

Abstract
Fibroblast growth factor receptor (FGFR) signaling is essential for nervous system development. We have shown that, in the normal postnatal brain, the spatial and temporal expression pattern of FGFR3 parallels the appearance of differentiated oligodendrocytes and that in culture FGFR3 is expressed maximally at the critical stage in the lineage at which oligodendrocyte late progenitors (Pro-OLs) enter terminal differentiation. Therefore, FGFR3 expression is positioned ideally to have an impact on oligodendrocyte differentiation. In support of this we show that, during the onset and active phase of myelination in FGFR3-deficient mice, there are reduced numbers of differentiated oligodendrocytes in the forebrain, cerebellum, hindbrain, and spinal cord. Furthermore, myelination is delayed in parallel. Delay of oligodendrocyte differentiation also is observed in primary cell culture from this mutant. On the other hand, no differences are observed in the survival or proliferation of oligodendrocyte progenitors. This suggests that the decrease in the number of differentiated oligodendrocytes is attributable to a delay in the timing of their differentiation process. Astrocytes also express FGFR3, and in mice lacking FGFR3 there is an enhancement of the astrocytic marker glial fibrillary acidic protein expression in a region-specific manner. Thus our findings suggest that there are cell type- and region-specific functions for FGFR3 signaling and in particular emphasize a prominent role for FGFR3 as part of a system regulating the onset of oligodendrocyte terminal differentiation.
Keywords: oligodendrocyte, myelin, FGF, astrocyte, cerebellar neuron, FGF receptor
Fibroblast growth factors (FGFs), a family of 23 known members, play central roles in nervous system development (Vaccarino et al., 1999; Ford-Perriss et al., 2001; Ornitz and Itoh, 2001). FGFs signal through four high-affinity tyrosine kinase receptors (FGFR1–FGFR4) (Johnson and Williams, 1993). FGFRs are expressed differentially during development and in the adult brain (Orr-Urtreger et al., 1991; Peters et al., 1992, 1993; Asai et al., 1993; Yazaki et al., 1994; Miyake et al., 1996; Ford-Perriss et al., 2001). FGFs modulate a variety of biological activities including proliferation, migration, and survival of neurons and glial cells (Vaccarino et al., 1999; Ford-Perriss et al., 2001). Furthermore, FGFs influence specification of neuronal and glial cell fate, regional patterning of neocortex and midbrain–hindbrain boundaries, cerebellar development, and cerebral cortex size (Crossley et al., 1996; Qian et al., 1997; Ye et al., 1998; Fukuchi-Shimogori and Grove, 2001). For example, mice lacking FGF-2 exhibit decreased numbers of neurons and glia, whereas the injection of FGF-2 into the embryonic subventricular zone produces the opposite effect (Vaccarino et al., 1999). In mice lacking both FGF-17 and one allele of FGF-18 the anterior lobe of the cerebellar vermis does not develop (Xu et al., 2000), whereas in mice lacking FGF-14 recent studies have identified a function for this molecule in neuronal signaling in the adult brain (Wang et al., 2002). However, the role of FGFs in early postnatal development is less well understood, especially with respect to oligodendrocyte and astrocyte differentiation.
Oligodendrocyte (OL) progenitors originate at specific locations in the ventral ventricular zones. As they migrate to their final destinations, they mature through a series of stages that include proliferative and migratory early progenitors (EPs) and proliferative and nonmigratory late progenitor or pro-oligodendrocytes (Pro-OLs). Ultimately, the Pro-OLs become postmitotic and enter terminal differentiation, leading to myelination (Warrington and Pfeiffer, 1992; Pfeiffer et al., 1993;Miller, 1996; Woodruff et al., 2001). OL development is regulated by a number of growth factors, including FGF-2 (McMorris and McKinnon, 1996). FGF-2 affects multiple responses including proliferation and migration of OL progenitors and the conversion of mature OLs to a novel phenotype (for review, see Oh and Yong, 1996; Bansal and Pfeiffer, 1997; Bansal, 2002). During OL maturation FGF receptor transcripts are expressed in a developmentally regulated manner that could account at least in part for the variety of responses of OL lineage cells to FGF-2. Specifically, although FGFR1 is expressed at all stages, FGFR2 appears only in differentiated OLs, and FGFR3 expression peaks in Pro-OLs and then is downregulated as cells enter terminal differentiation (Bansal et al., 1996).
On the basis of the temporal expression pattern of FGFR3, we hypothesize that FGFR3 transduces signals important for the regulation of the critical interface between proliferation and differentiation. We report here that, in mice lacking FGFR3, the appearance of terminally differentiated OLs is retarded initially, whereas the expression of the astrocytic marker glial fibrillary acidic protein is enhanced. Because neither survival nor proliferation of OL progenitors is altered, we conclude that FGFR3 signaling is involved in the regulation of the onset of terminal differentiation of Pro-OLs and in the negative regulation of astrocytic differentiation and/or function.
Materials and Methods
FGFR3 null mice
FGFR3 null mice exhibit a characteristic phenotype including crooked tail, curvature of vertebrae, and abnormal long bone growth (Colvin et al., 1996; Deng et al., 1996). FGFR3 null and wild-type mice were obtained from heterozygous crosses of breeders received from Dr. David Ornitz (Washington University, St. Louis, MO). Genotypes were determined by PCR of tail DNA by using three sets of primers: (1) 5′-GGGCTCCTTATTGGACTCGC-3′, (2) 5′-AGGTATAGTTGCCACCATCGGAGGG-3′, and (3) 5′-TGCTAAAGCGCATGCTCCAGACTGC-3′. Products of 322 bp (wild-type gene) and 221 bp (homozygote gene) were amplified by using primers 1 and 2 and 1 and 3, respectively (35 cycle PCR reactions: 2 min at 94°C, 30 sec at 94°C, 30 sec at 55°C, and 10 sec at 72°C). Initially, three groups of mice were studied: wild types (+/+), heterozygotes (+/−), and FGFR3 null mice (−/−). However, no differences were observed between +/+ and +/−, in agreement with previous studies (Colvin et al., 1996).
In situ hybridization
Mice older than postnatal day 7 (P7) were perfused with 4% paraformaldehyde (PFA), and the brains were removed, postfixed in 4% PFA at 4°C overnight, and cryoprotected by consecutive incubations in 10% sucrose and 30% sucrose overnight at 4°C (mice at P2 and P7 were not perfused but followed the same postfixation procedure). Brains were immersed in cryo-embedding media and were quick frozen at –80°C. Using a cryostat, we cut whole brain sections (with a small portion of the cervical spinal cord attached) parasagittally (15 μm thick) and collected them on RNase-free Superfrost glass slides (Fisher Scientific, Pittsburgh, PA), stored them at –20°C, and used them for in situ hybridization and immunohistochemistry.
A riboprobe specific for proteolipid protein (PLP) mRNA was designed to cover the entire coding region (a gift from B. Fuss and W. B. Macklin, Cleveland, OH). A platelet-derived growth factor receptor α (PDGF-Rα) mRNA probe was transcribed from a 1637 bp EcoRI cDNA fragment encoding most of the extracellular domain of mouse PDGF-Rα, and a FGFR3 probe was transcribed from a 900 bpEcoRI cDNA fragment encoding the extracellular domain and a part of intracellular tyrosine kinase domain (a gift from Bill Richardson and Nigel Pringle, London, UK). Hybridization for PLP mRNA was performed as described previously (Bansal et al., 1999). Briefly, sections were dried for 2 hr at room temperature and fixed with 3% PFA for 30 min. The sections were treated with 0.1 mHCl (5 min), followed by acetylation with 0.1 mtriethanolamine, pH 8, in acetic anhydride (10 min). After the sections were washed in 2× SSC and air-dried, hybridization was performed overnight at 50°C by using digoxigenin-labeled sense and antisense cRNA probes in hybridization solution [containing 50% formamide and (in μm) 350 NaCl, 10 dithiothreitol, 20 Tris-Cl, pH 7.5, 1 EDTA plus 1× Denhardt's, 500 μg/ml tRNA, and 100 μg/ml poly(A) RNA]. Then the sections were washed in 2× SSC three times and digested in RNase solution (20 μg/ml RNase and 1 U RNase T1 at 37°C for 30 min), followed by washing in 0.2× SSC at 50°C (5 min) and at room temperature (5 min). After equilibration in 100 mm Tris-HCl plus 150 mmNaCl (10 min) and blocking for nonspecific binding in 1% blocking buffer (Boehringer Mannheim, Indianapolis, IN) and 0.5% bovine serum albumin (BSA; 1 hr), the bound cRNA was detected via an alkaline phosphatase-coupled anti-digoxigenin antibody (1:1000 for 2 hr; Boehringer Mannheim). After a washing in equilibrium buffer containing (in mm) 100 Tris-HCl, pH 9.5, 100 NaCl, and 50 MgCl2, color development in the presence of 4-nitroblue tetrazolium chloride, 5-bromo-4-chloro-3-indolylphosphate, and levamisole was performed in the dark at room temperature. The sections were washed in PBS and incubated in Hoechst blue dye 33342 (1 μg/ml; Sigma, St. Louis, MO) to counterstain the nuclei, were air-dried, and were mounted with 90% glycerol.
In situ hybridization for PDGFR-α or FGFR3 mRNAs was performed with a slight modification. Briefly, air-dried and PFA-fixed sections were hybridized directly with these probes overnight at 65°C. Then the slides were rinsed and incubated in preheated wash buffer (1× SSC, 50% formamide, 0.1% Tween 20) at 65°C (15 min), washed twice with wash buffer at room temperature (30 min) and twice with MABT solution (30 min) [100 mm maleic acid, pH 7.5, 150 mm NaCl, and 0.1% (v/v) Tween 20], blocked in MABT containing 2% blocking reagent (Boehringer Mannheim) and 1% BSA (1 hr), incubated in alkaline phosphatase-coupled anti-digoxigenin antibody, and developed as described above. To increase sensitivity, we included 50% (w/v) polyvinyl alcohol in the final color reaction and developed it overnight at 37°C.
Immunohistochemistry
Tissue preparation and sectioning were performed as described above. For myelin basic protein (MBP) and glial fibrillary acidic protein (GFAP) immunolabeling, whole brain parasagittal cryosections were permeabilized in 100% ethanol (10 min), washed in PBS, and blocked for 1 hr in a buffer consisting of 10% normal goat serum (NGS), 5% BSA, 0.05% NaN3, and 0.1% gelatin in PBS. For NG2 immunolabeling the sections were blocked in 3% NGS/0.1% Triton X-100 in PBS. Sections were incubated overnight at 4°C in polyclonal rabbit anti-MBP (1:3000; Dr. S. E. Pfeiffer, Farmington, CT), monoclonal anti-rat GFAP (1:50; Dr. Virginia Lees, University of Pennsylvania, Philadelphia, PA), or polyclonal rabbit anti-NG2 (1:100; Chemicon, Temecula, CA). After being washed in PBS, the sections were incubated for 1 hr with either goat anti-rabbit IgG conjugated to Oregon green (1:100; Molecular Probes, Eugene, OR) or goat anti-rat IgG conjugated to fluorescein (1:100; Chemicon), washed in PBS, mounted in DABCO [1,4-diazobicyclo-(2,2,2)-octane in glycerol], and analyzed with an epifluorescent microscope (Axiovert microscope, Carl Zeiss, Thornwood, NY).
Detection of cell proliferation
So that we could identify cells that were in the S phase of the cell cycle, mice received an intraperitoneal injection of bromodeoxyuridine (BrdU; 100 μg/gm body weight) for incorporation into newly synthesized DNA and were killed 3 hr later. After perfusion, postfixation, and sectioning as described above, the sections were rinsed in PBS for 10 min, fixed with acid alcohol (95% ethanol/5% acetic acid, 2 min, –20°C), washed in PBS, denatured with 2N HCl (10 min), neutralized with 0.1 m sodium borate buffer, pH 8.5 (10 min), blocked with 3% NGS/PBS (1 hr), incubated in mouse monoclonal anti-BrdU antibody (overnight at 4°C; 1:50; Becton Dickinson, Lincoln Park, NJ), and then washed in PBS, followed by incubation in goat anti-mouse IgG conjugated to Cy3 (1:500;Jackson ImmunoResearch, West Grove, PA) and Hoechst Blue 33342 (1:1000; 1 hr). Slides were washed and mounted in DABCO. In double-labeling experiments the sections were immunolabeled first with anti-NG2 as described above, followed by anti-BrdU labeling.
Detection of apoptotic cells
Apoptotic cells were detected by using terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) assay (Apoptag kit; Intergen, Purchase, NY) according to the manufacturer's protocol. In brief, brain cryosections were incubated in 4% PFA for 30 min, treated with 0.3% H2O2 to quench endogenous peroxidase, washed in equilibrium buffer, and incubated in reaction buffer containing digoxygenin-dNTP and terminal deoxynucleotidyl transferase (30 min, 37°C). The sections were washed and incubated with anti-digoxygenin–peroxidase conjugate for 30 min. The TUNEL+ cells were identified by reaction with 3,3-diaminobenzidine (DAB; Research Genetics, Huntsville, AL) and analyzed by epifluorescence microscopy.
Immunoblotting
Forebrains, cerebella, hindbrains, and spinal cords were harvested and stored at −80°C before analysis. Mixed primary cultures were harvested in RIPA buffer (10 mm Tris-HCl, 150 mm NaCl, 0.1% SDS, 1% deoxycholate, and 1% NP-40, pH 7.4) with protease inhibitors (2 mm PMSF, 2 μg/ml leupeptin, 2 μg/ml aprotinin) on ice and were cup sonicated (30 sec; 4°C). Tissue samples were homogenized similarly. Then the homogenates were incubated (30 min, on ice) and centrifuged (15,000 ×g for 10 min at 4°C). The protein concentration was assayed with the DC Protein Assay kit (Bio-Rad, Hercules, CA). Aliquots of total protein were electrophoresed on 12% SDS polyacrylamide gels and transferred onto polyvinylidene difluoride membranes. The membranes were blocked for 1 hr (Tris-buffered saline, 5% powder milk, 0.2% Tween 20) and incubated for 1 hr in monoclonal anti-myelin oligodendrocyte glycoprotein (MOG; 1:5000; Dr. C. Linington, Max Planck Institute, Munich, Germany), polyclonal anti-MBP (1:10,000), polyclonal anti-2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNP; 1:5000; Dr. S. Pfeiffer, Farmington, CT), polyclonal anti-FGFR3 (1:1000; Dr. D. Ornitz, Washington University, St. Louis, WA), or polyclonal anti-GFAP (1:5000; Dako, Carpinteria, CA). Then the membranes were incubated for 30 min in either anti-rabbit IgG (1:10,000; Santa Cruz Biotech, Santa Cruz, CA) or anti-mouse IgG (1:10,000; Transduction Laboratories, Lexington, KY), both conjugated to horseradish peroxidase. The membranes were developed with the ECL Plus kit (Amersham, Arlington Heights, IL). The NIH Image analysis program (Bethesda, MD) was used for quantification of the bands.
Electron microscopy
Animals were anesthetized and perfused with 2% PFA/2.5% glutaraldehyde in 100 mm sodium phosphate buffer, pH 7.4. The brains were removed and postfixed overnight in the same fixative. The medial parts of corpus callosum were dissected, treated with 1% osmium tetroxide, stained in block in 0.5% uranyl acetate, dehydrated in ethanol, cleared in propylene oxide, and embedded in Polybed (Polysciences, Warrington, PA). The sections were stained in uranyl acetate and lead citrate and then mounted on a 200 mesh grid; fields were sampled randomly, and axons within these fields were chosen randomly for analysis. Axon diameters were determined by tracing the parameters, using the NIH Image program; myelin thickness was measured similarly and quantified.
Cell culture
Mixed primary cultures. Mixed primary cultures of neonatal (P2) mice telencephala were prepared as described previously (Bansal et al., 1999). Briefly, dissociated cells were plated in 10% fetal calf serum in DMEM (FCS/DMEM) at a density of 2.5 × 105 cells/cm2into poly-l-lysine-coated (50 μg/ml; Sigma) 35 mm tissue culture plates for protein isolation. After 1 d the cultures were changed to defined medium mN2 [DMEM with 4.5 gm/ld-glucose, 50 μg/ml human transferrin, 5 μg/ml bovine pancreatic insulin, and (in nm) 15 3,3,5-triiodo-l-thyronine, 30 sodium selenium, 10d-biotin, 10 hydrocortisone, plus 0.11 mg/ml sodium pyruvate, penicillin/streptomycin (10 IU/ml and 100 μg/ml, respectively), and 0.1% BSA (all ingredients from Sigma) plus 1% FCS and 1% horse serum].
Purified populations. Purified populations of developmentally synchronized OL lineage cells were prepared at three stages: EPs, Pro-OLs, and terminally differentiated OLs; we characterized the purity and phenotype of each population extensively by immunolabeling the cells with a panel of antibodies (Pfeiffer et al., 1993; Bansal et al., 1996). Briefly, progenitors were obtained from mixed primary cultures from neonatal rat telencephalon by overnight shaking, followed by differential adhesion and complement lysis with anti-galactocerebroside to remove astrocytes, macrophages, and terminally differentiated OLs. Cells were plated in 5% FCS/DMEM in tissue culture plates coated with 50 μg/ml poly-d,l-ornithine (Sigma). After cell attachment for 2–3 hr the medium was changed to serum-free defined medium mN2 (see above). Cultures were grown for 7 d to produce terminally differentiated OLs or were expanded and arrested at either the EP stage by growth in PDGF-BB plus FGF-2 (10 ng/ml each; Upstate Biotechnology, Lake Placid, NY) for 2 d or at the Pro-OL stage by growth in FGF-2. For some experiments the progenitors also were prepared without growth factor treatment (similar results were obtained with cells grown in both conditions).
Astrocyte cultures. Astrocyte cultures prepared from monolayer cultures remaining after releasing OL progenitors from mixed primary cultures (see above; Bansal et al., 1996) were 99% positive for the astrocytic marker GFAP.
Comparative analyses of wild-type and mutant mice. These assays were done as described previously (Bansal et al., 1999). Briefly, three to nine mice from at least three separate litters were analyzed from both control and mutant groups at each time point. Whole brains (plus the most rostral portion of the cervical spinal cord) were cut parasagittally (15-μm-thick sections) in the sampling plane, which was located ∼300 μm from the midline. Wild-type and FGFR3 null sections were matched by comparing landmarks observed by counterstaining with Hoechst dye so that they were equidistant from the midline and in the hippocampal region. Cell counts were obtained from sections from the sampling plane through the entire brain region (such as cortex, corpus callosum, cerebellar white matter, or cerebellar cortex), and all of the cells in each area were counted in these sections, except in cases for which specific matched subregions are specified. Cells in the sections were counted systematically with a grid and 20× objective. Cell counts and observations were made by double-blind analysis by at least two independent investigators.
Results
Developmental expression of FGFR3 in normal postnatal brain
OL differentiation and myelination in the mouse CNS occur in a caudal to rostral manner. For example, at birth small numbers of differentiated OL are already present in the spinal cord but only subsequently appear sequentially in the hindbrain, cerebellum, corpus callosum, and finally in the cerebral cortex. This spatial and temporal pattern of development can be visualized by immunohistochemical andin situ hybridization analyses of parasagittal sections of the whole brain. Although expression of FGFR3 has been demonstrated in multiple cell types in different regions of the CNS, including astrocytes in the adult brain (Yazaki et al., 1994; Miyake et al., 1996), OL progenitors in the adult spinal cord (Messersmith et al., 2000), spinal motor neurons (Philippe et al., 1998), embryonic neuroepithelium (Peters et al., 1992, 1993), forebrain-derived cultured OL progenitors and astrocytes (Bansal et al., 1996), and, more recently, spinal cord astrocytes and their neuroepithelial precursors (W. Richardson, personal communications), its expression pattern as a function of postnatal brain development had not been addressed. Because OL development in the brain takes place postnatally and because FGFR3 is expressed by OL progenitors in vitro (Bansal et al., 1996), we considered the possibility that FGFR3 signaling may be important for OL development. We first analyzed the spatiotemporal pattern of expression of FGFR3 mRNA in vivo and related it to the appearance of OL progenitors and differentiated OLs at P2, P4, P7, P9, P13, and P31. Typical results are shown for P2 and P9 schematically and as representative sections taken from the forebrain and hindbrain (Fig. 1).
Fig. 1.
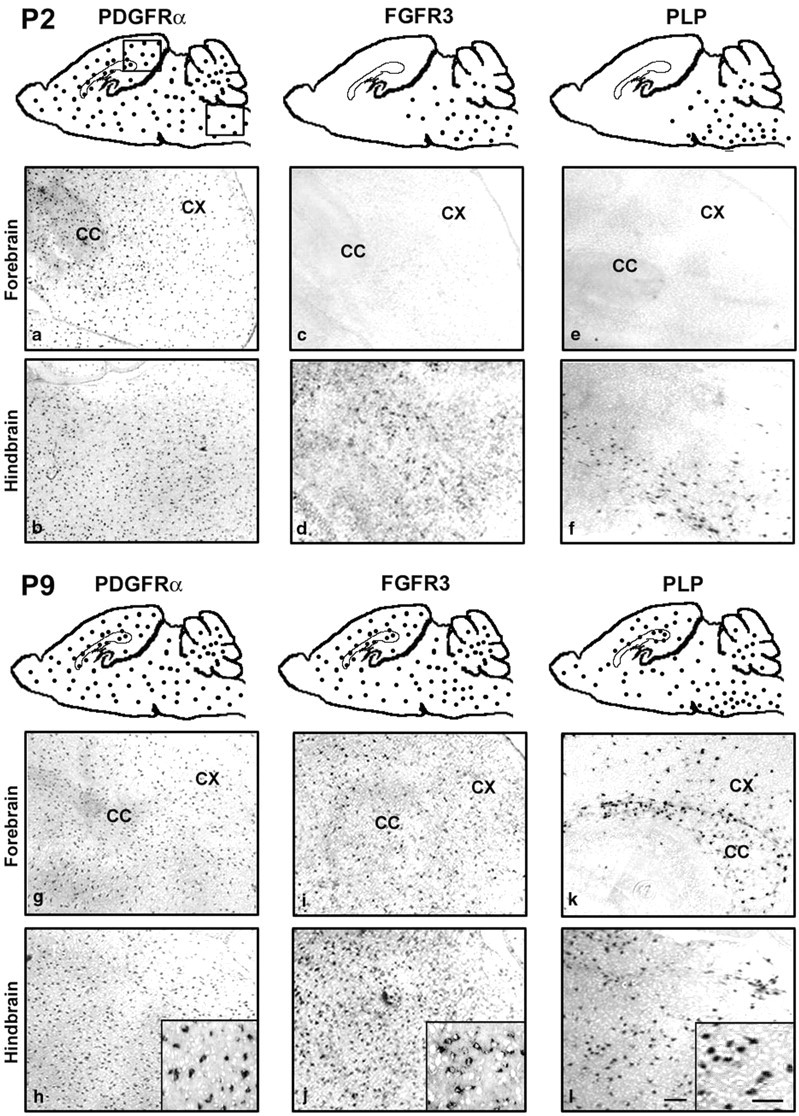
Spatiotemporal expression of FGFR3 mRNA in normal brain. Parasagittal sections of wild-type mouse brain were analyzed byin situ hybridization for PDGFRα, FGFR3, and PLP mRNA at P2 and P9. The regional distributions of positive cells are shown diagrammatically as dots and as representative sections taken from forebrain and hindbrain from regions marked by abox. Higher magnifications are shown asinsets. The temporal wave of FGFR3 mRNA expression moves from caudal to rostral brain in parallel to that for PLP but not PDGFRα. CC, Corpus callosum; CX, cortex. Scale bars: (in l) a–l, 100 μm; insets, 50 μm.
At P2, PDGFRα mRNA (a marker for OL early progenitors; Pringle et al., 1992) expression already was widespread over the entire brain, and the pattern remained the same at P9 (Fig.1P2a,P2b,P9g,P9h). In contrast, at P2, FGFR3 was expressed only in the spinal cord, pons, and medulla (hindbrain), with very little expression in the midbrain and none in the forebrain (Fig.1P2c,P2d). By P4 the FGFR3 mRNA expression had increased rostrally into the midbrain and cerebellum, and by P7 it appeared in the forebrain where it continued to be expressed until adulthood (Fig. 1P9i,P9j). The expression of PLP mRNA (a marker for differentiated OLs; Hudson et al., 1989) follows a caudal to rostral progression with a developmental age similar to FGFR3 (Fig.1P2e,P2f,P9k,P9l), although initially it was more restricted, appearing first in the corpus callosum by P7 before spreading progressively into the cerebral cortex with time. These results show that there is a wave of FGFR3 mRNA expression in the normal postnatal brain that spreads from the hindbrain to the forebrain in parallel to markers for terminally differentiated OLs rather than for OL early progenitors.
FGFR3 protein is expressed maximally by Pro-OLs and downregulated as the Pro-OLs differentiate into oligodendrocytes; FGFR3 protein also is expressed by astrocytes
Purified populations of OL early progenitors (EPs) from rat forebrain growing in culture express low levels of FGFR3 mRNA. This mRNA expression increases twofold as EPs mature to the Pro-OL stage and then is downregulated dramatically as the cells enter terminal differentiation (Bansal et al., 1996). Because protein expression does not necessarily follow mRNA expression resulting from post-transcriptional regulation, we examined FGFR3 protein expression from purified populations of EPs, Pro-OLs, OLs, and astrocytes by immunoblotting (Fig.2A). Consistent with the data for mRNA expression, FGFR3 protein was present in EPs but was increased in amount in Pro-OLs, and it was virtually absent in OLs. Astrocytes (AST) also expressed FGFR3 protein. Analyses of homogenates (2 μg) from transfected cell lines (3T3 and PC12) overexpressing FGFR1, FGFR2, or FGFR3 demonstrated the specificity of the antibody used for FGFR3. We conclude that FGFR3 protein is expressed maximally at the Pro-OL stage and that its expression is downregulated as the cells differentiate into OLs even more dramatically than at the mRNA level.
Fig. 2.

The expression of FGFR3 protein is regulated during the maturation of OL lineage cells. A, Purified cells from rat forebrain, at three stages of OL maturation and astrocytes, were analyzed by immunoblotting for FGFR3. Equal amounts of total protein (50 μg) were loaded in each lane. FGFR3 protein is expressed by early progenitors (EP), is increased substantially as EPs mature to Pro-OLs, and is downregulated dramatically to undetectable levels with the terminal differentiation of progenitors into OLs (OL). Astrocytes (AST) also express FGFR3 protein. Analyses of homogenates (2 μg) from transfected cell lines (3T3 and PC12) overexpressing FGFR1, FGFR2, or FGFR3 demonstrate the specificity of the antibody used for FGFR3. B, Immunoblots of homogenates (50 μg) from P9 wild types (+/+), heterozygotes (+/−), and FGFR3 null (−/−) hindbrains with anti-FGFR3 show the loss of FGFR3 protein to undetectable levels in FGFR3 null animals. Representative experiments of three are shown.
Myelination is delayed in FGFR3 null mice in vivo
Because the FGFR3 expression pattern in vivo andin vitro was consistent with a role in OL terminal differentiation, we postulated that FGFR3 signaling is important for myelination. We therefore analyzed mice lacking FGFR3 to evaluate the effect of eliminating FGFR3 function on myelin formation. Consistent with the null-genotype (Colvin et al., 1996), no FGFR3 protein was detected in the mutant mice (Fig. 2B); in contrast, the levels of FGFR1 and FGFR2 proteins were unchanged (data not shown). We examined MBP expression as a myelin marker by immunohistochemistry on parasagittal sections of whole brain from wild-type (+/+) and FGFR3 null (−/−) mice as a function of development (Fig.3A,B). Myelin formation in the corpus callosum of normal mice was initiated at approximately P7 when a few MBP+ fibers appeared (data not shown). At P9 (Fig. 3Aa) more apparent myelin developed, and myelination progressed rapidly by P13 (Fig. 3Ac). In contrast, in FGFR3 null mice there were no myelinated fibers at P7 (data not shown); at P9 there were fewer myelinated fibers, and they were arranged more loosely (Fig. 3Ab,Ad). These differences became progressively less marked with age, and by 1 month of age both normal and mutant mice appeared to be myelinated similarly (Fig.3Ae,Af). Reduced myelination also was observed in the cerebellum at these ages (P7 is shown) (Fig. 3B). This difference in myelination between the wild-type and mutant mice was confirmed by immunoblotting (Fig. 3C) for the myelin markers CNP and MBP in homogenates of forebrain at P13 (active myelination) and 5 months (myelination completed). Consistent with the immunohistochemical results, the expression of both proteins was reduced ∼1.5-fold in the FGFR3 null mice compared with wild type (P13) and became comparable in the adult (5 months). Although the myelin from adult FGFR3 mice appeared normal by immunohistochemistry, it did not ensure that the myelin formed was also normal ultrastructurally. Therefore, we further analyzed adult FGFR3 null mice by electron microscopy (Fig.4A–D). Cross sections from the corpus callosum of 2-month-old wild-type and FGFR3 null littermates showed no significant differences in the number of myelinated and unmyelinated axons (Fig. 4E) or in the thickness of myelin sheaths (Fig. 4F).
Fig. 3.
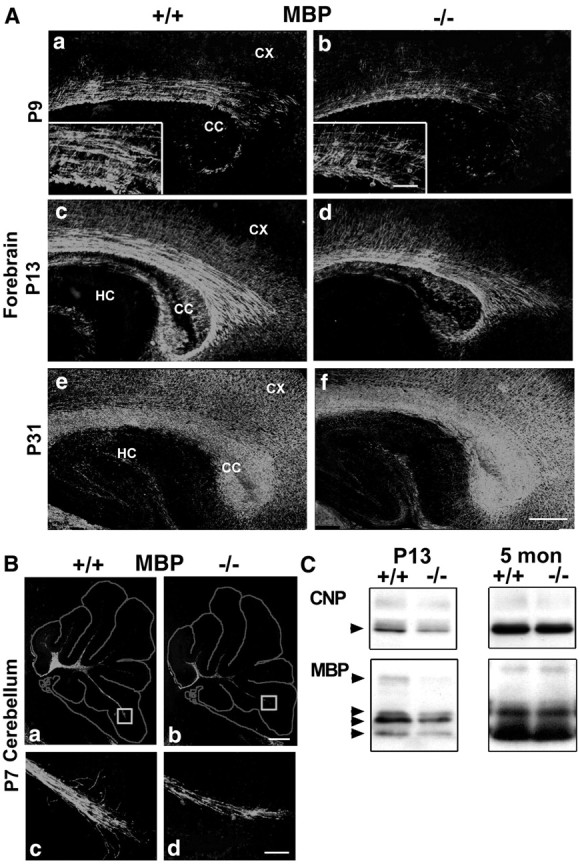
Myelin formation is delayed in FGFR3 null mice brain. Parallel parasagittal sections from forebrain (A) and cerebellum (B) of wild-type (+/+) and FGFR3 null (−/−) mice were analyzed by immunohistochemistry for MBP, a marker for myelinated fibers. In the forebrain of FGFR3 null mice, when compared with wild-type mice, fewer myelinated fibers developed initially, as seen at both P9 (Aa, Ab; higher magnifications are shown in insets) and P13 (Ac, Ad); this difference became comparable with wild type by P31 (Ae, Af). B, In cerebellar white matter there were also fewer myelinated fibers in the FGFR3 null mice when compared with the wild type, as shown in a representative section taken at P7 (Ba, Bb). Theboxed regions are shown at higher magnification (Bc, Bd). A, B, Two to four sections each from three to nine mice from each group and age were analyzed; similar results were obtained. CC, Corpus callosum;CX, cortex; HC, hippocampus. Scale bars:Af, 100 μm; insets (for A, Bc, Bd), 50 μm. C, Immunoblot analysis of forebrain homogenates from wild-type (+/+) and FGFR3 null (−/−) mice. Compared with wild-type mice, in FGFR3 null mice the levels of CNP and MBP isoforms were reduced at P13; however, they reached wild-type levels by adulthood.
Fig. 4.
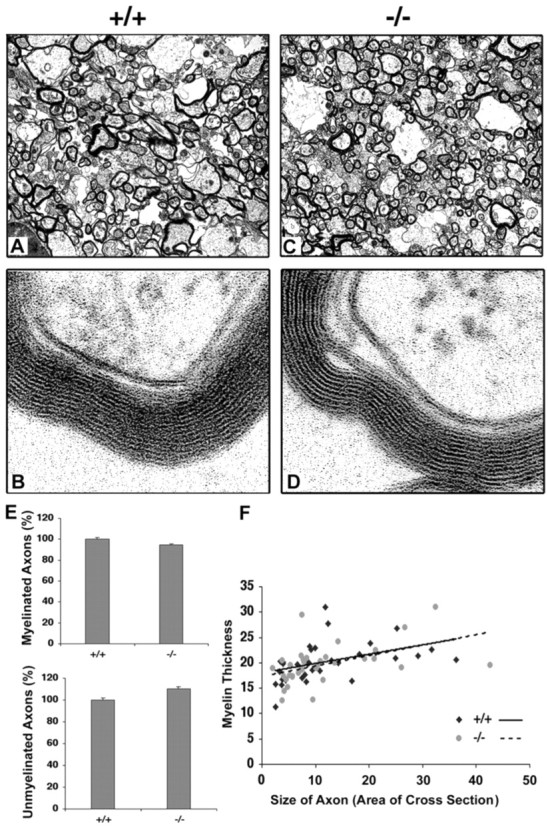
Ultrastructural analysis of myelinated axons in FGFR3 null mice. Cross sections of corpus callosum from 2-month-old littermate wild-type (+/+) and FGFR3 null (−/−) mice were analyzed by electron microscopy at low (A, C; 8230×) and high (B, D; 193,000×) magnification. E, Numbers of myelinated and unmyelinated axons were counted from the two groups (∼70 randomly chosen axons each). Wild-type number are set to 100%, and FGFR3 null levels are shown relative to that. Error bars indicate SEM. F, Thickness of myelin sheath and size of axon (arbitrary units) were measured from 30 randomly chosen myelinated axons from each group (NIH Image software). No apparent differences were observed in the ultrastructure of myelin between the two groups of mice.
These results show that myelination is delayed in FGFR3 null mice but appears structurally normal. We conclude that FGFR3 signaling is important for OLs to progress to the stage of myelination.
Reduced numbers of differentiated oligodendrocyte appear in FGFR3 null mice
We hypothesized that the reduced myelination at early postnatal ages is attributable to a reduction in the number of terminally differentiated OLs. To examine this possibility, we determined the time course of OL differentiation in parallel parasagittal sections from +/+ and −/− littermates, using in situ hybridization for PLP mRNA to identify and count the number of OLs (Fig.5; the estimation of OL cell numbers by immunohistochemistry with myelin protein markers is difficult because of the background of highly immunolabeled myelinated fibers). During the onset and active phase of myelination there were markedly fewer differentiated OLs in all regions of the brains from FGFR3 null mice, including spinal cord at P2 (Fig. 5A; the most dorsal cervical region is shown), and in the corpus callosum and cortex (Fig.5B) and cerebellum (Fig. 5C) at P9. Quantification of the data for the corpus callosum and cortex (Fig.5D) showed that there were twofold to threefold fewer OLs expressing PLP mRNA in the FGFR3 null than in wild types at P9 and P13. The difference between wild type and mutant continued to become progressively smaller with increasing age, and the numbers of PLP+ cells reached control levels with subsequent development (P31) (Fig. 5D), consistent with the normal level of myelination observed at this age (Fig.3Ae,Af). Similar differences in the number of OLs also were observed in spinal cords from wild-type and mutant mice (data not shown). These results suggest that, during the period when myelination is initiated and is progressing actively, fewer differentiated OLs appear in FGFR3 null brain compared with wild-type mice.
Fig. 5.
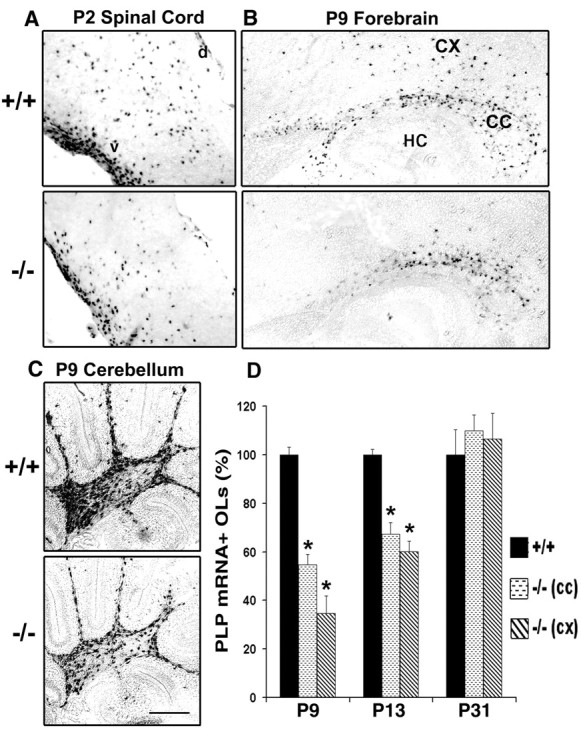
Oligodendrocyte differentiation is delayed in FGFR3 null mice in vivo. Parasagittal sections taken from parallel regions of wild-type (+/+) and FGFR3 null (−/−) mice from spinal cord at P2 (A), forebrain at P9, P13, and P31 (P9; B, D), and cerebellum at P9 (C) were analyzed by in situhybridization for the OL marker PLP mRNA. Fewer PLP mRNA+ cells were present in FGFR3 null compared with wild-type mice of the same age in all three regions of the brain (A–C); representative sections are shown. Sections hybridized with sense cRNA probe showed no labeling (data not shown). Scale bar, 100 μm. D, Quantification of the number of PLP mRNA+ cells that differentiated as a function of time in the corpus callosum (CC) and the cortex (CX) of wild-type and FGFR3 null mice. All PLP mRNA+ cells in the entire cortical or corpus callosum region were counted for each section. Two to four sections each from three to nine mice from each group and age were counted. Wild-type numbers (average of all +/+ animals) were set to 100%; FGFR3 null (−/−) numbers are shown relative to that. The numbers of PLP mRNA+ OLs were reduced at P9 and P13 in FGFR3 null corpus callosum and cerebral cortex, which became comparable with that of wild type by P31. HC, Hippocampus; d, dorsal region of the spinal cord; v, ventral region of the spinal cord. Error bars indicate SEM (n = 3–9); *p < 0.005, unpaired Student'st test.
The decrease in oligodendrocyte number in FGFR3 null mice is not attributable to either reduced proliferation or survival of oligodendrocyte progenitors
The reduced number of OLs expressing PLP mRNA observed in FGFR3 null mice during the active phase of myelination could be caused by a reduction in the number of OL progenitors available for differentiation to mature OLs, attributable in turn to reduced proliferation and/or increased cell death or, alternatively, to a decreased efficiency of terminal differentiation by OL progenitors. We investigated these mechanisms by analyzing whole brain parasagittal sections at P2, P7, P9, and P13 from wild-type (+/+) and FGFR3 null (−/−) mice.
The proliferative capacity of OL progenitors was studied by injecting BrdU intraperitoneally into these mice 2 hr before the brains were harvested for double-immunofluorescence microscopy with anti-BrdU and anti-NG2, a marker for OL progenitors (Fig.6A,B). No differences in the total number of BrdU+ cells were observed between wild-type and FGFR3 null mice in either the cortex or corpus callosum (Fig. 6Ab–Ad). Further, ∼40–50% of the BrdU+ cells in the cerebral cortex were also NG2+ at P9 (Fig.6Bd), consistent with previous observations (Dawson et al., 2000; Mallon et al., 2002), in both wild-type and FGFR3 null mice. These data suggest that the proliferation of OL progenitors is not affected in the forebrain of FGFR3 null mice and cannot account for the reduced number of differentiated OLs.
Fig. 6.

Proliferation and survival of OL progenitors is not altered in FGFR3 null mice. P9 sagittal sections taken from parallel regions of wild-type (+/+) and FGFR3 null (−/−) mice were analyzed. A, B, BrdU incorporation analyzed by immunohistochemistry was used as a measure of proliferation (Ab–Ad). The sections were counterstained with Hoechst dye (Aa) to show the region of the brain that was analyzed. Scale bar, 100 μm. The total number of BrdU+ cells (Ad) was obtained by counting all BrdU+ cells in either the entire cerebral cortex (CX) or splenium of the corpus callosum region (CC) for each section. B, Cells double-immunolabeled with NG2 (Ba) and BrdU (Bb) are shown as a merged image (Bc) at higher magnification than in A. The percentage of BrdU+ cells colabeled with anti-NG2 estimates the number of proliferating OL progenitors (Bd). Neither the total numbers of proliferating cells nor the number of NG2+/BrdU+ cells showed significant differences between wild-type and FGFR3 null forebrain.C, TUNEL assay was used as a measure of cell death. TUNEL+ cells in the subependymal germinal zone of P9 forebrain are shown. Inset, High magnification (Ca, Cb, arrows). Two to four sections from three animals from each group were counted. The total number of TUNEL+ cells in either the entire cortex plus corpus callosum regions (CC) or in the whole cerebellar white matter (WM/CB; Cc) was counted for each section. Wild-type numbers were set to 100%; FGFR3 null levels are shown relative to that. No statistically significant differences were observed between the two groups. Scale bars: 100 μm;insets, 30 μm. D, PDGFRα mRNAin situ hybridization was used to identify OL progenitors. No obvious differences were observed between the numbers of PDGFRα mRNA+ cells at P2 in the forebrain of the wild-type and FGFR3 null mice. Scale bar, 100 μm. Error bars indicate SEM (n = 3).
OL cell death was studied in the white matter of FGFR3 null and wild-type mice via the TUNEL assay (a measure of apoptotic cell death). In the normal mouse brain cell death occurs in the subependymal germinal zone in the forebrain and cerebellum at an early postnatal period; then it is reduced progressively to barely detectable levels in the adult brain (Levison et al., 2000). Although a few TUNEL+ cells were observed at P9 in the forebrain (Fig. 6Ca,Cb) and in the corpus callosum and white matter of the cerebellum (WM/CB) (Fig. 6Cc), the numbers did not differ between wild-type and FGFR3 null mice. TUNEL+ cells were not observed in the spinal cord or hindbrain (data not shown). We conclude that reduced survival of OL progenitors or newly formed OLs is not the cause of reduced numbers of differentiated OLs seen in FGFR3 null mice during early postnatal differentiation.
Consistent with the absence of any changes in the proliferative or survival capacities, the numbers of OL early progenitors, determined byin situ hybridization for the progenitor markers PDGFRα (Fig. 6D) or Olig-1 (data not shown), were similar in wild-type and FGFR3 null mice at P2. We conclude that changes in OL progenitor population size do not account for the reduced number of terminally differentiated OLs in FGFR3 null mice.
In the absence of changes in OL progenitor survival, proliferation, or cell number, we conclude that the decrease in the number of terminally differentiated OLs in the FGFR3 null mice is attributable to delays in the timing of their differentiation process.
Oligodendrocyte differentiation also is inhibited in mixed primary cultures from FGFR3 null forebrain
OL development in vivo is orchestrated by complex interactions among at least three major cell types, OLs, astrocytes, and neurons. We studied the roles of these interactions on the regulation of OL number by examining the timing and extent of OL differentiation in mixed primary cultures grown in defined medium initiated from P1 forebrains of either wild-type (+/+) or FGFR3 null (−/−) littermates (Fig. 7). These cultures are devoid of neurons, as indicated by the virtual absence of the neuron-specific marker tubulin III (data not shown). FGF2, a ligand for FGFR3, is present in these cultures (immunoblot analyses; data not shown), consistent with the reported expression of FGF2 by astrocytes (Araujo and Cotman, 1992). Immunoblot analyses of marker proteins for OL terminal differentiation, which are expressed sequentially as CNP, MBP, and MOG, were performed as a function of time in culture. In FGFR3 null cultures the expression of all three markers lagged behind that seen in normal control cultures at P13, P17, and P24 d in vitro (DIV). Although the expression of CNP and MBP reached control levels by 28 DIV (Fig. 7A,B), the late marker MOG (Fig. 7C) still lagged behind control levels at P28 (the latest point that was studied). As an example of the reduced expression of OL markers, quantification of MOG protein levels is shown, demonstrating an approximate threefold decrease at 17 DIV in FGFR3 null compared with control cells (Fig. 7C). This is consistent with a reduced number of MOG+cells observed by immunofluorescence microscopy (data not shown). It is noteworthy that the expression of even the earliest marker of terminal differentiation, CNP, was reduced. This indicates that the inhibition of differentiation occurs at or near the onset of terminal differentiation (a transient increase in the number of cells at a stage just before the CNP+ stage of the lineage would be predicted); however, this fleeting stage is difficult to observe under the conditions of these analyses [i.e., “pre-GalC stage” (Bansal and Pfeiffer, 1992)]. The levels of total protein and the astrocytic marker GFAP were comparable in the two groups (data not shown), suggesting that the decrease in OL markers was not attributable to overall changes in the culture system. Note that these cultures were initiated from forebrain, where no change in GFAP expression was observed in FGFR3 null in vivo (see below). We conclude that the lower levels of OL marker proteins are attributable to a reduction in the rate of OL differentiation and that a similar delay of OL differentiation in the absence of FGFR3 signaling observed in vivo also occurs in culture in the absence of neurons, effectively ruling out the possibility that the effect on OL differentiation occurs indirectly via neurons.
Fig. 7.

Analysis of OL differentiation in mixed primary cell cultures from FGFR3 null forebrain. Mixed primary cell cultures initiated from P2 forebrain were analyzed by immunoblotting as a function of time in culture for wild-type (+/+) and FGFR3 null (−/−) littermate mice for the OL differentiation markers. A, CNP; B, MBP; C, MOG. D, Representative examples for the time courses of CNP, MBP, and MOG and quantification for MOG (NIH Image analysis) at one time point (17 DIV) are shown (n = 3 for wild type;n = 5 for mutants); p < 0.05. Then 10 μg of total protein was loaded in each lane. Note that, similar to in vivo, the expression of OL differentiation markers was delayed in vitro in the FGFR3 null mice compared with wild type.
There is increased expression of the astrocytic marker GFAP in the hindbrain and cerebellum of FGFR3 null mice, but not in the forebrain
Astrocytes express FGFR3 (Fig. 2) (Bansal et al., 1996; Miyake et al., 1996) and proliferate in response to FGFs. During development the differentiation of astrocytes is accompanied by the expression of GFAP, a marker for astrocytic maturation (Goldman, 2001). In adults the astrocytes then downregulate GFAP expression in a region-specific manner (DiProspero et al., 1997; Reilly et al., 1998). GFAP null mice display abnormalities in myelination, suggesting a link between astrocytic function and myelination (Liedtke et al., 1996). Hypothesizing that the elimination of FGFR3 would have an impact on astrocytic function, we analyzed GFAP expression in astrocytes by immunohistochemistry on whole brain parasagittal sections (including the most rostral region of cervical spinal cord) at P7, P9, P13, and P31 (Fig. 8A–H; P9 and P31 are shown). In the FGFR3 null brain at P7 and P9 there was an increase in GFAP expression in the spinal cord, hindbrain, and cerebellum when compared with wild-type mice. This increase in the spinal cord and hindbrain persisted at all of the ages that were examined and continued into adulthood, unlike the wild type, in which GFAP normally is downregulated. In contrast, no increase was seen in the forebrain. GFAP immunoblot analysis of these regions confirmed these observations (Fig. 8I,J). It is noteworthy that FGF-2 mediated the downregulation of astrocytic gap junction communication, and upregulation of dopamine receptor also occurs in a region-specific manner (Reuss et al., 1998, 2000). The GFAP+ astrocytes appeared to be morphologically normal and did not exhibit the stocky, ramified processes characteristic of reactive glia. These results suggest that the downregulation of GFAP by FGF-2 in culture (Reilly et al., 1998) involves FGFR3 signaling and that during development the upregulation of GFAP that normally occurs transiently in the hindbrain and spinal cord is accelerated and persists in the absence of FGFR3 signaling. Therefore, FGFR3 signaling appears to regulate negatively the astrocytic differentiation and/or function.
Fig. 8.
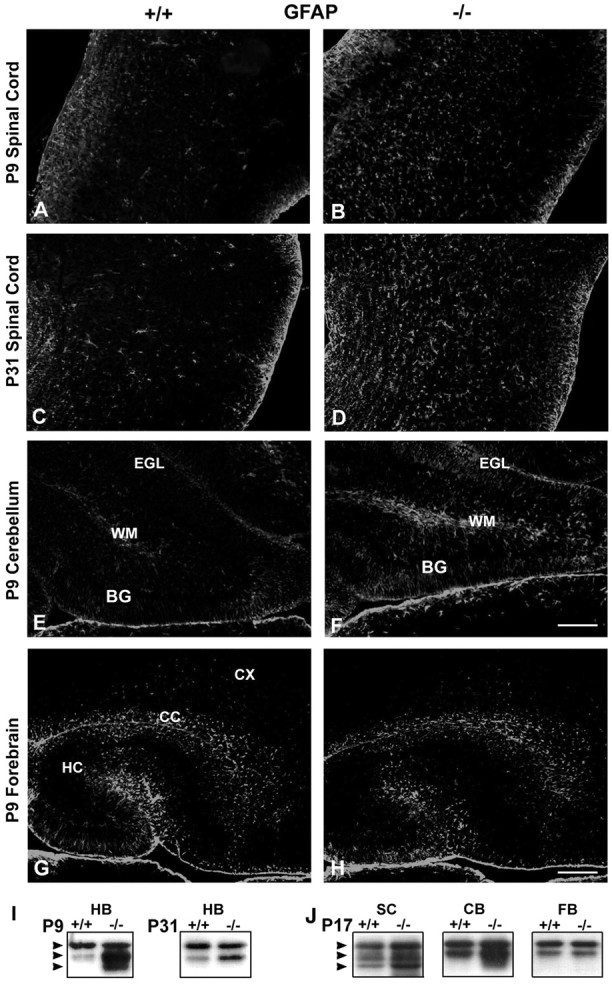
Astrocytic protein, GFAP, is increased in FGFR3 null mice. P9 and P31 parasagittal sections taken from parallel regions of wild-type (+/+) and FGFR3 null (−/−) spinal cord (A–D) at the most dorsal cervical region (just below the medulla), P9 cerebellum (E, F), and P9 forebrain (G, H) were immunolabeled with anti-GFAP. Two to four sections each from three to five mice from each group were analyzed. Representative sections are shown. Scale bars: (inF), A–F, 50 μm; (inH),G, H, 100 μm.BG, Bergmann glia; CX, cortex;CC, corpus callosum; EGL, external granular layer; HC, hippocampus; WM, white matter. A–D, Spinal cord orientation: dorsal (right), ventral (left), rostral (top), caudal (bottom). I, J, GFAP immunoblot analysis of homogenates from hindbrain (HB), spinal cord (SC), cerebellum (CB), and forebrain (FB) from wild-type (+/+) and FGFR3 null (−/−) mice is shown at P9, P17, and P31. Compared with wild type, in mutant mice there was an increased expression of GFAP in the spinal cord, cerebellum, and hindbrain. In contrast, no increase was seen in the forebrain. The increase in spinal cord and hindbrain GFAP immunolabeling continued into adulthood.
Increased cell death in cerebellar cortex of FGFR3 null mice
Because FGFs act as survival factors for many neuronal populationsin vitro, we asked whether FGF signaling via FGFR3 played a role in their survival, using TUNEL labeling of parallel whole brain parasagittal sections at P7, P9, P13, and P31 (Fig.9; P9 is shown). Increased TUNEL+ cells were, in fact, observed in the Purkinje, molecular, and external granular cell layers of the cerebellar cortex of FGFR3 null mice (Fig. 9A,B) (Fig.9C,D, sections counterstained with Hoechst dye). The cell death was particularly prominent in the first few folia, including many cells with the morphology of Purkinje neurons (Fig. 9D,inset). Quantification showed that the increase in the numbers of TUNEL+ cells was approximately twofold (Fig. 9E). FGFR3 mRNA expression was also present in the Purkinje cell layer (Fig. 9F). In contrast to the cerebellum, other areas, such as the hippocampus and cerebral cortex, did not show an increase in cell death in the FGFR3 null mice (data not shown). Moreover, general hippocampal development was normal (data not shown). We conclude that there is a region-specific increase in cell death in the absence of FGFR3.
Fig. 9.
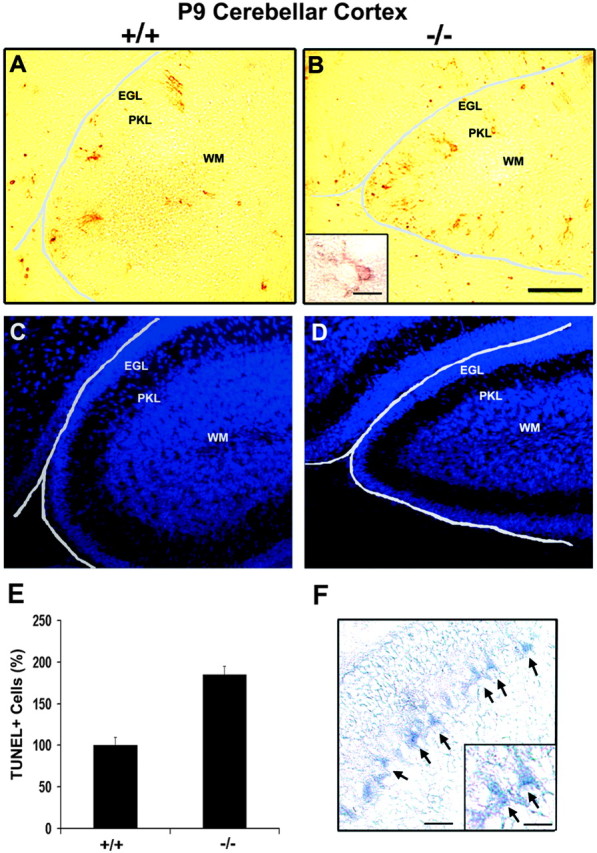
Cell death in the cerebellar cortex is increased in FGFR3 null mice. A, B, Parallel parasagittal sections of cerebella from P9 wild-type (+/+) and FGFR3 null (−/−) mice were analyzed by TUNEL assay. C, D, These sections were counterstained with Hoechst dye to show the regions analyzed inA and B. Scale bars: (inB), A–D, 100 μm; inset, 25 μm. E, TUNEL+ cells were counted from two to four sections each in the area covering the entire cerebellar cortex from three separate animals in each group. The wild-type values are set to 100%, and mutant numbers are plotted as a percentage of wild type. There was a significant increase in cell death in the Purkinje (PKL) and granule cell layers. Error bars indicate SEM (n = 3). *p< 0.1, unpaired Student's t test. F,In situ hybridization for FGFR3 mRNA shows a signal in Purkinje cell layer (arrows). Scale bars:F, 50 μm; inset, 25 μm.EGL, External germinal layer; WM, white matter.
Discussion
Three FGF receptors, FGFR1, FGFR2, FGFR3, are regulated differentially during the course of OL lineage progression. The main finding from this study is that FGFR3 signaling is needed for the timely generation of differentiated OLs. This conclusion is based on two principal observations. First, FGFR3 is positioned ideally both spatially and temporally to impact OL differentiation: that is, it is expressed by Pro-OLs at the brink of the onset of OL terminal differentiation and then is downregulated; further, in postnatal brain the spatial and temporal expression pattern of FGFR3 parallels the appearance of differentiated OLs. Second, the absence of FGFR3 expression in FGFR3 null mutants results in reduced numbers of differentiated OLs during the active phase of myelination; this deficit is corrected in mature animals, and there is no apparent abnormality in the ultrastructure of mature myelin. We therefore conclude that FGFR3 signaling is transient and part of a system regulating the onset of terminal differentiation of Pro-OL but that it is not needed for maintenance of the differentiated state of OLs or further events of myelination per se. This is consistent with the downregulation of FGFR3 in differentiated OLs.
The mechanism by which FGFR3 signaling influences the number of terminally differentiated OLs could involve the regulation of OL progenitor survival, proliferation, or differentiation. OL progenitor survival is influenced by a competition for survival factors. However, in contrast to other trophic factors such as PDGF, insulin-like growth factor-1 (IGF-1), ciliary neurotrophic factor (CNTF), neuregulin, and neurotrophin-3 (Barres et al., 1993; Trapp et al., 1997; Fernandez et al., 2000; Flores et al., 2000), there is conflicting evidence regarding the role of FGF in OL progenitor survival (McKinnon et al., 1991; Barres et al., 1993; Yasuda et al., 1995; Ebner et al., 2000). We did not observe a significant change in the survival capabilities of OL progenitors or OLs in FGFR3 null mice compared with wild-type mice. On the other hand, FGFs are known to be potent mitogens for OL progenitors (Bogler et al., 1990; McKinnon et al., 1990; Gard and Pfeiffer, 1993;Bansal and Pfeiffer, 1994; Fok-Seang and Miller, 1994; Ben-Hur et al., 1998; Baron et al., 2000). Therefore, the loss of one of the FGF receptors could affect proliferation adversely. However, we did not observe a change in the proliferative capacity of OL progenitors in FGFR3 null mice. These data suggest that FGFR3-mediated signaling is not required for either OL progenitor survival or proliferation in the postnatal brain; therefore, the decreased numbers of OLs are probably not attributable to alterations in either of these developmental parameters.
In the absence of effects on progenitor survival or proliferation, it seems likely that FGFR3 signaling is regulating the onset of terminal differentiation. Although proliferation and differentiation traditionally have been cast as opposing activities whereby progenitors need to cease proliferation to enter terminal differentiation, it is becoming apparent that cessation of proliferation is necessary but not sufficient to enable terminal differentiation. This predicts a transition state in which progenitors stop dividing but do not enter terminal differentiation automatically; that is, a second, temporally related but independent, signal is required. A stage with these characteristics has been observed (Bansal and Pfeiffer, 1992; Tang et al., 1998; Tikoo et al., 1998; Ghiani et al., 1999; Huang et al., 2002). Thus FGFR3 may provide one of the signals that promote OL terminal differentiation.
Because proliferation and survival of OL progenitors proceeded normally in FGFR3 null mice, they may be regulated instead by FGFR1 or FGFR2; alternatively, FGFR1 and/or FGFR2 could compensate for the absence of FGFR3 (FGFR4 is not expressed by OLs). These three receptors in fact do have many overlapping ligand-binding specificities and significant homology in their signaling domains. Nevertheless, receptor-specific differences in their signaling patterns, leading to different physiological responses, are also present. For example, in gene-targeted disruption of specific FGF receptors, signals mediated by one receptor are not rescued by those mediated by another (Deng et al., 1994; Arman et al., 1998); proliferation and Ras/MAP kinase pathway activation are promoted less effectively by activation of FGFR3 than of FGFR1 in cell lines (Wang et al., 1994; Shaoul et al., 1995) and OL progenitors (our unpublished observations), presumably because of differences in the levels of tyrosine kinase domain signaling, such as the absence in FGFR3 of one of the two specific tyrosine residues essential for the mitogenicity of FGFR1 (Wang and Goldfarb, 1994, 1997;Lin et al., 1996; Naski et al., 1996). On the other hand, FGFR3 does induce strong signals for promoting differentiation, e.g., via Ras/MAPK-independent signaling pathways (Choi et al., 2001;Rozenblatt-Rosen et al., 2002). In chondrocytes FGFR3 activates Stat-1 (Su et al., 1997; Sahni et al., 1999), which in turn upregulates the expression of the cell cycle protein p21CIP1 (Chin et al., 1996), leading to differentiation; consistent with this, OL differentiation is disrupted in p21 null mice (Zezula et al., 2001). Similarly, FGFR3 may activate pathways that promote OL terminal differentiation and not proliferation.
It seems likely that the inhibition of differentiation of OLs in FGFR3 null mice reflects signaling intrinsic to OL progenitors. Nevertheless, because astrocytes (Fig. 2) (Asai et al., 1993; Bansal et al., 1996;Miyake et al., 1996) and certain neurons (Philippe et al., 1998) also express FGFR3, the observed perturbation of OL development could result indirectly via these cells. However, because the inhibition of OL differentiation also is seen in neuron-free cultures, an indirect effect from neurons probably can be ruled out. Astrocytes, on the other hand, remain a possibility.
Could changes in glial cell fate account for the differences in the number of OLs? In FGFR3 null mice there is an increase in GFAP expression relative to wild-type littermates in the cerebellum, hindbrain, and spinal cord that continues into adulthood (Fig. 8). This increase in GFAP could reflect either an increase in the number of differentiated astrocytes expressing GFAP or to an increase in the amount of GFAP per astrocyte, such as is seen in reactive astrocytes after injury and cell death. There is, in fact, an elevated level of cell death in the cerebellum, so the latter mechanism is possible. However, in the hindbrain and spinal cord there is no apparent increase in cell death, and the morphology of FGFR3 null astrocytes remains normal (i.e., not reactive); therefore, the increase in the levels of GFAP in these regions may reflect an increase in the number of astrocytes. Thus in these regions there could be a relationship between the opposing effects of the numbers of OLs (decreased) and astrocytes (increased). For example, bone morphogenetic proteins (BMPs) have been shown to favor the differentiation, at least in culture, of certain progenitors into astrocytes rather than OLs (Mabie et al., 1997; Grinspan et al., 2000; Mehler et al., 2000). However, because lineage relationships between OLs and astrocytes in vivo remain a point of debate (Rao and Mayer-Proschel, 1997;Herrera et al., 2001; Lu et al., 2002; Rowitch et al., 2002; Zhou and Anderson, 2002) and evidence for the elevation of BMPs in FGFR3 null brain currently is lacking, the speculation that FGFR3 affects OL numbers by a mechanism related to glial cell fate decisions remains open.
FGF signaling also is involved in the regulation of glial migration. For example, disruption of FGF signaling inDrosophila blocks glial migration (Klambt et al., 1992), and OL progenitors expressing a dominant-negative FGFR1 transgene failed to migrate in a transplantation model (Osterhout et al., 1997). However, because heterodimerization of different FGFRs has been reported in cell lines (Bellot et al., 1991), these studies do not distinguish definitively between the relative importance of FGFR1 or FGFR3 (recall that FGFR2 is not expressed by OL progenitors) for migration of OL progenitors. Our current studies show that, in mice lacking FGFR3 signaling, OL progenitors still reach their final destinations, supporting the conclusion that OL progenitor migration is regulated by FGFR1 rather than by FGFR3.
Why does the absence of FGFR3 signaling lead to only a partial and transient inhibition of OL differentiation? A number of other mutant mice null for specific growth factors/receptors also exhibit defects in myelinogenesis, including mice null for CNTF (Barres et al., 1996), neuregulin receptor (erbB2) (Park et al., 2001), IGF-1 (Beck et al., 1995), PDGF-A (Fruttiger et al., 1999), and thyroid hormone (T3) receptors (Baas et al., 2002). These defects are also mostly partial or transient, involving reduced extents of differentiation and myelination. Thus there appears to be an orchestrated set of growth factors that regulate myelinogenesis. In a simple model two factors, both of which are stimulatory for OL differentiation, act together. Thus the loss of one merely reduces, rather than eliminates, terminal differentiation. Several studies suggest that FGF-2 does, in fact, act in concert with other trophic factors known to influence OL progenitors. For example, the mitogenic effect of FGF-2 on OL pre-progenitors and progenitors is enhanced by T3 (Ben-Hur et al., 1998) and IGF-1 (Jiang et al., 2001) and on astrocytes by CNTF (Albrecht et al., 2002); in addition, FGF-2 upregulates the expression by OL progenitors of PDGFRα (McKinnon et al., 1990), Notch-1 (Bongarzone et al., 2000), and the AMPA glutamate receptor GLUR4 (Gallo et al., 1994). How these interactions may relate to the regulation of OL differentiation in vivo is a challenging question.
In summary, we have demonstrated that, in the absence of FGFR3 signaling, the early events of the OL lineage progress normally, including proliferation, survival, and migration, but that the number of terminally differentiated OLs is reduced significantly. Combined with the “temporal location” of FGFR3 expression in the OL developmental pathway, these data suggest that FGFR3 signaling regulates the probability of terminal differentiation by Pro-OLs and thus specifically influences the timing of the onset of OL terminal differentiation. In addition, these studies take advantage of the full complement of signals present in vivo, allowing for the investigation of the overall effect of the loss of FGFR3 signaling on OL development within the context of the combinatorial interactions of other signals.
Footnotes
This work was supported by National Institutes of Health Grant NS 38878. We thank Drs. W. D. Richardson and N. P. Pringle (University College, London, UK) for communicating their unpublished data, for a critical reading of this manuscript, and for the PDGFRα and FGFR3 cRNA probe; we also thank Dr. W. B. Macklin (Cleveland Clinic, Cleveland, OH) for the generous gift of the PLP cRNA probe. We are pleased to acknowledge the contributions to cell culture by S. Winkler, manuscript processing by J. Seagren, and insightful manuscript reviewing by Drs. S. E. Pfeiffer, M. Rasband, and K. Morest (University of Connecticut Medical School). We especially appreciate the valuable advice and encouragement of Dr. Morest during the course of this work.
Correspondence should be addressed to Dr. Rashmi Bansal, Department of Neuroscience, University of Connecticut Medical School, 263 Farmington Avenue, Farmington, CT 06030-3401. E-mail: bansal@neuron.uchc.edu.
References
- 1.Albrecht PJ, Dahl JP, Stoltzfus OK, Levenson R, Levison SW. Ciliary neurotrophic factor activates spinal cord astrocytes, stimulating their production and release of fibroblast growth factor-2, to increase motor neuron survival. Exp Neurol. 2002;173:46–62. doi: 10.1006/exnr.2001.7834. [DOI] [PubMed] [Google Scholar]
- 2.Araujo DM, Cotman CW. Basic FGF in astroglial, microglial, and neuronal cultures: characterization of binding sites and modulation of release by lymphokines and trophic factors. J Neurosci. 1992;12:1668–1678. doi: 10.1523/JNEUROSCI.12-05-01668.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arman E, Haffner-Krausz R, Chen Y, Heath JK, Lonai P. Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc Natl Acad Sci USA. 1998;95:5082–5087. doi: 10.1073/pnas.95.9.5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asai T, Wanaka A, Kato H, Masana Y, Seo M, Tohyama M. Differential expression of two members of FGF receptor gene family, FGFR1 and FGFR2 mRNA, in the adult rat central nervous system. Brain Res Mol Brain Res. 1993;17:174–178. doi: 10.1016/0169-328x(93)90088-7. [DOI] [PubMed] [Google Scholar]
- 5.Baas D, Legrand C, Samarut J, Flamant F. Persistence of oligodendrocyte precursor cells and altered myelination in optic nerve associated to retina degeneration in mice devoid of all thyroid hormone receptors. Proc Natl Acad Sci USA. 2002;99:2907–2911. doi: 10.1073/pnas.052482299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bansal R. Fibroblast growth factors and their receptors in oligodendrocyte development: implications for demyelination and remyelination. Dev Neurosci. 2002;24:35–46. doi: 10.1159/000064944. [DOI] [PubMed] [Google Scholar]
- 7.Bansal R, Pfeiffer SE. Novel stage in the oligodendrocyte lineage defined by reactivity of progenitors with R-mAb prior to O-1 anti-galactocerebroside. J Neurosci Res. 1992;32:309–316. doi: 10.1002/jnr.490320303. [DOI] [PubMed] [Google Scholar]
- 8.Bansal R, Pfeiffer SE. Inhibition of protein and lipid sulfation in oligodendrocytes blocks biological responses to FGF-2 and retards cytoarchitectural maturation, but not developmental lineage progression. Dev Biol. 1994;162:511–524. doi: 10.1006/dbio.1994.1105. [DOI] [PubMed] [Google Scholar]
- 9.Bansal R, Pfeiffer SE. FGF-2 converts mature oligodendrocytes to a novel phenotype. J Neurosci Res. 1997;50:215–228. doi: 10.1002/(SICI)1097-4547(19971015)50:2<215::AID-JNR10>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 10.Bansal R, Kumar M, Murray K, Morrison RS, Pfeiffer SE. Regulation of FGF receptors in the oligodendrocyte lineage. Mol Cell Neurosci. 1996;7:263–275. doi: 10.1006/mcne.1996.0020. [DOI] [PubMed] [Google Scholar]
- 11.Bansal R, Winkler S, Bheddah S. Negative regulation of oligodendrocyte differentiation by galactosphingolipids. J Neurosci. 1999;19:7913–7924. doi: 10.1523/JNEUROSCI.19-18-07913.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baron W, Metz B, Bansal R, Hoekstra D, de Vries H. PDGF and FGF-2 signaling in oligodendrocyte progenitor cells: regulation of proliferation and differentiation by multiple intracellular signaling pathways. Mol Cell Neurosci. 2000;15:314–329. doi: 10.1006/mcne.1999.0827. [DOI] [PubMed] [Google Scholar]
- 13.Barres BA, Schmid R, Sendnter M, Raff MC. Multiple extracellular signals are required for long-term oligodendrocyte survival. Development. 1993;118:283–295. doi: 10.1242/dev.118.1.283. [DOI] [PubMed] [Google Scholar]
- 14.Barres BA, Burne JF, Holtmann B, Thoenen H, Sendtner M, Raff MC. Ciliary neurotrophic factor enhances the rate of oligodendrocyte generation. Mol Cell Neurosci. 1996;8:146–156. doi: 10.1006/mcne.1996.0053. [DOI] [PubMed] [Google Scholar]
- 15.Beck KD, Powell-Braxton L, Widmer HR, Valverde J, Hefti F. IGF1 gene disruption results in reduced brain size, CNS hypomyelination, and loss of hippocampal granule and striatal parvalbumin-containing neurons. Neuron. 1995;14:717–730. doi: 10.1016/0896-6273(95)90216-3. [DOI] [PubMed] [Google Scholar]
- 16.Bellot F, Crumley G, Kaplow JM, Schlessinger J, Jaye M, Dionne CA. Ligand-induced transphosphorylation between different FGF receptors. EMBO J. 1991;10:2849–2854. doi: 10.1002/j.1460-2075.1991.tb07834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ben-Hur T, Rogister B, Murray K, Rougon G, Dubois-Dalcq M. Growth and fate of PSA-NCAM+ precursors of the postnatal brain. J Neurosci. 1998;18:5777–5788. doi: 10.1523/JNEUROSCI.18-15-05777.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bogler O, Wren D, Barnett SC, Land H, Noble M. Cooperation between two growth factors promotes extended self-renewal and inhibits differentiation of oligodendrocyte-type-2 astrocyte (O-2A) progenitor cells. Proc Natl Acad Sci USA. 1990;87:6368–6372. doi: 10.1073/pnas.87.16.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bongarzone ER, Byravan S, Givogri MI, Schonmann V, Campagnoni AT. Platelet-derived growth factor and basic fibroblast growth factor regulate cell proliferation and the expression of notch-1 receptor in a new oligodendrocyte cell line. J Neurosci Res. 2000;62:319–328. doi: 10.1002/1097-4547(20001101)62:3<319::AID-JNR1>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 20.Chin YE, Kitagawa M, Su WC, You ZH, Iwamoto Y, Fu XY. Cell growth arrest and induction of cyclin-dependent kinase inhibitor p21 WAF1/CIP1 mediated by STAT1. Science. 1996;272:719–722. doi: 10.1126/science.272.5262.719. [DOI] [PubMed] [Google Scholar]
- 21.Choi DY, Toledo-Aral JJ, Lin HY, Ischenko I, Medina L, Safo P, Mandel G, Levinson SR, Halegoua S, Hayman MJ. Fibroblast growth factor receptor 3 induces gene expression primarily through Ras-independent signal transduction pathways. J Biol Chem. 2001;276:5116–5122. doi: 10.1074/jbc.M002959200. [DOI] [PubMed] [Google Scholar]
- 22.Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390–397. doi: 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- 23.Crossley PH, Martinez S, Martin GR. Midbrain development induced by FGF8 in the chick embryo. Nature. 1996;380:66–68. doi: 10.1038/380066a0. [DOI] [PubMed] [Google Scholar]
- 24.Dawson MR, Levine JM, Reynolds R. NG2-expressing cells in the central nervous system: are they oligodendroglial progenitors? J Neurosci Res. 2000;61:471–479. doi: 10.1002/1097-4547(20000901)61:5<471::AID-JNR1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 25.Deng CX, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P. Murine FGFR1 is required for early postimplantation growth and axial organization. Genes Dev. 1994;8:3045–3057. doi: 10.1101/gad.8.24.3045. [DOI] [PubMed] [Google Scholar]
- 26.Deng CX, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 27.DiProspero NA, Meiners S, Geller HM. Inflammatory cytokines interact to modulate extracellular matrix and astrocytic support of neurite outgrowth. Exp Neurol. 1997;148:628–639. doi: 10.1006/exnr.1997.6700. [DOI] [PubMed] [Google Scholar]
- 28.Ebner S, Dunbar M, McKinnon RD. Distinct roles for PI3K in proliferation and survival of oligodendrocyte progenitor cells. J Neurosci Res. 2000;62:336–345. doi: 10.1002/1097-4547(20001101)62:3<336::AID-JNR3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 29.Fernandez PA, Tang DG, Cheng L, Prochiantz A, Mudge AW, Raff MC. Evidence that axon-derived neuregulin promotes oligodendrocyte survival in the developing rat optic nerve. Neuron. 2000;28:81–90. doi: 10.1016/s0896-6273(00)00087-8. [DOI] [PubMed] [Google Scholar]
- 30.Flores AI, Mallon BS, Matsui T, Ogawa W, Rosenzweig A, Okamoto T, Macklin WB. Akt-mediated survival of oligodendrocytes induced by neuregulins. J Neurosci. 2000;20:7622–7630. doi: 10.1523/JNEUROSCI.20-20-07622.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fok-Seang J, Miller RH. Distribution and differentiation of A2B5+ glial precursors in the developing rat spinal cord. J Neurosci Res. 1994;37:219–235. doi: 10.1002/jnr.490370208. [DOI] [PubMed] [Google Scholar]
- 32.Ford-Perriss M, Abud H, Murphy M. Fibroblast growth factors in the developing central nervous system. Clin Exp Pharmacol Physiol. 2001;28:493–503. doi: 10.1046/j.1440-1681.2001.03477.x. [DOI] [PubMed] [Google Scholar]
- 33.Fruttiger M, Karlsson L, Hall AC, Abramsson A, Calver AR, Bostrom H, Willetts K, Bertold CH, Heath JK, Betsholtz C, Richardson WD. Defective oligodendrocyte development and severe hypomyelination in PDGF-A knock-out mice. Development. 1999;126:457–467. doi: 10.1242/dev.126.3.457. [DOI] [PubMed] [Google Scholar]
- 34.Fukuchi-Shimogori T, Grove EA. Neocortex patterning by the secreted signaling molecule FGF8. Science. 2001;294:1071–1074. doi: 10.1126/science.1064252. [DOI] [PubMed] [Google Scholar]
- 35.Gallo V, Wright P, McKinnon RD. Expression and regulation of a glutamate receptor subunit by bFGF in oligodendrocyte progenitors. Glia. 1994;10:149–153. doi: 10.1002/glia.440100209. [DOI] [PubMed] [Google Scholar]
- 36.Gard AL, Pfeiffer SE. Glial cell mitogens bFGF and PDGF differentially regulate development of O4+GalC− oligodendrocyte progenitors. Dev Biol. 1993;159:618–630. doi: 10.1006/dbio.1993.1269. [DOI] [PubMed] [Google Scholar]
- 37.Ghiani CA, Eisen AM, Yuan X, DePinho RA, McBain CJ, Gallo V. Neurotransmitter receptor activation triggers p27Kip1 and p21CIP1 accumulation and G1 cell cycle arrest in oligodendrocyte progenitors. Development. 1999;126:1077–1090. doi: 10.1242/dev.126.5.1077. [DOI] [PubMed] [Google Scholar]
- 38.Goldman JE. Developmental origins of astrocytes. In: Jessen KR, Richardson WD, editors. Glial cell development. Oxford UP; Oxford: 2001. pp. 55–69. [Google Scholar]
- 39.Grinspan JB, Edell E, Carpio DF, Beesley JS, Lavy L, Pleasure D, Golden JA. Stage-specific effects of bone morphogenetic proteins on the oligodendrocyte lineage. J Neurobiol. 2000;43:1–17. [PubMed] [Google Scholar]
- 40.Herrera J, Yang H, Zhang SC, Proschel C, Tresco P, Duncan ID, Luskin M, Mayer-Proschel M. Embryonic-derived glial-restricted precursor cells (GRP cells) can differentiate into astrocytes and oligodendrocytes in vivo. Exp Neurol. 2001;171:11–21. doi: 10.1006/exnr.2001.7729. [DOI] [PubMed] [Google Scholar]
- 41.Huang Z, Tang XM, Cambi F. Downregulation of the retinoblastoma protein (rb) is associated with rat oligodendrocyte differentiation. Mol Cell Neurosci. 2002;19:250–262. doi: 10.1006/mcne.2001.1077. [DOI] [PubMed] [Google Scholar]
- 42.Hudson LD, Friedrich VL, Jr, Behar T, Dubois-Dalcq M, Lazzarini RA. The initial events in myelin synthesis: orientation of proteolipid protein in the plasma membrane of cultured oligodendrocytes. J Cell Biol. 1989;109:717–727. doi: 10.1083/jcb.109.2.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang F, Frederick TJ, Wood TL. IGF-I synergizes with FGF-2 to stimulate oligodendrocyte progenitor entry into the cell cycle. Dev Biol. 2001;232:414–423. doi: 10.1006/dbio.2001.0208. [DOI] [PubMed] [Google Scholar]
- 44.Johnson DE, Williams LT. Structural and functional diversity in the FGF receptor multigene family. Adv Cancer Res. 1993;60:1–41. doi: 10.1016/s0065-230x(08)60821-0. [DOI] [PubMed] [Google Scholar]
- 45.Klambt C, Glazer L, Shilo BZ. breathless, a Drosophila FGF receptor homolog, is essential for migration of tracheal and specific midline glial cells. Genes Dev. 1992;6:1668–1678. doi: 10.1101/gad.6.9.1668. [DOI] [PubMed] [Google Scholar]
- 46.Levison SW, Rothstein RP, Brazel CY, Young GM, Albrecht PJ. Selective apoptosis within the rat subependymal zone: a plausible mechanism for determining which lineages develop from neural stem cells. Dev Neurosci. 2000;22:106–115. doi: 10.1159/000017432. [DOI] [PubMed] [Google Scholar]
- 47.Liedtke W, Edelmann W, Bieri PL, Chiu FC, Cowan NJ, Kucherlapati R, Raine CS. GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron. 1996;17:607–615. doi: 10.1016/s0896-6273(00)80194-4. [DOI] [PubMed] [Google Scholar]
- 48.Lin YZ, Yao SY, Hawiger J. Role of the nuclear localization sequence in fibroblast growth factor-1-stimulated mitogenic pathways. J Biol Chem. 1996;271:5305–5308. doi: 10.1074/jbc.271.10.5305. [DOI] [PubMed] [Google Scholar]
- 49.Lu QR, Sun T, Zhu Z, Ma N, Garcia M, Stiles CD, Rowitch DH. Common developmental requirement for OLIG function indicates a motor neuron/oligodendrocyte connection. Cell. 2002;109:75–86. doi: 10.1016/s0092-8674(02)00678-5. [DOI] [PubMed] [Google Scholar]
- 50.Mabie PC, Mehler MF, Marmur R, Papavasiliou A, Song Q, Kessler JA. Bone morphogenetic proteins induce astroglial differentiation of oligodendroglial–astroglial progenitor cells. J Neurosci. 1997;17:4112–4120. doi: 10.1523/JNEUROSCI.17-11-04112.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mallon BS, Shick HE, Kidd GJ, Macklin WB. Proteolipid promoter activity distinguishes two populations of NG2-positive cells throughout neonatal cortical development. J Neurosci. 2002;22:876–885. doi: 10.1523/JNEUROSCI.22-03-00876.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McKinnon RD, Matsui T, Dubois-Dalcq M, Aaronson SA. FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron. 1990;5:603–614. doi: 10.1016/0896-6273(90)90215-2. [DOI] [PubMed] [Google Scholar]
- 53.McKinnon RD, Matsui T, Aranda M, Dubois-Dalcq M. A role for fibroblast growth factor in oligodendrocyte development. Ann NY Acad Sci. 1991;638:378–386. doi: 10.1111/j.1749-6632.1991.tb49048.x. [DOI] [PubMed] [Google Scholar]
- 54.McMorris FA, McKinnon RD. Regulation of oligodendrocyte development and CNS myelination by growth factors: prospects for therapy of demyelinating disease. Brain Pathol. 1996;6:313–329. doi: 10.1111/j.1750-3639.1996.tb00858.x. [DOI] [PubMed] [Google Scholar]
- 55.Mehler MF, Mabie PC, Zhu G, Gokhan S, Kessler JA. Developmental changes in progenitor cell responsiveness to bone morphogenetic proteins differentially modulate progressive CNS lineage fate. Dev Neurosci. 2000;22:74–85. doi: 10.1159/000017429. [DOI] [PubMed] [Google Scholar]
- 56.Messersmith DJ, Murtie JC, Le TQ, Frost EE, Armstrong RC. Fibroblast growth factor 2 (FGF2) and FGF receptor expression in an experimental demyelinating disease with extensive remyelination. J Neurosci Res. 2000;62:241–256. doi: 10.1002/1097-4547(20001015)62:2<241::AID-JNR9>3.0.CO;2-D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miller RH. Oligodendrocyte origins. Trends Neurosci. 1996;19:92–96. doi: 10.1016/s0166-2236(96)80036-1. [DOI] [PubMed] [Google Scholar]
- 58.Miyake A, Hattori Y, Ohta M, Itoh N. Rat oligodendrocytes and astrocytes preferentially express fibroblast growth factor receptor-2 and -3 mRNAs. J Neurosci Res. 1996;45:534–541. doi: 10.1002/(SICI)1097-4547(19960901)45:5<534::AID-JNR3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 59.Naski MC, Wang Q, Xu J, Ornitz DM. Graded activation of fibroblast growth factor receptor 3 by mutations causing achondroplasia and thanatophoric dysplasia. Nat Genet. 1996;13:233–237. doi: 10.1038/ng0696-233. [DOI] [PubMed] [Google Scholar]
- 60.Oh LY, Yong VW. Astrocytes promote process outgrowth by adult human oligodendrocytes in vitro through interaction between bFGF and astrocyte extracellular matrix. Glia. 1996;17:237–253. doi: 10.1002/(SICI)1098-1136(199607)17:3<237::AID-GLIA6>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 61.Ornitz DM, Itoh N. Fibroblast growth factors. Genome Biol. 2001;2:3005. doi: 10.1186/gb-2001-2-3-reviews3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Orr-Urtreger A, Givol D, Yayon A, Yarden Y, Lonai P. Developmental expression of two murine fibroblast growth factor receptors, flg and bek. Development. 1991;113:1419–1434. doi: 10.1242/dev.113.4.1419. [DOI] [PubMed] [Google Scholar]
- 63.Osterhout DJ, Ebner S, Xu J, Ornitz DM, Zazanis GA, McKinnon RD. Transplanted oligodendrocyte progenitor cells expressing a dominant-negative FGF receptor transgene fail to migrate in vivo. J Neurosci. 1997;17:9122–9132. doi: 10.1523/JNEUROSCI.17-23-09122.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Park SK, Miller R, Krane I, Vartanian T. The erbB2 gene is required for the development of terminally differentiated spinal cord oligodendrocytes. J Cell Biol. 2001;154:1245–1258. doi: 10.1083/jcb.200104025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peters K, Werner S, Chen G, Williams LT. Two FGF receptor genes are differentially expressed in epithelial and mesenchymal tissues during limb formation and organogenesis in the mouse. Development. 1992;114:233–243. doi: 10.1242/dev.114.1.233. [DOI] [PubMed] [Google Scholar]
- 66.Peters K, Ornitz D, Werner S, Williams L. Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Dev Biol. 1993;155:423–430. doi: 10.1006/dbio.1993.1040. [DOI] [PubMed] [Google Scholar]
- 67.Pfeiffer SE, Warrington AE, Bansal R. The oligodendrocyte and its many processes. Trends Cell Biol. 1993;3:191–197. doi: 10.1016/0962-8924(93)90213-k. [DOI] [PubMed] [Google Scholar]
- 68.Philippe JM, Garces A, deLapeyiere O. FGF-R3 is expressed in a subset of chicken spinal motor neurons. Mech Dev. 1998;78:119–123. doi: 10.1016/s0925-4773(98)00158-0. [DOI] [PubMed] [Google Scholar]
- 69.Pringle NP, Mudhar HS, Collarini EJ, Richardson WD. PDGF receptors in the rat CNS: during late neurogenesis, PDGFα receptor expression appears to be restricted to glial cells of the oligodendrocyte lineage. Development. 1992;115:535–551. doi: 10.1242/dev.115.2.535. [DOI] [PubMed] [Google Scholar]
- 70.Qian X, Davis AA, Goderie SK, Temple S. FGF-2 concentration regulates the generation of neurons and glia from multipotent cortical stem cells. Neuron. 1997;18:81–93. doi: 10.1016/s0896-6273(01)80048-9. [DOI] [PubMed] [Google Scholar]
- 71.Rao MS, Mayer-Proschel M. Glial-restricted precursors are derived from multipotent neuroepithelial stem cells. Dev Biol. 1997;188:48–63. doi: 10.1006/dbio.1997.8597. [DOI] [PubMed] [Google Scholar]
- 72.Reilly JF, Maher PA, Kumari VG. Regulation of astrocyte GFAP expression by TGF-β1 and FGF-2. Glia. 1998;22:202–210. doi: 10.1002/(sici)1098-1136(199802)22:2<202::aid-glia11>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 73.Reuss B, Dermietzel R, Unsicker K. Fibroblast growth factor-2 (FGF-2) differentially regulates connexin (cx) 43 expression and function in astroglial cells from distinct brain regions. Glia. 1998;22:19–30. [PubMed] [Google Scholar]
- 74.Reuss B, Leung DS, Ohlemeyer C, Kettenmann H, Unsicker K. Regionally distinct regulation of astroglial neurotransmitter receptors by fibroblast growth factor-2. Mol Cell Neurosci. 2000;16:42–58. doi: 10.1006/mcne.2000.0857. [DOI] [PubMed] [Google Scholar]
- 75.Rowitch DH, Lu QR, Kessaris N, Richardson WD. An “oligarchy” rules neural development. Trends Neurosci. 2002;25:417–422. doi: 10.1016/s0166-2236(02)02201-4. [DOI] [PubMed] [Google Scholar]
- 76.Rozenblatt-Rosen O, Mosonego-Ornan E, Sadot E, Madar-Shapiro L, Sheinin Y, Ginsberg D, Yayon A. Induction of chondrocyte growth arrest by FGF: transcriptional and cytoskeletal alterations. J Cell Sci. 2002;115:553–562. doi: 10.1242/jcs.115.3.553. [DOI] [PubMed] [Google Scholar]
- 77.Sahni M, Ambrosetti DC, Mansukhani A, Gertner R, Levy D, Basilico C. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the STAT-1 pathway. Genes Dev. 1999;13:1361–1366. doi: 10.1101/gad.13.11.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shaoul E, Reich-Slotky R, Berman B, Ron D. Fibroblast growth factor receptors display both common and distinct signaling pathways. Oncogene. 1995;10:1553–1561. [PubMed] [Google Scholar]
- 79.Su WC, Kitagawa M, Xue N, Xie B, Garofalo S, Cho J, Deng C, Horton WA, Fu XY. Activation of STAT1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature. 1997;386:288–292. doi: 10.1038/386288a0. [DOI] [PubMed] [Google Scholar]
- 80.Tang XM, Strocchi P, Cambi F. Changes in the activity of cdk2 and cdk5 accompany differentiation of rat primary oligodendrocytes. J Cell Biochem. 1998;68:128–137. doi: 10.1002/(sici)1097-4644(19980101)68:1<128::aid-jcb13>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 81.Tikoo R, Osterhout DJ, Casaccia-Bonnefil P, Seth P, Koff A, Chao MV. Ectopic expression of p27Kip1 in oligodendrocyte progenitor cells results in cell cycle growth arrest. J Neurobiol. 1998;36:431–440. [PubMed] [Google Scholar]
- 82.Trapp BD, Nishiyama A, Cheng D, Macklin W. Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. J Cell Biol. 1997;137:459–468. doi: 10.1083/jcb.137.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vaccarino FM, Schwartz ML, Raballo R, Rhee J, Lyn-Cook R. Fibroblast growth factor signaling regulates growth and morphogenesis at multiple steps during brain development. Curr Top Dev Biol. 1999;46:179–200. doi: 10.1016/s0070-2153(08)60329-4. [DOI] [PubMed] [Google Scholar]
- 84.Wang JK, Goldfarb M. Amino acid residues which distinguish the mitogenic potentials of two FGF receptors. Oncogene. 1997;14:1767–1778. doi: 10.1038/sj.onc.1201021. [DOI] [PubMed] [Google Scholar]
- 85.Wang JK, Gao G, Goldfarb M. Fibroblast growth factor receptors have different signaling and mitogenic potentials. Mol Cell Biol. 1994;14:181–188. doi: 10.1128/mcb.14.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang Q, Bardgett ME, Wong M, Wozniak DF, Lou J, McNeil BD, Chen C, Nardi A, Reid DC, Yamada K, Ornitz DM. Ataxia and paroxysmal dyskinesia in mice lacking axonally transported FGF14. Neuron. 2002;35:25–38. doi: 10.1016/s0896-6273(02)00744-4. [DOI] [PubMed] [Google Scholar]
- 87.Warrington AE, Pfeiffer SE. Proliferation and differentiation of O4+ oligodendrocytes in postnatal rat cerebellum: analysis in unfixed tissue slices using anti-glycolipid antibodies. J Neurosci Res. 1992;33:338–353. doi: 10.1002/jnr.490330218. [DOI] [PubMed] [Google Scholar]
- 88.Woodruff RH, Tekki-Kessaris N, Stiles CD, Rowitch DH, Richardson WD. Oligodendrocyte development in the spinal cord and telencephalon: common themes and new perspectives. Int J Dev Neurosci. 2001;19:379–385. doi: 10.1016/s0736-5748(00)00083-6. [DOI] [PubMed] [Google Scholar]
- 89.Xu J, Liu Z, Ornitz DM. Temporal and spatial gradients of FGF8 and FGF17 regulate proliferation and differentiation of midline cerebellar structures. Development. 2000;127:1833–1843. doi: 10.1242/dev.127.9.1833. [DOI] [PubMed] [Google Scholar]
- 90.Yasuda T, Grinspan J, Stern J, Franceschini B, Bannerman P, Pleasure D. Apoptosis occurs in the oligodendroglial lineage, and is prevented by basic fibroblast growth factor. J Neurosci Res. 1995;40:306–317. doi: 10.1002/jnr.490400304. [DOI] [PubMed] [Google Scholar]
- 91.Yazaki N, Hosoi Y, Kawabata K, Miyake A, Minami M, Satoh M, Ohta M, Kawasaki T, Itoh N. Differential expression patterns of mRNAs for members of the fibroblast growth factor receptor family, FGFR1–FGFR4, in rat brain. J Neurosci Res. 1994;37:445–452. doi: 10.1002/jnr.490370403. [DOI] [PubMed] [Google Scholar]
- 92.Ye W, Shimamura K, Rubenstein JL, Hynes MA, Rosenthal A. FGF and Shh signals control dopaminergic and serotonergic cell fate in the anterior neural plate. Cell. 1998;93:755–766. doi: 10.1016/s0092-8674(00)81437-3. [DOI] [PubMed] [Google Scholar]
- 93.Zezula J, Casaccia-Bonnefil P, Ezhevsky SA, Osterhout DJ, Levine JM, Dowdy SF, Chao MV, Koff A. p21CIP1 is required for the differentiation of oligodendrocytes independently of cell cycle withdrawal. EMBO Rep. 2001;2:27–34. doi: 10.1093/embo-reports/kve008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109:61–73. doi: 10.1016/s0092-8674(02)00677-3. [DOI] [PubMed] [Google Scholar]