

Abstract
The aim of the present study was to investigate the role of dopamine D3 receptors in the rewarding effect and hyperlocomotion induced by a prototypical μ-opioid receptor agonist morphine using dopamine D3 receptor knock-out mice. The μ-opioid receptor in the brain determined by the [tylosil-3,5-3H(N)]-[d-Ala2,N-MePhe4,Gly-ol5]enkephalin binding assay was not significantly changed by a deletion of the dopamine D3 receptor gene. Furthermore, we found that no significant differences in G-protein activation by morphine in the limbic forebrain and lower midbrain were noted between the two genotypes. These results suggest that the function of the μ-opioid receptor itself was not affected by a deletion of the dopamine D3 receptor gene. To ascertain the morphine-induced rewarding effect in both genotypes, the conditioned place preference paradigm was performed. Deletion of the dopamine D3receptor gene resulted in a remarkable enhancement of the morphine-induced rewarding effect. Furthermore, knock-out mice with deletions of the dopamine D3 receptor revealed a dramatic potentiation of morphine-induced hyperlocomotion. Under these conditions, a loss of the dopamine D3 receptor gene had no effect on the basal levels of dopamine and the increased dopamine turnover by morphine in the limbic forebrain. These findings provide further evidence that dopamine D3 receptor contributes to the postsynaptically negative modulation of the mesolimbic dopaminergic pathway that is associated with the rewarding effect and hyperlocomotion through the stimulation of μ-opioid receptors induced by morphine in the mouse.
Keywords: dopamine D3 receptor, morphine, rewarding effect, hyperlocomotion, negative feedback system
Introduction
Many studies have suggested that the mesolimbic dopaminergic system that projects from the ventral tegmental area (VTA) to the nucleus accumbens is critical for the initiation of opioid reinforcement and hyperlocomotion (Stinus et al., 1985, 1986; Wise and Rompre, 1989; Koob, 1992). Use of the conditioned place preference paradigm and intra-VTA administration of the selective μ-opioid receptor agonist [d-Ala2,N-MePhe4,Gly-ol5]enkephalin (DAMGO) or a prototypical μ-opioid receptor agonist, morphine, produces the rewarding effect (Bals-Kubik et al., 1993; Narita et al., 2001). Either DAMGO- or morphine-induced place preference can be blocked by dopamine antagonists (Phillips et al., 1983; Shippenberg and Herz, 1988; Shippenberg et al., 1993). It has been recognized that the hyperlocomotion induced by morphine can be blocked by treatment with dopamine receptor antagonists into the nucleus accumbens (Maldonado et al., 1990; Funada et al., 1994). These findings indicate that the dopamine-containing neurons of the midbrain VTA, which has a high density of μ-opioid receptors, play a critical role in the rewarding effects by μ-opioids.
In humans, five dopamine receptor subtypes (D1–D5) have been identified by molecular cloning (Civelli et al., 1993; Gingrich and Caron, 1993). These five dopamine receptors are classified into two subfamilies according to their pharmacological profiles and sequence homologies. The D1-like receptor subtypes are composed of the D1 and D5receptors, which are coupled to stimulatory subsets of heterotrimeric G-proteins. In contrast, the D2-like subtypes consist of D2, D3, and D4 receptors, which are coupled to the inhibitory subsets of G-proteins and are major targets of the antipsychotics.
Among these receptors, the dopamine D3 receptor cloned by Sokoloff and colleagues (1990) has been extensively characterized. The dopamine D3 receptor shows a distinct distribution in limbic areas of the brain, including the nucleus accumbens and olfactory tubercle (Sokoloff et al., 1990). Several pharmacological studies with dopamine D3receptor-preferring agonists such as 7-hydroxy-N,N-di-n-propyl-2-aminotetralin (7-OH-DPAT) suggest that the dopamine D3 receptor regulates the inhibitory effect to produce hyperlocomotion in rodents (Suzuki et al., 1995; De Boer et al., 1997). Several investigators proposed the hypothesis that the dopamine D3receptor agonist is able to inhibit locomotion though a presynaptic autoreceptor mechanism by inhibiting the firing of dopaminergic cell bodies and dopamine release at nerve terminals (Meller et al., 1993;Devoto et al., 1995; Gainetdinov et al., 1996). However, the dopamine D3 receptor mRNAs cannot be detected in the rat midbrain area (Bouthenet et al., 1991; Landwehrmeyer et al., 1993;Richtand et al., 1995). It has been also proposed that the inhibition of spontaneous locomotion by dopamine D3 receptor agonists is independent of dopamine release at the terminal (Waters et al., 1993). The latter report indicates the possibility that the dopamine D3 receptor acts predominantly as the postsynaptic receptor.
It should be noted that these pharmacological approaches are not conclusive, because serious questions exist with respect to the selectivity of dopamine D3 receptor ligands. This problem can now be solved by the molecular biological technique of targeted gene deletion. It has been generally accepted that the use of mice lacking dopamine receptor subtypes can provide direct evidence for the physiological mechanisms of individual receptor subtypes (Baik et al., 1995; Rubinstein et al., 1997).
The dopamine D1 and D2receptors have been shown to play a substantial role in the rewarding and locomotor enhancing effect induced by μ-opioid receptor agonists (Shippenberg and Herz, 1987, 1988; Maldonado et al., 1997; Narita et al., 2001). Little is known, however, about the role of dopamine D3 receptors in the rewarding effect of μ-opioid receptor agonists. The present study was therefore designed to investigate the role of dopamine D3 receptors in the morphine-induced rewarding effect using dopamine D3 receptor knock-out mice.
Materials and Methods
The present study was conducted in accordance with the Guiding Principles for the Care and Use of Laboratory Animals, Hoshi University, as adopted by the Committee on Animal Research of Hoshi University, which is accredited by the Ministry of Education, Culture, Sports, Science and Technology of Japan. All efforts were made to minimize the number of animals used and their suffering.
Animals. The dopamine D3 receptor knock-out mice (C57BL/6J-Drd3tm1Dac) and their wild-type mice were used in the present study (The Jackson Laboratory, Bar Harbor, ME). The dopamine D3receptor knock-out mice used in our experiments were N5 congenic C57BL/6 mice genotyped and generated by a backcrossing strategy. Animals were housed in a room maintained at 22 ± 1°C with a 12 hr light/dark cycle (light on 8:00 A.M. to 8:00 P.M.). Food and water were available ad libitum.
RT-PCR assay. Total RNA in the whole brain was extracted using the SV Total RNA Isolation System (Promega, Madison, WI) following the manufacturer's instructions. The purified total RNA was quantified by spectrophotometer at A260. To prepare first-strand cDNA, 1 μg of RNA was incubated in 100 μl of buffer containing 10 mm dithiothreitol (DTT), 2.5 mm MgCl2, dNTP mix, 200 U of reverse transcriptase II (Invitrogen, Grand Island, NY), and 0.1 mm oligo (dT)12–18 (Invitrogen). The dopamine D3 receptor gene was amplified in a 50 μl PCR solution containing 0.8 mmMgCl2, dNTP mix, and DNA polymerase with the following synthesized primers: a sense primer of the dopamine D3 receptor at position 391–407 (5′-GCA GTG GTC ATG CCA GTT CAC TAT CAG-3′) of the receptor and an antisense primer at position 498–526 (5′-CCT GTT GTG TTG AAA CCA AAG AGG AGA GG-3′), which were designed according to sequence accession numbers U26915 in GenBank. The RT-PCR was performed under conditions used previously (Narita et al., 2002a,b). Briefly, samples were heated to 95°C for 2 min, 55°C for 2 min, and 72°C for 3 min and cycled 40 times through 95°C for 1 min, 55°C for 2 min, and 72°C for 3 min. The resulting 137 bp product amplified with the above primers was subcloned into pGEM-T vector (Invitrogen, San Diego, CA) by the T-A cloning method. DNA sequencing for the inserted region confirmed that the amplified nucleotides corresponded to those of murine dopamine D3 receptor cDNA. The mixture was run on 1% agarose gel electrophoresis with the indicated markers. The agarose gel was stained with ethidium bromide and photographed with UV transillumination.
Membrane preparations. Mice were killed by decapitation, and the whole brain except cerebellum, lower midbrain (containing the VTA), or limbic forebrain (containing the nucleus accumbens) was rapidly dissected for each purpose. The tissue was homogenized using a Potter-Elvehjem tissue grinder with a Teflon pestle in 20 vol (w/v) of ice-cold Tris buffer containing 50 mm Tris-HCl, pH 7.4, for the μ-opioid receptor binding assay, or in ice-cold Tris-Mg2+ buffer containing 50 mm Tris-HCl, pH 7.4, 5 mmMgCl2, and 1 mm EGTA for the [35S]GTPγS binding assay. The homogenate was centrifuged at 4°C for 10 min at 48,000 ×g. The pellet was resuspended in ice-cold Tris buffer or [35S]GTPγS binding assay buffer containing 50 mm Tris-HCl, pH 7.4, 5 mm MgCl2, 1 mm EGTA, and 100 mm NaCl and centrifuged at 4°C for 10 min at 48,000 × g. The resultant pellet was resuspended in ice-cold Tris buffer or [35S]GTPγS binding assay buffer and stored at −70°C until use.
μ-Opioid receptor binding assay. The μ-opioid receptor binding assays were performed in duplicate with [3H] DAMGO (specific activity, 67.0 Ci/mmol; Amersham Biosciences, Arlington Heights, IL) at 0.2–20 nm in a final volume of 1.0 ml that contained 50 mm Tris-HCl buffer, pH 7.4, and 0.1 ml of the homogenated membrane fraction. The amount of membrane proteins used in each assay was in the range of 90–140 μg, as determined by the method ofBradford (1976). The test tubes were incubated for 2 hr at 25°C. Specific binding was defined as the difference in bindings observed in the absence and presence of 10 μm unlabeled DAMGO. Incubation was terminated by collecting membranes on Whatman GF/B filters using a Brandel cell harvester. The filters were then washed three times with 5 ml Tris-HCl buffer, pH 7.4, at 4°C and transferred to scintillation vials. Then, 0.5 ml of Soluene-350 (Packard Instrument Company, Meriden, CT) and 4 ml of Hionic Fluor Cocktail (Packard Instrument Company) were added to the vials. After a 12 hr equilibration period, radioactivity in the samples was determined in a liquid scintillation analyzer.
[35S]GTPγS binding assay.The membrane homogenate (3–8 μg protein per assay) was incubated at 25°C for 2 hr in 1 ml of assay buffer with various concentrations of the agonist, 30 μm GDP, and 50 pm[35S]GTPγS (specific activity, 1000 Ci/mmol; Amersham Biosciences). The reaction was terminated by filtration using a Brandle cell harvester and Whatman GF/B glass filters presoaked in 50 mm Tris-HCl, pH 7.4, and 5 mm MgCl2 at 4°C for 2 hr. Filters were then washed three times with 5 ml of an ice-cold Tris-HCl buffer, pH 7.4, transferred to scintillation counting vials containing 0.5 ml of Soluene-350 and 4 ml of Hionic Fluor, and equilibrated for 12 hr, and the radioactivity in the samples was determined with a liquid scintillation analyzer. Nonspecific binding was measured in the presence of 10 μm unlabeled GTPγS. Comparable results were obtained from at least three independent sets of experiments.
Place conditioning. Place conditioning was conducted as described previously (Suzuki et al., 1990). The apparatus was a shuttle box (15 cm wide × 30 cm long × 15 cm high) that was made of acrylic resin board and divided into two equal-sized compartments. One compartment was white with a textured floor, and the other was black with a smooth floor to create equally preferred compartments. Only animals that did not exhibit a significant preference for either the white or black compartment were used and divided randomly into each separate group of 8–12 mice. Conditioning sessions (three for drug, three for vehicle) were conducted once daily for 6 d. Immediately after subcutaneous injection of morphine or vehicle, these animals were placed in the white or black compartment for 1 hr. On alternate days, the animals were given injections of saline or morphine and then placed in the other compartment. On day 7, tests of conditioning were performed as follows. The partition separating the two compartments was raised to 7 cm above on the floor, and the neutral platform was inserted along the seam separating the compartments. Mice that had not been treated with either drugs or saline were then placed on the platform. The time spent in each compartment during a 900 sec session was then recorded automatically using an infrared beam sensor (KN-80, Natume Seisakusyo Co., Tokyo, Japan). In a pretest, mice spent 458.6 ± 36.8 sec (mean ± SEM) in the white compartment and 444.2 ± 28.4 sec in the black compartment. The mean conditioning score represents the time spent in the morphine-conditioned compartment minus that spent in the saline-conditioned compartment. All sessions were conducted under conditions of dim illumination and masking white noise.
Locomotor activity. The locomotor activity of mice was measured by an activity monitoring system (NS-AS01; Neuroscience Inc., Tokyo) (Narita et al., 2002a,b). Briefly, the activity monitor is composed of the infrared ray sensor placed over an open-top box (23 cm wide × 33 cm long × 12.5 cm high), a signal amplification circuit, and a control circuit. The sensor can detect the movement of animals on the basis of released infrared rays associated with their temperature. Counts of locomotor activity were collected in 10 min intervals for 4 hr before treatment for habituation and for 3 hr after the treatment. The data were analyzed with a computer-associated analyzing system (Multidigital 32-port Counter System; Neuroscience Inc.).
Dopamine turnover. Using HPLC with electrochemical detection (HPLC-ECD), the concentrations of dopamine, 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillic acid (HVA) were determined as described previously (Narita et al., 1993). The mice were killed 30 min after subcutaneous injection of saline (10 ml/kg) or morphine (5 mg/kg). The brain was removed quickly, and the limbic forebrain was dissected on an ice-cold glass plate. The tissues were homogenized in 500 μl of 0.2 m perchloric acid containing 100 μm EDTA(2Na) and 100 ng isoproterenol as an internal standard. The homogenates were then centrifuged at 20,000 × g for 30 min at 4°C, and the supernatants were maintained at pH 3.0 using 1 msodium acetate. Samples were analyzed by HPLC-ECD. The HPLC system consisted of a delivery system (EP-10, Eicom Co., Kyoto, Japan), an analytical column (Eicompac, MA-5ODS, Eicom Co.), and a guard column (Eicom Co.). Dopamine and its metabolites were separated by a column with a mobile phase containing sodium acetate (0.1m), citric acid monohydrate (0.1m), sodium 1-octane sulfonate (170 mg/l), EDTA(2Na) (10 mg/l), and 15% methanol. The mobile phase was delivered at a flow rate of 1.0 ml/min. Identification of dopamine and its metabolites was determined according to the retention times of these standards, and the amounts were quantified by calculating peak area. The dopamine turnover, “DA ratio,” was calculated as (DOPAC + HVA)/dopamine.
Drug and injection procedure. Morphine (Sankyo, Tokyo, Japan) andd-Phe-Cys-Trp-d-Trp-Orn-Thr-Pen-Thr-NH2(CTOP) (Sigma, St. Louis, MO) were dissolved in saline. One day before the beginning of the drug or saline injection, mice were anesthetized with ether and a 2 mm double needle (tip, 27 gauge × 2 mm; base, 22 gauge × 10 mm; Natsume Seisakusho) attached to a 25 μl Hamilton microsyringe was inserted into the unilateral injection site to make the hole. The unilateral injection site was ∼2 mm from either side of the midline between the anterior roots of the ears. On the day for the drug injection, the head of the mouse was held against a V-shaped holder without any anesthetics, and the drugs were injected into the hole. The injection volume was 4 μl for each mouse. The placement for the injection was confirmed by cresyl violet injection after all experiments. CTOP (100 pmol per mouse, i.c.v.) was given to mice 10 min before the treatment with morphine or saline.
Statistical data analysis. The data are presented as the mean ± SEM. The statistical significance of differences between individual doses that produced significant conditioning was assessed with an ANOVA followed by Dunnett's multiple test. The comparison of dose–response curves was analyzed by a computer-associated program (Prism, GraphPad Inc., San Diego, CA). Data for the determination of the density (Bmax) and affinity (Kd) of binding sites were evaluated with the EDBA and LIGAND programs (Biosoft, Cambridge, UK). Student'st test was used for the statistical analysis of theBmax andKd values.
Results
Absence of dopamine D3 receptor mRNA in dopamine D3 receptor knock-out mice
To evaluate whether the mutation of the dopamine D3 receptor gene could result in a loss of expression of dopamine D3 mRNA, we isolated total RNA from the whole brain of wild-type and dopamine D3 knock-out mice to perform RT-PCR using dopamine D3 receptor-specific primers. The dopamine D3 receptor mRNA was readily identified in the whole brain of wild-type but not dopamine D3 knock-out mice (Fig.1). To determine whether the mutation could abolish dopamine D3 receptor and affect the levels of other dopamine receptors, we next measured the levels of dopamine D1, D2, and D3 receptor mRNAs in the forebrain of dopamine D3 receptor knock-out mice using each specific primer (Narita et al., 2002a,b). This mutation eliminated dopamine D3 receptors, with no significant changes in either dopamine D1 or D2 receptors (105.8 ± 2.5 and 89.9 ± 5.1% of wild-type, respectively), confirming the utility of these mice for studying the distinct role of dopamine D3receptors.
Fig. 1.
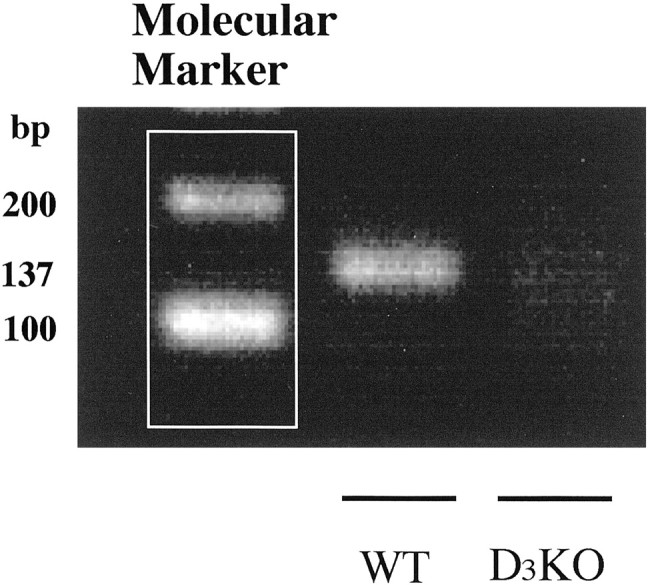
Analysis of dopamine D3 receptor mRNA expression by RT-PCR in the mouse whole brain from wild-type (WT) and dopamine D3 receptor knock-out (D3KO) mice. The dopamine D3 receptor gene was amplified in a 50 μl PCR solution with synthesized primers (137 bp): a sense primer of dopamine D3 receptor, which is at position 391–407 (5′-GCA GTG GTC ATG CCA GTT CAC TAT CAG-3′) of the receptor, and an antisense primer at position 498–526 (5′-CCT GTT GTG TTG AAA CCA AAG AGG AGA GG-3′). The PCR products were separated on 1% agarose gel and visualized after staining with ethidium bromide.
No change in μ-opioid receptor density as determined by [3H]DAMGO binding to the mouse brain membrane preparations in dopamine D3 receptor knock-out mice
To evaluate the population of μ-opioid receptors in the mouse brain, we performed a saturation-binding analysis using [3H]DAMGO. Saturation binding studies with [3H]DAMGO at different concentrations revealed a single high-affinity binding site that represents the μ-opioid receptor. There was no difference between the wild-type and dopamine D3 knock-out mice in either the Bmax orKd value of [3H]DAMGO binding (Table1).
Table 1.
μ-Opioid receptor density and affinity determined by [3H]DAMGO binding to the mouse brain membrane preparation from wild-type (WT) and dopamine D3 receptor knock-out (D3KO) mice
| Bmax (fmol/mg protein) | Kd (nm) | |
|---|---|---|
| WT | 70.12 ± 3.78 | 0.69 ± 0.04 |
| D3KO | 68.63 ± 2.31 | 0.64 ± 0.04 |
Each value represents the mean with SEM of three samples.
Comparison of the stimulation of [35S]GTPγS binding by morphine between dopamine D3 receptor knock-out and wild-type mice
We investigated whether deletion of the dopamine D3 receptor gene could affect G-protein activation through the stimulation of μ-opioid receptors. The ability of μ-opioid receptor agonists to activate G-proteins in the brain of wild-type and dopamine D3 knock-out mice was examined by monitoring binding to membranes of [35S]GTPγS. The μ-opioid receptor agonist morphine (0.1–10 μm) produced a concentration-dependent increase in [35S]GTPγS binding to membranes of the limbic forebrain (Fig.2A) and lower midbrain (Fig. 2B) from both wild-type and D3 knock-out mice. These effects were abolished by coincubating the membranes with the specific μ-opioid receptor antagonist CTOP (10 μm) in both wild-type and dopamine D3 knock-out mice (p < 0.001 vs 10 μmmorphine treatment). However, these effects did not differ significantly between the two genotypes.
Fig. 2.

Stimulation of [35S]GTPγS binding to membranes from the limbic forebrain (A) and lower midbrain (B) by the μ-opioid receptor agonist morphine in wild-type (WT) and dopamine D3 receptor knock-out (D3KO) mice. Membranes were incubated with [35S]GTPγS (50 pm) and GDP (30 μm) with morphine in the presence or absence of the selective μ-opioid receptor antagonist CTOP. The data are shown as the percentage of the basal [35S]GTPγS (50 pm) binding measured in the presence of GDP (30 μm) and the absence of morphine. Eachcolumn represents the mean with SEM of three samples.
Morphine-induced place preference in wild-type and dopamine D3 receptor knock-out mice
In both genotypes, the saline-conditioned group exhibited no preference for either place. The mean conditioning scores were 4.8 ± 41.3 sec for wild-type mice and −12.7 ± 46.6 sec for dopamine D3 knock-out mice. The dose–response curves for morphine-induced place preference in wild-type and dopamine D3 receptor knock-out mice are shown in Figure3. In wild-type mice, morphine (1–5 mg/kg, s.c.) produced a dose-dependent place preference for drug-associated place (144.7 ± 43.9 sec for 5 mg/kg morphine;F(1,24) = 5.38; p < 0.05; vs saline group) (Fig. 3A). In dopamine D3 receptor knock-out mice, lower doses of morphine (0.1–0.56 mg/kg, s.c.) produced a significant and dose-dependent place preference for drug-associated place (146.8 ± 35.2 sec for 0.56 mg/kg morphine;F(1,17) = 6.50; p < 0.05; vs saline group) (Fig. 3A). The dose relationship for the morphine-induced place preference in dopamine D3 receptor knock-out mice was significantly shifted to the left by 7.25-fold (p < 0.01 vs wild-type mice) (Fig. 3A). In addition, the place preference induced by 0.56 mg/kg morphine in dopamine D3receptor knock-out mice was completely blocked by intracerebroventricular treatment with CTOP (100 pmol per mouse; 11.3 ± 47.8 sec; F(1,16) = 7.24;p < 0.05; vs saline–morphine group) (Fig.3B).
Fig. 3.
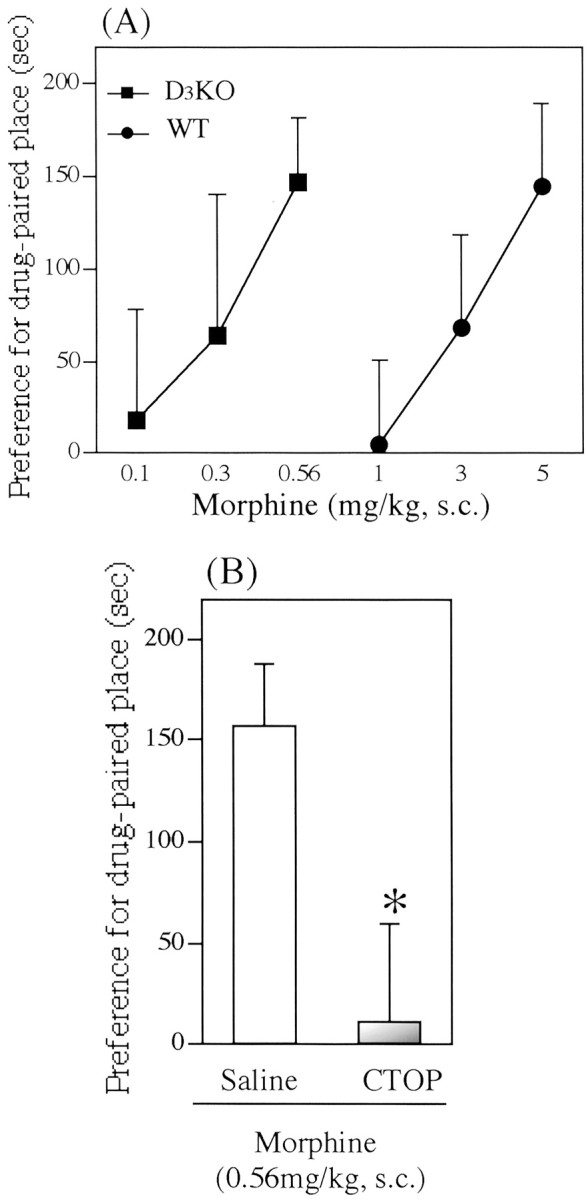
Morphine-induced place preference in wild-type (WT) and dopamine D3receptor knock-out (D3KO) mice.A, Dose–response curve for the morphine-induced place preference in WT and D3KO mice. Each pointrepresents the mean conditioning score with SEM of 8–10 mice.B, Effect of the selective μ-opioid receptor antagonist CTOP on morphine-induced place preference in D3KO mice. CTOP (100 pmol per mouse) or saline was pretreated intracerebroventricularly 10 min before subcutaneous administration of morphine. Each column represents the mean conditioning score with SEM of 8–10 mice. *p< 0.05 versus saline group.
Morphine-induced hyperlocomotion in wild-type and dopamine D3 knock-out mice
Figure 4 shows the morphine-induced hyperlocomotion in wild-type and dopamine D3knock-out mice. We first found that the mutant mice exhibited an enhanced locomotion in the novel environment (during 4 hr habituation; data not shown). This result was consistent with the report by Xu et al. (1997). After 4 hr habituation, saline treatment produced no differences in locomotion between wild-type and dopamine D3 knock-out mice; the mean total activity counts for 180 min were 215.2 ± 42.7 and 294.8 ± 91.3 counts per 180 min, respectively (Fig. 4B). Morphine (5 mg/kg, s.c.) produced a significant increase in locomotion in both genotypes [wild-type mice (F(1,189) = 13.3;p < 0.01 vs saline) and dopamine D3 knock-out mice (F(1,210) = 72.9; p < 0.001 vs saline)]. Under these conditions, the morphine-induced locomotor enhancing effect was significantly enhanced by a deletion of dopamine D3 receptor gene (Fig. 4).
Fig. 4.
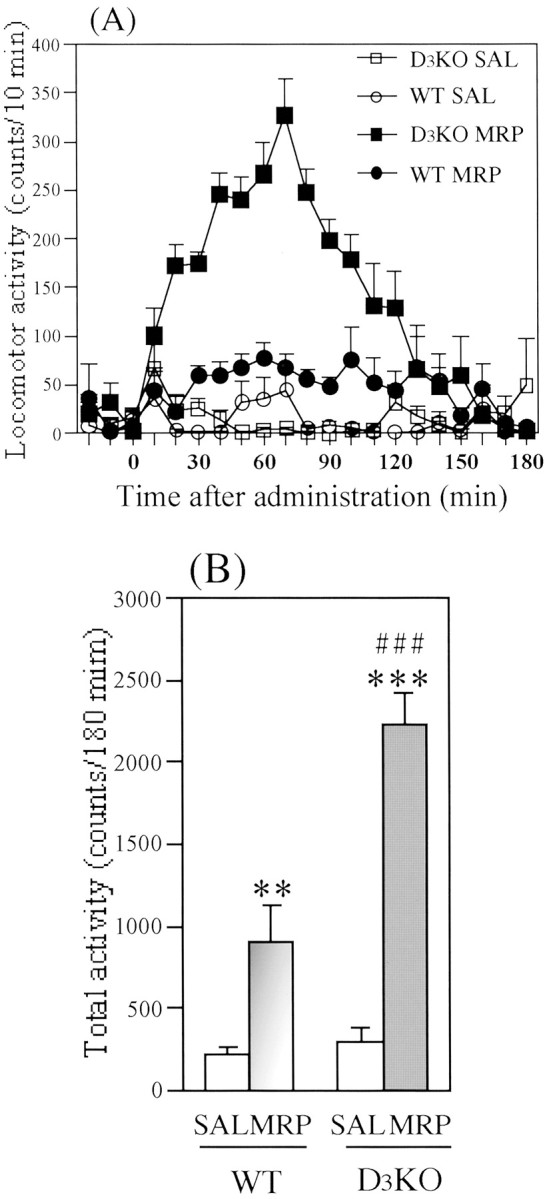
Morphine (MRP)-induced locomotor activity in wild-type (WT) and dopamine D3 receptor knock-out (D3KO) mice.A, Time course changes in the MRP-induced locomotor-enhancing effect in WT and D3KO mice. Eachpoint represents the mean activity counts for 10 min with SEM of 7–8 mice. B, Total locomotor activity of the MRP-induced locomotor-enhancing effect in WT and D3KO mice. Each column represents the mean activity for 180 min with SEM of 7–8 mice. **p < 0.01, ***p < 0.001 versus each saline group.### p < 0.001 versus MRP-treated WT group.
Levels of dopamine and its major metabolites in the limbic forebrain area from dopamine D3 receptor knock-out and wild-type mice
As shown in Table 2, the basal levels of dopamine, DOPAC and HVA, and the dopamine turnover in the limbic forebrain area were unchanged in dopamine D3 knock-out mice as compared with that in wild-type mice. Morphine (5 mg/kg, s.c.) produced a significant increase in levels of DOPAC (p < 0.01 vs saline) and HVA (p < 0.01 vs saline) and in dopamine turnover (p < 0.001 vs saline) in the limbic forebrain area in both wild-type and dopamine D3 knock-out mice. However, these effects did not differ significantly between the two genotypes (Table 2).
Table 2.
Levels of dopamine (DA) and its major metabolites (DOPAC and HVA) in the limbic forebrain area from wild-type (WT) and dopamine D3 receptor knock-out (D3KO) mice in the presence or absence of morphine (MRP)
| DA DOPAC HVA | DA ratio | |||
|---|---|---|---|---|
| (ng/gm wet tissue) | ||||
| WT | ||||
| Basal | 6918.1 ± 297.5 | 632.8 ± 40.4 | 461.3 ± 18.2 | 0.157 ± 0.004 |
| MRP | 6517.1 ± 278.6 | 1067.9 ± 43.82-160 | 817.3 ± 26.32-165 | 0.290 ± 0.0062-165 |
| D3KO | ||||
| Basal | 6525.7 ± 62.3 | 585.7 ± 1.4 | 434.7 ± 5.9 | 0.156 ± 0.002 |
| MRP | 6398.9 ± 532.3 | 1110.9 ± 85.92-160 | 714.2 ± 51.62-160 | 0.286 ± 0.0072-165 |
Each value represents the mean with SEM. The dopamine turnover (DA ratio) was calculated as follows: (DOPAC + HVA)/DA.
F2-160: p < 0.01,
F2-165: p< 0.001 versus basal group.
Discussion
The dopamine D3 receptor is highly distributed in the nucleus accumbens, the terminal site of the mesolimbic dopaminergic system (Sokoloff et al., 1990). The limbic system-selective expression of the dopamine D3receptor has led to particular interest in this receptor as a potential mediator of some of the psychoeffective functions of dopamine neurotransmission (Levant, 1997). Although pharmacological studies using dopamine D3 receptor-preferring agonists such as 7-OH-DPAT can generally support this view (Caine and Koob, 1993), the dubious selectivity of these agonists severely limits conclusions about the physiological functions of the dopamine D3 receptor.
Κnock-out mice with dopamine D3 receptor gene deletions have been successfully developed by homologous recombination (Accili et al., 1996; Xu et al., 1997). The availability of transgenic dopamine D3 receptor knock-out mice allows us to determine the physiological function mediated via the dopamine D3 receptor and the interaction between μ-opioidergic system and dopamine D3 receptors. Using dopamine D3 receptor knock-out mice, we confirmed that a deletion of the dopamine D3receptor had no effect on either dopamine D1 or D2 receptors, suggesting the utility of these mice for studying the distinct role of dopamine D3 receptors (Narita et al., 2002a,b; the present study). In the present study, we found that the mutation of the dopamine D3 receptor resulted in no detectable change in the μ-opioid receptor in the brain. Furthermore, the elimination of dopamine D3 receptor expression in mutant mice failed to affect morphine-stimulated [35S]GTPγS binding, indicating no significant change in the ability of the μ-opioid receptor-induced G-protein activation in this phenotype. These results suggest that deletion of the dopamine D3 receptor gene could not directly affect the μ-opioidergic function in the brain.
Under the present condition, we investigated the morphine-induced rewarding effect in mice lacking dopamine D3 receptor. A significant place preference for drug-associated place was observed in the mutant mice at 0.56 mg/kg morphine, whereas no preferences were seen in the wild-type mice at even 1–3 mg/kg. This enhanced effect was completely reversed by intracerebroventricular administration of a selective μ-opioid receptor antagonist, CTOP, indicating that the μ-opioidergic system is involved in the increased sensitivity to morphine in dopamine D3 receptor knock-out mice. Furthermore, the dose relationship for morphine-induced place preference in dopamine D3 receptor knock-out mice was significantly shifted to the left as compared with that in wild-type mice. A great deal of evidence suggests that tonic dopamine D1receptor activation is required for the rewarding effect of μ-opioid receptor agonists (Shippenberg and Herz, 1987, 1988). A recent genetic approach provides direct evidence for the implication of dopamine D2 receptor in the morphine-induced rewarding effect (Maldonado et al., 1997). Taken together, the data suggest the possibility that the dopamine D3 receptor may play a potential role in the negative modulation of the dopamine D1/D2 receptor-dependent rewarding effects induced by μ-opioid receptor agonists.
Various studies have demonstrated that the dopamine D3 receptor is implicated in motor behavior. A reduction in spontaneous locomotion is produced by the dopamine D3 receptor-preferring agonist 7-OH-DPAT (Daly and Waddington, 1993; Khroyan et al., 1995; De Boer et al., 1997). In the present study, we failed to find a significant change in saline-induced locomotion in mice lacking dopamine D3 receptor after long-term habituation. Under these conditions, morphine-induced hyperlocomotion was dramatically enhanced in mutant mice. It has been widely recognized that, as with the rewarding effect, the mesolimbic dopaminergic system plays a critical role in morphine-induced hyperlocomotion (Narita et al., 2001). Taken together, our results suggest that the dopamine D3 receptor may be essential for negative feedback on the mesolimbic dopaminergic pathway activated by μ-opioid receptor agonists.
We demonstrated previously that subcutaneous treatment with morphine at 5 mg/kg produced a significant increase in the levels of DOPAC and HVA without any changes in the levels of dopamine in the limbic forebrain area in mice (Narita et al., 1993). In the present study, the basal levels of dopamine, DOPAC and HVA in the limbic forebrain area were unchanged in dopamine D3 knock-out mice as compared with that in wild-type mice. We found that morphine (5 mg/kg, s.c.) produced a significant increase in levels of DOPAC and HVA and a dramatic enhancement of dopamine turnover in the limbic forebrain area in both wild-type and D3 knock-out mice. However, these levels did not differ significantly between the two genotypes. It has been proposed recently, using dopamine D2receptor knock-out mice, that dopamine release is suppressed by the stimulation of dopamine D2 receptor-like autoreceptors (Baik et al., 1995; L'hirondel et al., 1998). Like dopamine D2 receptors, the dopamine D3 receptor is considered to be a family of autoreceptors (Sokoloff et al., 1990; Gainetdinov et al., 1996). In contrast, a recent anatomical finding that few dopamine D3 receptors are detected in the VTA gives us the idea that dopamine D3 receptors could be distributed postsynaptically in the nucleus accumbens (Bouthenet et al., 1991; Diaz et al., 2000). Considerable evidence suggests that the postsynaptic D3 receptors are likely to negatively modulate the overexcitation of postsynaptic dopamine receptor-regulated actions (Waters et al., 1993; Xu et al., 1997;Koeltzow et al., 1998). Thus, a loss of postsynaptic dopamine D3 receptors may cause the sustained activation of postsynaptic dopamine receptor-mediated signaling. We propose here that the D3 receptor may be critical for postsynaptically negative modulation of the activated mesolimbic dopaminergic system by the stimulation of μ-opioid receptors located in the VTA of mice.
In conclusion, the present data provide direct evidence that a deletion of central dopamine D3 receptor enhances the rewarding effect and hyperlocomotion induced by morphine without directly affecting the μ-opioid receptor itself in the mouse brain.
Footnotes
This work was supported in part by grants from the Ministry of Health, Labour and Welfare, and the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Correspondence should be addressed to Dr. Tsutomu Suzuki, Department of Toxicology, Hoshi University School of Pharmacy and Pharmaceutical Sciences, 2-4-41 Ebara, Shinagawa-ku, Tokyo 142-8501, Japan. E-mail:suzuki@hoshi.ac.jp.
References
- 1.Accili D, Fishburn CS, Drago J, Steiner H, Lachowicz JE, Park BH, Gauda EB, Lee EJ, Cool MH, Sibley DR, Gerfen CR, Westphal H, Fuchs S. A targeted mutation of the D3 dopamine receptor gene is associated with hyperactivity in mice. Proc Natl Acad Sci USA. 1996;93:1945–1949. doi: 10.1073/pnas.93.5.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baik JH, Picetti R, Saiardi A, Thiriet G, Dierich A, Depaulis A, Le Meur M, Borrelli E. Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature. 1995;377:424–428. doi: 10.1038/377424a0. [DOI] [PubMed] [Google Scholar]
- 3.Bals-Kubik R, Ableitner A, Herz A, Shippenberg TS. Neuroanatomical sites mediating the motivational effects of opioids as mapped by the conditioned place preference paradigm in rats. J Pharmacol Exp Ther. 1993;264:489–495. [PubMed] [Google Scholar]
- 4.Bouthenet ML, Souil E, Martres MP, Sokoloff P, Giros B, Schwartz JC. Localization of dopamine D3 receptor mRNA in the rat brain using in situ hybridization histochemistry: comparison with dopamine D2 receptor mRNA. Brain Res. 1991;564:203–219. doi: 10.1016/0006-8993(91)91456-b. [DOI] [PubMed] [Google Scholar]
- 5.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 6.Caine SB, Koob GF. Modulation of cocaine self-administration in the rat through D-3 dopamine receptors. Science. 1993;260:1814–1816. doi: 10.1126/science.8099761. [DOI] [PubMed] [Google Scholar]
- 7.Civelli O, Bunzow JR, Grandy DK. Molecular diversity of the dopamine receptors. Annu Rev Pharmacol Toxicol. 1993;33:81–307. doi: 10.1146/annurev.pa.33.040193.001433. [DOI] [PubMed] [Google Scholar]
- 8.Daly SA, Waddington JL. Behavioural effects of the putative D-3 dopamine receptor agonist 7-OH-DPAT in relation to other “D-2-like” agonists. Neuropharmacology. 1993;32:509–510. doi: 10.1016/0028-3908(93)90177-5. [DOI] [PubMed] [Google Scholar]
- 9.De Boer P, Enrico P, Wright J, Wise LD, Timmerman W, Moor E, Dijkstra D, Wikstrom HV, Westerink BH. Characterization of the effect of dopamine D3 receptor stimulation on locomotion and striatal dopamine levels. Brain Res. 1997;758:83–91. doi: 10.1016/s0006-8993(96)01438-2. [DOI] [PubMed] [Google Scholar]
- 10.Devoto P, Collu M, Muntoni AL, Pistis M, Serra G, Gessa GL, Diana M. Biochemical and electrophysiological effects of 7-OH-DPAT on the mesolimbic dopaminergic system. Synapse. 1995;20:153–155. doi: 10.1002/syn.890200209. [DOI] [PubMed] [Google Scholar]
- 11.Diaz J, Pilon C, Le Foll B, Gros C, Triller A, Schwartz JC, Sokoloff P. Dopamine D3 receptors expressed by all mesencephalic dopamine neurons. J Neurosci. 2000;20:8677–8684. doi: 10.1523/JNEUROSCI.20-23-08677.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Funada M, Suzuki T, Misawa M. The role of dopamine D1-receptors in morphine-induced hyperlocomotion in mice. Neurosci Lett. 1994;169:1–4. doi: 10.1016/0304-3940(94)90342-5. [DOI] [PubMed] [Google Scholar]
- 13.Gainetdinov RR, Sotnikova TD, Grekhova TV, Rayevsky KS. In vivo evidence for preferential role of dopamine D3 receptor in the presynaptic regulation of release but not synthesis. Eur J Pharmacol. 1996;308:261–269. doi: 10.1016/0014-2999(96)00300-7. [DOI] [PubMed] [Google Scholar]
- 14.Gingrich JA, Caron MG. Recent advances in the molecular biology of dopamine receptors. Annu Rev Neurosci. 1993;16:299–321. doi: 10.1146/annurev.ne.16.030193.001503. [DOI] [PubMed] [Google Scholar]
- 15.Khroyan TV, Baker DA, Neisewander JL. Dose-dependent effects of the D3-preferring agonist 7-OH-DPAT on motor behaviors and place conditioning. Psychopharmacology. 1995;122:351–357. doi: 10.1007/BF02246265. [DOI] [PubMed] [Google Scholar]
- 16.Koeltzow TE, Xu M, Cooper DC, Hu XT, Tonegawa S, Wolf ME, White FJ. Alterations in dopamine release but not dopamine autoreceptor function in dopamine D3 receptor mutant mice. J Neurosci. 1998;18:2231–2238. doi: 10.1523/JNEUROSCI.18-06-02231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koob GF. Drugs of abuse: anatomy, pharmacology and function of reward pathways. Trends Pharmacol Sci. 1992;13:177–184. doi: 10.1016/0165-6147(92)90060-j. [DOI] [PubMed] [Google Scholar]
- 18.Landwehrmeyer B, Mengod G, Palacios JM. Differential visualization of dopamine D2 and D3 receptor sites in rat brain. A comparative study using in situ hybridization histochemistry and ligand binding autoradiography. Eur J Neurosci. 1993;5:145–153. doi: 10.1111/j.1460-9568.1993.tb00480.x. [DOI] [PubMed] [Google Scholar]
- 19.Levant B. The D3 dopamine receptor: neurobiology and potential clinical relevance. Pharmacol Rev. 1997;49:231–252. [PubMed] [Google Scholar]
- 20.L'hirondel M, Cheramy A, Godeheu G, Artaud F, Saiardi A, Borrelli E, Glowinski J. Lack of autoreceptor-mediated inhibitory control of dopamine release in striatal synaptosomes of D2 receptor-deficient mice. Brain Res. 1998;792:253–262. doi: 10.1016/s0006-8993(98)00146-2. [DOI] [PubMed] [Google Scholar]
- 21.Maldonado R, Dauge V, Feger J, Roques BP. Chronic blockade of D2 but not D1 dopamine receptors facilitates behavioural responses to endogenous enkephalins, protected by kelatorphan, administered in the accumbens in rats. Neuropharmacology. 1990;29:215–223. doi: 10.1016/0028-3908(90)90004-b. [DOI] [PubMed] [Google Scholar]
- 22.Maldonado R, Saiardi A, Valverde O, Samad TA, Roques BP, Borrelli E. Absence of opiate rewarding effects in mice lacking dopamine D2 receptors. Nature. 1997;388:586–589. doi: 10.1038/41567. [DOI] [PubMed] [Google Scholar]
- 23.Meller E, Bohmaker K, Goldstein M, Basham DA. Evidence that striatal synthesis-inhibiting autoreceptors are dopamine D3 receptors. Eur J Pharmacol. 1993;249:R5–R6. doi: 10.1016/0014-2999(93)90674-7. [DOI] [PubMed] [Google Scholar]
- 24.Narita M, Suzuki T, Funada M, Misawa M, Nagase H. Blockade of the morphine-induced increase in turnover of dopamine on the mesolimbic dopaminergic system by κ-opioid receptor activation in mice. Life Sci. 1993;52:397–404. doi: 10.1016/0024-3205(93)90153-t. [DOI] [PubMed] [Google Scholar]
- 25.Narita M, Funada M, Suzuki T. Regulations of opioid dependence by opioid receptor types. Pharmacol Ther. 2001;89:1–15. doi: 10.1016/s0163-7258(00)00099-1. [DOI] [PubMed] [Google Scholar]
- 26.Narita M, Mizuo K, Shibasaki M, Narita M, Suzuki T. Up-regulation of the G(q/11α) protein and protein kinase C during the development of sensitization to morphine-induced hyperlocomotion. Neuroscience. 2002a;111:127–132. doi: 10.1016/s0306-4522(01)00515-2. [DOI] [PubMed] [Google Scholar]
- 27.Narita M, Soma M, Tamaki H, Narita M, Suzuki T. Intensification of the development of ethanol dependence in mice lacking dopamine D3 receptor. Neurosci Lett. 2002b;324:129–132. doi: 10.1016/s0304-3940(02)00235-5. [DOI] [PubMed] [Google Scholar]
- 28.Phillips AG, LePiane FG, Fibiger HG. Dopaminergic mediation of reward produced by direct injection of enkephalin into the ventral tegmental area of rat. Life Sci. 1983;33:2505–2511. doi: 10.1016/0024-3205(83)90159-5. [DOI] [PubMed] [Google Scholar]
- 29.Richtand NM, Kelsoe JR, Segal DS, Kuczenski R. Regional quantification of D1, D2, and D3 dopamine receptor mRNA in rat brain using a ribonuclease protection assay. Mol Brain Res. 1995;33:97–103. doi: 10.1016/0169-328x(95)00112-6. [DOI] [PubMed] [Google Scholar]
- 30.Rubinstein M, Phillips TJ, Bunzow JR, Falzone TL, Dziewczapolski G, Zhang G, Fang Y, Larson JL, McDougall JA, Chester JA, Saez C, Pugsley TA, Gershanik O, Low MJ, Grandy DK. Mice lacking dopamine D4 receptors are supersensitive to ethanol, cocaine, and methamphetamine. Cell. 1997;90:991–1001. doi: 10.1016/s0092-8674(00)80365-7. [DOI] [PubMed] [Google Scholar]
- 31.Shippenberg TS, Herz A. Place preference conditioning reveals the involvement of D1-dopamine receptors in the motivational properties of mu- and kappa-opioid agonists. Brain Res. 1987;436:169–172. doi: 10.1016/0006-8993(87)91571-x. [DOI] [PubMed] [Google Scholar]
- 32.Shippenberg TS, Herz A. Motivational effects of opioids: influence of D-1 versus D-2 receptor antagonists. Eur J Pharmacol. 1988;151:233–242. doi: 10.1016/0014-2999(88)90803-5. [DOI] [PubMed] [Google Scholar]
- 33.Shippenberg TS, Bals-Kubik R, Herz A. Examination of the neurochemical substrates mediating the motivational effects of opioid: role of the mesolimbic dopamine system and D-1 vs. D-2 dopamine receptors. J Pharmacol Exp Ther. 1993;265:53–59. [PubMed] [Google Scholar]
- 34.Sokoloff P, Giros B, Martres M-P, Bouthnet M-L, Schwartz J-C. Molecular cloning and characterization of a novel dopamine receptor (D3) as a target for neuroleptics. Nature. 1990;347:146–151. doi: 10.1038/347146a0. [DOI] [PubMed] [Google Scholar]
- 35.Stinus L, Winnock M, Kelley AE. Chronic neuroleptic treatment and mesolimbic dopamine denervation induce behavioural supersensitivity to opiates. Psychopharmacology. 1985;85:323–328. doi: 10.1007/BF00428196. [DOI] [PubMed] [Google Scholar]
- 36.Stinus L, Nadaud D, Jauregui J, Kelley AE. Chronic treatment with five different neuroleptics elicits behavioral supersensitivity to opiate infusion into the nucleus accumbens. Biol Psychiatry. 1986;21:34–48. doi: 10.1016/0006-3223(86)90006-5. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki T, Masukawa Y, Misawa M. Drug interactions in the reinforcing effects of over-the-counter cough syrups. Psychopharmacology. 1990;102:438–442. doi: 10.1007/BF02247122. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki T, Maeda J, Funada M, Misawa M. The D3-receptor agonist (+/−)-7-hydroxy-N,N-di-n-propyl-2-aminotetralin (7-OH-DPAT) attenuates morphine-induced hyperlocomotion in mice. Neurosci Lett. 1995;187:45–48. doi: 10.1016/0304-3940(95)11334-s. [DOI] [PubMed] [Google Scholar]
- 39.Waters N, Svensson K, Haadsma-Svensson SR, Smith MW, Carlsson A. The dopamine D3-receptor: a postsynaptic receptor inhibitory on rat locomotor activity. J Neural Transm Gen Sect. 1993;94:11–19. doi: 10.1007/BF01244979. [DOI] [PubMed] [Google Scholar]
- 40.Wise RA, Rompre PP. Brain dopamine and reward. Annu Rev Psychol. 1989;40:191–225. doi: 10.1146/annurev.ps.40.020189.001203. [DOI] [PubMed] [Google Scholar]
- 41.Xu M, Koeltzow TE, Santiago GT, Moratalla R, Cooper DC, Hu XT, White NM, Graybiel AM, White FJ, Tonegawa S. Dopamine D3 receptor mutant mice exhibit increased behavioral sensitivity to concurrent stimulation of D1 and D2 receptors. Neuron. 1997;19:837–848. doi: 10.1016/s0896-6273(00)80965-4. [DOI] [PubMed] [Google Scholar]