

Abstract
Memory extinction refers to a gradual decrease of the previously acquired response when exposed to conditional stimulus without pairing with unconditional stimulus. Here we show for the first time that fear training-induced phosphorylation of specific substrates in the rat amygdala is reduced after extinction trials and is accompanied by an increase in the protein level and enzymatic activity of calcineurin. In parallel, calcineurin inhibitors prevented extinction-induced protein dephosphorylation as well as extinction of fear memory. Thus, extinction training increased phosphatase activity likely via an expression of calcineurin. Calcineurin then created a negative-feedback loop and directly or indirectly dephosphorylated specific substrates, which, in their phosphorylated state, were required for memory consolidation. Accordingly, in our experimental condition, extinction could be ascribed at least in part to a weakening of the original signaling.
Keywords: learning and memory, amygdala, calcineurin, synaptic plasticity, fear conditioning, fear-potentiated startle
Introduction
It is a general observation in animals that a cue [conditioned stimulus (CS)] comes to induce fear response when it is repeatedly paired with a noxious stimulus [unconditioned stimulus (US)], such as footshock (Davis, 2000;LeDoux, 2000). However, it is also known for some time that, if animals are exposed only to the CS without pairing with US, the previously acquired responses will gradually decrease, a phenomenon referred to as fear extinction (Rescorla, 2001). Converging evidence indicate that the amygdala plays an important role in both formation and extinction of fear memory (Falls et al., 1992; Maren, 1999; Nader et al., 2000; Lu et al., 2001). Experiments performed by various laboratories using different animal models indicated that long-term memory formation involved activation of the protein kinases such as calcium/calmodulin kinase II, cAMP-dependent protein kinase (PKA), and mitogen-activated protein kinase (MAPK) (Impey et al., 1998; Schafe et al., 2000;Josselyn et al., 2001). Once stimulated, these kinases could translocate to the nucleus and subsequently activate transcription factors to promote gene transcription and new protein synthesis. Much less is known about the cellular mechanisms leading to memory extinction. Previous studies have shown that experimental extinction was blocked by NMDA receptor antagonists and MAPK kinase (MEK) inhibitors (Falls et al., 1992; Lu et al., 2001) and facilitated byd-cycloserine, a partial NMDA agonist (Walker et al., 2002). Recently, it was found that extinction of fear conditioning was impaired in cannabinoid receptor 1-deficient (Marsicano et al., 2002) or protein phosphatase 1-inhibited (Genoux et al., 2002) mice.
This laboratory showed recently that acquisition of fear was associated with an activation of phosphatidylinositol 3-kinase (PI-3 kinase) and its downstream target Akt in the rat amygdala. PI-3 kinase and Akt were also activated in response to long-term potentiation (LTP)-inducing tetanic stimulation (TS). In parallel, PI-3 kinase inhibitors interfered with TS-induced LTP as well as long-term fear memory formation (Lin et al., 2001). Therefore, it is of particular interest to see whether extinction trials affect phosphorylated state of this protein kinase. Here we show for the first time that fear training-induced phosphorylation of Akt in the rat amygdala is reduced after extinction training and is accompanied by an increase in the protein level and enzymatic activity of calcineurin.
Materials and Methods
Surgery
Rats, anesthetized with sodium pentobarbital (50 mg/kg, i.p.), were mounted on a stereotaxic apparatus and two cannulas made of 22 gauge stainless steel tubing (C313G; Plastic Products) were implanted bilaterally into the lateral amygdala (LA) or basolateral amygdala (BLA). The coordinates were as follows (according to Paxinos and Watson, 1986): anteroposterior, −2.3 mm; mediolateral, ±4.5 mm; dorsoventral, −7.0 mm. Only rats with cannula tips within the boundaries of LA and BLA were included in the data analysis. Rats were monitored and handled daily and were given 7 d to recover. Calcineurin inhibitor FK-506 was dissolved in DMSO, and cypermethrin was dissolved in 95% ethanol. The drugs were administered bilaterally in a volume of 0.8 μl at a rate of 0.5 μl/min.
Behavioral apparatus and procedures
Fear conditioning was measured using potentiated startle paradigm adapted and modified from Lu et al. (2001). Rats were trained and tested in a stabilimeter device. A piezoelectric device mounted below stabilimeter detects and transduces the motion of the cylinder produced by the whole-body startle response of the rat (San Diego Instruments, San Diego, CA). The whole setup was enclosed in a ventilated, sound-attenuating cabinet (length of 38 cm, width of 38 cm, and height of 55 cm). The acoustic startle stimulus was a 50 msec white noise at the intensity of 95 dB. The visual CS was a 3.7 sec light produced by an 8 W fluorescent bulb attached to the back of stabilimeter. The US was a 0.6 mA footshock with a duration of 0.5 sec.
Acclimation. On 3 consecutive days, rats were placed in the startle test boxes for 10 min and returned to their home cages.
Matching. On 2 consecutive days, rats were placed in the startle box and, 3 min later, presented with 10 startle stimuli at a 2 min intertrial interval (ITI). On the basis of their mean startle amplitudes in the second of these two sessions, rats were matched into groups with similar response levels.
Training. Rats were placed in the startle boxes and received 10 light–footshock pairings with an ITI of 2 min. Unpaired controls received the same number of light and footshock presentation but in a pseudorandom manner in which the US could occur at anytime except at the 3.2 sec after the CS.
Preextinction test. Twenty-four hours after training, rats were tested for fear-potentiated startle. This involved 10 startle-eliciting noise bursts presented alone (noise-alone trial) and 10 noise bursts presented 3.2 sec after onset of the 3.7 sec light (light–noise trials). The two trial types were presented in a balanced mixed order (ITI, 30 sec). The percentage of fear-potentiated startle was computed as follows: [(startle amplitude on CS–noise minus noise-alone trials)/(noise-alone trials)] × 100.
Extinction training. Ten minutes after the pretests, rats were returned to the stabilimeter and given three sessions of 10 presentations of the 3.7 sec light in the absence of either shock or the startle-elicited noise burst (light-alone trials). Each session was separated by 10 min with an ITI of 1 min. The context control group remained in the stabilimeter for an equivalent period of time without receiving any stimulation.
Postextinction training. Immediately after extinction training, rats were tested for fear-potentiated startle in a procedure identical to pretests.
Western blot analysis
Rats were killed by decapitation, and the lateral and basolateral subregions of the amygdala were sonicated briefly in ice-cold buffer [50 mm Tris-HCl, pH 7.5, 0.3 msucrose, 5 mm EDTA, 2 mm sodium pyrophosphate, 1 mm sodium orthovanadate, 1 mmphenylmethylsulfonyl fluoride (PMSF), 20 μg/ml leupeptin, and 4 μg/ml aprotinin]. After sonication, the samples were centrifuged at 7500 rpm for 15 min, and the supernatant was obtained after pelleting the crude membrane fraction by centrifugation at 50,000 rpm for 1 hr at 4°C. Equivalent amounts of protein for each sample were resolved in 8.5% SDS-polyacrylamide gels, blotted electrophoretically to Immobilon, and blocked overnight in TBS buffer (50 mmTris-HCl, pH 7.5, and 150 mm NaCl) containing 3% bovine serum albumin. For detection of the phosphorylated forms of Akt or the protein level of calcineurin, blots were incubated with anti-phospho-Akt (1:1000; New England Biolabs, Beverly, MA) and anti-calcineurin (1:2000; BD Transduction Laboratories, Los Angeles, CA) antibodies, respectively. To assess for change in the activation of PI-3 kinase, total kinase levels were first normalized to total protein levels for each sample. Results were expressed by the ratio of activated kinase levels to total kinase levels. However, in some experiments, activated kinase levels in trained animals were normalized to total kinase levels and then were expressed as a percentage of those in unpaired controls.
Calcineurin activity assay
Light-alone trained rats were killed by decapitation immediately after trials. The LA and BLA were microdissected and frozen on dry ice. Phosphatase assay was performed according to the instructions of the calcineurin assay kit (Promega, Madison, WI). Pooled LA and BLA areas were homogenized in ice-cold buffer (50 mmTris-HCl, pH 7.5, 50 mm NaCl, 10 mm EGTA, 5 mm EDTA, 1 mm sodium orthovanadate, 1 mm PMSF, 20 μg/ml leupeptin, and 4 μg/ml aprotinin) and centrifuged at 100,000 rpm for 1 hr to remove particular matter. Supernatants were added to the reaction buffer from the kit, and the reaction was incubated at 30°C for 10 min. The reaction buffer contains 50 mm imidazole, pH 7.2, 0.2 mm EGTA, 10 mm MgCl2, 1 mmNiCl2, 50 μg/ml calmodulin, and 0.02% β-mercaptoethanol. FK-506 (1 μm) in the supernatant completely blocked inorganic phosphate (Pi) release, indicating that the measured phosphatase activity reflects calcineurin function. The enzyme activity was expressed in nanomoles of Pi released per minute per milligram of protein from the substrate. The calcineurin substrate sequence is RRA(pT)VA.
Results
Rats were given 10 pairings of light (CS) and footshock (US) and tested 24 hr later (pretest). After initial training, animals exhibited fear of the light manifesting as an increase in acoustic startle (195.9 ± 6.8% of potentiation; n = 54;p < 0.001). They were subsequently divided into three groups: light alone, context control, and light plus shock groups. The rats in the light-alone group were given three sessions of 10 presentations of light in the absence of either shock or startle-elicited noise burst. Group context control was placed in the testing apparatus for an equivalent amount of time without stimulation, and group light plus shock was given three sessions of light–shock pairs (Fig. 1A). As shown in Figure 1B, rats in the light-alone group displayed a significant reduction in startle amplitude relative to their preextinction levels. A two-way ANOVA with treatment (light-alone presentations vs context control) and sessions as between-subjects factors indicated a significant treatment effect (F(1,30) = 190.4; p < 0.0001) and also a significant treatment × sessions interaction (F(2,30) = 3.88; p < 0.05). As expected, rats in the light–shock group showed a slight increase after three sessions of training (t(5) = 2.82; p < 0.05). Together, these results indicate that fear extinction is attributable to repeated presentations of light in the absence of shock rather than to the exposure of experimental context or the desensitization to conditioning stimuli. It is important to note that, when presenting the light-alone protocol to unpaired rats that were exposed to the light and footshock in an unpaired, pseudorandom manner, startle responses were not decreased to below the baseline level. This result suggests a selective effect of extinction training on the learned association between CS and US but not on the startle reflex itself.
Fig. 1.

Effect of extinction training on the fear-potentiated startle. A, Timeline of behavioral procedures for experiments B. B, Percentage of startle potentiation before (pre-test), during, and after three sessions of extinction training, context exposure, or light–shock (post-test). The degree of potentiation was significantly reduced only in the light-alone group. It is noted that unpaired controls did not exhibit a decrease in startle responses. *p < 0.05, **p < 0.01, and ***p < 0.001 versus pretests.
We next examined whether the activated state of PI-3 kinase induced by fear conditioning is affected after extinction trials. Rats were exposed to the light repeatedly paired with footshock, and behavioral tests were performed 24 hr later (Fig.2A). Figure2B shows that conditioned rats exhibited an increase in Akt phosphorylation after test compared with those of naive and unpaired controls. The increase was transient and significant from 60 to 120 min after testing but not at other time points (F(6,35) = 24.32; p < 0.001). Post hoc comparisons (Newman–Keuls) revealed the differences between control and 60, 90, and 120 min time points (p < 0.01). No change was observed when blotted membrane was reprobed with an antibody that recognized Akt independently of its phosphorylated state. To determine whether the higher level of Akt phosphorylation in conditioned animals shown here is indicative of induction after behavioral tests or a persistent posttraining increase, we measured the level of Akt phosphorylation 24 hr after training but before test. Figure 2B also shows that the level of Akt phosphorylation in conditioned rats 24 hr after training is not significantly different from those of naive and unpaired controls. Thus, behavioral tests caused reactivation of protein kinase.
Fig. 2.

Effect of extinction training on conditioning-induced Akt phosphorylation. A, Behavioral procedure used in experiment B. B, Time course of PI-3 kinase activation in paired as opposed to those of unpaired and naive rats. Shown are the representative blots and mean ± SE ratios of P-Akt/Akt immunoreactivities from rats decapitated at 30, 60, 90, 120, 150, and 180 min (n = 6 rats in each time point) after pretests. The −10 min time point represents the level of Akt phosphorylation 24 hr after training but before test, whereas the 0 min time point is the value taken from naive rats. *p< 0.01 versus unpaired control. C, Behavioral procedure used in experiment D. D, Rats received fear training in a paired or unpaired manner and were tested 24 hr later (pretest). Shown are the representative blots and mean ± SE percentage of P-Akt immunoreactivities from unpaired (lane 1) and paired (lane 2) rats decapitated at 60 min after pretest. Ten minutes after pretest, paired rats were given light-alone trials (lane 3) or exposed to the context (lane 4), and the amygdala was removed for biochemical assay. The degree of Akt phosphorylation was significantly reduced in the light-alone group. Bilateral infusion of FK-506 (10 μg dissolved in 1.6 μl of DMSO, 0.8 μl per side) before light-alone trials blocked dephosphorylation (lane 5). *p < 0.01 versus pretest.
After been conditioned, rats were divided into two groups: light-alone and context control. Immunoblotting for phosphorylated Akt (P-Akt) was performed immediately after extinction training (Fig. 2C). Figure 2D indicates that the degree of Akt phosphorylation was significantly reduced in the light-alone group (t(10) = 3.75;p < 0.01). In contrast, Akt phosphorylation in the context control group was comparable with and not significantly different from the preextinction test (t(10) = 0.32; p = 0.75). These results suggest that memory testing-induced Akt phosphorylation was abrogated after light-alone trials. Given that synaptic plasticity may be governed by the balance between protein kinase and phosphatase activity (O'Dell and Kandel, 1994; Winder and Sweatt, 2001), a decrease in the phosphorylated state of Akt after extinction trials led us consider the possible involvement of calcium-dependent phosphatase calcineurin. We determined the involvement of calcineurin by infusing FK-506, a calcineurin inhibitor, bilaterally into the amygdala before extinction training. As shown in Figure 2D, FK-506 treatment reversed extinction-induced dephosphorylation.
We tested whether calcineurin inhibitors affected experimental extinction by giving cyclosporin A intravenously into the rats before light-alone trials. This drug, which inhibits calcineurin by forming complex with cyclophilin (Liu et al., 1991), dose-dependently blocked extinction (F(1,10) = 255.5;p < 0.001) (Fig. 3). Conversely, rats that received vehicle showed a normal decrease in the startle after extinction trials (F(2,15) = 82.8; p < 0.001). Additional support for an involvement of calcineurin came from the observation that direct injection of two other calcineurin inhibitors FK-506 (10 μg dissolved in 1.6 μl of DMSO, 0.8 μl per side) or cypermethrin (3 μg dissolved in 1.6 μl of ethanol, 0.8 μl per side) into the amygdala blocked extinction. Startle responses after light-alone trials in FK-506-treated (t(5) = 0.19; p = 0.85) or cypermethrin-treated (t(5) = 0.39; p = 0.71) rats were comparable with their pretests. These results suggest that FK-506 blocked memory extinction at the same dose that inhibited P-Akt dephosphorylation. However, the effects of FK-506 could reflect an enhancement of fear-potentiated startle rather than a blockade of extinction (e.g., FK-506 elevated the level of startle potentiation regardless of the extinction protocol). To differentiate these possibilities, additional rats were matched into four groups. Two groups (one group of vehicle-injected rats and one group of FK-506-injected rats) received fear training, whereas two other groups (one group of vehicle-injected rats and one group of FK-506-injected rats) were exposed to the CS and US in an unpaired, pseudorandom manner. We found that FK-506 did not affect startle potentiation in either the unpaired (t(10) = 0.48; p = 0.64) or paired (t(10) = 0.34;p = 0.74) groups because there was no difference in the degree of potentiation between vehicle- and FK-506 treated rats in both groups. In parallel to the startle results, FK-506 did not affect Akt phosphorylation. In both paired (t(10)= 0.15; p = 0.88) and unpaired (t(10) = 0.59; p = 0.57) rats, Akt phosphorylation in vehicle group was not different from that of FK-506 group.
Fig. 3.

Extinction training-induced reduction of fear memory is blocked by calcineurin inhibitors. Percentage of startle potentiation before (pretest) and after three sessions of extinction training in conditioned rats given intravenous administration of cyclosporin A (5 or 20 mg/kg), bilateral amygdala infusion of FK-506 (10 μg dissolved in 1.6 μl of DMSO, 0.8 μl per side), or cypermethrin (3 μg dissolved in 1.6 μl of ethanol, 0.8 μl per side) before light-alone trials. *p < 0.01 and **p < 0.001 versus pretests.
If calcineurin is involved in the extinction, then one might expect its activity to be regulated by extinction training. We assayed calcineurin activity by measuring the released Pi from the phosphopeptide substrate that was insensitive to okadaic acid but could be blocked by FK-506. In the first series of experiments, rats were assigned into three groups (naive, unpaired, and paired), and each individual group was trained according to its own protocol. Subsequently, the release of Pi from LA and BLA was measured in each group. There was no difference among these three groups. Next, conditioned rats were subjected to light-alone trials, and calcineurin activity was measured after training. Figure4A shows that, after extinction training, Pi release was enhanced from 5.0 ± 0.4 nmol Pi/min/mg (n = 6 rats) to 10.1 ± 0.3 nmol Pi/min/mg (n = 6; p < 0.001). Because it is possible that the observed increase may be attributable to some unrelated factors in the experimental procedure not specifically related to extinction, we repeated the experiments in the naive and unpaired animals. The results indicated that calcineurin activity was not altered in these animals (Fig. 4A).
Fig. 4.
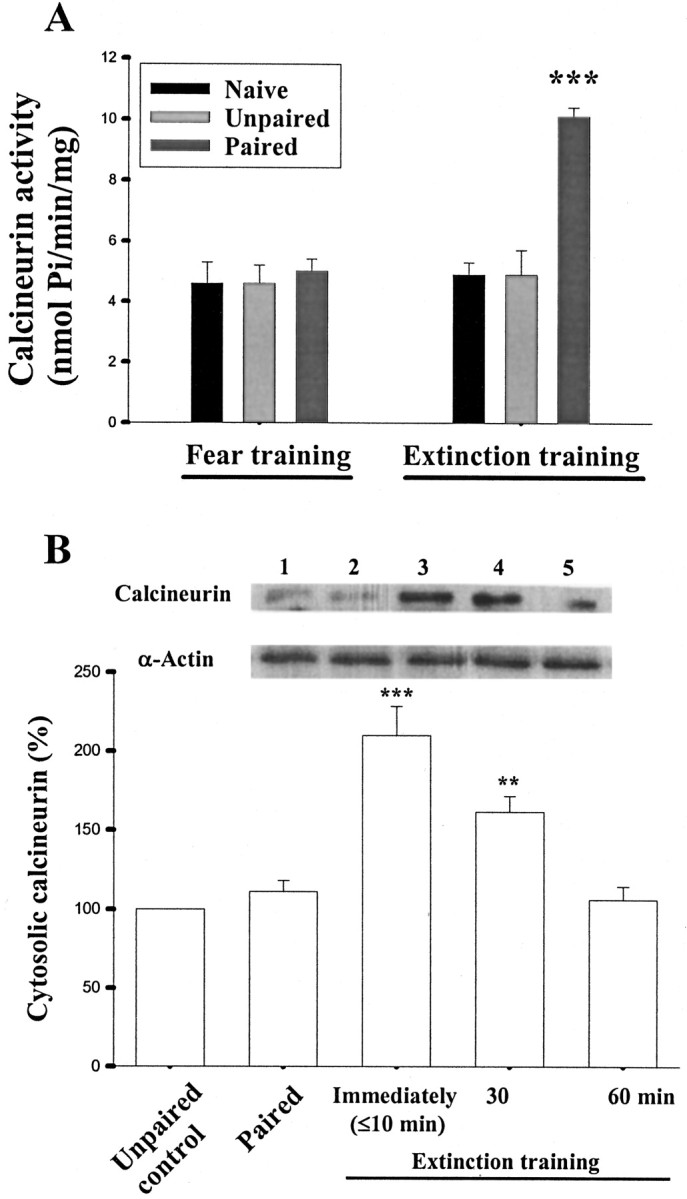
Increase in the enzymatic activity and protein expression of calcineurin after extinction training. A, Rats were assigned into three groups (naive, unpaired, and paired; 12 rats in each group), and each group was trained according to its own protocol. After training, six rats in each group were killed, and the release of Pi from LA and BLA was measured. Next, the remaining six rats in each group were subjected to extinction training, and calcineurin activity was measured after training. After extinction training, the release of Pi was enhanced only in the paired rats. ***p < 0.001 versus naive or unpaired groups.B, Time course of calcineurin expression induced by light-alone trials in cytosolic fraction. Shown are representative blots and mean ± SE of calcineurin immunoreactivities from rats decapitated at various time points (n = 6 rats in each time point) after extinction training (α-actin was used as internal control). **p < 0.01 and ***p < 0.001 versus paired.
We determined calcineurin protein levels after extinction training. Fear conditioning did not affect the cytosolic level of calcineurin (90.1 ± 6.8%; n = 6 rats; p > 0.1). However, after light-alone trials, calcineurin was significantly increased, which peaked at 10 min after training and subsided within 60 min (F(3,20) = 16.25;p < 0.001) (Fig. 4B). Newman–Keulst tests revealed that differences existed between control and 10 and 30 min time points (p < 0.001 andp < 0.01, respectively). As a control, the immunoreactivity for α-actin was checked and found unchanged.
Discussion
Previous studies have shown that infusion of anisomycin shortly after testing produced amnesia. This has led to the hypothesis that memory, when reactivated, became vulnerable and sensitive to disruption by protein synthesis inhibitors (Nader et al., 2000; Sara, 2000). In the present study, we found that behavioral tests (light plus noise) caused reactivation of kinase. The reactivation of kinase may lead to new protein synthesis that is crucial for memory reconsolidation. This could explain why memory became labile when retrieved. Conversely, extinction training (light-alone) resulted in dephosphorylation of Akt.
It is suggested that acquisition and extinction may share some common mechanisms (Corcoran and Maren, 2001; Vianna et al., 2001). For example, NMDA receptor and MEK inhibitors that blocked the acquisition of fear (Miserendino et al., 1990; Schafe et al., 2000) also blocked the extinction of conditioned fear (Falls et al., 1992; Lu et al., 2001). Consistent with these results, we found that wortmannin produced comparable effects on both acquisition and extinction of fear memory (Lin et al., 2001; our unpublished data). Furthermore, anisomycin, a protein synthesis inhibitor known to block acquisition of many memory tasks (Davis and Squire, 1984), also blocked extinction of an inhibitory avoidance task and the conditioned taste aversion (Berman and Dudai, 2001; Vianna et al., 2001) (but seeLattal and Abel, 2001). Thus, it is possible that extinction training initiates calcium influx through NMDA receptors in the amygdala. The increase in intracellular calcium results in the activation protein kinases such as PI-3 kinase and MAPK. Once stimulated, MAPK can translocate to the nucleus, in which they activate cAMP response element-binding protein (CREB) and unidentified transcriptional factors to reactivate original memory on one hand and promote calcineurin synthesis on the other hand. Calcineurin creates a negative-feedback loop to suppress phosphorylation and weakens the original memory. Thus, inhibition of protein synthesis during or immediately after retrieval can result in either enhancement or extinction of a learned response that is region or task dependent and also depends on the balance between protein kinase and phosphatase activity. When calcineurin activity induced by extinction protocol exceeds the activity of protein kinase, inhibition of protein synthesis would block extinction and thus strengthen memory. In contrast, if protein kinase activity dominates, then anisomycin produces amnesia after memory reactivation.
Calcineurin has long been implicated in the synaptic plasticity. In the hippocampus, calcineurin is involved in both long-term depression (Mulkey et al., 1994) and depotentiation (Zhuo et al., 1999) and negatively regulates the transition between early and late phases of LTP (Winder et al., 1998). Developmental shift of NMDA receptor-dependent dephosphorylation of CREB is mediated by calcineurin (Sala et al., 2000). Calcineurin also directly modulates AMPA (Banke et al., 2000), NMDA (Shi et al., 2000), as well as GABAA receptor-channel kinetics (Jones and Westbrook, 1997). In combination with behavioral and biochemical experiments, we show here that activation of calcineurin contributes to the extinction of fear memory. The identification of calcineurin as a molecular signal in memory liability suggests a potential new target for the treatment of anxiety and posttraumatic stress disorders.
Footnotes
This work was supported by National Science Council Grant NSC89-2320-B006-011 and Academic Excellence Program of the Ministry of Education of Taiwan Grant 89-B-FA08-1-4.
Correspondence should be addressed to Dr. Po-Wu Gean, Department of Pharmacology, College of Medicine, National Cheng-Kung University, Tainan, Taiwan 701. E-mail: powu@mail.ncku.edu.tw.
References
- 1.Banke TG, Bowie D, Lee HK, Huganir RL, Schousboe A, Traynelis SF. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci. 2000;20:89–102. doi: 10.1523/JNEUROSCI.20-01-00089.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berman DE, Dudai Y. Memory extinction, learning anew, and learning the new: dissociation in the molecular machinery of learning in cortex. Science. 2000;291:2417–2419. doi: 10.1126/science.1058165. [DOI] [PubMed] [Google Scholar]
- 3.Corcoran KA, Maren S. Hippocampal inactivation disrupts contextual retrieval of fear memory after extinction. J Neurosci. 2001;21:1720–1726. doi: 10.1523/JNEUROSCI.21-05-01720.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davis HP, Squire LR. Protein synthesis and memory: a review. Psychol Bull. 1984;96:518–559. [PubMed] [Google Scholar]
- 5.Davis M. The role of the amygdala in conditioned and unconditioned fear and anxiety. In: Aggleton JP, editor. The amygdala: a functional analysis. Oxford UP; New York: 2000. pp. 213–287. [Google Scholar]
- 6.Falls WA, Miserendino JD, Davis M. Extinction of fear-potentiated startle: blockade by infusion of an NMDA antagonist into the amygdala. J Neurosci. 1992;12:854–863. doi: 10.1523/JNEUROSCI.12-03-00854.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Genoux D, Haditsch U, Knobloch M, Michalon A, Storm D, Mansuy IM. Protein phosphatase 1 is a molecular constraint on learning and memory. Nature. 2002;418:970–975. doi: 10.1038/nature00928. [DOI] [PubMed] [Google Scholar]
- 8.Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- 9.Jones MV, Westbrook GL. Shaping of IPSCs by endogenous calcineurin activity. J Neurosci. 1997;17:7626–7633. doi: 10.1523/JNEUROSCI.17-20-07626.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Josselyn SA, Shi C, Carlezon WA, Neve RL, Jr, Nestler EJ, Davis M. Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci. 2001;21:2404–2412. doi: 10.1523/JNEUROSCI.21-07-02404.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lattal KM, Abel T. Different requirements for protein synthesis in acquisition and extinction of spatial preferences and context-evoked fear. J Neurosci. 2001;21:5773–5780. doi: 10.1523/JNEUROSCI.21-15-05773.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- 13.Lin CH, Yeh SH, Lin CH, Lu KT, Leu TH, Chang WC, Gean PW. A role for the PI-3 kinase signaling pathway in fear conditioning and synaptic plasticity in the amygdala. Neuron. 2001;31:841–851. doi: 10.1016/s0896-6273(01)00433-0. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, Farmker JD, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66:807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 15. Lu KT, Walker DL, Davis M. Mitogen-activated protein kinase cascade in the basolateral nucleus of amygdala is involved in extinction of fear-potentiated startle. J Neurosci 21 2001. RC162(1–5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maren S. Long-term potentiation in the amygdala: a mechanism for emotional learning and memory. Trends Neurosci. 1999;22:561–567. doi: 10.1016/s0166-2236(99)01465-4. [DOI] [PubMed] [Google Scholar]
- 17.Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Di Marzo V, Lutz B. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;418:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 18.Miserendino MJ, Sananes CB, Melia KR, Davis M. Blocking of acquisition but not expression on conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature. 1990;345:716–718. doi: 10.1038/345716a0. [DOI] [PubMed] [Google Scholar]
- 19.Mulkey RM, Endo S, Shenolikar S, Malenka RC. Involvement of a calcineurin/inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nature. 1994;369:486–488. doi: 10.1038/369486a0. [DOI] [PubMed] [Google Scholar]
- 20.Nader K, Schafe GE, LeDoux JE. Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature. 2000;406:722–726. doi: 10.1038/35021052. [DOI] [PubMed] [Google Scholar]
- 21.O'Dell T, Kandel E. Low-frequency stimulation erases LTP through an NMDA receptor-mediated activation of protein phosphatases. Learn Mem. 1994;1:129–139. [PubMed] [Google Scholar]
- 22.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1986. [DOI] [PubMed] [Google Scholar]
- 23.Rescorla RA. Experimental extinction. In: Mowrer RR, Klein S, editors. Handbook of contemporary learning theories. Erlbaum; Mahwah, NJ: 2001. pp. 119–154. [Google Scholar]
- 24.Sala C, Rudolph-Correia S, Sheng M. Developmentally regulated NMDA receptor-dependent dephosphorylation of cAMP response element-binding protein (CREB) in hippocampal neurons. J Neurosci. 2000;20:3529–3536. doi: 10.1523/JNEUROSCI.20-10-03529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sara SJ. Retrieval and reconsolidation: toward a neurobiology of remembering. Learn Mem. 2000;7:73–84. doi: 10.1101/lm.7.2.73. [DOI] [PubMed] [Google Scholar]
- 26.Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE. Activation of ERK/MAPK kinase in the amygdala is required for memory consolidation of Pavlovian fear conditioning. J Neurosci. 2000;20:8177–8187. doi: 10.1523/JNEUROSCI.20-21-08177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shi J, Townsend M, Constantine-Paton M. Activity-dependent induction of tonic calcineurin activity mediates a rapid developmental downregulation of NMDA receptor currents. Neuron. 2000;28:103–114. doi: 10.1016/s0896-6273(00)00089-1. [DOI] [PubMed] [Google Scholar]
- 28.Vianna MRM, Szapiro G, McGaugh JL, Medina JH, Izquierdo I. Retrieval of memory for fear-motivated training initiates extinction requiring protein synthesis in the rat hippocampus. Proc Natl Acad Sci USA. 2001;98:12251–12254. doi: 10.1073/pnas.211433298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walker DL, Ressler KJ, Lu KT, Davis M. Facilitation of conditioned fear extinction by systemic administration or intra-amygdala infusions of d-cycloserine as assessed with fear-potentiated startle in rats. J Neurosci. 2002;22:2343–2351. doi: 10.1523/JNEUROSCI.22-06-02343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winder DG, Sweatt JD. Roles of serine/threonine phosphatases in hippocampal synaptic plasticity. Nat Rev Neurosci. 2001;2:461–474. doi: 10.1038/35081514. [DOI] [PubMed] [Google Scholar]
- 31.Winder DG, Mansuy IM, Osman M, Moallem TM, Kandel ER. Genetic and pharmacological evidence for a novel, intermediate phase of long-term potentiation suppressed by calcineurin. Cell. 1998;92:25–37. doi: 10.1016/s0092-8674(00)80896-x. [DOI] [PubMed] [Google Scholar]
- 32.Zhuo M, Zhang W, Son H, Mansuy I, Sobel RA, Seidman J, Kandel ER. A selective role of calcineurin Aα in synaptic depotentiation in hippocampus. Proc Natl Acad Sci USA. 1999;96:4650–4655. doi: 10.1073/pnas.96.8.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]