

Abstract
Recently it was demonstrated that exposure of the developing brain during the period of synaptogenesis to drugs that block NMDA glutamate receptors or drugs that potentiate GABAA receptors can trigger widespread apoptotic neurodegeneration. All currently used general anesthetic agents have either NMDA receptor-blocking or GABAA receptor-enhancing properties. To induce or maintain a surgical plane of anesthesia, it is common practice in pediatric or obstetrical medicine to use agents from these two classes in combination. Therefore, the question arises whether this practice entails significant risk of inducing apoptotic neurodegeneration in the developing human brain. To begin to address this problem, we have administered to 7-d-old infant rats a combination of drugs commonly used in pediatric anesthesia (midazolam, nitrous oxide, and isoflurane) in doses sufficient to maintain a surgical plane of anesthesia for 6 hr, and have observed that this causes widespread apoptotic neurodegeneration in the developing brain, deficits in hippocampal synaptic function, and persistent memory/learning impairments.
Keywords: NMDA antagonists, GABA agonists, isoflurane, midazolam, nitrous oxide, apoptosis
Introduction
Advances in pediatric and obstetric surgery have resulted in an increased complexity, duration, and number of anesthesia procedures. To minimize risks, it is necessary to understand the effects of anesthetic drugs on the developing nervous system. Presently used anesthetics act by two principal mechanisms: (1) an increase in inhibition via GABAA receptors (e.g., benzodiazepines, barbiturates, propofol, etomidate, isoflurane, enflurane, and halothane) (Franks and Lieb, 1994), and (2) a decrease in excitation through NMDA receptors [e.g., ketamine, nitrous oxide (N2O), and xenon] (Lodge and Anis, 1982; Franks et al., 1998; Jevtovic-Todorovic et al., 1998; Mennerick et al., 1998). Recent findings indicate that drugs that act by either of these mechanisms induce widespread neuronal apoptosis in immature rat brain when administered during synaptogenesis (Ikonomidou et al., 1999, 2000;Ishimaru et al., 1999). In addition, ethanol, an agent with both NMDA antagonist and GABAmimetic properties, triggers a widespread pattern of apoptotic neurodegeneration in the developing rat brain, which is a composite of the patterns induced by NMDA antagonist and GABAmimetic drugs (Ikonomidou et al., 1999). These findings raise questions regarding the potential risk posed by currently used anesthesia protocols. To address this issue, we studied the histopathological, electrophysiological, and behavioral effects of exposure of 7-d-old rats to N2O, isoflurane, and midazolam, three agents commonly used in obstetric and pediatric anesthesia.
Materials and Methods
Animals. Seven-day-old male and female Sprague Dawley rats were used for all experiments. At postnatal day 7 (P7), experimental rats were exposed to 6 hr of anesthesia, and controls were exposed to 6 hr of mock anesthesia. They were then allowed to recover and were divided into three randomly selected groups. One group was used for histopathology studies at several acute postanesthesia intervals. The second group was used for behavioral studies, which involved evaluating the rats on several tests over a 160 d period. The third group was used to study long-term potentiation (LTP) in hippocampal slices at P29–P33. Daily inspection and weighing was performed on the latter two groups at P6–P21 to evaluate general health and development.
Anesthesia treatment. N2O and oxygen were delivered using a calibrated flowmeter. Midazolam was dissolved in 10% DMSO immediately before administration. For control experiments, 10% DMSO was used. To administer specific concentrations of N2O, both normobaric [1 atmosphere (atm), 100 vol%] and hyperbaric conditions were required. In experiments requiring hyperbaric conditions, the N2O/oxygen mixture was introduced at a pressure of 2.0 atm (200 vol% = 150 vol% N2O and 50 vol% oxygen) and sustained for the duration of the experiment (Mahmoudi et al., 1989; Gonsowski and Eger, 1994). A relief valve on the hyperbaric chamber allowed continuous escape of gases to avoid accumulation of carbon dioxide. Animals were kept normothermic throughout experiments. For N2O concentrations of <80 vol%, normobaric conditions were used. The N2O/oxygen mixture was delivered to the chamber while a relief valve was kept open so that pressure inside the chamber remained at 1 atm. For control experiments, air was substituted for the gas mixtures. For experiments with isoflurane, we used an agent-specific vaporizer that delivers a set percentage of anesthetic into the chamber. After initial equilibration of the N2O/oxygen/isoflurane or air/isoflurane atmosphere inside the chamber, the composition of the chamber gas was analyzed by mass spectrometry for N2O or nitrogen, isoflurane, carbon dioxide, and oxygen concentration. All experiments were approved by The Animal Use and Care Committees of the University of Virginia and Washington University School of Medicine.
Arterial blood gas analysis. To determine adequacy of ventilation, arterial blood was sampled at the end of anesthesia by obtaining a single sample (100 μl) from the left cardiac ventricle using a 32 gauge hypodermic needle. Bicarbonate concentration (millimoles per liter), oxygen saturation (%), pH, paCO2 (partial pressure of carbon dioxide in mmHg), and paO2 (partial pressure of oxygen in mmHg) were measured immediately after blood collection, using a Nova Biomedical blood gas apparatus. Control samples were obtained from air/DMSO-treated pups.
Histopathological studies. All pups were deeply anesthetized and perfused with aldehyde fixatives for histopathology studies of the brains. We have found that activated caspase-3 [immunocytochemical (ICC)] is an excellent method for marking neurons that are in an early stage of apoptosis (Olney et al., 2002a,b), and DeOlmos silver staining is very useful for mapping patterns of cell death in the developing brain (Ikonomidou et al., 1999, 2000). To confirm the apoptotic nature of the cell-death process, electron microscopy is the most reliable available means (Dikranian et al., 2001). Pups used for studying caspase-3 activation or for silver staining were perfused with a mixture of paraformaldehyde (4%) in Tris buffer, pH 7.4, either 2 hr (caspase) or 18 hr (silver) after cessation of anesthesia, whereas those used for electron microscopy studies were perfused with a mixture of paraformaldehyde (4%) and glutaraldehyde (1.5%) in cacodylate buffer, pH 7.4, at 10 hr after anesthesia.
For activated caspase-3 ICC, 50-μm-thick vibratome sections were processed by procedures described recently (Olney et al., 2002b), using a primary anti-active caspase-3 antiserum raised in rabbits (D175; Cell Signaling Technology, Beverly, MA). For silver staining, the fixed brains were cut by vibratome into 50 μm sections and stained by the DeOlmos cupric silver method, as described previously (DeOlmos and Ingram, 1971; Corso et al., 1997). For electron microscopy, ultrathin sections were stained with uranyl acetate/lead citrate and viewed in a 100C Jeol (Peabody, MA) electron microscope.
To determine the degree of neurodegeneration in a given brain region, we used the optical dissector and fractionator method (West, 1999). A counting frame (0.05 × 0.05 mm; dissector height, 0.05 mm) and a high numerical aperture objective lens were used to visualize neurons. Unbiased sampling of each brain region was performed by randomly selecting 10–12 viewing fields over which the counting frame was positioned for counting at different focal levels. The numerical density of degenerating neurons in any given region was determined by counting argyrophilic profiles in 50-μm-thick sections stained by the DeOlmos silver method. Differences between anesthetic-treated and control values for a given brain region were evaluated statistically by Student's t test. To obtain an estimate of the severity of induced degeneration in the anesthetic-treated brains, the numerical density values for the anesthetic-treated animals were divided by those for the controls. The result was expressed as a “fold” increase over baseline, baseline being the rate at which neuronal cell death was occurring as a natural phenomenon in control brains. Counting was done by an experienced histopathologist who was unaware of the treatment condition.
Electrophysiological studies. Rats exposed for 6 hr to anesthetic agents at P7 were killed at P29–P33 for preparation of hippocampal slices using standard methods (Zorumski et al., 1996). At the time of study, slices were placed in a submersion recording chamber at 30°C. Extracellular recordings were obtained from the apical dendritic region for analysis of population EPSPs using 2m NaCl electrodes. Evoked responses were elicited with 0.2 msec constant-current pulses through a bipolar electrode in the Schaffer collateral pathway (CA1 hippocampal field) every 30–60 sec at an intensity sufficient to elicit 50% maximal EPSPs. After establishing a stable baseline, LTP was induced by applying a single 100 Hz × 1 sec stimulus train using the same amplitude current.
Behavioral studies. Rats exposed to the anesthetic “triple cocktail” or DMSO vehicle on P7 were evaluated behaviorally at subsequent ages using the following measures [similar to those described in greater detail by Wozniak et al. (1989, 1990, 1991), Ho et al. (2000), and Hartman et al. (2001)]: (1) Ascent test at P10, P12, and P14 (to assess the acute response to drug treatment); (2) auditory/tactile startle and prepulse inhibition (PPI) of startle at P20; (3) a 1 hr locomotor activity test in a home-cage environment at P21; and (4) a sensorimotor battery of tests at P22, including walking initiation, ledge walking, inclined plane performance, and elevated platform performance. (5) Spatial reference memory was evaluated using the Morris water navigation test which included cued (P28), place (P32 and P131), and probe trials. (6) Spatial working memory was assessed at P53, using a win-shift spatial discrimination protocol in the radial arm maze.
Water navigation testing was conducted as two separate studies, each involving different groups of experimental and control rats (n = 9–11 per group). Except as is explained below, the same conditions were used for both studies, as follows: rats were tested in a small pool (100 cm inner diameter) as juveniles and in a larger pool (180 cm inner diameter) as adults. In the cued trials, rats were tested for their ability to swim to a visible platform that was switched to a new location for each trial. The place condition involved testing the rats' ability to learn the location of a platform (submerged, not visible) which remained in the same location for all trials. When subjected to place testing as juveniles (P32), the rats were given two blocks of trials (two trials per block) each day for 5 d, with an intertrial interval of 30 sec. When the rats were retested as adults (P131), using a new platform location, a protocol involving one block of trials (two trials per block) each day for 5 d was used in an effort to increase task difficulty and improve test sensitivity. Study 2 was conducted exactly like study 1, except that after 5 d of place testing as adults, rats in study 2 were tested for an additional 5 d to determine whether they could progressively improve their performance to an asymptotic level, to provide evidence of learning. Probe trials were conducted after the last place trial of juvenile testing and after the last place trials of blocks 5 and 10 during adult testing. Probe trials involved removing the platform and evaluating the rats' search patterns for the missing platform by quantifying time spent in the target quadrant and number of platform crossings.
The rats in both study 1 and study 2 were tested in the radial arm maze at P53 according to an identical win-shift spatial discrimination (working memory) protocol in each study.
Data analyses. The data from each behavioral test in both study 1 and study 2 were subjected to an ANOVA involving treatment and study as between-subjects variables. In no case was a treatment by study interaction found; therefore, for additional analysis, the data from studies 1 and 2 were combined (and the variable study was deleted), except for data pertaining to the last 5 d of place testing as adults, which were generated only in study 2. Data from the preweaning tests were typically analyzed using ANOVA models that involved treatment and litter as between-subjects variables and test sessions as a within-subjects variable. Data from postweaning tests were typically analyzed using ANOVA models with treatment and gender as between-subjects variables and blocks of trials as a within-subjects variable. Pairwise comparisons were conducted after significant effects of treatment or significant interactions involving treatment and other relevant variables, and p values exceeding Bonferroni corrected levels were noted when appropriate by the symbol †. The Huynh–Feldt statistic was used to adjust p values to help protect against violations of compound symmetry when more than two levels of a within-subjects variable were used in an ANOVA model.
Results
Neonatal anesthetic treatment does not induce metabolic or respiratory distress
To assess the effects of selected anesthetics on the developing brain, we exposed P7 rats to midazolam, isoflurane, and N2O, either individually or in clinically relevant combinations, for 6 hr. In animals exposed for 6 hr to these drugs, either individually or in combination, including the triple cocktail (isoflurane plus N2O plus midazolam) (Table 1), there were no signs of metabolic or respiratory distress. Oxygen saturation, paO2, paCO2, and pH did not differ significantly from control animals exposed to air plus vehicle for 6 hr. A slight increase in pH in both groups is attributable to transient hyperventilation caused by cardiac puncture.
Table 1.
Arterial blood gas analysis
| Arterial blood gas | Controls (n = 8) | Treated (n = 8) |
|---|---|---|
| pH | 7.52 ± 0.05 | 7.49 ± 0.02 |
| paCO2 (mmHg) | 30 ± 8 | 29 ± 2.7 |
| paO2 (mmHg) | 117 ± 13 | 93 ± 11 |
| HCO3− (mmol/l) | 23 ± 3 | 22 ± 1.5 |
| SaO2 (%) | 99 ± 0.3 | 96 ± 1.1 |
Neonatal anesthetic treatment induces extensive apoptotic neurodegeneration
In vehicle-treated animals (10% DMSO plus air), both cupric silver and activated caspase-3 ICC staining revealed a sparsely scattered pattern of baseline physiological cell death (Fig.1a,f,h,j). Animals treated with N2O alone (50, 75, or 150 vol%) or midazolam alone (3, 6, or 9 mg/kg, i.p.) showed no significant increase in apoptotic neurodegeneration compared with control animals (data not shown). However, animals that were treated with isoflurane alone (0.75, 1.0, or 1.5 vol%) exhibited dose-dependent neurodegeneration (p < 0.05). The most vulnerable brain regions were the laterodorsal and anteroventral thalamic nuclei, where even the lowest isoflurane concentration (0.75%) caused a significant increase in neuronal degeneration (16-fold and ninefold increase, respectively). The parietal cortex (layer II) was also affected in an apparently dose-dependent manner, although the damage was significantly greater than controls at only the highest isoflurane concentration (1.5 vol%) (p< 0.05).
Fig. 1.
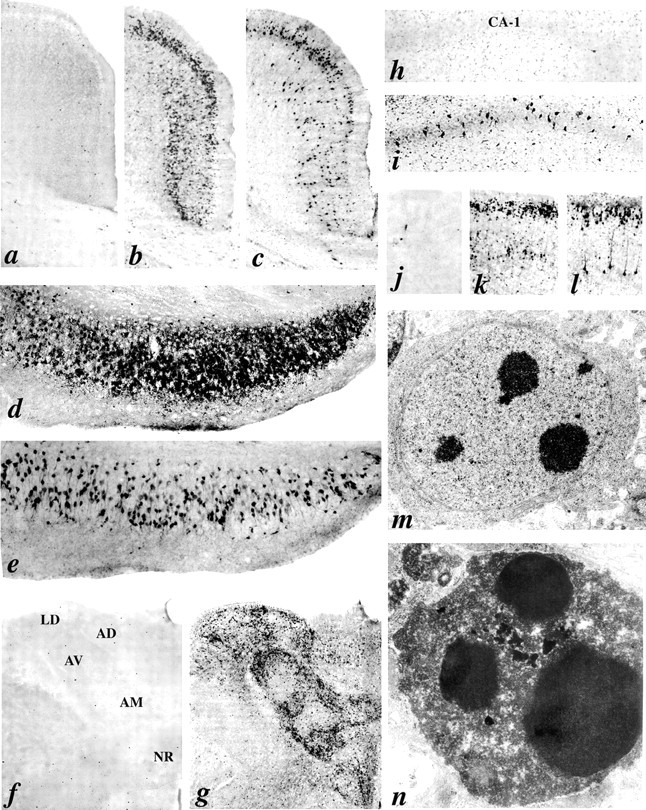
Triple anesthetic cocktail induces apoptotic neurodegeneration. a–l are light micrographic scenes from various brain regions of either a control rat (a,f, h, j) or a rat exposed to the triple anesthetic cocktail (b–e,g, i, k,l). Some sections are stained by the DeOlmos silver method (a, b, d,f, g, k), and the remainder are immunocytochemically stained to reveal caspase-3 activation (c, e, h–j,l). The regions illustrated are the posterior cingulate/retrosplenial cortex (a–c), subiculum (d, e), anterior thalamus (f, g), rostral CA1 hippocampus (h, i), and parietal cortex (j–l). The individual nuclei shown in the anterior thalamus (f, g) are laterodorsal (LD), anterodorsal (AD), anteroventral (AV), anteromedial (AM), and nucleus reuniens (NR).m and n are electron micrographic scenes depicting the ultrastructural appearance of neurons undergoing apoptosis. The cell in m displays a very early stage of apoptosis in which dense spherical chromatin balls are forming in the nucleus at a time when the nuclear membrane remains intact and very few changes are evident in the cytoplasm. The cell in nexhibits a much later stage of apoptosis in which the entire cell is condensed, the nuclear membrane is absent, and there is intermixing of nuclear and cytoplasmic constituents. These are hallmark characteristics of neuronal apoptosis as it occurs in the in vivo mammalian brain.
When a nontoxic dose of midazolam (9 mg/kg, i.p.) was followed by 6 hr of a low/minimally toxic concentration of isoflurane (0.75 vol%), there was a significant increase in apoptotic neurodegeneration compared with this concentration of isoflurane alone. Under this condition (double GABAmimetic cocktail), the damage was evident primarily in the laterodorsal and anterodorsal thalamus and parietal cortex (layer II). However, when 6 hr of N2O at a low/nontoxic concentration (75 vol%) was added to the above double cocktail, this triple cocktail (midazolam plus 6 hr of N2O plus isoflurane) resulted in a robust neurodegenerative reaction entailing more severe damage in the thalamus and parietal cortex than was caused by the double cocktail, and also producing moderate to severe damage in many other brain regions. It warrants emphasis that all triple cocktail-exposed pups invariably sustained brain damage, and that the pattern of damage was identical in each of these brains. We have compiled a comprehensive list of the damaged brain regions (Table 2), and for each region the severity of neurodegeneration in anesthesia-exposed brains is rated in terms of how many fold greater it is compared with the baseline rate in the same region of control brain (the baseline rate being that attributable to physiological cell death that occurs naturally in the developing brain). This list (Table 2) includes only those brain regions, or subfields within specific regions, where the rate of degeneration was at least 15-fold greater than the control rate. In all of these regions, the numerical density of degenerating neurons in the anesthetic cocktail rats was significantly greater than the control value at a level ofp < 0.001.
Table 2.
Brain regions in which anesthesia-induced neurodegeneration was most heavily concentrated
| Brain region | Severity (fold increase) |
|---|---|
| Medial septal nucleus | 27 |
| Diagonal band of Broca | 29 |
| Nucleus accumbens | 33 |
| Rostral caudate nucleus | 26 |
| Globus pallidus | 43 |
| Amygdaloid nuclei | |
| Basolateral | 22 |
| Medial | 25 |
| Cortical | 23 |
| Thalamic nuclei | |
| Paraventricular | 34 |
| Anterodorsal | 28 |
| Anteroventral | 57 |
| Anteromedial | 51 |
| Laterodorsal | 68 |
| Reuniens | 34 |
| Parafascicularis | 45 |
| Hippocampus, rostral CA1 | 21 |
| Subiculum | 58 |
| Cingulate cortex | 32 |
| Retrosplenial cortex | 53 |
| Neocortex (layers II and IV) | |
| Frontal | 18 |
| Parietal | 34 |
| Temporal | 22 |
| Occipital | 35 |
| Hypothalamus | |
| Anterior | 22 |
| Ventromedial | 34 |
| Dorsomedial | 21 |
| Mammillary complex | 33 |
Severity is expressed as the fold increase (i.e., how many times greater the density of degenerating neurons was in the experimental brain compared with the baseline rate of degeneration in the same region of control brain).
The two histological methods used (activated caspase-3 ICC and silver staining) to evaluate the neurodegenerative reaction in experimental brains compared with controls were mutually confirmatory regarding the pattern of neurodegeneration induced by the triple cocktail (Fig.1a–l). Electron microscopic evaluation confirmed that the degenerating neurons displayed classical morphological changes characteristic of apoptosis (Fig. 1m,n).
Triple anesthetic cocktail induces profound LTP suppression
To assess long-term electrophysiological effects of early anesthetic exposure, we examined synaptic function and LTP in hippocampal slices prepared at P29–P33 from rats treated at P7 with 10% DMSO (controls), N2O, isoflurane, midazolam, or a combination of the three agents. Baseline EPSPs elicited by single stimuli did not differ among the five groups (data not shown). LTP induction was unchanged in DMSO-treated controls compared with naive controls [changes in EPSP slope 60 min after tetanus in naive and DMSO animals were 144.1 ± 7.9% of baseline (n = 6) and 139.4 ± 6.3% of baseline (n = 10), respectively]. A less robust LTP response was observed in rats treated with N2O (129.1 ± 5.9%; n= 8), isoflurane (125.1 ± 3.1%; n = 6), or midazolam (120.9 ± 2.2%; n = 8), but only midazolam-treated animals differed significantly from controls (p = 0.022 by t test). In contrast, slices from rats treated with the triple anesthetic cocktail exhibited profound suppression of LTP (105.0 ± 6.5% of baseline;n = 10; p = 0.001), despite the presence of robust short-term potentiation (Fig.2).
Fig. 2.

Effects of anesthetic exposure on LTP in the CA1 region of rat hippocampal slices. a, The graph depicts the time course of change in field EPSP slope (± SEM) in hippocampal slices from control rats treated with DMSO (●) and rats exposed to the triple anesthetic combination (triple c., ○). A single 100 Hz × 1 sec tetanus was delivered at time 0 (arrow). b, Traces to theright of the graph show examples of EPSPs before (solid traces) and 60 min after (dashed traces) the tetanus in slices from animals treated with the various anesthetics. Hippocampal slices were prepared at P29–P33 from rats treated at P7.
Triple anesthetic cocktail induces long-term impairment in spatial learning/memory
Because the anesthetic triple cocktail produced the most profound neurodegenerative effects, we decided to determine whether developmental exposure to this treatment also had lasting effects on behavior. To accomplish this, we assessed sensorimotor and cognitive function from early stages of development into adulthood and found that the anesthetic cocktail rats did not differ from controls in (1) overall growth as indexed by daily body weight measurements, (2) sensorimotor ability as measured by the ascent test (preweaning) and sensorimotor battery (postweaning), (3) acoustic or tactile startle reactivity or PPI as measured by startle/PPI testing, or (4) various movement-related variables, as measured during a 1 hr locomotor activity test.
When tested in the Morris water maze on P28, anesthetic cocktail and control rats performed similarly in terms of path length and latency when learning to swim to a visible platform during cued trials (data not shown). However, when we examined spatial reference memory capabilities in the same animals during place trials (submerged platform, fixed location) on P32, anesthetic cocktail rats showed significant acquisition deficits (Fig.3a). The deficit was evidenced by the anesthetic cocktail rats showing slower acquisition rates during the middle blocks of trials, although they improved and performed like controls by the end of training and also exhibited control-like performance levels during probe trials (data not shown). When retested on the water maze as adults (P131), using a more difficult place-learning protocol, anesthetic cocktail rats again demonstrated impaired performance during place trials (Fig. 3b) but, unlike the earlier findings, they also demonstrated impaired retention performance during probe trials. Specifically, they spent significantly less time searching the pool quadrant where the platform had been (time in target quadrant) (Fig. 3c) and crossed over the former platform location significantly fewer times than controls (data not shown). Moreover, the control rats from study 2 that were subjected to an additional 5 d of testing improved their performance to asymptotic levels, suggesting that learning occurred, whereas the anesthetic cocktail group from study 2 showed no such improvement (Fig.3b). In addition, the control rats showed significantly higher levels of retention during the probe trials in terms of target quadrant time (Fig. 3c) and platform crossings (data not shown). Swimming speeds were also analyzed during cued and place trials, and no differences were observed, further suggesting that swimming performance deficits were not responsible for the place-learning impairments in the anesthetic cocktail rats (data not shown).
Fig. 3.

Effects of neonatal triple anesthetic cocktail treatment on spatial learning. a, Rats were tested at P32 for their ability to learn the location of a submerged (not visible) platform. An ANOVA of the escape path length data yielded a significant main effect of treatment (p = 0.032) and a significant treatment by blocks of trials interaction (p = 0.024), indicating that the performance of the rats that received the anesthetic cocktail was significantly inferior to that of control rats during place training. Subsequent pairwise comparisons indicated that differences were greatest during blocks 4, 5, and 6 (p = 0.003, 0.012, and 0.019, respectively). However, the rats receiving the anesthetic cocktail improved their performance to control-like levels during the last four blocks of trials. b, Rats were retested as adults (P131) for their ability to learn a different location of the submerged platform. The graph on the left represents the path length data from the first five place trials when all rats were tested. An ANOVA of these data yielded a significant main effect of treatment (p = 0.013), indicating that the control rats, in general, exhibited significantly shorter path lengths in swimming to the platform compared with anesthetic cocktail rats. Subsequent pairwise comparisons showed that differences were greatest during block 4 (p = 0.001). The graph on theright shows the data from study 2 rats that received 5 additional training days as adults. During these additional trials, the control group improved their performance and appeared to reach asymptotic levels, whereas the anesthetic cocktail rats showed no improvement. An ANOVA of these data yielded a significant main effect of treatment (p = 0.045) as well as a significant treatment by blocks of trials interaction (p = 0.001). Additional pairwise comparisons showed that group differences were greatest during blocks 7, 8, and 10 (p = 0.032, 0.013, and 0.017, respectively).c, Probe trial performance of anesthetic cocktail and control rats during adult testing. Search behavior of the rats was quantified when the submerged platform was removed from the pool after the last place trials in blocks 5 and 10. The histogram on theleft presents data for rats of both studies 1 and 2 combined after five blocks of place trials were completed. The histogram on the right presents data for rats of study 2 alone, after 10 blocks of place trials were completed. Thedotted line represents the amount of time that animals would be expected to spend in the target quadrant based on chance alone. Both histograms show that the control rats spent significantly more time in the target quadrant than the anesthesia-exposed rats, regardless of whether the probe tests were performed on both study groups after five blocks or only on the study 2 rats after 10 trials.d, e, Data from the radial arm maze test performed on P53 to evaluate spatial working memory capabilities are shown.d, A histogram showing that the anesthetic cocktail rats required significantly more days to reach a criterion demonstrating learning (8 correct responses out of the first 9 responses for 4 consecutive days) compared with controls. e, Plotting the days to criterion data as the cumulative percentage of rats reaching criterion in each group as a function of blocks of training days shows that the acquisition rate of the anesthetic cocktail rats began to slow around the fourth block of trials and remained slower throughout the rest of the experiment. Numbers inparentheses in each graph indicate sample sizes. *p < 0.05; Bonferroni corrected level:†p < 0.005 in a;†p < 0.01 in b.
When spatial working memory capabilities were tested in the radial arm maze at P53, anesthetic cocktail rats were significantly impaired relative to controls (Fig. 3d) in terms of days required to reach a criterion demonstrating learning. Graphing these data in terms of the cumulative percentage of rats reaching criterion as a function of blocks of trials (Fig. 3e) reveals that the acquisition rate of the anesthetic cocktail rats began to slow compared with controls by the fourth block of trials and remained substantially slower for the remainder of training. The anesthetic cocktail rats also made a larger number of errors in reaching criterion, although this difference was marginally nonsignificant (p = 0.065; data not shown).
Discussion
Our findings indicate that exposure of infant rats to an anesthetic cocktail (midazolam, isoflurane, N2O) that is commonly used in pediatric anesthesia triggers apoptosis in several major brain regions, resulting in deletion of many neurons from the developing brain and residual learning/memory deficits, coupled with dysfunction of hippocampal synaptic mechanisms putatively associated with memory.
These findings are consistent with other recent evidence that apoptotic neurodegeneration can be induced in the developing rodent brain by (1) drugs that block NMDA receptors, (2) drugs that hyperactivate GABAA receptors, or (3) ethanol, which has both NMDA antagonist and GABAmimetic properties. It appears that more profound neurodegeneration is induced if both NMDA and GABAA receptors are simultaneously altered, in that a more robust and widespread neurodegenerative response is seen after exposure to ethanol than to either an NMDA antagonist or GABAmimetic drug by itself. This principle is further corroborated by the present demonstration that combining a nontoxic concentration of the NMDA antagonist N2O with GABAmimetic agents induced a much more severe and widespread pattern of neurodegeneration than was induced by either drug category by itself, even at substantially higher doses.
Our data indicate that exposure of infant rats to a clinically relevant anesthesia protocol for 6 hr during synaptogenesis causes not only acute deletion of many neurons from the developing brain but also learning/memory disabilities that persist into adolescence and adulthood. Animals exposed to this anesthesia protocol displayed deficits in spatial reference memory capabilities as manifested by slower place learning acquisition as juveniles and by significant impairments in both spatial reference and working memory as adults. The lack of differences between groups during the cued trials or with regard to swimming speeds as well as the absence of performance differences on a wide array of behavioral tests suggest that the learning/memory deficits in the anesthetic cocktail rats did not result from nonassociative performance factors such as sensorimotor disturbances, altered emotionality, or changes in motivation. Our additional finding that the anesthesia-exposed rats had lasting deficits in hippocampal synaptic function may help explain the learning deficits, in that the hippocampus is well known to play an important role in memory and learning. However, it would probably be simplistic to attribute these deficits solely to hippocampal damage, in view of substantial evidence (Aggleton and Brown, 1999; Mitchell et al., 2002) that fundamental memory functions are not mediated by the hippocampus alone, but rather by a distributed network that includes, in addition to the hippocampus, anterior thalamic nuclei, mammillary bodies, and retrosplenial cortex. Each of the latter three structures was damaged in the anesthesia-exposed brains more severely than the hippocampus.
The triple anesthetic drug protocol used in these experiments is one that is commonly used in pediatric anesthesia practice. We selected a dose of midazolam that is considered sedating (sedative dose is between 1 and 10 mg/kg, i.p., in rats) (Kissin et al., 1990) and followed this with only those concentrations of N2O and isoflurane that are required to induce and maintain a surgical plane of anesthesia. The minimum alveolar anesthetic concentration that prevents purposeful movement to supramaximal noxious stimulation in 50% of subjects (MAC) for either N2O or isoflurane in humans is ∼67% of the MAC in rats (Hornbein et al., 1982; Mahmoudi et al., 1989; Stevens and Kingston, 1992; Orliaguet et al., 2001). Thus, the concentrations of N2O (75%) and isoflurane (0.75%) used in our rat study would be comparable with exposing a human to 50% N2O and 0.5% isoflurane, concentrations that are well within clinically used ranges.
It has been shown in previous studies (Ikonomidou et al., 1999, 2000) that peak vulnerability to the apoptogenic action of NMDA antagonists and GABAmimetics is during the synaptogenesis period, also known as the brain growth-spurt period. This is a readily recognized period during which the brain grows at an accelerated rate because newly differentiated neurons throughout the brain are rapidly expanding their dendritic arbors to provide the required surface area to accommodate new synaptic connections. The brain growth spurt occurs in different mammalian species at different times relative to birth. In rats, it begins a day or two before birth and ends ∼2 weeks after birth, whereas in humans it starts at the beginning of the third trimester and ends several years after birth (Dobbing and Sands, 1979). Comparing the brain growth curves for rats and humans, the period of peak brain growth occurs in rats between the fourth and tenth postnatal days and in humans between the last month of gestation and first 6 months after birth. Although neurodevelopmental age equivalencies between rats and humans cannot be specified with precision, our decision to use 7-d-old rats for the present study was based on the assumption that this neurodevelopmental age in the rat is equivalent to the human age from 0 to 6 months after a term birth, or perhaps from 0 to ≥12 months after a premature birth. It is quite common, often out of necessity, but sometimes on an elective basis, for surgical procedures requiring general anesthesia to be performed on human infants in this neurodevelopmental age range.
Various anesthesia protocols have been used in pediatric medicine for many decades without clear evidence linking anesthesia exposure to subsequent neurobehavioral disturbances. Establishing such a link is rendered difficult by many confounding variables. For example, human infants who undergo general anesthesia often have a history of prematurity and/or adverse peripartum events, including prolonged exposure to sedatives or anticonvulsants in an intensive care unit. Moreover, linking neurobehavioral disturbances to perinatal drug exposure is difficult unless gross signs of dysmorphogenesis are present. For example, researchers were first alerted to the fetal alcohol syndrome by conspicuous dysmorphogenic effects (craniofacial malformations), and this led to the subsequent recognition that fetal exposure to ethanol can cause a wide range of neurobehavioral disturbances in the absence of dysmorphogenic effects (Streissguth and O'Malley, 2000). Our findings indicate that an anesthesia protocol that does not alter somatic development or induce sensorimotor impairments in rats does result in learning/memory deficits that are subtle enough to be easily overlooked. Additional animal research will be needed to address the potential developmental neurotoxicity of other clinically used anesthetic protocols. In addition, well designed clinical investigations are needed to assess the potential relevance of the animal findings to obstetric or pediatric anesthesia.
Footnotes
This work was supported in part by National Institutes of Health Grants AG 11355, DA 05072, HD 37100, AG 18434, MH 45493, and AA 12951 and Career Development Award K08 DA 00406 (V.J.-T.). We thank Adam Myenberg, Anna Pieper, and Anthony Nardi for technical assistance.
Correspondence should be addressed to Vesna Jevtovic-Todorovic, Department of Anesthesiology, University of Virginia Health System, P.O. Box 800710, Charlottesville, VA 22908. E-mail: vj3w@virginia.edu.
References
- 1.Aggleton JP, Brown MW. Episodic memory, amnesia, and the hippocampal-anterior thalamic axis. Behav Brain Sci. 1999;22:425–489. [PubMed] [Google Scholar]
- 2.Corso TD, Sesma MA, Tenkova TI, Der TC, Wozniak DF, Farber NB, Olney JW. Multifocal brain damage induced by phencyclidine is augmented by pilocarpine. Brain Res. 1997;33:533–541. doi: 10.1016/s0006-8993(96)01347-9. [DOI] [PubMed] [Google Scholar]
- 3.DeOlmos JS, Ingram WR. An improved cupric-silver method for impregnation of axonal and terminal degeneration. Brain Res. 1971;33:523–529. doi: 10.1016/0006-8993(71)90130-2. [DOI] [PubMed] [Google Scholar]
- 4.Dikranian K, Ishimaru MJ, Tenkova T, Labruyere J, Qin YQ, Ikonomidou C, Olney JW. Apoptosis in the in vivo mammalian forebrain. Neurobiol Dis. 2001;8:359–379. doi: 10.1006/nbdi.2001.0411. [DOI] [PubMed] [Google Scholar]
- 5.Dobbing J, Sands J. The brain growth spurt in various mammalian species. Early Hum Dev. 1979;3:79–84. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- 6.Franks NP, Lieb WR. Molecular and cellular mechanism of general anesthesia. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- 7.Franks NP, Dickinson R, deSousa SLM, Hall AC, Lieb WR. How does xenon produce anesthesia? Nature. 1998;396:324. doi: 10.1038/24525. [DOI] [PubMed] [Google Scholar]
- 8.Gonsowski CT, Eger EI., II Nitrous oxide minimum alveolar anesthetic concentration in rats is greater than previously reported. Anesth Analg. 1994;79:710–712. doi: 10.1213/00000539-199410000-00016. [DOI] [PubMed] [Google Scholar]
- 9.Hartman RE, Wozniak DF, Nardi A, Olney JW, Sartorious L, Holtzman DM. (2002) Behavioral phenotyping of GFAP-apoE3 and -apoE4 transgenic mice: ApoE4 mice show profound working memory impairments in the absence of Alzheimer's-like neuropathology. Exp Neurol. 2001;170:326–344. doi: 10.1006/exnr.2001.7715. [DOI] [PubMed] [Google Scholar]
- 10.Ho N, Liauw JA, Blaeser F, Wei F, Hanissian S, Muglia LM, Wozniak DF, Nardi A, Linden DJ, Zhuo M, Muglia LJ, Chatila TA. Impaired synaptic plasticity and CREB activation in CaMKIV/Gr-deficient mice. J Neurosci. 2000;20:6459–6472. doi: 10.1523/JNEUROSCI.20-17-06459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hornbein TF, Eger EI, II, Winter PM, Smith G, Wetstone D, Smith KH. The minimum alveolar concentration of nitrous oxide in man. Anesth Analg. 1982;61:553–556. [PubMed] [Google Scholar]
- 12.Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–74. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 13.Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 14.Ishimaru MJ, Ikonomidou C, Tenkova TI, Der TC, Dikranian K, Sesma MA, Olney JW. Distinguishing excitotoxic from apoptotic neurodegeneration in the developing rat brain. J Comp Neurol. 1999;408:461–476. [PubMed] [Google Scholar]
- 15.Jevtovic-Todorovic V, Todorovic SM, Mennerick S, Powell S, Dikranian K, Benshoff N, Zorumski CF, Olney JW. Nitrous oxide (laughing gas) is an NMDA antagonist, neuroprotectant, and neurotoxin. Nat Med. 1998;4:460–463. doi: 10.1038/nm0498-460. [DOI] [PubMed] [Google Scholar]
- 16.Kissin I, Brown PT, Bradley EL. (1990) Sedative and hypnotic midazolam-morphine interactions in rats. Anesth Analg. 1990;71:137–143. doi: 10.1213/00000539-199008000-00005. [DOI] [PubMed] [Google Scholar]
- 17.Lodge D, Anis NA. Effects of phencyclidine on excitatory amino acid activation of spinal interneurons in the cat. Eur J Pharmacol. 1982;77:203–204. doi: 10.1016/0014-2999(82)90022-x. [DOI] [PubMed] [Google Scholar]
- 18.Mahmoudi NW, Cole DJ, Shapiro HM. Insufficient anesthetic potency of nitrous oxide in the rat. Anesthesiology. 1989;70:345–349. doi: 10.1097/00000542-198902000-00027. [DOI] [PubMed] [Google Scholar]
- 19.Mennerick S, Jevtovic-Todorovic V, Todorovic SM, Shen W, Olney JW, Zorumski CF. Effect of nitrous oxide on excitatory and inhibitory synaptic transmission in hippocampal cultures. J Neurosci. 1998;18:9716–9726. doi: 10.1523/JNEUROSCI.18-23-09716.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell AS, Dalrymple-Alford JC, Christie MA. Spatial working memory and the brainstem cholinergic innervation to the anterior thalamus. J Neurosci. 2002;22:1922–1928. doi: 10.1523/JNEUROSCI.22-05-01922.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olney JW, Tenkova TI, Dikranian K, Muglia LJ, Jermakowicz WJ, D'Sa C, Roth KA. Ethanol-induced apoptotic neurodegeneration in the developing C57BL/6 mouse brain. Brain Res Dev Brain Res. 2002a;133:115–126. doi: 10.1016/s0165-3806(02)00279-1. [DOI] [PubMed] [Google Scholar]
- 22.Olney JW, Tenkova TI, Dikranian K, Qin YQ, Labruyere J, Ikonomidou C. Ethanol-induced caspase-3 activation in the in vivo developing mouse brain. Neurobiol Dis. 2002b;9:205–219. doi: 10.1006/nbdi.2001.0475. [DOI] [PubMed] [Google Scholar]
- 23.Orliaguet G, Vivien B, Langeron O, Bouhemad B, Coriat P, Riou B. Minimum alveolar concentration of volatile anesthetics in rats during postnatal maturation. Anesthesiology. 2001;95:734–739. doi: 10.1097/00000542-200109000-00028. [DOI] [PubMed] [Google Scholar]
- 24.Stevens WC, Kingston HGG. Inhalation anesthesia. In: Barash PG, Cullen BF, Stoelting RK, editors. Clinical anesthesia. Lippincott; Philadelphia: 1992. pp. 385–412. [Google Scholar]
- 25.Streissguth AP, O'Malley K. Neuropsychiatric implications and long-term consequences of fetal alcohol spectrum disorders. Semin Clin Neuropsychiatry. 2000;5:177–190. doi: 10.1053/scnp.2000.6729. [DOI] [PubMed] [Google Scholar]
- 26.West MJ. Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci. 1999;22:51–61. doi: 10.1016/s0166-2236(98)01362-9. [DOI] [PubMed] [Google Scholar]
- 27.Wozniak DF, Stewart GR, Finger S, Olney JW. Comparison of behavioral effects of nucleus basalis magnocellularis lesions and somatosensory cortex ablation in the rat. Neuroscience. 1989;32:685–700. doi: 10.1016/0306-4522(89)90290-x. [DOI] [PubMed] [Google Scholar]
- 28.Wozniak DF, Olney JW, Kettinger L, III, Price M, Miller JP. Behavioral effects of MK-801 in the rat. Psychopharmacology. 1990;101:47–56. doi: 10.1007/BF02253717. [DOI] [PubMed] [Google Scholar]
- 29.Wozniak DF, Cicero TJ, Kettinger L, III, Meyer ER. Paternal alcohol consumption in the rat impairs spatial learning performance in male offspring. Psychopharmacology. 1991;105:289–302. doi: 10.1007/BF02244324. [DOI] [PubMed] [Google Scholar]
- 30.Zorumski CF, Mennerick S, Izumi Y. Assessment of synaptic effects of nitric oxide in hippocampal neurons. Methods Neurosci. 1996;31:283–299. [Google Scholar]