

Abstract
The robotic mouse is an autosomal dominant mutant that arose from a large-scale chemical mutagenesis program. It has a jerky, ataxic gait and develops adult-onset Purkinje cell loss in the cerebellum in a striking region-specific pattern, as well as cataracts. Genetic and physical mapping of the disease locus led to the identification of a missense mutation in a highly conserved region of Af4, a putative transcription factor that has been previously implicated in leukemogenesis. We demonstrate that Af4 is specifically expressed in Purkinje cells, and we hypothesize that the expression of mutant Af4 leads to neurodegeneration. This function was not identified through knock-out studies, highlighting the power of phenotype-driven mutagenesis in the mouse to identify new pathways involved in neurological disease.
Keywords: cerebellum, Purkinje cell, neurodegeneration, ENU mutagenesis, mouse models, positional cloning
Introduction
Mouse mutants are an important resource for understanding mammalian gene function and disease and will play an increasingly significant role as we attempt to assign functions to all human genes. Of the possible routes to mutagenizing the mouse genome, gene-trapping vectors and chemical mutagenesis are being used on a large scale to provide a significant number of new mutants for gene function analysis (Hrabe de Angelis et al., 2000; Nolan et al., 2000; Stanford et al., 2001). An advantage of phenotype-based mutagenesis is that no assumptions are made about the possible function of the genes involved; instead we rely on sensitive phenotype analysis of individual animals. This approach therefore might be more likely to identify novel genes and pathways in processes of interest than genotype-driven mutagenesis, which identifies the mutated gene before phenotype analysis. In addition to the loss-of-function phenotypes generally seen in knock-out and gene-trap mutated mice, chemical mutagenesis using a point mutagen such asN-ethyl-N-nitrosurea (ENU) can generate partial loss of function, gain of function, and dominant negative mutations, providing a wider range of mutants that may reveal different aspects of the function of a gene (Justice, 2000; Brown and Balling, 2001).
We have used the phenotype-driven chemical mutagenesis approach coupled with a sensitive phenotype analysis protocol known as SHIRPA to screen for motor mutants (Rogers et al., 1997; Nolan et al., 2000). During this screen, we identified a novel cerebellar mouse mutant that develops Purkinje cell loss, and we predict that a gene that was previously implicated in leukemia, Af4, is responsible.
Translocations involving the mixed lineage leukemia (MLL) gene on chromosome 11 and some 30 loci on various chromosomes are associated with 5–10% of human acute leukemias. Of these, the t(4;11) translocation is the most common, accounting for ∼50% of acute lymphoblastic leukemia (ALL) in infants under 1 year of age (Mitelman, 1994; Rowley, 1998). This translocation results in a fusion product coding for a chimeric protein derived from MLL and AF4 (ALL1-fused gene from chromosome 4) (Gu et al., 1992; Domer et al., 1993). However, the function of AF4 in leukemogenesis is not well understood, despite B and T cell development defects in the Af4knock-out mouse, suggesting a role in lymphopoeisis (Isnard et al., 2000). We present a potentially novel role for Af4 in neurodegeneration, demonstrating the power of phenotype-driven mutagenesis in the mouse for ascribing new functions to genes.
Materials and Methods
Histology and immunohistochemistry. Brains were removed from mice transcardially perfused with 4% paraformaldehyde and embedded in paraplast wax, and 10 μm sections were cut for histology and immunostaining. For cell counts, sections were stained in 0.5% aqueous cresyl violet, and Purkinje cells were counted in every 25th parasagittal section throughout the whole brain. Cells were only counted if part of their nucleus was visible, and the Hendry correction factor was used to account for double cell counting (Hendry, 1976). For individual cerebellar lobe counts, totals were calculated from a 40 μm region around the central vermis. For immunohistochemistry, sections were incubated with one of the following primary antibodies: rabbit anti-calbindin (SWant, Bellinzona, Switzerland) diluted 1:15,000 for 2 nights at 4°C; rabbit anti-parvalbumin (SWant) diluted 1:10,000 for 3 nights at 4°C; rabbit anti-GFAP (Dako, Glostrup, Denmark) diluted 1:400 overnight at room temperature; rabbit anti-ubiquitin (Novocastra, Newcastle-upon-Tyne, UK) diluted 1:4,500 overnight at room temperature. After incubation, sections were immunoperoxidase labeled using the Vectastain Elite ABC Kit (Vector Laboratories, Burlingame, CA), and peroxidase activity was detected using diaminobenzidine (Vector Laboratories) as the substrate.
Genetic and physical mapping. DNA for genotyping was extracted from mouse tail biopsies by standard procedures, and the PCR products derived from each marker were analyzed on ethidium bromide-stained 4% Nusieve GTG agarose (Flowgen, Ashby de la Zouch, UK) gels. For those markers that could not be resolved on conventional gels, one primer was radiolabeled before amplification, and the products were run on denaturing 6% polyacrylamide gels. The RPCI-23 bacterial artificial chromosome (BAC) library filters (obtained from the Human Genome Mapping Resource Centre, Hinxton, UK) were screened initially with combined radiolabeled 35-mer oligonucleotides specific for markersD5Mit93, D5Mit23, and D5Mit10. BAC end sequences were obtained from The Institute for Genomic Research database (http://www.tigr.org/tdb/bac_ends/mouse/bac_end_intro.html) or determined by direct sequencing. To construct the contig, PCR was used to confirm the presence of a marker in an individual BAC clone. New polymorphic markers were identified by cloning and sequencing products derived from Interspersed Repeat Element PCR (Detter et al., 1998) or by screening genomic sublibraries with a radiolabeled (CA)15 repeat probe. Exon trapping was performed using the Exon Trapping System (Invitrogen, San Diego, CA) with three BAC clones that spanned the robotic candidate region. Purified BAC DNA was completely digested with BamHI/BglII or PstI and shotgun cloned into the pSPL3 vector. COS-7 cell transfection, exon amplification, subcloning, and sequencing were performed according to the manufacturer's instructions.
Candidate gene analysis. Cerebellum and liver cDNA for candidate gene sequencing was generated from RNA extracted using RNeasy Midi columns (Qiagen, Hilden, Germany) or Tri reagent (Sigma, St. Louis, MO). Five overlapping PCR products were generated to span the 1887 bp coding region of Klhl8, and eight products were designed to cover the 3636 bp coding region ofAf4 from cerebellum cDNA. Two overlapping products were generated to span the 900 bp coding region of Hsd17b12 from liver cDNA. Primer sequences are available from K.E.D. RT-PCR products were purified using the Qiaspin gel extraction kit (Qiagen) before sequencing.
In situ hybridization. A 309 bp region of mouseAf4 (mAF4) (3194–3502 bp of GenBank accession number AF074266) was PCR amplified and cloned into the pCR4-TOPO vector (Invitrogen). Whole mouse brains were frozen in OCT (Merck, Darmstadt, Germany) on dry ice, and 14 μm parasagittal cryosections were cut and mounted on positively charged slides. Digoxigenin-labeled riboprobe synthesis and hybridization were performed essentially as described previously (Wilkinson, 1992).
Flow cytometric analysis. Single-cell suspensions of thymocytes from 3-week-old mice (n = 3) were obtained by gentle disruption of the thymus organ, and four-color staining with anti-CD8 Tricolor (Caltag, San Francisco, CA), anti-CD4 PE, anti-CD25 APC, and CD44 FITC (Becton Dickinson, Rutherford, NJ) was performed. Samples were analyzed on a Becton Dickinson FACS machine.
Results
Robotic mice develop region-specific Purkinje cell loss
The robotic mouse was identified from the progeny of an ENU-injected BALB/c male and a C3H/HeH female because of its jerky, ataxic gait, which is apparent from ∼3 weeks of age. Mutants are also generally 50% of the size of their littermates from this time and remain so into adulthood. Robotic animals also develop cataracts that become complete and bilateral.
Spinal cord, peripheral nerve, and brain were analyzed using histological stains, and no morphological abnormalities were observed in any tissue or brain region other than the cerebellum. Further analysis of the robotic cerebellum using calbindin and cresyl violet-stained sections at 3, 5, 10, 20, and 42 weeks of age showed that its lobular structure was unaltered at all time points (Figs.1a–d,2a,b). Purkinje cell loss was observed in a striking, region-specific pattern in the vermis, with the anterior cerebellum much more severely affected and lobe X spared throughout (Fig. 1a–d). Cell loss was first visible at 8 weeks of age (data not shown), although the compartmentalized Purkinje cell death was obvious by 20 weeks (Fig.1c). By 42 weeks of age, only 30% of Purkinje cells remained, with lobe X completely spared and almost no cells surviving in lobes I–IV (Fig. 1d). The total Purkinje cells numbers and lobe-specific counts for the vermis are shown in Table1.
Fig. 1.

Progression of Purkinje cell loss in the robotic cerebellum. a–d, Calbindin-stained vermal parasagittal sections at 3 (a), 10 (b), 20 (c), and 42 (d) weeks. Patchy Purkinje cell loss occurs at 10 weeks in the anterior lobes, which is more obvious by 20 weeks of age. At 42 weeks most of the Purkinje cells have been lost, although lobe X is spared, and the anterior lobes are more severely affected. The lobes of the cerebellum indicated (a,I–X) are morphologically unaltered at all time points. Scale bar, 1 mm. e,f, Calbindin-stained transverse sections from control littermate (e) and robotic (f) mice at 20 weeks of age. Parasagittal bands of resistant Purkinje cells in the robotic cerebellum are indicated (arrowheads). Scale bar, 1 mm.
Table 1.
Corrected Purkinje cell (PC) number in the cerebellum of robotic and littermate control mice at 10 weeks (n = 2) and 42 weeks (n = 3)
| Mouse line | Vermal PC number1-a | Total PC number1-b | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Lobe 1 | Lobe 2 | Lobe 3 | Lobe 4,5 | Lobe 6 | Lobe 7 | Lobe 8 | Lobe 9 | Lobe 10 | ||
| 10 week control | 1066 ± 125 | 2977 ± 319 | 3639 ± 19 | 6675 ± 588 | 3738 ± 350 | 1225 ± 250 | 2212 ± 206 | 3913 ± 238 | 2012 ± 313 | 99910 ± 2062 |
| 10 week robotic | 50 ± 19 | 1075 ± 25 | 1638 ± 6 | 3088 ± 150 | 3238 ± 88 | 963 ± 6 | 1988 ± 81 | 2900 ± 119 | 1925 ± 75 | 74673 ± 1744 |
| 42 week control | 1125 ± 189 | 2550 ± 95 | 3375 ± 160 | 5738 ± 331 | 3363 ± 259 | 912 ± 124 | 1938 ± 219 | 3963 ± 219 | 2275 ± 231 | 95731 ± 3792 |
| 42 week robotic | 25 ± 15 | 75 ± 37 | 100 ± 30 | 503 ± 140 | 1012 ± 110 | 350 ± 51 | 638 ± 137 | 1575 ± 155 | 2087 ± 260 | 26912 ± 7547 |
Mean number of Purkinje cells calculated for the cerebellar vermis ± SEM. Numbers were determined as described in Materials and Methods.
Mean total number calculated for the whole cerebellum ± SEM.
Analysis of calbindin immunostaining on transverse sections revealed parasagittal bands of surviving Purkinje cells in the anterior cerebellum at 20 weeks of age. The cell loss was mainly confined to the central vermis, and the surviving cells were in three clear bands: one at the midline with one on either side (Fig.1e,f) in a pattern similar to that of zebrin-positive parasagittal bands in the anterior cerebellar vermis (Hawkes and Herrup, 1995; Herrup and Kuemerle, 1997). At 42 weeks of age the banding was disrupted, indicating that at least a subset of the resistant cells at 20 weeks of age can only delay but not prevent cell death (data not shown).
Neurodegenerative changes in the robotic cerebellum
Other pathological features were also apparent using calbindin immunostaining. In a subset of mutants, swelling of Purkinje cell dendrites was apparent at 3 weeks of age. By 5 weeks, this feature was more pronounced, with additional swelling of the Purkinje cell bodies and focal swellings of Purkinje cell axons, known as axonal torpedoes (Fig. 2c). These were also apparent at 8 and 10 weeks of age. In addition, there was a marked decrease in size of the molecular layer of the cerebellum from 10 weeks onward, presumably caused by the loss of Purkinje cell dendrites that normally fill this region. This phenomenon is clearly demonstrated using parvalbumin immunostaining, which also revealed a concomitant increase in the density of basket and stellate cells that reside in the molecular layer (Fig.2d,e) and may account for the reduction in size of the cerebellum observed at 42 weeks of age (Figs. 1d,2b). Qualitative assessment of the granule cell population on cresyl violet-stained sections revealed no obvious loss in granule cells at any time point, although subtle cell loss in the granule cell population cannot be ruled out (Fig. 2a,b).
Fig. 2.

Neuropathological findings in the robotic cerebellum. Cresyl violet-stained parasagittal vermal sections at 3 (a) and 42 (b) weeks show shrinking of the robotic cerebellum over time attributable to a decrease in size of the molecular layer, whereas the general morphology of the cerebellar lobules remains intact. Scale bar, 1 mm.c, Calbindin immunostaining shows the Purkinje cell bodies, dendrites, and axons at 5 weeks of age. There is no Purkinje cell loss in robotic mice, but dendrites are thickened (arrowhead) and swellings of the axons are also apparent (arrow). Scale bar, 100 μm. Parvalbumin immunostaining from littermate control (d) and robotic (e) mice shows Purkinje cell loss from the Purkinje cell layer (PCL) at 10 and 20 weeks in parasagittal sections. The decrease in size of the molecular layer (ML) and the resulting increase in cell density are also apparent. Scale bar, 100 μm. f, GFAP immunostaining shows a marked increase in staining density in the robotic cerebellum at 10 weeks of age. The increase occurs in the Bergmann glia of the molecular layer (ML) and in astrocytes in the granule cell layer (GL). Scale bar, 400 μm. g, Ubiquitin immunostaining at 10 weeks of age reveals dot-like deposits in the cerebellum of robotic mice in the granule cell layer (arrowhead) and axon tracts (arrow).
GFAP immunostaining revealed marked upregulation of GFAP, indicative of gliosis, at 8 weeks onward (Fig. 2f). Ubiquitin immunostaining revealed no ubiquitinated aggregates in Purkinje cells that have been reported in human spinocerebellar ataxias (Kaytor and Warren, 1999); however, dot-like ubiquitin staining was observed in the molecular layer and axon tracts from 8 weeks of age, similar to that reported in a number of neurodegenerative conditions (Fig.2g) (Leigh et al., 1989). Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining was performed to identify single-stranded DNA termini indicative of an apoptotic mode of cell death, but no positive staining was identified other than on positive control sections (data not shown).
Additional features of the robotic phenotype
To investigate the effects of the robotic mutation on T lymphocyte development in relation to the Af4 knock-out phenotype (Isnard et al., 2000), the thymus was analyzed for cellularity, for weight, and by flow cytometry. Whole thymus weights were significantly lower in mutants compared with littermate controls, as was thymic cellularity (Table 2). Thymocyte development can be analyzed by quantifying changes in surface marker phenotypes as they move from the CD4−/CD8−coreceptor double-negative (DN) to the CD4+/CD8+double-positive (DP) stages, followed by selection and regulation of coreceptor levels to become CD4+ or CD8+ single-positive (SP) T cells. The surface markers CD25 and CD44 further characterize the DN stage, and a significant increase in the population of CD25+/CD44−as well as a decrease in CD25−/CD44+cells was observed (Fig. 3a). In addition, we found a significant increase of coreceptor expression on CD4+ and CD8+ SP thymocytes in robotic (Fig.3b). However, the proportion of the total thymic population at the DP and SP stages was not significantly different in mutant and control mice (Table 2).
Table 2.
Thymocyte cell count numbers in control and robotic mice
| Control | Robotic | |
|---|---|---|
| Weight (g) | 15.4 ± 0.36 | 11.2 ± 0.81* |
| Thymus (mg) | 83.3 ± 3.48 | 55.0 ± 7.37* |
| Thymocytes per lobe (106) | 67.5 ± 0.35 | 27.3 ± 1.782-160 |
| DP (%) | 77.3 ± 0.88 | 80.0 ± 3.0 |
| SP CD8+ (%) | 5.0 ± 0.01 | 4.3 ± 0.33 |
| SP CD4+ (%) | 9.7 ± 1.45 | 8.5 ± 0.89 |
Results are expressed as mean ± SEM. Two-tailed Student'st test:
p < 0.01;
F2-160: p < 0.001.
Fig. 3.

Thymocyte development abnormalities in robotic mice. Flow cytometric analysis of littermate control (white bars) and robotic (black bars) thymus at 3 week of age (n = 3). Data for immature T cell markers CD25/CD44 (a) and average SP CD4/CD8 expression (b) are shown as the mean + SEM. Two-tailed Student's t test: *p < 0.05, **p < 0.03, ***p < 0.01.
Robotic mice breed poorly, and in vitro fertilization has been required to maintain stocks for genetic mapping purposes. No females have been observed that carry pups to full term, and small litters result from the mating of mutant males to wild-type females. A skewed ratio of wild-type to robotic mice that reach weaning age reflects a prenatal and postnatal death rate of ∼20%; however, histological analysis of robotic mouse testis did not reveal any gross abnormalities.
A preliminary clinical biochemistry study was performed (n = 5) to determine possible defects in other organs in robotic mice (T. Hough, personal communication). A small but significant increase in creatinine (p< 0.01) levels was measured in mutant animals that may reflect renal dysfunction, although histologically the kidneys appeared normal. Levels of total cholesterol and high-density lipoprotein cholesterol in blood were also measured and were not elevated in robotic mutants.
To quantify the balance and motor coordination deficits of the robotic ataxic phenotype, latency before falling from an accelerating rotarod was measured at 5 weeks of age. Robotic and control mice both displayed improved performance over the trial period, although mutants showed a significant impairment in this task (data not shown). The observation that motor coordination is reduced before overt cell loss (which begins at ∼8 weeks of age) indicates that Purkinje cell function is impaired early in the disease process, consistent with the early neuropathological findings described above.
Genetic and physical mapping of the robotic locus identifies a point mutation in Af4
A genome scan had previously mapped the robotic mutation to a 15 cM region on mouse chromosome 5 (Nolan et al., 2000). A high-resolution genetic map was generated using 515 backcross animals, and a BAC contig was constructed spanning the 1.2 cM candidate region between flanking microsatellites D5Mit93 andD5Mit10.
Exon trapping isolated multiple clones representing three genes in the contig, confirming the distribution of genes predicted by annotation of the human and mouse genomic sequence from both the public and Celera (Celera Discovery System) databases. We cloned two novel mouse genes in the region, named hydroxysteroid 17-β dehydrogenase type 12 (Hsd17b12; GenBank accession number AY101768) and Kelch-like 8 (Klhl8; GenBank accession number AY101769), although no mutations were identified in either gene by sequencing the coding regions from robotic cDNA. Af4, the third gene in the robotic candidate region, was sequenced from cerebellum cDNA, and a T to C transition was identified in the second base of codon 280 leading to a valine to alanine amino acid substitution. Parental strains C3H/HeH and BALB/c were sequenced and shown to have a T at this position, implying that the mutation was a result of ENU administration. The mutation occurred in a highly conserved region ofAF4, within a run of 13 amino acids that share 100% identity with all known AF4 homologs, suggesting that it plays a critical role in the function of the protein (Fig.4) (Nilson et al., 1997; Taki et al., 1999; Britanova et al., 2002). No differences in expression level of any of the genes in the candidate region were observed between robotic and control animals by quantitative RT-PCR (data not shown).
Fig. 4.

Multiple amino acid sequence alignment of members of the AF4 protein family showing the highly conserved 18 amino acid region within which the robotic mutation lies (arrow). Mouse AF5q31 sequence was taken from an unpublished predicted mouse protein (GenBank accession numberAAG17126). Fugu rubripes sequence was taken from an Ensembl predicted protein of 1123 amino acids, SINFRUP00000087356 (www.ensembl.org/Fugu_rubripes).
Af4 expression in the cerebellum reflects the pathology of the mutant
In situ hybridization on robotic and control littermate sections showed that within the cerebellum Af4 was expressed solely in Purkinje cells (Fig.5a–e) and that the expression was clearly lower in lobe X of the cerebellum in both robotic and littermate control mice (Fig. 5d,e). Lobe X is completely spared from Purkinje cell loss in robotic mice even at 42 weeks of age, suggesting that higher levels of Af4expression are required to facilitate Purkinje cell death and implying that the robotic Af4 allele is acting via a gain-of-function mechanism. RT-PCR was used to demonstrate that Af4 is expressed in the lens of control animals (data not shown), consistent with a role in the cataract formation observed in robotic mutants.
Fig. 5.
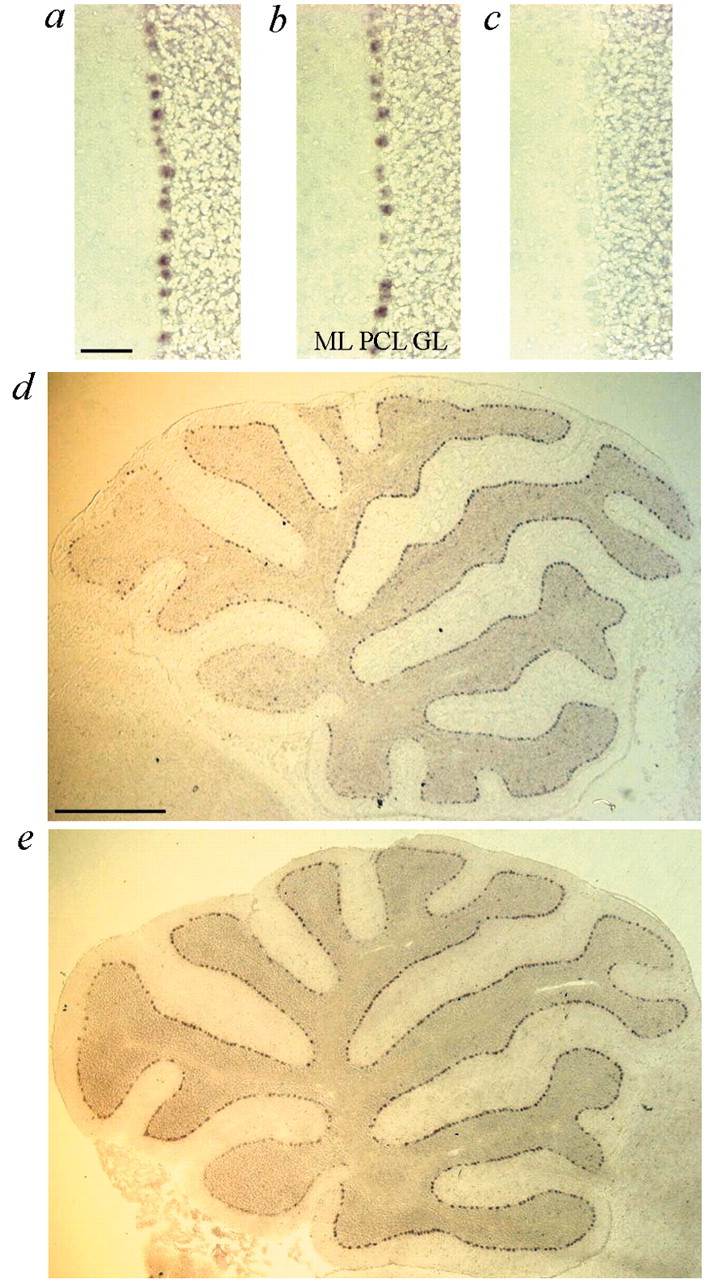
Expression of Af4 in the cerebellum. Bright-field images of in situ hybridization in littermate control (a) and robotic (b) animals at 5 weeks of age with an antisenseAf4 riboprobe. Expression is confined to the Purkinje cell layer (PCL) in both cases. The molecular layer (ML) and granule cell layer (GL) are marked. The corresponding sense probe is shown (c). Scale bar, 100 μm. d,e, Vermal parasagittal cerebellum sections from littermate control (d) and robotic (e) mice at 5 weeks using the antisenseAf4 riboprobe. Reduced levels of expression are observed in lobe X. Scale bar, 1 mm.
Discussion
The data presented here describe a new mutant mouse model for cerebellar ataxia that develops Purkinje cell loss in a stereotypical region-specific manner. High-resolution genetic and physical mapping reduced the robotic locus to a small region containing three genes, and sequencing revealed a point mutation in a highly conserved region ofAf4.
Although the possibility of there being a second mutation in the region genetically linked to the robotic phenotype cannot be ruled out absolutely, there is compelling evidence that Af4 is the causative gene. The robotic locus was reduced to a 0.5 Mb region that contained only three genes, as determined by exon trapping studies and analysis of genomic sequence annotation from both the public and Celera databases. All three genes in this region were sequenced, and no differences in their expression levels were observed between robotic and control animals; only one coding point mutation was isolated in the positional candidate Af4. Second, the Af4mutation occurred in the most highly conserved region of the AF4/LAF4/FMR2 (ALF) domain, suggesting that the residue mutated in robotic mice is of critical importance to the function of the protein (Nilson et al., 1997). The evolutionary conservation of this motif is demonstrated by the presence of an AF4-related homolog fromfugu rubripes containing the 13 amino acids that are identical to the mammalian proteins. Previously, the only nonmammalian homolog of this protein family described was the Drosophilaprotein Lilliputian, which shares identity only to the C-terminal homology domain of AF4 (Tang et al., 2001; Wittwer et al., 2001). Such evolutionary conservation is important given the conservative nature of the valine to alanine amino acid change that occurs in robotic Af4. An analysis of 10121 disease causing missense mutations in humans shows that valine to alanine substitutions do cause disease but that such a change is only 1.43 times more likely to be disease causing than a nonharmful substitution (Goodstadt and Ponting, 2001). Finally, we have shown that Af4 is expressed specifically in Purkinje cells in the cerebellum, the main site of pathology in the robotic mouse, and that expression is lower in the Purkinje cells that are not affected by the mutation.
AF4 is a member of a protein family containing FMR2, LAF4, and AF5q31, which are all nuclear proteins with transactivation activity, suggesting that they act as transcription factors (Prasad et al., 1995;Gu et al., 1996; Ma and Staudt, 1996; Baskaran et al., 1997; Gecz et al., 1997; Nilson et al., 1997; Isnard et al., 1998; Li et al., 1998;Taki et al., 1999). In addition, it has been demonstrated that LAF4 has nonspecific DNA binding properties (Ma and Staudt, 1996). Recent studies suggest that the ALF domain may act as a transcriptional repressor on the basis of increased levels of reporter gene expression from AF4, LAF4, and FMR2 constructs in which this region of the protein was deleted (Hillman and Gecz, 2001). A mutation in the ALF domain could therefore alter transcriptional regulation of downstream target genes caused by a gain-of-function or dominant-negative effect. This mechanism would be consistent with the fact that the lower levels ofAf4 expression observed in lobe X of the cerebellum are not sufficient to cause Purkinje cell death in robotic mice. However, without detailed functional or structural information on the AF4 family, it is still possible that the robotic mutation may influence protein–protein binding events specific to Purkinje cells in the brain.
A pattern of Purkinje cell loss similar to that observed in robotic mice has been described in a number of other cerebellar mouse mutants. Three central bands of resistant cells and preferential loss of Purkinje cells in the anterior cerebellum with sparing of lobe X have been described in the leaner mutant (Herrup and Wilczynski, 1982) and in the mouse models of Niemann-Pick disease types A/B [acid sphingomyelinase knock-out (ASMKO)] and C (Higashi et al., 1993; Sarna et al., 2001). These changes are preceded by granule cell loss in the leaner mouse (Herrup and Wilczynski, 1982; Heckroth and Abbott, 1994), distinguishing the pathological findings from those seen in robotic mutants. However, the similarity of the region-specific cell loss in robotic mice to the Niemann-Pick models is more striking, and dot-like ubiquitin staining has also been noted in the type C disease model (Higashi et al., 1993; Sarna et al., 2001).
Niemann–Pick disease is an autosomal recessive lysosomal storage disorder that is characterized by hepatosplenomegaly and lipid accumulation as well as retarded physical growth and severe neurologic disturbances, including ataxia in many cases (Pentchev et al., 1995;Schuchman and Desnick, 1995). Types A and B are caused by deficient acid sphingomyelinase activity (Levran et al., 1991), and type C is caused by mutations in NPC1, a gene thought to be involved in intracellular trafficking of cholesterol (Carstea et al., 1997). Mouse models of related lipid storage disorders such as Tay-Sachs disease also exhibit reproductive impairment (Trasler et al., 1998), and recent studies of the ASMKO line revealed defects in sperm physiology attributable to lipid-filled vacuoles present in the testis and epididymis (Butler et al., 2002).
Considering that a number of these pathological features are observed in robotic mice, it might be hypothesized that AF4 is involved in pathways important for cholesterol homeostasis and metabolism. However, we have found no direct evidence for cholesterol accumulation in the brain of robotic mice or other organs, including the kidney, spleen, liver, and testis. Moreover, the clinical biochemistry data indicate that levels of blood total cholesterol and HDL cholesterol were not elevated in mutants, as is characteristic of the ASMKO line (Horinouchi et al., 1995). Despite the neurodegenerative features linking the robotic phenotype to lysosomal storage disorders, expression levels ofSmpd1 and Npc1 were not altered in robotic mice compared with wild-type animals when examined by quantitative RT-PCR (data not shown), suggesting that the AF4 mutation may have an influence on related, but not identical, pathways. The stereotyped pattern of cell death in these mutants therefore may reflect a more general underlying susceptibility of biochemically distinct subsets of Purkinje cells to a range of CNS insults.
Although the role of AF4 in leukemogenesis is unclear, it is generally thought that the fusion protein derived from the N-terminal portion of MLL and the C-terminal portion of AF4 is responsible for causing the leukemic phenotype (Borkhardt et al., 1994; Downing et al., 1994). The mutation that we have described occurs upstream of known translocation breakpoints, suggesting that the function of Af4 in robotic mouse may not reflect its potential oncogenic role. The Af4 knock-out mouse (mAf4 −/−) displayed lymphoid development problems but was not reported to have any motor abnormalities indicative of a cerebellar defect, although 20% had a reduced size up to 6 weeks of age, a phenotype shown by all robotic mice throughout life (Isnard et al., 2000). Heterozygous (mAf4 +/−) animals showed no such abnormalities, suggesting that the robotic phenotype is unlikely to be caused by a loss-of-function mutation.
Consistent with a role for Af4 in lymphocyte maturation, we observed abnormalities in T cell development in robotic mice that were similar but distinct from those seen in the Af4 knock-out mice. A reduction in thymic cellularity and weight as observed in robotic mice was described as part of the Af4 knock-out phenotype, although no data from wild-type (mAf4 +/+) mice were reported (Isnard et al., 2000); however, we did discover distinct differences between the knock-out and robotic lines. The phenotypic changes in the DN population as indicted by the CD25 and CD44 marker profiles of robotic mice indicate that the seeding of prothymocytes at this stage is significantly affected (Godfrey and Zlotnick, 1993). In the knock-out line, however, the subsets of DN precursor thymocytes were normal (Isnard et al., 2000). At the DP stage, upregulation of coreceptors as well as intermediate expression of the CD3/T cell receptor complex facilitates positive and negative selection events. Before thymocytes then enter the periphery as mature T cells, downregulation of one coreceptor occurs, and they become either CD4+ or CD8+ SP. Because both coreceptors play an essential role in signaling via the T cell receptor, the higher levels of expression observed in robotic mice might indicate that cells have undergone a more stringent negative selection (Lee et al., 1992; Robey et al., 1992). Alternatively, it may be the direct or indirect result of the transcriptional regulatory activities of the mutant Af4 gene. This effect on thymocyte development in the robotic mouse is once again distinct from the Af4 knock-out phenotype. Taken together, these data are consistent with our hypothesis that the Af4allele is acting through a gain-of-function mechanism.
The identification of novel gene functions is inherent to the phenotype-driven mutagenesis strategy used here because no a priori assumptions are made concerning the underlying genetic defect. The hypothesis that a gain-of-function mutation inAf4 causes neurodegeneration may lead to the identification of new pathways that are important for neuronal survival and adds to the list of cerebellar mutants that have provided a wealth of information regarding CNS function (Heintz and Zoghbi, 2000).
A.M.I and P.L.O contributed equally to this work.
A.J. is supported by a Wellcome Trust Clinical Training Fellowship. We thank Andrew Weir for help with statistical analysis and Paul Denny and Matthew Cadman for assistance with BAC library resources. Certain data were generated through use of the Celera Discovery System and Celera Genomics' associated databases.
Correspondence should be addressed to Professor Kay E. Davies, Medical Research Council Functional Genetics Unit, Department of Human Anatomy and Genetics, University of Oxford, South Parks Road, Oxford, OX1 3QX, UK. E-mail: kay.davies@anat.ox.ac.uk.
References
- 1.Baskaran K, Erfurth F, Taborn G, Copeland NG, Gilbert DJ, Jenkins NA, Iannaccone PM, Domer PH. Cloning and developmental expression of the murine homolog of the acute leukemia proto-oncogene AF4. Oncogene. 1997;15:1967–1978. doi: 10.1038/sj.onc.1201365. [DOI] [PubMed] [Google Scholar]
- 2.Borkhardt A, Repp R, Haupt E, Brettreich S, Buchen U, Gossen R, Lampert F. Molecular analysis of MLL-1/AF4 recombination in infant acute lymphoblastic leukemia. Leukemia. 1994;8:549–553. [PubMed] [Google Scholar]
- 3.Britanova O, Lukyanov S, Gruss P, Tarabykin V. The mouse Laf4 gene: exon/intron organization, cDNA sequence, alternative splicing and expression during central nervous system development. Genomics. 2002;80:31–37. doi: 10.1006/geno.2002.6796. [DOI] [PubMed] [Google Scholar]
- 4.Brown SDM, Balling R. Systematic approaches to mouse mutagenesis. Curr Opin Gen Dev. 2001;11:268–273. doi: 10.1016/s0959-437x(00)00189-1. [DOI] [PubMed] [Google Scholar]
- 5.Butler A, He X, Gordon RE, Wu HS, Gatt S, Schuchman EH. Reproductive pathology and sperm physiology in acid sphingomyelinase-deficient mice. Am J Pathol. 2002;161:1061–1075. doi: 10.1016/S0002-9440(10)64267-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB, Nagle J, Polymeropoulos MH, Sturley SL, Ioannou YA, Higgins ME, Comly M, Cooney A, Brown A, Kaneski CR, Blanchette-Mackie EJ. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 1997;277:180–181. doi: 10.1126/science.277.5323.228. [DOI] [PubMed] [Google Scholar]
- 7.Detter JC, Nguyen QA, Kingsmore SF. Identification of novel simple sequence length polymorphisms (SSLPs) in mouse by interspersed repetitive element (IRE)-PCR. Nucleic Acids Res. 1998;26:4091–4092. doi: 10.1093/nar/26.17.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Domer PH, Fakharzadeh SS, Chen CS, Jockel J, Johansen L, Silverman GA, Kersey JH, Korsmeyer SJ. Acute mixed-lineage leukemia t(4;11) (q21;q23) generates an MLL-AF4 fusion product. Proc Natl Acad Sci USA. 1993;90:7884–7888. doi: 10.1073/pnas.90.16.7884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Downing JR, Head DR, Raimondi SC, Carroll AJ, Curcio-Brint AM, Motroni TA, Hulshof MG, Pullen DJ, Domer PH. The der(11)-encoded MLL/AF-4 fusion transcript is consistently detected in t(4;11)(q21;q23)-containing acute lymphoblastic leukemia. Blood. 1994;83:330–335. [PubMed] [Google Scholar]
- 10.Gecz J, Bielby S, Sutherland GR, Mulley JC. Gene structure and subcellular localization of FMR2, a member of a new family of putative transcription activators. Genomics. 1997;44:201–213. doi: 10.1006/geno.1997.4867. [DOI] [PubMed] [Google Scholar]
- 11.Godfrey DI, Zlotnick A. Control points in early T cell development. Immunol Today. 1993;14:547–553. doi: 10.1016/0167-5699(93)90186-O. [DOI] [PubMed] [Google Scholar]
- 12.Goodstadt L, Ponting CP. Sequence variation and disease in the wake of the draft human genome. Hum Mol Genet. 2001;10:2209–2214. doi: 10.1093/hmg/10.20.2209. [DOI] [PubMed] [Google Scholar]
- 13.Gu Y, Nakamura T, Alder H, Prasad R, Canaani O, Cimino G, Croce CM, Canaani E. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to Drosophila trithorax, to the AF-4 gene. Cell. 1992;71:701–708. doi: 10.1016/0092-8674(92)90603-a. [DOI] [PubMed] [Google Scholar]
- 14.Gu Y, Shen Y, Gibbs RA, Nelson DL. Identification of FMR2, a novel gene associated with the FRAXE CCG repeat and CpG island. Nat Genet. 1996;13:109–113. doi: 10.1038/ng0596-109. [DOI] [PubMed] [Google Scholar]
- 15.Hawkes R, Herrup K. Aldolase C/zebrin II and the regionalization of the cerebellum. J Mol Neurosci. 1995;6:147–158. doi: 10.1007/BF02736761. [DOI] [PubMed] [Google Scholar]
- 16.Heckroth JA, Abbott LC. Purkinje cell loss from alternating sagittal zones in the cerebellum of leaner mutant mice. Brain Res. 1994;658:93–104. doi: 10.1016/s0006-8993(09)90014-2. [DOI] [PubMed] [Google Scholar]
- 17.Heintz N, Zoghbi HY. Insights from mouse models into the molecular basis of neurodegeneration. Annu Rev Physiol. 2000;62:779–802. doi: 10.1146/annurev.physiol.62.1.779. [DOI] [PubMed] [Google Scholar]
- 18.Hendry IA. A method to correct adequately for the change in neuronal size when estimating neuronal numbers after nerve growth factor treatment. J Neurocytol. 1976;5:337–349. doi: 10.1007/BF01175119. [DOI] [PubMed] [Google Scholar]
- 19.Herrup K, Kuemerle B. The compartmentalization of the cerebellum. Annu Rev Neurosci. 1997;20:61–90. doi: 10.1146/annurev.neuro.20.1.61. [DOI] [PubMed] [Google Scholar]
- 20.Herrup K, Wilczynski SL. Cerebellar cell degeneration in the leaner mutant mouse. Neuroscience. 1982;7:2185–2196. doi: 10.1016/0306-4522(82)90129-4. [DOI] [PubMed] [Google Scholar]
- 21.Higashi Y, Murayama S, Pentchev PG, Suzuki K. Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol (Berl) 1993;85:175–184. doi: 10.1007/BF00227765. [DOI] [PubMed] [Google Scholar]
- 22.Hillman MA, Gecz J. Fragile XE-associated familial mental retardation protein 2 (FMR2) acts as a potent transcription activator. J Hum Genet. 2001;46:251–259. doi: 10.1007/s100380170074. [DOI] [PubMed] [Google Scholar]
- 23.Horinouchi K, Erlich S, Perl DP, Ferlinz K, Bisgaier CL, Sandhoff K, Desnick RJ, Stewart CL, Schuchman EH. Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nat Genet. 1995;3:288–293. doi: 10.1038/ng0795-288. [DOI] [PubMed] [Google Scholar]
- 24.Hrabe de Angelis MH, Flaswinkel H, Fuchs H, Rathkolb B, Soewarto D, Marschall S, Heffner S, Pargent W, Wuensch K, Jung M, Reis A, Richter T, Alessandrini F, Jakob T, Fuchs E, Kolb H, Kremmer E, Schaeble K, Rollinski B, Roscher A. Genome-wide, large-scale production of mutant mice by ENU mutagenesis. Nat Genet. 2000;25:444–447. doi: 10.1038/78146. [DOI] [PubMed] [Google Scholar]
- 25.Isnard P, Depetris D, Mattei MG, Ferrier P, Djabali M. cDNA cloning, expression and chromosomal localization of the murine AF-4 gene involved in human leukemia. Mamm Genome. 1998;9:1065–1068. doi: 10.1007/s003359900927. [DOI] [PubMed] [Google Scholar]
- 26.Isnard P, Core N, Naquet P, Djabali M. Altered lymphoid development in mice deficient for the mAF4 proto-oncogene. Blood. 2000;96:705–710. [PubMed] [Google Scholar]
- 27.Justice MJ. Capitalizing on large-scale mouse mutagenesis screens. Nat Rev Genet. 2000;1:109–115. doi: 10.1038/35038549. [DOI] [PubMed] [Google Scholar]
- 28.Kaytor MD, Warren ST. Aberrant protein deposition and neurological disease. J Biol Chem. 1999;274:37507–37510. doi: 10.1074/jbc.274.53.37507. [DOI] [PubMed] [Google Scholar]
- 29.Lee NA, Loh DY, Lacey E. CD8 surface levels alter the fate of alpha/beta T cell receptor-expressing thymocytes in transgenic mice. J Exp Med. 1992;174:1013–1025. doi: 10.1084/jem.175.4.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leigh PN, Probst A, Dale GE, Power DP, Brion JP, Dodson A, Anderton BH. New aspects of the pathology of neurodegenerative disorders as revealed by ubiquitin antibodies. Acta Neuropathol. 1989;79:61–72. doi: 10.1007/BF00308959. [DOI] [PubMed] [Google Scholar]
- 31.Levran O, Desnick RJ, Schuchman EH. Niemann-Pick type B disease. Identification of a single codon deletion in the acid sphingomyelinase gene and genotype/phenotype correlations in type A and B patients. J Clin Invest 1991. 1991;88:806–810. doi: 10.1172/JCI115380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Q, Frestedt JL, Kersey JH. AF4 encodes a ubiquitous protein that in both native and MLL-AF4 fusion types localizes to subnuclear compartments. Blood. 1998;92:3841–3847. [PubMed] [Google Scholar]
- 33.Ma C, Staudt LM. LAF-4 encodes a lymphoid nuclear protein with transactivation potential that is homologous to AF-4, the gene fused to MLL in t(4;11) leukemias. Blood. 1996;87:734–745. [PubMed] [Google Scholar]
- 34.Mitelman F. Catalog of chromosome aberrations in cancer, Ed 5. Wiley; New York: 1994. [Google Scholar]
- 35.Nilson I, Reichel M, Ennas MG, Greim R, Knorr C, Siegler G, Greil J, Fey GH, Marschalek R. Exon/intron structure of the human AF-4 gene, a member of the AF-4/LAF-4/FMR-2 gene family coding for a nuclear protein with structural alterations in acute leukaemia. Br J Haematol. 1997;98:157–169. doi: 10.1046/j.1365-2141.1997.1522966.x. [DOI] [PubMed] [Google Scholar]
- 36.Nolan PM, Peters J, Strivens M, Rogers D, Hagan J, Spurr N, Gray IC, Vizor L, Brooker D, Whitehill E, Washbourne R, Hough T, Greenaway S, Hewitt M, Liu X, McCormack S, Pickford K, Selley R, Wells C, Tymowska-Lalanne Z. A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nat Genet. 2000;25:440–443. doi: 10.1038/78140. [DOI] [PubMed] [Google Scholar]
- 37.Pentchev PG, Vanier MT, Suzuki K, Patterson MC. Niemann-Pick disease types C: a cellular cholesterol lipidosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. McGraw-Hill; New York: 1995. pp. 2625–2640. [Google Scholar]
- 38.Prasad R, Yano T, Sorio C, Nakamura T, Rallapalli R, Gu Y, Leshkowitz D, Croce CM, Canaani E. Domains with transcriptional regulatory activity within the ALL1 and AF4 proteins involved in acute leukemia. Proc Natl Acad Sci USA. 1995;92:12160–12164. doi: 10.1073/pnas.92.26.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robey EA, Ramsdell F, Kioussis D, Sha W, Loh D, Axel R, Fowlkes BJ. The level of CD8 expression can determine the outcome of thymic selection. Cell. 1992;69:1089–1096. doi: 10.1016/0092-8674(92)90631-l. [DOI] [PubMed] [Google Scholar]
- 40.Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioural and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- 41.Rowley JD. The critical role of chromosome translocations in human leukemias. Annu Rev Genet. 1998;32:495–519. doi: 10.1146/annurev.genet.32.1.495. [DOI] [PubMed] [Google Scholar]
- 42.Sarna J, Miranda SR, Schuchman EH, Hawkes R. Patterned cerebellar Purkinje cell death in a transgenic mouse model of Niemann Pick type A/B disease. Eur J Neurosci. 2001;13:1873–1880. doi: 10.1046/j.0953-816x.2001.01564.x. [DOI] [PubMed] [Google Scholar]
- 43.Schuchman EH, Desnick RJ. Niemann-Pick disease types A and B: acid sphingomyelinase deficiencies. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular bases of inherited disease. McGraw-Hill; New York: 1995. pp. 2601–2624. [Google Scholar]
- 44.Stanford WL, Cohn JB, Cordes SP. Gene-trap mutagenesis: past, present and beyond. Nat Rev Genet. 2001;2:756–768. doi: 10.1038/35093548. [DOI] [PubMed] [Google Scholar]
- 45.Taki T, Kano H, Taniwaki M, Sako M, Yanagisawa M, Hayashi Y. AF5q31, a newly identified AF4-related gene, is fused to MLL in infant acute lymphoblastic leukemia with ins(5;11)(q31;q13q23). Proc Natl Acad Sci USA. 1999;96:14535–14540. doi: 10.1073/pnas.96.25.14535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang AH, Neufeld TP, Rubin GM, Muller HA. Transcriptional regulation of cytoskeletal functions and segmentation by a novel maternal pair-rule gene, lilliputian. Development. 2001;128:801–813. doi: 10.1242/dev.128.5.801. [DOI] [PubMed] [Google Scholar]
- 47.Trasler J, Saberi F, Somani IH, Adamli HI, Huang JQ, Fortunato SR, Ritter G, Gu M, Aebersold R, Gravel RA, Hermo L. Characterisation of the testis and epididymis in mouse models of human Tay-Sachs and Sandhoff diseases and partial determination of accumulated gangliosides. Endocrinology. 1998;139:3280–3288. doi: 10.1210/endo.139.7.6117. [DOI] [PubMed] [Google Scholar]
- 48.Wilkinson DG. Whole mount in situ hybridisation of vertebrate embryos. In: Wilkinson DG, editor. In situ hybridisation: a practical approach. IRL; Oxford, UK: 1992. pp. 75–83. [Google Scholar]
- 49.Wittwer F, van der Straten A, Keleman K, Dickinson BJ, Haufen E. Lilliputian: an AF4/FMR2-related protein that controls cell identity and cell growth. Development. 2001;128:791–800. doi: 10.1242/dev.128.5.791. [DOI] [PubMed] [Google Scholar]