

Abstract
Humans with heterozygous inactivating mutations of theLis1 gene display type I lissencephaly, a severe form of cortical dysplasia hypothesized to result from abnormal neuronal migration. Previously we reported the construction of an allelic series of the Lis1 gene in mice to analyze the effects of graded reduction of LIS1 protein on the pathogenesis of this disorder and demonstrated a cell autonomous defect in neuronal migration (Hirotsune et al., 1998). Here we report the systematic examination of the consequences of dosage reduction of LIS1 on neocortical development using wild-type, null heterozygous (45% LIS1 protein), and compound null/hypomorphic (35% LIS1 protein) mice. The development of the preplate, Cajal-Retzius cells, and the radial glial scaffold appeared unaffected by LIS1 levels. However, a dose-dependent morphologic change in disorganization of the subplate was noted. LIS1 dose-dependent defects in neuronal migration were found in vivo andin vitro. The position and number of mitotic cells in the ventricular zone were more abnormal as LIS1 levels decreased, suggesting defects in interkinetic nuclear migration and neuroblast proliferation. LIS1 dose-dependent progressive thinning of the cortex and ventricular zone occurred by programmed cell death. Thus, in addition to its requirement for cell autonomous neuronal migration, LIS1 influences the generation and survival of cortical ventricular zone neuroblasts. These studies reveal the importance of LIS1 levels in orderly cerebral cortical morphogenesis and suggest new insights into the pathogenesis of type I lissencephaly.
Keywords: neuronal migration, LIS1, development, proliferation, interkinetic nuclear migration, cell death
Introduction
Haploinsufficiency of theLIS1 gene results in type I lissencephaly (Reiner et al., 1993; Lo Nigro et al., 1997). This developmental disorder results from defects in neuronal migration and is characterized by a smooth (lissencephalic) and disorganized cerebral cortex, mental retardation, and epilepsy (Barkovich et al., 1991; Dobyns and Truwit, 1995).LIS1 encodes a multifunctional 45 kDa protein with homologs in filamentous fungi (Xiang et al., 1995), yeast (Geiser et al., 1997), flies (Liu et al., 1999), and mice (Peterfy et al., 1995, 1998;Hirotsune et al., 1997). LIS1 is the noncatalytic subunit of the G-protein-like trimeric brain-specific enzyme, platelet-activating factor acetyl hydrolase (PAFAH) 1B; the formal gene name isPAFAH1B1 (Hattori et al., 1994; Ho et al., 1997). PAFAHs inactivate the signaling molecule, platelet-activating factor (PAF) (for review, see Stafforini et al., 1997). It is unknown whether the enzymatic function of LIS1 is important for neuronal migration, although several neuron-specific effects of PAF have been demonstrated (Hattori et al., 1996; Adachi et al., 1997; Bix and Clark, 1998). LIS1 is also a component of the cytoplasmic dynein/microtubule motor system (Faulkner et al., 2000; Smith et al., 2000). Studies inAspergillus and Drosophila demonstrated a role for the LIS1 and dynein homologs in nuclear translocation in fungal hyphae, developing egg chambers, neuroblast proliferation, dendritic elaboration, and axonal transport (Xiang et al., 1995; Liu et al., 1999, 2000); however, the mechanisms by which LIS1 protein reduction results in human lissencephaly are unknown.
To study the roles of reduced Lis1 expression on the pathogenesis of classical lissencephaly, two Lis1 alleles in the mouse were generated (Hirotsune et al., 1998). These alleles were used to produce a series of mice with reduced levels of LIS1 in the brain. Juvenile or adult animals demonstrated progressively more severe neuronal migration defects in the neocortex, hippocampus, olfactory bulb, and cerebellum, as well as cell autonomous in vitromigration defects (Hirotsune et al., 1998). The effects of LIS1 levels on other aspects of cortical development are unknown. The present investigation was undertaken to analyze the roles of LIS1 levels on several aspects of cerebral cortical morphogenesis.
Cortical neurogenesis begins in the pseudostratified epithelial layer of the telencephalic vesicles (for review, see Reid and Walsh, 1996). The preplate is the first organized laminar structure containing Cajal-Retzius cells, which produce Reelin, an extracellular signaling molecule. The preplate is split into a marginal zone and subplate by invading post-mitotic cortical plate neurons that will form the mature cortex in an inside-out manner (Angevine and Sidman, 1961). Radial glia provide the scaffold for the radially migrating neurons. We analyzed several of these aspects of cortical development and performed a more detailed analysis of postnatal lamination in heterozygous and compound heterozygous mice. These results show that in addition to cortical lamination, the subplate, interkinetic nuclear migration, and cell death are dependent on cellular levels of the LIS1 protein. Hence human lissencephaly may result from defects in multiple aspects of cerebral cortical development.
Materials and Methods
Brain lysates and immunoblotting. Brain lysates were prepared from three litters of P0 pups from three different mating pairs. Brains were homogenized in 2 ml of modified RIPA buffer (1% sodium phosphate, 150 mm NaCl, 1 mm MgCl2, 1 mm EGTA, 1.25 gm deoxycholate/500 ml, 10 μg/ml leupeptin, 1 μg/ml aprotinin, 1 mmNaVO4, 1 mm PMSF, and 20 mm β glycerol phosphate), incubated for 30 min on ice, and centrifuged at 14,000 rpm for 10 min at 4°C. Before electrophoresis, lysates were diluted in 2× sample buffer (2% SDS, 100 mm dithiothreitol, 60 mm Tris, pH 6.8, 0.01% bromophenol blue) and heated at 95°C for 5 min. For LIS1 protein quantitation, brain lysates from 10 wild-type, 10 Lis1+/KO and 9 Lis1cko/ko were analyzed. Proteins were separated by SDS-PAGE, transferred to Immobilon polyvinylidene difluoride membranes (Millipore, Bedford, MA), and immunoblotted with anti-LIS1 (1:250; Santa Cruz Biotechnology, Santa Cruz, CA) and anti-γ-tubulin (1:500; Sigma, St. Louis, MO). Secondary antibodies were horseradish peroxidase-conjugated anti-goat and anti-rabbit IgG (1:1000; Zymed, San Francisco, CA). The membranes were developed with SuperSignal Chemiluminescent Substrate (Pierce, Rockford, IL) and exposed to Kodak (Rochester, NY) BIOMAX ML film over a series of time points from 5 sec to 1 min. The films were digitized using a flatbed HP ScanJet 4C scanner. The digitized images were then quantitatively analyzed for total protein using Gel-Pro Analyzer software. The absolute integrated optical density of each band was compared with the exposure time to ensure that analysis was performed in the linear range for both antibodies. Each sample was run in duplicate and normalized to γ-tubulin. The samples were averaged by genotype, and the SEM was determined.
Mouse strains and genotyping. All mice were of a 129SvEvTac and NIH Black Swiss background and housed in a standard 12 hr light/dark cycle. For timed matings, the day of the vaginal plug was considered embryonic day 0.5 (E0.5). For all embryonic work, embryos were staged (Kaufman, 1992), and either the yolk sac or a piece of the embryo was used for genotyping. The Lis-Neo allele (referred to as KO in this paper) and the Lis1-loxP allele (referred to as CKO) were genotyped using the following primers: KO: forward 5′-GTGTGGGATT-ATGAGACTGG-3′, reverse 5′-GATCTCT-CGTGGGATCATTG-3′; CKO: forward 5′-TGAATGCATCAGAACCATGC-3′, reverse 5′-CCTCTACCACTAAAGCTTGTTC-3′.
Histology and immunohistochemistry. For embryonic day of birth (P0) and postnatal day 10 (P10) analysis, samples were collected on ice and drop fixed in 4% paraformaldehyde overnight and then either cryoprotected in 30% sucrose and embedded in OCT (Tissue-Tek) for frozen sectioning or dehydrated and embedded in paraffin. Paraffin sections were 5 μm thick. For RC2 staining, brains were fixed for 3 hr in 2% paraformaldehyde, cryoprotected, embedded in OCT, and cut into 80 μm sections on a freezing microtome. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) analysis was performed using the ApopTag kit (Intergen, Purchase, NY) according to the manufacturer's recommendations, with an additional 0.5% Triton X-100 incubation before labeling. For immunohistochemistry, paraffin sections were dewaxed in xylenes, rehydrated, and reacted with primary antibody overnight at 4°C. Secondary antibody was either fluorescently conjugated or biotinylated with subsequent use of the Vector ABC detection system (Vector Laboratories, Burlingame, CA). For Reelin and TuJ1 detection, the Vector Laboratories Mouse on Mouse kit was used. Primary antibodies were used at the following concentrations: anti-Reelin G10 monoclonal (generous gift from A. Goffinet, University of Louvain Medical School, Brussels, Belgium), 1:1000; anti-TuJ1, 1:500 (Babco, Richmond, CA); monoclonal anti-chondroitin sulfate, 1:100 (Sigma; Clone CS-56); monoclonal anti-bromodeoxyuridine (BrdU), 1:50 (Becton Dickinson, Mountain View, CA); anti-testis-1 polyclonal antisera, 1:1000 (generous gift of M. G. Rosenfeld, University of California San Diego School of Medicine, LaJolla, CA); anti-phospho-histone H3, 1:1000 (Upstate Biotechnology, Lake Placid, NY); monoclonal anti-RC2, 1:2 (Developmental Hybridoma Bank; developed under the auspices of the National Institute of Child Health and Human Development and maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA). Secondary antibodies were from the following sources: biotinylated goat anti-mouse IgG (Sigma), tetramethylrhodamine isothiocyanate goat anti-mouse IgG (Cappel, West Chester, PA), biotinylated goat anti-rabbit (Jackson ImmunoResearch, West Grove, PA), and biotinylated goat anti-mouse IgM (Jackson Immuno-Research). Images were captured with a SPOT2 digital camera (Diagnostic, Inc.) directly into Adobe Photoshop.
BrdU analysis. Pregnant dams were injected at the appropriate gestational age with 100 μg/gm BrdU (Sigma). For in vivo migration analysis, pregnancy and lactation were allowed to continue until the age at which the pups were killed (P0, P10). For proliferation analysis, pregnant dams were killed 30 min after BrdU injection, and embryos were isolated, staged, genotyped, and embedded in paraffin. Slides were dewaxed, microwaved in 10 mm citrate to expose epitopes, and reacted with 1:50 mouse monoclonal BrdU antibody (Becton Dickinson) at 4°C overnight. Biotinylated goat anti-mouse was used at 1:100, and Vector ABC elite was used for development with DAB plus metal enhancement (Sigma) as the chromogen.
Cell quantitation. The number of TuJ1-positive cells in the ventricular zone was determined from three well matched, serial parasagittal sections from each genotype. The number of TuJ1-positive cells (in a grid of 300 μm width by the thickness of the ventricular zone) was determined for each genotype, averaged, and reported as the mean ± SE. For the BrdU birth-dating experiments, four to seven serial coronal sections were analyzed for each genotype at each time point. To normalize for differences in cortical thickness, an equal length of cortex (300 μm) was divided into five equal sectors, which were numbered 1–5 from deepest to most superficial. BrdU-positive cells were counted in each layer, averaged, and reported as a mean ± SE. For proliferation analysis (E15.5 analysis: two embryos of each genotype; E13.5 analysis, three wild-type, two+/KO, and four CKO/KO), three to five matched parasagittal sections from each embryo were analyzed. For E13.5, blue (hematoxylin stained) and black (BrdU labeled) cells were counted in five randomly placed grids of 20 μm wide by the thickness of the developing caudal telencephalon. For E15.5, blue and black cells were counted in vertically stacked grids of 200 × 100 μm spanning the ventricular and intermediate zones of the caudal telencephalon. The number and position of M-phase cells in the developing cortex were determined by counting cells in 100 × 20 mm bins spanning the developing cortex. Two embryos of each genotype and three serial parasagittal sections from each embryo were analyzed. Data were represented as the percentage of labeled cells in each bin of the total number of labeled cells.
Granule cell reaggregate assay. Reaggregate assays were done in duplicate from preparations from two different litters. Cerebellar granule cells were purified from P5 or P6 mice as described previously (Bix and Clark, 1998). Cells (1 × 106) were diluted in granule cell medium (Basal Medium Eagle with 1× glutamine, 10% horse serum, 5% fetal bovine serum, 0.9% glucose) on glass-bottom chamber slides for 12 hr, resulting in uniform reaggregates (100–120 μm in diameter). Reaggregates were transferred by pipette to glass-bottom chamber slides treated overnight with poly-d-lysine (0.5 mg/ml) followed by laminin 25 mg/ml (Sigma). After 12 hr, each reaggregate was photographed using a 20× objective with phase to document neuronal position. Migration distances were separated into bins for visual representation. The number of measurements for each genotype was as follows: +/+, n = 598; +/KO, n = 582;CKO/KO, n = 305. Statistical significance among the means was determined using the Student–Newman–Keuls test for multiple comparisons of parametric data.
Results
LIS1 dosage
The level of LIS1 in the brains of the various mutants was determined. We prepared P0 brain lysates from multiple brains of each genotype and analyzed them for LIS1 protein by immunoblotting. There were decreasing levels of brain LIS1 in the +/KO(45 ± 3.7%) and CKO/KO (33 ± 2.2%) compared with wild type (100 ± 18%) as demonstrated by the lighter intensity of LIS1 bands (Fig.1A). These results are consistent with a null heterozygote (+/KO) and a hypomorph/null (compound heterozygote, CKO/KO) genotype.
Fig. 1.

A, LIS1 protein levels in P0 brain lysates. Lysates were separated by SDS-PAGE electrophoresis, transferred to a membrane, and probed with anti-LIS1 antibody. Note the decreasing levels of LIS1 in the mutant brains. γ-tubulin was used as a loading control. Analysis of preplate and Cajal-Retzius cells:Lane 1, Wild type; lane 2,+/KO; lane 3, CKO/KO.B–D, Hematoxylin and eosin staining of parasagittal sections from E12.5 embryos. Note the lighter staining superficial preplate. E–G, TuJ1 expression of preplate (black). H–J, Anti-Reelin immunohistochemistry demonstrating Cajal-Retzius cells (black) indicated with arrows. There were similar numbers and distribution in all genotypes. B,E, H, Wild type; C,F, I, heterozygote (+/KO);D, G, J, compound heterozygote (CKO/KO).B–D, Parasagittal E12.5 sections.E–J, Coronal E12.5 sections.PP, Preplate. Scale bar, 50 μm.
Development of the preplate, splitting of the cortical plate, and subplate development
One of the earliest stages in cortical development is the formation of the primordial plexiform layer or preplate (Marin-Padilla, 1971; Rickman et al., 1997). This early postmitotic structure formed normally in both Lis1 heterozygotes and compound heterozygotes as demonstrated by the lighter staining laminar structure in the hematoxylin and eosin (HE)-stained sections at E12.5 (Fig.1B–D). To better visualize the preplate, TuJ1 immunohistochemistry was performed. TuJ1 recognizes class III β-tubulin, which selectively marks the preplate at E12.5 (Menezes and Luskin, 1994). A similar darkly staining preplate layer was detected in all three genotypes (Fig. 1E–G). Cells of the preplate are thought to differentiate into Cajal-Retzius cells and subplate cells. Cajal-Retzius cells line the most superficial layer of the preplate and express the proteins Reelin and calretinin (D'Archangelo et al., 1995; del Rio et al., 1995). Cajal-Retzius cells are important for the proper laminar development of the cortical plate as well as the maintenance of the radial glial scaffold (Rice and Curran, 1999; Super et al., 2000). Analysis of Cajal-Retzius cells at E12.5 with the monoclonal antibody G10 (Bergeyck et al., 1998) demonstrated a normal pattern and number of Cajal-Retzius cells in all genotypes (Fig. 1H–J)
The preplate provides the foundation for the cortical plate. Migrating neurons invade the cortical plate and split the preplate into the superficial marginal zone and subplate (for review, see Super et al., 2000). We analyzed HE-stained sections of E15.5 embryos, which demonstrate abnormalities in several structures after the formation of the preplate. The marginal zone was formed in all three genotypes (Fig.2A–C), although the CKO/KO marginal zone was slightly thinner, with areas of clusters of cells in an otherwise cell-sparse zone (Fig.2C). A cortical plate was present in all three genotypes, but it became less well defined, particularly at its deep boundary from+/KO to CKO/KO. There was no obvious subplate inCKO/KO; the cortical plate gradually fused with the intermediate zone. The ventricular zone was smaller inCKO/KO, and the corresponding intermediate zone was more cellular. Preplate derivatives (marginal zone and subplate) are rich in chondroitin sulfated glycoproteins (CSGPs) (Sheppard and Pearlman, 1996). To further assess the resultant marginal zone and subplate formed from the splitting of the preplate by cortical plate cells, we analyzed chondroitin sulfate glycoprotein expression at E15.5 (Fig.2D–F). The preplate was split into a marginal zone and subplate as indicated by the CSGP signal in all genotypes. This event represents a clear distinction from thereeler phenotype in which the cortical plate neurons pile up underneath the preplate in a structure dubbed the superplate (Caviness, 1982). Interestingly, there was an increasing diffuseness and intensity of the CSGP signal in the marginal zone and subplate from wild type to+/KO to CKO/KO. A similar diffuseness was seen with anti-calretinin immunostaining, another marker specific for subplate derivatives (data not shown). Because subplate cells produce significant amounts of CSGPs, this diffuseness suggests that these cells have not migrated properly to their appropriate positions. There is evidence that CSGPs plays an important role in the proliferation, migration, and cell differentiation process (Letourneau et al., 1994;Moro Balbas et al., 1998). Hence the increase and diffuseness of the CSGP signal as Lis1 decreases have implications for subsequent neuronal migration and development.
Fig. 2.
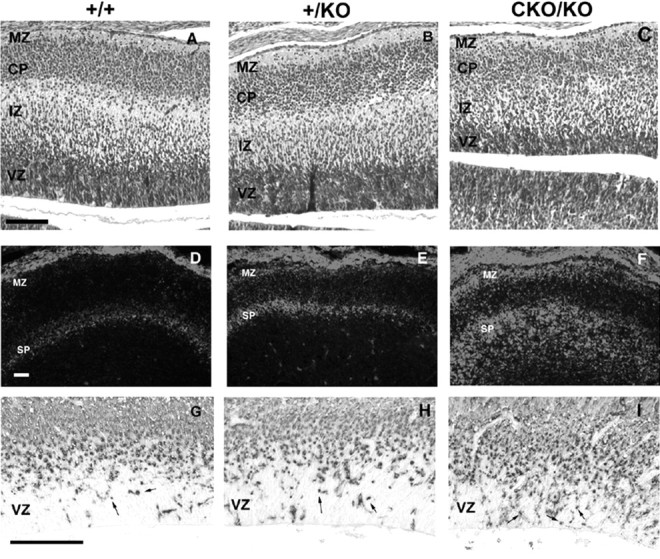
E15.5 analysis of cortical plate, subplate, and intermediate and ventricular zones. A–C, Hematoxylin and eosin-stained E15.5 parasagittal sections. Note poorly defined cortical plate (CP) in CKO/KO brain. The marginal and ventricular zones (MZ, VZ) as well as the entire cortical plate were thinner in theCKO/KO. D–F, Subplate and marginal zone analysis with anti-chondroitin sulfate monoclonal antibody. The expression of chondroitin sulfated proteins in the marginal zone and subplate was higher and more diffuse as LIS1 levels were reduced. G–I, TuJ1 immunohistochemistry. Note the larger number of TuJ1-positive cells (arrows) in the ventricular zone of CKO/KO(I). A, D,G, Wild type; B, E,H, heterozygote; C, F,I, compound heterozygote. MZ, Marginal zone; CP, cortical plate; SP, subplate;IZ, intermediate zone; VZ, ventricular zone. Scale bar, 50 μm.
At E15.5 most of the post-mitotic neurons in the intermediate zone and the cortical plate express TuJ1, with few TuJ1-positive neurons in the ventricular zone, which contains mainly neuroblasts and recently born neurons (Menezes and Luskin, 1994). When E15.5 brains were stained with TuJ1, we found few TuJ1-positive neurons in the ventricular zone of wild-type (85 ± 7 cells) and heterozygous (82 ± 8 cells) mice (Fig. 2G,H). In contrast, there were many more TuJ1-expressing cells in the ventricular zones ofCKO/KO mice (106 ± 3 cells) (Fig.2I). The presence of these cells in the ventricular zone also suggests either a delay in migration of post-mitotic neurons from the ventricular zone into the strongly TuJ1-positive intermediate zone and cortical plate or the misspecification of cells in the ventricular zone.
The radial glial scaffold is a crucial substrate for gliophilic radial migration (Rakic, 1972). We assessed radial glial morphology by immunostaining with RC2 monoclonal antibody (Misson et al., 1988). The general parallel radial arrangement of fibers projecting from the ventricular lumen to the pial surface was well preserved at low (data not shown) and high power (Fig. 3) Thus, abnormalities in the formation of the subplate, chondroitin sulfate glycoprotein expression and distribution, the thickness of the ventricular zone, and the number of differentiating neuroblasts in the ventricular lumen were affected as LIS1 dosage was reduced. The general morphology of the radial glia appeared to be spared.
Fig. 3.
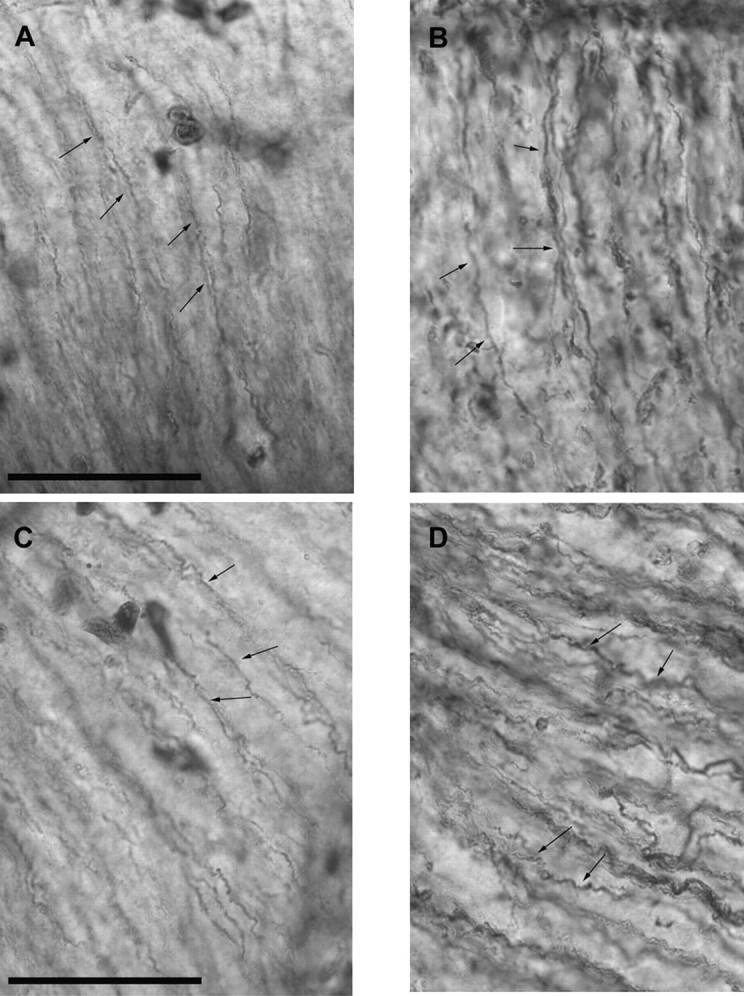
Radial glia analysis with RC2 immunohistochemistry. Note preservation of the parallel glial scaffold as well as the structure of the individual fibers (arrows). A, C, Wild type;B, D, CKO/KO. Scale bar, 50 μm.
In vivo migration and cortical layering
To assess the extent to which Lis1 dosage reduction affected cortical plate formation and laminar structure, BrdU birth-dating was performed. Pregnant dams were injected with BrdU at E12.5 and E15.5 to assess migration of early and late generated neurons, respectively. Pups were killed at P0 and P10, and brains were processed for BrdU immunohistochemistry. This resulted in a higher overall background staining for BrdU, because there were fewer cell divisions between labeling and analysis to dilute the BrdU signal in cells born after the time of injection. In the brains of E12.5 injected wild-type embryos (Figs. 4, 5,top panel), most labeled neurons were in the deep layers of the cortex destined to form layers 5 and 6, as described previously (Caviness, 1982). The few labeled cells in the marginal zone may represent late labeled preplate cells; preplate neurogenesis is believed to occur from E10 to E13 (Valverde et al., 1995). The pattern was similar in P0–BrdU–E12.5+/KO brain. In the CKO/KO (Fig. 4C), most of the labeled cells were similarly in the deeper layers of the developing cortex like wild type and +/KO, but fewer neurons were labeled and the most intensely labeled neurons were in the deepest layer of the cortex. These results suggest that there were fewer proliferating CKO/KO neuroblasts compared with wild type and +/KO at the time of BrdU injection or there was an increase in cell death, or both.
Fig. 4.
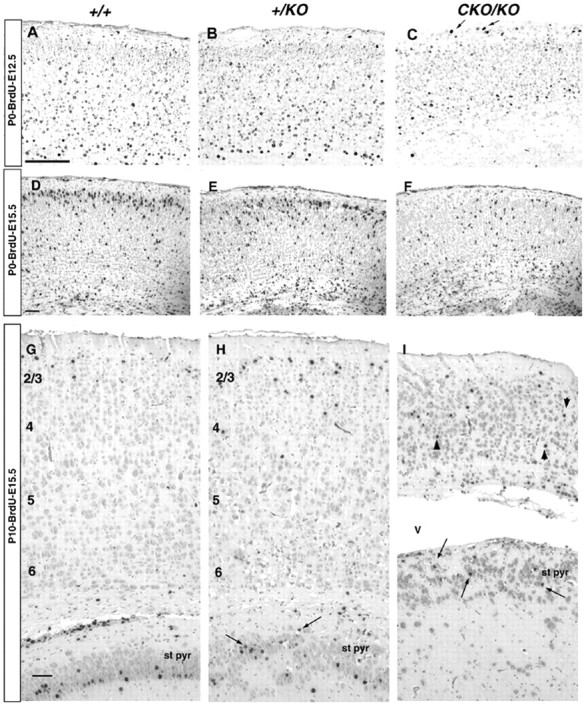
BrdU birth-dating analysis of caudal coronal sections. A–C, P0 brains, BrdU injection, E12.5. Note that most of the labeled cells are deep within the cortex, with occasional cells in the marginal zone (arrows). TheCKO/KO brains had fewer labeled cells in the deep cortex. D–F, P0 brains, BrdU injection, E15.5. Most labeled cells were in a well defined cortical layer 2/3 (cortical bin 5) in the wild type (D). This layer was more diffuse in the +/KO (E) and nonexistent in CKO/KO (F).G–I, P10 brains, BrdU injection, E15.5. Labeled cells were in the most superficial part of layer 2, and the deepest layer of the stratum pyramidale in the wild type representing late born neurons migrating the farthest (G). The+/KO brain (H) displayed a more diffuse labeling in layer 2 and labeled cells scattered throughout the stratum pyramidale (arrows). TheCKO/KO cortex (I) was markedly thinner, with labeled cells throughout the cortex (arrowheads). Labeled cells in stratum pyramidale were scattered throughout this layer (arrows). st pyr, Stratum pyramidale. Cortical layers are indicated withnumbers. Scale bar, 50 μm.
Fig. 5.
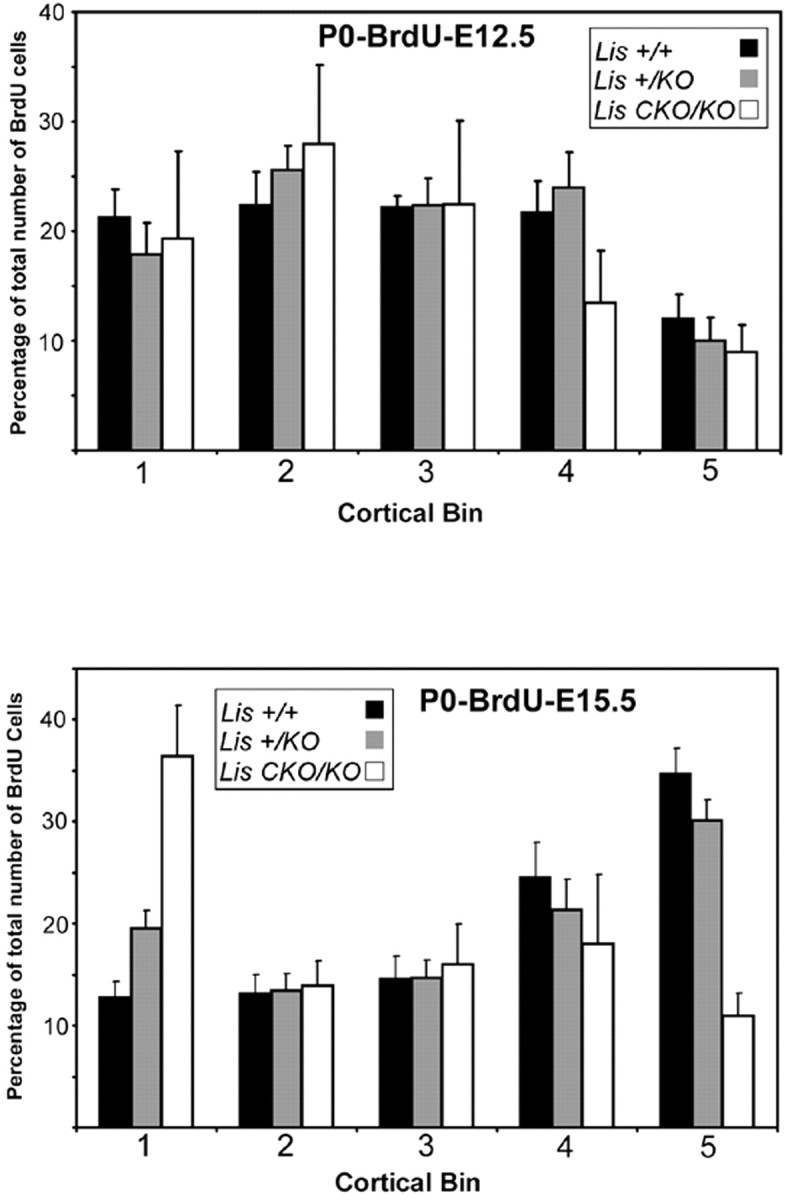
Histograms of E12.5 and E15.5 BrdU birth-dating.Top panel, Percentages of total number of BrdU-positive cells in cortical bins from P0 brains injected at E12.5. Cortical bin 1 is closer to the ventricular lumen, whereas bin 5 is the most superficial part of the cortex, near the pial surface. Note that most of the BrdU-labeled +/KO and CKO/KO cells are clustered in bins closer to the ventricular lumen as in the wild type, representing cells occupying deeper layers of the developing cortex. Bottom panel, Percentages of total number of BrdU-positive cells in cortical bins from P0 brains injected at E15.5. In wild type and +/KO, most of the BrdU-positive cells are in bins closer to the pial surface, whereas a significant number of BrdU-labeled CKO/KO cells remain deep in cortical bin 1, suggesting a migration defect.
In the P0–BrdU–E15.5 analysis, BrdU-labeled neurons in wild-type brains already formed a well demarcated layer destined to become layer 2/3, subjacent to the marginal zone (Figs. 4D, 5,bottom panel). There were BrdU-labeled neurons throughout the thickness of the cortex in accordance with postnatally migrating neurons, but most were in the well defined superficial layers. In the +/KO P0–BrdU–E15.5 brain, this superficial layer of labeled cells was less well defined, with labeled cells found in a broader distribution (Figs. 4E, 5, bottom panel). The histogram also demonstrates more labeled neurons in cortical bin 1 in +/KO compared with wild type, suggesting that more neurons had hardly migrated. CKO/KOdisplayed the most profound phenotype, with virtually no BrdU labeled cells near the pial surface. Labeled cells were scattered throughout the developing cortex and many remained deep in the subplate in bin 1 (Figs. 4F, 5, bottom panel). There were also fewer BrdU-labeled cells in the CKO/KO brain, suggesting the possibility of fewer proliferating cells or an increase in cell death, or both, as in the E12.5 injections.
It was shown previously that the +/KO mouse demonstrated a delay in neuronal migration at P0 but by P21 there was no significant difference in BrdU labeling in the neocortex, unlike the archicortex (hippocampus) (Hirotsune et al., 1998; Fleck et al., 2000). This suggested that the neurons in the +/KO mice eventually ended up in the appropriate location. To assess whether a similar scenario occurred in the CKO/KO mouse, we repeated the BrdU experiments and analyzed the brains at a later time point when neuronal migration would be more complete. We chose P10 because mostCKO/KO mice die before weaning (P21), and hydrocephalus becomes grossly deforming at older ages. In the P10–BrdU–E15.5 wild-type brains, most BrdU-labeled neurons were found in a tight layer representing the latest born neurons in layer 2 (Fig. 4G). The +/KO mouse brain still displayed a diffuseness of the BrdU labeling, consistent with a slowing of neuronal migration (Fig.4H). The CKO/KO brain displayed very few BrdU-labeled neurons (Fig. 4I). Those that were labeled were scattered throughout the cortex, with several remaining in the white matter, although there were several neurons that did appear to reach the presumptive layer 2 (Fig. 4I). There was also a marked reduction in thickness of the CKO/KO cortex. Similar to the results at P0, there is a marked reduction in the number of BrdU-labeled neurons consistent with a proliferative or cell death defect.
To assess the development of specific cortical layers, we performed immunohistochemistry using a polyclonal antibody to a POU domain transcription factor, Testis-1 (Tst-1). Anti-Tst-1 selectively labels cells in layers 2/3 and 5 (Bermingham et al., 1996). The P0 wild-type brain displayed a superficial layer of small Tst-1-positive neurons in layer 2/3 and a subjacent layer of larger labeled neurons in layer 5 (Fig.6A). By P10, cells in layer 2/3 and larger layer 5 neurons were distinctly labeled (Fig.6D,G). The +/KO brain displayed a very similar pattern of staining at P0 and P10 in the neocortex. By contrast the P0 CKO/KO brain contained labeled cells in superficial layers, but many labeled cells were found deep in the subplate. By P10, labeled cells were found throughout the cortex, demonstrating a severe disruption of laminar organization. In this respect the CKO/KO brain more closely resembles the human lissencephalic brain, which has poorly defined layers, suggesting that there are slight differences in dosage sensitivity to migration between mice and humans.
Fig. 6.
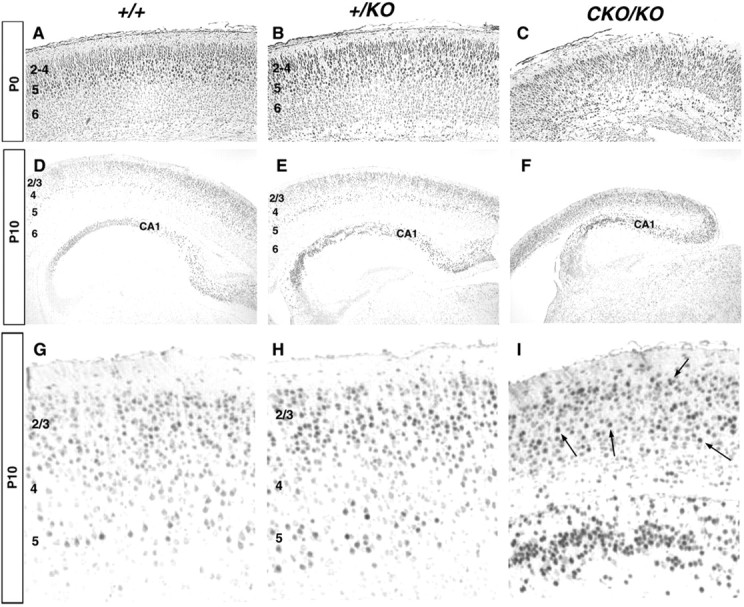
Tst-1 immunohistochemistry on caudal coronal sections. A–C, P0 brains. StrongTst-1 expression was present in layers 2–4 and 5 in both wild-type (A) and+/KO (B) brains with a slightly broader layer 5 in +/KO. Expression was diffusely detected throughout the cortex in CKO/KO(C). D–F, P10 brains, with higher magnification in G,H, and I. Expression was confined mainly to cortical layers 2–3 and 5 in both wild type (D, G) and +/KO (E, H). In theCKO/KO brains (F, I), labeled neurons were scattered throughout the cortex (arrows), with no obvious lamination pattern. Note the strongTst-1 expression in CA1 of the hippocampus in all genotypes.
During the migration of neurons to the hippocampus, cells migrate through the stratum oriens-alveus to eventually form the pyramidal cells layers of CA1, CA2, and CA3, with the latest born neurons occupying the innermost part of the pyramidal cell layer (for review, see Super et al., 2000). In the wild-type brain, most labeled neurons at P10–BrdU–E15.5 were deep in CA1 (Fig. 4G). In the+/KO brain there was marked disorganization of CA1, with labeled neurons scattered throughout the pyramidal cell layer (Fig.4H), as reported previously (Hirotsune et al., 1998;Fleck et al., 2000). In the CKO/KO brain there was a more disorganized CA1, with fewer labeled neurons (Fig.4I). None of the labeled cells resided in the deepest part of CA1. Similar results were found in other pyramidal layer regions (data not shown). Tst-1 expression was confined to the CA1 region in all genotypes (Fig.6D–F), suggesting that the disorganization in the mutants had little effect on the specification of this transcription factor. These results demonstrate a dependence of hippocampal development on LIS1 levels.
In vitro migration defects
To further support the in vivo LIS1 dosage sensitivity of neuronal migration, we performed a cerebellar reaggregate assay using granule cells prepared from wild-type, +/KO, andCKO/KO mice. The cerebellar reaggregate assay has been used to simulate neuronal migration in vitro. We previously used this assay to show that Lis1+/KO neurons migrated more slowly than wild-type neurons and found that there was aLis1 dose-dependent defect in cerebellar development (Hirotsune et al., 1998). Reaggregates extend neurites on which granule cells will migrate away from the cluster. A much greater percentage of wild-type cells migrated farther distances than both mutant cell preparations (+/+ vs +/KO, p < 0.01; +/+ vs CKO/KO, p < 0.01) (Fig.7A,B). Many heterozygous (Fig. 7C) and compound heterozygous (Fig.7D) cells migrated smaller distances, and many were nearly stationary. Of note, the mean migration distances reduced with decreasing LIS1 dosage (+/KO vs CKO/KO,p < 0.05). These results support the observations seenin vivo regarding dose-dependent defects in neuronal migration in the cerebral cortex.
Fig. 7.

Cerebellar granule cell reaggregate assay. Reaggregates were purified from each genotype, plated on a laminin substrate, and allowed to extend neurites and migrate for 12 hr. Graph depicts number of cells from each genotype found in a particular bin after 12 hr (see Materials and Methods). More wild-type cells (black bars) were found in bins farther from the center of the reaggregate than +/KO (gray bars) or CKO/KO (white bars) cells. More CKO/KO cells migrated shorter distances. Inset bar graph represents mean distances migrated from two different experiments. Note that the mean distances migrated were sensitive to LIS1 dosage.
Proliferation and interkinetic nuclear migration
Several observations suggested that reduction of LIS1 affected cell proliferation. The ventricular zone, containing the majority of proliferating neuroblasts, was thinner in Lis1 mutant mice than in wild type (Fig. 1). Birth-dating analysis suggested that there may have been fewer neurons generated in the compound heterozygous mice (Fig. 4). Moreover the size of the mutant brains was smaller than wild type (Hirotsune et al., 1998). To assess the effect of LIS1 levels on proliferation, we analyzed cells in S phase at two time points during neurogenesis, E13.5 and E15.5. Pregnant dams were labeled with BrdU for 30 min and killed, and embryos were isolated to determine the fraction of BrdU-labeled cells. Most S phase nuclei were found in the upper layers of the ventricular zone as demonstrated previously (Takahashi et al., 1992). In all genotypes many BrdU-labeled cells were found in the upper layers of the ventricular zone at E13.5 and E15.5 (Fig.8A–C,E–G). We performed cell counts at E13.5 (Fig. 8D), and no differences in the number of S phase cells in wild type versus+/KO (p > 0.8) or wild type versusCKO/KO (p > 0.8) were found. There was, however, a statistical difference in the total number of ventricular zone cells in the wild type versus CKO/KO(p < 0.001), suggesting an overall reduction of neuronal precursors. At E15.5 a significant reduction in S phase cells was observed in +/KO and CKO/KO mice compared with wild type (Fig. 8E–G). Cell counts revealed a 20% (p < 0.001) and 40% (p < 0.001) reduction of proliferating neuroblasts in the ventricular zones of the +/KO andCKO/KO mice compared with wild type, respectively (Fig.8H). There was a similar reduction in the total number of cells. The number of labeled cells in the subventricular/intermediate zones remained equal, whereas the total number of cells in these zones was increased in the Lis1mutants.
Fig. 8.

Proliferation analysis. Photomicrographs depict the caudal region of matched parasagittal sections from E13.5 and E15.5 embryos labeled with BrdU for 30 min. Black cellsrepresent BrdU-labeled cells in S phase. Sections were counterstained with hematoxylin. Note the equal numbers of BrdU-labeled cells in all genotypes at E13.5 (A–C). The histogram (D) demonstrates roughly equal numbers of BrdU-labeled cells in each genotype (bottom bars) but progressively decreasing total numbers of cells (topplus bottom bars) when quantitation was done. At E15.5 (E–G) there was a progressive decrease in the number of BrdU-labeled cells in +/KO(F) and CKO/KO(G) brains. H, Note the marked difference in BrdU-labeled cells in the ventricular zone (bottom bars). There was also a similar decrease in the total number of cells in this zone. These reductions were also LIS1 dosage sensitive. Histogram y-axis = mean number of cells.CP, Cortical plate; IZ, intermediate zone; VZ, ventricular zone. A,D, G, Wild type; B,E, H, +/KO;C, F, I,CKO/KO. Scale bar, 50 μm.
We also assessed cells in metaphase (M phase) with antibody to phosphorylated H3 histone (Hendzel et al., 1997). The nuclei of M phase cells are located predominantly at the ventricular lumen, with some cells cycling in the subventricular zone (Takahashi et al., 1996). This pattern of labeling was found in wild-type brains at the two embryonic ages analyzed (Fig.9A,D). However, the position of mitotic cells became progressively more ectopic between the ventricular lumen and the subventricular zone from the +/KO to the CKO/KO brains (Fig.9B,C,E–G). These results suggest a defect in interkinetic nuclear migration, with neurons critically deficient in LIS1 unable to migrate to the ventricular lumen to divide.
Fig. 9.

M phase analysis. Each figure represents the caudal region of matched parasagittal sections stained for the metaphase marker phosphorylated H3 histone (black) at E13.5 (A–C), E15.5 (D–F). In wild-type brains (A, D), two populations of M phase cells were demonstrated. There was a large population at the ventricular luminal surface and a smaller population in the subventricular zone, consistent with previous studies. Although similar populations were seen in the +/KO (B, E) and CKO/KO (C, F) brains, there was an increase in ectopically placed M phase cells, between the luminal surface and the subventricular zone (arrows). The histogram (G) demonstrates that more+/KO (gray bar) and CKO/KO(white bar) cells were located between the luminal surface and the subventricular zone. More ectopic cells correlated with lower LIS dosage. These misplaced cells suggest a defect in interkinetic nuclear migration or the length of M phase dependent on LIS1, or both. *p < 0.005 wild type versusCKO/KO; ∧ p < 0.05+/KO versus CKO/KO; two-tailed Student'st test. CP, Cortical plate;IZ, intermediate zone; VZ, ventricular zone. Scale bar, 50 μm.
Cell death
The decrease in proliferating cells at late embryonic time points in the mutant mice suggested that a pool of cells was lost from the cycling population. To assess whether LIS1 reduction resulted in a greater propensity for developing neurons to undergo programmed cell death, we performed TUNEL assays on sections from E12.5 (data not shown), E13.5, and E15.5 parasagittal sections (Fig.10). Occasional TUNEL-positive neurons were seen in wild-type brains, consistent with normal programmed cell death (Fig. 10A) (Thomaidou et al., 1997). However, there was an increase in the number of apoptotic neurons in the+/KO brain (Fig.10B,F) and significantly increased numbers in the brains from CKO/KO mice (Fig.10C,G). Interestingly, most of the apoptotic neurons were in the ventricular zone. 4′,6′-Diamidino-2-phenylindole (DAPI) counterstaining revealed many pyknotic micronuclei in the CKO/KO brains and also to a lesser extent in the +/KO brains (Fig. 10C,inset). Thus, the proliferative differences seen at E15.5 may be the combined result of loss of cycling neuroblasts as well as an intrinsic proliferation defect within the surviving cells.
Fig. 10.
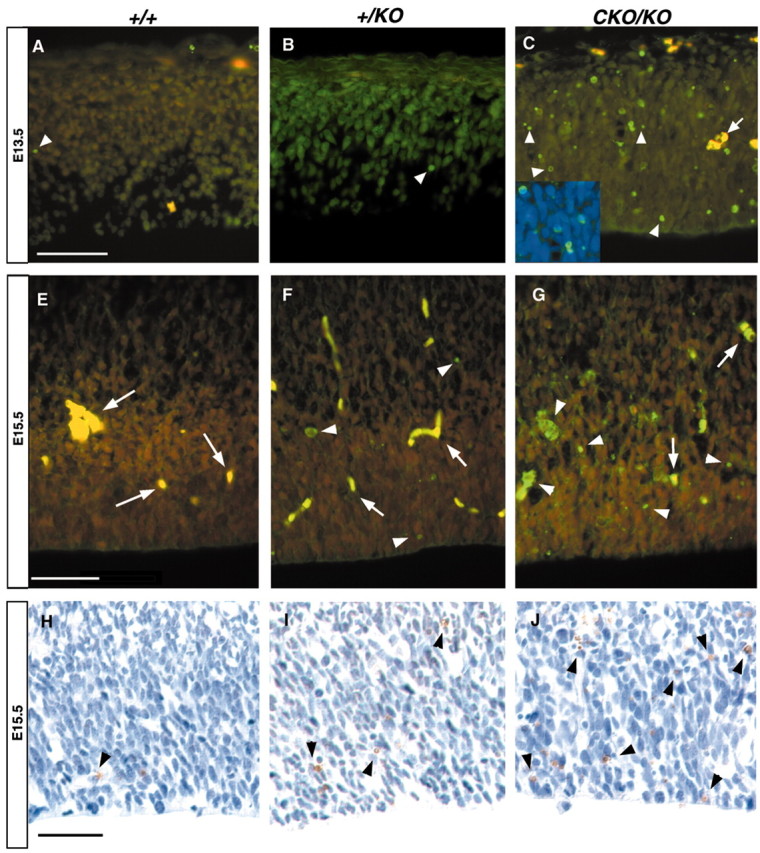
TUNEL analysis. Each figure represents the caudal region of matched parasagittal sections that have been analyzed for apoptotic cell death using both fluorescence (green cells) and DAB (brown cells) detection (arrowheads) at E13.5 (A–C) and E15.5 (E–J). Fluorescence images represent composites of FITC and rhodamine channels; yellow-staining cells represent nonspecific fluorescence caused by red blood cells/vessels (arrows). Note the marked increase in apoptotic cells in the +/KO and CKO/KObrains using both detection techniques (B,C, F, G, I,J). TUNEL-positive cells were found to be pyknotic nuclei when DAPI counterstained (inset C), a morphologic change consistent with apoptotic cell death. A, E, H, Wild type;B, F, I,+/KO; C, G,J, CKO/KO. Scale bar, 50 μm.
Discussion
We have performed a developmental analysis of the effect of reduced LIS1 levels on cortical development. In addition to confirming and extending our previous findings on the dosage dependence of LIS1 onin vivo and in vitro neuronal migration and cortical lamination (Hirotsune et al., 1998; Fleck et al., 2000), we have demonstrated that LIS1 is involved in the formation of the subplate. Furthermore, interkinetic nuclear migration was dependent on LIS1 levels, indicating a role for LIS1 in generating neuroblasts and postmitotic neurons. We have observed that LIS1-deficient cells have an increased susceptibility to cell death, demonstrating a role for LIS1 in cell survival. Thus, the cortical disorganization in LIS1-deficient mice, and presumably humans with type I lissencephaly, results from a combined effect of LIS1 reduction on neuronal migration in post-mitotic neurons and cell numbers in the ventricular zone by influencing cell proliferation and survival of neuroblasts. These phenotypes (proliferation and cell death) could be caused primarily by a direct effect of loss of Lis1 on cell physiology or secondarily by incomplete nuclear migration.
Cortical development prior to cortical plate formation appears to be insensitive to LIS1 levels. The preplate was of similar thickness in all three genotypes, with normal number and position of Cajal-Retzius cells. Preplate cells may be relatively insensitive to LIS1 dosage given their short migration distance (Walsh, 1998). Two modes of radial migration have been described: (1) locomotion on radial glia and (2) somal translocation (Nadarajah et al., 2001). It is possible thatLis1 has a minimal function in the process of somal translocation; this coupled with the short migration distances early in cortical development may render preplate formation relatively insensitive to LIS1 levels.
The radial glial scaffold in all genotypes at E14.5 displayed a parallel organization throughout the cortical plate (for review, seeRakic, 1988; Super et al., 2000). Interestingly, curvaceous radial glia were found in another mouse model of Lis1 deficiency (Cahana et al., 2001). Although we observed some curved radial glia in theCKO/KO brains, this appeared to be attributable to an overall distortion in the shape of the CKO/KO brains. RC2 staining clearly demonstrated a preserved parallel arrangement of the radial glial scaffold, suggesting that severe LIS1 reduction has a minimal effect of radial glial development.
Abnormalities among the different genotypes were apparent during cortical plate formation. Splitting of the preplate appeared more defective as LIS1 levels were reduced. Consequently a diffuse, ill-defined subplate was formed, with abnormally elevated expression of chondroitin sulfated glycoproteins, particularly in theCKO/KO. The marginal zone also showed increased CSGP expression with decreasing LIS1 dosage. These data are consistent with either a primary defect in the formation of the cortical plate and subplate or a secondary delay in neuronal migration and inefficient splitting of the preplate. In support of a migration defect, TuJ1 labeling at E15.5 demonstrated that many post-mitotic neurons remained in the ventricular zone. Although we cannot definitively distinguish these hypotheses, they both may contribute to the phenotype. The elevated levels of CSGP suggest a primary defect in the cortical plate, whereas the diffuse nature of the subplate could be secondary to the slowly migrating neurons that take longer to traverse it. The subplate is implicated in the maturation of thalamocortical and corticothalamic connections as well as the precursor of layer 6B (McConnell et al., 1989; Bicknese et al., 1994; McConnell et al., 1994). The increasing diffuseness of the subplate layer may disrupt these connections. In support of this, abnormal thalamocortical development was found in another mouse mutant with reducedLis1 dosage (Cahana et al., 2001). Because our heterozygous and compound heterozygous mutants were affected more severely than the mutant described by Cahana et al. (2001), aberrant thalamocortical connections might be predicted in our mice.
Birth-dating analysis demonstrated that the migration phenotype displayed by Lis1 mutants is different from several other mutants such as reeler, VLDL/ApoE2, andmDab1. In these mutants there is a clear cortical layer inversion with an outside-in pattern of BrdU labeling (Caviness, 1982;Chae et al., 1997; Howell et al., 1997; Trommsdorff et al., 1999), whereas reduction of LIS1 does not preclude later born neurons from migrating past earlier ones. Because LIS1 dosage was reduced, however, there was a progressive slowing of migration at all time points analyzed. The slowest neurons were found in the CKO/KObrains, as demonstrated by the broad distribution of BrdU-labeled cells. This was supported by immunohistochemistry with anti-Tst-1, which revealed a Lis1dosage-dependent perturbation of cortical lamination. Furthermore, analysis of cerebellar granule cell reaggregates demonstrates that the migration of granule cells was sensitive to the level of LIS1, because the CKO/KO reaggregates were the most severely affected in their ability to migrate.
Interkinetic nuclear migration was disrupted in the Lis1mutants in a dose-dependent manner. Normally, nuclei of neuroblasts move radially up and down the ventricular zone according to their phase of the cell cycle (Angevine and Sidman, 1961; Takahashi et al., 1995b). Most metaphase nuclei are located at the ventricular lumen, with a smaller population within the ventricular/subventricular zone (Takahashi et al., 1995a). Many M phase nuclei in the Lis1mutants were not at the ventricular luminal surface. It is possible that as nuclei rapidly move toward the ventricular lumen during G2, LIS1-deficient cells move more slowly and never reach the luminal surface before division begins. A second hypothesis is based on the observation that decreased LIS1 activity can cause mitotic delay (Faulkner et al., 2000). LIS1-deficient cells may leave the luminal surface before the completion of mitosis. There were more cells in M phase in early embryonic compound heterozygote brains than wild type, suggesting a possible mitotic delay (Fig.9C,F). These hypotheses are not mutually exclusive and together may participate to some degree in the observed interkinetic migration defects.
There are developmental implications of cell divisions not in contact with the ventricular lumen (Chenn and McConnell, 1995). Many LIS1-deficient neuroblasts actually divided within the ventricular zone, unattached to the lumen. Such misplaced cell divisions may affect neural cell fate caused by inappropriate extracellular signals, possibly abnormal symmetric/asymmetric (Chenn and McConnell, 1995;Zhong et al., 1996).
The birth-dating studies demonstrated fewer BrdU-labeled neurons in theCKO/KO brains at all times analyzed, suggesting reduced proliferation, increased cell death, or both. At E13.5 the number of proliferating cells was roughly equal; however, there was significant apoptosis at this time point, as well as at E12.5 (data not shown). Therefore the decrease in BrdU-labeled neurons in all of theCKO/KO brains is caused mainly by increased cell loss. It is estimated that in the mouse roughly 11 cell cycles contribute to the birth of cortical neurons (for review, see McConnell, 1995; Takahashi et al., 1995a). The last four cycles appear to contribute to the most dense cortical layers 2, 3, and 4. Because increased cell death occurs through the period of rapid proliferation in our mutants, it is likely that there are significantly fewer precursors to generate such neurons. This may explain the reduced cortical thickness in mutants, especiallyCKO/KO mice.
Programmed cell death is a normal aspect of cortical development (Oppenhein, 1991; Thomaidou et al., 1997). In the Lis1mutants cell death was increased, suggesting an important role forLis1 in neuronal survival. The neuronal sensitivity to LIS1 dosage may result from abnormalities in neuronal or interkinetic migration and proliferation. Most apoptotic cells were found within the proliferative zone, suggesting that cell death occurred within cycling cells. Previous studies found that LIS1 is associated with the centrosomes, microtubules, and the kinetochore (Sapir et al., 1997;Faulkner et al., 2000; Smith et al., 2000). LIS1 is also important in chromosome segregation (Faulkner et al., 2000), and consequently, reduced LIS1 dosage could result in chromosomal lag, aneuploidy, and apoptosis (for review, see Sorger et al., 1997).
The molecular mechanisms of neuronal migration are being elucidated (Walsh and Goffinet, 2000; Herz, 2001; Wynshaw-Boris and Gambello, 2001; Gupta et al., 2002). LIS1 and the proteins NUDEL and mNudE regulate cytoplasmic dynein motor function (Feng et al., 2000;Niethammer et al., 2000; Sasaki et al., 2000). This pathway is conserved in Aspergillus nidulans, in which it regulates nuclear migration (Morris, 2000). It is likely that this conserved pathway regulates nuclear migration in mammals. Dysregulation of cytoplasmic dynein in the Lis1 mutants is likely the cause of the neuronal and interkinetic nuclear migration defects and possibly the increase in cell death. A decrease in intracellular LIS1 concentration may reduce the number of LIS1/dynein complexes, compromising dynein function (LaMonte et al., 2002) and affecting migrating neurons and immature cells within the proliferative ventricular zone.
Our analysis has demonstrated new developmental aspects of LIS1 in mice. Reduced LIS1 levels result in slower migration of post-mitotic neurons as well as defects in the subplate, interkinetic nuclear migration, and cell death. Our data support the notion that reduced LIS1 levels result in broader developmental defects than simply defective cell autonomous neuronal migration. All of these defects may contribute to the severe brain pathology seen inLis1-deficient mice and humans with lissencephaly.
Footnotes
This work was supported by a Howard Hughes Institute Fellowship for Physicians (M.J.G.), a National Science Foundation Pre-Doctoral Fellowship (J.Y.), an institutional grant from the Howard Hughes Medical Institute (A.W.B.), grants from the National Institute of Neurological Disorders and Stroke (NINDS) (NS39404) and the National Institute of Mental Health (MH62821), as well as grants from NINDS (K12NS01701–06), the American Epilepsy Foundation Junior Investigator Research Grant, the John Merck Award in the Developmental Disabilities in Childhood, the Searle Scholars Program, the Klingenstein Foundation, and the Department of Neurosciences at University of California San Diego (J.G.G.). We thank Geoff Rosenfeld and Andre Goffinet for providing antibodies. We thank Jianbo Wang, David Rapaport, and David Tarin for critically reading this manuscript.
Correspondence should be addressed to Anthony Wynshaw-Boris, Departments of Pediatrics and Medicine, University of California, San Diego, School of Medicine, 9500 Gilman Drive, Mail Code 0627, LaJolla, CA 92093. E-mail: awynshawboris@ucsd.edu.
M. J. Gambello's present address: Department of Pediatrics, Division of Medical Genetics, University of Texas Health Sciences Center Houston, Medical School, 6431 Fannin Street, Medical School Building 3.144, Houston, TX 77030.
References
- 1.Adachi T, Aoki J, Manya H, Asou H, Araim H, Inoue K. PAF analogues capable of inhibiting PAF acetylhydrolase activity suppress migration of isolated rat cerebellar granule cells. Neurosci Lett. 1997;235:133–136. doi: 10.1016/s0304-3940(97)00742-8. [DOI] [PubMed] [Google Scholar]
- 2.Angevine J, Sidman R. Autoradiographic study of cell migration during histogenesis of cerebral cortex in the mouse. Nature. 1961;192:766–768. doi: 10.1038/192766b0. [DOI] [PubMed] [Google Scholar]
- 3.Barkovich A, Koch T, Carroll C. The spectrum of lissencephaly: report of ten patients analyzed by magnetic resonance imaging. Ann Neurol. 1991;30:139–146. doi: 10.1002/ana.410300204. [DOI] [PubMed] [Google Scholar]
- 4.Bergeyck V, Naerhuyzen B, Goffinet A, Lambert de Rouvroit C. A panel of monoclonal antibodies against reelin, the extracellular matrix protein defective in reeler mutant mice. J Neurosci Methods. 1998;82:17–24. doi: 10.1016/s0165-0270(98)00024-7. [DOI] [PubMed] [Google Scholar]
- 5.Bermingham J, Jr, Scherer S, O'Connell S, Arroyo E, Kalla K, Powell F, Rosenfeld M. Tst-1/Oct-6/SCIP regulates a unique step in peripheral myelination and is required for normal respiration. Genes Dev. 1996;10:1751–1762. doi: 10.1101/gad.10.14.1751. [DOI] [PubMed] [Google Scholar]
- 6.Bicknese A, Sheppard A, O'Leary D, Pearlman A. Thalamocortical axons extend along a chondroitin sulfate proteoglycan-enriched pathway coincident with neocortical subplate and distinct from the efferent path. J Neurosci. 1994;14:3500–3510. doi: 10.1523/JNEUROSCI.14-06-03500.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bix G, Clark G. Platelet-activating factor receptor stimulation disrupts neuronal migration in vitro. J Neurosci. 1998;18:307–318. doi: 10.1523/JNEUROSCI.18-01-00307.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cahana A, Escamez T, Nowakowski R, Hayes N, Giacobini M, von Holst A, Shmueli O, Sapir T, McCopnnell S, Wurst W, Martinez S, Reiner O. Targeted mutagenesis of Lis1 disrupts cortical development and Lis1 homodimerization. Proc Natl Acad Sci USA. 2001;98:6429–6434. doi: 10.1073/pnas.101122598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caviness V., Jr Neocortical histogenesis in normal and reeler mice: a developmental study based on [3H]thymidine autoradiography. Brain Res. 1982;256:293–302. doi: 10.1016/0165-3806(82)90141-9. [DOI] [PubMed] [Google Scholar]
- 10.Chae T, Kwon Y, Bronson R, Dikkes P, Li E, Tsai L. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18:29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- 11.Chenn A, McConnell S. Cleavage orientation and symmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell. 1995;82:631–641. doi: 10.1016/0092-8674(95)90035-7. [DOI] [PubMed] [Google Scholar]
- 12.D'Archangelo G, Miao G, Chen S, Soares H, Morgan J, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 13.del Rio J, Martinez A, Fonseca M, Auladell C, Soriano E. Glutamate-like immunoreactivity and fate of Cajal-Retzius cells in the murine cortex as identified with calretinin antibody. Cereb Cortex. 1995;5:13–21. doi: 10.1093/cercor/5.1.13. [DOI] [PubMed] [Google Scholar]
- 14.Dobyns W, Truwit C. Lissencephaly and other malformations of cortical development. Neuropediatrics. 1995;26:132–147. doi: 10.1055/s-2007-979744. [DOI] [PubMed] [Google Scholar]
- 15.Faulkner N, Dujardin D, Tai C-Y, Vaughan K, O'Connell C, Wang Y, Vallee R. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat Cell Biol. 2000;2:784–791. doi: 10.1038/35041020. [DOI] [PubMed] [Google Scholar]
- 16.Feng Y, Olson E, Stukenberg P, Flanagan L, Kirschner M, Walsh C. LIS1 regulates CNS lamination by interacting with mNudE, a central component of the centrosome. Neuron. 2000;3:665–679. doi: 10.1016/s0896-6273(00)00145-8. [DOI] [PubMed] [Google Scholar]
- 17.Fleck M, Hirotsune S, Gambello M, Phillips-Tansey E, Suares G, Mervis R, Wynshaw-Boris A, McBain C. Hippocampal abnormalities and enhanced excitability in a murine model of human lissencephaly. J Neurosci. 2000;20:2439–2450. doi: 10.1523/JNEUROSCI.20-07-02439.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Geiser J, Schott EJ, Kingsbury TJ, Cole NB, Totis LJ, Bhattacharyya G, He L, Hoyt MA. Saccharomyces cerevisiae genes required in the absence of the CIN8-encoded spindle motor act in functionally diverse mitotic pathways. Mol Biol Cell. 1997;8:1035–1050. doi: 10.1091/mbc.8.6.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta A, Tsai L, Wynshaw-Boris A. Life is a journey: a genetics look at neocortical development. Nat Rev Genet. 2002;3:342–355. doi: 10.1038/nrg799. [DOI] [PubMed] [Google Scholar]
- 20.Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. Miller-Dieker lissencephaly gene encodes a subunit of brain platelet-activating factor acetylhydrolase. Nature. 1994;370:216–218. doi: 10.1038/370216a0. [DOI] [PubMed] [Google Scholar]
- 21.Hattori M, Aoki J, Arai H, Inoue K. PAF and PAF acetylhydrolase in the nervous system. J Lipid Med Cell Signal. 1996;14:99–102. doi: 10.1016/0929-7855(96)00514-7. [DOI] [PubMed] [Google Scholar]
- 22.Hendzel M, Wei Y, Mancini M, Van Hooser A, Ranalli T, Brinkley B, Bazett-Jones D, Allis A. Mitosis-specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348–360. doi: 10.1007/s004120050256. [DOI] [PubMed] [Google Scholar]
- 23.Herz J. The LDL receptor gene family: (un)expected signal transducers in the brain. Neuron. 2001;29:571–581. doi: 10.1016/s0896-6273(01)00234-3. [DOI] [PubMed] [Google Scholar]
- 24.Hirotsune S, Pack S, Chong S, Robbins C, Pavan W, Ledbetter D, Wynshaw-Boris A. Genomic organization of the murine Miller-Dieker/lissencephaly region: conservation of linkage with the human region. Genome Res. 1997;7:625–634. doi: 10.1101/gr.7.6.625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirotsune S, Fleck M, Gambello M, Bix G, Chen A, Clark G, Ledbetter D, McBain C, Wynshaw-Boris A. Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat Genet. 1998;19:333–339. doi: 10.1038/1221. [DOI] [PubMed] [Google Scholar]
- 26.Ho Y, Swenson L, Derewenda U, Serre L, Wei Y, Dauter Z, Hattori M, Adachi T, Aoki J, Arai H, Inoue K, Derewenda Z. Brain acetylhydrolase that inactivates platelet-activating factor is a G-protein-like trimer. Nature. 1997;385:89–93. doi: 10.1038/385089a0. [DOI] [PubMed] [Google Scholar]
- 27.Howell B, Hawkes R, Soriano P, Cooper J. Neuronal position in the developing brain is regulated by mouse disabled-1. Nature. 1997;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- 28.Kaufman M. The atlas of mouse development. Academic; San Diego: 1992. [Google Scholar]
- 29.LaMonte B, Wallace K, Holloway B, Shelly S, Ascano J, Tokito M, Van Winkle T, Howland D, Holzbaur E. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- 30.Letourneau P, Condic M, Snow D. Interactions of developing neurons with extracellular matrix. J Neurosci. 1994;14:915–928. doi: 10.1523/JNEUROSCI.14-03-00915.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Z, Xie T, Steward R. Lis1, the Drosophila homolog of a human lissencephaly disease gene, is required for germline cell division and oocyte differentiation. Development. 1999;121:4477–4488. doi: 10.1242/dev.126.20.4477. [DOI] [PubMed] [Google Scholar]
- 32.Liu Z, Steward R, Luo L. Drosophila Lis1 is required for neuroblast proliferation, dendritic elaboration and axonal transport. Nat Cell Biol. 2000;2:776–783. doi: 10.1038/35041011. [DOI] [PubMed] [Google Scholar]
- 33.Lo Nigro C, Chong C, Smith A, Dobyns W, Carrozzo R, Ledbetter D. Point mutations and an intragenic deletion in LIS1, the lissencephaly causative gene in isolated lissencephaly sequence and Miller-Dieker syndrome. Hum Mol Genet. 1997;6:157–164. doi: 10.1093/hmg/6.2.157. [DOI] [PubMed] [Google Scholar]
- 34.Marin-Padilla M. Early prenatal ontogenesis of the cerebral cortex (neocortex) of the Felix domestica, a Golgi study. I. The primordial neocortical organization. Z Anat Entwicklungsgesch. 1971;134:117–145. doi: 10.1007/BF00519296. [DOI] [PubMed] [Google Scholar]
- 35.McConnell S. Constructing the cerebral cortex: neurogenesis and fate determination. Neuron. 1995;15:761–768. doi: 10.1016/0896-6273(95)90168-x. [DOI] [PubMed] [Google Scholar]
- 36.McConnell S, Ghosh A, Shatz C. Subplate neurons pioneer the first axon pathway from the cerebral cortex. Science. 1989;245:978–982. doi: 10.1126/science.2475909. [DOI] [PubMed] [Google Scholar]
- 37.McConnell S, Ghosh A, Shatz C. Subplate pioneers and the formation of descending connections from the cerebral cortex. J Neurosci. 1994;14:1892–1907. doi: 10.1523/JNEUROSCI.14-04-01892.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Menezes J, Luskin M. Expression of neuron-specific tubulin defines a novel population in the proliferative layers of the developing telencephalon. J Neurosci. 1994;14:5399–5416. doi: 10.1523/JNEUROSCI.14-09-05399.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Misson J, Edwards M, Yamamoto M, Caviness VS., Jr Identification of radial glial cells within the developing murine central nervous system: studies based upon a new immunohistochemical marker. Brain Res Dev Brain Res. 1988;44:95–108. doi: 10.1016/0165-3806(88)90121-6. [DOI] [PubMed] [Google Scholar]
- 40.Moro Balbas J, Gato A, Alonso M, Barboas E. Local increase level of chondroitin sulfate induces changes in the rhombencephalic neural crest migration. Int J Dev Biol. 1998;42:207–216. [PubMed] [Google Scholar]
- 41.Morris N. Nuclear migration: from fungi to the mammalian brain. J Cell Biol. 2000;148:1097–1101. doi: 10.1083/jcb.148.6.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nadarajah B, Brunstrom J, Grutzendler J, Wong R, Pearlman A. Two modes of radial migration in early development of the cerebral cortex. Nat Neurosci. 2001;4:143–150. doi: 10.1038/83967. [DOI] [PubMed] [Google Scholar]
- 43.Niethammer M, Smith D, Ayala R, Peng J, Ko J, Lee M, Morabito M, Tsai L. NUDEL is a novel Cdk5 substrate that associates with LIS1 and cytoplasmic dynein. Neuron. 2000;28:697–711. doi: 10.1016/s0896-6273(00)00147-1. [DOI] [PubMed] [Google Scholar]
- 44.Oppenhein R. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 45.Peterfy M, Hozier J, Hall B, Gyuris T, Peterfy K, Takecs L. Localization of the mouse lissencephaly-1 gene to mouse chromosome 11B3, in close proximity to D11Mit65. Somat Cell Mol Genet. 1995;21:345–349. doi: 10.1007/BF02257469. [DOI] [PubMed] [Google Scholar]
- 46.Peterfy M, Gyuris T, Grosshans D, Cuaresma C, Takacs L. Cloning and characterization of cDNAs and the gene encoding the mouse platelet-activating factor acetylhydrolase Ib alpha subunit/lissencephaly-1 protein. Genomics. 1998;47:200–206. doi: 10.1006/geno.1997.5121. [DOI] [PubMed] [Google Scholar]
- 47.Rakic P. Mode of cell migration to the superficial layers of fetal monkey cortex. J Comp Neurol. 1972;145:61–84. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- 48.Rakic P. Specification of cerebral cortical areas. Science. 1988;241:170–176. doi: 10.1126/science.3291116. [DOI] [PubMed] [Google Scholar]
- 49.Reid C, Walsh C. Early development of the cerebral cortex. In: Mize R, Erzurumlu R, editors. Progress in brain research. Elsevier; New York: 1996. pp. 17–30. [DOI] [PubMed] [Google Scholar]
- 50.Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns W, Caskey C, Ledbetter D. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- 51.Rice D, Curran T. Mutant mice with scrambled brains: understanding the signaling pathways that control cell positioning in the CNS. Genes Dev. 1999;13:2758–2773. doi: 10.1101/gad.13.21.2758. [DOI] [PubMed] [Google Scholar]
- 52.Rickman M, Chronwall B, Wolf J. On the development of nonpyramidal neurons and axons outside the cortical plate: the early marginal zone as a pallial anlage. Anat Embryol. 1997;151:285–307. doi: 10.1007/BF00318931. [DOI] [PubMed] [Google Scholar]
- 53.Sapir T, Elbaum M, Reiner O. Reduction of microtubule catastrophe events by LIS1, platelet-activating factor acetylhydrolase subunit. EMBO J. 1997;16:6977–6984. doi: 10.1093/emboj/16.23.6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sasaki S, Shionoya A, Ishida M, Gambello M, Yingling J, Wynshaw-Boris A, Hirotsune S. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 2000;28:681–696. doi: 10.1016/s0896-6273(00)00146-x. [DOI] [PubMed] [Google Scholar]
- 55.Sheppard A, Pearlman A. Extracellular matrix in early cortical development. Prog Brain Res. 1996;108:117–134. [PubMed] [Google Scholar]
- 56.Smith D, Niethammer M, Ayala R, Zhou Y, Gambello M, Wynshaw-Boris A, Tsai L. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat Cell Biol. 2000;2:767–775. doi: 10.1038/35041000. [DOI] [PubMed] [Google Scholar]
- 57.Sorger P, Dobles M, Tournebize R, Hyman A. Coupling cell division and cell death to microtubule dynamics. Curr Opin Cell Biol. 1997;9:807–814. doi: 10.1016/s0955-0674(97)80081-6. [DOI] [PubMed] [Google Scholar]
- 58.Stafforini D, McIntyre T, Zimmerman G, Prescott S. Platelet-activating factor acetylhydrolases. J Biol Chem. 1997;272:17859–17898. doi: 10.1074/jbc.272.29.17895. [DOI] [PubMed] [Google Scholar]
- 59.Super H, Del Rio J, Martinez A, Perez-Sust P, Soriano E. Disruption of neuronal migration and radial glia in the developing cerebral cortex following ablation of Cajal-Retzius cells. Cereb Cortex. 2000;10:602–613. doi: 10.1093/cercor/10.6.602. [DOI] [PubMed] [Google Scholar]
- 60.Takahashi T, Nowakowski R, Caviness VS., Jr BUdR as an S-phase marker for quantitative studies of cytokinetic behaviour in the murine cerebral ventricular zone. J Neurocytol. 1992;21:185–197. doi: 10.1007/BF01194977. [DOI] [PubMed] [Google Scholar]
- 61.Takahashi T, Nowakowski R, Caviness VS., Jr The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J Neurosci. 1995a;15:6046–6057. doi: 10.1523/JNEUROSCI.15-09-06046.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takahashi T, Nowakowski R, Caviness VS., Jr Early ontogeny of the secondary proliferative population of the embryonic murine cerebral wall. J Neurosci. 1995b;15:6058–6068. doi: 10.1523/JNEUROSCI.15-09-06058.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Takahashi T, Nowakowski R, Caviness VS., Jr Interkinetic and migratory behavior of a cohort of neocortical neurons arising in the early embryonic murine cerebral wall. J Neurosci. 1996;16:5762–5776. doi: 10.1523/JNEUROSCI.16-18-05762.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Thomaidou D, Mione M, Cavanagh J, Parnavelas J. Apoptosis and its relation to the cell cycle in the developing cerebral cortex. J Neurosci. 1997;17:1075–1085. doi: 10.1523/JNEUROSCI.17-03-01075.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer R, Richardson J, Herz J. Reeler/disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]
- 66.Valverde F, DeCarlos A, Lopez-Mascaraque L. Time of origin and early fate of preplate cells in the cerebral cortex of the rat. Cereb Cortex. 1995;5:483–493. doi: 10.1093/cercor/5.6.483. [DOI] [PubMed] [Google Scholar]
- 67.Walsh C. LISen up! Nat Genet. 1998;19:307–308. doi: 10.1038/1186. [DOI] [PubMed] [Google Scholar]
- 68.Walsh C, Goffinet A. Potential mechanisms of mutations that affect neuronal migration in man and mouse. Curr Opin Genet Dev. 2000;10:270–274. doi: 10.1016/s0959-437x(00)00076-9. [DOI] [PubMed] [Google Scholar]
- 69.Wynshaw-Boris A, Gambello M. LIS1 and dynein motor function in neuronal migration and development. Genes Dev. 2001;15:639–651. doi: 10.1101/gad.886801. [DOI] [PubMed] [Google Scholar]
- 70.Xiang X, Osmani A, Osmani S, Xin M, Morris N. NudF, a nuclear migration gene in Aspergillus nidulans, is similar to the human LIS-1 gene required for neuronal migration. Mol Biol Cell. 1995;6:297–310. doi: 10.1091/mbc.6.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhong W, Feder J, Jiang M, Jan L, Jan Y. Asymmetric localization of the mammalian numb homologue during mouse cortical neurogenesis. Neuron. 1996;17:43–53. doi: 10.1016/s0896-6273(00)80279-2. [DOI] [PubMed] [Google Scholar]