

Abstract
The Bad signaling pathway contributes to the regulation of apoptosis after a variety of cell death stimuli, and Bad plays a key role in determining cell death or survival. We have reported that overexpression of copper/zinc superoxide dismutase (SOD1) reduces apoptotic cell death after transient focal cerebral ischemia (tFCI). However, both the role of the Bad pathway after tFCI and the role of oxygen free radicals in the regulation of apoptosis remain unknown. To clarify these issues, we used an in vivo tFCI model of SOD1 transgenic mice and wild-type mice. Moreover, to examine the role of protein kinase A (PKA) in the Bad pathway after tFCI, we administered the PKA inhibitor, H89, into the mouse brain after tFCI. Immunohistochemistry and Western blot analysis showed that dephosphorylation and translocation of Bad were detected early after tFCI and that they were promoted by H89 treatment but prevented by SOD1. Coimmunoprecipitation revealed that the dimerization of Bad progressed with 14-3-3 (Bad/14-3-3) and with Bcl-xL(Bad/Bcl-xL) after tFCI. Moreover, Bad/14-3-3 was prevented by H89 treatment but promoted by SOD1. Bad/Bcl-xLwas prevented by SOD1 but promoted by H89 treatment. A cell death assay revealed that apoptotic-related DNA fragmentation was aggravated by H89 treatment but reduced by SOD1. These results suggest that the Bad pathway mediated by PKA is involved in apoptotic cell death after tFCI and that overexpression of SOD1 may attenuate this apoptotic cell death.
Keywords: cerebral ischemia, apoptosis, Bad, Bcl-xL, 14-3-3, superoxide
Introduction
The cell survival pathway has been the focus in clarifying the machinery of apoptotic neuronal cell death after cerebral ischemia. The Bcl-2 family of proteins and many kinase-signaling pathways play pivotal roles in regulation of the balance between cell death and survival (Adachi et al., 1997; Datta et al., 2000). The Bcl-2 family comprises proapoptotic and anti-apoptotic proteins; some proteins within this family, such as Bcl-2, Bcl-xL, and Bcl-w, inhibit apoptosis, whereas others, including Bad, Bax, Bid, and Bcl-xS, promote apoptosis (Yang and Korsmeyer, 1996; Kroemer, 1997). Dimerization of Bcl-2 family members involves interactions between amino acid sequences, known as Bcl-2 homology domains (Yang and Korsmeyer, 1996; Kroemer, 1997; Reed, 1997; Kelekar and Thompson, 1998). Bad, an important proapoptotic member of the Bcl-2 family, links signal transduction events to this family (Yang et al., 1995; Zha et al., 1996). Via the phosphorylation of four serine residues (Ser-112, 136, 155, 170) Bad resides in an inactive complex with the molecular chaperone 14-3-3 in in vitro studies (Zha et al., 1996;Datta et al., 2000; Lizcano et al., 2000; Dramsi et al., 2002). In the presence of apoptotic stimuli Bad is dephosphorylated, dissociated from 14-3-3, and translocated to the outer membrane of mitochondria, where it subsequently dimerizes with Bcl-xL (Zha et al., 1996; Hsu et al., 1997; Zhang et al., 1999). Ser-155 residue is critical to the direct interaction of Bad with Bcl-xL after apoptotic stimuli in many in vitro studies (Tan et al., 1999, 2000; Datta et al., 2000). Ser-155 phosphorylation is regulated by protein kinase A (PKA) or mitogen-activated protein kinase-activated protein kinase 1 (also called RSK) (Tan et al., 1999; Lizcano et al., 2000). However, Bad prophosphorylation and dephosphorylation after apoptotic stimuli remain unclear in vivo. Intraventricular injection of H89, a specific inhibitor of PKA, effectively suppresses the activity of PKA (Cervo et al., 1997; Kimura et al., 1998). To clarify the Bad signaling pathway and the role of PKA in vivo, we used a mouse transient focal cerebral ischemia (tFCI) model and administered H89 after tFCI.
Reactive oxygen species (ROS) has been implicated in the machinery of reperfusion injury after cerebral ischemia (Chan, 1994; Fujimura et al., 1999). The electron flow in isolated brain mitochondria produces superoxide anions, which are scavenged by superoxide dismutase (SOD) (Boveris and Chance, 1973). We have shown that copper/zinc-SOD (SOD1), a cytosolic antioxidant isoenzyme, is highly protective against ischemia and reperfusion injury after cerebral ischemia (Kinouchi et al., 1991; Chan, 1996; Kondo et al., 1997). In examining the mechanism of the proapoptotic pathway, our studies have demonstrated that SOD1 affected the early release of cytochrome c (Fujimura et al., 2000) and the second mitochondria-derived activator of caspases (Sugawara et al., 2002) from mitochondria to the cytosol after cerebral ischemia. However, whether SOD1 could affect the balance between cell survival and the apoptotic pathway has not been demonstrated. The present study was designed to clarify the role of SOD1, an endogenous antioxidant, in the Bad signaling pathway after tFCI.
Materials and Methods
SOD1 transgenic mice. Heterozygous SOD1 transgenic (Tg) mice of the SOD1 TGHS/SF-218–3 strain with a CD-1 background, carrying human SOD1 genes with a threefold increase in copper/zinc-SOD, were derived from the founder stock described previously (Chan, 1994). They were bred further with CD-1 wild-type mice to generate heterozygous mice. The SOD1 Tg mice were identified by quantitative demonstration of SOD1 with the use of nondenaturing gel electrophoresis, followed by nitroblue tetrazolium staining. There were no differences in the phenotypes, including the anatomy of the circle of Willis, between the SOD1 Tg mice and their wild-type littermates. There was no difference in the regional cerebral blood flow before and after FCI between the SOD1 Tg and wild-type mice.
Focal cerebral ischemia. Adult male mice (3 months old, 35–40 gm) were subjected to tFCI by intraluminal middle cerebral artery (MCA) blockade with a nylon suture as described previously. The mice were anesthetized with 1.5% isoflurane in 30% oxygen and 70% nitrous oxide, using a face mask. The rectal temperature was controlled at 37°C with a homeothermic blanket. Cannulation of a femoral artery allowed for the monitoring of blood pressure and arterial blood gases, samples for analysis being taken immediately after cannulation, 10 min after occlusion, and 10 min after reperfusion. Blood gas was analyzed with a pH/blood gas analyzer (Chiron Diagnostics, Essex, UK). After a midline skin incision the left external carotid artery was exposed, and its branches were electrocoagulated. A 11.0 mm 5-0 surgical monofilament nylon suture, blunted at the end, was introduced into the left internal carotid artery through the external carotid artery stump. After 60 min of MCA occlusion the blood flow was restored by the withdrawal of the nylon suture. To examine the role of PKA in the Bad signaling pathway, we intraventricularly injected a PKA inhibitor, H89, after tFCI. H89 [N-(2- [p-bromocinnamylamino]ethyl)-5-isoquinolinesulfonamide-2HCl] was purchased from Sigma (St. Louis, MO) and dissolved in MilliQ H2O (Millipore, Bedford, MA). The dose of the drug was decided by referring to a previous intraventricular injection study (Cervo et al., 1997). The scalp was incised on the midline, and the skull was exposed. H89 (4 μg/μl in MilliQ H2O) and the vehicle (MilliQ H2O alone) were injected intraventricularly (2 μl, bregma; 1.0 mm lateral, 0.2 mm posterior, 3.1 mm deep). H89 and the vehicle were injected 1 hr before MCA occlusion.
Immunohistochemistry. Anesthetized animals, as well as normal controls (n = 4 each), were perfused with 10 U/ml heparin and subsequently with 4% paraformaldehyde in 0.1m PBS, pH 7.4, at 1, 2, 4, 8, and 24 hr of reperfusion. The brains were removed, postfixed for 12 hr, sectioned at 50 μm on a vibratome, and processed for immunohistochemistry. The sections were incubated with blocking solution and reacted with rabbit polyclonal anti-Bad antibody, which detects total (phosphorylation state-independent) Bad levels (Cell Signaling Technology, Beverly, MA) at a dilution of 1:400, and rabbit polyclonal anti-phosphorylated Bad (Ser-155) antibody, which detects Bad only when phosphorylated at Ser-155 (Cell Signaling Technology) at a dilution of 1:400, plus rabbit polyclonal anti-Bcl-xL antibody (Cell Signaling Technology) at a dilution of 1:1000, rabbit polyclonal anti-14-3-3 antibody (Cell Signaling Technology) at a dilution of 1:400, and heat shock protein (HSP) 60 (Chemicon, Temecula, CA) at a dilution of 1:400. Immunohistochemistry was performed via the avidin–biotin technique, and then the nuclei were counterstained with methyl green solution for 2 min. For histological assessment alternate slices from each brain section were stained with cresyl violet.
Terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling staining. To clarify the spatial distribution of DNA fragmentation, we performed terminal deoxynucleotidyl transferase-mediated uridine 5′-triphosphate-biotin nick end labeling (TUNEL; n = 4 each). The sections fixed by 4% paraformaldehyde were prepared as described above and were incubated with NeuroPore (Trevigen, Gaithersburg, MD) for 30 min. They were placed in 1× terminal deoxynucleotidyl transferase (TdT) buffer (Invitrogen, Carlsbad, CA) for 30 min, followed by reaction with TdT enzyme (Invitrogen) and biotinylated 16-dUTP (Roche Diagnostics, Indianapolis, IN) at 37°C for 90 min. The sections were washed two times in saline-sodium citrate (150 mm sodium chloride, 15 mm sodium citrate, pH 7.4) for 15 min, followed by washing in PBS two times for 15 min. The avidin–biotin technique was applied, and then the nuclei were counterstained with methyl green solution for 2 min.
Immunofluorescent double-labeling staining. To evaluate colocalization of Bad and neuron-specific nuclear protein (NeuN) and Bad and HSP 60, we performed double immunofluorescent staining for these proteins (n = 4 each). The sections fixed by 4% paraformaldehyde were immunostained with anti-Bad antibody as described above, with biotinylated goat anti-rabbit immunoglobulin G (IgG; Vector Laboratories, Burlingame, CA), followed by fluorescein avidin DCS (Vector Laboratories). Then the sections were incubated with a blocking solution and reacted with anti-NeuN or HSP 60 antibody (Cell Signaling Technology) as described above, followed by Texas Red-conjugated donkey anti-mouse IgG antibody (Jackson ImmunoResearch, West Grove, PA) at a dilution of 1:400. The sections were placed on slides, which then were covered with Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories). Fluorescence of fluorescein was observed at excitation (Ex) of 495 nm and emission (Em) of >515 nm, and fluorescence of Texas Red was observed at Ex of 510 nm and Em of >580 nm. Fluorescence of DAPI also was observed at Ex of 360 nm and Em of >460 nm.
Immunofluorescent double labeling with phosphorylated Bad immunohistochemistry and TUNEL. To clarify the spatial relationship between phosphorylated Bad at Ser-155 (pBad) expression and DNA fragmentation, we performed double staining for pBad and TUNEL, using a fluorescent method (n = 4 each). The sections fixed by 4% paraformaldehyde were immunostained with the pBad antibody as described above with biotinylated goat anti-rabbit IgG (Vector Laboratories), followed by fluorescein avidin DCS (Vector Laboratories). Then the sections were incubated with NeuroPore (Trevigen) for 30 min and placed in 1× TdT buffer (Invitrogen) for 30 min, followed by reaction with TdT enzyme (Invitrogen) and biotinylated 16-dUTP (Roche Diagnostics) at 37°C for 90 min. The sections were washed two times in saline-sodium citrate (150 mm sodium chloride, 15 mm sodium citrate, pH 7.4) for 15 min, followed by washing in PBS two times for 15 min. Texas Red avidin DCS (Vector Laboratories) was applied to the sections for 30 min. Subsequently, the slides were covered with Vectashield mounting medium with DAPI (Vector Laboratories). Fluorescence was assessed as described previously.
Western blot analysis. Protein extraction of the cytosol fraction was performed as described previously with some modification (Fujimura et al., 1999). Samples were obtained from the MCA territory brain tissue on the ischemic sides and from nonischemic controls (n = 4 each). Fresh brain tissue was removed after 1, 2, 4, 8, and 24 hr (n = 4 each) of reperfusion and homogenized by gently douncing 35 times in a glass tissue grinder (Wheaton, Millville, NJ) in 7 vol of cold suspension buffer [containing (in mm) 20 HEPES-KOH, pH 7.5, 250 sucrose, 10 KCl, 1.5 MgCl2, 1 EDTA, and 1 EGTA plus 0.7% protease and phosphatase inhibitor mixtures (Sigma)]. The homogenate was centrifuged at 750 ×g for 10 min at 4°C and then at 8000 × gfor 20 min at 4°C. The 8000 × g pellets were used to obtain the mitochondrial fraction. The supernatant was centrifuged further at 100,000 × g for 60 min at 4°C. This supernatant was used for the cytosolic analysis. After adding the same volume of Tris-glycine sodium dodecyl sulfate sample buffer (Invitrogen) to the supernatant, we loaded equal amounts of the samples per lane. The primary antibodies were 1:600 dilution of rabbit polyclonal antibody against Bad (Cell Signaling Technology), 1:1000 dilution of mouse polyclonal antibody against Bcl-xL (Cell Signaling Technology), 1:1000 dilution of mouse polyclonal antibody against 14-3-3 (Cell Signaling Technology), 1:10,000 dilution of anti-β-actin monoclonal antibody (Sigma), and 1:2000 dilution of anti-cytochrome oxidase IV monoclonal antibody (Molecular Probes, Eugene, OR). Western blots were performed with horseradish peroxidase-conjugated anti-rabbit IgG (Cell Signaling Technology) or anti-mouse IgG (Chemicon) by using enhanced chemiluminescence Western blotting detection reagents (Amersham Biosciences, Buckinghamshire, UK). The film was scanned with a GS-700 imaging densitometer (Bio-Rad Laboratories, Hercules, CA) and the results were quantified with Multi-Analyst software (Bio-Rad).
Coimmunoprecipitation. The protein extraction of the cytosol and the mitochondrial fraction was performed as described previously with some modification (Fujimura et al., 1999). The procedure for precipitation was performed as described previously with some modification (Springer et al., 2000). Samples were obtained from the MCA territory brain tissue on the ischemic side and from nonischemic controls (n = 4 each). Fresh brain tissue was removed after 1, 2, 4, 8, and 24 hr (n = 4 each) of reperfusion and was prepared for collection of the cytosolic fraction and the mitochondrial fraction in the same manner as for Western blotting. Protein concentrations were determined by the Bradford method (Bio-Rad). Three hundred micrograms of protein from the cytosol fraction and 15 μg from the mitochondrial fraction were used for coimmunoprecipitation. Whole brain extract was included as a positive control. The protein sample was incubated with 50% slurry of protein G-Sepharose (Amersham Biosciences, Uppsala, Sweden) for 1 hr at 4°C, and this mixed sample was centrifuged at 12,000 × g for 1 min. The supernatant was incubated with 2 μg of polyclonal rabbit anti-Bcl-xLantibody (Cell Signaling Technology) or polyclonal mouse anti-14-3-3 antibody (Cell Signaling Technology) and 15 μl of protein G-Sepharose (50% slurry) for 1 hr at 4°C. The negative control was prepared with protein G-Sepharose without antibody. The 14,000 × g pellets were washed three times and used as the samples bound to each antibody. After adding the same volume of Tris-glycine sodium dodecyl sulfate sample buffer (Invitrogen) to the sample, we boiled these samples to remove Sepharose beads. After centrifugation at 14,000 ×g for 1 min, the supernatant was immunoblotted with a 1:600 dilution of anti-Bad antibody (Cell Signaling Technology) as described in the Western blotting method.
In situ detection of superoxide anion production. The early production of superoxide anions (O2–) in cerebral ischemia was investigated by using hydroethidine (HEt) via a previously described method. HEt is diffusible into the CNS parenchyma after an intravenous injection and is oxidized to ethidium selectively by O2–, but not by other ROS such as hydrogen peroxide, hydroxyl radical, or peroxynitrite. HEt solution (200 μl; 1 mg/ml in PBS) was administered intravenously 15 min before ischemia induction as described. In the brains of the animals intravenously injected with HEt, fluorescence was assessed microscopically at Ex = 355 nm and Em > 415 nm for HEt detection or at Ex = 510–550 nm and Em > 580 nm for ethidium detection. Animals were killed 1 hr after tFCI by transcardial perfusion as described (n = 4 each). After fixation with 4% paraformaldehyde for 12 hr the brains were sectioned at 50 μm on a vibratome. Subsequently, the slides were covered with Vectashield mounting medium with DAPI (Vector Laboratories). These sections were observed with a microscope under fluorescent light.
Cell death assay. For quantification of apoptotic-related DNA fragmentation we used a commercial enzyme immunoassay to determine cytoplasmic histone-associated DNA fragments (Roche Molecular Biochemicals, Mannheim, Germany), which detect apoptotic but not necrotic cell death (Leist et al., 1998). Samples were obtained from the entire MCA territory on the ischemic side and from nonischemic controls (n = 4 each). Fresh brain tissue was cut into pieces after 1, 2, 4, 8, and 24 hr of reperfusion and homogenized with a Teflon homogenizer in 5 vol of ice-cold buffer (50 mmKH2PO4, 0.1 mm EDTA, pH 7.8) and spun for 10 min at 750 × g. The supernatant was collected and spun for 20 min at 10,000 × g and was centrifuged further at 100,000 × g for 60 min at 4°C. The resulting supernatant was collected, and the protein concentration was determined. A cytosolic volume containing 20 μg of protein was used for the enzyme-linked immunosorbent assay by following the manufacturer's protocol.
Quantification and statistical analysis. The data are expressed as mean ± SD. Comparisons among multiple groups were performed with a one-way ANOVA with appropriate post hoctests (SigmaStat software, Jandel, San Rafael, CA). Comparisons between two groups were achieved with Student'st test. Significance was accepted with p < 0.05.
Results
Physiological data and cerebral infarction
Physiological data demonstrated no significant differences in body temperature, mean arterial blood pressure, and arterial blood gas analysis between the groups. The preischemic physiological values were as follows (in mmHg): 36.4 ± 0.3°C body temperature, 83 ± 3.1 mean arterial blood pressure, 7.3 ± 0.1 pH, 164.9 ± 19 PaO2, 30 ± 10 PaCO2(values are mean ± SD; n = 4). There was no deviation from these values over the period of assessment. An ischemic lesion of the core of the caudate–putamen was visible as a pale, slightly stained area in the ischemic hemisphere as early as 1 hr after reperfusion and extended to the entire MCA territory at 4 hr by cresyl violet staining (data not shown). The time-dependent increase in infarction in the mouse brains with the intraluminal suture blockade is consistent with previous reports that used the same FCI model in mice (Yang et al., 1994; Kondo et al., 1997).
14-3-3 protein and Bcl-xL expression were not different among the time courses and between SOD1 Tg mice and wild-type mice after tFCI
14-3-3 protein immunoreactivity was evident as bands with a molecular mass of 30 and 33 kDa in the cytosolic fraction from the MCA territory (Fig. 1A). 14-3-3 expression showed no difference among all time courses after tFCI (Fig. 1A). 14-3-3 protein-immunopositive cells were seen in the cortex and the caudate–putamen of both the nonischemic and ischemic hemispheres, and there was no conspicuous difference in the reactivity between the hemispheres (data not shown). Double immunofluorescent staining for 14-3-3 and NeuN demonstrated that 14-3-3 protein expression colocalized with neurons in the ischemic cortex 24 hr after reperfusion (Fig. 1B). Bcl-xL was expressed as a single band with a molecular mass of 30 kDa in the cytosolic fraction (Fig.1A). Bcl-xL expression did not change time dependently (Fig. 1A). Bcl-xL-immunopositive cells were seen in the cortex and the caudate–putamen of both the nonischemic and the ischemic sides, and there was no difference in the reactivity between the hemispheres (data not shown). Double immunofluorescent staining for Bcl-xL and NeuN demonstrated that Bcl-xL expression colocalized with neurons in the ischemic cortex 24 hr after reperfusion (Fig. 1B). Western blot analysis demonstrated that neither 14-3-3 protein nor Bcl-xL immunoreactivity was different between the SOD1 Tg mice and the wild-type mice 24 hr after reperfusion (Fig.1C). These results suggest that the expression of both 14-3-3 and Bcl-xL was neither increased nor reduced after tFCI and that it also was not affected by intrinsic SOD1.
Fig. 1.
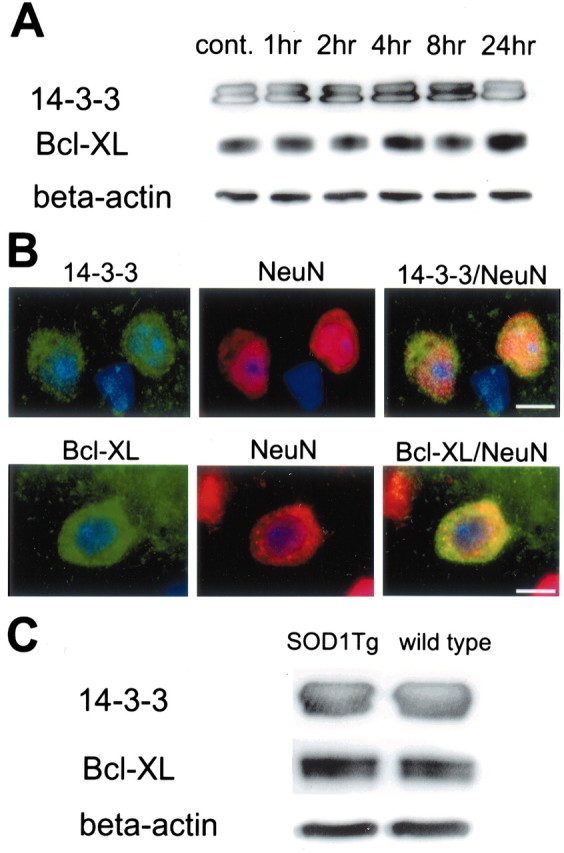
A, Western blot analysis of 14-3-3, Bcl-xL, and β-actin after tFCI. Shown are 14-3-3, Bcl-xL, and β-actin from the cytosolic fraction samples in the nonischemic control brains (cont. lane) and ischemic brains (lanes 1–24 hr;n = 4 each). 14-3-3 immunoreactivity is evident as bands with a molecular mass of 30 and 33 kDa in the cytosolic fraction from the MCA territory in mouse brains (top row). Bcl-xL immunoreactivity is evident as a band with a molecular mass of 30 kDa (middle row). Both 14-3-3 and Bcl-xL were expressed constitutively in the control brain (top, middle rows, cont. lane) and showed no prominent increase or decrease after ischemia (top, middle rows, lanes 1–24 hr). The results of the β-actin analysis are shown as an internal control (bottom row). B, Representative photomicrographs show double immunofluorescent staining for 14-3-3, Bcl-xL, and NeuN 4 hr after tFCI (n = 4 each). Expression of 14-3-3 was observed in the ischemic cortex (top left panel, green) and showed no prominent change in any of the time courses. NeuN immunoreactivity (red) showed the distribution of neurons in the same view (top middle panel). Overlapped image of 14-3-3 and NeuN immunoreactivity demonstrated that 14-3-3 expression colocalized with neurons in the same view (top right panel, yellow). Expression of Bcl-xL was observed in the ischemic cortex (bottom left panel, green) and showed no prominent change in any of the time courses. NeuN immunoreactivity (red) showed the distribution of neurons in the same view (bottom middle panel). Overlapped image of Bcl-xL and NeuN immunoreactivity demonstrated that Bcl-xL expression colocalized with neurons in the same view (bottom right panel, yellow). Scale bars, 4 μm.C, Western blot analysis of 14-3-3, Bcl-xL, and β-actin 24 hr after tFCI in SOD1 Tg mice and wild-type mice (n = 4 each). No difference was observed between the SOD1 Tg mice and the wild-type mice in either 14-3-3 or Bcl-xL expression (top, middle rows). β-Actin was used as an internal control, and no difference was observed between the samples (bottom row).
Phosphorylated Bad at Ser-155 was diminished after tFCI and did not colocalize with TUNEL-positive cells
pBad immunoreactivity was evident as a single band with a molecular mass of 23 kDa (Fig.2A). pBad expression decreased 1 hr after reperfusion and was diminished significantly 8 hr after reperfusion (Fig. 2A) (p< 0.05). TUNEL-positive cells were seen in the caudate–putamen and the cortex of the MCA territory surrounding the ischemic core, most obviously 24 hr after reperfusion (data not shown). These results are consistent with previous reports that used the same FCI model in mice (Fujimura et al., 1999). The pBad-positive cells were seen in the nonischemic hemisphere, and fewer positive cells were seen in the ischemic cortex (data not shown). In the area in which TUNEL-positive cells were observed, many fewer pBad-positive cells were seen, and most TUNEL-positive cells did not colocalize with pBad immunoreactivity (Fig. 2B). These results suggest that the dephosphorylation of Bad at Ser-155 progressed during the very early period after tFCI and that the decrease in pBad might be related to the beginning of apoptotic cell death after tFCI.
Fig. 2.

A, Western blot analysis of phosphorylated Bad at serine-155 residue (pBAD) after tFCI. The expression of pBad was detected as a band with a molecular mass of 23 kDa (n = 4 each). The immunoreactivity of pBad decreased gradually in a time-dependent manner (top row). The quantitative analysis showed that pBad expression was decreased significantly 8 hr after tFCI (bottom panel; *p < 0.05). β-Actin was used as an internal control, and no difference was observed between the samples (middle row). B, Representative photomicrographs show double immunofluorescent staining for pBad and TUNEL 24 hr after tFCI (n = 4 each). The pBad was expressed strongly in the nonischemic control area and expressed weakly in the ischemic cortex of the MCA territory and the partial caudate–putamen surrounding the ischemic core (left panel, red). TUNEL-positive cells were observed in the ischemic cortex of the MCA territory in the same view (middle panel, green). Overlapped image of pBad and TUNEL demonstrated that most pBad-immunopositive cells were not colocalized with TUNEL-positive cells (right panel). Scale bar, 10 μm.C, D, Western blot analyses of Bad in the cytosolic fraction (C) and in the mitochondrial fraction (D) after tFCI (n = 4 each). The expression of Bad was detected as a band with a molecular mass of 23 kDa (C, D, top row). The cytosolic Bad immunoreactivity decreased in a time-dependent manner (C). In particular, Bad expression 8 and 24 hr after tFCI decreased significantly compared with the nonischemic control sample (cont.) (C, top row, 8 hr, p > 0.05;24 hr, p > 0.05). Mitochondrial Bad immunoreactivity gradually increased after 1 hr of tFCI (D, top row). At 8 hr after tFCI the Bad expression decreased significantly (D, top row; *p < 0.05). β-Actin was used as an internal control for the cytosolic fraction, and cytochrome oxidase (COX) was used as an internal control for the mitochondrial fraction (C, D, middle rows). No difference was observed between the fraction samples (C, D, middle rows). E, Representative photomicrographs show double immunofluorescent staining for Bad and NeuN 4 hr after tFCI (n = 4 each). Expression of Bad was observed in the ischemic cortex (left panel, green) and showed no prominent change in any of the time courses. NeuN immunoreactivity (red) showed the distribution of neurons in the same view (middle panel, red). Overlapped image of Bad and NeuN immunoreactivity demonstrated that Bad expression colocalized with neurons in the same view (right panel, yellow). Scale bar, 15 μm.F, Representative photomicrographs show double immunofluorescent staining for Bad and heat shock protein 60 (HSP60) at 8 hr after tFCI (n = 4 each). HSP 60 expression was used as a marker for the distribution of mitochondria. Expression of Bad was observed in the ischemic cortex and showed particle-like punctate stains in the cell body (left panel, green). This particle-like immunoreactivity was observed in the HSP 60 immunoreactivity (middle panel, red). Overlapped image of Bad and HSP 60 immunoreactivity demonstrated that punctate Bad expression colocalized with the distribution in mitochondria in the same view (right panel, yellow). Scale bar, 10 μm. OD, Optical density.
Western blot analysis and immunofluorescent double staining demonstrated translocated Bad after tFCI
Bad expression was detected as a band with a molecular mass of 23 kDa (Fig. 2C,D). Bad expression decreased as early as 2 hr after reperfusion, and at 8 hr after reperfusion a significant decrease was seen (Fig. 2C). In contrast, mitochondrial Bad expression gradually increased after tFCI (Fig. 2D). One hour after reperfusion the band was barely visible, but after 4 hr of reperfusion it was obvious (Fig. 2D). A significant increase was observed 8 hr after reperfusion (Fig.2D). Double immunofluorescent staining for Bad and NeuN demonstrated that Bad immunoreactivity colocalized with neurons in the cortex of the ischemic hemisphere 24 hr after reperfusion (Fig.2E). The immunohistochemical expression of HSP 60 is reported to be an effective marker for the localization of mitochondria and is used to examine the colocalization or the translocation related to mitochondria (Springer et al., 2000). Double immunofluorescence for Bad and HSP 60 demonstrated that the punctate Bad immunoreactivity was obvious in the cortex surrounding the ischemic core after 8 hr of reperfusion and that the particle-like immunoreactivity colocalized with mitochondrial expression (Fig. 2F). These results suggest that Bad translocated from the cytosol to mitochondria during the early period after tFCI and that the timing of Bad translocation was consistent with that of the dephosphorylation of Bad. We could not find the relationship between Bad expression and glial fibrillary acidic protein in the mouse brains after transient focal ischemia (data not shown). However, we cannot exclude completely the possibility that such a relationship exists between the Bad signaling reaction and astrocytes after ischemic damage.
Coimmunoprecipitation demonstrated the direct binding of Bad to 14-3-3 and Bcl-xL
To investigate the direct interaction of Bad with 14-3-3 and Bcl-xL, we performed coimmunoprecipitation. In the cytosolic fraction a robust band of Bad precipitated by 14-3-3 protein (Bad/14-3-3) was detected in the sham control, and the bands gradually decreased by 24 hr after reperfusion (Fig.3A,B). In contrast, Bad expression precipitated by Bcl-xL(Bad/Bcl-xL) in the mitochondrial fraction increased time dependently, and a significant increase was observed 8 hr after reperfusion (Fig. 3A,B). These results suggest that with a nonischemic stimulus Bad sequestered with 14-3-3 in the cytosol, but it dissociated from 14-3-3 after tFCI and that the translocated Bad dimerized with Bcl-xL concomitant with the dissociation from 14-3-3.
Fig. 3.

A, B, Coimmunoprecipitation analysis for Bad immunoreactivity precipitated by 14-3-3 in the cytosolic fraction (BAD/14-3-3) (A,top row; B, left panel) and Bad immunoreactivity precipitated by Bcl-xL in the mitochondrial fraction (BAD/Bcl-xL) (A,bottom row; B, right panel) after tFCI (n = 4 each). Bad/14-3-3 expression gradually decreased in a time-dependent manner (A, top row; B,left panel). At 24 hr after tFCI the Bad/14-3-3 expression significantly decreased (B, left panel; *p < 0 0.05). Bad/Bcl-xL expression gradually increased (A, bottom row; B,right panel). At 8 hr after tFCI the Bad/Bcl-xL expression significantly increased (B, right panel; *p< 0.05). p.c., Positive control; n.c., negative control; cont., nonischemic control sample.
Cell death assay demonstrated that DNA fragmentation after tFCI significantly decreased in SOD1 Tg mice compared with wild-type mice and that it increased with intraventricular administration of H89
Apoptotic-related DNA fragmentation after tFCI was analyzed with a commercial cell death assay kit. As shown in Figure4A, DNA fragmentation was increased significantly in the ischemic MCA territory 24 hr after reperfusion. It was reduced significantly in the SOD1 Tg mice compared with the wild-type mice 24 hr after tFCI (Fig. 4B) (p < 0.05). However, it was increased significantly in the H89-treated mice compared with the vehicle-treated mice at the same time point (Fig. 4C) (p < 0.05). We performed the intraventricular injection of H89 in the mouse brains as reported previously and confirmed that the administration of H89 effectively reduced PKA activity (Cervo et al., 1997; Kimura et al., 1998). TUNEL-positive cells were seen in the caudate–putamen and in the cortex of the MCA territory surrounding the ischemic core and were observed most obviously 24 hr after reperfusion (data not shown). TUNEL-positive cells were reduced in the SOD1 Tg mice compared with the wild-type mice 24 hr after tFCI (Fig. 4D, top panels). However, TUNEL-positive cells were increased in the H89-treated mice compared with the vehicle-treated mice at the same time point (Fig.4D, bottom panels). To confirm whether free radical induction after reperfusion was promoted by H89, we examined O2– production by using the H89-treated mice and the vehicle-treated mice 1 hr after reperfusion. Production of O2– was determined by using HEt as described previously (Murakami et al., 1997). Oxidized HEt signals were observed most strongly in the ischemic cortex 1 hr after reperfusion and were reduced in the SOD1 Tg mice compared with the wild-type mice (Fig. 4E, top panels). These results are consistent with previous reports that used the same tFCI model in mice (Murakami et al., 1997; Fujimura et al., 2000;Noshita et al., 2002). There was no conspicuous difference in the oxidized HEt signals between the H89-treated mice and the vehicle-treated mice (Fig. 4E, bottom panels). These results suggest that overexpression of SOD1 attenuates apoptotic cell death after tFCI and that inhibition of PKA activity aggravated apoptotic cell death after tFCI. Moreover, data on the detection of O2–show that SOD1 prevented the early production of O2– after tFCI and that inhibition of PKA did not affect O2– production after tFCI, suggesting that ROS might be upstream of activation of the Bad signaling pathway, which may be mediated by PKA.
Fig. 4.
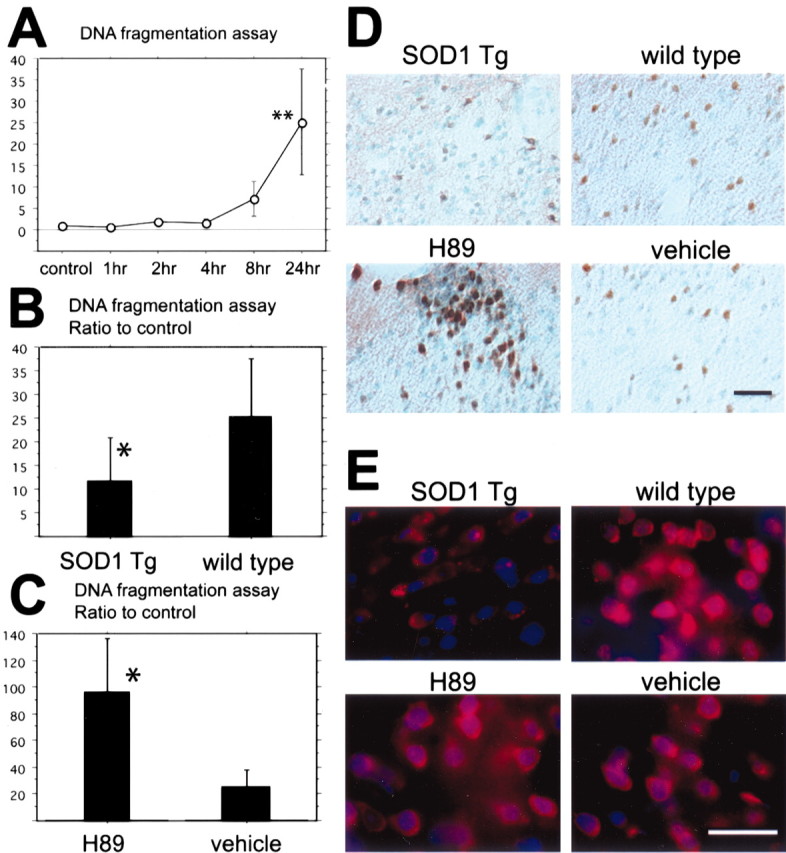
A, Apoptotic-related DNA fragmentation after tFCI was analyzed with a commercial cell death detection kit (n = 4 each). DNA fragmentation increased significantly in the entire MCA territory lesion 24 hr after tFCI (**p < 0.01). B, In the SOD1 Tg mice DNA fragmentation significantly decreased compared with the wild-type mice 24 hr after tFCI (*p < 0.05;n = 4 each). C, In the H89-treated mice, as a protein kinase A inhibitor sample, DNA fragmentation significantly increased compared with the vehicle-treated mice 24 hr after tFCI (*p < 0.05; n = 4 each). D, Representative photomicrographs show TUNEL staining in SOD1 Tg mice (top left panel) and in the H89-treated mice (bottom left panel) and, as a control, in the wild-type mice (top right panel) and the vehicle-treated mice (bottom right panel) 24 hr after tFCI (n = 4 each). TUNEL-positive cells were observed in the ischemic cortex of the MCA territory and the partial caudate–putamen surrounding the ischemic core 24 hr after tFCI. TUNEL reactivity was strongly observed in the wild-type mice (top right panel), whereas in the SOD1 Tg mice TUNEL reactivity decreased (top left panel). TUNEL reactivity in the H89-treated mice was much stronger compared with the vehicle-treated mice (bottom panels). Scale bar, 50 μm. E, Representative photomicrographs show superoxide production in the H89-treated mice (bottom left panel) and the vehicle-treated mice (bottom right panel) 1 hr after tFCI (n = 4 each). Superoxide production was observed by oxidized HEt signals (red), and nuclei were stained by DAPI (blue) 1 hr after reperfusion injury. The oxidized HEt signal was reduced in the SOD1 Tg mice compared with the wild-type mice (top panels). No conspicuous difference was observed between the H89-treated mice and the vehicle-treated mice (bottom panels). Scale bar, 25 μm.
H89 administration affected phospho-Bad and translocated Bad expression and dimerization with 14-3-3 and Bcl-xLafter tFCI
We examined the role of PKA in the Bad signaling pathway after tFCI by using the tFCI model with H89-treated mice. Western blot analysis demonstrated that pBad immunoreactivity was reduced significantly in the H89-treated mice compared with the vehicle-treated mice 24 hr after reperfusion (Fig.5A) (p< 0.05). In the wild-type mice pBad expression was barely visible in the ischemic core, and its weak expression was observed in the caudate–putamen and the cortex of the MCA territory surrounding the ischemic core after 8 hr of reperfusion (data not shown). Western blot analysis demonstrated that translocated Bad expression in the mitochondrial fraction was significantly greater in the H89-treated mice compared with the vehicle-treated mice 24 hr after reperfusion (Fig. 5B) (p < 0.05). Immunohistochemistry also showed that the expression of pBad in the penumbra regions was diminished in the H89-treated mice compared with the vehicle-treated mice (Fig. 5C). Coimmunoprecipitation demonstrated that Bad/14-3-3 immunoreactivity in the cytosolic fraction was reduced significantly in the H89-treated mice compared with the vehicle-treated mice 24 hr after reperfusion (Fig. 5D) (p < 0.05), whereas Bad/Bcl-xL immunoreactivity in the mitochondrial fraction significantly increased in the H89-treated mice compared with the vehicle-treated mice 24 hr after reperfusion (Fig. 5E) (p < 0.05). These results suggest that the inhibition of PKA caused progression of dephosphorylation and translocation of Bad and that it also caused progression of the dissociation from 14-3-3 and the dimerization with Bcl-xL after tFCI.
Fig. 5.
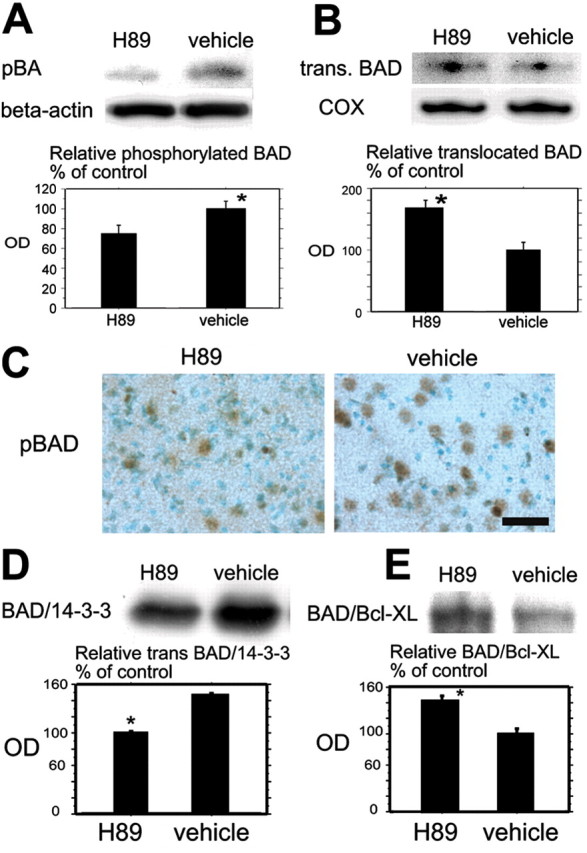
A, Western blot analysis for pBad in the cytosolic fraction in the H89-treated mice and the vehicle-treated mice 24 hr after tFCI. pBad expression was significantly stronger in the vehicle-treated mice compared with the H89-treated mice at the same time course (top row, bottom panel; *p < 0.05). β-Actin was used as an internal control for the cytosolic fraction, and no difference was seen between the samples (middle row). B, Western blot analysis for translocated Bad (trans.BAD) in the mitochondrial fraction in the H89-treated mice and the vehicle-treated mice 24 hr after tFCI. Translocated Bad expression was significantly stronger in the H89-treated mice compared with the vehicle-treated mice at the same time course (top row, bottom panel; *p < 0.05). Cytochrome oxidase (COX) expression was used as an internal control for the mitochondrial fraction, and no difference was seen between the samples (middle row). C, Representative photomicrographs show the immunohistochemistry for pBad in the H89-treated mice (left) and the vehicle-treated mice (right) in the cortex of the MCA territory 24 hr after tFCI. The immunoreactivity of pBad was stronger in the vehicle-treated mice compared with the H89-treated mice at the same time course. Scale bar, 20 μm. D, Coimmunoprecipitation analysis of Bad/14-3-3 in the cytosolic fraction in the H89-treated mice and the vehicle-treated mice 24 hr after tFCI. The expression of Bad/14-3-3 was significantly stronger in the vehicle-treated mice compared with the H89-treated mice (*p < 0.05). E, Coimmunoprecipitation analysis of Bad/Bcl-xL in the mitochondrial fraction in the H89-treated mice and the vehicle-treated mice 24 hr after tFCI. Expression of Bad/Bcl-xL was significantly stronger in the H89-treated mice compared with the vehicle-treated mice (*p < 0.05).OD, Optical density.
Overexpression of SOD1 increased phospho-Bad, diminished the translocated Bad, and affected the dimerization with 14-3-3 and Bcl-xL after tFCI
We examined the relationship between the Bad signaling pathway after tFCI and oxidative stress by using the tFCI model with SOD1 Tg mice. Western blot analysis demonstrated that pBad immunoreactivity was detected as a significantly stronger band in the SOD1 Tg mice compared with the wild-type mice 24 hr after reperfusion (Fig.6A) (p < 0.05). Western blot analysis that used the mitochondrial fraction demonstrated that translocated Bad immunoreactivity was diminished significantly in the SOD1 Tg mice compared with the wild-type mice 24 hr after reperfusion (Fig.6B) (p < 0.05). In the penumbra areas of the MCA territory surrounding the ischemic core, immunoreactivity of pBad was also greater in the SOD1 Tg mice compared with the wild-type mice 24 hr after reperfusion (Fig. 6C). Coimmunoprecipitation demonstrated that the expression of Bad/14-3-3 in the cytosolic fraction significantly increased in the SOD1 Tg mice compared with the wild-type mice 24 hr after reperfusion (Fig.6D) (p < 0.05) and that Bad/Bcl-xL immunoreactivity was diminished significantly in the SOD1 Tg mice compared with the wild-type mice (Fig. 6E) (p < 0.05). These results suggest that overexpression of SOD1 prevented dephosphorylation and translocation of Bad and that it also prevented the dissociation from 14-3-3 and the dimerization with Bcl-xLafter tFCI.
Fig. 6.
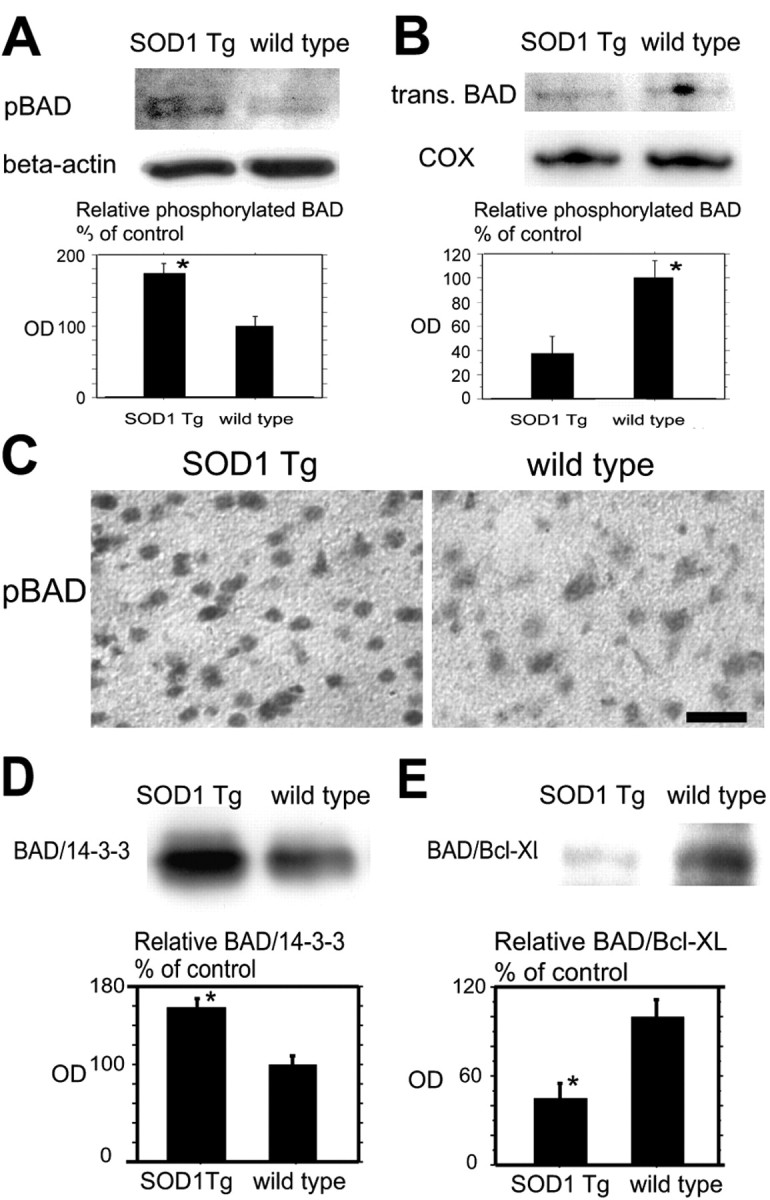
A, Western blot analysis for pBad in the cytosolic fraction in the SOD1 Tg mice and the wild-type mice 24 hr after tFCI. pBad expression was significantly stronger in the SOD1 Tg mice compared with the wild-type mice at the same time course (top row, bottom panel; *p < 0.05). β-Actin was used as an internal control for the cytosolic fraction, and no difference was seen between the samples (middle row). B, Western blot analysis for translocated Bad in the mitochondrial fraction in SOD1 Tg mice and wild-type mice 24 hr after tFCI. Translocated Bad (trans. BAD) expression was significantly stronger in the wild-type mice compared with the SOD1 Tg mice at the same time course (top row, bottom panel; *p < 0.05). Cytochrome oxidase (COX) expression was used as an internal control for the mitochondrial fraction, and no difference was seen between the samples (middle row). C, Representative photomicrographs show immunohistochemistry for pBad in the SOD1 Tg mice (left) and the wild-type mice (right) in the cortex of the MCA territory 24 hr after tFCI. Immunoreactivity of pBad was observed more strongly in the SOD1 Tg mice compared with the wild-type mice at the same time course. Scale bar, 20 μm.D, Coimmunoprecipitation analysis for Bad/14-3-3 in the cytosolic fraction in the SOD1 Tg mice and the wild-type mice 24 hr after tFCI. Expression of Bad/14-3-3 was significantly stronger in the SOD1 Tg mice compared with the wild-type mice (*p< 0.05). E, Coimmunoprecipitation analysis for Bad/Bcl-xL in the mitochondrial fraction in the SOD1 Tg mice and the wild-type mice 24 hr after tFCI. Expression of Bad/Bcl-xL was significantly stronger in the wild-type mice compared with the SOD1 Tg mice (*p < 0.05).OD, Optical density.
Discussion
There is clear evidence that neuronal apoptotic cell death after cerebral ischemia involves factors that determine the balance of cell death and survival (Noshita et al., 2002; Zhu et al., 2002). In recent studies cell survival factors have been the focus as critical regulators of the progress of apoptosis; these include serine-threonine kinase, the Akt cell signaling pathway, mitogen-activated protein kinase (MAPK) and extracellular signal-regulated kinase (ERK) kinase (MEK) and the ERK pathway (Hetman et al., 1999; Rommel et al., 1999;Zimmermann and Moelling, 1999). Phosphorylation of serine or threonine residues interacts with these signaling factors and plays a pivotal role in the regulation of the balance between cell death and cell survival (Burgering and Coffer, 1995; Flanke et al., 1999). As an executor downstream of these signaling factors, Bad is reported to have an important role and act dynamically via the phosphorylation of its four serine residues in response to different stimuli (Burgering and Coffer, 1995; Datta et al., 2000; Lizcano et al., 2000; Dramsi et al., 2002). As for Ser-112, one of the serine residues of Bad, it has been demonstrated that transforming growth factor-β1 suppresses Bad expression and increases phosphorylation via the activation of the MAPK/ERK pathway in both in vivo and in vitrocerebral ischemia models (Zhu et al., 2002). An in vitrostudy showed that phosphorylation of Bad at Ser-155 was affected by some kinases, such as RSK and PKA, and that the ERK pathway is upstream of RSK (Bonni et al., 1999; Pastorino et al., 1999; Tan et al., 1999;Shimamura et al., 2000). In particular, Ser-155 residue is known as the most critical site for the translocation of Bad and dimerization with Bcl-xL, which is the turning point for the regulation of apoptosis (Lizcano et al., 2000; Tan et al., 2000). However, little is known about the relationship between Bad phosphorylation at Ser-155 and the Bad signaling pathway in vivo. In particular, nothing is known about the link between the Bad signaling pathway and ischemic stimuli in vivo. In the present study we demonstrated for the first time the following points. (1) The translocation of Bad from the cytosol to the mitochondria was detected during the early period after tFCI. The expression of phosphorylated Bad at Ser-155 residue decreased in the cytosol, which was related concomitantly to the translocation of Bad. This suggests that, in the absence of ischemic stimuli, Bad localized in the cytosol and that after reperfusion injury phosphorylated Bad at Ser-155 residue was diminished to translocate from the cytosol to the mitochondria. (2) The direct binding of Bad to 14-3-3 in the cytosol and to Bcl-xL in the mitochondria was detected after tFCI. Whereas Bad/14-3-3 binding gradually increased, in contrast Bad/Bcl-xL binding decreased in a time-dependent manner after tFCI. These results demonstrate that after reperfusion injury Bad dissociated from 14-3-3 and the translocated Bad dimerized with Bcl-xL in the mitochondria. (3) A PKA-specific inhibitor, H89, decreased the phosphorylated Bad at Ser-155 and increased the translocated Bad. Moreover, it also decreased Bad/14-3-3 binding and increased Bad/Bcl-xLbinding after tFCI. Quantitative analysis showed that the PKA inhibitor apparently aggravated apoptotic neuronal cell death after tFCI. These results reveal that the inhibition of PKA accelerated the apoptotic cell death signaling pathway after tFCI by blocking phosphorylation of Bad at Ser-155. PKA seems to phosphorylate Bad, at least at Ser-155, and to affect cell survival in reperfusion injury after tFCI. There is a possibility that the PKA inhibitor is related to the dephosphorylation of Bad not only at Ser-155 but also at Ser-112, as reported in an in vitro study. However, in our study we did not elucidate the exact role of Ser-112 residue on Bad after tFCI. Further study is required to clarify the difference in function among other serine residues on Bad. PKA inhibitor is thought to affect phosphorylation-related mechanisms globally. We cannot exclude completely the possibility that PKA inhibition might affect the cell death pathway in a manner unrelated to the Bad reaction after ischemic insult. (4) Overexpression of intrinsic SOD1 diminished dephosphorylation of Bad at Ser-155 and the translocation of Bad. Moreover, it increased Bad/14-3-3 binding and decreased Bad/Bcl-xL to protect the neurons against apoptotic cell death after tFCI. In an in situ superoxide detection analysis H89 did not affect the production of superoxide after reperfusion injury, suggesting that the increase in production of superoxide after reperfusion injury is upstream of the Bad phosphorylation pathway mediated by PKA.
In our mouse tFCI model such dynamic activities as dephosphorylation, dissociation, translocation, and dimerization of Bad after reperfusion injury were observed, as they have been in many in vitrostudies. However, other important links between Bad and other kinase signaling pathways, for example MEK/ERK and phosphatidylinositol 3-kinase/Akt upstream of the Bad signaling pathway or Bax and other Bcl-2 family member pathways downstream of that, remain unknown, especially in in vivo ischemia models (Noshita et al., 2002;Zhu et al., 2002). As for Bax, it translocates from the cytosol to the mitochondria after reperfusion injury in mouse tFCI and induces mitochondrial membrane permeabilization by interacting with proteins of the mitochondrial permeability transition pore complex, such as voltage-dependent anion channel and adenine nucleotide translocator (Cao et al., 2001). The translocation of Bax is suggested to result in the release of apoptosis-promoting factors, including cytochromec (Jürgensmeier et al., 1998; Narita et al., 1998). However, the interaction between Bax translocation and Bad remains unclear. It was reported that Bcl-xL resides constitutively bound to Bax in the cytosol and that Bad displaces and releases Bax from Bcl-xL (Yang et al., 1995). Thereafter, Bax translocates from the cytosol to mitochondria (Li et al., 1997; Jürgensmeier et al., 1998). Our data show that Bad/Bcl-xL binding was detected in the mitochondrial fraction, and expression of the binding increased in a time-dependent manner after reperfusion injury, suggesting that mitochondria are the main site at which Bad interacts with Bcl-xL. Similarly, Bad/Bcl-xL binding was detected in mitochondria after traumatic spinal cord injury in rats, and the binding contrasted with Bad/14-3-3 binding in the cytosol fraction (Springer et al., 2000). Because Bcl-xL, which could bind to Bax in mitochondria, decreases as Bad/Bcl-xL binding increases after reperfusion injury, we suggest that Bcl-xL unbound to Bad is also important for activation downstream of the Bad signaling pathway.
Our previous studies demonstrated that overexpression of intrinsic SOD1 has protective effects on ischemic damage (Kinouchi et al., 1991; Chan, 1996; Kondo et al., 1997; Fujimura et al., 1999, 2000). In this study we focused on the role of the Bad signaling pathway on the neuroprotective effect of SOD1. Our results demonstrate that overexpression of SOD1 prevented the Bad cell death pathway after reperfusion injury, and it shifted the balance between survival and death to cell survival. The ERK pathway is reported to be one of the kinase pathways for Bad phosphorylation in in vitro studies (Pastorino et al., 1999; Lizcano et al., 2000). It is reported that ROS can induce MEK/ERK cell death pathway activation in vitro(Baas and Berk, 1995; Guyton et al., 1996; Seo et al., 2001). We also have demonstrated that oxidative stress plays a crucial role in ERK cell death pathway activation after tFCI (Noshita et al., 2002). It is not clear whether the ERK pathway regulates phosphorylation of Badin vivo, including reperfusion injury after tFCI. Moreover, transforming growth factor (TGF) also has been suggested to contribute to activation of the MAPK/ERK pathway and PKA (Wang et al., 1998; Zhu et al., 2002). There is also a possibility that ROS affect TGF activation or production via reduction in oxidation-sensitive transcription factors, such as NF-κB and hypoxia-inducible factor-1α after apoptotic stimuli (Haddad, 2002; Ishida et al., 2002). It remains unclear how oxidative stress is involved with the Bad signaling pathway after tFCI, but it is obvious that oxidative stress plays an important role in the activation of the Bad cell death pathway. Further study is required to clarify the link between the Bad signaling pathway and oxidative stress after tFCI.
Conclusion
Our results imply that the Bad cell death signaling pathway is activated in neurons after tFCI and that SOD1 contributes to the inhibition of apoptosis induced by FCI by reducing the early formation of superoxide radicals and preventing the dephosphorylation of Bad. We suggest that SOD1 may play a critical role in the Bad signaling pathway.
Footnotes
This work was supported by National Institutes of Health Grants P50NS14543, R0125372, R0136147, and R0138653 and an American Heart Association Bugher Foundation award. P.H.C. is a recipient of the Jacob Javits Neuroscience Investigator Award. We are grateful to Dr. Charles J. Epstein (Department of Pediatrics, University of California, San Francisco, School of Medicine) for the breeding pairs of SOD1 transgenic mice. We thank Cheryl Christensen for editorial assistance, Liza Reola and Bernard Calagui for technical assistance, and Elizabeth Hoyte for figure preparation.
Correspondence should be addressed to Dr. Pak H. Chan, Neurosurgical Laboratories, Stanford University School of Medicine, 1201 Welch Road, MSLS #p314, Stanford, CA 94305-5487. E-mail:phchan@leland.stanford.edu.
References
- 1.Adachi S, Cross AR, Babior BM, Gottlieb RA. Bcl-2 and the outer mitochondrial membrane in the inactivation of cytochrome c during Fas-mediated apoptosis. J Biol Chem. 1997;272:21878–21882. doi: 10.1074/jbc.272.35.21878. [DOI] [PubMed] [Google Scholar]
- 2.Baas AS, Berk BC. Differential activation of mitogen-activated protein kinases by H2O2 and O2– in vascular smooth muscle cells. Circ Res. 1995;77:29–36. doi: 10.1161/01.res.77.1.29. [DOI] [PubMed] [Google Scholar]
- 3.Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- 4.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol 3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- 6.Cao G, Minami M, Pei W, Yan C, Chen D, O'Horo C, Graham SH, Chen J. Intracellular Bax translocation after transient cerebral ischemia: implications for a role of the mitochondrial apoptotic signaling pathway in ischemic neuronal death. J Cereb Blood Flow Metab. 2001;21:321–333. doi: 10.1097/00004647-200104000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Cervo L, Mukherjee S, Bertaglia A, Samanin R. Protein kinases A and C are involved in the mechanisms underlying consolidation of cocaine place conditioning. Brain Res. 1997;775:30–36. doi: 10.1016/s0006-8993(97)00866-4. [DOI] [PubMed] [Google Scholar]
- 8.Chan PH. Oxygen radicals in focal cerebral ischemia. Brain Pathol. 1994;4:59–65. doi: 10.1111/j.1750-3639.1994.tb00811.x. [DOI] [PubMed] [Google Scholar]
- 9.Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- 10.Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME. 14-3-3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell. 2000;6:41–51. [PubMed] [Google Scholar]
- 11.Dramsi S, Scheid MP, Maiti A, Hojabrpour P, Chen X, Schubert K, Goodlett DR, Aebersold R, Duronio V. Identification of a novel phosphorylation site, Ser-170, as a regulator of Bad pro-apoptotic activity. J Biol Chem. 2002;277:6399–6405. doi: 10.1074/jbc.M109990200. [DOI] [PubMed] [Google Scholar]
- 12.Flanke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1999;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 13.Fujimura M, Morita-Fujimura Y, Kawase M, Copin J-C, Calagui B, Epstein CJ, Chan PH. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome c and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci. 1999;19:3414–3422. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujimura M, Morita-Fujimura Y, Noshita N, Sugawara T, Kawase M, Chan PH. The cytosolic antioxidant copper/zinc-superoxide dismutase prevents the early release of mitochondrial cytochrome c in ischemic brain after transient focal cerebral ischemia in mice. J Neurosci. 2000;20:2817–2824. doi: 10.1523/JNEUROSCI.20-08-02817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem. 1996;271:4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- 16.Haddad JJ. Antioxidant and pro-oxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cell Signal. 2002;14:879–897. doi: 10.1016/s0898-6568(02)00053-0. [DOI] [PubMed] [Google Scholar]
- 17.Hetman M, Kanning K, Cavanaugh JE, Xia Z. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:22569–22580. doi: 10.1074/jbc.274.32.22569. [DOI] [PubMed] [Google Scholar]
- 18.Hsu SY, Kaipia A, Zhu L, Hsueh AJW. Interference of BAD (Bcl-xL/Bcl-2-associated death promoter)-induced apoptosis in mammalian cells by 14-3-3 isoforms and P11. Mol Endocrinol. 1997;11:1858–1867. doi: 10.1210/mend.11.12.0023. [DOI] [PubMed] [Google Scholar]
- 19.Ishida A, Fujita N, Kitazawa R, Tsuruo T. Transforming growth factor-β induces expression of receptor activator of NF-κB ligand in vascular endothelial cells derived from bone. J Biol Chem. 2002;277:26217–26224. doi: 10.1074/jbc.M111093200. [DOI] [PubMed] [Google Scholar]
- 20.Jürgensmeier JM, Xie Z, Deveraux Q, Ellerby L, Bredesen D, Reed JC. Bax directly induces release of cytochrome c from isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:4997–5002. doi: 10.1073/pnas.95.9.4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelekar A, Thompson CB. Bcl-2 family proteins: the role of the BH3 domain in apoptosis. Trends Cell Biol. 1998;8:324–330. doi: 10.1016/s0962-8924(98)01321-x. [DOI] [PubMed] [Google Scholar]
- 22.Kimura S, Uchiyama S, Takahashi HE, Shibuki K. cAMP-dependent long-term potentiation of nitric oxide release from cerebellar parallel fibers in rats. J Neurosci. 1998;18:8551–8558. doi: 10.1523/JNEUROSCI.18-21-08551.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kinouchi H, Epstein CJ, Mizui T, Carlson E, Chen SF, Chan PH. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing Cu/Zn superoxide dismutase. Proc Natl Acad Sci USA. 1991;88:11158–11162. doi: 10.1073/pnas.88.24.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kondo T, Reaume AG, Huang T-T, Carlson E, Murakami K, Chen SF, Hoffman EK, Scott RW, Epstein CJ, Chan PH. Reduction of Cu/Zn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J Neurosci. 1997;17:4180–4189. doi: 10.1523/JNEUROSCI.17-11-04180.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat Med. 1997;3:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- 26.Leist M, Kuhnle S, Single B, Nicotera P. Differentiation between apoptotic and necrotic cell death by means of the BM cell death detection ELISA or annexin V staining. Biochemica. 1998;2:25–28. [Google Scholar]
- 27.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 28.Lizcano JM, Morrice N, Cohen P. Regulation of BAD by cAMP-dependent protein kinase is mediated via phosphorylation of a novel site, Ser-155. Biochem J. 2000;349:547–557. doi: 10.1042/0264-6021:3490547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murakami K, Kondo T, Epstein CJ, Chan PH. Overexpression of Cu/Zn-superoxide dismutase reduces hippocampal injury after global ischemia in transgenic mice. Stroke. 1997;28:1797–1804. doi: 10.1161/01.str.28.9.1797. [DOI] [PubMed] [Google Scholar]
- 30.Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H, Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc Natl Acad Sci USA. 1998;95:14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noshita N, Sugawara T, Hayashi T, Lewén A, Omar G, Chan PH. Copper/zinc superoxide dismutase attenuates neuronal cell death by preventing extracellular signal-regulated kinase activation after transient focal cerebral ischemia in mice. J Neurosci. 2002;22:7923–7930. doi: 10.1523/JNEUROSCI.22-18-07923.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pastorino JG, Tafani M, Farber JL. Tumor necrosis factor induces phosphorylation and translocation of Bad through a phosphatidylinositide-3-OH kinase-dependent pathway. J Biol Chem. 1999;274:19411–19416. doi: 10.1074/jbc.274.27.19411. [DOI] [PubMed] [Google Scholar]
- 33.Reed JC. Double identity for proteins of the Bcl-2 family. Nature. 1997;387:773–776. doi: 10.1038/42867. [DOI] [PubMed] [Google Scholar]
- 34.Rommel C, Clarke BA, Zimmermann S, Nuñez L, Rossman R, Reid K, Moelling K, Yancopoulos GD, Glass DJ. Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science. 1999;286:1738–1741. doi: 10.1126/science.286.5445.1738. [DOI] [PubMed] [Google Scholar]
- 35.Seo SR, Chong SA, Lee S-I, Sung JY, Ahn YS, Chung KC, Seo JT. Zn2+-induced ERK activation mediated by reactive oxygen species causes cell death in differentiated PC12 cells. J Neurochem. 2001;78:600–610. doi: 10.1046/j.1471-4159.2001.00438.x. [DOI] [PubMed] [Google Scholar]
- 36.Shimamura A, Ballif BA, Richards SA, Blenis J. Rsk1 mediates a MEK–MAP kinase cell survival signal. Curr Biol. 2000;10:127–135. doi: 10.1016/s0960-9822(00)00310-9. [DOI] [PubMed] [Google Scholar]
- 37.Springer JE, Azbill RD, Nottingham SA, Kennedy SE. Calcineurin-mediated BAD dephosphorylation activates the caspase-3 apoptotic cascade in traumatic spinal cord injury. J Neurosci. 2000;20:7246–7251. doi: 10.1523/JNEUROSCI.20-19-07246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sugawara T, Noshita N, Lewén A, Gasche Y, Ferrand-Drake M, Fujimura M, Morita-Fujimura Y, Chan PH. Overexpression of copper/zinc superoxide dismutase in transgenic rats protects vulnerable neurons against ischemic damage by blocking the mitochondrial pathway of caspase activation. J Neurosci. 2002;22:209–217. doi: 10.1523/JNEUROSCI.22-01-00209.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan Y, Ruan H, Demeter MR, Comb MJ. p90RSK blocks Bad-mediated cell death via a protein kinase C-dependent pathway. J Biol Chem. 1999;274:34859–34867. doi: 10.1074/jbc.274.49.34859. [DOI] [PubMed] [Google Scholar]
- 40.Tan Y, Demeter MR, Ruan H, Comb MJ. BAD Ser-155 phosphorylation regulates BAD/Bcl-xL interaction and cell survival. J Biol Chem. 2000;275:25865–25869. doi: 10.1074/jbc.M004199200. [DOI] [PubMed] [Google Scholar]
- 41.Wang L, Zhu Y, Sharma K. Transforming growth factor-β1 stimulates protein kinase A in mesangial cells. J Biol Chem. 1998;273:8522–8527. doi: 10.1074/jbc.273.14.8522. [DOI] [PubMed] [Google Scholar]
- 42.Yang E, Korsmeyer SJ. Molecular thanatopsis: a discourse on the BCL2 family and cell death. Blood. 1996;88:386–401. [PubMed] [Google Scholar]
- 43.Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- 44.Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H. Human copper/zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165–170. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]
- 45.Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3, not Bcl-xL. Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 46.Zhang L, Chen J, Fu H. Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14-3-3 proteins. Proc Natl Acad Sci USA. 1999;96:8511–8515. doi: 10.1073/pnas.96.15.8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu Y, Yang G-Y, Ahlemeyer B, Pang L, Che X-M, Culmsee C, Klumpp S, Krieglstein J. Transforming growth factor-β1 increases Bad phosphorylation and protects neurons against damage. J Neurosci. 2002;22:3898–3909. doi: 10.1523/JNEUROSCI.22-10-03898.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science. 1999;286:1741–1744. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]