

Abstract
Cluster I neurons of the suprachiasmatic nucleus (SCN), which are thought to be pacemakers supporting circadian activity, fire spontaneous action potentials that are followed by a monophasic afterhyperpolarization (AHP). Using a brain slice preparation, we have found that the AHP has a shorter duration in cells firing at higher frequency, consistent with circadian modulation of the AHP. The AHP is supported by at least three subtypes of KCa channels, including apamin-sensitive channels, iberiotoxin-sensitive channels, and channels that are insensitive to both of these antagonists. The latter KCa channel subtype is involved in rate-dependent regulation of the AHP. Voltage-clamped, whole-cell Ca2+ channel currents recorded from SCN neurons were dissected pharmacologically, revealing all of the major high-voltage activated subtypes: L-, N-, P/Q-, and R-type Ca2+channel currents. Application of Ca2+ channel antagonists to spontaneously firing neurons indicated that predominantly L- and R-type currents trigger the AHP. Our findings suggest that apamin- and iberiotoxin-insensitive KCachannels are subject to diurnal modulation by the circadian clock and that this modulation either directly or indirectly leads to the expression of a circadian rhythm in spiking frequency.
Keywords: suprachiasmatic nucleus, calcium channel, calcium-activated potassium channel, action potential, spontaneous activity, afterhyperpolarization, circadian rhythm
Introduction
The suprachiasmatic nuclei (SCN) of the hypothalamus contain the primary circadian clock that controls various physiological and behavioral rhythms in mammals, including rhythms in body temperature and sleep-wake behavior (Moore and Eichler, 1972; Klein et al., 1991). Single SCN neurons are autonomous pacemaker cells that harbor the fundamental molecular workings of the circadian clock, an interconnected set of transcriptional/translational negative and positive feedback loops that produce coordinated, rhythmic changes in expression of clock genes (Dunlap, 1999; Reppert and Weaver, 2001). In keeping with the role of a 24 hr pacemaker, firing of SCN neurons follows the circadian clock, with higher spike activity during the day and lower activity at night (Inouye and Kawamura, 1979; Gillette, 1991; Jagota et al., 2000). Even an isolated SCN neuron in culture, lacking any synaptic input, will sustain spontaneous and rhythmic firing (Welsh et al., 1995). Such circadian oscillation in firing frequency is known to be critical for transmittal of time information because suppression of action potentials via selective application of tetrodotoxin to the SCN abolishes the circadian rhythm of many organismal behaviors (Schwartz et al., 1987; Earnest et al., 1991; Schwartz, 1991; Newman et al., 1992). Little is known, however, regarding the mechanism that links the molecular clockworks to rhythmic electrophysiological output.
A prerequisite for understanding how the rhythm of the core of the clock is transduced into a rhythm in spike rate is the identification of ion channels responsible for spontaneous, rhythmic firing. Some of these have been identified. Thus it is known that a slowly inactivating Na+ current generates an interspike depolarization that brings the membrane potential to firing threshold (Pennartz et al., 1997). Additional ion channels, including T-type Ca2+ channels, L-type Ca2+ channels, and hyperpolarization-activated cation channels (Ih), may assist in generating the interspike depolarization (Akasu et al., 1993; Jiang et al., 1995; Pennartz et al., 2002).
Other channels involved in spontaneous, rhythmic firing have remained uncharacterized. After action potential repolarization, SCN neurons exhibit an intermediate-duration afterhyperpolarization (AHP) that may contribute to regulation of firing rate. Because the AHP in other spontaneously active central neurons is a key determinant of cellular excitability (Aizenman and Linden, 1999; Bevan and Wilson, 1999) and is subject to modulation by intracellular messengers (Sah, 1996), the AHP represents a potential target for regulation by the circadian clock.
In this study, we have found that the duration of the AHP in SCN neurons firing at higher frequencies is significantly shorter than in more slowly firing neurons, consistent with circadian modulation. To better understand the nature of the changes in the AHP, we undertook a pharmacological analysis of the ion channels supporting the AHP in cluster I SCN neurons, which are the most abundant neuronal type in the SCN and are thought to be important in the output of these nuclei (Pennartz et al., 1998). This analysis classifies KCa channels that underlie the AHP, identifies the Ca2+ channels that trigger the AHP, and highlights particular subtypes of KCa and Ca2+ channels as candidates for circadian regulation.
Materials and Methods
Preparation of brain slices. Hypothalamic slices containing the SCN were obtained from Fisher 344 or Sprague Dawley rats (17–25 d postnatal) that had been maintained on a 12 hr light/dark cycle for at least 1 week before they were killed. Animals were anesthetized with halothane and decapitated, and the brain was quickly removed and placed in ice-cold artificial CSF (ACSF) of the following composition (in mm): 126 NaCl, 3 KCl, 1.5 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 11 glucose, 25.9 NaHCO3, continuously gassed with 95% O2–5% CO2, pH 7.4. Coronal hypothalamic slices (250 μm) were cut on a vibratome and incubated in ACSF at room temperature (∼23–25°C) for at least 45 min before recording. Experiments were performed primarily on neurons taken from rats killed during their subjective day, although a subset of experiments was performed with neurons from subjective night rats. Recordings were made from all regions of the SCN.
Current-clamp recordings. Slices were placed in a recording chamber mounted on the stage of an upright microscope (Nikon Eclipse E600 FN). Single SCN neurons in slices were visualized using a water-immersion objective lens [40×, 0.8 numerical aperture (NA)], differential interference contrast optics, and a 0.9 NA condenser. An infrared-sensitive video camera was used to display the field of view on a video monitor. To measure the AHP, action potential firing rate, and other properties of membrane potential changes in SCN neurons, recordings were made using the current-clamp mode of the Axopatch 200B amplifier. The bathing solution, ACSF, was bubbled continuously with 95% O2–5% CO2. Recordings were obtained at room temperature (20–23°C). For Co2+ substitution experiments, a HEPES-based ACSF was used that contained (in mm): 126 NaCl, 3 KCl, 2.4 CaCl2 (or CoCl2), 1.5 MgCl2, 26 HEPES, 11 glucose, pH 7.4, bubbled with 100% O2. The pipette solution contained (in mm): 135 potassium gluconate, 10 KCl, 10 HEPES, 0.5 EGTA, 2 MgATP, pH 7.4. The pipette was mounted on a piezoelectric manipulator bolted onto the fixed stage of the microscope, and whole-cell recordings were obtained at room temperature using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA). Data were filtered at 2 kHz (−3 dB; four-pole low-pass Bessel filter integral to the Axopatch 200B amplifier) and sampled at 5 kHz using an ITC-16 analog-to-digital converter (Instrutech, Great Neck, NY) and Pulse software (HEKA, distributed by Instrutech). In a few experiments, 10 mm BAPTA was substituted for EGTA. A liquid junction potential of −12.5 mV was measured for the solutions used in current-clamp recording (Neher, 1992); data were not adjusted for this junction potential.
For experiments examining the contribution of ryanodine receptors to the control of AHP amplitude, perforated-patch recording with amphotericin B (Sigma, St. Louis, MO) was used. The perforated-patch method was used in these experiments because it facilitated the long-duration, stable recordings that were needed to study the effects of the slowly acting, bath-applied ryanodine (Vilchis et al., 2000). A stock solution of amphotericin B (1 mg/ml, in dimethylsulfoxide) was diluted into a potassium gluconate-based pipette solution (1:200) that was otherwise identical to that used in the ruptured-patch current-clamp recordings. The patch pipette tip was filled with amphotericin B-free potassium gluconate-based solution via tip immersion, and then the pipette was back-filled with the amphotericin B-containing solution. After obtaining a high-resistance seal between pipette and cell membrane, access resistance was monitored by regular examination of the current response to a voltage step (+10 mV). Because amphotericin B partitioned into the pipette-encircled patch, access resistance dropped within 10 min to a stable level of ∼40 MΩ.
Voltage-clamp of Ca2+ channel currents. For recording whole-cell Ba2+ currents, the extracellular solution contained (in mm): 100 NaCl, 3 KCl, 0.15 MgCl2, 5 BaCl2, 17 tetraethylammonium chloride (TEA-Cl), 0.5 4-aminopyridine, 5 CsCl, 26 HEPES, 10 glucose, 0.03 bicuculline methiodide, and 0.001 tetrodotoxin, pH 7.4. A reservoir containing this solution was bubbled continuously with 100% oxygen, and a gravity-driven system was used to perfuse the oxygenated solution through the recording chamber at 3–5 ml/min. Whole-cell recording pipettes pulled from borosilicate glass (GC120F-10; Warner Instruments, Hamden, CT) had resistances of 4–6 MΩ when filled with a solution containing (in mm): 120 TEA-Cl, 9 EGTA, 2 MgATP, and 9 HEPES, pH 7.4. After seal formation (Rseal > 3 GΩ), neurons were allowed to stabilize for 300–500 sec before data were recorded. Series resistance compensation and capacitance transient cancellation were performed using the circuitry of the amplifier. Data were filtered at 2 kHz and sampled at 4 kHz. Linear leak and residual capacitance currents were removed on-line by subtracting scaled and inverted current responses to hyperpolarizing voltage steps of one-quarter the amplitude of the depolarizing test pulse. A liquid junction potential of +1.4 mV was measured for the solutions used in voltage-clamp recording (Neher, 1992); the reported data have not been corrected for this junction potential. All experiments were performed at room temperature.
In all SCN whole-cell voltage-clamp recordings, a series resistance of typically 20–60 MΩ was compensated by ∼80%. Whole-cell Ba2+ current amplitude ranged up to ∼300 pA, so in the well clamped compartment of the cell the maximal error in command voltage attributable to pipette series resistance was estimated as 1.2–3.6 mV for the largest (300 pA) currents. Although SCN neurons are electrotonically compact compared with many central neurons, Ba2+ currents originating from distal processes of SCN neurons were unlikely to have been adequately clamped. An indication of this was provided by examination of the kinetics of Ca2+ channel currents. Comparison of activation and deactivation rates of whole-cell Ba2+ currents obtained from slice recordings with those recorded from dissociated SCN neurons [prepared as described by Huang (1993)] showed that activation times measured in slices (τact = 2.4 ± 0.2 msec;n = 12; −10 mV) were longer than those measured in dissociated neurons (τact = 1.1 ± 0.3 msec; n = 5; −10 mV). Measured deactivation times were also significantly longer in duration for slices: τdeact = 1.7 ± 0.2 msec in slices versus 0.4 ± 0.3 msec in dissociated neurons. As a consequence of the limited quality of voltage clamp in the slice recording configuration, we restricted our analysis of Ca2+ channel currents in SCN slices to peak current amplitudes: the relatively slow rates of activation and inactivation for Ca2+ channels make peak current amplitude somewhat less sensitive to clamp quality than other parameters.
Pharmacological analysis of channels. Channel antagonists used to identify KCa channels in SCN neurons are in general highly specific (Gribkoff et al., 1997; Vergara et al., 1998; Kaczorowski and Garcia, 1999; Sah and Davies, 2000). Iberiotoxin (dose = 100 nm; IC50 = 1–5 nm) (Galvez et al., 1990; Candia et al., 1992) is a very selective blocker of type I, but not type II, BKCa channels (Gribkoff et al., 1997; Meera et al., 2000). Apamin (dose = 100 nm; IC50 = 0.06–1 nm) (Hughes et al., 1982; Blatz and Magleby, 1986) blocks many, but perhaps not all, neuronal SKCa channels. Charybdotoxin (dose = 100 nm; IC50 = 10 nm) (Anderson et al., 1988), another blocker of type I but not type II BKCa channels, was tested in some experiments. Interpretation of the effects of this latter blocker was complicated by the fact that it is, in addition, a potent antagonist of some intermediate conductance KCa channels (IKCa) and some voltage-gated potassium channels. At the doses used for iberiotoxin and apamin, essentially complete block of sensitive channels was expected, whereas insensitive KCasubtypes and other ion channels would not have been affected.
The stability of Ca2+ channel currents recorded from slices was advantageous for pharmacological analysis of Ca2+ channel types present in SCN neurons. During the first 300–500 sec after breakthrough, voltage-clamped Ba2+ currents carried by Ca2+ channels usually increased in amplitude and then became nearly constant; peak current amplitude diminished by only 2 ± 0.02% over the next 420 sec (n = 9 neurons). Consequently, Ca2+ channel antagonists were applied after a stabilization period of generally 300–500 sec after the start of whole-cell recording. To achieve a steady level of block, channel blocking agents were usually applied for 180–300 sec via the bath perfusion system (3–5 ml/min). Delays in onset of antagonist action of up to ∼50 sec were often observed, reflecting the slow solution exchange time and the time needed for antagonists to diffuse into the tissue. Considering the time needed to achieve steady block relative to the rate of run-down in peak current amplitude, run-down had only a minor effect on our measurements.
Although the Ca2+ channel antagonists used here are not ideal in their specificity, these blockers were nonetheless effective in identifying Ca2+channel isoforms that activate the action potential AHP. The classical L channel antagonist nimodipine (dose = 10 μm; IC50 = 50 nm under our recording conditions) (Marchetti et al., 1995), the N channel antagonist ω-conotoxin-GVIA (ω-CTx-GVIA) (dose = 3 μm; IC50 ≤ 0.7 nm) (Boland et al., 1994), and the P channel antagonist ω-agatoxin-IVA (ω-Aga-IVA) (dose = 200 nm; IC50 = 1.5 nm) (Mintz et al., 1992) were applied by bath perfusion at concentrations 133–4300× IC50 values and therefore were expected to block at least 99% of current carried by these Ca2+ channel subtypes. Nonclassical L channels based on the CaV1.3 (α1D) subunit may not have been fully blocked by 10 μm nimodipine (Xu and Lipscombe, 2001). Q-type channels are blocked by ω-Aga-IVA, but these channels are less sensitive to this antagonist, so that ∼70% of Q-type current was expected to be blocked at the dose used (dose = 200 nm; IC50 = 90 nm) (Randall and Tsien, 1995). However, there was no significant difference between a mixture of blockers containing ω-Aga-IVA and one containing ω-conotoxin-MVIIC (ω-CTx-MVIIC) (dose = 3 μm) suggesting that the contribution of Q-type current is not large in SCN neurons. R-type current was defined as the Ba2+ current not blocked by a combination of nimodipine, ω-CTx-GVIA, ω-Aga-IVA, and mibefradil. We further dissected the R-type current according to block by the toxin SNX-482, which blocks α1E-based Ca2+ channels (CaV2.3; dose = 200 nm; IC50 = 15 nm) (Newcomb et al., 1998) and the less specific blocker Ni2+(dose = 30 μm; IC50 = 4–30 μm) (Tottene et al., 1996; Zamponi et al., 1996). In this concentration range, Ni2+ would not be expected to inhibit N- or P/Q-type currents but would block a portion of L-type currents (IC50 = 60 μm) (Zamponi et al., 1996). Ni2+ also blocks one of the three T-type Ca2+channel isoforms, but SCN neurons do not express the Ni2+-sensitive T channel isoform CaV3.3 (α1H) (Lee et al., 1999; Talley et al., 1999). Because of the difficulty in accurately voltage clamping SCN neurons, we did not attempt to estimate the contribution of T channel current in these cells. The modestly selective blocker mibefradil was included in the mixture that was used to estimate the contribution of R-type Ca2+ channels in whole-cell Ba2+ currents and to examine the contribution of Ca2+ channel subtypes to AHP activation. By using interpolated IC50 values appropriate for our recording conditions (∼100–800 nm), a 500 nm dose of mibefradil was expected to block 38–83% of T current in SCN neurons (McDonough and Bean, 1998; Martin et al., 2000). Higher doses of mibefradil were not used because this drug interacts with other Ca2+ channel subtypes, albeit with lower potency (Bezprozvanny and Tsien, 1995;Martin et al., 2000).
Stock solutions of nimodipine, ryanodine, and picrotoxin were made by dissolving these ion channel antagonists in 100% ethanol. Stock solutions for all other ion channel antagonists were made using distilled water. Stocks were stored at −20°C and in the dark. (−)-Bicuculline methiodide was not prepared as a stock solution because of its chemical instability; instead this agent was dissolved in the bath solution on a daily basis, before the start of experiments.
Channel antagonists were obtained from the following sources: picrotoxin, (−)-bicuculline methiodide, ω-CTx-GVIA, apamin, iberiotoxin, charybdotoxin, and ryanodine from Sigma; ω-Aga-IVA, ω-CTx-MVIIC, and SNX-482 from Peptide Institute (Osaka, Japan); nimodipine from Research Biochemicals (Natick, MA); and mibefradil was a generous gift from Dr. Jean-Paul Cloze and Dr. Eric Ertel (F. Hoffmann-La Roche, Basel, Switzerland).
Mean values are reported together with their SEs, and all error bars indicate SEs of the mean. The number of experiments, n, indicates in all cases the number of neurons studied. Student'st test was used to determine statistical significance.
Results
Spontaneous neuronal firing rate correlates with AHP duration
SCN neurons spontaneously fire action potentials over a range of frequencies (0–15 Hz), with average firing rates being higher during the daytime and lower during the nighttime (Inouye and Kawamura, 1979;Gillette, 1991; Jagota et al., 2000). Clock cells are not tightly synchronized with one another, however, and the circadian time at which peak firing occurs can be considerably out of phase among SCN neurons. We therefore used comparisons of action potential waveforms recorded from cells firing spontaneously at different rates, regardless of circadian time, as a strategy to identify components of the action potential that might be regulated in a circadian manner. From these comparisons, we identified an altered AHP waveform as a major factor regulating firing frequency: the AHP became shorter in duration as spike frequency increased (Fig.1A). In addition, the action potential width was significantly broader in slower firing neurons, but other action potential properties remained unchanged: the depolarizing ramp preceding the action potential upstroke, interspike membrane potential, action potential height, and maximum hyperpolarization after the action potential were not significantly different between fast and slow firing neurons (Table1).
Fig. 1.
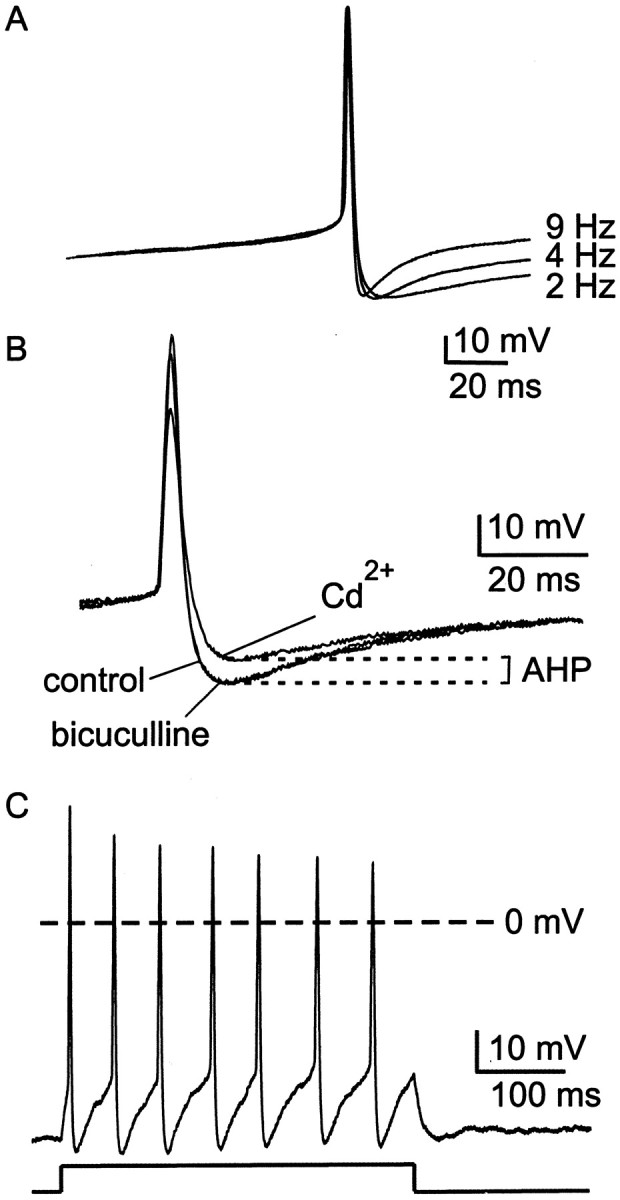
Firing properties of cluster I SCN neurons under whole-cell current clamp. A, Action potential waveforms, averaged from four to six spikes, from three different cells firing at the rates indicated. Note that in cells with faster firing rates, the duration of the AHP was briefer. Records of action potentials were shifted along the voltage axis to superimpose action potential threshold. B, Single action potentials recorded in control conditions, in the presence of 30 μm bicuculline methiodide and in the presence of 30 μmCd2+ are superimposed. Cd2+ was used as a measure of the Ca2+-dependent AHP. Bicuculline did not cause a reduction in the amplitude of the AHP.C, Positive current injection (20 pA, as indicated below current-clamp record), evoked a train of action potentials that did not exhibit spike frequency adaptation. In many, but not all neurons, there was a progressive diminution in the amplitude of action potentials that followed the initial spike.
Table 1.
Membrane properties of slow- and fast-firing cluster I neurons
| Depolarizing ramp (mV/msec) | Interspike potential (mV) | Spike width (msec) | Spike amplitude (mV) | Maximum hyperpolarization (mV) | AHP t½(msec) | Cd2+-subtracted AHP | IBTX-subtracted AHP | |||
|---|---|---|---|---|---|---|---|---|---|---|
| t½ (msec) | Amplitude (mV) | t½ (msec) | Amplitude (mV) | |||||||
| Number of cells (n) | 9 | 9 | 9 | 9 | 9 | 9 | 6 | 6 | 5 | 5 |
| Slow rate (<3 Hz) | 0.23 ± 0.01 | −41.3 ± 1.9 | 4.0 ± 0.1 | 82.8 ± 1.7 | −53.8 ± 1.6 | 37.3 ± 5.0 | 7.1 ± 0.9 | 11.0 ± 3.8 | 2.8 ± 0.8 | 17.4 ± 3.3 |
| Fast rate (>6 Hz) | 0.25 ± 0.03 | −44.7 ± 2.3 | 3.0 ± 0.1 | 77.7 ± 3.6 | −58.1 ± 2.2 | 17.8 ± 0.9 | 3.1 ± 0.4 | 26.0 ± 2.9 | 3.1 ± 0.7 | 10.4 ± 2.0 |
| p | NS | NS | 0.0001 | NS | NS | 0.001 | 0.003 | 0.01 | NS | NS |
Membrane properties were compared between slow (<3 Hz) and fast (>6 Hz) spontaneously firing cluster I SCN neurons. The depolarizing ramp was measured as the rate of voltage change (mV/msec) over the 50 msec preceding action potential threshold. Interspike potential (VIP) was estimated as the plateau region between action potentials. For fast-spiking neurons in which interspike plateaus were mostly brief, VIP was estimated using the longer interspike intervals recorded from these irregularly firing neurons. Spike width was measured at action potential threshold. Spike amplitude was measured from VIP to maximum depolarization. Maximum hyperpolarization reached during the AHP was measured from 0 mV. t½ for the AHP and Cd2+-subtracted AHP was estimated as the half-decay time. For measurements of Cd2+-subtracted AHPs, slow-firing cells included those firing at <5 Hz. Statistical significance (pvalues) for differences between fast- and slow-firing neurons is indicated at the bottom of each column, with p ≥ 0.05 considered not significant (NS).
Under current-clamp conditions, action potential discharge was followed by a monophasic AHP in the majority of neurons (98 of 110) studied (Fig. 1A,B). These kinds of neurons have been classified previously as cluster I neurons (Pennartz et al., 1998). Cells with a biphasic AHP (cluster II and III neurons) were not included in our analysis. Identification of cluster I neurons was confirmed by testing for the absence of spike frequency adaptation during a train of action potentials evoked by injection of steady positive current (Fig. 1C) (Pennartz et al., 1998).
In this work, we defined AHP as the Ca2+-dependent component of the hyperpolarization that followed each action potential (Fig.1B). We measured the Ca2+-dependent component by blocking Ca2+ entry with 30 μm Cd2+, which blocks virtually all Ca2+ channel current in SCN neurons (93 ± 2%; n = 10). The average difference between maximum hyperpolarization before and after Cd2+ application was 10.0 ± 2.1 mV (n = 10). We compared this result with experiments in which external Ca2+ was replaced with Co2+. Using this method, the amplitude of the Ca2+-dependent AHP was 11.3 ± 1.0 mV (n = 5), which was not significantly different from measurements using Cd2+ (p > 0.1). We also tested the ability of a Ca2+ chelator (10 mm BAPTA, included in the whole-cell patch pipette solution) to occlude the effect of Cd2+. Consistent with expectation, Cd2+ had a negligible effect on the AHP in cells loaded with BAPTA (average difference between maximum hyperpolarization before and after Cd2+application was 2.6 ± 0.6 mV; n = 9), indicating that the Ca2+-dependent AHP was abolished by high intracellular BAPTA. These results indicate that 30 μm Cd2+ blocks the entry of Ca2+ required for activation of KCa channels and that Cd2+ block provides a practical index of AHP amplitude.
In most experiments, bicuculline methiodide (30 μm) or picrotoxin (100 μm) was used to block GABAA receptors because SCN neurons make extensive GABAergic connections with one another (Strecker et al., 1997). Although bicuculline methiodide is known to block some kinds of KCa channels (Khawaled et al., 1999), in the work described here, no significant reduction in AHP amplitude was observed after application of bicuculline methiodide (Fig. 1B) or picrotoxin. In the presence of bicuculline methiodide, the AHP averaged 7.8 ± 0.4 mV (n = 7), and in the presence of picrotoxin the AHP averaged 6.7 ± 0.8 mV (n = 5). These values were not significantly different from the AHP amplitude measured in the absence of GABAA receptor antagonists (p > 0.1). In reporting the results of subsequent experiments, we have therefore combined experimental results from slices treated with bicuculline methiodide, with picrotoxin, or with no GABAA receptor antagonist.
To begin probing for factors controlling AHP duration and firing frequency, we examined the effects of blocking Ca2+ influx on AHP waveform in neurons firing at different spontaneous rates (Fig.2). In the example shown in Figure2A, block of Ca2+ influx by Cd2+ had a much greater effect on the AHP in a rapidly firing neuron (11 Hz, right) than in a slowly firing neuron (1.3 Hz, left). Subtraction of the action potential waveform recorded in the presence of Cd2+ from that recorded in control conditions (Fig. 2A, inset) revealed that the Ca2+-dependent AHP decayed more quickly in the faster firing neuron. Paradoxically, the Ca2+-dependent AHP was also larger in amplitude in the faster firing neuron. Among neurons firing at different spontaneous rates, the duration and amplitude of their Cd2+-subtracted AHP waveforms were highly correlated with neuronal firing rate (Fig. 2B,C).
Fig. 2.

Changes in the Ca2+-dependent AHP over a range of firing frequencies. A, Comparison of two cells, one firing at 1.3 Hz (left panel) and one firing at 11 Hz (right panel). After several minutes of spontaneous firing in control conditions, 30 μm Cd2+ was applied to slices. Ensemble-averaged action potential waveforms (4–6 action potentials for each condition) are shown superimposed. Insets show subtraction of waveforms recorded in 30 μmCd2+ from control waveforms (Cd2+-subtracted AHP). Solid line in the inset indicates 0 mV. Calibration: 10 mV, 10 msec.B, Half-time for decay of the Cd2+-subtracted AHP as a function of spontaneous firing frequency. For each neuron analyzed, decay half-time (t1/2) was measured as the time from maximum hyperpolarization to 50% of that value, and the measuredt1/2 value was plotted versus the spontaneous firing frequency in that particular neuron. Thet1/2 versus frequency data were fit with a linear regression, using a maximum likelihood estimate. Correlation coefficient was r = −0.67. C, Amplitude of the Cd2+-subtracted AHP as a function of spontaneous firing frequency. Amplitude of the subtracted AHP was measured from maximum hyperpolarization to 0 mV. Correlation coefficient for the regression fit was r = 0.66.
Ca2+-sensitive K+ channels underlying the AHP of cluster I neurons
Subtype-specific antagonists were used to identify the kinds of KCa channels that produce the AHP in cluster I SCN neurons. In this procedure, blockers of various kinds of KCa channels were applied to spontaneously firing SCN neurons, and after establishment of a stable reduction in AHP amplitude, the AHP was completely blocked by applying 30 μm Cd2+. Percentage reduction in AHP amplitude was calculated according to the equation: percentage reduction = [(AHPcontrol − AHPKCa-antagonist)/(AHPcontrol− AHPCd)] × 100%. As shown in Figure3A, iberiotoxin (100 nm), a blocker of certain large conductance KCa channels (BKCachannels), reduced AHP amplitude by ∼40%. A peptide antagonist similar to iberiotoxin in action, charybdotoxin (100 nm), reduced AHP amplitude by a roughly similar amount (28 ± 12%; range, 0–58%; n = 6; data not shown). Also illustrated in Figure 3A is the effect on AHP amplitude of apamin (100 nm), a blocker of certain small conductance KCa channels (SKCa channels): apamin reduced AHP amplitude by ∼20%. Coapplication of apamin and iberiotoxin reduced AHP amplitude by only ∼55%, suggesting that SCN neurons possess some KCa channels that are insensitive to both apamin and iberiotoxin. Because 30 μmCd2+ may also have actions on channels other than voltage-gated Ca2+ channels (Bekkers, 2000), however, we tested the prediction that block of the AHP by apamin and iberiotoxin would be smaller than block by a mixture designed to block Ca2+ channels, including L-, N-, P/Q-, R-, and T-type Ca2+channels. Figure 3B shows that the Ca2+ channel mixture further reduced AHP amplitude after application of the combination of apamin and iberiotoxin, consistent with the idea that the AHP of SCN neurons is supported by at least three subtypes of KCachannels: apamin-sensitive channels, iberiotoxin-sensitive channels, and channels insensitive to both antagonists. In addition, the wide range in percentage block by iberiotoxin or apamin suggests that there is considerable cell-to-cell variability in the expression of KCa channels (Fig. 3, legend).
Fig. 3.
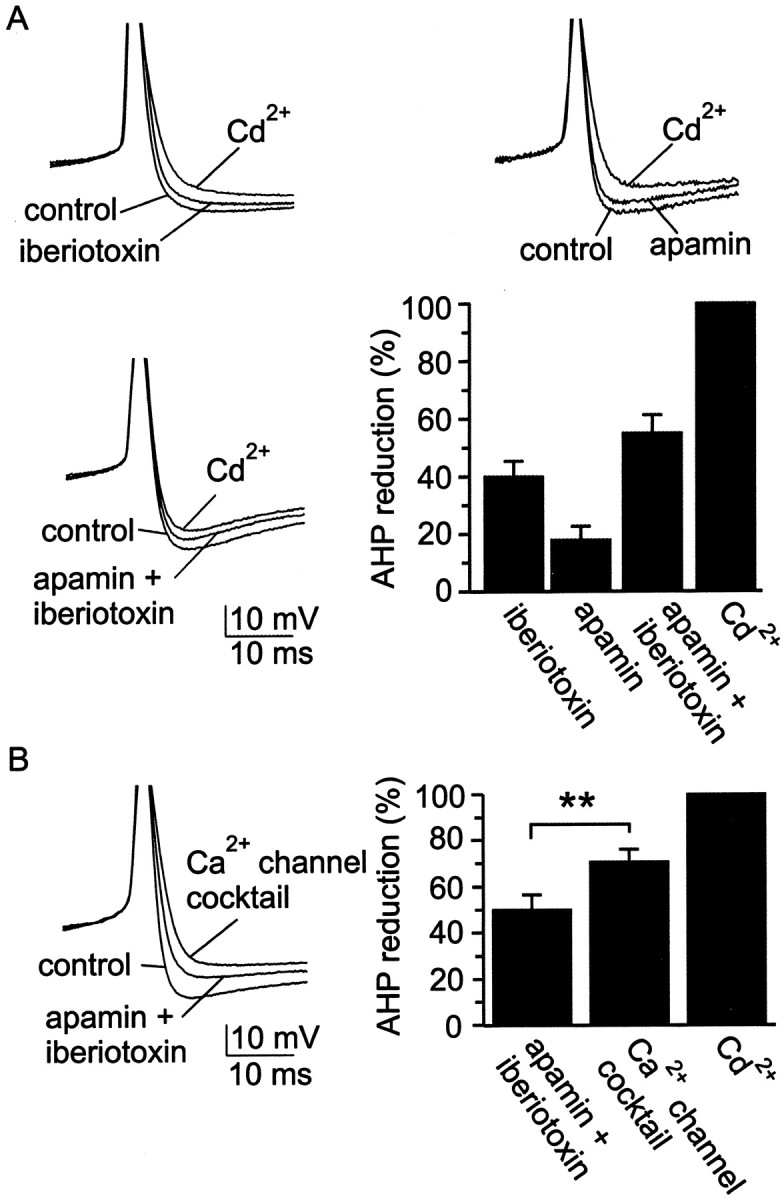
SCN neurons possess at least three classes of KCa channels. A, Example records and group data from experiments using blockers of KCa channels. Spontaneous action potentials were recorded under control conditions and in the presence of iberiotoxin (100 nm), apamin (100 nm), or both of these channel blockers. Cd2+ (30 μm) was added at the end of each experiment to fully block the Ca2+-dependent AHP, thus establishing a baseline from which to estimate the contribution to the AHP of KCa channels that were sensitive to apamin, sensitive to iberiotoxin, or insensitive to both blockers. Superimposed records were always obtained from the same neuron, but each panel of records was recorded from a different neuron. Iberiotoxin reduced AHP amplitude by 40 ± 5%, with a range of 18–55% (n = 6); apamin reduced AHP amplitude by 18 ± 5%, with a range of 8–37% (n = 6); and the combination of apamin and iberiotoxin reduced AHP amplitude by 54 ± 10%, with a range of 32–91% (n = 5).B, Further diminution in the AHP by the Ca2+ channel blocker mixture after application of apamin and iberiotoxin. The Ca2+ channel mixture contained 10 μm nimodipine, 3 μmω-CTx-GVIA, 200 nm ω-Aga-IVA, 30 μmNi2+, and 500 nm mibefradil, which will block most of the Ca2+ entry into SCN neurons (see also Fig. 5). Example action potentials in the various conditions are superimposed on the left, and a bar chartshowing the percentage reduction in AHP amplitude is shown on theright. The reductions in AHP amplitude produced by the two sets of blockers (apamin + iberiotoxin,Ca2+channel cocktail) were statistically different from one another at the p < 0.05 level.
We examined whether the iberiotoxin-sensitive portion of the AHP changed with firing frequency, as observed for the Cd2+-subtracted AHP (Fig. 2). When we applied 100 nm iberiotoxin to SCN neurons firing spontaneously between 0.2 and 8.2 Hz (n = 16) and measured the amplitude and decay kinetics of the iberiotoxin-subtracted potential, we found no correlation between firing rate and either decay half-time (correlation coefficient r = 0.08; Table 1) or amplitude (r = −0.25).
Ca2+ channel subtypes present in neurons of the SCN
Subtypes of Ca2+ channels present in SCN neurons were identified using antagonists effective against particular Ca2+ channel subtypes. In these voltage-clamp experiments, Ba2+ was substituted for Ca2+ as the permeant ion to suppress Ca2+-sensitive currents. Figure 4A–Dillustrates the action of subtype-selective Ca2+ channel antagonists on whole-cell Ba2+ current recorded from voltage-clamped SCN neurons in hypothalamic slices. As shown in Figure4A, the L channel antagonist nimodipine partially blocked Ba2+ current, indicating that L-type Ca2+ channels carried a fraction of whole-cell Ca2+ channel current. For a series of control neurons, total Ba2+current remained stable over the typical duration of experiments (5–15 min), as shown by the data points and dashed line in Figure4A. N-type and P/Q-type Ca2+ channels were also identified in SCN neurons, on the basis of partial block of whole-cell Ba2+ current by high concentrations of ω-CTx-GVIA and ω-Aga-IVA (Fig. 4B,C).
Fig. 4.

Pharmacological identification of Ca2+ channel currents in SCN neurons.A, Plot of peak inward Ba2+ current versus time for a single SCN neuron recorded in a hypothalamic slice. The L channel antagonist nimodipine (10 μm) was applied during the period indicated by the black bar. Nimodipine blocked 29% of total Ba2+ current in this neuron (filled circles). Asterisksconnected by a dotted line mark the mean Ba2+ current recorded from seven neurons to which no antagonist was applied. Inset shows current records obtained at the times indicated by the numbers. The slow time course of the tail currents reflects in part the speed of voltage clamp for SCN neurons in slices. B, Application of the N channel antagonist ω-CTx-GVIA (3 μm) to a neuron in another slice blocked 30% of total Ba2+ current in this example. C, Application of the P/Q channel antagonist ω-Aga-IVA (200 nm) to a neuron in another slice blocked 47% of total Ba2+ current in this example. D, A combination of nimodipine (nimod.) (10 μm), ω-CTx-GVIA (3 μm), ω-Aga-IVA (200 nm), and mibefradil (mibef.) (500 nm) was added to block L-, N-, P/Q-, and T-type Ca2+ channels, revealing a prominent component of antagonist-resistant current (R-type Ca2+ current). In all examples currents were activated every 5 sec by 50 msec voltage steps from −80 to −10 mV (voltage at which peak inward current was obtained).
Figure 4D shows that application of a combination of antagonists designed to block L-, N-, P/Q-, and T-type channels (10 μm nimodipine, 3 μmω-CTx-GVIA, 200 nm ω-Aga-IVA, and 500 nm mibefradil) blocked only half of the total Ba2+ current in an SCN neuron. At the antagonist concentrations used, classical L-, N-, and P-type channels were expected to be fully blocked. Approximately 70% of any Q-type current present was expected to have been blocked by 200 nm ω-Aga-IVA. We tested whether the current remaining in the presence of L-, N-, P-, and T-type channel blockers was carried by Q-type channels by substituting ω-CTx-MVIIC (3 μm) for ω-Aga-IVA in the mixture. This concentration of ω-CTx-MVIIC would be expected to fully block both P- and Q-type Ca2+ channels over the time that it was applied (>10 min) (Randall and Tsien, 1995). We found that the mixture containing ω-CTx-MVIIC did not block significantly more Ba2+ current (52 ± 6%;n = 4) than the mixture containing ω-Aga-IVA (51 ± 4%; n = 12; p > 0.1). Thus, most of the current resistant to block by the combination of nimodipine, ω-CTx-GVIA, ω-Aga-IVA (or ω-CTx-MVIIC), and mibefradil was, by definition, R type (Zhang et al., 1993; Randall and Tsien, 1995).
In some neuronal cell types, R-type current is carried, at least in part, by α1E-based Ca2+ channels (Wang et al., 1999; Foehring et al., 2000; Tottene et al., 2000; Lee et al., 2002). We therefore tested the effect of SNX-482, a toxin selective for α1E-based Ca2+channels, on whole-cell Ba2+current. Figure 5A illustrates a cell in which a large portion of Ba2+ current (77%) was blocked by a combination of nimodipine, ω-CTx-GVIA, ω-Aga-IVA, and mibefradil. The remaining current was not affected by application of SNX-482 (200 nm) but could be blocked by addition of Cd2+ (30 μm), a nonselective blocker of all voltage-gated Ca2+ channel subtypes. In seven cells tested, SNX-482 blocked <10% of current remaining in the presence of L-, N-, P/Q-, and T-type channel blockers, suggesting that α1E-based Ca2+channels are not significantly expressed in SCN neurons.
Fig. 5.

SCN neurons possess a large R-type Ca2+ current. A, Plot of peak inward Ba2+ current versus time for an experiment designed to test the sensitivity of R-type Ca2+ current to the α1E channel blocker SNX-482. A combination of nimodipine (10 μm), ω-CTx-GVIA (3 μm), ω-Aga-IVA (200 nm), and mibefradil (500 nm) was added to block L-, N-, P/Q-, and T-type Ca2+channels. After a washout period, SNX-482 (200 nm) was applied but had no effect on the current. At the end of the experiment, complete block of Ca2+ channels was produced by applying 30 μm Cd2+. Example records obtained at various times during the experiment are illustrated in theinset. B, In a similar experiment, the R current antagonist Ni2+ (30 μm) was applied, which blocked 75% of the current remaining after application of blockers of L-, N-, P/Q-, and T-type channels. C, Summary of average percentage block by Ca2+ channel antagonists (same concentrations as described in A). InC1, the various antagonists were applied either alone or in various combinations. As a percentage of total Ba2+ current, nimodipine blocked 18 ± 3%, with a range of 0–33% (n= 18); ω-CTx-GVIA blocked 27 ± 1%, with a range of 24–30% (n= 6); ω-Aga-IVA blocked 24 ± 4%, with a range of 15–47% (n= 8). Combined application of nimodipine, ω-CTx-GVIA, ω-Aga-IVA, and mibefradil blocked 51 ± 4%, with a range of 31–77% (n = 12); substitution of ω-Aga-IVA with ω-CTx-MVIIC (3 μm) blocked 52 ± 6%, with a range of 36–62% (n = 4). Combined application of nimodipine, ω-CTx-GVIA, ω-Aga-IVA, mibefradil, and Ni2+ (cocktail) blocked 76 ± 4% (n= 6) of total Ba2+ current. At 30 μm, Cd2+ applied alone blocked 93 ± 2% (n= 10) of total Ba2+current in SCN neurons. In C2, nimodipine, ω-CTx-GVIA, ω-Aga-IVA, and mibefradil were applied together to neurons first, followed by SNX-482 and then Cd2+. The combination of nimodipine, ω-CTx-GVIA, ω-Aga-IVA, and mibefradil blocked 57 ± 6%, with a range of 44–77% (n= 6) of total Ba2+ current. After application of this combination of blockers, addition of SNX-482 blocked only 6 ± 2% (n = 6) of total Ba2+ current, and subsequent application of 30 μm Cd2+ blocked the remaining current (25 ± 5%; n = 6). In C3, the experiment was conducted in an identical manner, with the exception that application of the L-, N-, P/Q-, and T-type blockers was followed by Ni2+ and then Cd2+. The combination of nimodipine, ω-CTx-GVIA, ω-Aga-IVA, and mibefradil blocked 44 ± 7%, with a range of 31–50% (n= 6) of total Ba2+ current. After application of this combination of blockers, addition of Ni2+ blocked 52 ± 3% (n = 6) of total Ba2+ current, and subsequent application of 30 μm Cd2+ blocked the remaining current (15 ± 5%; n = 6). Ni2+ added alone to SCN neurons blocked 45 ± 6%, with a range of 29–66% (n=6).
R-type current can also be blocked with some selectivity (see Materials and Methods) by the divalent cation Ni2+(Zamponi et al., 1996; N′Gouemo and Rittenhouse, 2000; Tottene et al., 2000), and so we tested Ni2+ on the current resistant to block by the combination of nimodipine, ω-CTx-GVIA, ω-Aga-IVA, and mibefradil. As illustrated in Figure5B, Ni2+ (30 μm)blocked ∼75% of the resistant Ba2+current, confirming the identification of R-type current in these neurons. In the presence of nimodipine, ω-CTx-GVIA, ω-Aga-IVA, and mibefradil, Ni2+ blocked 40 ± 6% (n = 7) of the original total Ba2+ current, a percentage not significantly different from Ni2+ block in the absence of the other four Ca2+ channel antagonists (45 ± 6%; n = 6;p > 0.1). This observation suggests that 30 μm Ni2+ does not block Ca2+ currents other than R type in SCN neurons. A summary of the Ca2+ channel antagonist data is illustrated in Figure 5C.
Effect of Ca2+ channel blockers on the AHP
In some neurons, tight coupling exists between specific classes of Ca2+ channels and KCa channels (Davies et al., 1996; Marrion and Tavalin, 1998; Pineda et al., 1998). We therefore tested the effect of individual Ca2+ channel antagonists on the AHP amplitude in spontaneously firing SCN neurons (Fig.6). The N-, P/Q-, and T-type Ca2+ channel blockers ω-CTx-GVIA (3 μm), ω-Aga-IVA (200 nm), and mibefradil (500 nm) had few or negligible effects on AHP amplitude (<10%; n = 6 for each), suggesting minimal participation of these kinds of channels in activating the AHP. In contrast, the L-type Ca2+ channel antagonist nimodipine (10 μm) had a significant effect on peak AHP, reducing the amplitude by ∼30%.
Fig. 6.

Action of Ca2+ channel blockers on AHP amplitude. A, Ca2+ channel antagonists were applied to spontaneously firing neurons in SCN slices. Examples from individual experiments are shown, superimposing three records from each experiment (control, block by an isoform-specific antagonist, and block byCd2+). B, A summary of the results from these experiments is presented as a bar chart. Doses used for individual antagonists were 10 μm nimodipine, 3 μm ω-CTx-GVIA, 200 nm ω-Aga-IVA, and 500 nmmibefradil. Nimodipine reduced AHP amplitude by 31 ± 2%, with a range of 17–51% (n = 6); the blockers ω-CTx-GVIA, ω-Aga-IVA, and mibefradil reduced AHP amplitude by <10% (n = 6 for each).
We also tested whether R-type current was involved in triggering the AHP (Fig. 7A,B). A combination of antagonists that block non-R-type Ca2+current (nimodipine, ω-CTx-GVIA, ω-Aga-IVA, and mibefradil) only partially suppressed the AHP (46 ± 5%; n = 5), suggesting that R-type current activated KCachannels responsible for the remainder of the AHP. Indeed, block of R current by Ni2+ (30 μm) partially suppressed the AHP (∼20%). Coapplication of the L- and R-type blockers nimodipine and Ni2+ had effects that were additive, reducing the AHP by ∼50%. This block was not significantly different from that produced by application of a mixture containing all Ca2+ channel blockers (nimodipine, ω-CTx-GVIA, ω-Aga-IVA, mibefradil, Ni2+; 63% ± 5%; n = 7;p > 0.1).
Fig. 7.

Participation of R-type current in AHP activation.A, Coapplication of antagonists for L-, N-, P/Q-, and T-type Ca2+ channels blocked ∼50% of the AHP, suggesting that R-type Ca2+ channels must also participate in activation of KCa channels. Application of Ni2+ (30 μm) reduced AHP amplitude and was additive with nimodipine (10 μm). A mixture of antagonists that included nimodipine, ω-CTx-GVIA, ω-Aga-IVA, mibefradil, and Ni2+ blocked the AHP by a similar amount as Ni2+ and nimodipine applied together.B, Summary of AHP reduction by combinations of Ca2+ channel blockers. A combination of nimodipine (10 μm), ω-CTx-GVIA (3 μm), ω-Aga-IVA (200 nm), and mibefradil (500 nm) inhibited the AHP by 46 ± 5%, with a range of 31–56% (n= 5). Ni2+ added alone (30 μm) reduced AHP amplitude by 20 ± 2%, with a range of 10–25% (n= 6). Inhibition of the AHP by coapplication of nimodipine plus Ni2+ (50 ± 8%; range, 17–90%; n = 8) was comparable with application of a mixture containing nimodipine, ω-CTx-GVIA, ω-Aga-IVA, mibefradil, and Ni2+ (63 ± 5%; range, 49–78%; n = 7). C, Plot of normalized AHP amplitude versus time for four cells in which ryanodine (10 μm), a blocker of Ca2+-induced Ca2+ release, was applied for >30 min. Long-term recordings were made using the perforated-patch method to prevent rundown of the AHP.
Because Ca2+ release via ryanodine receptors triggers the AHP in some kinds of neurons, we tested the effect of ryanodine, an inhibitor of Ca2+-induced Ca2+ release (Sutko et al., 1985; Akita and Kuba, 2000), on the AHP of cluster I SCN neurons (Fig.7C). Measured from spike threshold, maximum hyperpolarization after a spike was diminished by 1.4 ± 0.5 mV (n = 4) after 30 min of exposure to 10 μm ryanodine. This was not statistically different from the reduction in afterhyperpolarization magnitude observed after 30 min in control conditions (1.4 ± 1.0 mV;n = 3). Thus release of Ca2+ from ryanodine-sensitive stores appears to play a minimal or no role in generating the AHP in cluster I SCN neurons, despite the importance of ryanodine receptors in mediating light-induced phase delays in the SCN (Ding et al., 1998).
Effects on firing rate resulting from pharmacological antagonism of the AHP
In many kinds of neurons, the magnitude and duration of the AHP are important factors in setting interspike interval and, thereby, neuronal firing rate. For example, reduction in the AHP by the action of neurotransmitters can increase firing rate (Madison and Nicoll, 1982; Pedarzani and Storm, 1995). The relationship between firing frequency of SCN neurons and duration of the Ca2+-dependent AHP (Fig.2B) predicts that reducing AHP duration would increase firing rate. We therefore examined how pharmacological antagonism of the AHP affected spike frequency in spontaneously active cluster I SCN neurons (Fig.8A). In these experiments, Ca2+ channel blockers and KCa channel blockers were tested. Slices were bathed in bicuculline methiodide (30 μm) or picrotoxin (100 μm) to preclude secondary effects of Ca2+ or KCa channel blockers on firing rate, effects that could otherwise have arisen via alteration of Ca2+-dependent release of GABA. In control experiments, spontaneous firing rates were found to be stable during the initial 15 min after breakthrough, and we therefore confined our measurements to this time period.
Fig. 8.

Effect on firing rate of Ca2+channel and KCa channel antagonists. A, Records (2 sec duration) of spontaneous firing in an SCN neuron under control conditions and after application of a mixture of Ca2+ channel blockers in the bath solution. The Ca2+ channel mixture contained nimodipine (10 μm), ω-CTx-GVIA (3 μm), ω-Aga-IVA (200 nm), mibefradil (500 nm), and Ni2+ (30 μm). B, Normalized firing rate in the presence of various channel blockers.Black bars represent results for Ca2+channel antagonists, and gray bars represent results for KCa channel antagonists. All firing rates with antagonist were compared with a baseline firing rate in that neuron measured just before antagonist application. In control recordings, firing frequency did not significantly change over the first 15 min of whole-cell recording; at 15 min, firing frequency was the same as that measured just after establishment of whole-cell recording (striped bar labeled control; p > 0.1; n = 5). The duration of experiments testing antagonist action on firing frequency was in all cases <15 min. Individual selective Ca2+ channel antagonists were applied at a dose equal to that used in the Ca2+channel blocker mixture. Apamin and iberiotoxin were applied at 100 nm. Cd2+ was applied at 30 μm. Firing rate experiments were performed in the presence of 30 μm bicuculline methiodide. Sample size ranged from n = 6–10, except for experiments with Cd2+, for which n = 16. Paired Student's t test; **p < 0.01.
KCa channel antagonists did not alter firing rate. Neither block of SKCa channels by apamin (100 nm) nor block of BKCa channels by iberiotoxin (100 nm) had a statistically significant effect on spike frequency (Fig. 8B), nor did individual subtype-selective blockers of Ca2+ channel subtypes alter firing rate. Applied alone, nimodipine, ω-CTx-GVIA, ω-Aga-IVA, mibefradil, or Ni2+ did not significantly affect firing rate (Fig. 8B). However, application of a combination of nimodipine plus Ni2+ had a significant effect on firing rate (p < 0.01;n = 6), although the effect was, unexpectedly, a decrease in rate. Simultaneous application of all five subtype-selective Ca2+ channel blockers also slowed firing rate (p < 0.01;n = 7). In contrast, complete blockade of Ca2+ entry by Cd2+ (30 μm) increased firing rate ∼1.5-fold (see Discussion).
Effects of channel blockers on spike parameters
Blockers of Ca2+ channels and KCa channels were examined for their effects on spike parameters other than the AHP: interspike potential (VIP), determined as the plateau region of membrane potential between action potentials; spike amplitude, measured from VIP to peak depolarization; and spike width, measured at half amplitude. None of the blockers tested significantly affectedVIP, indicating that changes in firing rate could not be attributed to changes inVIP (Table2). This was confirmed for Cd2+ by the fact that membrane potential was not affected when this blocker was applied, whereas spiking was suppressed by tetrodotoxin (1 μm):VIP was −40 ± 3.1 mV before Cd2+ application and −39 ± 2.4 mV during Cd2+ treatment (p > 0.1; n = 6).
Table 2.
Effect of blockers of Ca2+ channels and of KCa channels on membrane properties of cluster I SCN neurons
| Control | Cd2+ | ω-CTx-GVIA | ω-Aga-IVA | Nimodipine | Mibefradil | Ni2+ | ICamixture | Apamin | Iberiotoxin | |
|---|---|---|---|---|---|---|---|---|---|---|
| Number of neurons (n) | 60 | 10 | 6 | 6 | 6 | 6 | 6 | 8 | 6 | 6 |
| Interspike potential (mV) | −41 ± 0.8 | −41 ± 1.6 | −37 ± 1.9 | −38 ± 1.4 | −39 ± 2.7 | −40 ± 1.3 | −37 ± 2.1 | −38 ± 2.6 | −41 ± 1.5 | −37 ± 3.1 |
| Spike amplitude (mV) | 75 ± 1.7 | 60 ± 4.72-160 | 69 ± 1.8 | 71 ± 5.2 | 75 ± 4.5 | 81 ± 4.9 | 78 ± 5.1 | 72 ± 4.3 | 80 ± 3.5 | 73 ± 6.2 |
| Spike width (msec) | 2.4 ± 0.1 | 4.2 ± 0.52-160 | 3.1 ± 0.2* | 3.0 ± 0.3 | 2.6 ± 0.3 | 2.3 ± 0.2 | 2.4 ± 0.2 | 3.6 ± 0.42-160 | 2.4 ± 0.2 | 4.0 ± 0.42-160 |
Membrane properties were measured as described in Table 1. Concentrations of blockers used are as given in Materials and Methods. ICa blocker mixture represents the combination of 10 μm nimodipine, 3 μmω-CTx-GVIA, 200 nm ω-Aga-IVA, 30 μmNi2+, and 500 nm mibefradil.
p < 0.05,
F2-160: p < 0.01, compared with control.
Action potential amplitude was not affected by KCa channel blockers nor by Ca2+ channel blockers except for Cd2+, which substantially attenuated spike height (Table 2). A possible explanation for the Cd2+ effect is that block of the AHP, complete for Cd2+ but only partial for the other blockers (even the Ca2+ channel blocker mixture) (Fig. 7), prevented normal recovery from inactivation of voltage-gated Na+ channels. In contrast to VIP and spike amplitude, spike width was more sensitive to blockers (Table 2). Action potentials were broadened by the BKCa channel blocker iberiotoxin, consistent with the known role of BKCa channels in action potential repolarization. Action potentials were also broadened by the N-channel blocker ω-CTx-GVIA, by the five-component Ca2+channel blocker mixture (nimodipine, ω-CTx-GVIA, ω-Aga-IVA, Ni2+, and mibefradil), and by the nonselective Ca2+ channel blocker, Cd2+, presumably because BKCa channel activity was reduced consequent to Ca2+ channel block.
Discussion
Firing frequency of neurons is commonly modulated by spike afterhyperpolarizations (Barrett and Barrett, 1976; Yarom et al., 1985;Madison and Nicoll, 1986; Sah, 1996). For SCN neurons, in which rhythmic modulation of firing communicates circadian information to the organism, the role of the AHP in modulating spike rate has previously received little attention. Here we have focused on cluster I neurons of the SCN, which are the most abundant cells present in SCN (Pennartz et al., 1998) and exhibit rhythmic modulation of firing frequency (Welsh et al., 1995). Our examination of action potential waveforms from cluster I neurons firing at different rates revealed that the AHP duration shortens as spike frequency increases.
Three classes of KCa channels in SCN neurons
Evidence that KCa channels regulate rhythmic firing of SCN neurons has been sparse. Injection of apamin into brain ventricles has been found to disrupt normal circadian behaviors (Gandolfo et al., 1996), and charybdotoxin and apamin block serotonin-induced phase shifting of SCN firing (Prosser et al., 1994). Our pharmacological dissection of KCa channel current shows that cluster I neurons express three major groups of KCa channels. SKCa and BKCa channels were identified on the basis of partial block of the AHP by apamin and iberiotoxin. Apamin selectively blocks SK2 and SK3 subtypes of SKCa channels, whereas iberiotoxin selectively blocks type I BKCa channels. Coapplication of nearly saturating concentrations of apamin and iberiotoxin reduced the amplitude of the AHP by only ∼55%, however, indicating that a large fraction of the channels underlying the AHP are insensitive to these blockers. SK1-based channels, which under some conditions have been found to be insensitive to apamin and iberiotoxin (Kohler et al., 1996; Sah, 1996;Vergara et al., 1998; Kaczorowski and Garcia, 1999; Sah and Davies, 2000), are not expressed in SCN neurons (Stocker and Pedarzani, 2000). Other channels that might support the apamin- and iberiotoxin-insensitive KCa channel current in SCN neurons include iberiotoxin-insensitive BKCachannels (Meera et al., 2000), intermediate conductance IKCa channels, or the essentially uncharacterized channels that underlie the apamin- and iberiotoxin-insensitive slow afterhyperpolarization in hippocampal pyramidal neurons (IsAHP) (Sah and Faber, 2002).
Four types of high voltage-gated Ca2+ channels in SCN neurons
On the basis of sensitivity to Ca2+channel antagonists, our work shows that SCN neurons possess four principal components of high-voltage activated Ca2+ channel current: L-, N-, and P/Q-currents, which had been previously identified in these neurons (Huang, 1993; Chen and van den Pol, 1998), and also R-type current. The precision in our measurement of relative current magnitude for the various Ca2+ channel subtypes was compromised by the imperfect selectivity of the specific channel antagonists, the incomplete block of individual Ca2+ channel subtypes, and the inability to voltage clamp distal processes of SCN neurons. Nonetheless, our pharmacological dissection of components of Ca2+ channel current yielded an estimate for the size of the L-type component that is similar to that reported for dissociated SCN neurons (Huang, 1993).
Our analysis revealed that R-type current was the largest current component in SCN neurons from slices. The majority of R current was blocked by low concentrations of Ni2+, although a proportion (∼15% of total Ca2+ channel current) remained that was insensitive to this concentration of Ni2+. The nonblocked fraction might represent Ni2+-insensitive R current (Schramm et al., 1999) or other Ca2+ channel components incompletely blocked at the antagonist doses used. Because SNX-482 did not block R current, α1E-based Ca2+ channels are not likely to support the R current of SCN neurons. R current identified here may correspond to the large component of Ca2+ current that was found in previous studies to be insensitive to the combination of nimodipine and ω-CTx-GVIA (Huang, 1993).
Coupling of Ca2+ channels to KCachannels in SCN neurons
In some neurons, tight coupling exists between specific Ca2+ channel subtypes and KCa channel subtypes (Davies et al., 1996;Marrion and Tavalin, 1998; Borde et al., 2000; Martinez-Pinna et al., 2000), whereas in other neurons, multiple Ca2+ channel subtypes contribute to AHP activation (Williams et al., 1997; Pineda et al., 1998). In cluster I neurons of the SCN, both L- and R-type Ca2+ currents are involved in AHP activation. N and T channels make small contributions as well, and P/Q channels appear not to contribute. That large-amplitude N and P/Q currents contributed little or nothing to AHP activation suggests that Ca2+ channel subtypes are differentially distributed on SCN neurons, with L- and R-type channels located in closest proximity to KCa channels.
Physiological significance
Neurons studied here were in the daytime phase of the circadian cycle, but we suggest that the relationship between firing frequency and AHP waveform may contribute to circadian control of firing frequency in SCN neurons. This proposed mechanism for circadian regulation of KCa channel activity and the AHP might arise from circadian changes in the expression, splicing, post-translational modification, or regulation of specific KCa channels.
In faster firing SCN neurons, a shortened interspike interval appears to rely in part on speeded decay of the Cd2+-sensitive component of the AHP, yet the Cd2+-sensitive component of the AHP was larger and action potentials were narrower in faster firing neurons, suggesting that peak KCa channel activity is augmented in faster firing neurons. Thus in faster firing neurons the number of KCa channels open at the peak of the AHP is increased, but the AHP terminates more rapidly. These two changes in the AHP might be accomplished in various ways. For example, the kinetics of KCa channel gating might be speeded globally such that the maximal number of channels open simultaneously is increased (greater AHP magnitude) and yet the duration of the AHP is shortened. Alternatively, the increased magnitude of the AHP in faster firing neurons might be attributable to an increase in the number of KCa channels available to open, and, in addition, SCN channels active in the range −50 to −60 mV and with a reversal potential positive to that of the AHP might exhibit increased activity (e.g., hyperpolarization-activated cation channels (Akasu et al., 1993) or Na+ channels (Pennartz et al., 1997). Increased activity of these latter kinds of channels might mask the full duration of KCa channel activity and terminate the AHP more rapidly. The fact that faster firing neurons did not hyperpolarize more than slower firing neurons, although the net size of the Cd2+-subtracted AHP was greater in faster neurons, is consistent with the idea that these channels might compensate for the hyperpolarizing influence of KCa channels. In this case, circadian dependence on the activity of these channels, as well as of KCa channels, may be involved in determining the duration of the AHP and the frequency of firing.
In spontaneously firing SCN neurons, full suppression of the Ca2+-dependent AHP by application of Cd2+ increased firing rate (Fig.8B), whereas partial suppression of the AHP by mixtures of specific Ca2+ channel blockers had the opposite effect. This unexpected contrast could arise if voltage-gated Ca2+ channels participate both in bringing neurons above firing threshold and in triggering the AHP. In this scenario, the net effect on firing rate of block of Ca2+ channels would reflect the balance between a slowed trajectory through firing threshold (decreased firing rate) versus a reduced AHP duration (increased rate). In the case of Cd2+ block, we speculate that the effect of full suppression of the AHP dominates over the slowed trajectory through threshold, so that neurons fire faster. With the Ca2+ channel blocker mixtures, we speculate that slowed trajectory through threshold dominates over the effect of partial suppression of the AHP, resulting in a net decrease in neuronal firing.
The molecular mechanism of the frequency-dependent change in AHP waveform appears to be based in part on KCachannels that are insensitive to either apamin or iberiotoxin, because application of these specific antagonists did not alter firing frequency. Additionally, there was no correlation between parameters of the iberiotoxin-sensitive component of the AHP and firing rate. We conclude that other KCa channel types, perhaps iberiotoxin-insensitive BKCa channels (Meera et al., 2000) or IKCa channels, are modulated in a manner that diurnally alters the firing rate of SCN neurons. For these KCa channels, circadian modulation might occur via second messenger-mediated modification of channel function, via changes in the coupling of Ca2+ entry (Pennartz et al., 2002) to channel activation, or by altered expression of channels or associated regulatory proteins. A notable recent finding regarding this latter point is that transcription of Slob, which encodes a Ca2+-binding protein that interacts with and regulates the activity of the SloKCa channel (Schopperle et al., 1998), is under circadian control in Drosophila (Claridge-Chang et al., 2001; McDonald and Rosbash, 2001). More recently, expression of the mammalian ortholog of the BK-type Slo channel,Kcnma1, has also been found to be under circadian control (Panda et al., 2002). The convergence on KCachannels of molecular and electrophysiological approaches underscores the likely importance of these channels in the mechanism of circadian firing of SCN neurons.
Footnotes
This work was supported by National Institutes of Health Grants NS35245 and NS07083.
Correspondence should be addressed to William A. Sather, Neuroscience Center, Box B-138, University of Colorado Health Sciences Center, 4200 East Ninth Avenue, Denver, CO 80262. E-mail:william.sather@UCHSC.edu.
References
- 1.Aizenman CD, Linden DJ. Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J Neurophysiol. 1999;82:1697–1709. doi: 10.1152/jn.1999.82.4.1697. [DOI] [PubMed] [Google Scholar]
- 2.Akasu T, Shoji S, Hasuo H. Inward rectifier and low-threshold calcium currents contribute to the spontaneous firing mechanism in neurons of the rat suprachiasmatic nucleus. Pflügers Arch. 1993;425:109–116. doi: 10.1007/BF00374510. [DOI] [PubMed] [Google Scholar]
- 3.Akita T, Kuba K. Functional triads consisting of ryanodine receptors, Ca2+ channels and Ca2+-activated K+ channels in bullfrog sympathetic neurons: plastic modulation of action potential. J Gen Physiol. 2000;116:697–720. doi: 10.1085/jgp.116.5.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson CS, MacKinnon R, Smith C, Miller C. Charybdotoxin block of single Ca2+-activated K+ channels. Effects of channel gating, voltage, and ionic strength. J Gen Physiol. 1988;91:317–333. doi: 10.1085/jgp.91.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrett EF, Barrett JN. Separation of two voltage-sensitive potassium currents, and demonstration of a tetrodotoxin-resistant calcium current in frog motoneurones. J Physiol (Lond) 1976;255:737–774. doi: 10.1113/jphysiol.1976.sp011306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bekkers JM. Properties of voltage-gated potassium currents in nucleated patches from large layer 5 cortical pyramidal neurons of the rat. J Physiol (Lond) 2000;525:593–609. doi: 10.1111/j.1469-7793.2000.t01-1-00593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bevan MD, Wilson CJ. Mechanisms underlying spontaneous oscillation and rhythmic firing in rat subthalamic neurons. J Neurosci. 1999;19:7617–7628. doi: 10.1523/JNEUROSCI.19-17-07617.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bezprozvanny I, Tsien RW. Voltage-dependent blockade of diverse types of voltage-gated Ca2+ channels expressed in Xenopus oocytes by the Ca2+ channel antagonist mibefradil (Ro 40–5967). Mol Pharmacol. 1995;48:540–549. [PubMed] [Google Scholar]
- 9.Blatz A, Magleby KL. Single apamin-blocked Ca-activated K+ channels of small conductance in cultured rat muscle cells. Nature. 1986;323:718–720. doi: 10.1038/323718a0. [DOI] [PubMed] [Google Scholar]
- 10.Boland LM, Morril JA, Bean BP. ω-conotoxin block of N-type calcium channels in frog and rat sympathetic neurons. J Neurosci. 1994;14:5011–5027. doi: 10.1523/JNEUROSCI.14-08-05011.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borde M, Bonansco C, Fernandez de Sevilla D, Le Ray D, Bruno W. Voltage-clamp analysis of the potentiation of the slow Ca2+-activated K+ current in hippocampal pyramidal neurons. Hippocampus. 2000;10:198–206. doi: 10.1002/(SICI)1098-1063(2000)10:2<198::AID-HIPO9>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 12.Candia S, Garcia ML, Latorre R. Mode of action of iberiotoxin, a potent blocker of the large conductance Ca2+-activated K+ channel. Biophys J. 1992;63:583–590. doi: 10.1016/S0006-3495(92)81630-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen G, van den Pol AN. Presynaptic GABAB autoreceptor modulation of P/Q-type calcium channels and GABA release in rat suprachiasmatic nucleus neurons. J Neurosci. 1998;18:1913–1922. doi: 10.1523/JNEUROSCI.18-05-01913.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Claridge-Chang A, Wijnen H, Naef F, Boothroyd C, Rajewsky N, Young MW. Circadian regulation of gene expression systems in the Drosophila head. Neuron. 2001;32:657–671. doi: 10.1016/s0896-6273(01)00515-3. [DOI] [PubMed] [Google Scholar]
- 15.Davies PJ, Ireland DR, McLachlan EM. Sources of Ca2+ for different Ca2+-activated K+ conductances in neurones of the rat superior cervical ganglion. J Physiol (Lond) 1996;494:353–366. doi: 10.1113/jphysiol.1996.sp021599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding JM, Buchanan GF, Tischkau SA, Chen D, Kuriashkina L, Faiman LE, Alster JM, McPherson PS, Campbell KP, Gillette MU. A neuronal ryanodine receptor mediates light-induced phase delays of the circadian clock. Nature. 1998;394:381–384. doi: 10.1038/28639. [DOI] [PubMed] [Google Scholar]
- 17.Dunlap JC. Molecular bases for circadian clocks. Cell. 1999;96:271–290. doi: 10.1016/s0092-8674(00)80566-8. [DOI] [PubMed] [Google Scholar]
- 18.Earnest DJ, Digiorgio SM, Sladek CD. Effects of tetrodotoxin on the circadian pacemaker mechanism in suprachiasmatic explants in vitro. Brain Res Bull. 1991;26:677–682. doi: 10.1016/0361-9230(91)90160-l. [DOI] [PubMed] [Google Scholar]
- 19.Foehring RC, Mermelstein PG, Song WJ, Ulrich S, Surmeier DJ. Unique properties of R-type calcium currents in neocortical and neostriatal neurons. J Neurophysiol. 2000;84:2225–2236. doi: 10.1152/jn.2000.84.5.2225. [DOI] [PubMed] [Google Scholar]
- 20.Galvez A, Gimenez-Gallego G, Reuben JP, Roy-Contancin L, Feigenbaum P, Kaczorowski GJ, Garcia ML. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J Biol Chem. 1990;265:11083–11090. [PubMed] [Google Scholar]
- 21.Gandolfo G, Schweitz H, Lazdunski M, Gottesmann C. Sleep cycle disturbances induced by apamin, a selective blocker of Ca2+-activated K+ channels. Brain Res. 1996;736:344–347. doi: 10.1016/0006-8993(96)00690-7. [DOI] [PubMed] [Google Scholar]
- 22.Gillette MU. SCN electrophysiology in vitro: rhythmic activity and endogenous clock properties. In: Klein DC, Moore RY, Reppert SM, editors. Suprachiasmatic nucleus: the mind's clock. Oxford UP; New York: 1991. pp. 125–143. [Google Scholar]
- 23.Gribkoff VK, Starrett JE, Dworetzky SI. The pharmacology and molecular biology of large-conductance calcium-activated (BK) potassium channels. Adv Pharmacol. 1997;37:319–348. doi: 10.1016/s1054-3589(08)60954-0. [DOI] [PubMed] [Google Scholar]
- 24.Huang RC. Sodium and calcium currents in acutely dissociated neurons from rat suprachiasmatic nucleus. J Neurophysiol. 1993;70:1692–1703. doi: 10.1152/jn.1993.70.4.1692. [DOI] [PubMed] [Google Scholar]
- 25.Hughes M, Romey G, Duval D, Vincent J, Lazdunski M. Apamin as a selective blocker of the calcium-dependent potassium channel in neuroblastoma cells. Voltage-clamp and biochemical characterization of the toxin receptor. Proc Natl Acad Sci USA. 1982;79:1308–1312. doi: 10.1073/pnas.79.4.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inouye S-IT, Kawamura H. Persistence of circadian rhythmicity in a mammalian hypothalamic “island” containing the suprachiasmatic nucleus. Proc Natl Acad Sci USA. 1979;76:5962–5966. doi: 10.1073/pnas.76.11.5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jagota A, de la Iglesia HO, Schwartz WJ. Morning and evening circadian oscillations in the suprachiasmatic nucleus in vitro. Nat Neurosci. 2000;3:372–376. doi: 10.1038/73943. [DOI] [PubMed] [Google Scholar]
- 28.Jiang ZG, Nelson CS, Allen CN. Melatonin activates an outward current and inhibits Ih in rat suprachiasmatic nucleus neurons. Brain Res. 1995;687:125–132. doi: 10.1016/0006-8993(95)00478-9. [DOI] [PubMed] [Google Scholar]
- 29.Kaczorowski GJ, Garcia ML. Pharmacology of voltage-gated and calcium-activated potassium channels. Curr Opin Chem Biol. 1999;3:448–458. doi: 10.1016/S1367-5931(99)80066-0. [DOI] [PubMed] [Google Scholar]
- 30.Khawaled R, Bruening-Wright A, Adelman JP, Maylie J. Bicuculline block of small-conductance calcium-activated potassium channels. Pflügers Arch. 1999;438:314–321. doi: 10.1007/s004240050915. [DOI] [PubMed] [Google Scholar]
- 31.Klein DC, Moore RY, Reppert SM. Suprachiasmatic nucleus: the mind's clock. Oxford UP; New York: 1991. [Google Scholar]
- 32.Kohler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–1714. doi: 10.1126/science.273.5282.1709. [DOI] [PubMed] [Google Scholar]
- 33.Lee JH, Gomora JC, Cribbs LL, Perez-Reyes E. Nickel block of three cloned T-type calcium channels: low concentrations selectively block α1H. Biophys J. 1999;77:3034–3042. doi: 10.1016/S0006-3495(99)77134-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee SC, Choi S, Lee T, Kim HL, Chin H, Shin HS. Molecular basis of R-type calcium channel in central amygdala neurons of the mouse. Proc Natl Acad Sci USA. 2002;99:3276–3281. doi: 10.1073/pnas.052697799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madison DV, Nicoll RA. Noradrenaline blocks accommodation of pyramidal cell discharge in the hippocampus. Nature. 1982;299:636–638. doi: 10.1038/299636a0. [DOI] [PubMed] [Google Scholar]
- 36.Madison DV, Nicoll RA. Actions of noradrenaline recorded intracellularly in rat hippocampal CA1 pyramidal neurones, in vitro. J Physiol (Lond) 1986;372:221–244. doi: 10.1113/jphysiol.1986.sp016006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marchetti C, Amico C, Usai C. Functional characterization of the effects of nimodipine on the calcium current in rat cerebellar granule cells. J Neurophysiol. 1995;73:1169–1180. doi: 10.1152/jn.1995.73.3.1169. [DOI] [PubMed] [Google Scholar]
- 38.Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- 39.Martin RL, Lee JH, Cribbs LL, Perez-Reyes E, Hanck DA. Mibefradil block of cloned T-type calcium channels. J Pharmacol Exp Ther. 2000;295:302–308. [PubMed] [Google Scholar]
- 40.Martinez-Pinna J, McLachlan EM, Gallego R. Distinct mechanisms for activation of Cl– and K+ currents by Ca2+ from different sources in mouse sympathetic neurones. J Physiol (Lond) 2000;527:249–264. doi: 10.1111/j.1469-7793.2000.00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDonald MJ, Rosbash M. Microarray analysis and organization of circadian gene expression in Drosophila. Cell. 2001;107:567–578. doi: 10.1016/s0092-8674(01)00545-1. [DOI] [PubMed] [Google Scholar]
- 42.McDonough SI, Bean BP. Mibefradil inhibition of T-type calcium channels in cerebellar Purkinje neurons. Mol Pharmacol. 1998;54:1080–1087. doi: 10.1124/mol.54.6.1080. [DOI] [PubMed] [Google Scholar]
- 43.Meera P, Wallner M, Toro L. A neuronal beta subunit (KCNMB4) makes the large conductance, voltage- and Ca2+-activated K+ channel resistant to charybdotoxin and iberiotoxin. Proc Natl Acad Sci USA. 2000;97:5562–5567. doi: 10.1073/pnas.100118597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mintz IM, Venema VJ, Swiderek KM, Lee TD, Bean BP, Adams ME. P-type calcium channels blocked by the spider toxin ω-Aga-IVA. Nature. 1992;355:827–829. doi: 10.1038/355827a0. [DOI] [PubMed] [Google Scholar]
- 45.Moore RY, Eichler VB. Loss of circadian adrenal corticosterone rhythm following suprachiasmatic lesions in the rat. Brain Res. 1972;42:201–206. doi: 10.1016/0006-8993(72)90054-6. [DOI] [PubMed] [Google Scholar]
- 46.Neher E. Correction for liquid junction potentials in patch clamp experiments. In: Rudy B, Iverson LE, editors. Methods in enzymology. Academic; San Diego: 1992. pp. 123–131. [DOI] [PubMed] [Google Scholar]
- 47.Newcomb R, Szoke B, Palma A, Wang G, Chen XH, Hopkins W, Cong R, Miller J, Urge L, Tarczy-Hornoch K, Loo JA, Dooley DJ, Nadasdi L, Tsien RW, Lemos J, Miljanich G. Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry. 1998;37:15353–15362. doi: 10.1021/bi981255g. [DOI] [PubMed] [Google Scholar]
- 48.Newman GC, Hospod FE, Patlak CS, Moore RY. Analysis of in vitro glucose utilization in a circadian pacemaker model. J Neurosci. 1992;12:2015–2021. doi: 10.1523/JNEUROSCI.12-06-02015.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.N′Gouemo P, Rittenhouse AR. Biophysical and pharmacological characterization of voltage-sensitive calcium currents in neonatal rat inferior colliculus neurons. Neuroscience. 2000;96:753–765. doi: 10.1016/s0306-4522(00)00006-3. [DOI] [PubMed] [Google Scholar]
- 50.Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell. 2002;109:307–320. doi: 10.1016/s0092-8674(02)00722-5. [DOI] [PubMed] [Google Scholar]
- 51.Pedarzani P, Storm JF. Dopamine modulates the slow Ca2+-activated K+ current IAHP via cyclic AMP-dependent protein kinase in hippocampal neurons. J Neurophysiol. 1995;74:2749–2753. doi: 10.1152/jn.1995.74.6.2749. [DOI] [PubMed] [Google Scholar]
- 52.Pennartz CMA, Bierlaagh MA, Geurtsen AMS. Cellular mechanisms underlying spontaneous firing in rat suprachiasmatic nucleus: involvement of a slowly inactivating component of sodium current. J Neurophysiol. 1997;78:1811–1825. doi: 10.1152/jn.1997.78.4.1811. [DOI] [PubMed] [Google Scholar]
- 53.Pennartz CMA, de Jeu MTG, Geurtsen AMS, Sluiter AA, Hermes MLHJ. Electrophysiological and morphological heterogeneity of neurons in slices of rat suprachiasmatic nucleus. J Physiol (Lond) 1998;506:775–793. doi: 10.1111/j.1469-7793.1998.775bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pennartz CMA, de Jeu MTG, Bos NP, Schaap J, Geurtsen AMS. Diurnal modulation of pacemaker potentials and calcium current in the mammalian circadian clock. Nature. 2002;416:286–290. doi: 10.1038/nature728. [DOI] [PubMed] [Google Scholar]
- 55.Pineda JC, Waters RS, Foehring RC. Specificity in the interaction of HVA Ca2+ channel types with Ca2+-dependent AHPs and firing behavior in neocortical pyramidal neurons. J Neurophysiol. 1998;79:2522–2534. doi: 10.1152/jn.1998.79.5.2522. [DOI] [PubMed] [Google Scholar]
- 56.Prosser RA, Heller HC, Miller JD. Serotonergic phase advances of the mammalian circadian clock involve protein kinase A and K+ channel opening. Brain Res. 1994;644:67–73. doi: 10.1016/0006-8993(94)90348-4. [DOI] [PubMed] [Google Scholar]
- 57.Randall AD, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reppert SM, Weaver DR. Molecular analysis of mammalian circadian rhythms. Annu Rev Physiol. 2001;63:647–676. doi: 10.1146/annurev.physiol.63.1.647. [DOI] [PubMed] [Google Scholar]
- 59.Sah P. Ca2+-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci. 1996;19:150–154. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- 60.Sah P, Davies P. Calcium-activated potassium currents in mammalian neurons. Clin Exp Pharmacol Physiol. 2000;27:657–663. doi: 10.1046/j.1440-1681.2000.03317.x. [DOI] [PubMed] [Google Scholar]
- 61.Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–353. doi: 10.1016/s0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- 62.Schopperle WM, Holmqvist MH, Zhou Y, Wang J, Wang Z, Griffith LC, Keselman I, Kusinitz F, Dagan D, Levitan IB. Slob, a novel protein that interacts with the Slowpoke calcium-dependent potassium channel. J Neurosci. 1998;20:565–573. doi: 10.1016/s0896-6273(00)80995-2. [DOI] [PubMed] [Google Scholar]
- 63.Schramm M, Vajna R, Preverzev A, Tottene A, Klockner U, Pietrobon D, Hescheler J, Schneider T. Isoforms of α1E voltage-gated calcium channels in rat cerebellar granule cells: detection of major calcium channel α1 transcripts by reverse transcription-polymerase chain reaction. Neuroscience. 1999;92:565–575. doi: 10.1016/s0306-4522(99)00013-5. [DOI] [PubMed] [Google Scholar]
- 64.Schwartz WJ. Further evaluation of the tetrodotoxin-resistant circadian pacemaker in the suprachiasmatic nuclei. J Biol Rhythms. 1991;6:149–158. doi: 10.1177/074873049100600205. [DOI] [PubMed] [Google Scholar]
- 65.Schwartz WJ, Gross RA, Morton MT. The suprachiasmatic nuclei contain a tetrodotoxin-resistant circadian pacemaker. Proc Natl Acad Sci USA. 1987;84:1694–1698. doi: 10.1073/pnas.84.6.1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stocker M, Pedarzani P. Differential distribution of three Ca2+-activated K+ channel subunits, SK1, SK2, and SK3, in the adult rat central nervous system. Mol Cell Neurosci. 2000;15:476–493. doi: 10.1006/mcne.2000.0842. [DOI] [PubMed] [Google Scholar]
- 67.Strecker GJ, Wuarin JP, Dudek FE. GABAA-mediated local synaptic pathways connect neurons in the rat suprachiasmatic nucleus. J Neurophysiol. 1997;78:2217–2220. doi: 10.1152/jn.1997.78.4.2217. [DOI] [PubMed] [Google Scholar]
- 68.Sutko JL, Ito K, Kenyon JL. Ryanodine: a modifier of sarcoplasmic reticulum calcium release in striated muscle. Fed Proc. 1985;44:2984–2988. [PubMed] [Google Scholar]
- 69.Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tottene A, Moretti A, Pietrobon D. Functional diversity of P-type and R-type calcium channels in rat cerebellar neurons. J Neurosci. 1996;16:6353–6363. doi: 10.1523/JNEUROSCI.16-20-06353.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tottene A, Volsen S, Pietrobon D. α1E subunits form the pore of three cerebellar R-type calcium channels with different pharmacological and permeation properties. J Neurosci. 2000;20:171–178. doi: 10.1523/JNEUROSCI.20-01-00171.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vergara C, Latorre R, Marrion NV, Adelman JP. Calcium-activated potassium channels. Curr Opin Neurobiol. 1998;8:321–329. doi: 10.1016/s0959-4388(98)80056-1. [DOI] [PubMed] [Google Scholar]
- 73.Vilchis C, Bargas J, Ayala GX, Galvan E, Galarraga E. Ca2+ channels that activate Ca2+-dependent K+ currents in neostriatal neurons. Neuroscience. 2000;95:745–752. doi: 10.1016/s0306-4522(99)00493-5. [DOI] [PubMed] [Google Scholar]
- 74.Wang G, Dayanithi G, Newcomb R, Lemos JR. An R-type Ca2+ current in neurohypophysial terminals preferentially regulates oxytocin secretion. J Neurosci. 1999;19:9235–9241. doi: 10.1523/JNEUROSCI.19-21-09235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Welsh DK, Logothetis DE, Meister M, Reppert SM. Individual neurons dissociated from rat suprachiasmatic nucleus express independently phased circadian firing rhythms. Neuron. 1995;14:697–706. doi: 10.1016/0896-6273(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 76.Williams S, Serafin M, Mühlethaler M, Bernheim L. Distinct contributions of high- and low-voltage-activated calcium currents to afterhyperpolarizations in cholinergic nucleus basalis neurons of the guinea pig. J Neurosci. 1997;17:7307–7315. doi: 10.1523/JNEUROSCI.17-19-07307.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu W, Lipscombe D. Neuronal Ca(V)1.3α(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci. 2001;21:5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yarom Y, Sugimori M, Llinas R. Ionic currents and firing patterns of mammalian vagal motoneurons in vitro. Neuroscience. 1985;16:719–737. doi: 10.1016/0306-4522(85)90090-9. [DOI] [PubMed] [Google Scholar]
- 79.Zamponi GW, Bourinet E, Snutch TP. Nickel block of a family of neuronal calcium channels: subtype- and subunit-dependent action at multiple sites. J Membr Biol. 1996;151:77–90. doi: 10.1007/s002329900059. [DOI] [PubMed] [Google Scholar]
- 80.Zhang JF, Randall AD, Ellinor PT, Horne WA, Sather WA, Tanabe T, Schwarz TL, Tsien RW. Distinctive pharmacology and kinetics of cloned neuronal Ca2+ channels and their possible counterparts in mammalian CNS neurons. Neuropharmacology. 1993;32:1075–1088. doi: 10.1016/0028-3908(93)90003-l. [DOI] [PubMed] [Google Scholar]