

Abstract
We showed previously that cAMP is a survival-promoting stimulus for cultured postnatal rat spiral ganglion neurons (SGNs) and that depolarization promotes SGN survival in part via recruitment of cAMP signaling. We here investigate the subcellular locus of cAMP prosurvival signaling. Transfection of GPKI, a green fluorescent protein (GFP)-tagged cAMP-dependent protein kinase (PKA) inhibitor, inhibits the ability of the permeant cAMP analog cpt-cAMP [8-(4-chlorophenylthio)-cAMP] to promote survival, indicating that PKA activity is necessary. Transfection of GFP-tagged PKA (GPKA) is sufficient to promote SGN survival, but restriction of GPKA to the nucleus by addition of a nuclear localization signal (GPKAnls) almost completely abrogates its prosurvival effect. In contrast, GPKA targeted to the extranuclear cytoplasm by addition of a nuclear export signal (GPKAnes) promotes SGN survival as effectively as does GPKA. Moreover, GPKI targeted to the nucleus lacks inhibitory effect on SGN survival attributable to cpt-cAMP or depolarization. These data indicate an extranuclear target of PKA for promotion of neuronal survival. Consistent with this, we find that dominant-inhibitory CREB mutants inhibit the prosurvival effect of depolarization but not that of cpt-cAMP. SGN survival is compromised by overexpression of the proapoptotic regulator Bad, previously shown to be phosphorylated in the cytoplasm by PKA. This Bad-induced apoptosis is prevented by cpt-cAMP or by cotransfection of GPKA or of GPKAnes but not of GPKAnls. Thus, cAMP prevents SGN death through a cytoplasmic as opposed to nuclear action, and inactivation of Bad proapoptotic function is a mechanism by which PKA can prevent neuronal death.
Keywords: membrane depolarization, spiral ganglion neuron, cell survival, cAMP, cAMP-dependent protein kinase, PKA, apoptosis, Bad, intracellular signal, CREB
Introduction
In neurons, the second messenger cAMP mediates prosurvival signaling, evidenced by the ability of permeant cAMP analogs, e.g., cpt-cAMP [8-(4-chlorophenylthio)-cAMP], to maintain neuronal survival in the absence of other trophic factors (Rydel and Greene, 1988; Michel and Agid, 1996; Hanson et al., 1998; Nakao, 1998). In particular, we showed that neonatal rat spiral ganglion neuron (SGN) survival is promotedin vitro by cpt-cAMP and by membrane depolarization, with the latter being more effective (Hansen et al., 2001). This complements other studies showing that direct electrical stimulation in vivo reduces the death of SGNs that would otherwise occur after the loss of hair cells (Leake et al., 1999; Miller, 2001). Ca2+ influx consequent to membrane depolarization leads to increased intracellular cAMP (Kalix and Roch, 1976; Iuvone et al., 1991; Nakao, 1998; Shen et al., 1999), and promotion of the survival of SGNs (Hansen et al., 2001) and CNS neurons (Meyer-Franke et al., 1995; Hanson et al., 1998) in vitro by membrane depolarization is reduced by a cAMP antagonist. Thus, cAMP is a prosurvival signal and mediates part of the prosurvival effect of depolarization.
The cAMP-dependent protein kinase (PKA) is an effector of cAMP signaling, and we show here that PKA is the major effector of cAMP prosurvival signaling. After elevation of intracellular cAMP concentration, the inactive PKA complex dissociates, releasing catalytic subunits, which then phosphorylate substrate proteins (Francis and Corbin, 1994). Although initially released in the cytoplasm, catalytic subunits translocate to the nucleus and so can phosphorylate and regulate transcription factors in addition to cytoplasmic effectors (Bacskai et al., 1993). An important nuclear target of PKA is the Ca2+–cAMP-responsive element binding protein (CREB) family of transcription factors (De Cesare and Sassone-Corsi, 2000). CREB, in particular, is phosphorylated on serine-133 (Ser133), which allows it to recruit the coactivator CREB binding protein (CBP) and activate transcription. CREB is a mediator of the prosurvival effect of neurotrophins in sympathetic and cerebellar granule neurons (Bonni et al., 1999; Riccio et al., 1999) and plays a critical role in transcriptional activation of prosurvival genes such as BDNF (Shieh et al., 1998; Tao et al., 1998) and Bcl-2 (Wilson et al., 1996; Riccio et al., 1999). PKA can also exert a prosurvival effect by phosphorylating cytoplasmic targets such as the proapoptotic regulator Bad, a BH3 domain-only Bcl-2 family member (Harada et al., 1999; Lizcano et al., 2000; Virdee et al., 2000). PKA is one of several protein kinases capable of functionally inactivating Bad and thus inhibiting apoptosis (Datta et al., 1997; del Peso et al., 1997; Bonni et al., 1999).
Because PKA can act both in the nucleus and in the cytoplasm, PKA could contribute to neuronal survival through regulation of transcription, e.g., activation of CREB-dependent transcription, or by posttranslational modification of apoptotic regulators, e.g., inhibition of the proapoptotic Bad protein. To determine the relative contribution of nuclear and cytoplasmic PKA function, we used green fluorescent protein (GFP)-tagged PKA catalytic subunits (GPKA) and GFP-tagged PKA inhibitor protein (GPKI) that were restricted to the nucleus or cytoplasm, respectively, by insertion of a nuclear localization signal (nls) or nuclear export signal (nes). [In the case of PKI, the nes is endogenous but was removed in adding the nls (Wen et al., 1994).] Expression of these constructs in transfected spiral ganglion neurons in vitro shows that cytoplasmic activity of PKA is necessary and sufficient for its prosurvival effect but that nuclear activity is dispensable. Consistent with this, we observed that, although CREB is phosphorylated by cAMP signaling in SGNs, CREB activity is not necessary for the prosurvival effect of cAMP. Conversely, cAMP signaling effectively inactivates the proapoptotic function of Bad, indicating a prosurvival role for PKA in posttranslational control of cytoplasmic apoptotic regulators.
Materials and Methods
Spiral ganglion neuron culture and transfection. Our basic culture medium consisted of high-glucose (4.5 mg/ml) DMEM, 0.1 mg/ml penicillin, 0.1 mg/ml streptomycin, N2 supplement, and 10 μg/ml insulin and is designated here as “5K” because in this medium, [K+] = 5.4 mm. We also used a depolarizing medium in which Na+ was replaced with equimolar K+ to raise [K+] to 25 or 30 mm while maintaining osmolarity. These media are designated here as “25K” or “30K,” respectively. (Survival is equivalent in either medium.) Before and during transfection, the cultures were maintained in 25K to which 5% fetal bovine serum was added (“25K+S”). The DMEM, antibiotics, and serum were obtained from the University of Iowa Diabetes and Endocrinology Research Core, and the other supplements were from Sigma (St. Louis, MO) or Invitrogen (Carlsbad, CA).
Dissociated cultures of SGNs were prepared from postnatal day 4–5 rats and maintained by a modification of the methods described previously (Hegarty et al., 1997). Briefly, rat cochleae were removed from the temporal bone and placed in ice-cold PBS. Each spiral ganglion was isolated from the cochlea by sequential removal of the bony cochlear capsule, the spiral ligament, and the organ of Corti, leaving the spiral ganglion within the modiolus. These were collected in HBSS on ice and then transferred to Ca2+–Mg2+-free HBSS with 0.1% trypsin and 0.1% collagenase at 37°C for 20 min to enzymatically dissociate the cells. After three washes with culture medium, an additional mechanical dissociation was performed by trituration using either fire-polished glass Pasteur pipettes or a mechanical pipettor with 1 ml pipette tips. The dissociated cells were plated in 25K+S. Neurons were plated in tissue culture dishes (Falcon or Corning, purchased from Fisher Scientific, Houston, TX) or slides (Lab-Tek, Rochester, NY) previously coated sequentially with polyornithine (Sigma) (0.1 mg/ml in 10 mm borate buffer, pH 8.4) for 1 hr at room temperature, followed by laminin (Invitrogen) (20 μg/ml in HBSS) overnight at 4°C. Typically, the cells were plated in 48-well culture plates at a density of approximately one ganglion per well. Thus, there are ∼5000 neurons per well (∼28% plating efficiency), and these are ∼10% of the total number of cells. The low plating efficiency for SGNs is presumably a result of the extensive dissection and treatment required to separate the ganglion from the bone.
Transfection was performed when the neurons had firmly attached to the substrate, ∼6 hr after plating. We used a calcium phosphate-based protocol modified from that by Gabellini et al. (1992) as we described previously (Zha et al., 2001). Briefly, plasmids (see below for description of plasmids used) were mixed with 1.25 mCaCl2 solution, and then sterile deionized water was added to bring the CaCl2 concentration to 0.25 m. An equal volume of 2× depolarizing HEPES buffer (in mm: 50 HEPES, 220 NaCl, 1.5 Na2HPO4, and 60 KCl, pH 7.1) was added slowly and with agitation. The DNA–Ca2+–PO4mixture was left for 20 min at room temperature to allow precipitates to form and then added to the culture medium (25K+S) on the cells at a 1:10 (v/v) ratio, the final concentration of DNA being 9.1 μg/ml. After 6 hr, the culture medium containing DNA mixture was washed once with DMEM and replaced with 25K medium. We found that the presence of serum before and during the transfection improves the viability of the neurons but that removal of the serum after the incubation with DNA–Ca2+–PO4improves the transfection efficiency. Typically, 5–10% of the SGNs were transfected. In some cases, multiple plasmids were cotransfected. In experiments performed using two plasmids that both encode a detectable product, we found that >95% of the transfected cells expressed both gene products.
Immunocytochemistry. After culture for the times indicated, the cells were fixed for 15 min with 4% paraformaldehyde in PBS, washed with PBS, and incubated with blocking buffer (PBS, 2% BSA, 5% normal goat serum, and 0.1% NaN3) for 1 hr at 37°C, then with primary antibodies in blocking buffer for 1 hr at 37°C or overnight at 4°C, and then with fluorescently labeled secondary antibodies in blocking buffer for 1 hr at 37°C. After washing with PBS, the nuclei were stained with Hoechst 33342 (10 μg/ml in PBS) for 15 min, washed again with PBS, and viewed with a Leica Leitz DMR microscope equipped for fluorescence optics (Leica, Nussloch, Germany). The images were captured using a Photometrics CH250 cooled CCD camera with IPLab Spectrum software (Signal Analytics, Vienna, VA). The images were prepared for publication using Adobe Photoshop and Illustrator (Adobe Systems, San Jose, CA). NIH Image on a Macintosh computer or the equivalent Image J software on a Windows computer were used to quantify phospho-CREB immunofluorescence. In the program, a circle was drawn just inside the boundary of each neuronal nucleus, and the intensity of the phospho-CREB immunofluorescence was determined as the average pixel density within the circle. Background fluorescence was determined as the average pixel density within a circle of equal diameter just outside of the nucleus, and this background was subtracted from the value obtained for nuclear fluorescence. The scale used was arbitrary but linear and consistent among all experiments.
The primary antibodies used in this study were rabbit anti-phospho-CREB antibody, which specifically detects CREB phosphorylated at Ser133 (Upstate Biotechnology, Waltham, MA) (1:1000 dilution) and mouse anti-neurofilament 200 kDa isoform (NF200) antibody (Sigma) (1:400 dilution) to identify neurons. Secondary antibodies used were Alexa Fluor 488 goat anti-rabbit IgG and Alexa Fluor 350 or 568 goat anti-mouse IgG antibodies (Molecular Probes, Eugene, OR) (1:400 dilution).
Assay of neuronal survival–apoptosis. For survival assays, replicate wells were prepared for each plasmid or combination of plasmids transfected. Two wells were fixed 18 hr after the start of transfection, by which time expression of the introduced genes was already apparent, and were used to establish the transfection efficiency of that combination of plasmids. The remaining wells (in separate multiwell plates) were washed three times with PBS and then maintained for 48 hr under the indicated experimental conditions: 5K medium, 25K medium, or 5K plus 1 mm cpt-cAMP (Calbiochem, La Jolla, CA). The cells were then fixed, permeabilized, and stained with anti-NF200 antibody, as described above. All transfected (GFP-expressing) neuron (NF200-positive) cells in each well were counted to assess survival. This number was corrected for differing transfection efficiencies among different combinations of plasmids. As noted above, a significant number of neurons die rapidly within the first 24 hr after plating (our unpublished observations) (Kim et al., 2001). This is presumably because of the trauma of removing the ganglion from the bone, because it occurs regardless of the conditions in which the cultures are maintained. Therefore, the survival of SGNs maintained in 25K for 48 hr is actually ∼25% of those that survive plating. However, to allow for comparison among experiments, the number of SGNs transfected with GFP only and maintained in 25K was arbitrarily set to 100%, to which relative survival in other experimental conditions was normalized.
For apoptosis assays, SGNs were cotransfected with combinations of plasmids, described below, including those encoding wild-type or mutant Bad and those encoding GFP or GFP-tagged PKA constructs. After recovery from transfection, the cultures were maintained for 48 hr in 5K plus 1% FBS, fixed, and stained with anti-NF200 antibody and Hoechst 33342. SGN apoptosis was established by the presence of condensed nuclei and of collapsed cytoskeletal organization (see Fig. 8).
Fig. 8.
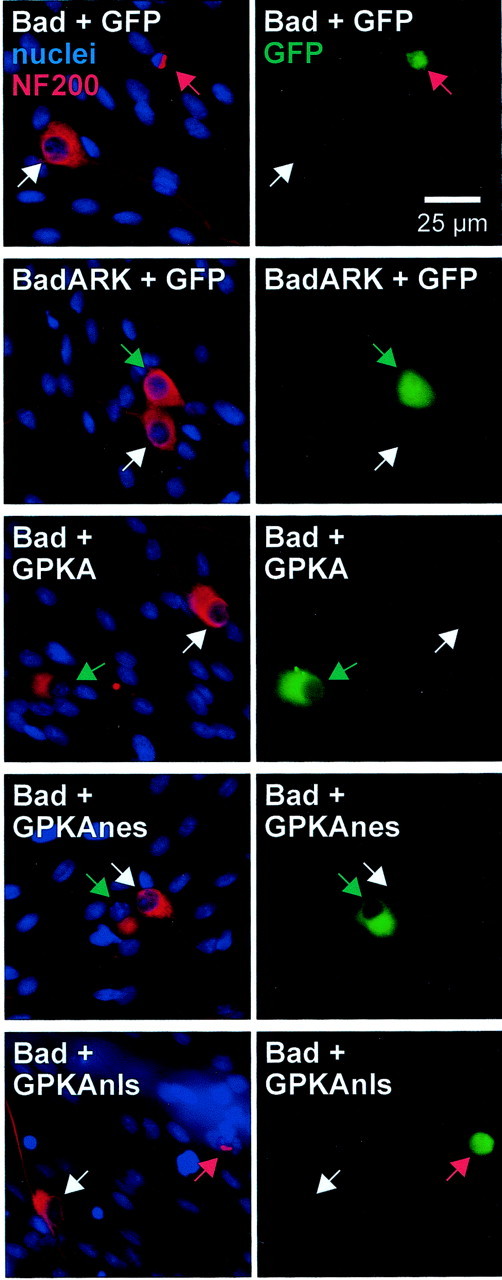
Expression of cytoplasmic GPKA rescues SGNs from apoptosis caused by Bad overexpression. SGNs were transfected with wild-type Bad or inactive mutant Bad plasmids in a 1:1 mixture with GFP-tagged PKA or control GFP plasmids, as indicated. Eighteen hours after transfection, the cultures were switched to culture medium containing 1% horse serum and, 48 hr later, were fixed and stained as in Figure 2. Transfected cells were identified by green GFP fluorescence, shown in the right panel of each pair of images. Neurons were identified by NF200 immunoreactivity (red), and nuclei were stained with Hoechst 33342 (blue). These images were superimposed and are shown in the left panel of each pair. Arrows point to neuronal nuclei and indicate identical positions in each pair of images. The images were chosen so that each shows a typical transfected and a typical untransfected neuron. Untransfected neurons are indicated bywhite arrows. Transfected neurons that are apoptotic (identified by their condensed nuclei and collapsed cytoplasm) are indicated by red arrows. Transfected nonapoptotic neurons are indicated by green arrows. Shown in the image pairs from top to bottom: transfection of wild-type Bad results in apoptosis but BadARK does not. Cotransfection of GPKA or GPKAnes with Bad results in nonapoptotic cells, but cotransfection of GPKAnls with Bad does not prevent Bad-induced apoptosis.
Expression constructs. Enhanced green fluorescent protein-C1 (EGFP-C1) was purchased from Clontech (Cambridge, UK). Wild-type CREB, CREBm1, and KCREB expression vectors were kindly provided by Drs. Marc Montminy (University of California, San Diego, Salk Institute, La Jolla, CA) (Montminy et al., 1990) and Richard Goodman [Oregon Health Sciences University (OHSU), Vollum Institute, Portland, OR] (Walton et al., 1992). The PKI and PKInls expression plasmids were kindly provided by Dr. Philip Stork (OHSU, Vollum Institute, Portland, OR) (Billiard et al., 2001). The wild-type BAD expression vector was kindly provided by Dr. Gabriel Nuñez (University of Michigan, Ann Arbor, MI) (del Peso et al., 1997).
A cAMP-dependent protein kinase (PKA) catalytic subunit expression vector (RSV-PKA-Cα) was kindly provided by Dr. Richard Maurer (OHSU, Portland, OR) (Howard et al., 1991). This was used as a template for PCR amplification of the PKA-C using the following primers: 5′-AAGTCCGGAGGCGGCACCATGGGCAACGCCG-3′ (forward) and 5′-TTGAGCTCGAGAAAACTCAGTAAACTCCTTGCCACACTTCT-3′ (reverse).
After PCR amplification, the PKA catalytic subunit cDNA was subcloned into pEGFP-C1 between BspEI and XhoI, which places the PKA catalytic subunit at the C-terminus of enhanced GFP. This construct was called GPKA. GPKAnls was constructed by adding a double-stranded oligonucleotide encoding the SV40 large T antigen nls (PPKKKRKVPG) (Kalderon et al., 1984) at the C-terminus of GPKA between HindIII and BamHI. The oligonucleotides used were as follows: 5′-AGCTGGCGGCACGCCACCTAAGAAGAAGAGAAAGGTGG3′ and 5′-GATCCCACCTTTCTCTTCTTCTTAGGTGGGCTGCCGCC-3′.
To make GPKAnes, a double-stranded oligonucleotide encoding the MEK (mitogen-activated protein kinase kinase) nes (EALQKKLEELELDE) (Wu et al., 1992) was added to the C-terminus of the GPKA between HindIII and BamHI. The oligonucleotides used were as follows: 5′-AGCTTTGCAGAAGAAGCTGGAGGAGCTGGAGCTGGACGAGGGATC-3′ and 5′-GATCCCTCGTCCAGCTCCAGCTCCTCCAGCTTCTTCTGCAA-3′.
BadARK, an inactive Bad mutant, was constructed by mutating three conserved amino acids at the BH3 domain of wild-type Bad: leucine 151 to alanine (A), aspartate 156 to arginine (R), and glutamate 157 to lysine (K). This was done by PCR-mediated mutagenesis. The primers used were 5′-CCCCAATCTCTGGGCAGCGCAGCGCTACGGCCGTGAGGCCCGAAGGATGAGCCGTAAGTTTGAGGG-3′ (forward) and 5′-TCTAGCGAATTCACTGGGAGGGGGT-3′ (reverse). The italicized nucleotides indicate the mutated sequences. The mutated PCR product contained unique restriction sites, PflMI andEcoRI. The wild-type Bad sequence between PflMI and EcoRI was replaced by the PCR product. Unlike wild-type Bad, overexpression of BadARK did not induce apoptosis in the transfected SGNs (see Results).
Results
Targeting of the PKA catalytic subunits to specific subcellular locations
Addition of permeant cAMP analogs, such as cpt-cAMP, to cultured neurons, including SGNs, is sufficient to promote their survival in the absence of other trophic support (Rydel and Greene, 1988; Michel and Agid, 1996; Hanson et al., 1998; Nakao, 1998; Hansen et al., 2001). This implies that PKA, a major downstream effector of cAMP, exerts a prosurvival effect. To test this hypothesis, we transfected SGNs with a construct encoding GPKA, causing overexpression of PKA activity in the neurons and identifying the transfected cells with GFP fluorescence. Because PKA exerts distinct actions in both the nucleus and the cytoplasm, we distinguished between them by targeting GPKA to the nucleus or cytoplasm exclusively. To target GPKA to the cytoplasm, we added an MEK nes to the C-terminus of GPKA (GPKAnes). To target GPKA to the nucleus, we added a SV40 nuclear localization signal to the C-terminus of GPKA (GPKAnls). The construction of these cDNAs is described in Materials and Methods.
The presence of GFP in the encoded proteins allowed verification of both their expression in transfected cells and their desired subcellular localization, as shown in Figure1. Hoechst was used to mark the nuclei, and anti-NF200 antibody was used to mark the cytoplasm of the neurons in the culture. As shown in Figure 1, GPKA, like GFP, was present in both the nucleus and cytoplasm, coinciding with Hoechst fluorescence and NF200 immunofluorescence. In some neurons, GPKA was preferentially distributed in the cytoplasm rather than in the nucleus (see Fig. 8). In contrast, GPKAnls was present almost exclusively in the nucleus, primarily coinciding with Hoechst fluorescence, whereas GPKAnes was present almost exclusively in the cytoplasm, not overlapping with Hoechst staining and coinciding with NF200 immunoreactivity.
Fig. 1.
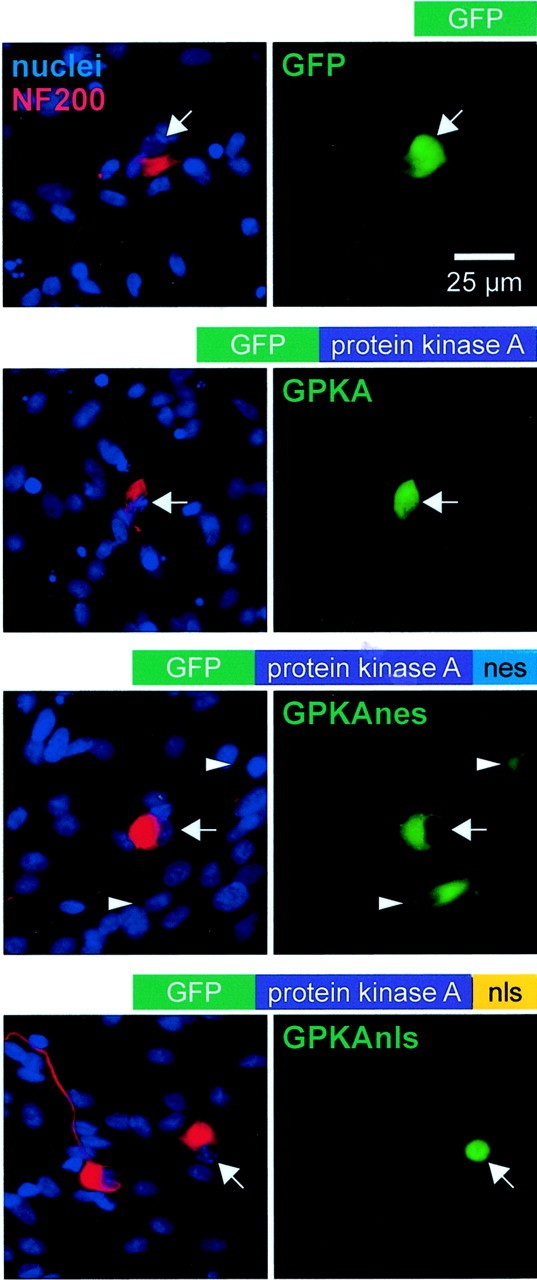
Expression of subcellularly targeted GFP-tagged PKA catalytic subunit in SGNs. SGNs were transfected with GFP, GPKA, GPKAnes, a GFP-tagged PKA to which a nuclear export signal was added, or GPKAnls, a GFP-tagged PKA to which a nuclear localization signal was added, as indicated. After fixation, neurons were incubated with anti-NF200 antibody, followed by Alexa Fluor 568-conjugated secondary antibody. The nuclei were labeled by staining with Hoechst 33342. The cultures were photographed sequentially with a red filter to detect NF200 immunofluorescence, a blue filter to detect Hoechst 33342 fluorescence, and a green filter to detect GFP fluorescence. The diagrams above each pair of images show the general structure of the construct transfected. The left panel of each pair shows a composite of the NF200 and nucleus images. The corresponding GFP or GPKA images are shown at right. Thearrows identify the location of neuronal nuclei and indicate identical positions in both panels of each pair. Green fluorescence is evident in the nucleus and cytoplasm of neurons transfected with GFP or GPKA but is evident primarily in the cytoplasm of the GPKAnes-transfected neuron and primarily in the nucleus of the GPKAnls-transfected neuron. The arrowheads indicate the position of two non-neuronal cells fortuitously transfected with GPKAnes, showing primarily cytoplasmic localization of GPKAnes in this glial cell and in the neuron.
Cytoplasmic but not nuclear PKA promotes SGN survival
SGNs were transfected in serum-containing medium, with GFP, GPKA, or targeted GPKA expression plasmids, and after a recovery period of 18–20 hr, the serum was washed out and the culture medium was replaced with one devoid of neurotrophic factors and containing nondepolarizing 5K. As a control, SGNs were also maintained in depolarizing culture medium containing 25K, previously shown to maintain SGN survival. SGN survival was determined 48 hr later by counting only transfected neurons (green fluorescent and NF200-expressing cells) to determine directly the consequence of expression of GPKA constructs. GPKA expression significantly enhanced the survival of SGNs over that of SGNs expressing only GFP (Fig. 2). This indicates that PKA activity is sufficient to promote neuronal survival, implying that PKA may be responsible for the prosurvival effect of cAMP. We show below (see Fig. 4) that PKA activity is necessary for the prosurvival effect of cAMP.
Fig. 2.

Expression of cytoplasmic but not nuclear PKA is sufficient to promote SGN survival. SGNs were transfected with GFP plasmid or with a 1:4 mixture of GFP and GPKA, GPKAnls, or GPKAnes plasmids and maintained in 5K or 25K culture conditions, as indicated, for 48 hr. The cultures were then fixed and immunolabeled with anti-NF200 antibody and fluorescent secondary antibody. Surviving transfected SGNs (i.e., cells that were both GFP-expressing and NF200-immunoreactive) were counted. To allow combining of results from multiple experiments, SGN survival was normalized to and expressed as a percentage of survival in the 25K condition, which was arbitrarily assigned a value of 100%. The survival of SGNs maintained in 5K and transfected with GPKA or GPKAnes was significantly (p < 0.005) different from survival of SGNs maintained in 5K and transfected with GFP or GPKAnls. Results shown are means of three or more separate experiments, each performed in duplicate. In this and all figures showing quantification of survival–apoptosis experiments, the error bars show SD.
Fig. 4.

Expression of cytoplasmic but not nuclear GFP-tagged PKA inhibitor protein inhibits SGN survival. SGNs were transfected with GFP (open bars) or with a 1:1 mixture of GFP and GPKI (light gray bars) or GPKInls (dark gray bars) and maintained in 5K, 1 mmcpt-cAMP, or 25K culture conditions, as indicated, for 48 hr. Also, SGNs were transfected with a 1:1 mixture of GPKAnes and GFP, GPKI, GPKInls. Surviving transfected SGNs (i.e., cells that were both GFP-expressing and NF200-immunoreactive) were counted as in Figure 2and normalized, in each experiment, to the number of surviving SGNs in the control 25K condition. The ability of cpt-cAMP, GPKAnes, or 25K to promote SGN survival was significantly (p < 0.005) reduced by GPKI. GPKInls did not significantly affect SGN survival under these conditions: survival was comparable with that of SGNs transfected with GFP only. Results shown are means of three or more separate experiments, each performed in duplicate.
We next determined survival of SGNs expressing subcellularly targeted GPKAs. Expression of GPKAnes significantly enhanced SGN survival over that of SGNs expressing GFP (Fig. 2) to an extent comparable with that of expression of GPKA. This indicates that cytoplasmic PKA activity alone is sufficient for the survival-promoting effect of PKA. In contrast, transfection of GPKAnls failed to promote SGN survival; the survival of SGNs expressing GPKAnls was not significantly different from that of SGNs expressing GFP alone (Fig. 2). Thus, nuclear PKA activity does not promote neuronal survival, implying an essential role for cytoplasmic PKA activity.
In this study of the promotion of survival by cAMP–PKA signaling, we scored SGN survival after 48 hr in experimental conditions. This time point was chosen because cAMP–PKA resulted in shrinkage of the SGN somata and neurites, increasing with time, making it more difficult to identify neurons after 48 hr. However, because the process of cell death was still continuing at this time, SGN survival in the control 5K condition was somewhat greater in these experiments than what we observed previously (Hegarty et al., 1997; Hansen et al., 2001). Also, these experiments all involved transfection, and, as noted in Materials and Methods, this required maintaining the cultures in serum-containing medium to improve viability. The inclusion of serum early in the culture process reduces the rate of SGN death after subsequent transfer to serum-free conditions, presumably because we are unable to wash sufficiently vigorously to completely remove serum components. This further increased SGN survival in the control 5K condition, reducing the difference between minimal and maximal survival. However, the differences obtained were significant in all cases, withp < 0.001.
Cytoplasmic but not nuclear PKI inhibits the ability of cAMP and of depolarization to promote survival
We showed previously that the membrane-permeant, specific cAMP antagonist Rp-cAMPS prevents promotion of SGN survival by cpt-cAMP and reduces promotion of SGN survival by depolarization (Hansen et al., 2001). Although use of this reagent established cAMP as a mediator of the survival-promoting effect of depolarization, it did not distinguish between cAMP signaling via PKA and cAMP signaling via other effectors, e.g., Rap1 (Vossler et al., 1997; Grewal et al., 2000). To determine whether PKA is the principal mediator of the survival-promoting effect of cAMP and to determine where in the cell PKA activity is required, we used the PKA inhibitor protein PKI (Walsh et al., 1971). PKI is an endogenous protein that binds to the PKA catalytic subunit and inhibits its kinase activity with great specificity and efficacy (Walsh et al., 1971; Whitehouse and Walsh, 1983). PKI contains an endogenous nes (Wen et al., 1994), causing it to be generally excluded from the nucleus. This also allows it to prevent entry of the PKA catalytic subunit into the nucleus (Wiley et al., 1999), thus blocking nuclear and cytoplasmic PKA activity (Wiley et al., 1999; Billiard et al., 2001). In particular, cAMP-dependent CCAAT/enhancer-binding protein δ (C/EBPδ) translocation to the nucleus is prevented by cytoplasmic PKI but not by nuclear PKI, but nuclear PKA does prevent C/EBPδ-dependent transcription (Billiard et al., 2001).
A construct consisting of PKI fused to the C-terminus of GFP (GPKI) was transfected into SGNs. The GFP tag allowed us to verify the non-nuclear location of the GPKI (Fig.3A). Because PKI prevents both cytoplasmic and nuclear PKA activity, we also used a modified GPKI, one with the nes replaced with a dual nls (GPKInls). Nuclear-restricted PKI blocks nuclear but not cytoplasmic actions of PKA (Billiard et al., 2001). GPKInls was transfected into SGNs, and its nuclear location was verified by means of the GFP tag (Fig.3A). To verify that the expressed PKI constructs blocked PKA function in transfected SGNs, we transfected GPKI or GPKInls into SGNs, exposed the cultures to 1 mm cpt-cAMP for 30 min to activate PKA, fixed them, and then determined phospho-CREB immunoreactivity using an anti-phospho-CREB antibody. Because cells with active PKA phosphorylate CREB in response to cpt-cAMP, lack of phospho-CREB immunoreactivity indicates PKA inhibition. We compared phospho-CREB immunoreactivity in adjacent transfected (GFP-positive) and untransfected SGNs in 10 pairs of SGNs in GPKI-transfected cultures and 10 pairs of SGNs in GPKInls-transfected cultures. In all cases, the two PKI constructs blocked CREB phosphorylation; representative examples are shown in Figure 3B. These data confirm the efficacy of the PKI constructs. Moreover, this extends the previous study of C/EBPδ (Billiard et al., 2001) to show that CREB activation by cAMP is blocked by either inhibition of PKA in the nucleus or prevention of PKA translocation to the nucleus.
Fig. 3.
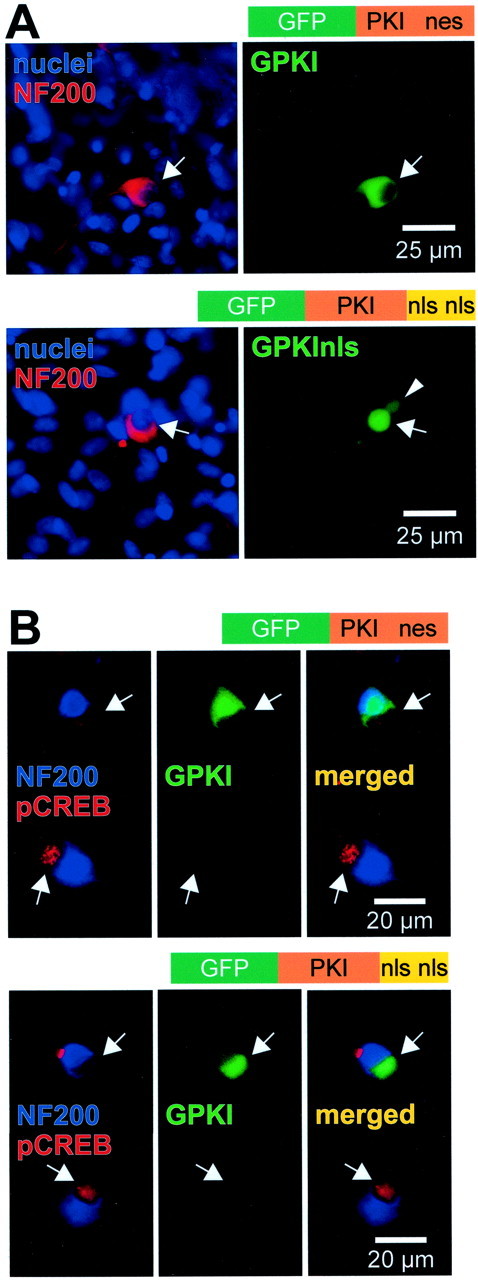
Expression and function of subcellularly targeted GFP-tagged PKI constructs in SGNs. SGNs were transfected with GPKI, which contains an intrinsic nuclear export signal, or with GPKInls, in which the nuclear export signal was deleted and replaced with two nuclear localization signals, as indicated. A, After allowing 24 hr for expression, the cultures were fixed, and nuclei and neuronal cytoplasm were labeled with Hoechst 33342 and anti-NF200, respec- tively, as in Figure 1. The diagrams above each pair of images show the general structure of the construct transfected. Theleft panel of each pair shows a composite of the nuclei and NF200 images. The corresponding GPKI or GPKInls images are shown atright. The arrows identify the location of neuronal nuclei and indicate identical positions in bothpanels of each pair. The arrowheadindicates the nucleus of a fortuitously transfected glial cell. Green fluorescence is evident primarily in the cytoplasm of the GPKI-transfected neuron and primarily in the nucleus of the GPKInls-transfected neuron. B, Cultures transfected with GPKI (top) or GPKInls (bottom), as inA, were treated for 30 min with 1 mmcpt-cAMP before fixation. Two neighboring SGNs are shown in each set ofpanels, one (top) transfected and one (bottom) untransfected. In each set,left, superimposed blue (NF200) and red (phospho-CREB) images identifying SGNs and showing phospho-CREB immunoreactivity.Center, Same field using a green filter to detect GFP and shows the upper neuron of each pair expressing the indicated GFP-tagged PKI construct. Right, Mergedleft and center images. For either construct, the transfected neuron exhibits greatly reduced phospho-CREB immunoreactivity relative to the untransfected.
After verifying the subcellular localization and function of GPKI and GPKInls in SGNs, we determined their effect on the survival of SGNs maintained by cAMP signaling. As shown in Figure4, GPKI but not GPKInls inhibited the survival-promoting effect of cpt-cAMP. This result shows that the prosurvival effect of cAMP signaling is mediated principally by PKA but not by other putative cAMP effectors, which would not have been inhibited by PKI. Moreover, because inhibition of any nuclear PKA activity by nuclear-restricted GPKInls had no effect on survival, PKA activity in the nucleus must not be necessary for neuronal survival. To verify that the GPKInls had no effect on cytoplasmic PKA signaling, we cotransfected it with GPKAnes, and, predictably, there was no significant reduction of SGN survival (Fig. 4).
We noted previously that cAMP signaling mediates part of the survival-promoting effect of depolarization (Hansen et al., 2001). We therefore asked whether this is a consequence of PKA activation and, if so, whether nuclear or cytoplasmic PKA was required. As shown in Figure4, GPKI but not GPKInls partially inhibited the prosurvival effect of depolarization (i.e., culture in 25K medium). This supports previous studies implicating PKA as a mediator of depolarization-promoted survival and indicates that nuclear PKA activity, in particular, is not involved. Blockade of PKA activity throughout the cell inhibits the survival-promoting effect of cAMP and of depolarization, but blockade only in the nucleus has no effect. This indicates a cytoplasmic locus of PKA action in promotion of neuronal survival, consistent with the results described above showing that overexpression of PKA catalytic subunit in the cytoplasm but not in the nucleus promotes survival.
CAMP and depolarization induce CREB phosphorylation in SGNs
Previous studies (Gonzalez and Montminy, 1989) have shown that PKA activates CREB-dependent transcription by directly phosphorylating CREB in the nucleus. This phosphorylation is on serine 133 (Gonzalez and Montminy, 1989), so we used the antibody that specifically recognizes CREB phosphorylated on serine 133 to determine immunocytochemically whether CREB is phosphorylated by cAMP in SGNs. Phospho-CREB immunoreactivity was detected with a fluorescent secondary antibody. Both depolarization and cpt-cAMP caused increased CREB phosphorylation (Fig. 5). CREB phosphorylation caused by cpt-cAMP was blocked by the cAMP antagonist Rp-cAMPS, but CREB phosphorylation was still evident in depolarized neurons incubated with Rp-cAMPS (Fig. 5). Thus, although cAMP-dependent signaling partially accounts for the prosurvival effect of depolarization, a cAMP-independent mechanism must also exist to mediate CREB phosphorylation in response to depolarization.
Fig. 5.

CREB phosphorylation on Ser133 in SGNs treated with cpt-cAMP or depolarized. SGNs were cultured in control medium containing no trophic additions (5K) or exposed for 15 min to 1 mm cpt-cAMP or depolarizing (30K) medium, as indicated. Additional cultures included Rp-cAMPS, a cAMP antagonist, which was added 30 min before and throughout the 15 min incubation with cpt-cAMP or 30K. The cultures were fixed, and neurons were identified by NF200 immunoreactivity (red). Nuclei of all cells were labeled with Hoechst 33342 (blue). Phosphorylation of CREB on Ser133 was detected by immunofluorescence (green fluorescence superimposed on the blue nuclear Hoechst 33342 fluorescence appearscyan). Increased phospho-CREB immunoreactivity is evident in nuclei in the cpt-cAMP and 30K conditions relative to the 5K condition. The increase in phospho-CREB immunoreactivity caused by cpt-cAMP but not that caused by depolarization was blocked by Rp-cAMPS, indicating that a cAMP-independent pathway must exist for phosphorylation of CREB by depolarization.
Phospho-CREB immunofluorescence was quantified (using NIH Image) for SGNs transfected with GPKA constructs or control GFP, allowed 18 hr to express the proteins, and then maintained for 12 hr in 25K or 5K culture medium. The immunofluorescence level was determined separately for transfected and untransfected neurons in the cultures and was expressed as a value on a scale that was arbitrary but consistent among all of the experiments. The data were then plotted in the form of histograms (Fig. 6). Expression of GPKA and culture in 25K both resulted in a large increase in the percentage of neurons exhibiting higher levels of phospho-CREB immunoreactivity. Transfection by itself did not cause an increase in CREB phosphorylation, because transfection of GFP or of inactive kinases resulted in no change in phospho-CREB immunoreactivity (data not shown). Thus, depolarization and cAMP signaling both cause a significant increase in CREB phosphorylation in SGNs.
Fig. 6.

Quantification of CREB phosphorylation caused by depolarization or PKA transfection. SGNs were transfected with GFP or GPKA, as indicated, and allowed 18 hr to express the proteins. After 12 hr incubation in control 5K or depolarizing 25K culture conditions, the cultures were fixed and stained for nuclei, NF200, and phospho-CREB immunoreactivity as in Figure 5. The NF200 immunoreactivity and Hoechst 33342 images were used to identify the positions of neuronal nuclei on the corresponding phospho-CREB image. The intensity of the phospho-CREB immunoreactivity was determined as the average pixel density within the circle. Background fluorescence was determined and corrected for background using NIH Image or Image J as described in Materials and Methods and plotted here as a histogram of the number of neuronal nuclei displaying each intensity value. The histograms for neurons cultured in depolarizing medium and for neurons transfected with GPKA show a shift from a condition in which most SGNs have low levels of phospho-CREB immunoreactivity to one in which all SGNs have varying but much higher levels of phospho-CREB immunoreactivity.
CREB mediates part of the prosurvival effect of depolarization but is dispensable for the prosurvival effect of cAMP
It has been shown previously (Bonni et al., 1999; Riccio et al., 1999) that CREB is required for promotion of neuronal survival by peptide neurotrophic factors. Because we found that both depolarization and cAMP caused increased CREB phosphorylation in SGNs, we asked whether CREB is also required for all or part of the prosurvival effect of these stimuli. To test for a requirement for CREB function, we inhibited endogenous CREB by transfecting into the SGN cDNAs encoding one of two types of dominant-inhibitory CREB mutants, CREBm1 (Struthers et al., 1991) and KCREB (Walton et al., 1992). These two mutants were chosen because they inhibit CREB function by different mechanisms. Also, they inhibit the function of CREB family members other than CREB itself (Walton et al., 1992; Jean et al., 1998), including factors such as CREM (cAMP response element modulator) or ATF-1 (activating transcription factor 1) that can also be regulated by cAMP. Both mutants blocked expression from a CRE reporter in response to cpt-cAMP in PC12 cells (data not shown).
After transfection, the cultures were maintained in nondepolarizing (5K) medium, in 5K medium containing cpt-cAMP, or in 25K medium. After 48 hr, the cultures were fixed, and transfected neurons were counted as before. Inhibition of CREB function by either CREBm1 or KCREB significantly reduced the prosurvival effect of 25K (Fig.7). In contrast, inhibition of CREB function had little effect on cAMP-promoted survival (Fig. 7). Thus, although cAMP causes CREB activation, this appears to be dispensable for the prosurvival effect of cAMP. This is consistent with the observations described above showing that nuclear PKA activity fails to promote survival and that blockade of PKA activity in the nucleus, which prevents it from activating CREB, does not significantly affect neuronal survival.
Fig. 7.

Expression of dominant-inhibitory CREB mutants inhibits the prosurvival effect of depolarization but not that of cpt-cAMP. SGNs were transfected with a 1:3 mixture of GFP plasmid and plasmid encoding either wild-type CREB (open bars), CREBm1 (light gray bars), or KCREB (dark gray bars); the latter two are dominant-inhibitory CREB mutants. The cultures were maintained in control (5K), depolarizing (25K), or 1 mm cpt-cAMP culture conditions, as indicated, for 48 hr. Surviving transfected SGNs were counted as in Figure 2 and normalized, in each experiment, to the number of surviving SGNs in the control 25K condition. Results shown are means of three separate experiments, each performed in duplicate. The ability of 25K to promote SGN survival was significantly (p < 0.005) reduced by either CREB mutant but was not affected by wild-type CREB. In contrast, the survival-promoting effect of cpt-cAMP was unaffected by the CREB mutants.
Cytoplasmic activity of PKA rescues SGNs from apoptosis caused by Bad overexpression
The data described above indicate that nuclear actions of PKA are not necessary for its prosurvival effect. This is consistent with the observation that CREB family transcription factors are dispensable for the prosurvival effect of PKA, although they are important nuclear targets of PKA and are implicated in neuronal survival mediated by other stimuli. The data implicate a cytoplasmic effector for the prosurvival effect of PKA, and we next sought to identify at least one candidate. The proapoptotic regulator Bad has been shown to be phosphorylated by PKA, and this phosphorylation can inactivate the proapoptotic function of Bad (Harada et al., 1999; Lizcano et al., 2000; Virdee et al., 2000). Although we could show that GPKA or GPKAnes expression or treatment with cpt-cAMP all caused Bad phosphorylation in PC12 cells (X.-M. Zha, D. Li, J. Bok, and S. H. Green, unpublished observations), the small number of cells and the presence of non-neuronal cells and neurons precluded such a biochemical determination using these spiral ganglion cultures. Nevertheless, we were able to ask whether PKA could prevent cell death specifically by functionally inactivating Bad. To do this, we maintained spiral ganglion cultures in 5K medium containing 1% FBS, which marginally supports SGN survival, delaying their death. Under these conditions, overexpression of wild-type Bad caused a large and rapid increase in the number of apoptotic SGNs (Figs. 8,9), whereas higher concentrations of FBS (e.g., 5–10%) prevented SGN death regardless of Bad overexpression and lower FBS concentrations resulted in rapid apoptosis in all cells. The use of 1% FBS allowed us to readily assess the ability of various agents to suppress the Bad-induced increase in SGN apoptosis and use this as a measure of their ability to inactivate Bad.
Fig. 9.

Quantification of rescue of SGNs by PKA from apoptosis caused by Bad overexpression. SGNs were transfected with GFP plasmid (first column) or a 1:1 mixture of GFP with either Bad or BadARK (second and third columns) or a 1:1 mixture of wild-type Bad with GFP-tagged PKA constructs (fourth through sixth columns). Eighteen hours after transfection, the cultures were switched to culture medium containing 1% horse serum and, 48 hr later, were fixed and stained as in Figure 8 to distinguish apoptotic and nonapoptotic transfected SGNs. Randomly selected neurons were scored, and the percentage of apoptotic SGNs in each condition was calculated. Each column shows the mean and SD of at least three separate determinations, each performed in duplicate. The total number of SGNs counted is shown above each bar; thenumber in parentheses is the number of separate experiments pooled to obtain this number of SGNs. Expression of Bad but not of BadARK caused a significant (p < 0.001) increase in the number of apoptotic SGNs. This increase, in turn, was prevented by transfection of GPKA or GPKAnes but not by transfection of GPKAnls: the percentage of apoptotic transfected neurons was significantly lower (p < 0.001 and p < 0.01, respectively) in cultures transfected with Bad and GPKA or GPKAnes than in those transfected with Bad and GFP or GPKAnls. There was no significant difference in the percentage of apoptotic transfected neurons between cultures transfected with Bad plus GFP and those transfected with Bad plus GPKAnls. There was no significant difference in the percentage of apoptotic transfected neurons among cultures transfected with GFP only, BadARK plus GFP, Bad plus GPKA, or Bad plus GPKAnes.
Bad-transfected cells were identified by cotransfecting GFP. Apoptosis was determined in transfected neurons by staining with Hoechst 33342 and observing nuclear pyknosis and by staining with anti-NF200 and observing collapse of the cytoskeleton. Typical examples are shown in Figure 8. Nuclear and cytoskeletal condensation was apparent in Bad-transfected SGNs but was uncommon in untransfected cells in the same culture (Fig. 8A). Transfection of GFP alone (data not shown) or of BadARK, an inactivated Bad mutant, did not cause increased apoptosis (Fig. 8B,C). Increased apoptosis was not observed in SGNs cotransfected with Bad and GPKA or GPKAnes (Fig. 8D,E) but was observed in SGNs cotransfected with Bad and GPKAnls (Fig.8F).
These data are quantified in Figure 9. For cultures maintained in 1% FBS, transfection of wild-type Bad increased the percentage of apoptotic SGNs from ∼30 to ∼70%. This increase was entirely a result of overexpression of Bad, because transfection of an inactivated mutant Bad (BadARK) resulted in no change in the percentage of apoptotic SGNs. This increase in apoptosis was almost entirely reversed by cotransfection of GPKA or GPKAnes but was unaffected by cotransfection of GPKAnls. Thus, cytoplasmic PKA but not nuclear PKA activity results in functional inactivation of Bad, which suggests that Bad inactivation is a means by which PKA can promote neuronal survival.
Discussion
cAMP promotes survival through cytoplasmic PKA activity
The second messenger cAMP has been shown to be a prosurvival signal, suggesting that stimuli, e.g., hormones or neurotransmitters, that raise intracellular cAMP levels could use cAMP to promote survival. This seems to be the case for at least one critically important prosurvival stimulus, membrane depolarization: the Ca2+ increase that is the immediate consequence of membrane depolarization can increase cytosolic cAMP levels via Ca2+–calmodulin-dependent adenylyl cyclase (Antoni, 2000). This has been observed in the promotion of motor neuron survival by depolarization (Hanson et al., 1998). Blockade of cAMP signaling prevents or reduces the ability of depolarization to promote the survival of retinal ganglion cells (Meyer-Franke et al., 1995), motor neurons (Hanson et al., 1998), and SGNs (Hansen et al., 2001), indicating that cAMP signaling is necessary, at least in part, for depolarization to promote the survival of CNS and PNS neurons.
CAMP can act via both PKA-dependent and -independent pathways, and PKA itself can act in both the cytoplasm and the nucleus. To distinguish among these alternative pathways by which cAMP could promote survival, we used a specific PKA inhibitor protein (PKI) directed to either a nuclear or cytoplasmic location and catalytic PKA subunits likewise directed to either a nuclear or cytoplasmic location. The effect of these constructs on SGN survival indicates that cAMP exerts its survival-promoting effect via PKA and more specifically via cytoplasmic PKA. This is not consistent with a role for CREB in cAMP prosurvival signaling, in contrast to the CREB requirement for prosurvival signaling by neurotrophins (Bonni et al., 1999; Riccio et al., 1999). Indeed, we find that direct blockade of CREB with dominant-negative mutants did not compromise the ability of cAMP to promote survival. That PKA acts in the cytoplasm to promote survival is consistent with previous studies, discussed below, that have identified means by which cAMP signaling can promote survival through various actions that seem to take place in the cytoplasm. One of these is phosphorylation of the BH3 domain-only Bcl-2 family member Bad by PKA (Harada et al., 1999; Lizcano et al., 2000; Virdee et al., 2000). Indeed, we found that cytoplasmic but not nuclear PKA causes Bad phosphorylation in a neuronal cell line (Zha, Li, Bok, and Green, unpublished observations), and we show here that cytoplasmic but not nuclear PKA inactivates the proapoptotic effect of Bad in spiral ganglion neurons.
CREB activity is not required for the prosurvival effect of cAMP
An unexpected result of these studies is that blockade of CREB activity with dominant-negative mutants did not affect the ability of cAMP to maintain neuronal survival, indicating that CREB activity is not required. This is surprising in view of previous studies showing that CREB is required for the prosurvival effect of neurotrophins (Bonni et al., 1999; Riccio et al., 1999) and of our present observation that CREB is required for at least part of the prosurvival effect of depolarization (Fig. 7). The inability of these CREB dominant-negative mutants to inhibit the prosurvival effect of cAMP is unlikely to be caused by incomplete inhibition of CREB, because the same mutants did inhibit the prosurvival effect of depolarization in this study and the prosurvival effect of neurotrophins in other studies (Bonni et al., 1999; Riccio et al., 1999). The lack of a CREB requirement for the survival-promoting effect of cAMP is not caused by a lack of CREB activation by cAMP: cAMP and PKA cause CREB phosphorylation in SGNs comparable with that caused by depolarization (Figs. 5, 6) and, in a neuronal cell line, induce transcription from a CRE reporter comparable with that induced by depolarization or neurotrophins (Bok, Li, and Green, unpublished observations).
In SGNs, the ability of depolarization to activate CREB does not require cAMP (Fig. 5) and may depend primarily on Ca2+/calmodulin-dependent protein kinase (CaMK) signaling (Hansen et al., 2003). Both cAMP and CaMKIV phosphorylate CREB, allowing it to recruit CBP and so promote transcription (Chrivia et al., 1993; Kwok et al., 1994). However, recruitment of CBP may not be sufficient: although a VP16-CREB fusion protein, which directly activates transcription, could promote survival of CNS neurons (Bonni et al., 1999; Riccio et al., 1999) and SGNs (Bok, J. Huang, and Green, unpublished observations), a mutant that constitutively binds CBP (Cardinaux et al., 2000) could not promote SGN survival (Bok, Huang, and Green, unpublished observations). This suggests that additional posttranslational modification of CBP is necessary for prosurvival function of CREB-dependent transcription, and this may distinguish cAMP and CaMKIV signaling.
Both CaMKIV and cAMP signaling potentiate CREB-dependent transcription (Chawla et al., 1998; Cardinaux et al., 2000; Impey et al., 2002) at the level of CBP or downstream of CBP. However, although CaMKIV has been shown to directly phosphorylate CBP on Ser301 (Impey et al., 2002), the region of CBP containing this site is entirely dispensable for transcription induction by PKA (Zanger et al., 1999). Moreover, although Xu et al. (1998) implicated the consensus PKA phosphorylation site in CBP, Ser1772, in transcriptional activation by PKA, Zanger et al. (1999) showed transcriptional activation by PKA in CBP mutants lacking this site. Therefore, depolarization and CaMKIV may differ from PKA in the mechanism by which they potentiate CREB-dependent transcription, and this could result in distinct patterns of CREB-dependent transcription in which CaMKIV-activated but not PKA-activated genes contribute to survival. Alternatively, the pattern of CREB-dependent transcription is the same between CaMKIV and cAMP, but CaMKIV might activate, in addition to CREB, effectors that allow CREB-dependent transcription to contribute to neuronal survival. Thus, CREB would be necessary in the context of promotion of survival by depolarization but not in the context of promotion of survival by cAMP.
CREB knock-out compromises the survival of certain peripheral neurons (Lonze et al., 2002), e.g., sympathetic neurons, at a developmental stage at which they are dependent on target-derived neurotrophic factors. This is consistent with the observed CREB requirement for support by NGF of cultured neonatal sympathetic neurons (Riccio et al., 1999). As neurons mature and lose their dependence on peptide neurotrophic factors, stimuli such as electrical activity or neurotransmitters that use cAMP signaling may become more important for maintenance of survival. We speculate that this also involves a shift away from a completely CREB-dependent prosurvival mechanism, which may be essential in allowing a role for CREB in neuronal plasticity and other functions in mature neurons.
Cytoplasmic effectors of cAMP prosurvival signaling
Protein kinase B (PKB, Akt) is a multifunctional prosurvival signal (Datta et al., 1999). Because cAMP can activate PKB (Sable et al., 1997; Filippa et al., 1999), it might represent a possible cytoplasmic target of PKA for prosurvival signaling. Another potential prosurvival signaling pathway, activated in the cytoplasm, is the MEK–ERK (extracellular signal-regulated kinase) pathway, and this pathway has been shown to be activated by PKA (Impey et al., 1998). However, inhibition of PI-3-OH kinase, an upstream activator of PKB, or of PKB itself does not significantly reduce cAMP-promoted survival in sympathetic (Crowder and Freeman, 1999), cerebellar granule (Miller et al., 1997; Li et al., 2000), or spiral ganglion neurons (Hansen et al., 2001), and inhibition of MEK also does not significantly reduce cAMP-promoted survival in these diverse types of neurons (Creedon et al., 1996; Hansen et al., 2001). Thus, cAMP signaling can promote survival without recruiting these signal pathways recruited by peptide neurotrophic factors. This is in contrast to retinal ganglion neurons in which cAMP has been shown to promote survival by inducing translocation of a neurotrophin receptor (TrkB) to the cell membrane, thus enhancing the responsiveness of the neurons to exogenous BDNF (Meyer-Franke et al., 1998). Moreover, the ability of cAMP to promote SGN survival is not affected by using TrkB-IgG or TrkC-IgG fusion proteins to block neurotrophin binding to neurotrophin receptors on SGNs (Hansen et al., 2001). Thus, the mechanism operating in retinal ganglion neurons for response to cAMP may be a distinctive feature of those cells and, possibly, other types of CNS neurons, as has been suggested (Meyer-Franke et al., 1995, 1998).
The data presented here suggest that cAMP, acting through PKA, can directly affect the apoptotic regulatory apparatus. Here, we identified the proapoptotic regulator Bad as a target of cAMP signaling that allows cAMP, via PKA, to affect the apoptotic decision in neurons. Specifically, we show that PKA can rescue SGNs from apoptotic death by functionally inactivating Bad that has been overexpressed in the neurons. This is consistent with previous studies (Harada et al., 1999;Lizcano et al., 2000; Tan et al., 2000; Virdee et al., 2000; Zhou et al., 2000) that have shown that PKA can phosphorylate Bad in a manner that potentially interferes with the ability of Bad to interact with other Bcl-2 family apoptotic regulatory proteins on the mitochondrion and perform a proapoptotic function. Although they indicate that PKA can promote survival via Bad inactivation, these data do not rule out other mechanisms by which PKA can, in parallel, suppress cell death by targeting cytoplasmic effectors. For example, glycogen synthase kinase 3 is a proapoptotic effector that has been shown to be phosphorylated and inactivated by PKA in cerebellar granule neurons (Li et al., 2000). Moreover, although the data argue against a nuclear role for PKA in maintaining neuronal survival, they do not completely exclude transcription-dependent mechanisms; PKA might regulate transcription while in the cytoplasm. One example of this is activation of nuclear factor (NF)-κB, which can act as a prosurvival effector (Foehr et al., 2000; Yang et al., 2000; Sarmiere and Freeman, 2001). PKA can act in the cytoplasm to stimulate the DNA-binding ability of NF-κB and its translocation from cytoplasm to nucleus (Shirakawa and Mizel, 1989). Cytoplasmic PKA activity is sufficient to cause nuclear translocation of C/EBPδ, although nuclear PKA is required for its transcriptional activity (Billiard et al., 2001). Additional studies, focused on such cytoplasmic targets of PKA, should define the mechanisms by which cAMP promotes neuronal survival.
Additivity among survival-promoting factors
Neurons are normally exposed to multiple potential survival-promoting factors, e.g., peptide neurotrophic factors and stimuli, such as membrane depolarization, that can raise cAMP levels. These can cooperate to promote survival, and we showed that this is indeed the case for spiral ganglion neurons for which cAMP, depolarization, and neurotrophins promote survival in an additive manner (Hegarty et al., 1997). Neurotrophins promote survival, at least in part, via a nuclear mechanism, recruitment of CREB (Bonni et al., 1999; Riccio et al., 1999), whereas cAMP apparently promotes survival via cytoplasmic targets. We suggest that the additivity observed is not a result of their convergence on a common target but rather of their targeting different points in the sequence of molecular events that control survival. Neurotrophins and CaMKIV control the synthesis of apoptotic regulators, whereas cAMP posttranslationally controls the activity of apoptotic regulators in the cytoplasm.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant DC02961 (S.H.G.), and facilities and services were provided by the University of Iowa Diabetes and Endocrinology Research Core, funded by NIH Grant DK25295. We thank Dr. Stefan Strack for comments on this manuscript. We also thank Drs. Richard Goodman, Richard Maurer, Marc Montminy, Gabriel Nuñez, and Philip Stork for generously providing plasmids used in these studies.
Correspondence should be addressed to Steven H. Green, Department of Biological Sciences, University of Iowa, 138 Biology Building, Iowa City, IA 52242-1324. E-mail: steven-green@uiowa.edu.
References
- 1.Antoni FA. Molecular diversity of cyclic AMP signalling. Front Neuroendocrinol. 2000;21:103–132. doi: 10.1006/frne.1999.0193. [DOI] [PubMed] [Google Scholar]
- 2.Bacskai BJ, Hochner B, Mahaut-Smith M, Adams SR, Kaang BK, Kandel ER, Tsien RY. Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science. 1993;260:222–226. doi: 10.1126/science.7682336. [DOI] [PubMed] [Google Scholar]
- 3.Billiard J, Grewal SS, Lukaesko L, Stork PJS, Rotwein P. Hormonal control of insulin-like growth factor-I gene transcription in human osteoblasts: dual actions of cAMP-dependent protein kinase on CCAAT/enhancer binding protein δ. J Biol Chem. 2001;276:31238–31246. doi: 10.1074/jbc.M103634200. [DOI] [PubMed] [Google Scholar]
- 4.Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 5.Cardinaux JR, Notis JC, Zhang Q, Vo N, Craig JC, Fass DM, Brennan RG, Goodman RH. Recruitment of CREB binding protein is sufficient for CREB-mediated gene activation. Mol Cell Biol. 2000;20:1546–1552. doi: 10.1128/mcb.20.5.1546-1552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chawla S, Hardingham GE, Quinn DR, Bading H. CBP: a signal-regulated transcriptional coactivator controlled by nuclear calcium and CaM kinase IV. Science. 1998;281:1505–1509. doi: 10.1126/science.281.5382.1505. [DOI] [PubMed] [Google Scholar]
- 7.Chrivia JC, Kwok RPS, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 8.Creedon DJ, Johnson EM, Jr, Lawrence JC., Jr Mitogen-activated protein kinase-independent pathways mediate the effects of nerve growth factor and cAMP on neuronal survival. J Biol Chem. 1996;271:20713–20718. doi: 10.1074/jbc.271.34.20713. [DOI] [PubMed] [Google Scholar]
- 9.Crowder RJ, Freeman RS. The survival of sympathetic neurons promoted by potassium depolarization, but not by cyclic AMP, requires phosphatidylinositol 3-kinase and Akt. J Neurochem. 1999;73:466–475. [PubMed] [Google Scholar]
- 10.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 11.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 12.De Cesare D, Sassone-Corsi P. Transcriptional regulation by cyclic AMP-responsive factors. Prog Nucleic Acid Res Mol Biol. 2000;64:343–369. doi: 10.1016/s0079-6603(00)64009-6. [DOI] [PubMed] [Google Scholar]
- 13.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nuñez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 14.Filippa N, Sable CL, Filloux C, Hemmings B, Van Obberghen E. Mechanism of protein kinase B activation by cyclic AMP-dependent protein kinase. Mol Cell Biol. 1999;19:4989–5000. doi: 10.1128/mcb.19.7.4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foehr ED, Lin X, O'Mahony A, Geleziunas R, Bradshaw RA, Greene WC. NF-κB signaling promotes both cell survival and neurite process formation in nerve growth factor-stimulated PC12 cells. J Neurosci. 2000;20:7556–7563. doi: 10.1523/JNEUROSCI.20-20-07556.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Francis SH, Corbin JD. Structure and function of cyclic nucleotide-dependent protein kinases. Annu Rev Physiol. 1994;56:237–272. doi: 10.1146/annurev.ph.56.030194.001321. [DOI] [PubMed] [Google Scholar]
- 17.Gabellini N, Minozzi M-C, Leon A, Dal Toso R. Nerve growth factor transcriptional control of c-fos promoter transfected in cultured spinal sensory neurons. J Cell Biol. 1992;118:131–138. doi: 10.1083/jcb.118.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–682. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- 19.Grewal SS, Fass DM, Yao H, Ellig CL, Goodman RH, Stork PJ. Calcium and cAMP signals differentially regulate cAMP-responsive element-binding protein function via a Rap1-extracellular signal-regulated kinase pathway. J Biol Chem. 2000;275:34433–34441. doi: 10.1074/jbc.M004728200. [DOI] [PubMed] [Google Scholar]
- 20.Hansen MR, Zha X-M, Bok J, Green SH. Multiple distinct signal pathways, including an autocrine neurotrophic mechanism, contribute to the survival-promoting effect of depolarization on spiral ganglion neurons. J Neurosci. 2001;21:2256–2267. doi: 10.1523/JNEUROSCI.21-07-02256.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen MR, Bok J, Devaiah AK, Zha X-M, Green SH (2003) Ca2+/calmodulin-dependent protein kinases II and IV both promote survival but differ in their effects on axon growth in spiral ganglion neurons. J Neurosci Res, in press. [DOI] [PubMed]
- 22.Hanson MG, Jr, Shen S, Wiemelt AP, McMorris FA, Barres BA. Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro. J Neurosci. 1998;18:7361–7371. doi: 10.1523/JNEUROSCI.18-18-07361.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harada H, Becknell B, Wilm M, Mann M, Huang LJ, Taylor SS, Scott JD, Korsmeyer SJ. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol Cell. 1999;3:413–422. doi: 10.1016/s1097-2765(00)80469-4. [DOI] [PubMed] [Google Scholar]
- 24.Hegarty JL, Kay AR, Green SH. Trophic support of cultured spiral ganglion neurons by depolarization exceeds and is additive with that by neurotrophins or cyclic AMP, and requires elevation of [Ca2+]i within a set range. J Neurosci. 1997;17:1959–1970. doi: 10.1523/JNEUROSCI.17-06-01959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Howard P, Day KH, Kim KE, Richardson J, Thomas J, Abraham I, Fleischmann RD, Gottesman MM, Maurer RA. Decreased catalytic subunit mRNA levels and altered catalytic subunit mRNA structure in a cAMP-resistant Chinese hamster ovary cell line. J Biol Chem. 1991;266:10189–10195. [PubMed] [Google Scholar]
- 26.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB- dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 27.Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, Wayman GA, Storm DR, Soderling TR, Goodman RH. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- 28.Iuvone PM, Gan J, Avendano G. K+-evoked depolarization stimulates cyclic AMP accumulation in photoreceptor-enriched retinal cell cultures: role of calcium influx through dihydropyridine-sensitive calcium channels. J Neurochem. 1991;57:615–621. doi: 10.1111/j.1471-4159.1991.tb03792.x. [DOI] [PubMed] [Google Scholar]
- 29.Jean D, Harbison M, McConkey DJ, Ronai Z, Bar-Eli M. CREB and its associated proteins act as survival factors for human melanoma cells. J Biol Chem. 1998;273:24884–24890. doi: 10.1074/jbc.273.38.24884. [DOI] [PubMed] [Google Scholar]
- 30.Kalderon D, Roberts BL, Richardson WD, Smith AE. A short amino acid sequence able to specify nuclear location. Cell. 1984;39:499–509. doi: 10.1016/0092-8674(84)90457-4. [DOI] [PubMed] [Google Scholar]
- 31.Kalix P, Roch P. Evidence of depolarization-induced cAMP increase in the superior cervical ganglion of several mammalian species. Gen Pharmacol. 1976;7:267–270. doi: 10.1016/0306-3623(76)90087-2. [DOI] [PubMed] [Google Scholar]
- 32.Kim MS, Chung W-H, Cho Y-S, Shin D-B, Bang E-G, Lyoo SH, Hong SH. Effect of neurotrophins and depolarization on survival of spiral ganglion neurons in dissociated culture. Korean J Otolaryngol. 2001;44:1133–1139. [Google Scholar]
- 33.Kwok RPS, Lundblad JR, Chrivia JC, Richards JP, Bächinger HP, Brennan RG, Roberts SGE, Green MR, Goodman RH. Nuclear protein CBP is a co-activator for the transcription factor CREB. Nature. 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- 34.Leake PA, Hradek GT, Snyder RL. Chronic electrical stimulation by a cochlear implant promotes survival of spiral ganglion neurons after neonatal deafness. J Comp Neurol. 1999;412:543–562. doi: 10.1002/(sici)1096-9861(19991004)412:4<543::aid-cne1>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 35.Li M, Wang X, Meintzer MK, Laessig T, Birnbaum MJ, Heidenreich KA. Cyclic AMP promotes neuronal survival by phosphorylation of glycogen synthase kinase 3. Mol Cell Biol. 2000;20:9356–9363. doi: 10.1128/mcb.20.24.9356-9363.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lizcano JM, Morrice N, Cohen P. Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser155. Biochem J. 2000;349:547–557. doi: 10.1042/0264-6021:3490547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lonze BE, Riccio A, Cohen S, Ginty DD. Apoptosis, axonal growth defects, and degeneration of peripheral neurons in mice lacking CREB. Neuron. 2002;34:371–385. doi: 10.1016/s0896-6273(02)00686-4. [DOI] [PubMed] [Google Scholar]
- 38.Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–819. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- 39.Meyer-Franke A, Wilkinson GA, Kruttgen A, Hu M, Munro E, Hanson MG, Jr, Reichardt LF, Barres BA. Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron. 1998;21:681–693. doi: 10.1016/s0896-6273(00)80586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Michel PP, Agid Y. Chronic activation of the cyclic AMP signaling pathway promotes development and long-term survival of mesencephalic dopaminergic neurons. J Neurochem. 1996;67:1633–1642. doi: 10.1046/j.1471-4159.1996.67041633.x. [DOI] [PubMed] [Google Scholar]
- 41.Miller AL. Effects of chronic stimulation on auditory nerve survival in ototoxically deafened animals. Hear Res. 2001;151:1–14. doi: 10.1016/s0378-5955(00)00226-4. [DOI] [PubMed] [Google Scholar]
- 42.Miller TM, Tansey MG, Johnson EM, Jr, Creedon DJ. Inhibition of phosphatidylinositol 3-kinase activity blocks depolarization- and insulin-like growth factor I-mediated survival of cerebellar granule cells. J Biol Chem. 1997;272:9847–9853. doi: 10.1074/jbc.272.15.9847. [DOI] [PubMed] [Google Scholar]
- 43.Montminy MR, Gonzalez GA, Yamamoto KY. Regulation of cAMP-inducible genes by CREB. Trends Neurosci. 1990;13:184–188. doi: 10.1016/0166-2236(90)90045-c. [DOI] [PubMed] [Google Scholar]
- 44.Nakao N. An increase in intracellular levels of cyclic AMP produces trophic effects on striatal neurons developing in culture. Neuroscience. 1998;82:1009–1020. doi: 10.1016/s0306-4522(97)00349-7. [DOI] [PubMed] [Google Scholar]
- 45.Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- 46.Rydel RE, Greene LA. cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc Natl Acad Sci USA. 1988;85:1257–1261. doi: 10.1073/pnas.85.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sable CL, Filippa N, Hemmings B, Van Obberghen E. cAMP stimulates protein kinase B in a wortmannin-insensitive manner. FEBS Lett. 1997;409:253–257. doi: 10.1016/s0014-5793(97)00518-8. [DOI] [PubMed] [Google Scholar]
- 48.Sarmiere PD, Freeman RS. Analysis of the NF-κB and PI 3-kinase/Akt survival pathways in nerve growth factor-dependent neurons. Mol Cell Neurosci. 2001;18:320–331. doi: 10.1006/mcne.2001.1021. [DOI] [PubMed] [Google Scholar]
- 49.Shen S, Wiemelt AP, McMorris FA, Barres BA. Retinal ganglion cells lose trophic responsiveness after axotomy. Neuron. 1999;23:285–295. doi: 10.1016/s0896-6273(00)80780-1. [DOI] [PubMed] [Google Scholar]
- 50.Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- 51.Shirakawa F, Mizel SB. In vitro activation and nuclear translocation of NF-κB catalyzed by cyclic AMP-dependent protein kinase and protein kinase C. Mol Cell Biol. 1989;9:2424–2430. doi: 10.1128/mcb.9.6.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Struthers RS, Vale WW, Arias C, Sawchenko PE, Montminy MR. Somatotroph hypoplasia and dwarfism in transgenic mice expressing a non-phosphorylatable CREB mutant. Nature. 1991;350:622–624. doi: 10.1038/350622a0. [DOI] [PubMed] [Google Scholar]
- 53.Tan Y, Demeter MR, Ruan H, Comb MJ. BAD Ser-155 phosphorylation regulates BAD/Bcl-XL interaction and cell survival. J Biol Chem. 2000;275:25865–25869. doi: 10.1074/jbc.M004199200. [DOI] [PubMed] [Google Scholar]
- 54.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 55.Virdee K, Parone PA, Tolkovsky AM. Phosphorylation of the pro-apoptotic protein BAD on serine 155, a novel site, contributes to cell survival. Curr Biol. 2000;10:1151–1154. doi: 10.1016/s0960-9822(00)00702-8. [DOI] [PubMed] [Google Scholar]
- 56.Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- 57.Walsh DA, Ashby CD, Gonzalez C, Calkins D, Fischer EH. Krebs EG: purification and characterization of a protein inhibitor of adenosine 3′,5′-monophosphate-dependent protein kinases. J Biol Chem. 1971;246:1977–1985. [PubMed] [Google Scholar]
- 58.Walton KM, Rehfuss RP, Chrivia JC, Lochner JE, Goodman RH. A dominant repressor of cyclic adenosine 3′, 5′-monophosphate (cAMP)-regulated enhancer-binding protein activity inhibits the cAMP-mediated induction of the somatostatin promoter in vivo. Mol Endocrinol. 1992;6:647–655. doi: 10.1210/mend.6.4.1350057. [DOI] [PubMed] [Google Scholar]
- 59.Wen W, Harootunian AT, Adams SR, Feramisco J, Tsien RY, Meinkoth JL, Taylor SS. Heat-stable inhibitors of cAMP-dependent protein kinase carry a nuclear export signal. J Biol Chem. 1994;269:32214–32220. [PubMed] [Google Scholar]
- 60.Whitehouse S, Walsh DA. Inhibitor protein of the cAMP-dependent protein kinase: characteristics and purification. Methods Enzymol. 1983;99:80–93. doi: 10.1016/0076-6879(83)99044-4. [DOI] [PubMed] [Google Scholar]
- 61.Wiley JC, Wailes LA, Idzerda RL, McKnight GS. Role of regulatory subunits and protein kinase inhibitor (PKI) in determining nuclear localization and activity of the catalytic subunit of protein kinase A. J Biol Chem. 1999;274:6381–6387. doi: 10.1074/jbc.274.10.6381. [DOI] [PubMed] [Google Scholar]
- 62.Wilson BE, Mochon E, Boxer LM. Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol. 1996;16:5546–5556. doi: 10.1128/mcb.16.10.5546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu J, Michel H, Rossomando A, Haystead T, Shabanowitz J, Hunt DF, Sturgill TW. Renaturation and partial peptide sequencing of mitogen-activated protein kinase (MAP kinase) activator from rabbit skeletal muscle. Biochem J. 1992;285:701–705. doi: 10.1042/bj2850701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu L, Lavinsky RM, Dasen JS, Flynn SE, McInerney EM, Mullen TM, Heinzel T, Szeto D, Korzus E, Kurokawa R, Aggarwal AK, Rose DW, Glass CK, Rosenfeld MG. Signal-specific co-activator domain requirements for Pit-1 activation. Nature. 1998;395:301–306. doi: 10.1038/26270. [DOI] [PubMed] [Google Scholar]
- 65.Yang CH, Murti A, Pfeffer SR, Basu L, Kim JG, Pfeffer LM. IFNα/β promotes cell survival by activating NF-κB. Proc Natl Acad Sci USA. 2000;97:13631–13636. doi: 10.1073/pnas.250477397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zanger K, Cohen LE, Hashimoto K, Radovick S, Wondisford FE. A novel mechanism for cyclic adenosine 3′,5′-monophosphate regulation of gene expression by CREB-binding protein. Mol Endocrinol. 1999;13:268–275. doi: 10.1210/mend.13.2.0245. [DOI] [PubMed] [Google Scholar]
- 67.Zha X-M, Bishop JF, Hansen MR, Victoria L, Abbas PJ, Mouradian MM, Green SH. BDNF synthesis in spiral ganglion neurons is constitutive and CREB-dependent. Hear Res. 2001;156:53–68. doi: 10.1016/s0378-5955(01)00267-2. [DOI] [PubMed] [Google Scholar]
- 68.Zhou XM, Liu Y, Payne G, Lutz RJ, Chittenden T. Growth factors inactivate the cell death promoter BAD by phosphorylation of its BH3 domain on ser155. J Biol Chem. 2000;275:25046–25051. doi: 10.1074/jbc.M002526200. [DOI] [PubMed] [Google Scholar]