

Abstract
Norepinephrine (NE) transporters (NETs) terminate noradrenergic synaptic transmission and represent a major therapeutic target for antidepressant medications. NETs and related transporters are under intrinsic regulation by receptor and kinase-linked pathways, and clarification of these pathways may suggest candidates for the development of novel therapeutic approaches. Syntaxin 1A, a presynaptic soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein, interacts with NET and modulates NET intrinsic activity. NETs colocalize with and bind to syntaxin 1A in both native preparations and heterologous systems. Protein kinase C activation disrupts surface NET/syntaxin 1A interactions and downregulates NET activity in a syntaxin-dependent manner. Syntaxin 1A binds the NH2 terminal domain of NET, and a deletion of this domain both eliminates NET/syntaxin 1A associations and prevents phorbol ester-triggered NET downregulation. Whereas syntaxin 1A supports the surface trafficking of NET proteins, its direct interaction with NET limits transporter catalytic function. These two contradictory roles of syntaxin 1A on NET appear to be linked and reveal a dynamic cycle of interactions that allow for the coordinated control between NE release and reuptake.
Keywords: catecholamine, norepinephrine, antidepressant, transport, syntaxin 1A, phorbol ester, surface trafficking
Introduction
The catecholamine neurotransmitter norepinephrine (NE) modulates multiple cognitive and emotional circuits in the mammalian brain including those subserving alertness, attention, learning and memory, and mood (Foote et al., 1983). NE also regulates autonomic function at brainstem sites and via its role as a neurotransmitter in postganglionic sympathetic synapses (Axelrod and Kopin, 1969). NE is released predominantly at axonal varicosities in which the availability of extracellular NE is limited by NE transporters (NETs), presynaptic transporters that catalyze neurotransmitter reuptake via coupling to transmembrane Na+ and Cl−gradients (Iversen, 1971; Graefe and Bönisch, 1988). NETs are targets of psychostimulants, including cocaine and amphetamine (Ritz et al., 1990; Chen and Reith, 1994; Wall et al., 1995), and have been recognized as important sites of action for tricyclic antidepressants (Schildkraut, 1965). Although more selective NET antagonists [norepinephrine serotonin reuptake inhibitors (NSRIs)] appear promising in the treatment of mood disorders (Tatsumi et al., 1997; Burrows et al., 1998; Gorman and Sullivan, 2000), strategies for manipulating catecholamine reuptake remain limited to the identification of agents capable of occluding NE binding or translocation. Dysfunction of NE clearance or NET density has been associated with attention and mood disorders (Hadley et al., 1995;Delgado and Moreno, 2000; Arnsten, 2001), suicide (Klimek et al., 1997), and cardiovascular disease (Esler et al., 1981; Liang et al., 1989; Merlet et al., 1992; Bohm et al., 1995; Shannon et al., 2000). Notably, altered regulatory mechanisms that might disrupt NET function in disease states are unknown.
NETs belong to a gene family (SLC6A) of Na+/Cl−-dependent transporters (Pacholczyk et al., 1991), the other members of which include dopamine, serotonin, glycine, and GABA transporters (DAT, SERT, GLYT, and GAT, respectively) (Barker and Blakely, 1995). These transporters exhibit a predicted topology of 12 transmembrane domains (TMDs) with cytoplasmic N and C termini. Increasing evidence indicates that both extrinsic signals and intracellular kinase-linked pathways regulate biogenic amine transporters, with effects seen on transporter cell surface trafficking and/or intrinsic activity (Blakely and Bauman, 2000; Zahniser and Doolen, 2001). For example, NE transport capacity in noradrenergic SK-N-SH cells can be diminished rapidly by muscarinic receptor activation (Apparsundaram et al., 1998a), a process linked to protein kinase C (PKC) activation and a loss of carriers from the cell surface. Direct activation of PKC with phorbol esters also redistributes NETs in heterologous expression systems as visualized by confocal microscopy (Apparsundaram et al., 1998b). Analogous findings of cell surface redistribution after receptor stimulation have been reported for DAT, SERT, and GAT1 proteins (Qian et al., 1997; Beckman et al., 1999; Ramamoorthy and Blakely, 1999; Saunders et al., 2000) and may involve phosphorylation of transporters as well as the coordinated association of protein kinases, phosphatases, and scaffolding proteins (Blakely and Bauman, 2000; Deken et al., 2001). Recently, we identified a trafficking-independent pathway for NET regulation linked to PI-3 kinase and p38 mitogen-activated protein (MAP) kinase activation (Apparsundaram et al., 2001), suggesting that neurons likely have multiple pathways to modulate NE clearance capacity intrinsically. A greater understanding of these mechanisms will clarify how the processes of neurotransmitter release and reuptake are coordinated in space and time and extend the range of targets for drug development.
Neurotransmitter transporters, like other membrane proteins involved in cell signaling, appear to be organized as multiprotein complexes. For example, several PDZ [postsynaptic density-95 (PSD-95)/Discs large (Dlg)/zona occludens-1 (ZO-1)] domain proteins have been identified that physically interact with C termini of glutamate, GABA, and biogenic amine transporters (Perego et al., 1999; Jackson et al., 2001; Torres et al., 2001) and may help to establish targeted expression to discrete membrane domains, although their role in acute transporter regulation is unclear. Several studies recently have drawn explicit attention to the physical and functional interaction of transporters with the t-soluble N-ethylmaleimide-sensitive factor attachment protein receptor (t-SNARE) protein syntaxin 1A (Beckman et al., 1998; Geerlings et al., 2000; Haase et al., 2001). These studies with the transporters for inhibitory amino acids suggest that syntaxin 1A may control neurotransmission not only via the regulated fusion of neurotransmitter vesicles but also via the delivery of transporters that support neurotransmitter inactivation (Deken et al., 2000; Geerlings et al., 2001). Whether regulated catecholamine transporter trafficking and/or intrinsic activity are/is supported similarly by syntaxin 1A interactions and whether these processes are linked are unknown. In the present report we used botulinum toxin C1 (BoNT/C1) and syntaxin 1A antisense treatments to establish a tonic requirement for the SNARE protein on NE transport capacity in neuronal preparations. Moreover, we find that NET colocalizes, and forms stable associations, with syntaxin 1A in vivo. NET/syntaxin 1A interactions are direct and mediated by the NET NH2 terminus, and stimuli known to trigger NET redistribution destabilize NET/syntaxin 1A interactions. Whereas syntaxin 1A supports surface expression of NETs, we find that the SNARE protein limits NET catalytic activity, measured as an elimination of NET-associated currents and a dissociation of surface NET density from NE uptake activity. We discuss our findings in the context of a model whereby vesicular NE release and the trafficking and intrinsic activity of NETs are both linked to syntaxin 1A availability, allowing enhanced coordination of catecholamine release and reuptake. We suggest that physical interactions of syntaxin 1A with NET provide a means to elaborate tight control of NE clearance capacity via modulation of both surface trafficking and intrinsic activity in parallel with signals impinging on NE release.
Materials and Methods
Antibodies and other reagents. Polyclonal antibody 43411, generated against the C-terminal sequences of mouse NET and recognizing rat, mouse, and human NET (hNET) proteins, has been described previously (Schroeter et al., 2000). Polyclonal NET antibody 43408, raised against the peptide TKYSKYKFTPAAEFY (amino acids 99–214) located in the second extracellular loop of human and mouse NET, was used at 1:500 and will be described more fully (V. Savchenko, U. Sung, and R. D. Blakely, unpublished data). Monoclonal anti-hNET antibody (catalog number NET 17-1, Mab Technologies, Atlanta, GA) was used at a dilution of 1:1000 for immunoblotting. Immunoblotting of syntaxin 1A was performed by using anti-syntaxin antibody (HPC-1, Sigma, St. Louis, MO) at a dilution of 1:2000. Immunohistochemistry of syntaxin 1A was performed with anti-syntaxin antibody from Chemicon (Temecula, CA) at a dilution of 1:1000. Monoclonal anti-histidine antibody (Clontech, Palo Alto, CA) was used at 1 μg for each immunoprecipitation. Polyclonal anti-histidine antibody (Santa Cruz Biotech, Santa Cruz, CA) was used at a dilution of 1:200 for immunoblots. Anti-hemagglutinin (HA) antibody (3F10) conjugated with peroxidase (Boehringer Mannheim, Mannheim, Germany) was used at a dilution of 1:200–1:500 for immunoblots. Phorbol 12-myristate 13-acetate (β-PMA) was from Calbiochem (La Jolla, CA) or from Alexis (San Diego, CA); okadaic acid (OA) was also from Alexis. Methacholine, carbachol, desipramine, and cocaine were obtained from Sigma.
Constructs. HA-tagged hNET in pcDNA3 (Invitrogen, Carlsbad, CA) has been described previously (Bauman and Blakely, 2002). His-hNET in pcDNA3 was prepared by inserting HHHHHHG between the translation initiation site and the second residue (L) of hNET. Insertion of tags and deletion and point mutations of hNET (hNETΔ2–42, hNETΔ43–64, hNETΔ2–64, hNET D51A, D53A, E58A) in pcDNA3 were made by site-directed mutagenesis with the Quick Change site-directed mutagenesis kit (Stratagene, La Jolla, CA). Syntaxin 1A in pCMV5 and Munc18-1 in pGEX KG were generous gifts from Dr. T. Sudhof (Howard Hughes Medical Institute, University of Texas Southwestern Medical Center, Dallas, TX). The cytoplasmic domain of syntaxin 1A in pCMV5 (SynΔTM) was made by inserting a stop codon just before the transmembrane domain. Munc18-1 was moved to pcDNA3 (Invitrogen). pGEX5X-1-SynΔTM (GST-syntaxin 1A bearing a deletion of the transmembrane domain, GST-SynΔTM), pMAL NET-N [ maltose-binding protein (MBP) fused to the N-terminal domain of NET, residues 1–63], and pMAL-NET-C (MBP fused to the C-terminal domain of NET, residues 576–617) were constructed by the subcloning of PCR products of the corresponding sequences into pGEX (Pharmacia, Peapack, NJ) or pMAL cRI (New England Biolabs, Beverly, MA).
Primary neuronal cultures and immunohistochemistry. C57BL/6 mouse pups, 2 d old, were anesthetized with Nembutal (50 mg/kg). Superior cervical ganglia (SCG) were dissected, treated with 0.3% collagenase and 0.1% trypsin for 30 min, plated on culture dishes, and incubated in F-14 medium containing 5% FCS and 20 ng/ml NGF for 2 hr at 37°C to purify SCG neurons from fibroblasts. Floating cells were replated on poly-d-lysine and laminin-coated coverslips and incubated further. After 24 hr the cultures were treated with 10 μm 5-fluoro-5′-deoxyuridine and grown for 5–14 d before staining. For double staining of total NET and syntaxin 1A, SCG cells were fixed with 3% paraformaldehyde, blocked with 2% normal donkey serum (NDS) and 0.2% NP-40 in PBS, and incubated with NET and syntaxin 1A antibodies in the same solution for 1 hr at room temperature (RT), followed by an incubation with donkey anti-rabbit conjugated with CY3 (1:1000) and donkey anti-mouse conjugated with CY2 (1:500;Jackson ImmunoResearch, West Grove, PA). To detect surface labeling of NET, we incubated live cells with antibody 43408 in PBS/1% NDS for 1 hr at RT, fixed them with PBS/3% paraformaldehyde, blocked them with PBS/2% NDS/0.2% NP-40, and incubated them with syntaxin 1A antibody in the same buffer for 2 hr at RT. Methods for tissue preparation and immunofluorescence localization of NET with antibody 43411 and syntaxin were implemented on rat vas deferens as described previously (Schroeter et al., 2000). Briefly, vas deferens was obtained from perfused Sprague Dawley rats (Harlan, Indianapolis, IN), cryoprotected, frozen, and then sectioned at 14 μm with a cryostat and collected on slides. Sections were blocked and permeabilized for 30 min in 4% normal goat serum/0.3% Triton X-100/TBS (50 mm Tris, 90 mmNaCl, pH 7.4) and incubated with antibodies. All specimens were examined with a Zeiss (Oberkochen, Germany) LSM 410 confocal imaging system equipped with internal He/Ne and external Ar/Kr lasers (Vanderbilt University Medical Center Cell Imaging Core Resource). Z-series images were collected by optical sectioning at intervals of 1 μm. Image processing and montage assembly were performed with Adobe Photoshop.
Transport assays. NE transport assays on rat brain synaptosomes or minced rat vas deferens (Bauman et al., 2000) and uptake activity of cells (Apparsundaram et al., 1998a) were performed as described previously. All uptake assays were performed by using [3H]NE (1-[7,8-3H]noradrenaline; Pharmacia) at 50 nm final concentration, 37°C for 10 min. Nonspecific uptake was defined by using 1 μm desipramine (Sigma). Mean values for specific uptake (pmol/mg protein ± SEM) were determined from at least three experiments. BoNT/C1 (Calbiochem) was applied before uptake assays by incubating the synaptosomes or minced vas deferens with the toxin (10 ng/ml or as noted in the figures) for 1 hr at 37°C as described previously (Beckman et al., 1998; Deken et al., 2000). For experiments with drugs the synaptosomes or tissue slices were incubated with vehicle, β-PMA at 1 μm, or OA at 1 μm for 30 min at 37°C before transport assays. Effects of drugs and toxin on transport activity versus vehicle controls were evaluated with a two-tailed Student's t test, withp < 0.05 considered significant.
Cell culture and transfection. CAD cells (Qi et al., 1997) were a generous gift from Dr. D. M. Chikaraishi (Duke University Medical Center, Durham, NC) and were maintained in DMEM/F-12 medium supplemented with 8% fetal bovine serum (FBS), 2 mml-glutamine (l-Glu), 100 IU/ml penicillin, and 100 μg/ml streptomycin (pen/strep). CAD-hNET cells were generated by stable transfection of hNET in pcDNA3 with Lipofectin (Invitrogen, San Diego, CA) and selected/maintained in the same medium with the addition of 200 μg/ml of G418 (Mediatech, Herndon, VA). SK-N-SH cells (ATCC, Manassas, VA) were maintained in RPMI 1640, 10% FBS, l-Glu, and pen/strep. Human embryonic kidney-293 (HEK-293) hNET cells were described previously (Galli et al., 1995). Chinese hamster ovary (CHO) cells and COS-7 cells were maintained in DMEM, 10% FBS, l-Glu, and pen/strep. CHO-M3 cells were maintained in Ham's F-12, 10% FBS, l-Glu, and pen/strep. Transfections, unless otherwise mentioned, were performed with Fugene 6 (Roche, Indianapolis, IN) according to the manufacturer's instruction. Typically, 1 μg cDNA was added to 300,000 cells in each well of six-well plates, or 200 ng of cDNA was added to 50,000 cells per well in 24-well plates. All cells were incubated for 24–48 hr before assay. Amounts of syntaxin cDNAs that were transfected were adjusted after plasmid titration experiments to diminish nonspecific effects on NET protein synthesis (reported in the figure legends). Transfection of oligonucleotides into CADhNET cells was performed as described by Beckman et al. (1998) with minor modification. The sense and antisense oligonucleotides, corresponding to bases –1 to 18 bp of mouse syntaxin 1A (sense strand 5′-CATGAAGGACCGAACCCAG-3′), were synthesized in the Vanderbilt University Medical Center DNA Chemistry Core Facility. A mixture of oligonucleotides, 1% serum-containing medium, and Lipofectamine (Invitrogen) was added on cells plated on poly-d-lysine (Boehringer Mannheim). Cells were incubated for 2 hr, supplemented with 5× volume of normal media, and further incubated for 42 hr before assays.
Biochemical analysis. Glutathione S-transferase (GST) or MBP fusion proteins were expressed in Escherichia coli BL21pLysS (Invitrogen) and induced with 1 mm IPTG for 4 hr. Fusion proteins were purified by one-step affinity chromatography, using glutathione beads (Amersham Biosciences, Uppsala, Sweden) or amylose resin (New England Biolabs) according to the manufacturers' instructions. All gel analyses were performed by using 10% SDS-PAGE. Pull-down experiments were performed as described previously (Deken et al., 2000). Briefly, ∼150 pmol of fusion proteins was bound to 15 μl of amylose or glutathione resin before the experiments. Protein-coated beads were incubated with GST-SynΔTM in PBS or with cell lysates expressing hNET in PBS/1% Triton X-100, containing 0.5 mm phenylmethylsulfonyl fluoride (PMSF; Sigma) at 4°C for 1–2 hr. Immunoprecipitation of the extracts from rat vas deferens was performed by using 43411 antisera against NET as described by Bauman et al. (2000). For immunoprecipitation of His-hNET the transfected cells in six-well plates were washed with (in mm) 50 NaH2PO4, 10 Tris, 100 NaCl, 0.5 PMSF, pH 8.0, and incubated in 400 μl/well of lysis buffer [containing (in mm) 50 NaH2PO4, 10 Tris, 100 NaCl, 0.5 PMSF, pH 8.0, plus 1% Triton X-100] for 1 hr at 4°C. Cell lysates were recovered by centrifugation at 20,000 × gfor 30 min at 4°C and incubated with anti-histidine antibody for 1 hr to overnight at 4°C. Complexes were retrieved by the addition of 15 μl of protein G-Sepharose (Amersham Biosciences), followed by three washes with the same lysis buffer. For drug treatments the cells were preincubated in serum-free medium overnight and further incubated with serum-free medium containing β-PMA at 0.1–1 μm or OA at 1 μmfor 30 min. Cell surface biotinylation was performed as described in detail previously (Apparsundaram et al., 1998b), using EZ-link NHS-sulfo-S-S-biotin (Pierce, Rockford, IL), followed by streptavidin bead capture. Bound proteins were eluted by using Laemmli sample buffer containing 3% β-mercaptoethanol. For the immunoprecipitation of surface proteins the cells were biotinylated with EZ-link NHS-sulfo-S-S-biotin (Pierce) and lysed as described above. Monomeric avidin beads (15–30 μl of beads/cell lysates from one well; Pierce) were preblocked with 10 mg/ml BSA in lysis buffer before use for the capture of biotinylated proteins. Avidin beads were washed five times with lysis buffer, and bound proteins were eluted by using three washes (total 600 μl) with lysis buffer containing 2 mm biotin (Sigma). Anti-histidine antibody was added to the eluted proteins and processed for immunoprecipitation as described above. For estimation of relative amounts of proteins in immunoblots, exposed films of immunoblots were scanned with an Agfa Duoscan T1200, and the captured images were processed in Adobe Photoshop and quantitated with NIH Image. Multiple films were exposed for each immunoblot to insure linearity of detection.
Electrophysiology. Cells stably transfected with the hNET were plated at a density of 105 per 35 mm culture dish. Before electrical recordings the attached cells were washed three times with the bath solution of (in mm) 137 NaCl, 2.7 KCl, 1.5 KH2PO4, and 9.7 NaHPO4 pH-adjusted to 7.4 and 276 mOsm. The recording pipette, in the cell-detached inside-out configuration, was filled with a solution containing the following (in mm): 130 NaCl, 1.3 KH2PO4, 0.5 MgSO4, 1.5 CaCl2, 10 HEPES, and 34 dextrose pH-adjusted to 7.35 and 300 mOsm. NE (30 μm) was added to the patch electrode solution to activate NET channel-like activity along with ascorbic acid (100 μm) to prevent NE oxidation. GST, GST-SynΔTM, and cocaine were dissolved in the bath solution used to perfuse the cytoplasmic face of the plasma membrane of the detached patch. Quartz electrodes (10 MΩ) were pulled with a programmable puller (P-2000, Sutter Instruments, Novato, CA). An Axopatch 200B amplifier band-limited at 1000 Hz was used to measure NET channel activity. Single channel events were recorded at RT with a membrane patch potential set to –80 mV. Data were stored digitally on a VCR and analyzed with a Nicolet Integra Model 20 oscilloscope and a DELL computer, using instrumentation and programs written by W. N. Goolsby (Emory University, Atlanta, GA). Multiple 8 sec traces captured for each condition (GST, GST-SynΔΤΜ, cocaine) were analyzed to calculate the mean open time (NPo). Comparisons were performed by a one-way ANOVA, followed by Tukey's test, with p < 0.05 taken as significant.
Results
NET-mediated NE transport requires syntaxin 1A
To evaluate the syntaxin 1A dependence of NET-mediated NE transport, we established hNET stably transfected CAD cells (CADhNET) and examined whether antisense oligonucleotide-mediated suppression of syntaxin 1A synthesis would influence NE transport. CAD cells are catecholaminergic neuroblastoma cells (Qi et al., 1997) that natively express syntaxin 1A protein. We found that a 2 d treatment with syntaxin 1A antisense oligonucleotides, but not sense oligonucleotides, markedly diminished syntaxin 1A levels (Fig.1A, top). In parallel, we measured a significant, dose-dependent reduction in desipramine-sensitive NE transport activity (Fig.1A). Because CAD cells lack other pathways for NE accumulation at the concentrations that have been used, these findings suggest that syntaxin 1A-dependent processes linked to NET surface expression or intrinsic activity were modified. An independent, and more rapid, paradigm for inactivation of syntaxin 1A involves cleavage of the SNARE protein with BoNT/C1. BoNT/C1 cleaves syntaxin 1A near the plasma membrane and destroys its activity in mediating SNARE-dependent vesicular fusion (Schiavo et al., 2000). Efficient cleavage of syntaxin 1A was achieved with BoNT/C1 after 1 hr incubations of rat cortical synaptosomes, slices from rat vas deferens, human SK-N-SH cells (express native hNET), or hNET stably transfected CAD cells, as revealed by syntaxin 1A immunoblots (Fig. 1B). Assays of NET activity performed on these preparations revealed a significant 50–75% loss of desipramine-sensitive NE transport activity (Fig.1B). The reduction of NE transport mediated by BoNT/C1 transport in SK-N-SH cells was dose-dependent and saturated at 10 ng/ml toxin (data not shown), with ∼25% activity retained after 1 hr of treatment.
Fig. 1.
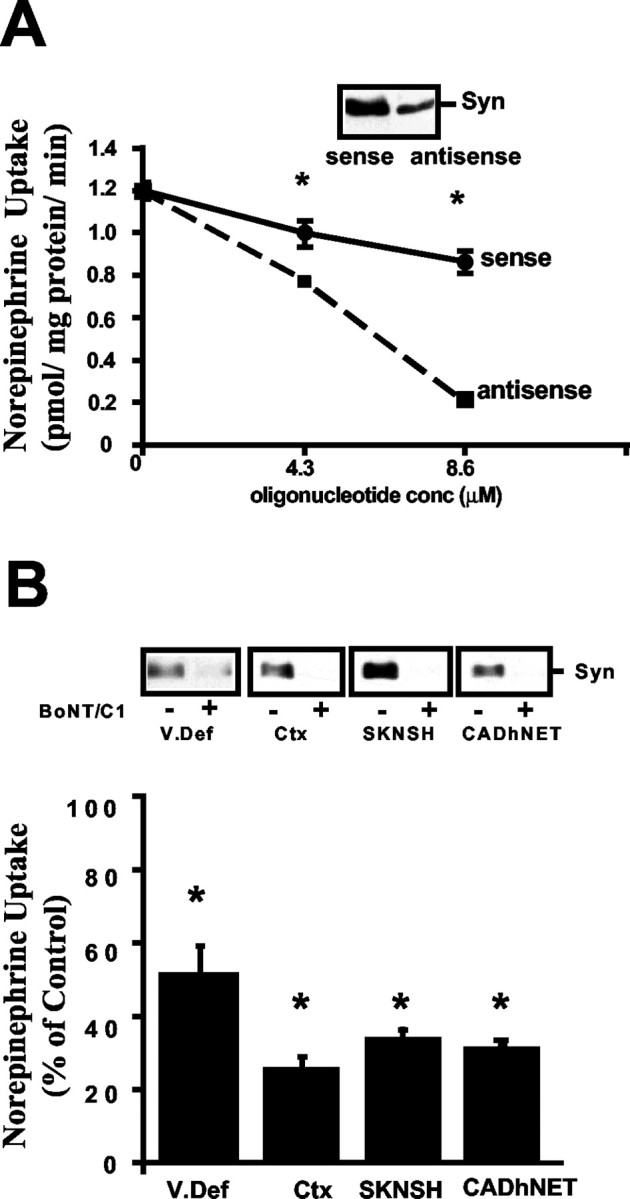
Toxin or antisense disruption of syntaxin 1A diminishes NE transport activity. A, Antisense suppression of syntaxin 1A expression reduces NE transport in CADhNET cells. CADhNET cells were transfected with mouse sense or antisense syntaxin 1A oligonucleotides and assayed for syntaxin content or NE transport activity 2 d later. The cells in one well for each transfection were lysed in PBS/1% Triton X-100, assayed for protein, and then immunoblotted for syntaxin 1A content. Cells treated with antisense syntaxin 1A oligonucleotides (8.6 μm) displayed a reduction of syntaxin 1A protein, compared with the cells treated with sense oligonucleotides. In parallel, a dose-dependent effect of syntaxin 1A antisense oligonucleotides on NE transport activity was observed. Results are mean values ± SEM (n = 3); *p < 0.05; Student's t test.B, Treatments with BoNT/affect syntaxin cleavage and reduce NE transport in native rat tissues. Minced rat vas deferens and synaptosomes from rat brain cortex as well as SK-N-SH cells and CADhNET cells were incubated with BoNT/C1 for 1 hr at 37°C before NE transport assay. Aliquots of tissue extracts or cell lysates treated with BONT/C1 (+) or vehicle (−) were immunoblotted for syntaxin 1A content. BONT/C1-treated tissues or cells displayed weak or no immunoreactivity for syntaxin 1A in contrast to vehicle-treated cells. Values reported are mean transport activities ± SEM (n = 3); *p < 0.05; Student'st test.
Syntaxin 1A colocalizes and associates with NET
The effects on NE transport activity after syntaxin 1A oligonucleotide and toxin treatments could represent long-range or indirect effects of SNARE manipulation or, alternatively, could arise from disruptions of more intimate associations. In the latter case, NET and syntaxin 1A proteins would be expected to colocalize in neurons. Syntaxin 1A is expressed on axonal membranes and at membrane terminals in the CNS (Bennett et al., 1993; Sesack and Snyder, 1995) as well as on noradrenergic varicosities in the periphery (Brain et al., 1997). NET proteins also are enriched on sympathetic noradrenergic axonal membranes (Schroeter et al., 2000). To establish whether syntaxin 1A and NET proteins are colocalized, we double-labeled cultured sympathetic neurons prepared from mouse SCG. After differentiation the SCG cultures elaborate neurites with periodic, varicose enlargements that label with FM1-43, a dye taken up after synaptic vesicle recycling (data not shown). Staining of these cultures with syntaxin 1A and NET antibodies revealed colocalization at the varicosities (Fig.2A–F). Using permeabilized cells, we obtained similar results with either an intracellular NET-directed antibody (43411) or a surface epitope-directed antibody (43408). By using the surface epitope-directed NET antibody on living cells, however, followed by fixation and permeabilization to detect the cytoplasmically directed syntaxin 1A epitope, we could determine that this colocalization was evident with plasma membrane-inserted NET and not a consequence of close apposition between NET transport vesicles and the plasma membrane. To verify these findings in native tissues, we immunolabeled the rat vas deferens, a peripheral preparation rich in noradrenergic axons and high-affinity desipramine-binding sites (Raisman et al., 1982), and found again NET and syntaxin 1A labeling to be strikingly discontinuous and colocalized, consistent with the spacing and size of sympathetic varicosities (Fig. 2G–L).
Fig. 2.

Colocalization of NET and syntaxin 1A in sympathetic axons. Mouse superior cervical ganglion cultures were cultured for 5 d and stained for NET and syntaxin 1A as described in Materials and Methods. In A–C, double staining was performed by using permeabilized, fixed cells to reveal total NET (antibody 43408; A) and syntaxin immunoreactivity (B); a merged image is shown in C. In D, labeling of live, nonpermeabilized cells was achieved with 43408 antibody to detect surface NET protein, followed by permeabilization to detect cytoplasmic labeling of syntaxin 1A (E). Note the overlap in labeling apparent as yellow fluorescence in the merged image (F), particularly evident at varicosities (arrows). Colocalization of NET and syntaxin 1A in rat vas deferens is shown in G–L. Frozen sections of rat vas deferens were double labeled with anti-NET (43411 antibody) and anti-syntaxin 1A as described in Materials and Methods, and the immunofluorescence was detected by confocal microscopy. The merged images (I, L) demonstrate the discontinuous and colocalized expression of NET and syntaxin 1A along sympathetic axonsin vivo. Scale bars: A–C, 15 μm;D–F, 7 μm, G–L, 5 μm.
Our evidence that NET and syntaxin 1A colocalize at noradrenergic varicosities could reflect coexpression of the two proteins at synaptic membranes in separate complexes. However, the results of native tissue coimmunoprecipitation experiments support a more direct interaction (Fig. 3A,B). NET immune serum (the same serum used for immunocolocalization studies), but not preimmune serum, precipitated syntaxin 1A from the rat vas deferens (Fig. 3A). In contrast, we detected no 25 kDa synaptosome-associated protein (SNAP-25) above background (data not shown). Similar results were found with coimmunoprecipitation experiments that used the mouse vas deferens (Fig. 3B). Importantly, this preparation allowed us to check the specificity of our immunoprecipitations with tissue derived from NET knock-out mice (Xu et al., 2000). Indeed, we recovered little or no syntaxin 1A in NET immunoprecipitations by using homozygous (−/−) vas deferens despite normal levels of syntaxin 1A protein (Fig. 3B).
Fig. 3.
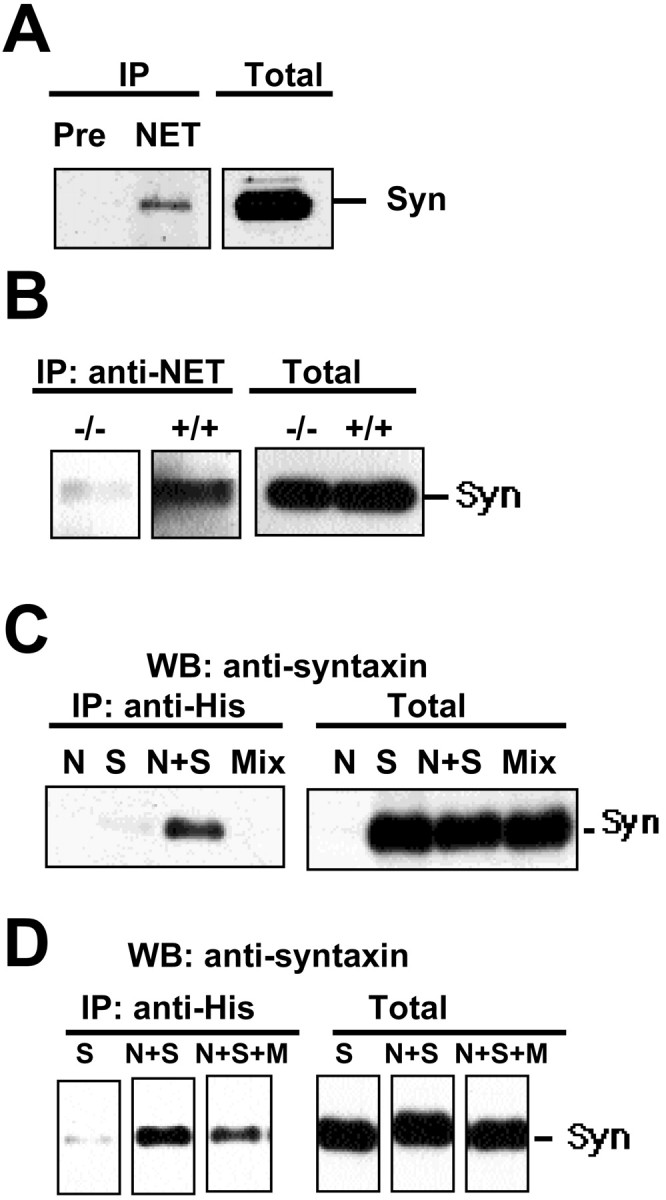
Coimmunoprecipitation of NET and syntaxin 1A.A, Syntaxin 1A coimmunoprecipitates with NET. Solubilized rat vas deferens membranes were immunoprecipitated with NET antisera 43411 or preimmune serum, and complexes were resolved by SDS-PAGE, followed by immunoblotting for syntaxin 1A. An aliquot of the total extracts was blotted in parallel. B, Coimmunoprecipitation of NET and syntaxin 1A is diminished in vas deferens extracts from NET knock-out mice. Extracts were prepared from wild-type C57BL/6 (+/+) and homozygous null (−/−) NET knock-out mice as described for rat preparations and were immunoprecipitated with NET antibody 43411 before syntaxin 1A immunoblot. Total extracts were blotted for syntaxin 1A in parallel and showed no loss of syntaxin as a result of NET deficiency. C, Coimmunoprecipitation of syntaxin 1A and His-NET in cotransfected CHO cells. As noted by others (Bittner et al., 1996; Rowe et al., 1999), reduced concentrations of syntaxin 1A cDNA were required in cotransfection studies to limit the suppression of hNET biosynthetic progression, observed as diminished N-glycosylated cell surface transporters (see below). CHO cells grown in six-well plates were singly or cotransfected with His-tagged hNET (670 ng) and full-length syntaxin 1A (45 ng). Cell lysates were immunoprecipitated with anti-His, resolved on SDS-PAGE, and blotted for syntaxin 1A. Immunoprecipitations also were performed with extracts mixed from separately transfected cells (Mix). Aliquots of total cell lysates show equal expression of syntaxin 1A in each transfection (Total) except for lysates derived from cells transfected with only hNET. CHO cells do not express endogenous syntaxin 1A. D, Coexpression of Munc18 diminishes recovery of syntaxin 1A from NET immunoprecipitates. CHO cells were transfected with syntaxin 1A alone (S, 42 ng), His-hNET (640 ng) and syntaxin 1A (42 ng) (N+S), or His-hNET (640 ng), syntaxin 1A (42 ng), and Munc18 (318 ng) (N+S+M). pcDNA3 was used to adjust transfections to 1 μg of total DNA for N and N+S. Extracts were immunoprecipitated with anti-HIS before SDS-PAGE and syntaxin 1A immunoblots. Blots of total cell lysates reveal equivalent expression of syntaxin 1A. Results presented in A–D are representative of two to six experiments for each condition.
Next we sought to reconstitute a NET/syntaxin 1A interaction in cotransfected mammalian cells to provide a model system suitable for a structural and functional characterization of transporter/SNARE interactions. For these studies CHO cells were transiently transfected with either, or both, syntaxin 1A and His-tagged hNET cDNAs. We found that NH2 terminal His or HA-tagged hNET cDNAs express equivalently to hNET cDNAs (data not shown). We found that NET antibodies failed to immunoprecipitate syntaxin 1A from cells transfected with either hNET or syntaxin 1A alone. In contrast, syntaxin 1A was readily detectable in immunoprecipitates of dually transfected cells (Fig. 3C). To control for the possibility that our coimmunoprecipitation results arise from nonspecific aggregation of solubilized proteins, we mixed detergent extracts prepared from separately transfected cells and then repeated our immunoprecipitation experiments but found no evidence of association (Fig. 3C). As an additional test of specificity, we sought to compete for hNET/syntaxin 1A interactions with the high-affinity syntaxin-binding protein Munc18 (Hata et al., 1993; Pevsner et al., 1994). Transfected Munc18 cDNA had no effect on the amount of hNET (data not shown) or syntaxin 1A protein evident in total extracts. However, Munc18 transfection significantly reduced the amount of syntaxin 1A recovered from NET immunoprecipitates (Fig. 3D). Therefore, NET and syntaxin 1A form a stable complex in intact cells that, like the assembly of SNARE complexes engaged in vesicular fusion (Jahn and Sudhof, 1999), can be influenced by the availability of additional cellular syntaxin 1A binding partners.
Syntaxin 1A binds directly to the hNET N terminus
Possibly, NET/syntaxin 1A complexes observed in immunoprecipitates of cotransfected CHO cells could reflect indirect associations. Because we also were able to achieve similar results by using cotransfected COS-7 and CAD cells (data not shown), we suspected that the lack of host cell specificity to these interactions might indicate direct interactions as observed for GAT1, CFTR, sodium, potassium, and calcium channels (Bezprozvanny et al., 1995; Naren et al., 1997; Beckman et al., 1998; Saxena et al., 1999; Yang et al., 1999; Fili et al., 2001). To explore this question, we first tested a GST fusion protein containing the cytoplasmic domain of syntaxin 1A (GST-SynΔTM) (Naren et al., 1997; Deken et al., 2000) for its ability to extract NET proteins from detergent extracts of NET transfected CHO cells. Whereas GST protein recovered little or no NET in pull-down experiments, GST-SynΔTM fusion bound NET significantly above background (Fig. 4A). NETs exist in transfected cells in both 60 kDa immature forms and 90 kDa mature forms, differing the extent of N-glycosylation (Melikian et al., 1996). GST-SynΔTM was able to extract both forms of NET from detergent extracts, indicating that interactions are likely independent of transporter carbohydrate modifications.
Fig. 4.
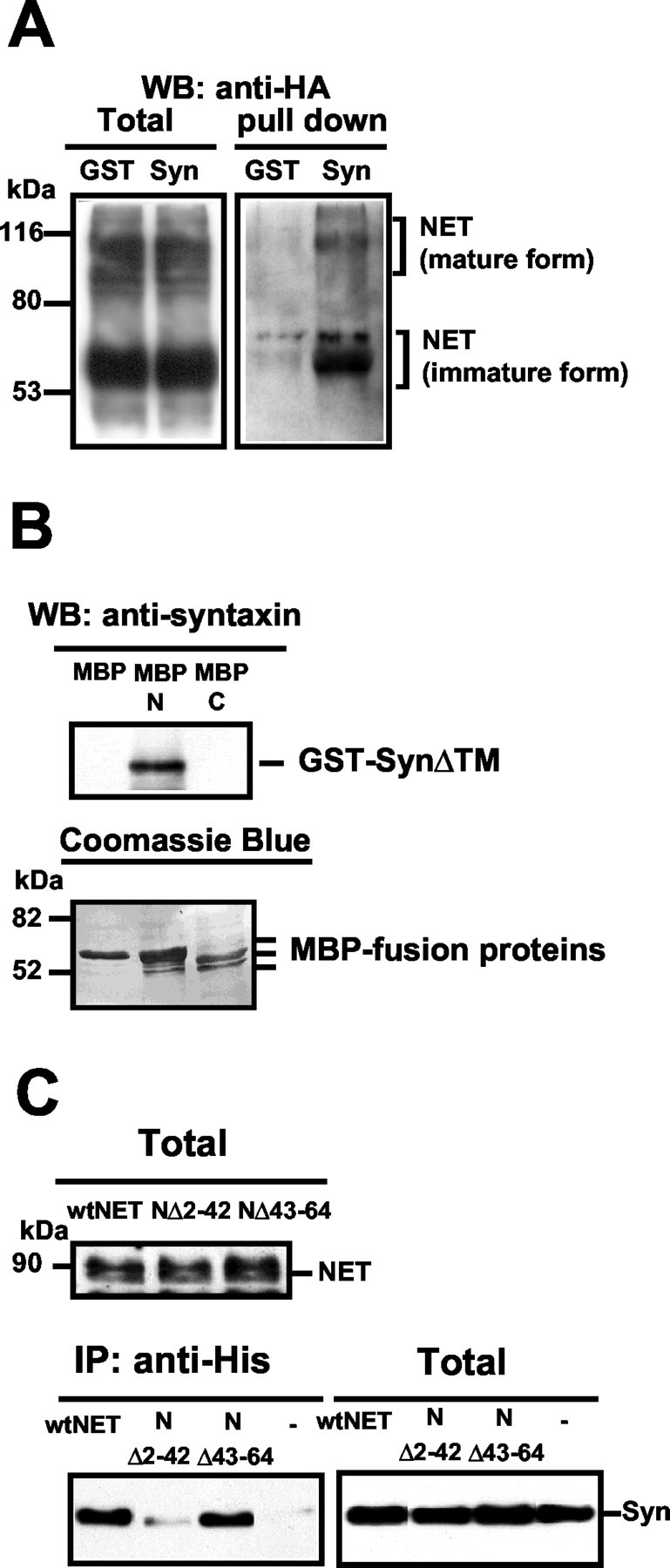
Syntaxin 1A binds NET directly via sequences in the NH2 terminus of the transporter. A, GST-SynΔTM pull-down of NET protein. COS-7 cells were transfected with HA-tagged hNET, and detergent lysates were incubated with glutathione beads precoated with either GST (GST) or GST-SynΔTM (Syn). Proteins bound to the beads were eluted and subjected to SDS-PAGE, followed by immunoblotting with anti-HA. Unlike GST beads, GST-SynΔTM beads retrieved NET proteins both in the immature and mature forms. B, Direct binding of syntaxin 1A cytoplasmic domain to the hNET NH2 terminus. Amylose resins, precoated with equimolar MBP, MBP-hNET NH2 terminal protein (MBP-N), or MBP-hNET COOH terminal protein (MBP-C), were incubated with GST-SynΔTM as described in Materials and Methods, followed by elution of bound material, SDS-PAGE, and immunoblotting for syntaxin 1A. Only MBP-N retained GST-SynΔTM. Membranes subsequently were stained with Coomassie brilliant blue to reveal equivalent amounts of MBP fusion proteins used in the experiments. C, An hNET NH2 terminal deletion disrupts NET/syntaxin 1A coimmunoprecipitation. The top panel shows an immunoblot of hNET along with the NH2 terminal deletion mutants that were used. CHO cells were transfected with either His-hNET (wtNET) or His-hNET mutants NΔ2–42 or NΔ43–64. Aliquots of extracts were analyzed in SDS-PAGE and probed with polyclonal anti-His antibody to reveal equivalent expression. The bottom panel shows results of cotransfection of N or NET mutants (670 ng) with syntaxin 1A (45 ng)/coimmunoprecipitation experiments, immunoprecipitating with anti-His and probing for syntaxin 1A. The NΔ2–42 mutant significantly diminished syntaxin 1A recovery relative to wt hNET or NΔ43–64. Syntaxin 1A expression was equivalent in all samples as assessed with syntaxin 1A immunoblots of total cell extracts. Results presented in A–C are representative of three to six experiments for each condition.
We next considered whether a discrete domain of NET supported syntaxin 1A associations. Although the GAT1 NH2 terminus has been reported to serve as a direct binding partner for syntaxin 1A (Deken et al., 2000), this region exhibits limited conservation with hNET and other members of the gene family. To explore the possibility that the equivalent hNET domain harbors a structurally analogous docking site for syntaxin 1A, we synthesized MBP fusions incorporating either the hNET N or C terminus (MBP-N and MBP-C, respectively) and tested their ability to bind to GST-SynΔTM in vitro. MBP-N, but not MBP-C nor MBP, could be recovered in GST-SynΔTM pull-down assays (Fig. 4B). If the latter findings are important for NET/syntaxin 1A interactions in intact cells, we expect that mutation of NH2 terminal NET sequences should abolish interactions in intact cells. A full deletion of the hNET N terminus (Δ2–64) fails to support normal levels of transporter protein expression and could not be examined further. However, hNET deleted from amino acids 2–42 (hNETΔ2–42) or from amino acids 43–64 (hNETΔ43–64) is expressed at levels equivalent to hNET (Fig.4C, top). Importantly, the hNETΔ43–64 deletion spans the area implicated in syntaxin 1A/GAT1 interactions (Deken et al., 2000). Remarkably, coimmunoprecipitation experiments revealed no impact of the hNETΔ43–64 deletion on syntaxin 1A recovery, relative to full-length hNET (Fig. 3C, bottom). Moreover, because this region harbors several acidic residues (51D, 53D, 58E) that could be homologous to the sites of charge–charge pairing proposed to stabilize syntaxin 1A and GAT1 interactions (Deken et al., 2000), we mutated these sites in hNET to alanine but also failed to disrupt hNET/syntaxin 1A associations (data not shown). In contrast, the Δ2–42 mutation effectively abolished coimmunoprecipitation of syntaxin 1A. These findings demonstrate that, although both GAT1 and NET bind syntaxin 1A, distinct domains within the NH2 termini of these transporters support stable interactions with the SNARE protein.
Acute regulation of the NET/syntaxin 1A interaction
As noted previously (Bauman et al., 2000), acute (1 μm, 30 min, 37°C) treatment of rat vas deferens slices with the phorbol ester β-PMA significantly diminishes desipramine-sensitive NE transport activity (Fig.5A), an effect attributable to a diminished NE transport capacity (Vmax control = 6.9 ± 0.7 pmol/mg per min, Vmax β-PMA = 4.8 ± 0.47 pmol/mg per min; Kmcontrol = 416 ± 32 nm,Km β-PMA = 391 ± 22 nm). Given our findings of NET/syntaxin 1A colocalization and complex formation, we next examined the sensitivity of these associations to phorbol ester treatment and found a significant loss of the SNARE protein in coimmunoprecipitation experiments (Fig. 5B). Recently, we also have shown that PP1/2A phosphatase antagonists also diminish NET activity in the vas deferens in vitro (Fig. 5A) and in vivo (Bauman et al., 2000). Okadaic acid treatments (1 μm, 30 min, 37°C) that diminish NE transport capacity also significantly diminish syntaxin 1A content in NET immunoprecipitates (Fig. 5B). Finally, we ascertained whether syntaxin 1A participates in phorbol ester-mediated NET regulation by measuring transport activity in rat synaptosomes exposed to BoNT/C1. As shown in Figure 5C, whereas β-PMA triggered a reduction of NE transport activity in untreated synaptosomes, this regulation was lost in BoNT/C1-treated preparations. These findings indicate that syntaxin 1A/NET associations are responsive to phorbol esters and that they are required to elaborate β-PMA-triggered NET downregulation.
Fig. 5.
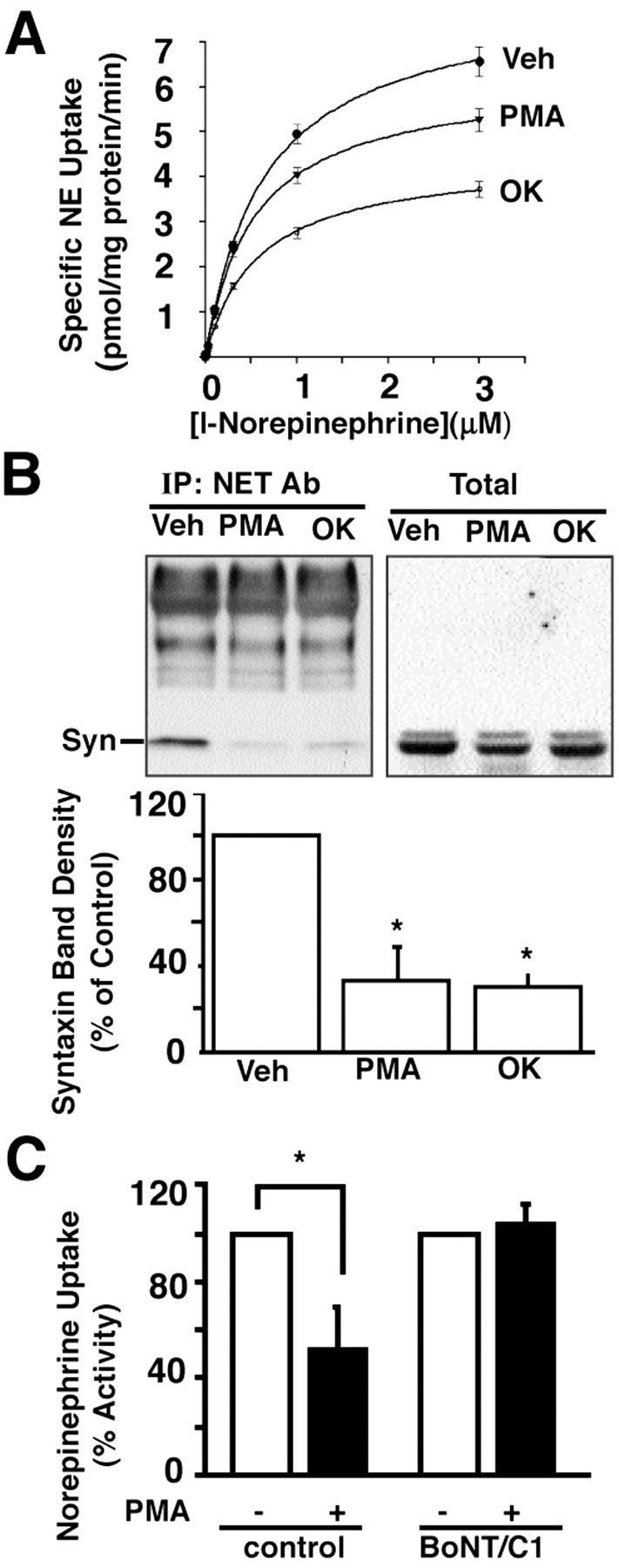
Phorbol ester and okadaic acid modulate NE transport and levels of NET/syntaxin 1A complexes in rat vas deferens.A, NE transport activity of rat vas deferens. Minced vas deferens was pretreated with DMSO (Veh), 1 μm β-PMA (PMA), or 1 μmokadaic acid (OK) for 30 min at 37°C and subjected to evaluation of NE transport activity. MeanKm and Vmaxvalues were obtained from three separate experiments; forVmax, control = 6.9 ± 0.7 pmol/mg per min, PMA = 4.8 ± 0.47 pmol/mg per min, and OK = 4.1 ± 0.55 pmol/mg per min.Vmax values of PMA and OK are different from the control value in the analysis of one-way ANOVA, followed by Tukey's test;p < 0.05. The mean Kmvalues were control = 416 ± 32 nm, PMA = 391 ± 22 nm, and OK = 364 ± 41 nm. The differences of Km values are not statistically significant. B,Top, Evaluation of NET/syntaxin coimmunoprecipitation after phorbol ester or okadaic acid treatment. Minced rat vas deferens was pretreated with DMSO (Veh), 1 μm PMA (PMA), or 1 μm okadaic acid (OK), as described in A, before extraction, immunoprecipitation with anti-NET sera 43411, SDS-PAGE, and immunoblotting for syntaxin 1A. Total extracts from all treatments were blotted in parallel for syntaxin 1A and revealed equivalent levels.Bottom, Average syntaxin band density ± SEM from three different immunoprecipitation experiments conducted as intop panel were quantitated by densitometric scanning; the values obtained after phorbol ester or okadaic acid treatments are expressed as a percentage of syntaxin 1A levels found in the vehicle-treated sample. *Significant loss of syntaxin 1A from NET immunoprecipitates as assessed by one-way ANOVA, followed by Tukey's comparisons of group means; p < 0.05.C, Phorbol ester-induced downregulation of NET activity in rat synaptosomes is lost after BONT/C1 pretreatment. Rat cortical synaptosomes, prepared as described in Materials and Methods, were pretreated with BONT/C1 (100 nm) 1 hr before treatment with either vehicle or 1 μm PMA for 30 min, followed by assay of [3H]NE transport as described in Materials and Methods. Results reflect the mean of three experiments ± SEM; *p < 0.05; Student's t test.
Consistent with our findings of a regulated NET/syntaxin 1A complex in native tissues, we also found that syntaxin1A/hNET complexes could be destabilized in cotransfected CHO cells after acute treatments with phorbol esters or okadaic acid (Fig.6A). The PKC inhibitor staurosporine, which blocks phorbol ester-triggered downregulation of NET (Apparsundaram et al., 1998a,b), reverses phorbol ester-induced dissociation of NET/syntaxin 1A complexes in this model (data not shown). M3 muscarinic receptors are coupled to phospholipase C and PKC activation and have been shown to trigger rapid NET downregulation and internalization in the noradrenergic neuroblastoma SK-N-SH (Apparsundaram et al., 1998a). We cotransfected His-hNET and syntaxin 1A into M3-CHO cells and found that the muscarinic agonists methacholine and carbachol destabilized NET/syntaxin 1A complexes (Fig.6B). Together, these findings indicate that the NET/syntaxin 1A interaction is not constitutive but, rather, can be affected by stimuli known to alter NET trafficking.
Fig. 6.
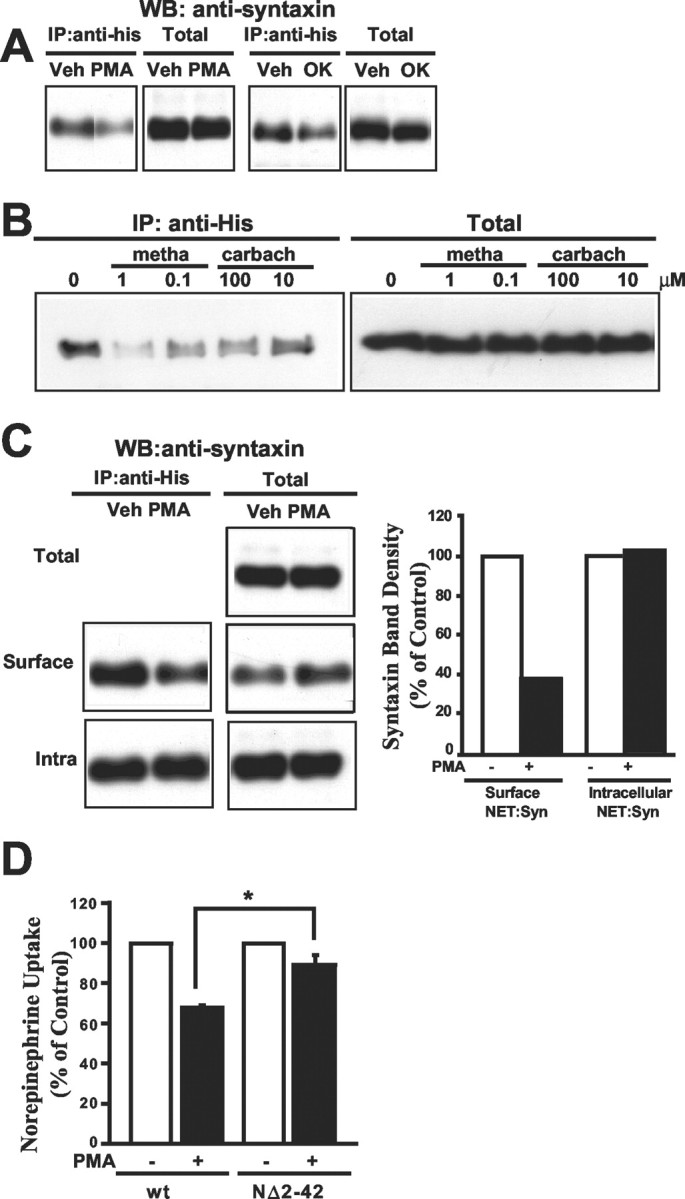
Regulated association of NET and syntaxin 1A in cotransfected CHO cells. A, Phorbol ester or okadaic acid treatments diminish recovery of syntaxin 1A from NET immunoprecipitations. CHO cells, cotransfected with His-hNET and syntaxin 1A, were preincubated with DMSO (Veh), 1 μm β-PMA (PMA), or 1 μmokadaic acid (OK) for 30 min at 37°C before immunoprecipitation with anti-His, SDS-PAGE, and immunoblotting for syntaxin 1A. Total cell extracts for each condition were blotted for syntaxin 1A in parallel. Both PMA and okadaic acid diminished recovery of syntaxin 1A relative to vehicle-treated samples. B, Muscarinic receptor activation diminishes recovery of syntaxin 1A from NET immunoprecipitates. Stable M3 muscarinic receptor-transfected CHO cells that had been cotransfected transiently with His-hNET and syntaxin 1A were treated with the indicated concentrations (in μm) of the muscarinic agonists methacholine or carbachol (30 min) before extraction and immunoprecipitation of complexes with anti-His, SDS-PAGE, and blotting for syntaxin 1A. In parallel, syntaxin 1A was blotted from total cell extracts and is evident at equivalent levels in all conditions. C, Phorbol ester regulation of the interaction between NET and syntaxin 1A occurs with plasma membrane-localized complexes. CHO cells, cotransfected with His-hNET and syntaxin 1A, were treated with DMSO (Veh) or 1 μm β-PMA for 30 min at 37°C. Surface proteins were labeled with NHS-sulfo-biotin at 4°C before cell lysis and recovery of surface complexes (Surface) on avidin beads. Bound proteins were eluted with 2 mm biotin, immunoprecipitated with anti-His, resolved on SDS-PAGE, and immunoblotted for syntaxin 1A. Nonbound (Intra) extracts were immunoprecipitated and blotted in parallel. Phorbol ester-induced reduction in syntaxin 1A in NET immunoprecipitates is evident in surface fractions, but not in intracellular complexes. Blots of total cell extracts (Total) and nonbiotinylated, intracellular samples (Intra Total) show no impact of phorbol ester on syntaxin 1A content. PMA increased syntaxin 1A contents in total biotinylated pools (Surface Total). The bar graph on the right is a quantitation of syntaxin 1A recovery in the immunoprecipitates. D, hNET NH2 terminal deletion that disrupts NET/syntaxin 1A interactions diminishes phorbol ester-mediated NET downregulation. CHO cells were transfected with His-hNET (wt) or hNET NΔ2–42 as described in Figure 4, followed by treatment of cells with 1 μm β-PMA or vehicle for 30 min. Cells receiving hNET NΔ2–42 were significantly less sensitive to phorbol ester treatment with respect to NE transport activity (n = 3; *p < 0.05; Student's ttest).
Access to a cell culture model supporting regulated NET/syntaxin associations allowed us to implement biotinylation techniques to explore whether NET/syntaxin complexes are evident on the plasma membrane and whether these complexes are regulated selectively by β-PMA. We repeated our coimmunoprecipitation experiments as above, except that before immunoprecipitation we enriched the pool of plasma membrane NET protein complexes by surface biotinylation and subsequent capture on immobilized avidin (Fig. 6C). Analysis of the avidin retained and nonretained complexes demonstrates that NET/syntaxin 1A complexes are recovered from both cell surface-enriched and intracellular fractions. However, evaluation of β-PMA sensitivity of these complexes differed because only the surface pool of NET/syntaxin 1A complexes was destabilized by β-PMA treatments. These findings suggest that the destabilization of the NET/syntaxin 1A complex may be a step in the pathway toward cell surface NET downregulation. In support of this idea, we found that loss of syntaxin 1A interactions incurred by the hNET Δ2–42 mutation limited the ability of phorbol ester (Fig. 6D) and methacholine (data not shown) treatments to downregulate NET activity.
Syntaxin 1A inhibits NET intrinsic activity
To this point our studies are consistent with an influence of syntaxin 1A on NET surface expression in both the SNARE-mediated fusion of vesicles harboring transporters as well as via direct associations that might be disassembled to permit NET internalization. An additional role for NET/syntaxin 1A associations is in the modulation of NET intrinsic activity, an idea supported by multiple studies showing direct actions of syntaxin 1A on transport and gating properties of other ion channels and transporters (Bezprozvanny et al., 1995; Naren et al., 1997; Beckman et al., 1998; Saxena et al., 1999; Yang et al., 1999; Fili et al., 2001). To ascertain whether syntaxin 1A modulates NET function, we took advantage of our ability to monitor NET-mediated channel activity in detached patches pulled from stably transfected HEK-293 hNET cells (Galli et al., 1995, 1998). These currents are recorded in the inside-out configuration with patch pipettes filled with 30 μm NE (Fig. 7) and defined as NET-dependent through (1) their absence from nontransfected cells, (2) their blockade by desipramine or cocaine, and (3) their correlation with amperometric spikes recorded on a separate catecholamine sensor placed beneath the patch (Galli et al., 1995,1998). Traces were isolated with well resolved inward current transients to focus attention on NET-dependent events separate from background channel activity. Figure 7A presents representative current traces of detached patches in response to sequential application of GST, GST-SynΔTM (syntaxin), GST (recovery), and cocaine. The average NPo of the NET channel measured from four independent experiments was 10.2 ± 2.8%. Notably, the application of 3 μmGST-SynΔTM decreased the channel-like activity by 83% (Fig.7B), comparable with that achieved with local perfusion of 20 μm cocaine (NPo reduced by 86%). Because NET channel activity could be recovered by perfusion of the membrane patch with GST, the interaction of syntaxin with the NET protein is a reversible process.
Fig. 7.
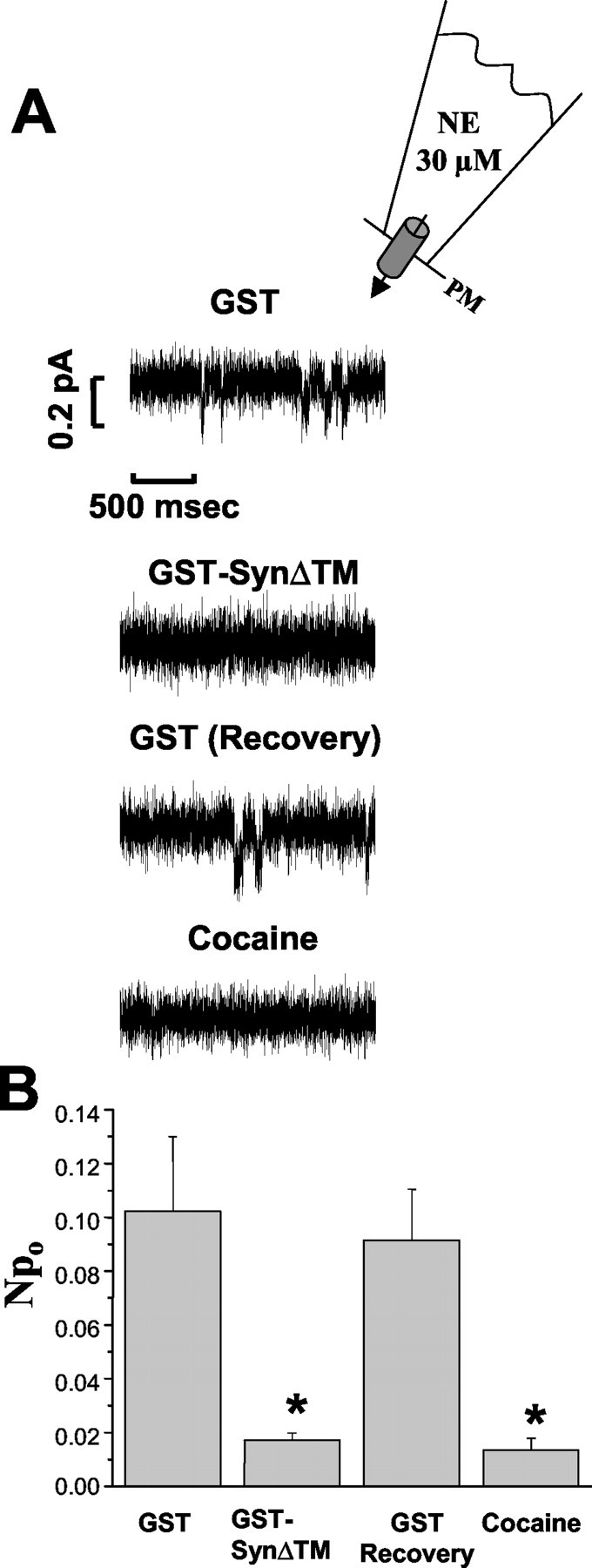
Syntaxin reversibly inhibits NET-mediated single channel currents. A, Four representative current traces of single channel activity measured from an individual inside-out membrane excised patch held at –80 mV with sequential application of GST (3 μm), GST-SynΔTM (3 μm), and cocaine (20 μm). The patch pipette was filled with 30 μm NE (inset) to open maximally the NET-dependent channels. B, Amplitude histograms for each trace were analyzed to calculate the cumulative open probabilityNPo for the NET-mediated channel-like events. Statistical analysis of the NPo determined from four independent experiments is shown. TheNPo of the channel decreased by 83% after perfusion of the membrane patch with GST-syntaxin 1AΔTM, compared with GST alone; NPo = 10.2 ± 2.8 to 1.7 ± 0.3%, respectively. The channel-like activity was recovered by perfusion with GST alone (NPo = 9.2 ± 1.9%). Cocaine sensitivity, tested after completion of the reversal experiments, revealed the hNET origin of the channel-like activity, reducingNPo to 1.4 ± 0.4%. *p < 0.05 by one-way ANOVA, followed by Tukey's test.
Syntaxin 1A cytoplasmic domain differentially affects NET surface expression and NE transport activity
A striking indication of the ability of syntaxin 1A to impact the intrinsic activity of NET proteins was found in transfection studies of the cytoplasmic domain of syntaxin 1A (SynΔTM) into CADhNET cells. Although SynΔTM cannot support SNARE-mediated fusion (McNew et al., 2000), we found that SynΔTM significantly increased NET surface expression (Fig. 8A) with no change in NET protein levels as measured in total cell extracts. We suspect that this activity arises from displacement of syntaxin 1A from host cell syntaxin-binding proteins (e.g., Munc18, NET) that normally would limit the availability of syntaxin 1A for membrane fusion of NET-containing vesicles (Jahn and Sudhof, 1999). Remarkably, we found that this increase in NET surface expression was not paralleled by a commensurate increase in NE transport activity (Fig. 8B). In multiple experiments we achieved an average surface recovery of NET proteins of 232 ± 22% versus controls after SynΔTM cotransfection, whereas the NE transport activity was unaffected, averaging 96 ± 2% of activity measured in the absence of SynΔTM cotransfection. This discrepancy is not attributable to saturation of surface capacities for NET function because, when cell surface expression is titrated across the same range simply by variation of transfected NET cDNA, an increase in NE transport activity is evident (Fig. 8C). We also found that cotransfection of full-length syntaxin 1A with hNET suppressed NE transport activity (data not shown). Together with our electrophysiological studies, these findings indicate that transport rates of NET proteins are not governed solely by NET surface expression and support the contention that the cytoplasmic domain of syntaxin 1A can influence NET activity.
Fig. 8.
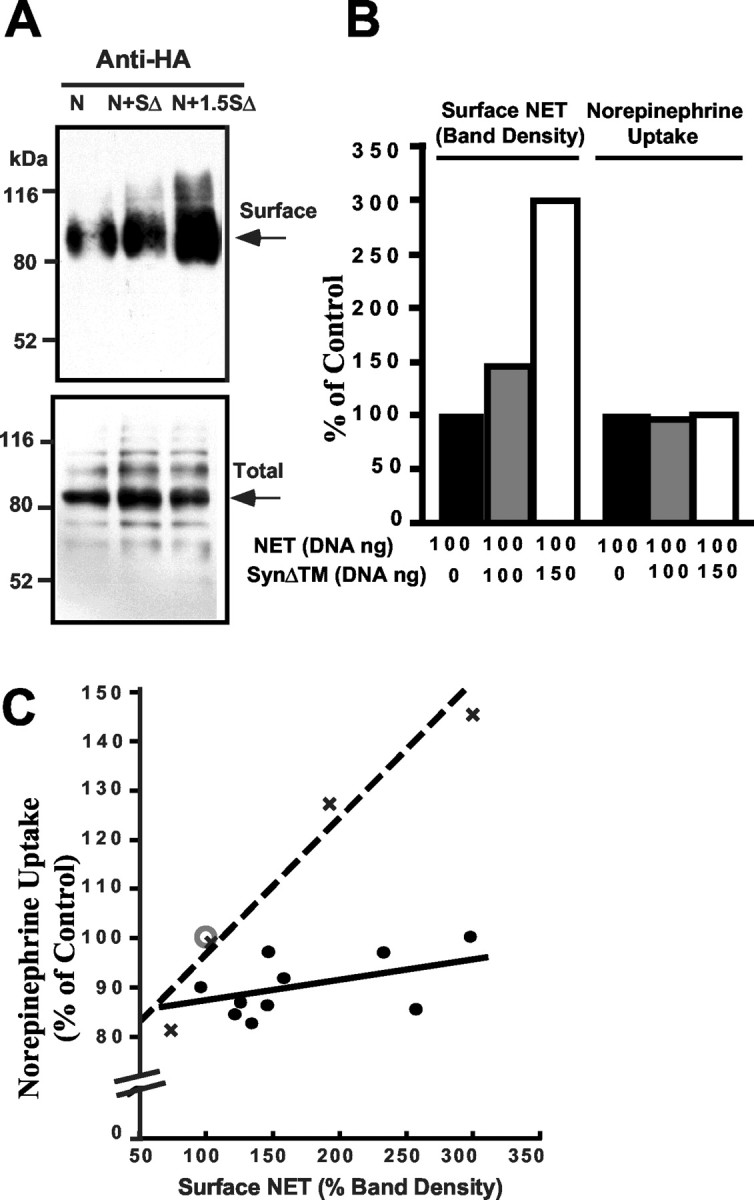
Fusion-incompetent form of syntaxin (SynΔTM) increases NET surface expression in CAD cells without an increase in NE transport activity. A, HA-hNET-transfected (100 ng) CAD cells (N) were transfected additionally with 100 ng SynΔTM (N+SΔ) or 150 ng SynΔTM (N+1.5SΔ). Surface proteins of transfected cells were labeled with NHS-sulfo-S-S-biotin, extracted, and isolated on streptavidin beads. Bound proteins were eluted, resolved by SDS-PAGE, and immunoblotted for NET by using anti-HA. SynΔTM increases recovery of surface biotinylated NET (top). Probing for NET in the whole extracts reveals no impact of SynΔTM on total NET levels (bottom). B, Impact of SynΔTM on NET activity in cells monitored for surface NET expression in parallel. Transfection, biotinylation, and NE transport assays were performed as described above, with one set of samples used for surface biotinylation/immunoblot with anti-HA and the other used for NE transport determinations. SynΔTM increases surface NET but does not increase NE transport activity. C, Analysis of the impact of SynΔTM on NET surface expression and NE transport activity in CAD cells. For cotransfection of HA-NET with SynΔTM (line with filled circles), CAD cells were transfected with 100–200 ng of HA-hNET and variable amounts of SynΔTM (50–300 ng). Surface NET proteins in immunoblots were quantitated and normalized to the amount of surface NET obtained in the absence of SynΔTM (set at 100%; open circle withX). Similarly, NE transport activity in these assays was compared with the activity of NET in the absence of SynΔTM cotransfection. The dotted line represents a comparison of surface NET density and transport activity when surface density was varied by varying the concentration of HA-hNET cDNA. Note that, whereas a comparable range of surface abundance is achieved via either of these two methods, transfection with SynΔTM blunts the ability of increasing hNET surface protein to result in an increase in NE transport activity.
Discussion
Norepinephrine inactivation after vesicular fusion is mediated predominantly by reuptake via cocaine- and antidepressant-sensitive NET proteins (Axelrod and Kopin, 1969; Iversen, 1971). Like all functionally characterized members of the Na+/Cl−-coupled neurotransmitter transporter gene family, a single cDNA suffices to confer high-affinity NE transport sites after heterologous expression. Recent studies suggest, however, that the surface expression and intrinsic activity of NETs and homologs are regulated by G-protein- and tyrosine kinase-linked receptor pathways (Apparsundaram et al., 1998a,b, 2001), bringing into focus the presence and activities of transporter-associated proteins. We recently have established that the catalytic subunit of PP2A forms a phorbol ester-sensitive complex with NETs, DATs, and SERT proteins (Bauman et al., 2000), suggesting that the phosphatase may coordinate transporter trafficking and/or intrinsic activity via phosphorylation-associated mechanisms (Blakely and Bauman, 2000). Torres and coworkers (2001) have shown that the PDZ domain protein PICK1 colocalizes and coimmunoprecipitates with NET, and α-synuclein has been reported to form a complex with the cocaine-sensitive DAT protein (Lee et al., 2001), although their participation in catecholamine transporter regulation is unclear.
Recently, we obtained direct evidence for the localization of NETs at sympathetic varicosities (Schroeter et al., 2000) and were struck by reports of a similarly restricted localization of the SNARE protein syntaxin 1A in the same membrane domain (Brain et al., 1997). Moreover, evidence has been gathered that syntaxin 1A forms physical complexes with the NET homologues GAT1 and GLYT1 (Beckman et al., 1998; Geerlings et al., 2000). In particular, the GAT1 GABA transporters interact with syntaxin 1A in a phorbol ester-sensitive manner, with evidence that multiple PKC-linked receptors can trigger GAT1 redistribution and syntaxin 1A/GAT1 complex disassembly. Because syntaxin 1A is required for vesicular release (Jahn and Sudhof, 1999), it seemed possible that the SNARE protein might play an unrecognized role in linking the opposing processes of catecholamine release and reuptake. We thus sought evidence for a functional role of syntaxin 1A in establishing NE transport capacity.
In initial tests of the syntaxin 1A requirement for NET activity, we found that both antisense treatments and syntaxin cleavage with BoNT/C1 significantly reduced desipramine-sensitive NET activity. Based on current models for syntaxin 1A participation in vesicular fusion (Jahn and Sudhof, 1999), these findings appear consistent with a requirement for syntaxin 1A to deliver NET-containing vesicles to the cell. Because syntaxin 1A is enriched at noradrenergic varicosities, delivery of NETs via a syntaxin 1A-mediated membrane fusion event would assist in localization of transporters near sites of NE release for efficient reuptake. Indeed, evidence has been provided that NET even may be transported on a population of catecholamine secretory vesicles (Kippenberger et al., 1999). Regardless, the existence of NET as one of several cargo proteins in trafficking vesicles would not predict a stable association with syntaxin 1A nor that this association should be modulated by G-protein-coupled receptors (GPCRs) and other cellular stimuli. Possibly, the interactions we identify could arise artifactually via nonspecific associations that syntaxin might makein vivo or during extraction. However, all tests that we could perform to document specificity with native tissues, including loss of syntaxin 1A coimmunoprecipitations from NET knock-out mice, and our ability to document and map direct interactions in vitrosupport the idea that NET/syntaxin 1A interaction occurs in vivo and supports NE uptake. Interestingly, we found no evidence for SNAP-25 within NET/syntaxin 1A complexes, unlike observations with calcium and potassium channels (Rettig et al., 1996; Ji et al., 2002). These findings indicate that the composition of transporter/syntaxin 1A complexes is distinct from the channel/syntaxin 1A complexes that coordinate vesicular release of neurotransmitter, although it remains possible that the two pools communicate via syntaxin 1A availability.
NET binds syntaxin 1A directly through sequences within the cytoplasmic NH2 terminus of the transporter. Evidence to support this contention includes the ability of GST-SynΔTM to pull down full-length NET from solubilized transfected cells, findings that GST-SynΔTM binds selectively MBP fusions of the NET NH2 terminus in vitro, and loss of coimmunoprecipitation of syntaxin 1A by an hNET NH2 terminal truncation. Although the NH2 terminus of GAT1 also supports syntaxin 1A associations, the GAT1 interaction is mediated by more membrane proximal sequences (Deken et al., 2000) in which the deletion or point mutation in NET fails to disrupt associations. Ongoing studies are exploring the specific contact sites required for direct NET/syntaxin 1A interactions and should illuminate better how the NET NH2 terminus participates in transport function. Of note are studies from our lab and others that point to important roles for TMD1 and juxta-TMD1 sequences in ligand recognition and transport (Mager et al., 1996; Barker et al., 1999; Bennett et al., 2000). We found that cotransfection of the high-affinity syntaxin 1A binding protein Munc18 (Hata et al., 1993; Pevsner et al., 1994) reduced recovery of syntaxin 1A in NET immunoprecipitates. Munc18 binds syntaxin 1A via the cytoplasmic domain of the SNARE protein and therefore suggests that NET/syntaxin 1A interactions in cells are unlikely to be reliant on the syntaxin 1A transmembrane domain. The antagonistic activity of Munc18 also highlights the idea that complexes containing NET and syntaxin 1A can be regulated in cells by other SNARE-associated proteins. One class of proteins of particular note is UNC-13 homologs, for which the ability to bind phorbol esters and modulate syntaxin 1A/SNARE associations (Betz et al., 1998) suggests greater complexity to phorbol ester-mediated transporter regulation than PKC activation. Regardless, phorbol ester modulation of NET activity appears to require intact syntaxin 1A, suggesting an intimate role of the SNARE protein in both constitutive transporter trafficking and acute regulation.
NETs not only support antidepressant-sensitive NE uptake but also support substantial NE-gated channel activity (Galli et al., 1995). Amperometric recordings reveal bursts of NE translocated across the plasma membrane that are correlated significantly with NET channel openings (Galli et al., 1998). The existence of NET channel activity permits the analysis of putative cytoplasmic modulators by using inside-out patch recordings, here achieved for the first time by using perfused peptide modulators. We found that GST-SynΔTM, but not GST, effectively could eliminate NET currents with an effect resembling that of the competitive NET antagonist cocaine. We recognize that our detached patch experiments with heterologously expressed NET likely represent a highly artificial situation, because other NET-associated proteins and syntaxin-binding proteins are absent. However, as we identified syntaxin/NET complexes in vivo, it is tempting to speculate that a population of “inactive” carriers resides in the plasma membrane (Ramsey and DeFelice, 2002), awaiting activation via the elimination of syntaxin 1A associations. Such a model would be similar to that suggested for the GAT1 GABA transporter (Deken et al., 2000) and bears similarity to the notion that syntaxins negatively modulate the intrinsic activities of calcium channels and other membrane proteins (Bezprozvanny et al., 1995; Naren et al., 1997;Saxena et al., 1999).
Transfection of the cytoplasmic domain of syntaxin 1A enhanced NET surface expression, but this enhancement was without consequence for NE transport activity. This is not attributable to assay limitations because we clearly were able to monitor increased transport activity by using hNET cDNA titration across the same surface abundance range. Rather, our findings support the contention that, like transporter-associated currents, the intrinsic transport activity of surface NET proteins can be controlled by syntaxin 1A interactions. It is perhaps surprising that SynΔTM transfection increases NET surface density when SynΔTM cannot support SNARE-mediated fusion (McNew et al., 2000). We believe this is a consequence of SynΔTM displacement of endogenous syntaxin 1A from nonproductive complexes that indirectly limit fusion capacity. Such a model underscores two modes for syntaxin 1A in establishing NE clearance capacity, one controlling transporter trafficking and the other controlling intrinsic activity, connected by a limited number of syntaxin 1A molecules and the opportunity for coordination between the two modes. Physical linkages between these two facets of the activities of syntaxin 1A would offer tighter coordination between transmitter release and reuptake. The ability of syntaxin 1A antisense and BoNT/C1 to diminish NE transport activity in native cells and tissues suggests that surface trafficking of NETs is a dominant mode by which NE clearance capacity is supported by syntaxin 1A under basal conditions. A large reserve pool of carriers may await translocation to the plasma membrane to support neurotransmitter reuptake, a surmise in keeping with electron microscopic studies that show intracellular pools of biogenic amine transporters in vivo (Nirenberg et al., 1997; Miner et al., 2000; Schroeter et al., 2000) (L. H. Miner, S. Schroeter, Blakely, and S. R. Sesack, unpublished data). In contrast to our findings with NET, treatment of neurons with BoNT/C1 actually elevates GABA uptake (Deken et al., 2000). Because BoNT/C1 significantly diminishes GAT1 surface levels, a greater fraction of total GAT1 proteins may be inhibited constitutively at the plasma membrane by syntaxin 1A, whereas NET resides mainly intracellular, awaiting SNARE-dependent trafficking to the plasma membrane. Acute regulatory processes, in contrast, may target NET1 and GAT1 interactions similarly, modulating selectively the pool of transporter proteins already inserted in the plasma membrane. Thus phorbol esters and GPCR stimulation trigger NET/syntaxin 1A disassembly and NET downregulation in much the same manner as is seen with GAT1, and both transporters require syntaxin 1A-interacting sequences to support regulation (Beckman et al., 1998). Although downregulation of NETs coincides with a loss of syntaxin 1A from NET complexes, alternative pathways for regulating NET intrinsic activity in the absence of changes in NET surface expression also can be envisioned so long as NETs are not targeted for redistribution by the same stimuli. For example, we recently have described that NETs in SK-N-SH cells can be activated by a calcium and p38 MAP kinase-linked pathway that elevates NE uptake without alterations in NET surface expression (Apparsundaram et al., 2001). Okadaic acid, which we show disassembles the syntaxin 1A/NET complex, prevents activation by the Ca2+/p38 MAP kinase pathway (Apparsundaram et al., 2001). Because PP2A has been found to be a NET-associated protein, signals impinging on preassembled syntaxin 1A/NET complexes that do not alter surface trafficking of NET vesicles may do so via PP2A-linked pathways.
Our model for NET regulation by syntaxin 1A also provides an opportunity for an integrated control of NE release and reuptake. The provision of syntaxin 1A for fusion of NE secretory vesicles may have a parallel cycle in the production of syntaxin-free NET proteins. Indeed, NETs could provide a reservoir for syntaxin 1A proteins such that dissociation from NETs and transporter activation occurs in concert with the provision of free SNARE protein to enhance vesicular NE release. In turn, transporters thus may be able to keep pace with the release process by increased surface abundance and increased intrinsic activity. Otherwise, stimuli that enhance NE release would generate asynchrony between release and reuptake. Time-resolved methods that permit the evaluation of NE transport activity and its regulation on a time scale similar to techniques used to monitor secretion events are under development (Schwartz et al., 2002) and should be beneficial in evaluating this model. Given the tight control of extracellular NE by NETs and the critical role of NETs in cardiovascular, cognition, and mood circuits (Axelrod and Kopin, 1969;Iversen, 1971), a further understanding of how to manipulate the syntaxin 1A/NET complex could yield novel therapeutic strategies for the treatment of autonomic disorders and mental illness. Although the clinical utility of NET-specific antagonists is well documented (Burrows et al., 1998; Gorman and Sullivan, 2000), pharmaceutical development focused on biogenic amine transporters has uncovered no new modes of therapeutic manipulation since the first generation of antidepressant compounds were found to block NE transport more than four decades ago (Axelrod et al., 1961; Hertting et al., 1961; Iversen, 1965). Our findings may provide a path to novel drug design via the selective inhibition of NET/syntaxin 1A interactions. Furthermore, because SERT proteins, the target of SSRI medications, also appear to interact with syntaxin 1A (Haase et al., 2001), this idea may be more broadly applicable to multiple classes of novel antidepressant agents.
U.S. and S.A. contributed equally to this work.
U.S., S.A., V.S., S.S., and R.D.B. were supported by National Institutes of Health (NIH) Award MH58923. K.M.K. and A.G. were supported by NIH Award DA 14684. M.Q. was supported by NIH Award MH61468. We gratefully acknowledge the support provided by Denise Malone in the Center for Molecular Neuroscience (CMN) Neurogenomics Core for DNA sequencing, Jane Wright in the CMN Neurohistology and Imaging Core, and Sam Wells in the Vanderbilt Cell Imaging Resource for assistance with microscopy studies. We also thank Marc Caron (Duke University) for the gift of NET knock-out mice, Mark Brann (Acadia Pharmaceuticals) for the gift of M3 muscarinic receptor-transfected CHO cells, Bruce Carter (Vanderbilt University) for advice with SCG cultures, Pat Bauman (Vanderbilt University) for construction of tagged NET cDNA, and Lou DeFelice (Vanderbilt University) for scientific advice.
Correspondence should be addressed to Dr. Randy D. Blakely, Center for Molecular Neuroscience, 7140 Medical Research Building III, Vanderbilt University School of Medicine, Nashville, TN 37232-8548. E-mail:randy.blakely@vanderbilt.edu.
S. Apparsundaram's present address: Department of Anatomy and Neurobiology, University of Kentucky, Lexington, KY 40504.
S. Schroeter's present address: Pharmacia Corporation, Chesterfield, MO 63017.
A. Galli's and K. M. Kahlig's present address: Department of Molecular Physiology and Biophysics, Center for Molecular Neuroscience, Vanderbilt University School of Medicine, Nashville, TN 37232-8548.
M. Quick's present address: Department of Biological Sciences, University of Southern California, Hedco Neurosciences Building 228, 3641 Watt Way, Los Angeles, CA 90089-2520.
References
- 1.Apparsundaram S, Galli A, DeFelice LJ, Hartzell HC, Blakely RD. Acute regulation of norepinephrine transport. I. Protein kinase C-linked muscarinic receptors influence transport capacity and transporter density in SK-N-SH cells. J Pharmacol Exp Ther. 1998a;287:733–743. [PubMed] [Google Scholar]
- 2.Apparsundaram S, Schroeter S, Giovanetti E, Blakely RD. Acute regulation of norepinephrine transport. II. PKC-modulated surface expression of human norepinephrine transporter proteins. J Pharmacol Exp Ther. 1998b;287:744–751. [PubMed] [Google Scholar]
- 3.Apparsundaram S, Sung U, Price RD, Blakely RD. Trafficking-dependent and -independent pathways of neurotransmitter transporter regulation differentially involving p38 mitogen-activated protein kinase revealed in studies of insulin modulation of norepinephrine transport in SK-N-SH cells. J Pharmacol Exp Ther. 2001;299:666–677. [PubMed] [Google Scholar]
- 4.Arnsten AF. Modulation of prefrontal cortical–striatal circuits: relevance to therapeutic treatments for Tourette syndrome and attention-deficit hyperactivity disorder. Adv Neurol. 2001;85:333–341. [PubMed] [Google Scholar]
- 5.Axelrod J, Kopin IJ. The uptake, storage, release, and metabolism of noradrenaline in sympathetic nerves. Prog Brain Res. 1969;31:21–32. doi: 10.1016/S0079-6123(08)63224-0. [DOI] [PubMed] [Google Scholar]
- 6.Axelrod J, Whitby LG, Hertting G. Effect of psychotropic drugs on the uptake of H3-norepinephrine by tissues. Science. 1961;133:383–384. doi: 10.1126/science.133.3450.383. [DOI] [PubMed] [Google Scholar]
- 7.Barker EL, Blakely RD. Norepinephrine and serotonin transporters: molecular targets of antidepressant drugs. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: the fourth generation of progress. Raven; New York: 1995. pp. 321–333. [Google Scholar]
- 8.Barker EL, Moore KR, Rakhshan F, Blakely RD. Transmembrane domain I contributes to permeation pathways for serotonin and ions in the serotonin transporter. J Neurosci. 1999;19:4705–4717. doi: 10.1523/JNEUROSCI.19-12-04705.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bauman AL, Apparsundaram S, Ramamoorthy S, Wadzinski BE, Vaughan RA, Blakely RD. Cocaine and antidepressant-sensitive biogenic amine transporters exist in regulated complexes with protein phosphatase 2A. J Neurosci. 2000;20:7571–7578. doi: 10.1523/JNEUROSCI.20-20-07571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bauman PA, Blakely RD. Determinants within the C terminus of the human norepinephrine transporter dictate trafficking, stability, and activity. Arch Biochem Biophys. 2002;404:80–91. doi: 10.1016/s0003-9861(02)00232-1. [DOI] [PubMed] [Google Scholar]
- 11.Beckman ML, Bernstein EM, Quick MW. Protein kinase C regulates the interaction between a GABA transporter and syntaxin 1A. J Neurosci. 1998;18:6103–6112. doi: 10.1523/JNEUROSCI.18-16-06103.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Beckman ML, Bernstein EM, Quick MW. Multiple G-protein-coupled receptors initiate protein kinase C redistribution of GABA transporters in hippocampal neurons. J Neurosci 19 1999. RC9(1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bennett ER, Su H, Kanner BI. Mutation of arginine 44 of GAT-1, a (Na+ + Cl−)-coupled γ-aminobutyric acid transporter from rat brain, impairs net flux but not exchange. J Biol Chem. 2000;275:34106–34113. doi: 10.1074/jbc.M004229200. [DOI] [PubMed] [Google Scholar]
- 14.Bennett MK, Garcia-Arraras JE, Elferink LA, Peterson K, Fleming AM, Hazuka CD, Scheller RH. The syntaxin family of vesicular transport receptors. Cell. 1993;74:863–873. doi: 10.1016/0092-8674(93)90466-4. [DOI] [PubMed] [Google Scholar]
- 15.Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Sudhof TC, Rettig J, Brose N. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- 16.Bezprozvanny I, Scheller RH, Tsien RW. Functional impact of syntaxin on gating of N-type and Q-type calcium channels. Nature. 1995;378:623–626. doi: 10.1038/378623a0. [DOI] [PubMed] [Google Scholar]
- 17.Bittner MA, Bennett MK, Holz RW. Evidence that syntaxin 1A is involved in storage in the secretory pathway. J Biol Chem. 1996;271:11214–11221. doi: 10.1074/jbc.271.19.11214. [DOI] [PubMed] [Google Scholar]
- 18.Blakely RD, Bauman AL. Biogenic amine transporters: regulation in flux. Curr Opin Neurobiol. 2000;10:328–336. doi: 10.1016/s0959-4388(00)00088-x. [DOI] [PubMed] [Google Scholar]
- 19.Bohm M, La Rosee K, Schwinger RHG, Erdmann E. Evidence for reduction of norepinephrine uptake sites in the failing human heart. J Am Coll Cardiol. 1995;25:146–153. doi: 10.1016/0735-1097(94)00353-r. [DOI] [PubMed] [Google Scholar]
- 20.Brain KL, Cottee LJ, Bennett MR. Varicosities of single sympathetic nerve terminals possess syntaxin zones and different synaptotagmin N terminus labeling following stimulation. J Neurocytol. 1997;26:491–500. doi: 10.1023/a:1018533524643. [DOI] [PubMed] [Google Scholar]
- 21.Burrows GD, Maguire KP, Norman TR. Antidepressant efficacy and tolerability of the selective norepinephrine reuptake inhibitor reboxetine: a review. J Clin Psychiatry. 1998;59:4–7. [PubMed] [Google Scholar]
- 22.Chen NH, Reith ME. Effects of locally applied cocaine, lidocaine, and various uptake blockers on monoamine transmission in the ventral tegmental area of freely moving rats: a microdialysis study on monoamine interrelationships. J Neurochem. 1994;63:1701–1713. doi: 10.1046/j.1471-4159.1994.63051701.x. [DOI] [PubMed] [Google Scholar]
- 23.Deken SL, Beckman ML, Boos L, Quick MW. Transport rates of GABA transporters: regulation by the N-terminal domain and syntaxin 1A. Nat Neurosci. 2000;3:998–1003. doi: 10.1038/79939. [DOI] [PubMed] [Google Scholar]
- 24.Deken SL, Beckman ML, Quick MW. PICKing on transporters. Trends Neurosci. 2001;24:623–625. doi: 10.1016/s0166-2236(00)01933-0. [DOI] [PubMed] [Google Scholar]
- 25.Delgado PL, Moreno FA. Role of norepinephrine in depression. J Clin Psychiatry. 2000;61:5–12. [PubMed] [Google Scholar]
- 26.Esler M, Jackman G, Bobik A, Leonard P, Kelleher D, Skews H, Jennings G, Korner P. Norepinephrine kinetics in essential hypertension. Defective neuronal uptake of norepinephrine in some patients. Hypertension. 1981;3:149–156. doi: 10.1161/01.hyp.3.2.149. [DOI] [PubMed] [Google Scholar]
- 27.Fili O, Michaelevski I, Bledi Y, Chikvashvili D, Singer-Lahat D, Boshwitz H, Linial M, Lotan I. Direct interaction of a brain voltage-gated K+ channel with syntaxin 1A: functional impact on channel gating. J Neurosci. 2001;21:1964–1974. doi: 10.1523/JNEUROSCI.21-06-01964.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foote SL, Bloom FE, Aston-Jones G. Nucleus locus ceruleus: new evidence of anatomical and physiological specificity. Physiol Rev. 1983;63:844–914. doi: 10.1152/physrev.1983.63.3.844. [DOI] [PubMed] [Google Scholar]
- 29.Galli A, DeFelice LJ, Duke BJ, Moore KR, Blakely RD. Sodium-dependent norepinephrine-induced currents in norepinephrine transporter-transfected HEK-293 cells blocked by cocaine and antidepressants. J Exp Biol. 1995;198:2197–2212. doi: 10.1242/jeb.198.10.2197. [DOI] [PubMed] [Google Scholar]
- 30.Galli A, Blakely RD, DeFelice LJ. Patch-clamp and amperometric recordings from norepinephrine transporters: channel activity and voltage-dependent uptake. Proc Natl Acad Sci USA. 1998;95:13260–13265. doi: 10.1073/pnas.95.22.13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geerlings A, Lopez-Corcuera B, Aragon C. Characterization of the interactions between the glycine transporters GLYT1 and GLYT2 and the SNARE protein syntaxin 1A. FEBS Lett. 2000;470:51–54. doi: 10.1016/s0014-5793(00)01297-7. [DOI] [PubMed] [Google Scholar]
- 32.Geerlings A, Nunez E, Lopez-Corcuera B, Aragon C. Calcium- and syntaxin 1-mediated trafficking of the neural glycine transporter GLYT2. J Biol Chem. 2001;276:17584–17590. doi: 10.1074/jbc.M010602200. [DOI] [PubMed] [Google Scholar]
- 33.Gorman JM, Sullivan G. Noradrenergic approaches to antidepressant therapy. J Clin Psychiatry. 2000;61:13–16. [PubMed] [Google Scholar]
- 34.Graefe KH, Bönisch H. The transport of amines across the axonal membranes of noradrenergic and dopaminergic neurons. In: Trebdelenburg U, Weiner N, editors. Handbook of experimental pharmacology, Vol 90, Catecholamine, Pt I. Springer; Heidelberg: 1988. pp. 193–245. [Google Scholar]
- 35.Haase J, Killian AM, Magnani F, Williams C. Regulation of the serotonin transporter by interacting proteins. Biochem Soc Trans. 2001;29:722–728. doi: 10.1042/0300-5127:0290722. [DOI] [PubMed] [Google Scholar]
- 36.Hadley D, Hoff M, Holik J, Reimherr F, Wender P, Coon H, Byerley W. Manic depression and the norepinephrine transporter gene. Hum Hered. 1995;45:165–168. doi: 10.1159/000154279. [DOI] [PubMed] [Google Scholar]
- 37.Hata Y, Slaughter CA, Sudhof TC. Synaptic vesicle fusion complex contains UNC-18 homologue bound to syntaxin. Nature. 1993;366:347–351. doi: 10.1038/366347a0. [DOI] [PubMed] [Google Scholar]
- 38.Hertting G, Axelrod J, Whitby LG. Effect of drugs on the uptake and metabolism of 3H-norepinephrine. J Pharmacol Exp Ther. 1961;134:146–153. [PubMed] [Google Scholar]
- 39.Iversen LL. Inhibition of noradrenaline uptake by drugs. J Pharm Pharmacol. 1965;17:62–64. doi: 10.1111/j.2042-7158.1965.tb07572.x. [DOI] [PubMed] [Google Scholar]
- 40.Iversen LL. Role of transmitter uptake mechanisms in synaptic neurotransmission. Br J Pharmacol. 1971;41:571–591. doi: 10.1111/j.1476-5381.1971.tb07066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jackson M, Song W, Liu MY, Jin L, Dykes-Hoberg M, Lin CI, Bowers WJ, Federoff HJ, Sternweis PC, Rothstein JD. Modulation of the neuronal glutamate transporter EAAT4 by two interacting proteins. Nature. 2001;410:89–93. doi: 10.1038/35065091. [DOI] [PubMed] [Google Scholar]
- 42.Jahn R, Sudhof TC. Membrane fusion and exocytosis. Annu Rev Biochem. 1999;68:863–911. doi: 10.1146/annurev.biochem.68.1.863. [DOI] [PubMed] [Google Scholar]
- 43.Ji J, Tsuk S, Salapatek AM, Huang X, Chikvashvili D, Pasyk EA, Kang Y, Sheu L, Tsushima R, Diamant N, Trimble WS, Lotan I, Gaisano HY. The 25 kDa synaptosome-associated protein (SNAP-25) binds and inhibits delayed rectifier potassium channels in secretory cells. J Biol Chem. 2002;277:20195–20204. doi: 10.1074/jbc.M201034200. [DOI] [PubMed] [Google Scholar]
- 44.Kippenberger AG, Palmer DJ, Comer AM, Lipski J, Burton LD, Christie DL. Localization of the noradrenaline transporter in rat adrenal medulla and PC12 cells: evidence for its association with secretory granules in PC12 cells. J Neurochem. 1999;73:1024–1032. doi: 10.1046/j.1471-4159.1999.0731024.x. [DOI] [PubMed] [Google Scholar]
- 45.Klimek V, Stockmeier C, Overholser J, Meltzer HY, Kalka S, Dilley G, Ordway GA. Reduced levels of norepinephrine transporters in the locus ceruleus in major depression. J Neurosci. 1997;17:8451–8458. doi: 10.1523/JNEUROSCI.17-21-08451.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee FJ, Liu F, Pristupa ZB, Niznik HB. Direct binding and functional coupling of α-synuclein to the dopamine transporters accelerate dopamine-induced apoptosis. FASEB J. 2001;15:916–926. doi: 10.1096/fj.00-0334com. [DOI] [PubMed] [Google Scholar]
- 47.Liang C, Fan THM, Sullebarger JT, Sakamoto S. Decreased adrenergic neuronal uptake activity in experimental right heart failure. J Clin Invest. 1989;84:1267–1275. doi: 10.1172/JCI114294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mager S, Kleinberger-Doron N, Keshet GI, Davidson N, Kanner BI, Lester HA. Ion binding and permeation at the GABA transporter GAT1. J Neurosci. 1996;16:5405–5414. doi: 10.1523/JNEUROSCI.16-17-05405.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McNew JA, Weber T, Parlati F, Johnston RJ, Melia TJ, Sollner TH, Rothman JE. Close is not enough: SNARE-dependent membrane fusion requires an active mechanism that transduces force to membrane anchors. J Cell Biol. 2000;150:105–117. doi: 10.1083/jcb.150.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Melikian HE, Ramamoorthy S, Tate CG, Blakely RD. Inability to N-glycosylate the human norepinephrine transporter reduces protein stability, surface trafficking, and transport activity but not ligand recognition. Mol Pharmacol. 1996;50:266–276. [PubMed] [Google Scholar]
- 51.Merlet P, Dubois-Rande JL, Adnot S, Bourguignon MH, Benvenuti C, Loisance D, Valette H, Castaigne A, Syrota A. Myocardial β-adrenergic desensitization and neuronal norepinephrine uptake function in idiopathic dilated cardiomyopathy. J Cardiovasc Pharmacol. 1992;19:10–16. doi: 10.1097/00005344-199201000-00002. [DOI] [PubMed] [Google Scholar]
- 52.Miner LH, Schroeter S, Blakely RD, Sesack SR. Ultrastructural localization of the serotonin transporter in superficial and deep layers of the rat prelimbic prefrontal cortex and its spatial relationship to dopamine terminals. J Comp Neurol. 2000;427:220–234. doi: 10.1002/1096-9861(20001113)427:2<220::aid-cne5>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 53.Naren AP, Nelson DJ, Xie W, Jovov B, Pevsner J, Bennett MK, Benos DJ, Quick MW, Kirk KL. Regulation of CFTR chloride channels by syntaxin and Munc18 isoforms. Nature. 1997;390:302–305. doi: 10.1038/36882. [DOI] [PubMed] [Google Scholar]
- 54.Nirenberg MJ, Chan J, Pohorilee A, Vaughan RA, Uhl GR, Kuhar MJ, Pickel VM. The dopamine transporter: comparative ultrastructure of dopaminergic axons in limbic and motor compartments of the nucleus accumbens. J Neurosci. 1997;17:6899–6907. doi: 10.1523/JNEUROSCI.17-18-06899.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pacholczyk T, Blakely RD, Amara SG. Expression cloning of a cocaine- and antidepressant-sensitive human noradrenaline transporter. Nature. 1991;350:350–354. doi: 10.1038/350350a0. [DOI] [PubMed] [Google Scholar]
- 56.Perego C, Vanoni C, Villa A, Longhi R, Kaech SM, Frohli E, Hajnal A, Kim SK, Pietrini G. PDZ-mediated interactions retain the epithelial GABA transporter on the basolateral surface of polarized epithelial cells. EMBO J. 1999;18:2384–2393. doi: 10.1093/emboj/18.9.2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pevsner J, Hsu SC, Scheller RH. n-Sec1: a neural-specific syntaxin-binding protein. Proc Natl Acad Sci USA. 1994;91:1445–1449. doi: 10.1073/pnas.91.4.1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qi Y, Wang JKT, McMilian M, Chikaraishi DM. Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J Neurosci. 1997;17:1217–1225. doi: 10.1523/JNEUROSCI.17-04-01217.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qian Y, Galli A, Ramamoorthy S, Risso S, DeFelice LJ, Blakely RD. Protein kinase C activation regulates human serotonin transporters in HEK-293 cells via altered cell surface expression. J Neurosci. 1997;17:45–47. doi: 10.1523/JNEUROSCI.17-01-00045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Raisman R, Sette M, Pimoule C, Briley M, Langer SZ. High-affinity [3H]desipramine binding in the peripheral and central nervous system: a specific site associated with the neuronal uptake of noradrenaline. Eur J Pharmacol. 1982;78:345–351. doi: 10.1016/0014-2999(82)90036-x. [DOI] [PubMed] [Google Scholar]
- 61.Ramamoorthy S, Blakely RD. Phosphorylation and sequestration of serotonin transporters differentially modulated by psychostimulants. Science. 1999;285:763–766. doi: 10.1126/science.285.5428.763. [DOI] [PubMed] [Google Scholar]
- 62.Ramsey IS, DeFelice LJ. Serotonin transporter function and pharmacology are sensitive to expression level: evidence for an endogenous regulatory factor. J Biol Chem. 2002;277:14475–14482. doi: 10.1074/jbc.M110783200. [DOI] [PubMed] [Google Scholar]
- 63.Rettig J, Sheng ZH, Kim DK, Hodson CD, Snutch TP, Catterall WA. Isoform-specific interaction of the α1A subunits of brain Ca2+ channels with the presynaptic proteins syntaxin and SNAP-25. Proc Natl Acad Sci USA. 1996;93:7363–7368. doi: 10.1073/pnas.93.14.7363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ritz MC, Cone EJ, Kuhar MJ. Cocaine inhibition of ligand binding at dopamine, norepinephrine, and serotonin transporters: a structure–activity study. Life Sci. 1990;46:635–645. doi: 10.1016/0024-3205(90)90132-b. [DOI] [PubMed] [Google Scholar]
- 65.Rowe J, Corradi N, Malosio ML, Taverna E, Halban P, Meldolesi J, Rosa P. Blockade of membrane transport and disassembly of the Golgi complex by expression of syntaxin 1A in neurosecretion-incompetent cells: prevention by rbSEC1. J Cell Sci. 1999;112:1865–1877. doi: 10.1242/jcs.112.12.1865. [DOI] [PubMed] [Google Scholar]
- 66.Saunders C, Ferrer JV, Shi L, Chen J, Merrill G, Lamb ME, Leeb-Lundberg LM, Carvelli L, Javitch JA, Galli A. Amphetamine-induced loss of human dopamine transporter activity: an internalization-dependent and cocaine-sensitive mechanism. Proc Natl Acad Sci USA. 2000;97:6850–6855. doi: 10.1073/pnas.110035297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saxena S, Quick MW, Tousson A, Oh Y, Warnock DG. Interaction of syntaxins with the amiloride-sensitive epithelial sodium channel. J Biol Chem. 1999;274:20812–20817. doi: 10.1074/jbc.274.30.20812. [DOI] [PubMed] [Google Scholar]
- 68.Schiavo G, Matteoli M, Montecucco C. Neurotoxins affecting neuroexocytosis. Physiol Rev. 2000;80:717–766. doi: 10.1152/physrev.2000.80.2.717. [DOI] [PubMed] [Google Scholar]
- 69.Schildkraut JJ. The catecholamine hypothesis of affective disorders: a review of supporting evidence. Am J Psychiatry. 1965;122:509–522. doi: 10.1176/ajp.122.5.509. [DOI] [PubMed] [Google Scholar]
- 70.Schroeter S, Apparsundaram S, Wiley RG, Miner LH, Sesack SR, Blakely RD. Immunolocalization of the cocaine- and antidepressant-sensitive l-norepinephrine transporter. J Comp Neurol. 2000;420:211–232. [PubMed] [Google Scholar]
- 71.Schwartz JW, Blakely RD, DeFelice LJ (2002) Binding and transport in norepinephrine transporters: real-time, spatially resolved analysis in single cells using a fluorescent substrate. J Biol Chem, in press. [DOI] [PubMed]
- 72.Sesack SR, Snyder CL. Cellular and subcellular localization of syntaxin-like immunoreactivity in the rat striatum and cortex. Neuroscience. 1995;67:993–1007. doi: 10.1016/0306-4522(95)00087-y. [DOI] [PubMed] [Google Scholar]
- 73.Shannon JR, Flattem NL, Jordan J, Jacob G, Black BK, Biaggioni I, Blakely RD, Robertson D. Orthostatic intolerance and tachycardia associated with norepinephrine transporter deficiency. N Engl J Med. 2000;342:541–549. doi: 10.1056/NEJM200002243420803. [DOI] [PubMed] [Google Scholar]
- 74.Tatsumi M, Groshan K, Blakely RD, Richelson E. Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol. 1997;340:249–258. doi: 10.1016/s0014-2999(97)01393-9. [DOI] [PubMed] [Google Scholar]
- 75.Torres GE, Yao WD, Mohn RR, Quan H, Kim K, Levey AI, Staudinger J, Caron MG. Functional interaction between monoamine plasma membrane transporters and the synaptic PDZ domain-containing protein PICK1. Neuron. 2001;30:121–134. doi: 10.1016/s0896-6273(01)00267-7. [DOI] [PubMed] [Google Scholar]
- 76.Wall SC, Gu H, Rudnick G. Biogenic amine flux mediated by cloned transporters stably expressed in cultured cell lines: amphetamine specificity for inhibition and efflux. Mol Pharmacol. 1995;47:544–550. [PubMed] [Google Scholar]
- 77.Xu F, Gainetdinov RR, Wetsel WC, Jones SR, Bohn LM, Miller GW, Wang YM, Caron MG. Mice lacking the norepinephrine transporter are supersensitive to psychostimulants. Nat Neurosci. 2000;3:465–471. doi: 10.1038/74839. [DOI] [PubMed] [Google Scholar]
- 78.Yang SN, Larsson O, Branstrom R, Bertorello AM, Leibiger B, Leibiger IB, Moede T, Kohler M, Meister B, Berggren PO. Syntaxin 1 interacts with the LD subtype of voltage-gated Ca2+ channels in pancreatic β-cells. Proc Natl Acad Sci USA. 1999;96:10164–10169. doi: 10.1073/pnas.96.18.10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zahniser NR, Doolen S. Chronic and acute regulation of Na+/Cl−-dependent neurotransmitter transporters: drugs, substrates, presynaptic receptors, and signaling systems. Pharmacol Ther. 2001;92:21–55. doi: 10.1016/s0163-7258(01)00158-9. [DOI] [PubMed] [Google Scholar]