur J Pain. 2000;4:247–257. doi: 10.1053/eujp.2000.0177. [DOI] [PubMed] [Google Scholar]
- 67.Ridley SH, Sarsfield SJ, Lee JC, Bigg HF, Cawston TE, Taylor DJ, DeWitt DL, Saklatvala J. Actions of IL1 are selectively controlled by p38 mitogen-activated protein kinase: regulation of prostaglandin H synthase-2, metalloproteinases, and IL6 at different levels. J Immunol. 1997;158:3165–3173. [PubMed] [Google Scholar]
- 68.Sato W, Enzan K, Masaki Y, Kayaba M, Suzuki M. The effect of isoflurane on the secretion of TNF-alpha and IL1 beta from LPS-stimulated human peripheral blood monocytes. Masui. 1995;44:971–975. [PubMed] [Google Scholar]
- 69.Seltzer Z, Dubner R, Shir Y. A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain. 1990;43:205–218. doi: 10.1016/0304-3959(90)91074-S. [DOI] [PubMed] [Google Scholar]
- 70.Shigemoto-Mogami Y, Koizumi S, Tsuda M, Ohsawa K, Kohsaka S, Inoue K. Mechanisms underlying extracellular ATP-evoked interleukin-6 release in mouse microglial cell line, MG-5. J Neurochem. 2001;78:1339–1349. doi: 10.1046/j.1471-4159.2001.00514.x. [DOI] [PubMed] [Google Scholar]
- 71.Shir Y, Seltzer Z. Effects of sympathectomy in a model of causalgiform pain produced by partial sciatic nerve injury in rats. Pain. 1991;45:309–320. doi: 10.1016/0304-3959(91)90056-4. [DOI] [PubMed] [Google Scholar]
- 72.Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain. 1999;83:389–400. doi: 10.1016/S0304-3959(99)00154-2. [DOI] [PubMed] [Google Scholar]
- 73.Sinnott CJ, Garfield JM, Strichartz GR. Differential efficacy of intravenous lidocaine in alleviating ipsilateral versus contralateral neuropathic pain in the rat. Pain. 1999;80:521–531. doi: 10.1016/S0304-3959(98)00245-0. [DOI] [PubMed] [Google Scholar]
- 74.Slart R, Yu AL, Yaksh TL, Sorkin LS. An animal model of pain produced by systemic administration of an immunotherapeutic anti-ganglioside antibody. Pain. 1997;69:119–125. doi: 10.1016/s0304-3959(96)03247-2. [DOI] [PubMed] [Google Scholar]
- 75.Sweitzer SM, Martin D, DeLeo JA. Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience. 2001;103:529–539. doi: 10.1016/s0306-4522(00)00574-1. [DOI] [PubMed] [Google Scholar]
- 76.Takahashi H, Suguro T, Okazima Y, Motegi M, Okada Y, Kakiuchi T. Inflammatory cytokines in the herniated disc of the lumbar spine. Spine. 1996;21:218–224. doi: 10.1097/00007632-199601150-00011. [DOI] [PubMed] [Google Scholar]
- 77.Tal M, Bennett GJ. Extra-territorial pain in rats with a peripheral mononeuropathy: mechano-hyperalgesia and mechano-allodynia in the territory of an uninjured nerve. Pain. 1994;57:375–382. doi: 10.1016/0304-3959(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 78.Tas PW, Kress HG, Koschel K. General anesthetics can competitively interfere with sensitive membrane proteins. Proc Natl Acad Sci USA. 1987;84:5972–5975. doi: 10.1073/pnas.84.16.5972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Traub RJ. The spinal contribution of substance P to the generation and maintenance of inflammatory hyperalgesia in the rat. Pain. 1996;67:151–161. doi: 10.1016/0304-3959(96)03076-X. [DOI] [PubMed] [Google Scholar]
- 80.Treutwein B, Strasburger H. Fitting the psychometric function. Percept Psychophys. 1999;61:87–106. doi: 10.3758/bf03211951. [DOI] [PubMed] [Google Scholar]
- 81.Verge G, Milligan ED, Twining C, Maier SF, Watkins LR, Foster AC, Naeve GS. Mapping fractalkine and its receptor (CX3CR1) in a rat model of inflammatory neuropathy, p 124. 10th World Congress on Pain Abstracts. IASP; Seattle: 2002. [Google Scholar]
- 82.Watkins LR. Glial activation as a mediator of pathological pain: implications for clinical pain control, p 123. 10th World Congress on Pain Abstracts. IASP; Seattle: 2002. [Google Scholar]
- 83.Watkins LR, Maier SF. The case of the missing brain: arguments for a role of brain-to-spinal cord pathways in pain facilitation. Behav Brain Sci. 1997;20:469–470. [Google Scholar]
- 84.Watkins LR, Maier SF. The pain of being sick: implications of immune-to-brain communication for understanding pain. Annu Rev Psychol. 2000;51:29–57. doi: 10.1146/annurev.psych.51.1.29. [DOI] [PubMed] [Google Scholar]
- 85.Watkins LR, Maier SF. Beyond neurons: evidence that immune and glial cells contribute to pathological pain states. Physiol Rev. 2002;82:981–1011. doi: 10.1152/physrev.00011.2002. [DOI] [PubMed] [Google Scholar]
- 86.Watkins L, Wiertelak EP, Martin D, Ulrich P, Tracey K, Maier S. Evidence for involvement of spinal cord glia in subcutaneous formalin induced hyperalgesia in the rat. Pain. 1997;71:225–235. doi: 10.1016/s0304-3959(97)03369-1. [DOI] [PubMed] [Google Scholar]
- 87.Watkins LR, Hansen MK, Nguyen KT, Lee JE, Maier SF. Dynamic regulation of the proinflammatory cytokine, interleukin-1 beta: molecular biology for non-molecular biologists. Life Sci. 1999;65:449–481. doi: 10.1016/s0024-3205(99)00095-8. [DOI] [PubMed] [Google Scholar]
- 88.Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- 89. Watkins LR, Milligan ED, Maier SF. Immune and glial involvement in physiological and pathological exaggerated pain states. Advances in pain research and therapy Dostrovsky JO, Carr DB, Koltzenburg M. 2003. IASP; Seattle, in press. [Google Scholar]
- 90.Wiertelak EP, Furness LE, Horan R, Martinez J, Maier SF, Watkins LR. Subcutaneous formalin produces centrifugal hyperalgesia at a non-injected site via the NMDA-nitric oxide cascade. Brain Res. 1994;649:19–26. doi: 10.1016/0006-8993(94)91044-8. [DOI] [PubMed] [Google Scholar]
- 91.Winkelstein BA, Rutkowski MD, Sweitzer SM, Pahl JL, DeLeo JA. Nerve injury proximal or distal to the DRG induces similar spinal glial activation and selective cytokine expression but differential behavioral responses to pharmacologic treatment. J Comp Neurol. 2001;439:127–139. [PubMed] [Google Scholar]
- 92.Woda A, Pionchon P. A unified concept of idiopathic orofacial pain: pathophysiologic features. J Orofacial Pain. 2000;14:196–212. [PubMed] [Google Scholar]
- 93.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 94.Wu L, Cao Z, Zhu X. Effects of glutamate and ATP on interleukin 6 production by cultured neonatal rat astrocyte. Hunan I Ko Ta Hsueh Hsueh Pao. 1997;22:116–118. [PubMed] [Google Scholar]
- 95.Zimmermann M. Pathobiology of neuropathic pain. Eur J Pharmacol. 2001;429:23–37. doi: 10.1016/s0014-2999(01)01303-6. [DOI] [PubMed] [Google Scholar]