

Abstract
Nitric oxide (NO) functions in several types of synaptic plasticity, including hippocampal long-term potentiation (LTP), in which it may serve as a retrograde messenger after postsynaptic NMDA receptor activation. In accordance with a prediction of this hypothesis, and with previous findings using guinea pig tissue, exogenous NO, when paired with a short tetanus (ST) to afferent fibers, generated a stable NMDA receptor-independent potentiation of rat CA1 hippocampal synaptic transmission that occluded LTP. Contrary to predictions, however, the pairing-induced potentiation was abolished in the presence of NO synthase inhibitors, indicating that endogenous NO is required for exogenous NO to facilitate LTP. Periodic application of NO while endogenous NO synthesis was blocked indicated that a tonic low level is necessary on both sides of the NO–ST pairing for the plasticity to occur. A similar dependence on tonic NO seems to extend to LTP, because application of an NO synthase inhibitor 5 min after tetanic stimulation blocked LTP as effectively as adding it beforehand. The posttetanus time window during which NO operated was restricted to <15 min. Inhibition of the guanylyl cyclase-coupled NO receptor indicated that the potentiation resulting from NO–ST pairing and the NO signal transduction pathway during early LTP are both through cGMP. We conclude that NO does not function simply as an acute signaling molecule in LTP induction but has an equally important role outside this phase. The results resonate with observations concerning the role of the hippocampal NO–cGMP pathway in certain types of learning behavior.
Keywords: nitric oxide, hippocampus, long-term potentiation, guanylyl cyclase, cGMP, synaptic plasticity
Introduction
Nitric oxide (NO) participates in several types of synaptic plasticity, including long-term depression (LTD) in the cerebellum and striatum and long-term potentiation (LTP) in the hippocampus and cerebral cortex (Garthwaite and Boulton, 1995;Daniel et al., 1998; Hawkins et al., 1998; Centonze et al., 1999). In cerebellar and striatal LTD, NO seems to be generated presynaptically or in interneurons and to act postsynaptically, whereas in hippocampal and cortical LTP, NO has been regarded as a retrograde messenger that is synthesized postsynaptically and acts on presynaptic terminals. Postsynaptic actions of NO in the hippocampus have also been documented (Ko and Kelly, 1999; Lu et al., 1999). In both LTP and LTD, NO signal transduction involves activation of the guanylyl cyclase-coupled receptor (NOGCR) (Garthwaite and Boulton, 1995). The ensuing accumulation of cGMP may then engage protein kinases to initiate phosphorylation cascades, leading, for example, to transcription factor activation (Lu et al., 1999). A few instances of cGMP-independent effects of NO contributing to synaptic plasticity also exist (Kleppisch et al., 1999; Jacoby et al., 2001; Lev-Ram et al., 2002).
In the hippocampal CA1 region, LTP is induced by brief tetanic stimulation of afferent glutamatergic fibers and is typically dependent on activation of postsynaptic NMDA receptors (Bliss and Collingridge, 1993). These receptors are physically associated with the Ca2+–calmodulin-dependent NO synthase through postsynaptic density-95 protein (Brenman and Bredt, 1997), an arrangement that allows Ca2+ influx through NMDA receptor channels to couple to NO formation. The rapid rates of diffusion of NO in lipid and aqueous environments enable it to function as a fast intercellular signaling molecule, and, according to the retrograde messenger hypothesis, synaptic specificity would be conferred by NO modifying only those terminals that have recently been active (Hawkins et al., 1998).
The finding that application of exogenous NO timed to coincide with weak tetanic stimulation of afferent fibers elicited an NMDA receptor-independent persistent potentiation of hippocampal synaptic transmission (Zhuo et al., 1993, 1994b; Malen and Chapman, 1997) is consistent with the retrograde messenger hypothesis. However, these studies did not consider a possible involvement of endogenous NO. This is an important omission, for two reasons. First, measurements of cGMP have shown that a biologically active level of NO exists even in unstimulated hippocampal slices (Chetkovich et al., 1993). Strictly speaking, therefore, the key prediction that exogenous NO should be able to substitute for endogenous NO in LTP has not been tested. Second, exogenous NO can elicit an enduring potentiation of CA1 hippocampal neurotransmission during low-frequency stimulation that occludes tetanus-induced LTP but that, paradoxically, requires the endogenous NO–cGMP pathway (Bon and Garthwaite, 2001a,b). This result raises the possibility that endogenous NO may play unsuspected roles in other forms of plasticity elicited by exogenous NO. Accordingly, in the present study, we sought to determine whether the endogenous NO–cGMP pathway contributes to the facilitation of LTP by exogenous NO and, in the light of the results, to reexamine the role of the endogenous pathway in LTP.
Materials and Methods
Tissue preparation. Experiments were performed on hippocampal slices from 6- to 8-week-old male Sprague Dawley rats. The slices were prepared as described previously (Bon and Garthwaite, 2001a) and maintained in oxygenated (95% O2 and 5% CO2) artificial CSF (aCSF) of the following composition (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 2 CaCl2, and 10d-glucose.
Electrophysiological recordings. Extracellular recordings of field EPSPs (fEPSPs) were made at 30°C from the stratum radiatum of the CA1 area after electrical stimulation of the Schaffer collateral–commissural pathway at a baseline frequency of 0.033 Hz as described previously (Bon and Garthwaite, 2001a). The short tetanus (ST) consisted of five pulses delivered at 50 Hz at the baseline stimulation voltage, whereas LTP was induced by a train of 100 shocks delivered at 100 Hz, also at the baseline voltage. The slope of the EPSP was measured in the region between 20 and 40% of the maximum, and the values were normalized relative to the mean values obtained during the first 15 min of recording in the absence of any treatment.
Stock solutions of drugs were made up as follows before being diluted at least 100-fold into the aCSF:d(−)-2-amino-5-phosphonopentanoic acid (d-AP-5) (Tocris Cookson, Bristol, UK), NMDA (Sigma, Poole, UK), and l-nitroarginine (l-NOArg) (Tocris Cookson) in 1 equivalent of NaOH; 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) (Tocris Cookson) in DMSO; andl-arginine (Sigma) andl-N5-(1-iminoethyl)-ornithine (l-NIO) (Alexis Corporation, Nottingham, UK) in distilled water. The NO donor 1,1-diethyl-2-hydroxy-2-nitroso-hydrazine sodium (DEA/NO) (Alexis Corporation) was prepared in 10 mm NaOH on the day of the experiment, kept on ice, and diluted at least 1000-fold into the aCSF immediately before use.
Measurement of cGMP. The levels of cGMP were determined as described previously (Boulton et al., 1994). Briefly, hippocampal slices were allowed to recover for 1–2 hr in gassed aCSF maintained at 30°C in a shaking water bath. The slices were then transferred to a fresh solution containing the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX) (1 mm). After 15 min, NMDA (300 μm) was added, and the slices were inactivated 2 min later by immersion in boiling hypotonic buffer for 2–3 min. When used, NO synthase inhibitors were present from 5 min before the IBMX until the end of the experiment. Control slices were treated identically except for the exposure to NMDA. The inactivated slices were homogenized by sonication, and the protein content was determined by the bicinchoninic acid method (Pierce, Rockford, IL) with bovine serum albumin as standard. After centrifugation, the cGMP content of the supernatant was determined by radioimmunoassay.
Statistics. Data are expressed as means ± SEM and were analyzed for statistical significance by the two-tailed, paired or unpaired t test; a value of p < 0.05 was considered statistically significant.
Results
Facilitation of LTP by NO
Initial experiments were performed in an attempt to reproduce results showing that NO could facilitate hippocampal LTP (Zhuo et al., 1993, 1994b). These experiments were performed in detail, partly because of apparent disagreements between different laboratories in analogous studies on the facilitation of LTP by cGMP derivatives (Zhuo et al., 1994a; Selig et al., 1996; Son et al., 1998) and partly because they are critical to the aims of the present work. Moreover, the previous results were obtained in guinea pig hippocampal slices rather than those of the rat, which were used here.
To supply NO, we perfused slices with the NONOate DEA/NO, which has quite a short half-life (∼6 min at 30°C) and delivers the authentic NO radical (Morley and Keefer, 1993). We chose to apply DEA/NO at a concentration of 3 μm, which elevates hippocampal slice cGMP levels to ∼70% of the maximum (Bon and Garthwaite, 2001b) but has no effect on the baseline fEPSPs elicited at 0.2 Hz (Bon and Garthwaite, 2001a). This concentration of DEA/NO also did not affect synaptic transmission at the lower baseline frequency used in the present experiments (0.033 Hz) (Fig.1A), and 30 min after washout, the slices were able to sustain normal LTP (144 ± 3% of baseline fEPSP slope) in response to brief high-frequency stimulation (HFS) (Fig. 1A).
Fig. 1.

Potentiation of CA1 hippocampal synaptic transmission by pairing exogenous NO with an ST.A, Lack of effect of DEA/NO (5 min perfusion) on baseline synaptic transmission (0.033 Hz stimulation). In this and subsequent figures, the administration of DEA/NO is indicated by ahorizontal bar, the progressive thinning of which reflects the exponential decline in DEA/NO concentration (half-life, 6 min). Thirty minutes later, the slices were given HFS (atarrow; n = 5). B, Facilitation of LTP by combined ST and DEA/NO. In each slice, an ST was delivered to the presynaptic fibers, and, 30 min later, a second ST was given in the absence or presence of DEA/NO, as indicated (n = 5–6). HFS was subsequently applied (atarrow) to all slices. C, Lack of potentiation in response to DEA/NO delivered 5 min after the ST (n = 4). The 40–50 sec delay in the perfusion system has not been corrected for in this and subsequent figures. Theinsets show representative fEPSPs (average of 4 consecutive traces) recorded in the presence or absence of DEA/NO at the times indicated by the letters.
An ST protocol (five pulses at 50 Hz) on its own produced only a short-term potentiation (STP). The same result was obtained when the protocol was repeated 30 min later (Fig. 1B). When slices were exposed to DEA/NO during the second ST, however, the fEPSP slope subsequently increased, to reach a plateau within 15 min (Fig.1B). Measured 30 min after the NO–ST pairing, the facilitation (137 ± 9% of control fEPSP slope) was significantly different from the preceding baseline value (104 ± 3%). After a subsequent HFS, no additional significant increase in fEPSP slope was observed (146 ± 3% 45 min later; p < 0.4 by two-tailed paired t test), signifying that the potentiation resulting from NO–ST pairing occluded LTP. In addition, the final level of potentiation was the same as that obtained in slices that had previously undergone ST twice without exposure to DEA/NO (145 ± 5%). Synergy between NO and the ST was required because, if DEA/NO was delivered 5 min after the ST, there was no change in fEPSP slope (102 ± 2% 30 min after washout) compared with the control value (101 ± 2%), although subsequent LTP was normal (143 ± 1%) (Fig. 1C).
The NMDA antagonist d-AP-5 (50 μm) inhibited the STP induced by ST, leaving only a transient posttetanic potentiation (Fig. 2A). This is as predicted (Malenka, 1991), but, bearing in mind analogous studies with cGMP (Son et al., 1998), the result provides an important positive control for the effectiveness of the antagonist under the conditions used. When the NO–ST pairing protocol was conducted in the presence of d-AP-5, the potentiation 30 min later (137 ± 6%) (Fig. 2B) was comparable with that obtained in the absence of d-AP-5 (137 ± 9%).
Fig. 2.

Facilitation of LTP by exogenous NO is independent of NMDA receptors. A, Positive control showing that the NMDA antagonist d-AP-5 inhibits the short-term potentiation induced by the ST (n = 3). B, Similar administration of d-AP-5 did not affect the potentiation of synaptic transmission brought about by giving an ST in the presence of DEA/NO (n = 4). Controls (−d-AP-5) were taken from Figure 1B. The insets show representative fEPSPs in the presence ofd-AP-5 (average of 4 consecutive traces) recorded in the presence of d-AP-5 at the times indicated by theletters.
Role of endogenous NO in LTP facilitation by exogenous NO
All the foregoing results accurately replicate those obtained in guinea pig hippocampal slices (Zhuo et al., 1993, 1994b). As a first step toward testing the role of endogenous NO in the LTP facilitation, slices were exposed to the standard NO synthase inhibitorl-NOArg at the concentration most often adopted for studies of synaptic plasticity (100 μm). In the presence of the inhibitor, the enduring potentiation produced by NO–ST pairing was abolished, whereas STP was preserved (Fig.3A). After 45 min of drug washout, a stable LTP occurred after HFS, although the final amplitude was less (122 ± 6%) than normal (146 ± 3%). Considering subsequent results (see below), the lower level of LTP would be explained by NO synthase inhibition by l-NOArg being only very slowly reversible (Dwyer et al., 1991), and thus, only the decaying NO-independent component would be available.
Fig. 3.
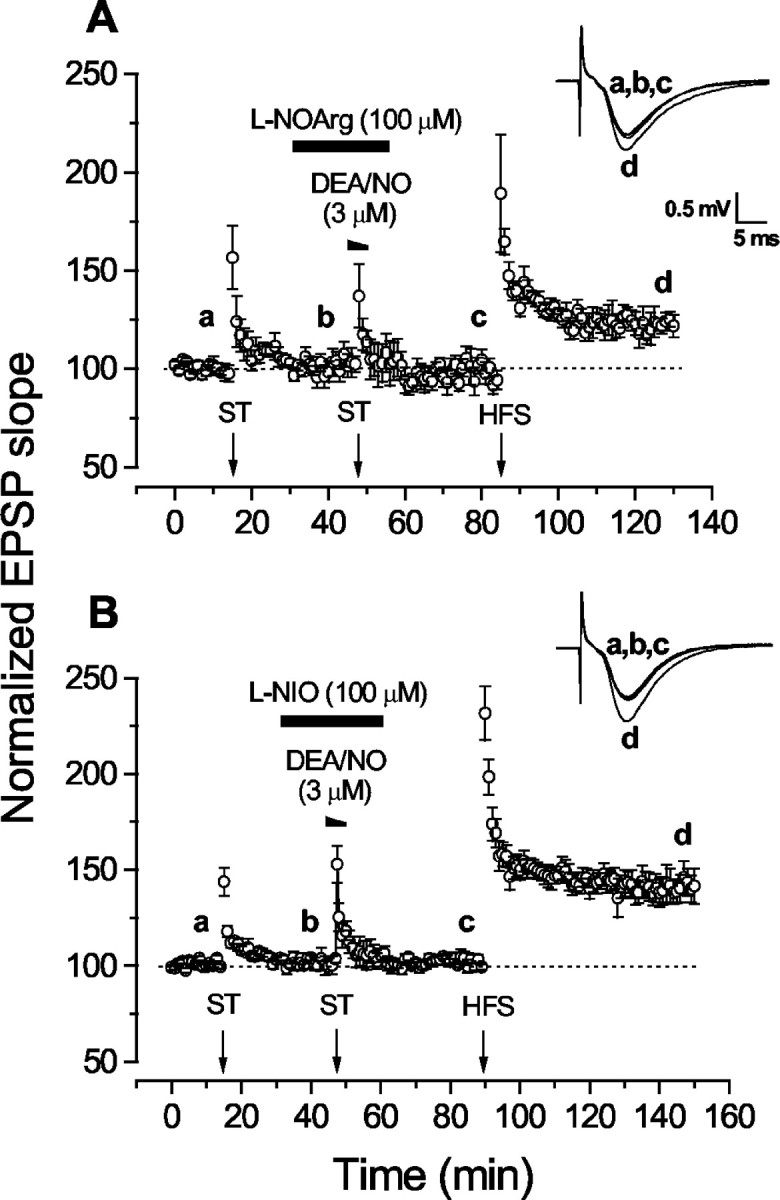
Effect of NO synthase inhibitors on LTP facilitation by exogenous NO. A, Block of facilitation by l-NOArg (n = 5). B, Block by l-NIO (n = 6). Theinsets show representative fEPSPs (average of 4 consecutive traces) recorded in the presence of l-NOArg orl-NIO at the times indicated by theletters.
Inhibition of the potentiation resulting from exogenous NO–ST pairing by an NO synthase inhibitor is contrary to the predictions of the simplest form of the retrograde messenger hypothesis or of any other hypothesis according to which NO functions only as an acute signal during the initiation of synaptic plasticity. One explanation would be that l-NOArg possesses another unknown pharmacological activity that inhibits synaptic plasticity. Accordingly, several experiments were performed to test the veracity of the result.
Another nonselective NO synthase inhibitor, l-NIO, was first tested. To the best of our knowledge, this inhibitor has not been used previously in brain slices, so we checked its effectiveness by measuring NMDA-evoked cGMP accumulation in hippocampal slices. The cGMP level found on exposing slices to 300 μm NMDA (12 ± 2 pmol/mg protein) was reduced by l-NIO (100 μm) to 4.0 ± 0.2 pmol/mg protein, a level similar to that found when 100 μml-NOArg was used as the inhibitor (3.6 ± 0.2 pmol/mg protein) and not significantly different from values in control slices not exposed to NMDA (3.4 ± 0.2 pmol/mg protein with l-NIO; 3.9 ± 0.2 pmol/mg with l-NOArg; n = 6 for all data). Thus, at this concentration, l-NIO completely blocked NMDA-evoked NO synthase activity. In electrophysiological experiments,l-NIO duplicated the effect ofl-NOArg in that the DEA/NO-induced facilitation of LTP was abolished, but the ST-induced STP was preserved (Fig.3B). After washout of l-NIO, the HFS-induced LTP had a normal amplitude (149 ± 9%), suggesting that inhibition by l-NIO reverses more rapidly than with l-NOArg.
As a additional test, we examined the ability of the NO synthase substrate l-arginine to neutralize the effect ofl-NOArg. This antagonist was chosen in preference tol-NIO to avoid possible complications arising from inhibition of l-arginine uptake by l-NIO (Bogle et al., 1992). Biochemical measurements in hippocampal slices have shown that even a 100-fold excess of l-arginine over l-NOArg provides only partial relief of the inhibition (East and Garthwaite, 1991); thus, to render the test feasible, the l-NOArg concentration was lowered to 1 μm. This concentration has been shown previously to be adequate to near-maximally inhibit NMDA-stimulated cGMP accumulation in slices of hippocampus (East and Garthwaite, 1991) and cerebellum (East and Garthwaite, 1990) in the absence of added l-arginine. Consistent with the biochemical data, l-NOArg at the lower concentration still blocked the facilitation of LTP induced by NO–ST pairing (Fig.4). Application of l-arginine (1 mm) throughout the experiment did not influence baseline synaptic transmission, nor did it convert the ST-induced STP into an enduring potentiation. However, it lessened the inhibitory effect ofl-NOArg (Fig. 4) such that the LTP facilitation was partially restored (121 ± 7%). A additional significant potentiation could be obtained after HFS (146 ± 9%;p < 0.03 by two-tailed paired t test). The rescue by l-arginine signifies thatl-NOArg was exerting its effects through NO synthase inhibition, and the fact that it was incomplete may be attributable to the difficulty in outcompetingl-NOArg with enzyme substrate (East and Garthwaite, 1990, 1991). The subsequent full restoration of LTP, unlike when 100 μml-NOArg was used in the absence of l-arginine (Fig.3A), is consistent with l-arginine hastening the recovery of NO synthase after washout ofl-NOArg (Boeckxstaens et al., 1991).
Fig. 4.

Excess l-arginine (L-Arg) reversesl-NOArg-induced inhibition of LTP facilitation by NO. Slices were subjected to ST–DEA/NO pairing in the presence ofl-NOArg, with (●; n = 4) or without (○; n = 6) l-arginine. Theinsets show representative fEPSPs (average of 4 consecutive traces) recorded in the absence (left) or presence (right) of l-arginine at the times indicated by the letters.
When is endogenous NO required for LTP facilitation?
If endogenously generated NO were required for LTP facilitation, as the above evidence suggests, it should be possible to overcome the effects of NO synthase inhibition by administering NO exogenously. Such experiments would also allow the time window over which endogenous NO operates in the phenomenon to be probed. To address these issues, a low concentration of DEA/NO was applied during the different periods during which endogenous NO synthesis was inhibited by l-NIO (100 μm). For this purpose, DEA/NO was used at a starting concentration of 0.3 μm, which is just above threshold for raising cGMP levels in hippocampal slices after a 10 min exposure (Bon and Garthwaite, 2001b). When the low DEA/NO concentration was superfused during the 10 min preexposure to l-NIO, the pairing of 3 μm DEA/NO with ST produced only a transient enhancement of synaptic transmission, the baseline synaptic strength being restored within 30 min (Fig.5A). If the low DEA/NO concentration was instead present after washout of the higher concentration (after the ST), only STP was observed (Fig.5B). When the two exposures were combined, so that the low DEA/NO concentration was present before and after the pairing protocol, fEPSP slopes increased and remained stable for ≥30 min (130 ± 2%) (Fig. 5C). A small additional potentiation was observed after HFS (147 ± 4%; p < 0.05 by two-tailed paired t test). The lack of complete restoration of the potentiation by exogenous NO administration may be because the timing and/or concentration applied was suboptimal. For example, the local NO concentration will inevitably have varied as the donor decayed, and, on the basis of the effect of exogenous cGMP derivatives, inappropriate exposure to NO could be inhibitory to LTP facilitation (Son et al., 1998). New ways of supplying NO in known, constant concentrations will be needed to conduct the experiment in a more controlled manner.
Fig. 5.
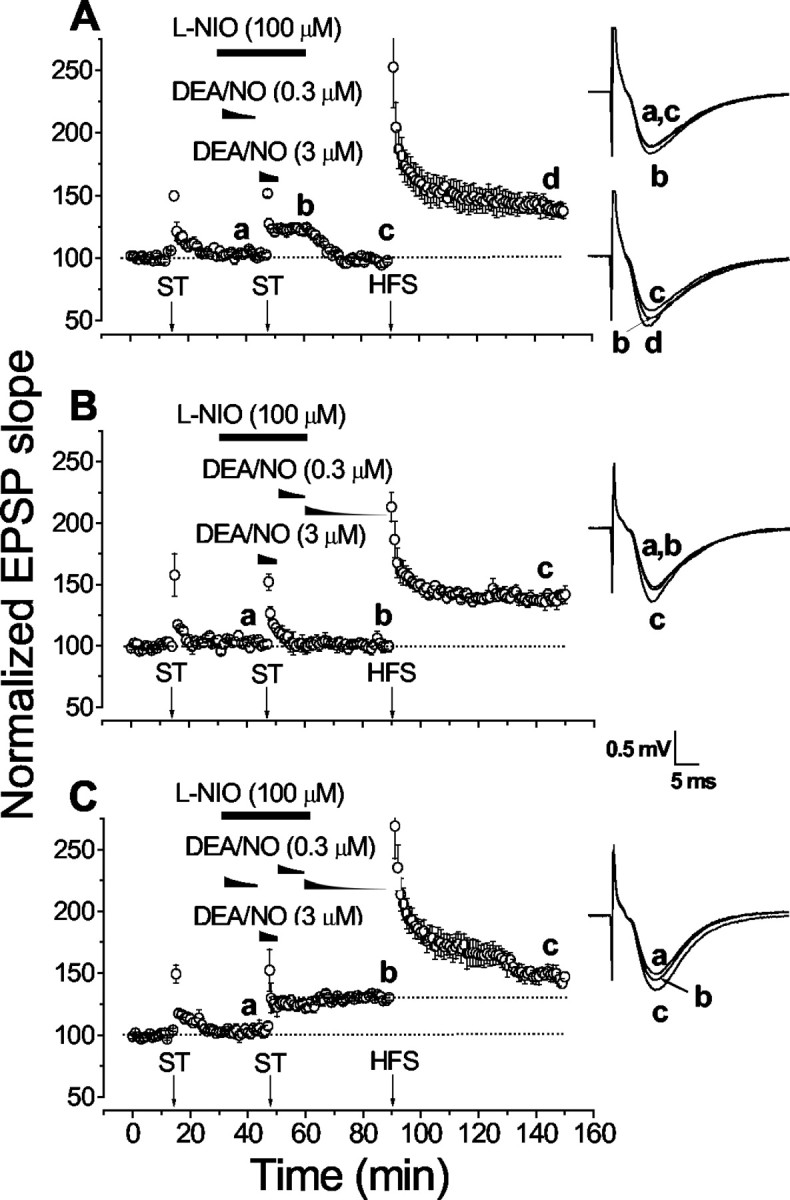
Timing of the participation of endogenous NO in the facilitation of LTP by exogenous NO. In all cases, the NO synthase inhibitor l-NIO was present (as indicated) during the pairing of DEA/NO (applied at 3 μm) and an ST, to inhibit the potentiation that would normally result; a lower DEA/NO concentration (initially 0.3 μm) was added (as indicated) in an attempt to overcome the effect of NO synthase inhibition.A, The low DEA/NO concentration was added together with the l-NIO 15 min before pairing (n = 4). B, Two fresh solutions of DEA/NO at the low concentration were superfused successively after washout of the higher concentration used in the pairing protocol (n = 4).C, Combined administration of the low DEA/NO concentration before and after the pairing protocol (n = 4). The insets show representative fEPSPs (average of 4 consecutive traces) recorded at the time indicated by the letters.
The effectiveness of the low DEA/NO concentrations in overcoming the effect of NO synthase inhibition raises the question of whether the higher DEA/NO concentration is in fact needed to generate the pairing-induced potentiation. In the absence of NO synthase inhibitor, however, pairing the ST with 0.5 μm DEA/NO gave a smaller potentiation (120 ± 7% 30 min after pairing; n = 3) than with 3 μm DEA/NO (137 ± 9%), although subsequent HFS-induced LTP was normal (146 ± 12%; complete data not shown).
Relevance to LTP
If the facilitation of LTP by NO–ST pairing is relevant to events occurring during normal LTP, then tetanus-induced LTP should show the same dependence on tonic NO synthase activity. This prediction was tested by applying an NO synthase inhibitor after tetanic stimulation (its application solely during the pretetanus period not being feasible). First, to check that l-NIO was able to inhibit LTP, the inhibitor was applied 15 min before HFS and for the remainder of the recording (Fig.6A). In control slices, HFS led to a persistent and stable potentiation amounting to 148 ± 6% of the baseline fEPSP slope at 75 min after tetanus. In interleaved experiments using slices from the same rats, treatment withl-NIO resulted in a gradually decaying potentiation, such that, by 75 min after tetanus, only a small amount (115 ± 12%) was left. Thus, like the findings withl-NOArg (Boulton et al., 1995; Lu et al., 1999),l-NIO inhibits primarily late-phase LTP. Thenl-NIO was applied 5 min after LTP induction and for a total of 30 min (Fig. 6B). This had the same effect as applying the inhibitor throughout the experiment in that the potentiation slowly declined back toward baseline. At 115 min after tetanus, the fEPSP slope was 113 ± 8% of the baseline value, whereas in paired control slices, it was 147 ± 4%. Finally, when administration of l-NIO was delayed until 15 min after tetanus (Fig. 6C), LTP was sustained (145 ± 3% compared with 152 ± 5% in control slices, both measured 100 min after HFS). This result agrees with those of Haley et al. (1992), who reported that l-nitroarginine methyl ester, which degrades to produce l-NOArg, did not affect LTP when delivered 15 min after the tetanus.
Fig. 6.

Role of NO synthase in LTP. A,l-NIO applied from 15 min before HFS until the end of the experiment resulted in a gradual loss of LTP compared with interleaved controls (n = 6). B,l-NIO superfused for 30 min starting 5 min after HFS also inhibited late-phase LTP (n = 5; 4 interleaved with controls). C, When the l-NIO application was delayed until 15 min after HFS, LTP was sustained (n = 4–5). The insets show representative fEPSPs (average of 4 consecutive traces) froml-NIO-treated slices recorded at the times indicated by theletters.
Role of the guanylyl cyclase-coupled NO receptor
The best-recognized target for physiological NO signal transduction is the NOGCR, a metabotropic type of receptor in which NO binding is coupled to the generation of cGMP from GTP in the catalytic domain of the protein. The standard inhibitor of NOGCR activity is the compound ODQ, which has been found to block the NO-dependent component of hippocampal LTP in this and other laboratories (Boulton et al., 1995; Son et al., 1998; Lu et al., 1999; Bon and Garthwaite, 2001b) (but see Kleppisch et al., 1999). To test whether the NOGCR was necessary for the potentiation induced by NO–ST pairing, ODQ (10 μm) was perfused from 15 min before until 5 min after the pairing protocol. The long-lasting enhancement of fEPSP slopes was completely blocked (Fig. 7A). After subsequent HFS, the level of LTP was slightly less (137 ± 3%) than in controls not treated with ODQ (146 ± 3%;p < 0.05, two-tailed unpaired t test), probably reflecting the slow recovery of the receptor after ODQ treatment (Bellamy and Garthwaite, 2002). Inhibition by ODQ does not distinguish between a requirement for NOGCR activity during the pairing itself or during the periods on either side when endogenous NO operates. To examine the latter possibility, ODQ was added for 30 min starting 5 min after a tetanic stimulation that normally generated LTP. As seen previously withl-NIO in the same paradigm (Fig. 6C), the fEPSP slopes decreased gradually in ODQ-treated slices (Fig.7B), such that, at the end of the recording period (115 min after tetanus), the potentiation (125 ± 8%) was ∼50% of that seen in interleaved controls (158 ± 6%).
Fig. 7.

Involvement of the guanylyl cyclase-coupled NO receptor in NO-induced LTP facilitation and LTP. A, Pairing of DEA/NO with ST fails to elicit a potentiation in the presence of ODQ (n = 4). B, Late delivery of ODQ (5 min after HFS) inhibits late-phase LTP (interleaved experiment; n = 5). The insets show representative fEPSPs (average of 4 successive traces) from ODQ-treated slices.
Discussion
The experiments addressed a fundamental prediction of the hypothesis that NO serves as a messenger in CA1 hippocampal LTP, namely, that exogenous NO should substitute for endogenous NO in the phenomenon. Although the results as a whole provide good support for the hypothesis, they also indicate that the role of NO is more complex than previously supposed, in that there is an unforeseen requirement for a tonic level of endogenous NO for exogenous NO to facilitate LTP. The findings extrapolate to LTP itself and echo observations of the role played by NO in various types of learning behavior.
Role of endogenous NO in LTP facilitation by exogenous NO
In agreement with results using guinea pig hippocampal slices (Zhuo et al., 1993, 1994b), exogenous NO paired with an ST generated a long-lasting potentiation of rat CA1 hippocampal neurotransmission that was independent of NMDA receptors and that occluded tetanus-induced LTP. Two compounds that can be degraded to yield NO, hydroxylamine and S-nitrosocysteine, were also reported to facilitate LTP in rat hippocampal slices similarly (Malen and Chapman, 1997). Despite other unwanted effects of these reactive chemicals (Bon and Garthwaite, 2001a), the results with the authentic molecule support the authors' proposal that NO release was responsible. In neither of these two previous cases, however, was the possible role of endogenous NO considered. Our results showed that LTP facilitation was completely blocked by two different NO synthase inhibitors and that the inhibition could be relieved by adding excess NO synthase substrate or by concomitant administration of low concentrations of exogenous NO. Together, therefore, the results merit the conclusion that endogenous NO is required for exogenous NO to facilitate LTP.
Administration of a low DEA/NO concentration while NO synthase was inhibited indicated that there is not one specific period before or after the NO–ST pairing when endogenous NO is needed. The transient potentiation observed when DEA/NO was given only before the pairing (Fig. 5A) could be explained by NO inevitably also being present for a time afterward as the 10-fold higher DEA/NO concentration used in the pairing protocol washed out. Rather, the results suggest that a continuous low level of NO is necessary before and after the pairing. Furthermore, because LTP facilitation was unaffected by blocking NMDA receptors, the NO synthase normally generating the tonic level of NO must be derived through another pathway. Evidence for tonic NO generated independently of NMDA receptors comes from data on hippocampal and striatal slices showing that most of the basal cGMP (augmented by a phosphodiesterase inhibitor) was sustained by NO synthase but not by NMDA receptors (Chetkovich et al., 1993; Griffiths et al., 2002). Of possible relevance here is the fact that the endothelial NO synthase isoform, which is a putative participant in LTP (Kantor et al., 1996; Son et al., 1996; Wilson et al., 1997), can become tonically active in response to phosphorylation cascades (Michell et al., 2002).
Roles of NO in LTP
The DEA/NO–ST pairing combines two events presumed to occur normally during tetanic stimulation leading to LTP: NO formation and presynaptic activity. In the past, the inhibition of LTP by NO synthase inhibitors, NO scavengers, or NO synthase gene deletion has been interpreted to reflect loss of the NO signaling pathway during LTP induction, which is when NMDA receptors are activated (Hawkins et al., 1998). We found, however, that LTP could be inhibited by administration of an NO synthase inhibitor 5 min after the tetanus, whereas, confirming the results of Haley et al. (1992), the same treatment delayed until 15 min after tetanus was ineffective. This indicates that, as with the NO–ST pairing protocol, tonic NO synthase activity is necessary for LTP and, moreover, that there is a ≤15 min time window after tetanus when it is required. From the NO–ST pairing experiments, endogenous NO in the pretetanus period is also expected to be important, but this was not explicitly tested.
Inhibition of LTP by blocking of NO synthesis after tetanus could indicate that only the tonic NO is needed for LTP. However, there is evidence for increased NO release after tetanic stimulation (Chetkovich et al., 1993), and our experiments indicated that a higher NO concentration is needed to facilitate LTP than to maintain tonic NO at active levels. Furthermore, in some studies, NO synthase inhibition has been found to reduce the early LTP observed during the first 20 min after tetanus, as expected for loss of an acute facilitation resembling the one occurring in response to NO–ST pairing. Under otherwise comparable conditions, the contribution of NO to early LTP in rat CA1 hippocampus appears greater at low temperature (22–24°C) (Schuman and Madison, 1991; Haley et al., 1992; Boulton et al., 1995) than at 30–32°C (Chetkovich et al., 1993; Boulton et al., 1995; Ko and Kelly, 1999; present report), indicating that early LTP may be a mixture of NO-dependent and NO-independent processes and that the NO-dependent component may be masked by a “ceiling” effect imposed by the NO-independent component at the more physiological temperatures. Hence, the most parsimonious conclusion to draw at this stage is that LTP requires both a tonic NO level and a phasic NO signal arising from the tetanic stimulation.
Possible mechanisms
The pathway for NO signal transduction in most forms of synaptic plasticity is through NOGCR activation, leading to cGMP accumulation (Garthwaite and Boulton, 1995; Daniel et al., 1998; Hawkins et al., 1998; Centonze et al., 1999). In the present experiments, the finding that the NOGCR antagonist ODQ abolished the potentiation induced by NO–ST pairing implicates a cGMP-dependent mechanism but cannot distinguish whether this pathway is engaged by exogenous NO, endogenous NO, or both. Favoring the first possibility are data showing that exogenous cGMP derivatives can, like NO, generate an enduring potentiation of hippocampal synaptic transmission if paired with an ST (Zhuo et al., 1994a,b) (but see Selig et al., 1996; Son et al., 1998). Favoring the second possibility is our finding that ODQ administered after the tetanus mimicked the inhibitory effect of l-NIO (also delivered after tetanus) on LTP. Consequently, both phasic and tonic NO signal transduction may be through the NOGCR–cGMP pathway. Consistent with this possibility, continuous perfusion with a cGMP analog was found to overcome the effect of NO synthase inhibition in hippocampal LTP (Haley et al., 1992).
With respect to the phasic component, there is direct evidence from hippocampal cultures that NO can potentiate synaptic transmission through a presynaptic mechanism involving cGMP and cGMP-dependent protein kinase (Arancio et al., 2001). At the same time, data from hippocampal slices indicate that NO can act postsynaptically to potentiate neurotransmission (Ko and Kelly, 1999) and that its involvement in late LTP is mediated by cGMP-dependent activation of the transcription factor CREB (cAMP response element-binding protein) in postsynaptic neurons (Lu et al., 1999). Also pertinent to a postsynaptic site of action, in situ hybridization has suggested that the dominant subtype of the NOGCR expressed in the hippocampal pyramidal neurons is the α2β1 isoform (Gibb and Garthwaite, 2001), and this isoform associates with the postsynaptic density-95 protein (Russwurm et al., 2001), providing the substrate necessary for NO to act postsynaptically through cGMP. Thus, although a presynaptic action may be responsible for an acute enhancement of synaptic efficacy, a postsynaptic action may mediate the longer-term changes.
The function fulfilled by tonic NO awaits investigation, but several possibilities exist. For instance, through cGMP, NO can influence cytoskeletal dynamics (Sauzeau et al., 2000) or mitogen-activated protein (MAP) kinase signaling (Komalavilas et al., 1999), and, in the CA1 hippocampus, inhibition of actin dynamics (Krucker et al., 2000) or MAP kinase (Rosenblum et al., 2002) immediately after tetanic stimulation results in a dwindling potentiation, much like that seen in the present experiments after a similar administration of inhibitors of NO synthase or NOGCR activity.
Parallels with learning behavior
Synaptic plasticity is commonly considered relevant to learning and memory formation. If so, and should tonic NO be necessary for some types of plasticity, disruption of the NO signaling pathway outside the acquisition phase should disrupt some types of memory. Behavioral studies on various species, including rats (Bernabeu et al., 1995), mice (Baratti and Kopf, 1996), fish (Xu et al., 2001), birds (Edwards et al., 2002), and snails (Kemenes et al., 2002), found that interference with NO and/or cGMP signaling after completion of the training period disrupts memory formation. The time window during which NO–cGMP signaling is required for memory formation is long (5 hr after training) for appetitive conditioning in a snail (Kemenes et al., 2002) but is shorter for inhibitory avoidance in rats, in which intrahippocampal injection of l-NOArg produced amnesia when given immediately after training but not 60 min later (Bernabeu et al., 1995). In the same model, hippocampal application of a cGMP analog directly after training, but not 180 min later, enhanced memory retention (Bernabeu et al., 1996; Prickaerts et al., 2002). The similar timing over which the NO–cGMP pathway operates in these memory paradigms and in LTP is consistent with a mechanistic link between the two.
Footnotes
This study was supported by The Wellcome Trust.
Correspondence should be addressed to J. Garthwaite, The Wolfson Institute for Biomedical Research, University College London, Gower Street, London WC1E 6BT, UK. E-mail: john.garthwaite@ucl.ac.uk.
References
- 1.Arancio O, Antonova I, Gambaryan S, Lohmann SM, Wood JS, Lawrence DS, Hawkins RD. Presynaptic role of cGMP-dependent protein kinase during long-lasting potentiation. J Neurosci. 2001;21:143–149. doi: 10.1523/JNEUROSCI.21-01-00143.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baratti CM, Kopf SR. A nitric oxide synthase inhibitor impairs memory storage in mice. Neurobiol Learn Mem. 1996;65:197–201. doi: 10.1006/nlme.1996.0023. [DOI] [PubMed] [Google Scholar]
- 3.Bellamy TC, Garthwaite J. Pharmacology of the nitric oxide receptor, soluble guanylyl cyclase, in cerebellar cells. Br J Pharmacol. 2002;136:95–103. doi: 10.1038/sj.bjp.0704687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernabeu R, de Stein ML, Fin C, Izquierdo I, Medina JH. Role of hippocampal NO in the acquisition and consolidation of inhibitory avoidance learning. NeuroReport. 1995;6:1498–1500. doi: 10.1097/00001756-199507310-00008. [DOI] [PubMed] [Google Scholar]
- 5.Bernabeu R, Schmitz P, Faillace MP, Izquierdo I, Medina JH. Hippocampal cGMP and cAMP are differentially involved in memory processing of inhibitory avoidance learning. NeuroReport. 1996;7:585–588. doi: 10.1097/00001756-199601310-00050. [DOI] [PubMed] [Google Scholar]
- 6.Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 7.Boeckxstaens GE, Pelckmans PA, Bogers JJ, Bult H, De Man JG, Oosterbosch L, Herman AG, Van Maercke YM. Release of nitric oxide upon stimulation of nonadrenergic noncholinergic nerves in the rat gastric fundus. J Pharmacol Exp Ther. 1991;256:441–447. [PubMed] [Google Scholar]
- 8.Bogle RG, Moncada S, Pearson JD, Mann GE. Identification of inhibitors of nitric oxide synthase that do not interact with the endothelial cell l-arginine transporter. Br J Pharmacol. 1992;105:768–770. doi: 10.1111/j.1476-5381.1992.tb09053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bon CLM, Garthwaite J. Nitric oxide-induced potentiation of CA1 hippocampal synaptic transmission during baseline stimulation is strictly frequency-dependent. Neuropharmacology. 2001a;40:501–507. doi: 10.1016/s0028-3908(00)00193-3. [DOI] [PubMed] [Google Scholar]
- 10.Bon CLM, Garthwaite J. Exogenous nitric oxide causes potentiation of hippocampal synaptic transmission during low-frequency stimulation via the endogenous nitric oxide-cGMP pathway. Eur J Neurosci. 2001b;14:585–594. doi: 10.1046/j.0953-816x.2001.01680.x. [DOI] [PubMed] [Google Scholar]
- 11.Boulton CL, Irving AJ, Southam E, Potier B, Garthwaite J, Collingridge GL. The nitric oxide-cyclic GMP pathway and synaptic depression in rat hippocampal slices. Eur J Neurosci. 1994;6:1528–1535. doi: 10.1111/j.1460-9568.1994.tb00543.x. [DOI] [PubMed] [Google Scholar]
- 12.Boulton CL, Southam E, Garthwaite J. Nitric oxide-dependent long-term potentiation is blocked by a specific inhibitor of soluble guanylyl cyclase. Neuroscience. 1995;69:699–703. doi: 10.1016/0306-4522(95)00349-n. [DOI] [PubMed] [Google Scholar]
- 13.Brenman JE, Bredt DS. Synaptic signaling by nitric oxide. Curr Opin Neurobiol. 1997;7:374–378. doi: 10.1016/s0959-4388(97)80065-7. [DOI] [PubMed] [Google Scholar]
- 14.Centonze D, Gubellini P, Bernardi G, Calabresi P. Permissive role of interneurons in corticostriatal synaptic plasticity. Brain Res Brain Res Rev. 1999;31:1–5. doi: 10.1016/s0165-0173(99)00018-1. [DOI] [PubMed] [Google Scholar]
- 15.Chetkovich DM, Klann E, Sweatt JD. Nitric oxide synthase-independent long-term potentiation in area CA1 of hippocampus. NeuroReport. 1993;4:919–922. doi: 10.1097/00001756-199307000-00020. [DOI] [PubMed] [Google Scholar]
- 16.Daniel H, Levenes C, Crepel F. Cellular mechanisms of cerebellar LTD. Trends Neurosci. 1998;21:401–407. doi: 10.1016/s0166-2236(98)01304-6. [DOI] [PubMed] [Google Scholar]
- 17.Dwyer MA, Bredt DS, Snyder SH. Nitric oxide synthase: irreversible inhibition by l-NG-nitroarginine in brain in vitro and in vivo. Biochem Biophys Res Commun. 1991;176:1136–1141. doi: 10.1016/0006-291x(91)90403-t. [DOI] [PubMed] [Google Scholar]
- 18.East SJ, Garthwaite J. Nanomolar N(G)-nitroarginine inhibits NMDA-induced cyclic GMP formation in rat cerebellum. Eur J Pharmacol. 1990;184:311–313. doi: 10.1016/0014-2999(90)90623-e. [DOI] [PubMed] [Google Scholar]
- 19.East SJ, Garthwaite J. NMDA receptor activation in rat hippocampus induces cyclic GMP formation through the L-arginine-nitric oxide pathway. Neurosci Lett. 1991;123:17–19. doi: 10.1016/0304-3940(91)90147-l. [DOI] [PubMed] [Google Scholar]
- 20.Edwards TM, Rickard NS, Ng KT. Inhibition of guanylate cyclase and protein kinase G impairs retention for the passive avoidance task in the day-old chick. Neurobiol Learn Mem. 2002;77:313–326. doi: 10.1006/nlme.2001.4021. [DOI] [PubMed] [Google Scholar]
- 21.Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annu Rev Physiol. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- 22.Gibb BJ, Garthwaite J. Subunits of the nitric oxide receptor, soluble guanylyl cyclase, expressed in rat brain. Eur J Neurosci. 2001;13:539–544. doi: 10.1046/j.1460-9568.2001.01421.x. [DOI] [PubMed] [Google Scholar]
- 23.Griffiths C, Garthwaite G, Goodwin DA, Garthwaite J. Dynamics of nitric oxide during simulated ischaemia-reperfusion in rat striatal slices measured using an intrinsic biosensor, soluble guanylyl cyclase. Eur J Neurosci. 2002;15:962–968. doi: 10.1046/j.1460-9568.2002.01930.x. [DOI] [PubMed] [Google Scholar]
- 24.Haley JE, Wilcox GL, Chapman PF. The role of nitric oxide in hippocampal long-term potentiation. Neuron. 1992;8:211–216. doi: 10.1016/0896-6273(92)90288-o. [DOI] [PubMed] [Google Scholar]
- 25.Hawkins RD, Son H, Arancio O. Nitric oxide as a retrograde messenger during long-term potentiation in hippocampus. Prog Brain Res. 1998;118:155–172. doi: 10.1016/s0079-6123(08)63206-9. [DOI] [PubMed] [Google Scholar]
- 26.Jacoby S, Sims RE, Hartell NA. Nitric oxide is required for the induction and heterosynaptic spread of long-term potentiation in rat cerebellar slices. J Physiol (Lond) 2001;535:825–839. doi: 10.1111/j.1469-7793.2001.t01-1-00825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kantor DB, Lanzrein M, Stary SJ, Sandoval GM, Smith WB, Sullivan BM, Davidson N, Schuman EM. A role for endothelial NO synthase in LTP revealed by adenovirus-mediated inhibition and rescue. Science. 1996;274:1744–1748. doi: 10.1126/science.274.5293.1744. [DOI] [PubMed] [Google Scholar]
- 28.Kemenes I, Kemenes G, Andrew RJ, Benjamin PR, O'Shea M. Critical time-window for NO-cGMP-dependent long-term memory formation after one-trial appetitive conditioning. J Neurosci. 2002;22:1414–1425. doi: 10.1523/JNEUROSCI.22-04-01414.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleppisch T, Pfeifer A, Klatt P, Ruth P, Montkowski A, Fassler R, Hofmann F. Long-term potentiation in the hippocampal CA1 region of mice lacking cGMP-dependent kinases is normal and susceptible to inhibition of nitric oxide synthase. J Neurosci. 1999;19:48–55. doi: 10.1523/JNEUROSCI.19-01-00048.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ko GY, Kelly PT. Nitric oxide acts as a postsynaptic signaling molecule in calcium/calmodulin-induced synaptic potentiation in hippocampal CA1 pyramidal neurons. J Neurosci. 1999;19:6784–6794. doi: 10.1523/JNEUROSCI.19-16-06784.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Komalavilas P, Shah PK, Jo H, Lincoln TM. Activation of mitogen-activated protein kinase pathways by cyclic GMP and cyclic GMP-dependent protein kinase in contractile vascular smooth muscle cells. J Biol Chem. 1999;274:34301–34309. doi: 10.1074/jbc.274.48.34301. [DOI] [PubMed] [Google Scholar]
- 32.Krucker T, Siggins GR, Halpain S. Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci USA. 2000;97:6856–6861. doi: 10.1073/pnas.100139797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lev-Ram V, Wong ST, Storm DR, Tsien RY. A new form of cerebellar long-term potentiation is postsynaptic and depends on nitric oxide but not cAMP. Proc Natl Acad Sci USA. 2002;99:8389–8993. doi: 10.1073/pnas.122206399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lu YF, Kandel ER, Hawkins RD. Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J Neurosci. 1999;19:10250–10261. doi: 10.1523/JNEUROSCI.19-23-10250.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malen PL, Chapman PF. Nitric oxide facilitates long-term potentiation, but not long-term depression. J Neurosci. 1997;17:2645–2651. doi: 10.1523/JNEUROSCI.17-07-02645.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malenka RC. Postsynaptic factors control the duration of synaptic enhancement in area CA1 of the hippocampus. Neuron. 1991;6:53–60. doi: 10.1016/0896-6273(91)90121-f. [DOI] [PubMed] [Google Scholar]
- 37.Michell BJ, Harris MB, Chen ZP, Ju H, Venema VJ, Blackstone MA, Huang W, Venema RC, Kemp BE. Identification of regulatory sites of phosphorylation of the bovine endothelial nitric-oxide synthase at serine 617 and serine 635. J Biol Chem. 2002;277:42344–42351. doi: 10.1074/jbc.M205144200. [DOI] [PubMed] [Google Scholar]
- 38.Morley D, Keefer LK. Nitric oxide/nucleophile complexes: a unique class of nitric oxide-based vasodilators. J Cardiovasc Pharmacol. 1993;22 [Suppl 7]:S3–S9. [PubMed] [Google Scholar]
- 39.Prickaerts J, de Vente J, Honig W, Steinbusch HW, Blokland A. cGMP, but not cAMP, in rat hippocampus is involved in early stages of object memory consolidation. Eur J Pharmacol. 2002;436:83–87. doi: 10.1016/s0014-2999(01)01614-4. [DOI] [PubMed] [Google Scholar]
- 40.Rosenblum K, Futter M, Voss K, Erent M, Skehel PA, French P, Obosi L, Jones MW, Bliss TV. The role of extracellular regulated kinases I/II in late-phase long-term potentiation. J Neurosci. 2002;22:5432–5441. doi: 10.1523/JNEUROSCI.22-13-05432.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Russwurm M, Wittau N, Koesling D. Guanylyl cyclase/PSD-95 interaction: targeting of the NO-sensitive α2β1 guanylyl cyclase to synaptic membranes. J Biol Chem. 2001;276:44647–44652. doi: 10.1074/jbc.M105587200. [DOI] [PubMed] [Google Scholar]
- 42.Sauzeau V, Le Jeune H, Cario-Toumaniantz C, Smolenski A, Lohmann SM, Bertoglio J, Chardin P, Pacaud P, Loirand G. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem. 2000;275:21722–21729. doi: 10.1074/jbc.M000753200. [DOI] [PubMed] [Google Scholar]
- 43.Schuman EM, Madison DV. A requirement for the intercellular messenger nitric oxide in long-term potentiation. Science. 1991;254:1503–1506. doi: 10.1126/science.1720572. [DOI] [PubMed] [Google Scholar]
- 44.Selig DK, Segal MR, Liao D, Malenka RC, Malinow R, Nicoll RA, Lisman JE. Examination of the role of cGMP in long-term potentiation in the CA1 region of the hippocampus. Learn Mem. 1996;3:42–48. doi: 10.1101/lm.3.1.42. [DOI] [PubMed] [Google Scholar]
- 45.Son H, Hawkins RD, Martin K, Kiebler M, Huang PL, Fishman MC, Kandel ER. Long-term potentiation is reduced in mice that are doubly mutant in endothelial and neuronal nitric oxide synthase. Cell. 1996;87:1015–1023. doi: 10.1016/s0092-8674(00)81796-1. [DOI] [PubMed] [Google Scholar]
- 46.Son H, Lu YF, Zhuo M, Arancio O, Kandel ER, Hawkins RD. The specific role of cGMP in hippocampal LTP. Learn Mem. 1998;5:231–245. [PMC free article] [PubMed] [Google Scholar]
- 47.Wilson RI, Yanovsky J, Godecke A, Stevens DR, Schrader J, Haas HL. Endothelial nitric oxide synthase and LTP. Nature. 1997;386:338. doi: 10.1038/386338a0. [DOI] [PubMed] [Google Scholar]
- 48.Xu X, Russell T, Bazner J, Hamilton J. NMDA receptor antagonist AP5 and nitric oxide synthase inhibitor 7-NI affect different phases of learning and memory in goldfish. Brain Res. 2001;889:274–277. doi: 10.1016/s0006-8993(00)03216-9. [DOI] [PubMed] [Google Scholar]
- 49.Zhuo M, Small SA, Kandel ER, Hawkins RD. Nitric oxide and carbon monoxide produce activity-dependent long-term synaptic enhancement in hippocampus. Science. 1993;260:1946–1950. doi: 10.1126/science.8100368. [DOI] [PubMed] [Google Scholar]
- 50.Zhuo M, Hu Y, Schultz C, Kandel ER, Hawkins RD. Role of guanylyl cyclase and cGMP-dependent protein kinase in long-term potentiation. Nature. 1994a;368:635–639. doi: 10.1038/368635a0. [DOI] [PubMed] [Google Scholar]
- 51.Zhuo M, Kandel ER, Hawkins RD. Nitric oxide and cGMP can produce either synaptic depression or potentiation depending on the frequency of presynaptic stimulation in the hippocampus. NeuroReport. 1994b;5:1033–1036. doi: 10.1097/00001756-199405000-00004. [DOI] [PubMed] [Google Scholar]